
Abstract
The maintenance of the stability of genetic material is an essential feature of every living organism. Organisms across all kingdoms have evolved diverse and highly efficient repair mechanisms to protect the genome from deleterious consequences of various genotoxic factors that might tend to destabilize the integrity of the genome in each generation. One such group of proteins that is actively involved in genome surveillance is the RecQ helicase family. These proteins are highly conserved DNA helicases, which have diverse roles in multiple DNA metabolic processes such as DNA replication, recombination and DNA repair. In humans, five RecQ helicases have been identified and three of them namely, WRN, BLM and RecQL4 have been linked to genetic diseases characterized by genome instability, premature aging and cancer predisposition. This helicase family plays important roles in various DNA repair pathways including protecting the genome from illegitimate recombination during chromosome segregation in mitosis and assuring genome stability. This review mainly focuses on various roles of human RecQ helicases in the process of recombination-based DNA repair to maintain genome stability and physiological consequences of their defects in the development of cancer and premature aging.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The stability of the genetic material from one generation to another is an important and essential feature for every living organism. The failure to do so efficiently can lead to chromosomal abnormalities, developmental abnormalities, progression of cancer and premature aging. Living organisms encounter different kinds of stresses provided by genotoxic elements from within (products of normal cellular metabolism i.e., ROS) or outside the cell (environmental factors such as radiation, chemicals, etc.) during the faithful transmission of their genetic material. To counteract these stress responses, organisms have evolved diverse and highly efficient DNA repair pathways. The repair proteins function in a highly coordinated fashion with other DNA metabolic processes during the different stages of the cell cycle to maintain genome integrity in each generation. An overview of the relationship between DNA repair, cellular stress responses, genome stability and aging has been presented in Fig. 1.

Overview of relationship between cellular stress responses, DNA repair and genome stability. During the course of their life span, organisms tolerate multiple stresses which might have deleterious effects on their genome. These deleterious effects are encountered by continuous genome surveillance by various DNA repair pathways such as BER, NER, DSB, MMR and mitochondrial DNA repair. Efficient DNA repair machinery in an organism leads to genomic stability, and protects the genome from harmful effects of stress. On the other hand, defective DNA repair machinery would ultimately lead to chromosomal abnormalities which might cause genetic and physiological defects, cellular death, cancer and/or premature aging
One major family of proteins that is actively involved in maintaining genome stability is the RecQ helicases. The RecQ protein family is a highly conserved group of DNA helicases, which have diverse roles in multiple DNA metabolic processes such as DNA replication, recombination and repair (see review by Brosh and Bohr 2007; Hanada and Hickson 2007; Bachrati and Hickson 2008; Sharma et al. 2006). The RecQ protein is evolutionarily conserved from bacteria, yeast and humans to plants. There is only one RecQ homolog in Escherichia coli (RecQ) and yeast (namely, Sgs1 and Rqh1 in Saccharomyces cerevisiae and Schizosaccharomyces pombe, respectively), but five RecQ homologs have been identified in both human and mouse. The largest number of RecQ helicases has been reported in plants, with a total of seven RecQ helicases. Therefore, the function of RecQ helicases seems to have adapted to the complexity of genomes present in higher eukaryotes by increasing their number. The domain architecture of RecQ helicase family members from different organisms is shown in Fig. 2. Five human RecQ homologs, called RECQL1, BLM, WRN, RECQL4 and RECQL5, have been identified so far, and three of them have been shown to be associated with autosomal recessive disorders characterized by premature aging, genome instability, and cancer predisposition. Werner syndrome (WS) is associated with defects in WRN protein, Bloom syndrome (BS) is associated with defects in BLM helicase, and Rothmund Thomson syndrome (RTS), RAPADILINO syndrome, Baller Gerold syndrome are all associated with defects in RECQL4.

