
Abstract
p53 is a tumor suppressor protein that prevents oncogenic transformation and maintains genomic stability by blocking proliferation of cells harboring unrepaired or misrepaired DNA. A wide range of genotoxic stresses such as DNA damaging anti-cancer drugs and ionizing radiation promote nuclear accumulation of p53 and trigger its ability to activate or repress a number of downstream target genes involved in various signaling pathways. This cascade leads to the activation of multiple cell cycle checkpoints and subsequent cell cycle arrest, allowing the cells to either repair the DNA or undergo apoptosis, depending on the intensity of DNA damage. In addition, p53 has many transcription-independent functions, including modulatory roles in DNA repair and recombination. This chapter will focus on the role of p53 in regulating or influencing the repair of DNA double-strand breaks that mainly includes homologous recombination repair (HRR) and non-homologous end joining (NHEJ). Through this discussion, we will try to establish that p53 acts as an important linchpin between upstream DNA damage signaling cues and downstream cellular events that include repair, recombination, and apoptosis.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- DNA damage response
- ATM kinase
- ATR kinase
- Nonhomologous end joining
- Homologous recombination repair
- Rad51
Introduction
A number of mutagenic and cytotoxic agents pose a major threat to genomic integrity and cellular homeostasis. Different cellular events that occur endogenously such as the reaction of DNA with oxygen and water lead to the formation of a myriad of DNA lesions, primarily involving chemical modifications of bases including oxidation (e.g. 8-oxoguanine) and hydrolysis (e.g. uracil). In addition, replication errors lead to base mismatches, while exogenous agents like ultraviolet light, industrial chemicals, or ionizing radiation produce diverse bulky adducts, alkylated bases and oxidized bases, all of which are potentially cytotoxic and mutagenic.
The tumor suppressor, p53, is a sequence-specific transcription factor, involved in the activation of many signaling molecules. Mutations in the p53 gene are found in ~50 % of tumors, providing a survival advantage to those cells. During normal conditions, the level of wild-type p53 in cells is kept under check by the E3 ubiquitin ligase, HDM2 (human homolog of the mouse double-minute 2 protein), which blocks p53’s interaction with other co-activators. Eventually, it ubiquitinates p53 and targets it for proteasomal degradation [21]. As an alternative mechanism, loss of wild-type p53 function has been shown to be due to the dominant-negative effect of mutant p53 [9]. The role of p53 has been viewed as a double-edged sword depending on the severity of damage. Early in the DNA damage response, p53 relays a wide range of pro-survival signals like cell cycle arrest allowing the cells to repair the damage. But if damage continues to accumulate, p53 is seen to shift gears and promote apoptosis or senescence. Over the past decade, p53 has also been shown to positively or negatively regulate autophagy, which is deemed to be another mode of programmed cell death.
The stabilization of p53 is caused by various cellular stresses such as irradiation, exposure to genotoxic chemicals, oncogenic activation, hypoxia, nutrient deprivation, etc. Since most of these processes damage DNA, there are various DNA repair mechanisms to correct the damage incurred and p53 has been shown to play an important role in several of these repair mechanisms including nucleotide excision repair (bulky DNA adducts) [3, 49], base excision repair (base modifications) [56, 100], mismatch repair (base mismatch due to replication errors) [15, 41], homologous recombination repair and non-homologous end joining (DNA double strand breaks). However, this chapter will focus on the role of p53 in the response to DSBs, and its involvement in the repair of DSBs. In this context, p53 primarily plays an “integrator-relayer” function, integrating upstream signaling events and relaying them downstream to activate various cellular events like apoptosis, senescence, or differentiation. Although this role is accomplished primarily by p53-mediated transcriptional activation/repression, p53 has transcription-independent functions in many pathways, including HRR.
DSBs and interstrand crosslinks (ICLs) constitute the most toxic DNA lesions because they involve both the DNA strands. DSBs are mainly induced by ionizing radiation (X-rays and gamma rays) [12, 34, 63] and radiomimetic drugs (bleomycin and neocarzinostatin) [58]. The defective processing of DNA DSBs result in chromosomal translocations, deletions, insertions etc., [18, 74] leading to genomic instability and subsequently malignancy.
In order to circumvent these effects, cells have evolved two major pathways for repairing DNA DSBs; homologous recombination repair (HRR) and non-homologous end joining (NHEJ). In addition to these, a third mechanism, single-strand annealing (SSA), utilizes components from both HRR and NHEJ [8]. The major difference between HRR and NHEJ lies in the fact that HRR is an error-free pathway that plays a pivotal role during meiosis and during S and G2 phases of the cell cycle when sister chromatids are available. On the other hand, NHEJ is an error-prone pathway that occurs throughout the cell cycle and is shown to be important in mitotic cells. Also, it has been shown that in spite of HRR being an error-free pathway of DNA DSB repair, NHEJ competes effectively for the DSBs even when HRR is available. Moreover, it appears that the initial recruitment of repair factors is crucial in selecting one pathway over the other.
Structure of p53
Structural studies of p53 show that it is a tetramer in its active form, containing four identical chains. The N-terminal region of p53 is a disordered, natively unfolded region [7] containing the acidic TAD (transactivation domain), which is a binding site for some of the p53 interacting proteins such as the transcriptional machinery proteins [44], and MDM2 [35]. This is followed by the proline-rich region (PRR) that links the TAD to the DNA binding domain (DBD) in p53 [89]. The central DNA binding domain of p53 is one of the crucial components of p53 wherein most of the cancer-related mutations occur (shown in Fig. 17.1). Most of these mutations are missense mutations [31] wherein not only the wild-type function is lost, but novel oncogenic functions are acquired along with a dominant-negative phenotype that can inactivate the normal protein functions through heterotetramerization [51], which is elicited via the tetramerization domain that follows the DNA binding domain. The C-terminal domain is also a disordered domain, but undergoes structured transitions on interaction with other proteins [66, 92]. This domain is where most of the post-translational modifications of p53 occur, such as phosphorylation, acetylation, sumoylation, neddylation, etc., which eventually regulate p53 levels and function. In addition to these domains, there is a bipartite nuclear localization signal located between the DBD and TET domain that is required for the nuclear import of p53 [39]. There is also a NES (nuclear export sequence) in the C-terminal region that allows the nuclear export of p53 into the cytoplasm [57].

Domain structure of p53. The N-terminal region consists of a transactivation domain followed by a proline-rich region, the central DNA binding domain, the tetramerization domain, and the C-terminal region. The hotspot mutations in the DNA binding domains are indicated. PTMs Post-translational modifications (phosphorylation, neddylation, sumoylation, acetylation, methylation etc.)
Activation of p53
In response to various cellular stresses that involve cellular DNA damage, p53 is activated and stabilized. It has been shown to be highly sensitive to small gaps and breaks in DNA that could ultimately lead to an early DNA damage response. However, the primary event in the p53 activation cascade is the disruption of its interaction with its negative regulators, MDM2 or MDM4. One of the mechanisms involved in disrupting this interaction is the activation of p53 by the ataxia-telengiectasia mutated (ATM) and ataxia-telengiectasia and Rad3-related (ATR) protein kinases, which are the major DNA damage sensors mediating the rapid degradation of MDM2 and MDM4 [81]. Both these kinases belong to the phosphatidylinositol-3-kinase-like kinase family and are involved in initiating a myriad of cellular signaling events following different forms of DNA damage.
The activation of p53 is also induced by the tumor suppressor, ARF. The INK/ARF locus produces two proteins, p16Ink4A and p19ARF [73], that are mainly involved in regulating the tumor suppressor functions of the retinoblastoma protein and p53. It has been shown that during oncogenic stimulation or replicative senescence, ARF binds to MDM2 and sequesters it in the nucleolus, thus stabilizing p53 levels [91].
A third mode of p53 activation is via its interaction with CBP (CREB binding protein) and p300, which are important regulators of eukaryotic transcription [27]. They act as co-activators wherein they bind to sequence-specific transcription factors and assist in the initiation of transcription from the target genes. They promote the interaction of the transcription factors with pol II holoenzyme [27] and acetylate the neighboring histones to allow an open configuration of the chromatin [75]. It was shown that there are two docking sites for p53 on p300/CBP; one in the C/H3 and one in the C-terminal domain [4, 29]. Following γ-irradiation, the interaction of p300/CBP with p53 increases due to the phosphorylation of p53 on S15 by the ATM kinase [37]. Furthermore, acetylation of p53 on the C-terminus by p300/CBP and P/CAF histone acetyltransferases (HAT) results in DNA binding activity and transactivation functions of p53 [33].
It is also important to mention that the other members of the p53 family which include p63 and p73 also play an important role in activating p53. Earlier reports using p63- and p73-null MEFs showed that both p63 and p73 are essential for p53-induced apoptosis following DNA damage [19].
DNA Damage Response and p53
ATM is primarily involved in detecting and perhaps binding to DSBs, leading ultimately to the activation of cell cycle checkpoints and modulation of DNA repair pathways. On the other hand, ATR mainly recognizes replication defects or disruption of replication by DNA lesions such as DNA-DNA/DNA-protein crosslinks arising either endogenously (e.g., malondialdehyde from lipid peroxidation) or from exposure to bifunctional DNA-damaging agents such as cisplatin and mitomycin C. During the DNA damage response, ATM and ATR induce a wide variety of post-translational modifications in p53 that promote its activation and stabilization. For instance, it is seen that during DNA damage, ATM phosphorylates the checkpoint kinase Chk2 [46], which in turn phosphorylates serine 20 of p53 [13, 30]. This residue is within the major site for MDM2 attachment [55] and as a result of its phosphorylation, p53-MDM2 interaction is disrupted leading to p53 stabilization. This cascade culminates in the G1 phase checkpoint wherein activated p53 induces the expression of its direct transcriptional target, p21, causing cell cycle arrest. A p53-independent activity of p21 in inducing both G1 and G2 phase cell cycle arrest has also been demonstrated in earlier work. MDM2, like p53, is also subjected to post-translational modifications following DNA damage. The p53 binding domain and the RING finger domains of MDM2 are the hotspots for these modifications. It was shown that following DNA damage, the serine 395 (S395) residue on MDM2 is phosphorylated by ATM both in vivo and in vitro [47]. This was corroborated recently by the demonstration that ATM-induced MDM2 phosphorylation at S395 increased the interaction of MDM2 with p53 mRNA, leading to increased p53 translation [23]. Furthermore, Gannon et al. [24] showed that ATM-mediated phosphorylation of S394 on MDM2 is important for the increase in p53 activity and subsequent activation of downstream p53 targets. These findings show that ATM plays a key role in regulating the DNA damage response by modifying both p53 and MDM2 in a way that allows activation and stabilization of p53.
In addition to the above regulatory role of ATM via Chk2 and MDM2, ATM directly phosphorylates serine 15 on p53 following exposure to ionizing radiation [36]. This effect was only partially suppressed in AT cells, suggesting that other kinases can also phosphorylate S15 on p53 [5, 77]. The role of ATR in phosphorylating S15 on p53 was shown in γ-irradiated fibroblasts that were transfected with a vector expressing a catalytically inactive mutant of ATR, designated ATRki. Overexpression of ATRki abrogated UV-induced p53 S15 phosphorylation [86]. Herein, it was shown that ATR phosphorylates p53 on S37 (also a phosphorylation site for DNA-PK) in vitro. Thus, although ATM and ATR are structurally similar, they have both common and unique substrates.
Mutant p53: Gain-of-Function and the DNA Damage Response
The wild-type activity of p53 plays a crucial role in the induction of multiple signaling processes such as cell cycle arrest, apoptosis, senescence, etc. However, in ~50 % of tumors, p53 is mutated [32, 88]. Numerous reports have shown that in addition to losing the tumor suppressive activity, certain p53 mutants acquire a “gain-of-function” (GOF) phenotype leading to oncogenesis and drug resistance. For example, mutant p53 (D281G) activates the expression of MDR1 gene (normally suppressed by wild-type p53) which encodes P-glycoprotein, an energy-dependent drug efflux pump [69, 85].