Domain architecture of RecQ helicase family. RecQ helicases from different organisms are shown. All the members have a conserved helicase domain in the central region of the protein (yellow). The nuclear localization signal (depicted in red) is present at the C-terminus in most of the family members, except in RECQL4 where it resides at the N-terminus. The WRN protein is unique among human RecQ helicase members in having an exonuclease domain (green) at the N-terminus
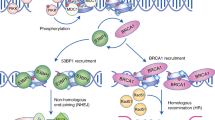
RecQ helicases play important roles in various DNA repair pathways including double-strand break (DSB) repair and protecting the genome from illegitimate recombination during chromosome segregation in mitosis (Chakraverty and Hickson 1999; Shen and Loeb 2000; van Brabant et al. 2000). In higher eukaryotes, homologous recombination (HR) is essential for segregation of homologous chromosomes during meiosis, generation of genetic diversity and maintenance of telomeres (Hoeijmakers 2001; West 2003; Krogh and Symington 2004). In addition, recombination-related processes also function in the repair of DNA double-strand breaks (DSBs), interstrand cross-links (ICLs), and recovery of stalled or broken replication forks in DNA replication through a series of interrelated pathways (Fig. 3). Two major recombination pathways have been identified in eukaryotes that are distinct with respect to mechanism and DNA homology requirements. The non-homologous DNA end-joining (NHEJ) pathway joins two ends of the DSB via a process that is largely independent of terminal DNA sequence homology. Therefore, NHEJ is error prone and can produce deletions, insertions and translocations (Thompson and Schild 2002). Homologous recombination (HR) corrects strand breaks using homologous sequences primarily from the sister chromatid and, to a lesser extent, from the homologous chromosome (Johnson and Jasin 2000; Liang et al. 1998). Therefore, it is a high fidelity repair mechanism. These repair pathways involve many proteins whose deficiency results in genome instability, cancer predisposition and premature aging (Ouyang et al. 2008; Li and Heyer 2008). The NHEJ plays a dominant role during G1 to early S-phase of the cell cycle, whereas HR is preferentially used in the late S–G2 phase (Takata et al. 1998), because of the presence of a suitable template (i.e., sister chromatid). The possible roles of different RecQ helicases at the multiple steps of major recombination pathways are represented in Fig. 4.

RecQ helicases play important roles in various DNA metabolic and repair pathways involving homologous recombination. The RecQ helicases are involved in (i) Resolving aberrant structures encountered at the replication fork during normal DNA replication, (ii) Replication restart at collapsed replication fork sites which arise due to the presence of a nick within the leading strand ahead of replication fork. When the progressing replication fork encounter these nicks (which mimic double strand breaks), it leads to replication arrest. RecQ helicases helps in the replication recovery at the arrested replication fork by promoting fork regression and homologous recombination, (iii) Lesion bypass when the base error is present in the lagging strand. The nick is created due to removal of an incorrectly incorporated nucleotide in the lagging strand which is filled by recombination-mediated gap repair. This is followed by RecQ helicase-mediated resolution of recombinogenic structures (iv) Telomeric maintenance by promoting intra- or inter-strand invasion of 3′ tail of the telomere followed by homologous recombination, (v) Resolution of Holliday junctions by branch migration activity, (vi) Homologous recombination and dissolution of Holliday junctions during the process of meiotic segregation and preventing illegitimate recombination during mitosis

RecQ helicases are involved in multiple steps of major recombination pathways. The members of the RecQ helicase family interacts with various proteins involved in different steps of the major recombination pathways i.e., error free homologous recombination (HR) pathway and error prone non-homologous end-joining (NHEJ) pathway. (See text for details)
One of the prominent functions of RecQ helicases is to prevent aberrant and potentially recombinogenic DNA structures that arise as intermediates during DNA replication, repair, or recombination. RecQ helicases in association with other repair proteins might properly resolve such recombinogenic structures to prevent illegitimate and deleterious crossover recombination. Various RecQ-deficient eukaryotic cells display elevated levels of recombination, suggesting their anti-recombination functions (Bugreev et al. 2007; Hu et al. 2007). This review mainly focuses on the roles of various RecQ helicases in the process of DNA recombination to maintain the integrity of the genome during the faithful segregation and transmission from one generation to the next, and the genetic and physiological abnormalities associated with their defects.