One of the key mechanisms of mutant p53 GOF is the ability of certain mutant forms of p53 to induce expression of genes that are not induced by wild-type p53. In addition to the MDR1 gene, other genes activated by mutant p53 include VEGFR and EGFR, whose products induce angiogenesis and cellular proliferation, respectively [60]. Mutant p53 also interacts with other proteins, modifying their functions so as to provide a survival advantage to the cells. For example, mutant p53 disrupts the tumor suppressive functions of both p63 and p73 [16, 22, 38] and increases DNA non-homologous recombination by increasing topoisomerase-I activity in cells [2].
However, a more dramatic effect of mutant p53 is seen in the DNA damage response. Earlier work showed an interaction of the R248W and R273H mutant forms of p53 with Mre11, which is a part of the MRN complex (Mre11/Rad50/NBS1) that is involved in the initial sensing of DNA DSBs and subsequent recruitment of ATM [94]. This interaction disrupts the ability of Mre11 to recruit ATM to DSBs, ultimately leading to inter-chromosomal translocations as a result of defective ATM-dependent cell cycle checkpoints [80]. While it is known that ATM-deficient cells are radiosensitive [76, 95], cells harboring mutant p53 usually do not exhibit radiosensitivity. This could be due to the presence of a partially functional ATM in the p53 mutant cells that could still confer some radioresistance despite the lack of interaction with Mre11. Alternatively, mutant p53 could inhibit the function of p73 which is a mediator of apoptosis in the absence of wild-type p53. Also, since mutant p53 has the ability of deregulating the expression of genes involved in cell survival, it could prevent radiosensitivity in ATM null cells [6].
As mentioned, p53 has been shown to play a role in myriad of cellular activities from DNA damage response to gene transcription to differentiation. The following sections will focus on its role specifically in the DSB repair pathways.
Homologous Recombination Repair and p53
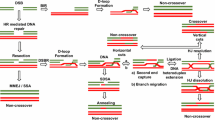
Homologous recombination repair (HRR) is an error-free DSB repair pathway, most active during late S and G2 phases of the cell cycle [83], and is primarily governed by the presence of homologous sister chromatids, homologous chromosomes, or DNA repeats. It is evolutionarily conserved and plays an important role in maintaining genomic integrity. HRR is believed to initiate with ATM sensing and localizing to DSBs, where it phosphorylates H2AX in the surrounding chromatin, which in turn recruits BRCA1 and NBS1, repair proteins that are also phosphorylated by ATM. The DNA is resected in a process that requires the MRN complex [87] and gives rise to single-strand DNA overhangs bound by RPA, which is subsequently replaced by RAD51. Since the MRN complex possesses only 3′ → 5′ but not 5′ → 3′ exonuclease activity, other nucleases such as CtIP [70] and Exo1 [11] may also be involved in the resection step. The polymerization of RAD51 on the overhangs takes place with the agency of RAD52. RAD51 then searches for DNA homology with the help of RAD54 which binds to RAD51. The ATPase activity of RAD54 helps unwind DNA and facilitates strand invasion [48]. RAD51 then forms a heteroduplex following the acquisition of a homologous duplex. This step is then followed by heteroduplex extension and branch migration. After this, either a non-crossing over event takes place wherein Holliday junctions are disengaged and DNA strands pair followed by gap filling or a crossing-over event takes place resulting from the resolution of Holliday junctions followed by gap filling.
Cells subjected to oxidative stress or to anti-cancer agents undergo a DNA damage response which is characterized by the activation and stabilization of p53, leading to cell cycle arrest in G1 phase, which requires the transactivation ability of p53. However, a plethora of studies have shown that some of the genotoxic agents including the replication elongation inhibitors hydroxyurea and aphidicolin, cause p53 accumulation independent of its role in G1 checkpoint [28, 67]. Likewise, some mutations in p53 (143, 175, 248, 273, and 281) do not affect the G1 phase cell cycle arrest but stimulate both spontaneous and radiation-induced recombination [1, 10, 50, 68].
Although p53 can by itself check the fidelity of homologous recombination by mismatch recognition of heteroduplex intermediates [17, 43], the inhibition of recombination by p53 is primarily mediated by its interaction with RAD51 (Fig. 17.2) and also RAD54. Inhibition of p53 activity promotes spontaneous and radiation-induced homologous recombination between both direct and inverted repeats [68] wherein the latter mainly involves a RAD51-dependent gene conversion process. This interaction of p53 with RAD51 and subsequent inhibition of recombination was further confirmed by overexpressing mutant L186PRAD51 that prevented p53 binding to RAD51 [43]. A 2–3 fold increase in homologous recombination was seen following the overexpression of this p53 non-binding mutant of RAD51. The interaction of p53 with RAD54 mainly occurs via the extreme C-terminal domain of p53 [43] which is involved in sensing mispaired homologous recombination intermediates. In vitro experiments have shown that RAD51 stimulates the 3′ → 5′ exonuclease activity of p53 that targets heteroduplexes containing base mismatches [82]. Also, the p53-RAD51 complex inhibits branch migration after the crossing-over or postsynaptic phase of recombination [96]. Further, the regulation of homologous recombination by p53 was found to be biased, with p53 depletion promoting both intra- and extrachromosomal recombination but not homologous DNA integration or gene targeting [97]. This is interesting because gene targeting or gene disruption could be considered a genome-destabilizing process. Although most of these mechanistic studies involved measurements of spontaneous recombination between ostensibly undamaged loci, other work [1, 10] demonstrates that recombination between a locus containing a site-specific I-SceI-induced DSB and an undamaged homologous sequence, presumably reflecting HRR, is likewise suppressed by both wild-type and transactivation-defective p53.

Inhibitory role of p53 in homologous recombination repair
While all the above studies are in general agreement that HRR suppression by p53 is largely independent of its transcriptional activation ability, this view has recently been challenged [59]. In this study, a wide variety of p53 manipulations were carried out, and in all cases (even those involving transactivation-deficient p53 mutants) the extent of HRR for DSBs induced by the meganuclease I-SceI was found to closely correlate with the fraction of cells in S/G2. Thus, these authors argue that most of the reported effects of p53 on HRR can be explained by transactivation-dependent cell cycle perturbations.
During DNA replication, p53 is found at replication sites [93] and also is transported into the nucleus during S-phase [45]. During replicative stress (resulting from treatment with replication inhibitors or DNA crosslinking agents), p53 inhibits homologous recombination and this effect is dependent on the S15 phosphorylation of p53 and the interaction of p53 with the ssDNA binding protein RPA [61]. As discussed earlier, the phosphorylation of p53 on S15 is mediated by both the ATM and ATR kinases. Inhibition of ATM with caffeine or the ATM specific inhibitor KU-55933 still allowed the formation of RAD51 foci, indicative of an active recombination in the absence of ATM. Subsequently, following ATR inhibition using RNA interference, the fraction of RAD51 foci-positive cells was reduced in the presence of either wild-type p53 or the transactivation mutant, p53QS [79]. A recent study [72] has shown that the trio: DNA-PK, ATM, and ATR together downregulate p53-RPA binding. DNA-PK phosphorylates RPA at S46, and ATM/ATR phosphorylates p53 at S37 (preceded by S15 phosphorylation as discussed above) causing the release of p53 and RPA from the p53-RPA complex and thereby allowing RPA to fulfill its normal role in facilitating HRR. DNA-PK is also involved in the other major DNA DSB repair pathway, the non-homologous end joining (NHEJ, discussed in the next section).
Also, an important potential role of p53 in HRR that is not discussed here in detail is its interaction with BLM (Bloom syndrome) and WS (Werner syndrome) proteins that belong to the class of RecQ helicases involved in homologous recombination [78]. It was shown that BLM and p53 show co-localization at sites of stalled replication forks [71]. Although BLM localizes to these sites in a p53-independent manner, it eventually enhances the p53 accumulation at these sites. Sengupta et al. [71] showed that the interaction of BLM with p53 is enhanced by the localization of 53BP1 at these sites, independent of γ-H2AX. This event was dependent on active Chk1 kinase (possibly phosphorylated by ATR), leading to BLM stabilization and ultimately p53 accumulation at stalled replication sites.
To summarize, p53 plays multiple roles in the regulation of the HRR pathway and the mechanism of some of these regulatory roles is still being elucidated.
Non-homologous End Joining and p53
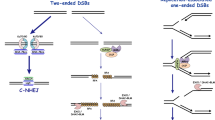
The NHEJ pathway is the predominant pathway for the repair of DNA DSBs, occurring throughout the cell cycle [65]. It is a dynamic process that does not require sequence homology as in HR [64] and has been shown to utilize a wide variety of DNA substrates converting them into joined products. Since this process can lead to the joining of incorrect ends, NHEJ is an error-prone pathway. Radiation-induced DNA damage via the production of oxygen radicals leads to deoxyribose fragmentation, as well as producing modified DNA bases such as 8-oxoguanine and thymine glycols. DNA DSBs induced by deoxyribose oxidation are characterized by both 5′- and 3′-staggered termini with chemically modified ends. Moreover, clusters of localized radicals can produce complex DNA lesions consisting of terminally blocked DSBs flanked by nearby damaged bases. DSBs of widely diverse terminal structure can be repaired via NHEJ irrespective of the sequence or DNA homology.
The first step in NHEJ is the binding of the Ku heterodimers to DSBs [52]. This is followed by DNA-PK binding to Ku and this complex serves as a beacon for nucleases, polymerases, and ligases to bind. However, the formation of a stable complex between Ku and DNA-PK requires conformational changes in Ku which occur only in the presence of DNA ends [40], and promote the interaction of Ku with DNA polymerases μ and λ, and with the XRCC4-DNA ligase IV complex [14]. Interaction between two such complexes on two DNA ends tethers the ends and triggers the kinase activity of DNA-PK which then phosphorylates various repair proteins. Artemis, along with DNA-PK, gains a 3′-endonucleolytic activity that has been shown earlier to act specifically near 3′ DNA termini and resolve noncanonical DNA DSB ends such as 3′-phosphoglycolate moieties. This endonuclease activity is essential for Artemis’ role in promoting radioresistance and repair proficiency in mammalian cells [53]. In addition to Artemis and DNA-PK, other proteins such as TDP1 and Metnase have been shown to function in a similar fashion to resolve damaged DNA overhangs. TDP1 removes glycolate residues from 3′ ends followed by additional processing by PNKP to remove the resulting 3′-phosphates [99]. Recently, Metnase was shown to endonucleolytically trim 3′-overhangs greater than 3 bp on DNA duplexes, although its lack of activity when added to NHEJ-competent extracts casts some doubt on such a role in vivo [54]. The various enzymes involved in the NHEJ pathway have a high flexibility in binding to DNA lesions, allowing them to interact or bind with a plethora of DNA end structures.
The role of p53 in NHEJ is not clearly understood although p53 has been shown to regulate NHEJ by itself or in association with other NHEJ proteins. Early studies with mice harboring knockouts of the NHEJ factors XRCC4 and Ligase IV have shown that in the absence of NHEJ, DNA DSBs remain unrepaired, and that these eventually trigger apoptosis in a p53-dependent manner [20, 26, 25]. Thus, p53 deficiency can rescue the otherwise embryonic lethal phenotype of Xrcc4−/− or Lig4−/− mice.
In an episomal reactivation assay, p53 was shown to enhance DSB rejoining of transfected linearized plasmids in γ-irradiated cells [84]. This enhancement was found to be dependent on the carboxy terminal domain of p53 (which harbors nonspecific DNA-binding activity), but independent of transcriptional activation ability. Interestingly, only the repair of DNA DSBs with short cohesive ends but not blunt ends was enhanced by p53. In cells harboring an integrated DSB substrate containing tandem sites for meganuclease I-SceI, the p53 inhibitor pifithrin-α reduced precise end-joining while having little or no effect on end joining overall, suggesting a role for p53 in enforcing end-joining fidelity [42]. In a different I-SceI-based NHEJ assay, expression of wild-type p53 inhibited NHEJ events that required trimming of noncomplementary overhangs from the DSB ends. In this case, it was speculated that p53 plays a role in NHEJ either by inhibiting exonucleolytic proofreading or by recognizing heterologies and inhibiting NHEJ [1].