Roles of Werner protein in maintaining genomic stability
Werner syndrome (WS) is an autosomal recessive disease characterized by premature aging associated with genome instability and an elevated risk of cancer. One of the earliest features recognized in primary fibroblasts cell derived from WS patients is limited replicative capacity and reduced ability to proliferate. The WS fibroblasts undergo premature entry into senescence compared to normal cells, which could be one of the reasons for premature aging in WRN patients (Epstein et al. 1965). However, not all cell types derived from WS patients undergo premature senescence. Human T lymphocytes represent a well-characterized example of such a cell type. These cells show no significant reduction in growth capacity compared to normal controls (James et al. 2000), but they are hypersensitive to DNA damaging agents, with aberrations consistent with WRN deficiency (Fukuchi et al. 1990). Cytogenetic observations of WS cells show various types of chromosomal aberrations including deletions, translocations, and rearrangements as well as increased spontaneous mutation (Hoehn et al. 1975; Salk et al. 1985; Fukuchi et al. 1989). Various studies have observed that WS cells show selective sensitivity towards many different types of DNA damaging agents such as 4-nitroquinoline-N-oxide (4-NQO, causing replication fork stalling), camptothecin (CPT) (causing replication fork collapse), ICLs, and ionizing radiation (IR) (Okada et al. 1998; Poot et al. 1999, 2002; Ogburn et al. 1997; Pichierri et al. 2000). Thus, the WS cellular phenotype is dependent on the sensitivities of WS cells to different DNA damaging agents and efficiency of DNA repair mechanisms including HR to counteract these damages.
Roles of Werner in genetic recombination
Cellular studies have shown marked reduction in cell proliferation following mitotic recombination in WS fibroblasts (Prince et al. 2001). These findings suggested a role of WRN in mitotic recombination. Prince et al. (2001), showed spontaneous genome instability of WS cells by measuring the capacity for mitotic recombination of the WS fibroblasts cell lines. This group observed that WS fibroblast cell lines failed to resolve recombinant products. Thus, in the absence of WRN, unresolved or disrupted gene conversion products may lead to gene rearrangement or loss mediated by other processes and result in recombination-initiated mitotic arrest, and cell death (Prince et al. 2001).
Several lines of evidence suggest that WRN is actively involved in the homologous recombination pathway. Saintigny et al. (2002), have postulated that the physiological role of WRN protein in the cell is to resolve RAD51-mediated homologous recombination (HR) products, and its failure to do so in WS leads to WS cellular phenotypes such as defective recombination resolution, mitotic arrest, cell death, or genomic instability (Saintigny et al. 2002). In accordance with this hypothesis, it has also been shown that WRN interacts physically and functionally with the homologous recombination mediator/single strand annealing protein RAD52 which has been found at arrested replication forks. Biochemical studies have shown that RAD52 both inhibits and enhances WRN helicase activity in a DNA structure dependent manner. WRN, in turn, stimulates RAD52-mediated homologous strand annealing between complementary sequences. Thus, the coordinated activities of WRN and RAD52 may be involved in replication fork rescue after DNA damage (Baynton et al. 2003).
Another major recombination pathway is the NHEJ pathway that is error-prone rejoining process of DSBs. A defect in this pathway consequently results in a loss or gain of genetic information. Oshima et al. (2002) found extensive deletions at non homologous joining ends of the linear plasmids with incompatible ends when introduced into WS cells. Thus, WRN might suppress extensive nucleotide loss during NHEJ and prevent aberrant DNA repair potentially by stabilizing the broken DNA ends or by direct competition with other helicases or exonucleases (Oshima et al. 2002). Thus, in the absence of WRN, regulatory processes controlling NHEJ may be disrupted and relatively large and potentially oncogenic deletions would be generated, leading to accelerated decline in the fidelity of DSB repair. In a very recent study, it has been shown that WRN physically and functionally interacts with the major NHEJ factor XRCC4-DNA ligase IV complex (X4L4) which stimulates WRN exonuclease activity but not WRN helicase activity. Further, X4L4 is able to ligate a substrate processed by WRN exonuclease, suggesting the functional importance of this interaction (Kusumoto et al. 2008).
Roles of Werner in repair of interstrand cross-links
DNA interstrand cross-links (ICLs) covalently bind the complementary strands of the double helix, thus blocking DNA replication and transcription. ICLs are some of the most cytotoxic and genotoxic DNA lesions known (Akkari et al. 2000). ICLs cause replication forks to stall, eventually leading to generation of one-sided DSBs near the ICL site. In eukaryotes, ICL repair is poorly understood, but is thought to involve a combination of nucleotide excision repair (NER), translesion synthesis (TLS) and/or recombination (De Silva et al. 2000; Zheng et al. 2003). Since cells lacking WRN are hypersensitive to DNA interstrand cross-links (ICLs), WRN is likely involved in the repair of ICLs to restore normal replication forks in the cell (Pichierri and Rosselli 2004).