The Artermis endonuclease involved trimming of damaged or noncomplementary ends for NHEJ has been shown to interact with p53 in the suppression of oncogenic N-Myc in progenitor B-cells [62]. Other reports have also shown Artemis to be a negative regulator of p53 activity in response to oxidative stress. Artemis knockdown in U2-OS cells induced p53 accumulation, cell cycle arrest, and apoptosis [98]. This reflects on a DNA repair-independent role of Artemis and its subsequent effect on p53 activity.
To summarize, the exact regulatory role of p53 in NHEJ is still poorly understood. Certainly, p53 does have genetic interactions with the players of the NHEJ pathway, eliciting DNA repair-dependent and -independent downstream effects on cell cycle progression and cell survival. However, early suggestions of more direct biochemical effects of p53 on NHEJ itself have been neither refuted nor further elucidated.
Conclusions
Overall, p53 acts as an important link between upstream signaling and activation of downstream signaling cascades depending on the extent of DNA damage. It can activate cell cycle arrest and allow the damage to be repaired or it could transactivate genes involved in the apoptotic machinery.
The role of p53 in regulating cell cycle checkpoints following DNA damage is a pivotal event toward maintaining genomic stability. DNA damage leads to the activation of cell cycle checkpoints in different phases of the cell cycle. The G1/S phase checkpoint that is mainly triggered by DNA DSBs (detected by the presence of γ-H2AX or 53BP1, [90]) involves the ATM kinase that detects DSBs and phosphorylates p53 either directly or indirectly leading to its stabilization. As the repair of some DSBs by NHEJ in G1 requires several hours, this arrest provides a crucial opportunity for the cell to restore the integrity of the genome before it can be replicated. The intra-S-phase checkpoint is mainly activated by stalled replication forks arising as a result of replication defects or DNA damaging agents. It likewise allows for repair of replication-associated DSBs by HRR before new replication forks are initiated. Thus, in either case, cell cycle arrest will serve to enhance genomic stability, and is the primary function of p53 in DSB repair.
More direct regulatory effects of p53 on DSB repair are more difficult to rationalize in terms of genomic stability. It might be expected that the role of p53 in regulating these pathways could be bimodal, either inhibiting these pathways to maintain genomic stability or activating them in response to genotoxic stress or DNA damage cues. While DSBs must be rejoined if genomic integrity is to be preserved, inaccurate repair by the same or very similar mechanisms will lead to rearrangements and instability. It is ostensibly surprising that the primary direct effect of p53 on DSB repair appears to be suppression of HRR, which is usually accurate and certainly more accurate than NHEJ or other alternative DSB repair pathways. Ideally, a genomic surveillance system would evolve so as to specifically detect and suppress events that are likely to be associated with inaccurate repair. There is some suggestion of such a bias in the finding that p53 most strongly inhibits HRR between substrates with a limited extent of homology (<200 bp) – events that could reflect illegitimate recombination between repetitive sequences that could lead to rearrangements [1]. Similarly, p53 reportedly suppresses NHEJ that requires resection of mismatched overhangs (such as might occur in the joining of exchanged ends of two DSBs), while promoting cohesive-end joins [1, 84]. Finally, in cases of extensive damage, the proliferation of cells with unstable genomes might be most efficiently prevented by blocking repair entirely and thereby driving those damaged cells toward apoptosis or senescence.
References
Akyuz N, Boehden GS, Susse S, Rimek A, Preuss U, Scheidtmann KH, Wiesmuller L (2002) DNA substrate dependence of p53-mediated regulation of double-strand break repair. Mol Cell Biol 22(17):6306–6317
Albor A, Kaku S, Kulesz-Martin M (1998) Wild-type and mutant forms of p53 activate human topoisomerase I: a possible mechanism for gain of function in mutants. Cancer Res 58(10):2091–2094
Amundson SA, Patterson A, Do KT, Fornace AJ Jr (2002) A nucleotide excision repair master-switch: p53 regulated coordinate induction of global genomic repair genes. Cancer Biol Ther 1(2):145–149
Avantaggiati ML, Ogryzko V, Gardner K, Giordano A, Levine AS, Kelly K (1997) Recruitment of p300/CBP in p53-dependent signal pathways. Cell 89(7):1175–1184
Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, Ziv Y (1998) Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 281(5383):1674–1677
Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, Wynshaw-Boris A (1996) Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell 86(1):159–171
Bell S, Klein C, Muller L, Hansen S, Buchner J (2002) P53 contains large unstructured regions in its native state. J Mol Biol 322(5):917–927
Bennardo N, Cheng A, Huang N, Stark JM (2008) Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet 4(6):e1000110
Blagosklonny MV (2000) P53 from complexity to simplicity: mutant P53 stabilization, gain-of-function, and dominant-negative effect. FASEB J 14(13):1901–1907
Boehden GS, Akyuz N, Roemer K, Wiesmuller L (2003) P53 Mutated in the transactivation domain retains regulatory functions in homology-directed double-strand break repair. Oncogene 22(26):4111–4117