It has been shown that WRN relocates to the sites of arrested replication induced by ICLs, where it physically and functionally interacts with RAD52 (Baynton et al. 2003). Molecular studies by Cheng et al. showed that WRN cooperates physically and functionally with BRCA1 in cellular response to ICLs. WRN helicase activity, but not its exonuclease activity is required to process the DNA ICLs. The BRCA1/BARD1 complex also associates with WRN and stimulates WRN helicase activity on forked and Holliday junction substrates (Cheng et al. 2006). WRN also cooperates with MRN complex both in vivo and in vitro via its association with Nbs1 (Cheng et al. 2004). Further, Otterlei et al. showed that WRN participates in a multiprotein complex containing RAD51, RAD54, RAD54B and ATR in cells where replication has been arrested by ICLs. These findings suggest that WRN plays a role in the recombination step of ICL repair (Otterlei et al. 2006).
One of the steps in HR is the formation of Holliday junctions (HJs) as a recombination intermediate. Although WRN is able to unwind Holliday junctions, it is unclear whether HJs accumulate in WS cells. Recently, Rodriguez-Lopez et al. (2007) created isogenic WS cell lines expressing a nuclear targeted bacterial HJ endonuclease, RusA and showed that Holliday junction resolution by RusA restores DNA replication capacity in primary WS fibroblasts and enhances their proliferation. Furthermore, RusA expression rescued the hypersensitivity of the WS fibroblasts cell to camptothecin and 4-NQO inducing the formation of a double-strand break and fork collapse (Rodriguez-Lopez et al. 2007). This finding suggests that HJs may persist in the absence of WRN (i.e., in WS cells), leading to illegitimate recombination. Thus, WRN is important in vivo in preventing accumulation of HJs. WRN also promotes the ATP-dependent translocation of Holliday junctions which are consistent with the model in which WRN prevents aberrant recombination events at sites of stalled replication forks by dissociating recombination intermediates (Constantinou et al. 2000). The role of WRN in resolving steps of recombination is supported by the observation that WRN contains an enzymatic property which unwinds Holliday junction structures. In vitro studies have demonstrated that the WRN helicase activity is able to unwind HJs through a branch migration-like activity (Constantinou et al. 2000; Shen and Loeb 2001; Bachrati and Hickson 2003; Khakhar et al. 2003 Lee et al. 2005).
Roles of Werner in stalled replication forks
WRN also plays an important role in the response to replication fork arrest and its recovery during the S-phase of the cell cycle. Cultured WS cells show poor S-phase progression with much lower levels of DNA synthesis activity and an apparent G1 DNA content (Rodriguez-Lopez et al. 2002). These phenotypes account for the loss of proliferative capacity of WS cells and appear to be responsible for early onset of cellular senescence. It has been further observed that there is significant asymmetry in bidirectional replication fork in the absence of functional WRN protein which suggests that WRN might acts to prevent collapse of replication forks or to resolve DNA junctions at stalled replication fork in normal cells (Rodriguez-Lopez et al. 2002; Sidorova et al. 2008). Thus, the WRN protein is important in the elongation stage of DNA replication. Further, as mentioned earlier WRN deficient cells are hypersensitive to clastogens (induces replication fork blockage), DNA interstrand cross-linking agents, camptothecin, and hydroxyurea (HU) (Okada et al. 1998; Poot et al. 2001; Pichierri et al. 2001; Bohr et al. 2001). These findings lead us to propose that the WRN helicase/exonuclease normally acts in the sites of stalled or collapsed replication forks.
Although the trigger(s) recruiting WRN to stalled replication sites are largely unknown, available evidence suggest that WRN involvement in response to replication stress is an ATM/ATR-dependent (Pichierri et al. 2003). In addition, several lines of evidence support the view that WRN might play an upstream role in response to DSBs at replication forks. WRN is required for activation of ATM as well as phosphorylation of downstream ATM substrates in cells with collapsed replication forks (Cheng et al. 2008). WRN also associates and colocalizes with the MRN complex following DNA replication arrest (Franchitto and Pichierri 2002, 2004).
Replication fork stalling or collapse is dependent on whether the fork-blocking lesion is on the leading strand or lagging strand. If the polymerase is kept in the coupled replication complex and skips ahead to the next primer to synthesis a new Okazaki fragment, leaving behind a single strand gap containing the lesion, HR is the preferred gap repair pathway. Another pathway to repair the gap is the generation of Displacement loop (D-loop) structures (recombination intermediates) by invasion of the blocked nascent lagging strand into its sister chromatid and extension of the invading strand by DNA polymerase. The extended D-loop structures are excellent substrates for WRN (and BLM helicase) and the dissociated D-loops would be used as a substrate for replication restart. A recent biochemical study suggests that WRN protein catalyses fork regression and HJ formation on a model replication fork in an ATP dependent manner (Machwe et al. 2007). Further, WRN exonuclease activity enhances regression of forks with smaller gaps on the leading arm. This finding suggests that WRN might regress replication forks in vivo, proposing a role for WRN in the recovery of replication arrest (Machwe et al. 2007).
Roles of Werner and other RecQ helicases in telomeric maintenance and preservation
Increasing evidence suggests that telomeric dysfunction is likely to be an important factor for premature senescence and decreased replicative capacity observed in WS cells. Consistent with roles of other RecQ helicases at the telomeres, human BS cells also show telomere defects (Lillard-Wetherell et al. 2004). Du et al. (2004) found that Wrn and Blm mutations, introduced in a telomerase null mice, accelerates the onset of the pathological phenotype, which includes increased telomeric loss and chromosomal end fusion, which is normally observed very late in telomerase null mice (Du et al. 2004). These findings suggest important roles played by RecQ helicases in telomere maintenance.
It has been shown that most human somatic cells do not possess sufficient telomerase to maintain telomere length intact through successive generations and in the absence of telomerase, telomeres progressively become shortened in each generation and eventually become dysfunctional, leading to genomic instability, growth arrest, and apoptosis (Harley et al. 1990; Blasco 2005; Opresko 2008). The primary role of telomeres in the cell is to protect the ends of linear chromosomes and prevent them from being recognized as double strand breaks (DSBs) by the cellular damage response machinery. The telomere protects the ends of the linear chromosome by forming a “telosome complex” consisting of six member core proteins and telomeric DNA (de Lange 2005). In humans, telomeres contain 2–10 kb of TTAGGG tandem repeats and end in a 3′ single strand G-rich tail that serves as the substrate for telomerase. The telomeric proteins remodel the telomere ends into a structure that sequesters the 3′ tail and protects them from degradation by nucleases, or prevents them from forming aberrant structures. Existing evidence supports a model whereby telomeric proteins modulate t-loop formation (lasso structure) in which the 3′ tail invades the telomeric duplex and forms a recombination-like D-loop (Griffith et al. 1999, see also Fig. 3 iv). Loss of telomere structure and function induces a DNA damage response that involves several proteins that normally respond to the process of DSBs (de Lange 2005). Moreover, WRN and BLM proteins interact physically and functionally with at least three critical members of telosome core complex, namely TRF2 and TRF1 which bind duplex telomeric DNA, and POT1 which binds single stranded TTAGGG repeats and protects the 3′ end of the telomere (Opresko et al. 2002; Stavropoulos et al. 2002; Machwe et al. 2004; Lillard-Wetherell et al. 2004).
RecQ helicases have been implicated in the telomere-based DNA damage response that is induced by mimics of telomeric ssDNA tails (Eller et al. 2006). Studies in yeast have shown the involvement of RecQ helicase protein in homologous recombination pathways at the telomeres. In budding yeast, RecQ helicase (Sgs1) functions in an alternative pathway for lengthening of telomeres (ALT) that occur via recombination in type II survivors of telomerase-negative mutants (Johnson et al. 2001; Huang et al. 2001). Evidence for an ALT-like pathway has also been detected in telomerase-negative mammalian cells from tumors or upon SV40 transformation (Yeager et al. 1999). WRN protein colocalize with telomeric DNA in human cell lines that maintain telomeres by ALT (Johnson et al. 2001; Opresko et al. 2004. The precise mechanism of the ALT pathway in human cells is poorly understood, but several models have proposed the involvement of intra- and inter-telomeric D-loop formation. For example, the 3′ telomeric tail invades telomeric duplex DNA in the same telomere (t-loop/D-loop), the sister chromatid, or in a separate chromosome (Neumann and Reddel 2002). A DNA polymerase initiates synthesis at the 3′-OH of the invading strand and increases the length of the telomere, followed by dissociation of the recombination intermediate. It has been postulated that the initiation of telomeric recombination may signal WRN recruitment to either suppress recombination or to dissociate intermediates to ensure proper separation of the recombining telomeric strand. In yeast, RecQ helicase Sgs1 acts in the resolution of recombination intermediates in telomerase deficient yeast strains near senescence, when critically shortened telomeres undergo recombination in an effort to restore telomere length (Lee et al. 2007). Failure to resolve these recombinant structures results in rapid senescence. Further data support the suggestion that Sgs1 is involved in the resolution of telomeric recombination intermediates rather than preventing its formation at the initiation stage (Lee et al. 2007).