Bolderson E, Tomimatsu N, Richard DJ, Boucher D, Kumar R, Pandita TK, Burma S, Khanna KK (2010) Phosphorylation of Exo1 modulates homologous recombination repair of DNA double-strand breaks. Nucleic Acids Res 38(6):1821–1831
Bradley MO, Kohn KW (1979) X-ray induced DNA double strand break production and repair in mammalian cells as measured by neutral filter elution. Nucleic Acids Res 7(3):793–804
Chehab NH, Malikzay A, Stavridi ES, Halazonetis TD (1999) Phosphorylation of Ser-20 mediates stabilization of human p53 in response to DNA damage. Proc Natl Acad Sci U S A 96(24):13777–13782
Chen L, Trujillo K, Sung P, Tomkinson AE (2000) Interactions of the DNA ligase IV-XRCC4 complex with DNA ends and the DNA-dependent protein kinase. J Biol Chem 275(34):26196–26205
Degtyareva N, Subramanian D, Griffith JD (2001) Analysis of the binding of p53 to DNAs containing mismatched and bulged bases. J Biol Chem 276(12):8778–8784
Di Como CJ, Gaiddon C, Prives C (1999) P73 function is inhibited by tumor-derived P53 mutants in Mammalian cells. Mol Cell Biol 19(2):1438–1449
Dudenhoffer C, Rohaly G, Will K, Deppert W, Wiesmuller L (1998) Specific mismatch recognition in heteroduplex intermediates by p53 suggests a role in fidelity control of homologous recombination. Mol Cell Biol 18(9):5332–5342
Ferguson DO, Alt FW (2001) DNA double strand break repair and chromosomal translocation: lessons from animal models. Oncogene 20(40):5572–5579
Flores ER, Tsai KY, Crowley D, Sengupta S, Yang A, McKeon F, Jacks T (2002) p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature 416(6880):560–564
Frank KM, Sharpless NE, Gao Y, Sekiguchi JM, Ferguson DO, Zhu C, Manis JP, Horner J, DePinho RA, Alt FW (2000) DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol Cell 5(6):993–1002
Freedman DA, Wu L, Levine AJ (1999) Functions of the MDM2 oncoprotein. Cell Mol Life Sci 55(1):96–107
Gaiddon C, Lokshin M, Ahn J, Zhang T, Prives C (2001) A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol Cell Biol 21(5):1874–1887
Gajjar M, Candeias MM, Malbert-Colas L, Mazars A, Fujita J, Olivares-Illana V, Fahraeus R (2012) The p53 mRNA-Mdm2 interaction controls Mdm2 nuclear trafficking and is required for p53 activation following DNA damage. Cancer Cell 21(1):25–35
Gannon HS, Woda BA, Jones SN (2012) ATM phosphorylation of Mdm2 Ser394 regulates the amplitude and duration of the DNA damage response in mice. Cancer Cell 21(5):668–679
Gao Y, Ferguson DO, Xie W, Manis JP, Sekiguchi J, Frank KM, Chaudhuri J, Horner J, DePinho RA, Alt FW (2000) Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature 404(6780):897–900
Gao Y, Sun Y, Frank KM, Dikkes P, Fujiwara Y, Seidl KJ, Sekiguchi JM, Rathbun GA, Swat W, Wang J, Bronson RT, Malynn BA, Bryans M, Zhu C, Chaudhuri J, Davidson L, Ferrini R, Stamato T, Orkin SH, Greenberg ME, Alt FW (1998) A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell 95(7):891–902
Goodman RH, Smolik S (2000) CBP/p300 in cell growth, transformation, and development. Genes Dev 14(13):1553–1577
Gottifredi V, Shieh S, Taya Y, Prives C (2001) p53 accumulates but is functionally impaired when DNA synthesis is blocked. Proc Natl Acad Sci U S A 98(3):1036–1041
Gu W, Shi XL, Roeder RG (1997) Synergistic activation of transcription by CBP and p53. Nature 387(6635):819–823
Hirao A, Kong YY, Matsuoka S, Wakeham A, Ruland J, Yoshida H, Liu D, Elledge SJ, Mak TW (2000) DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science 287(5459):1824–1827
Hollstein M, Rice K, Greenblatt MS, Soussi T, Fuchs R, Sorlie T, Hovig E, Smith-Sorensen B, Montesano R, Harris CC (1994) Database of p53 gene somatic mutations in human tumors and cell lines. Nucleic Acids Res 22(17):3551–3555
Hollstein M, Sidransky D, Vogelstein B, Harris CC (1991) P53 mutations in human cancers. Science 253(5015):49–53
Hupp TR, Meek DW, Midgley CA, Lane DP (1992) Regulation of the specific DNA binding function of p53. Cell 71(5):875–886
Kraxenberger F, Weber KJ, Friedl AA, Eckardt-Schupp F, Flentje M, Quicken P, Kellerer AM (1998) DNA double-strand breaks in mammalian cells exposed to gamma-rays and very heavy ions. Fragment-size distributions determined by pulsed-field gel electrophoresis. Radiat Environ Biophys 37(2):107–115
Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP (1996) Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 274(5289):948–953
Lakin ND, Jackson SP (1999) Regulation of p53 in response to DNA damage. Oncogene 18(53):7644–7655
Lambert PF, Kashanchi F, Radonovich MF, Shiekhattar R, Brady JN (1998) Phosphorylation of p53 serine 15 increases interaction with CBP. J Biol Chem 273(49):33048–33053
Li Y, Prives C (2007) Are interactions with p63 and p73 involved in mutant p53 gain of oncogenic function? Oncogene 26(15):2220–2225
Liang SH, Clarke MF (1999) A bipartite nuclear localization signal is required for p53 nuclear import regulated by a carboxyl-terminal domain. J Biol Chem 274(46):32699–32703
Lieber MR (2008) The mechanism of human nonhomologous DNA end joining. J Biol Chem 283(1):1–5
Lin X, Ramamurthi K, Mishima M, Kondo A, Howell SB (2000) p53 interacts with the DNA mismatch repair system to modulate the cytotoxicity and mutagenicity of hydrogen peroxide. Mol Pharmacol 58(6):1222–1229
Lin Y, Waldman BC, Waldman AS (2003) Suppression of high-fidelity double-strand break repair in mammalian chromosomes by pifithrin-alpha, a chemical inhibitor of p53. DNA Repair 2(1):1–11