An elevated level of sister chromatid exchange at the telomere (T-SCEs) has been observed in WS, indicating hyperrecombination at the telomere in the absence of WRN. Cells from late generation (G5) mTerc −/− Wrn −/− mice with shortened telomeres show elevated T-SCEs compared to cells from G5 mTerc −/− Wrn ± heterozygotes, which could be suppressed with WRN helicase activity (Laud et al. 2005). This increased propensity towards telomeric recombination correlates with an increase in emergence of immortalized clones from G5 mTerc −/− Wrn −/− cells which necessarily maintains telomeres by ALT, since telomerase is absent (Laud et al. 2005). These findings suggest that WRN normally suppresses sister chromatid exchanges between telomeres in the shortened or dysfunctional telomeres. Indeed, late passage embryonic stem cells from mTerc −/− mice display an increased propensity towards T-SCEs as the telomeres become critically short (Wang et al. 2005). Thus, a requirement for WRN and regulation of recombination at the telomeres to suppress ALT may become more crucial as the telomeres shorten.
The proteins of the NHEJ pathway of recombination, i.e., the Ku70/80 heterodimer, also localize to telomeres and function in telomere maintenance. Ku suppresses recombination at telomeres that are made dysfunctional through the loss of telomeric protein TRF2. Deletion of TRF2 in Ku70−/− mouse cells results in an elevated levels of T-SECs (Celli et al. 2006). WRN physically interacts with both POT1 and the Ku heterodimer, which stimulate WRN helicase and exonuclease activities, respectively (Cooper et al. 2000; Opresko et al. 2005). Very recently, it has been shown that POT1 promotes the apparent processivity of WRN helicase by maintaining partially unwound DNA strands in a melted state, rather than preventing WRN dissociation from the substrate (Sowd et al. 2008). Thus, WRN may function in pathways with Ku or POT1 to suppress recombination at telomeric ends.
Physiological consequences of WRN loss and its relation to aging
Taken together, the above findings support a multidimensional role of WRN in protecting the genome from aberrations and instability. The loss of WRN in cells leads to proliferative defects, limited replicative capacity, and premature cellular senescence. These phenotypes could be due to global genomic damage which results in the rapid exit from the cell cycle of WRN defective cells compared to normal control cells (Faragher et al. 1993).
Another important aspect of the aging phenotype is telomeric erosion or dysfunction. Telomeres play an important role in the genome stability and various theories have been put forward suggesting that telomeric dysfunction is directly linked to cellular senescence and the aging process. As discussed above, WRN protein, in co-ordination with other telomeric proteins like TRF1 and TRF2, is directly involved in preventing telomeric erosion and genome instability. The loss of WRN in the cell leads to elevated telomeric deletions and dysfunction. Therefore, another reason for premature senescence behaviors of WRN fibroblasts could be the telomeric driven senescence due to WRN deficiency (Cox and Faragher 2007). In accordance with this hypothesis, ectopic expression of telomerase (hTERT) prevents WRN fibroblasts from undergoing premature senescence and the WS cells become immortalized (Wyllie et al. 2000; Choi et al. 2001). However, there are some significant differences in the gene expression patterns between WS and normal hTERT-immortalized cells, indicating that telomerase expression does not prevent the phenotypic drift, or destabilized genotype, resulting from the WRN defect (Choi et al. 2001). However, immortalization of WRN cells by hTERT suggests that telomere effects are the predominant trigger of premature senescence in WRN cells. Contrary to this hypothesis, work of Baird et al. (2004) using single telomere length analysis (STELA) showed that WS dermal fibroblasts display normal rates of telomere erosion, suggesting that accelerated replicative decline seen in WS fibroblasts does not result primarily from accelerated telomere erosion (Baird et al. 2004).
Therefore, it is likely that the senescence observed in WS cells is a consequence of the combined effects of irreversible cell cycle exit due to genome instability and accelerated telomere-driven senescence, and that there is complex interplay between the two phenomena.