Linke SP, Sengupta S, Khabie N, Jeffries BA, Buchhop S, Miska S, Henning W, Pedeux R, Wang XW, Hofseth LJ, Yang Q, Garfield SH, Sturzbecher HW, Harris CC (2003) p53 interacts with hRAD51 and hRAD54, and directly modulates homologous recombination. Cancer Res 63(10):2596–2605
Lu H, Levine AJ (1995) Human TAFII31 protein is a transcriptional coactivator of the p53 protein. Proc Natl Acad Sci U S A 92(11):5154–5158
Martinez J, Georgoff I, Martinez J, Levine AJ (1991) Cellular localization and cell cycle regulation by a temperature-sensitive p53 protein. Genes Dev 5(2):151–159
Matsuoka S, Rotman G, Ogawa A, Shiloh Y, Tamai K, Elledge SJ (2000) Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc Natl Acad Sci U S A 97(19):10389–10394
Maya R, Balass M, Kim ST, Shkedy D, Leal JF, Shifman O, Moas M, Buschmann T, Ronai Z, Shiloh Y, Kastan MB, Katzir E, Oren M (2001) ATM-dependent phosphorylation of Mdm2 on serine 395: role in p53 activation by DNA damage. Genes Dev 15(9):1067–1077
Mazin AV, Alexeev AA, Kowalczykowski SC (2003) A novel function of Rad54 protein. Stabilization of the Rad51 nucleoprotein filament. J Biol Chem 278(16):14029–14036
McKay BC, Ljungman M, Rainbow AJ (1999) Potential roles for p53 in nucleotide excision repair. Carcinogenesis 20(8):1389–1396
Mekeel KL, Tang W, Kachnic LA, Luo CM, DeFrank JS, Powell SN (1997) Inactivation of p53 results in high rates of homologous recombination. Oncogene 14(15):1847–1857
Milner J, Medcalf EA (1991) Cotranslation of activated mutant p53 with wild type drives the wild-type p53 protein into the mutant conformation. Cell 65(5):765–774
Mimori T, Hardin JA (1986) Mechanism of interaction between Ku protein and DNA. J Biol Chem 261(22):10375–10379
Mohapatra S, Kawahara M, Khan IS, Yannone SM, Povirk LF (2011) Restoration of G1 chemo/radioresistance and double-strand-break repair proficiency by wild-type but not endonuclease-deficient Artemis. Nucleic Acids Res 39(15):6500–6510
Mohapatra S, Yannone SM, Lee SH, Hromas RA, Akopiants K, Menon V, Ramsden DA, Povirk LF (2013) Trimming of damaged 3′ overhangs of DNA double-strand breaks by the Metnase and Artemis endonucleases. DNA Repair 12(6):422–432
Moll UM, Petrenko O (2003) The MDM2-p53 interaction. Mol Cancer Res 1(14):1001–1008
Offer H, Wolkowicz R, Matas D, Blumenstein S, Livneh Z, Rotter V (1999) Direct involvement of p53 in the base excision repair pathway of the DNA repair machinery. FEBS Lett 450(3):197–204
O’Keefe K, Li H, Zhang Y (2003) Nucleocytoplasmic shuttling of p53 is essential for MDM2-mediated cytoplasmic degradation but not ubiquitination. Mol Cell Biol 23(18):6396–6405
Povirk LF (1996) DNA damage and mutagenesis by radiomimetic DNA-cleaving agents: bleomycin, neocarzinostatin and other enediynes. Mutat Res 355(1–2):71–89
Rieckmann T, Kriegs M, Nitsch L, Hoffer K, Rohaly G, Kocher S, Petersen C, Dikomey E, Dornreiter I, Dahm-Daphi J (2013) p53 modulates homologous recombination at I-SceI-induced double-strand breaks through cell-cycle regulation. Oncogene 32(8):968–975
Roemer K (1999) Mutant p53: gain-of-function oncoproteins and wild-type p53 inactivators. Biol Chem 380(7–8):879–887
Romanova LY, Willers H, Blagosklonny MV, Powell SN (2004) The interaction of p53 with replication protein A mediates suppression of homologous recombination. Oncogene 23(56):9025–9033
Rooney S, Sekiguchi J, Whitlow S, Eckersdorff M, Manis JP, Lee C, Ferguson DO, Alt FW (2004) Artemis and p53 cooperate to suppress oncogenic N-myc amplification in progenitor B cells. Proc Natl Acad Sci U S A 101(8):2410–2415
Roots R, Kraft G, Gosschalk E (1985) The formation of radiation-induced DNA breaks: the ratio of double-strand breaks to single-strand breaks. Int J Radiat Oncol Biol Phys 11(2):259–265
Roth DB, Porter TN, Wilson JH (1985) Mechanisms of nonhomologous recombination in mammalian cells. Mol Cell Biol 5(10):2599–2607
Rothkamm K, Kruger I, Thompson LH, Lobrich M (2003) Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol 23(16):5706–5715
Rustandi RR, Baldisseri DM, Weber DJ (2000) Structure of the negative regulatory domain of p53 bound to S100B(betabeta). Nat Struct Biol 7(7):570–574
Saintigny Y, Lopez BS (2002) Homologous recombination induced by replication inhibition, is stimulated by expression of mutant p53. Oncogene 21(3):488–492
Saintigny Y, Rouillard D, Chaput B, Soussi T, Lopez BS (1999) Mutant p53 proteins stimulate spontaneous and radiation-induced intrachromosomal homologous recombination independently of the alteration of the transactivation activity and of the G1 checkpoint. Oncogene 18(24):3553–3563
Sampath J, Sun D, Kidd VJ, Grenet J, Gandhi A, Shapiro LH, Wang Q, Zambetti GP, Schuetz JD (2001) Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1. J Biol Chem 276(42):39359–39367
Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP (2007) Human CtIP promotes DNA end resection. Nature 450(7169):509–514
Sengupta S, Linke SP, Pedeux R, Yang Q, Farnsworth J, Garfield SH, Valerie K, Shay JW, Ellis NA, Wasylyk B, Harris CC (2003) BLM helicase-dependent transport of p53 to sites of stalled DNA replication forks modulates homologous recombination. EMBO J 22(5):1210–1222
Serrano MA, Li Z, Dangeti M, Musich PR, Patrick S, Roginskaya M, Cartwright B, Zou Y (2013) DNA-PK, ATM and ATR collaboratively regulate p53-RPA interaction to facilitate homologous recombination DNA repair. Oncogene 32(19):2452–2462