Roles of BLM protein in genomic stability
Bloom syndrome (BS) is an autosomal recessive disorder characterized by growth retardation, sunlight sensitivity and predisposition to the development of cancer (German 1995; Luo et al. 2000). Cellular investigations show that BS is associated with inherent genomic instability (Bachrati and Hickson 2003; Hickson 2003). BS cells show an elevated level of several types of chromosomal aberrations, including breaks, quadriradials and translocations. The hallmark feature of BS cells is highly elevated levels of the frequency of sister chromatid exchange (SCEs), exchanges between homologous chromosomes, and loss of heterozygosity, which can be used as a molecular diagnostic for this disease (Chaganti et al. 1974; German 1995). These reciprocal DNA exchanges arise primarily as part of HR events that occur during repair of DNA damage in the S or G2 phases of the cell cycle.
Roles of BLM in homologous recombination
BLM forms a part of multienzyme complex that appears to play roles both in the disruption of alternative DNA structures such as quadruplexes and in the resolution of DNA intermediates that arises during homologous recombination. Consistent with its roles in HR, BLM physically interacts with HR proteins RAD51 and Rad51D, as well as with several other proteins involved in DNA repair and DNA damage signaling such as Mus81, MLH1, MSH6, RPA and ATM (Wu et al. 2001; Braybrooke et al. 2003; Beamish et al. 2002; Sharma et al. 2006; Pedrazzi et al. 2003).
The repair of DSBs by HR is a multistep process. One of the key steps in the HR pathway is the formation of nucleoprotein filaments by RAD51 binding to ssDNA resected ends of DSBs (Sung et al. 2003; West 2003). These RAD51 nucleoprotein filaments possess the interesting ability to “search” the entire genome for a homologous duplex sequence and then catalyses the ssDNA strand exchange reaction with the identical strand in the homologous duplex through complementary base pairing, resulting in the formation of a displacement loop (D-loop). This structure facilitates repair synthesis using the intact homologous sequence as the template strand and invading ssDNA as a primer for DNA polymerase during DNA repair synthesis.
Recent studies showed two novel pro- and anti-recombination activities of the human BLM helicase at different stages (Bugreev et al. 2007). In the early phase of HR, BLM disrupts the Rad51-ssDNA filament by dislodging human Rad51 protein from ssDNA in an ATPase dependent manner, thus preventing the formation of D-loop. These data are consistent with the established role of BLM in suppression of HR at an early stage (Bugreev et al. 2007). Evidence also suggests that BLM may act downstream of D-loop formation (Wu and Hickson 2006). HR can proceed down several pathways. Two pathways, known as synthesis-dependent strand-annealing (SDSA) and double Holliday junction dissolution (DJD), result exclusively in the formation of non-crossover products. BLM has been implicated in effecting both SDSA and DJD (Adams et al. 2003; Wu and Hickson 2003). In one case, a D-loop may eventually convert to a double Holliday junction, and is then processed by dissolution of the Holliday junction. However, SDSA requires the dissociation of a D-loop allowing complementary 3′ ssDNA tails of the broken chromosome to anneal and be ligated following DNA repair gap filling. Bugreev et al. (2007) showed that BLM may promote SDSA by facilitating D-loop mediated DNA repair synthesis.
Studies have also shown that BLM interacts physically and functionally with the type IA topoisomerase Topo IIIα, and catalyses a novel reaction in the resolution of recombination intermediates involving Double Holliday junctions (DHJs), termed as “Holliday junction dissolution” (Hu et al. 2001; Wu and Hickson 2003). This reaction gives rise exclusively to non-cross-over products, which fits very well with the role of BLM as a suppressor of SCEs. The BLM-Topo IIIα pair is tightly associated with a third protein called BLAP75. Attenuation of BLAP75 levels by RNA interference destabilizes both BLM and Topo IIIα (Yin et al. 2005). Biochemical analyses have revealed specific and direct interactions of BLAP75 with BLM and Topo IIIα and a strong enhancement of the BLM-Topo IIIα-mediated DHJ dissolution reaction by this novel protein (Wu et al. 2006; Raynard et al. 2006). Recently, Bussen et al. (2007) demonstrated that BLAP75 in conjunction with Topo IIIα greatly enhances the HJ unwinding activity of BLM. This functional interaction is highly specific, as the BLAP75-Topo IIIα pair has no effect on either WRN or Escherichia coli RecQ helicase activity, nor can E. coli Top3 substitute for Topo IIIα in the enhancement of the BLM helicase activity (Bussen et al. 2007).