Sharpless NE, DePinho RA (1999) The INK4A/ARF locus and its two gene products. Curr Opin Genet Dev 9(1):22–30
Sharpless NE, Ferguson DO, O’Hagan RC, Castrillon DH, Lee C, Farazi PA, Alson S, Fleming J, Morton CC, Frank K, Chin L, Alt FW, DePinho RA (2001) Impaired nonhomologous end-joining provokes soft tissue sarcomas harboring chromosomal translocations, amplifications, and deletions. Mol Cell 8(6):1187–1196
Shiama N (1997) The p300/CBP family: integrating signals with transcription factors and chromatin. Trends Cell Biol 7(6):230–236
Shiloh Y, Kastan MB (2001) ATM: genome stability, neuronal development, and cancer cross paths. Adv Cancer Res 83:209–254
Siliciano JD, Canman CE, Taya Y, Sakaguchi K, Appella E, Kastan MB (1997) DNA damage induces phosphorylation of the amino terminus of p53. Genes Dev 11(24):3471–3481
Singh DK, Ahn B, Bohr VA (2009) Roles of RECQ helicases in recombination based DNA repair, genomic stability and aging. Biogerontology 10(3):235–252
Sirbu BM, Lachmayer SJ, Wulfing V, Marten LM, Clarkson KE, Lee LW, Gheorghiu L, Zou L, Powell SN, Dahm-Daphi J, Willers H (2011) ATR-p53 restricts homologous recombination in response to replicative stress but does not limit DNA interstrand crosslink repair in lung cancer cells. PLoS One 6(8):e23053
Song H, Hollstein M, Xu Y (2007) p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat Cell Biol 9(5):573–580
Stommel JM, Wahl GM (2004) Accelerated MDM2 auto-degradation induced by DNA-damage kinases is required for p53 activation. EMBO J 23(7):1547–1556
Susse S, Janz C, Janus F, Deppert W, Wiesmuller L (2000) Role of heteroduplex joints in the functional interactions between human Rad51 and wild-type p53. Oncogene 19(39):4500–4512
Takata M, Sasaki MS, Sonoda E, Morrison C, Hashimoto M, Utsumi H, Yamaguchi-Iwai Y, Shinohara A, Takeda S (1998) Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J 17(18):5497–5508
Tang W, Willers H, Powell SN (1999) p53 directly enhances rejoining of DNA double-strand breaks with cohesive ends in gamma-irradiated mouse fibroblasts. Cancer Res 59(11):2562–2565
Thottassery JV, Zambetti GP, Arimori K, Schuetz EG, Schuetz JD (1997) p53-dependent regulation of MDR1 gene expression causes selective resistance to chemotherapeutic agents. Proc Natl Acad Sci U S A 94(20):11037–11042
Tibbetts RS, Brumbaugh KM, Williams JM, Sarkaria JN, Cliby WA, Shieh SY, Taya Y, Prives C, Abraham RT (1999) A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev 13(2):152–157
van den Bosch M, Bree RT, Lowndes NF (2003) The MRN complex: coordinating and mediating the response to broken chromosomes. EMBO Rep 4(9):844–849
Vogelstein B, Lane D, Levine AJ (2000) Surfing the p53 network. Nature 408(6810):307–310
Walker KK, Levine AJ (1996) Identification of a novel p53 functional domain that is necessary for efficient growth suppression. Proc Natl Acad Sci U S A 93(26):15335–15340
Ward IM, Minn K, Jorda KG, Chen J (2003) Accumulation of checkpoint protein 53BP1 at DNA breaks involves its binding to phosphorylated histone H2AX. J Biol Chem 278(22):19579–19582
Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar-Sagi D (1999) Nucleolar Arf sequesters Mdm2 and activates p53. Nat Cell Biol 1(1):20–26
Weinberg RL, Freund SM, Veprintsev DB, Bycroft M, Fersht AR (2004) Regulation of DNA binding of p53 by its C-terminal domain. J Mol Biol 342(3):801–811
Wilcock D, Lane DP (1991) Localization of p53, retinoblastoma and host replication proteins at sites of viral replication in herpes-infected cells. Nature 349(6308):429–431
Xu Y (2006) DNA damage: a trigger of innate immunity but a requirement for adaptive immune homeostasis. Nat Rev Immunol 6(4):261–270
Xu Y, Baltimore D (1996) Dual roles of ATM in the cellular response to radiation and in cell growth control. Genes Dev 10(19):2401–2410
Yoon D, Wang Y, Stapleford K, Wiesmuller L, Chen J (2004) P53 inhibits strand exchange and replication fork regression promoted by human Rad51. J Mol Biol 336(3):639–654
Yun S, Lie-A-Cheong C, Porter AC (2004) Discriminatory suppression of homologous recombination by p53. Nucleic Acids Res 32(22):6479–6489
Zhang X, Zhu Y, Geng L, Wang H, Legerski RJ (2009) Artemis is a negative regulator of p53 in response to oxidative stress. Oncogene 28(22):2196–2204
Zhou T, Akopiants K, Mohapatra S, Lin PS, Valerie K, Ramsden DA, Lees-Miller SP, Povirk LF (2009) Tyrosyl-DNA phosphodiesterase and the repair of 3′-phosphoglycolate-terminated DNA double-strand breaks. DNA Repair 8(8):901–911
Zurer I, Hofseth LJ, Cohen Y, Xu-Welliver M, Hussain SP, Harris CC, Rotter V (2004) The role of p53 in base excision repair following genotoxic stress. Carcinogenesis 25(1):11–19
Acknowledgment
The preparation of this review was supported in part by grants CA40615 and CA166264 from the National Cancer Institute, US DHHS.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Menon, V., Povirk, L. (2014). Involvement of p53 in the Repair of DNA Double Strand Breaks: Multifaceted Roles of p53 in Homologous Recombination Repair (HRR) and Non-Homologous End Joining (NHEJ). In: Deb, S., Deb, S. (eds) Mutant p53 and MDM2 in Cancer. Subcellular Biochemistry, vol 85. Springer, Dordrecht. https://doi.org/10.1007/978-94-017-9211-0_17
Download citation
DOI: https://doi.org/10.1007/978-94-017-9211-0_17
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-017-9210-3
Online ISBN: 978-94-017-9211-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)