Role of BLM in rescuing stalled replication fork
BLM also plays an important role in the repair of stalled or collapsed replication fork during the S-phase of the cell cycle. BS cells exhibit abnormal replication intermediate formation, delayed Okazaki fragment maturation and hypersensitivity to various inhibitors of replication (Davies et al. 2004; Lonn et al. 1990). In response to hydroxyurea-induced replicative stress, BLM localizes to repair centers at collapsed replication forks, which are dependent on stress-activated kinases ATM and ATR (Davalos et al. 2004).
When the replication fork encounters lesions on the leading strand, it causes the replication fork to stall. One potential role of BLM at the stalled replication fork is to promote the fork regression after which the nascent leading and lagging strands anneal to create a structure know as a “chicken foot” (Ralf et al. 2006). This structure will facilitate a process known as “template switching” in which the nascent lagging strand is used as a template and the leading strand extends further to bypass the lesion which is later processed by HR. However, the mechanism by which BLM catalyses regression of the replication fork requires further investigation.
As a result of HR-mediated restart/repair of a damaged replication fork, sister chromatids become covalently linked by Holliday junctions, which need to be resolved prior to mitosis. BLM is able to both bind and branch migrate synthetic Holliday junctions. It has been shown in S. cerevisiae that loss of Sgs1 results in the accumulation of HR-dependent replication intermediates that resemble Holliday junctions (Liberi et al. 2005) suggesting that BLM might function in resolving Holliday junctions in a TopIIIα and BLAP75 dependent manner (Karow et al. 2000; Johnson et al. 2000).
Roles of RECQL4 in maintaining genome stability
Defects in the RECQL4 gene are the cause of three rare autosomal recessive diseases, namely Rothmund Thomson syndrome (RTS), RAPADILINO syndrome and Baller–Gerold (BGS) syndrome (Kitao et al. 1999; Siitonen et al. 2003; Van Maldergem et al. 2006). Rothmund Thomson syndrome is an unusual disorder characterized by poikiloderma, growth deficiency, juvenile cataracts, premature aging and predisposition to malignant tumors especially osteosarcomas (Vennos et al. 1992; Stinco et al. 2008). Most of the RTS patients show mutations in the RECQL4 helicase domain, resulting in truncated protein due to premature termination of protein synthesis (Lindor et al. 2000). Cytological investigations of various cell types derived from RTS patients show genomic instability and chromosomal abnormalities such as trisomy, aneuploidy and chromosomal rearrangements (Vennos et al. 1992; Der Kaloustian et al. 1990; Orstavik et al. 1994; Durand et al. 2002; Anbari et al. 2000). The different cell types derived from RECQL4-knockout mice display an overall aneuploidy phenotype and a significant increase in the frequency of premature centromere separation (Mann et al. 2005). These results suggest a role of RECQL4 gene in preventing tumorigenesis and maintenance of genome integrity in humans.
There have been contradictory reports of sensitivity to different genotoxic agents of patient-derived RECQL4-deficient fibroblasts. In two independent studies, RTS cells showed sensitivity to H2O2, which creates oxidative damage, and ionizing radiation (Werner et al. 2006; Vennos and James 1995), resulting in irreversible growth arrest, decreased DNA synthesis and concomitant reduction of cells in S-phase compared to normal fibroblasts (Werner et al. 2006). However, in another recent study, primary RTS fibroblasts showed no sensitivity to wide variety of genotoxic agents including ionizing or UV radiation, nitrogen mustard, 4-NQO, 8-MOP, Cis-Pt, MMC, H2O2, HU, or UV plus caffeine, suggesting the complexity of various RTS cells towards genotoxic responses (Cabral et al. 2008). In another interesting report, it has been shown that, compared to wild type (wt) fibroblasts, primary fibroblasts carrying two deleterious RECQL4 mutations have increased sensitivity to HU, CPT, and doxorubicin (DOX), which exert their effects primarily during S-phase, suggesting a major role of RECQL4 protein in DNA replication (Jin et al. 2008). Further, RTS cells showed modest sensitivity to other DNA damaging agents including ultraviolet (UV) irradiation, ionizing radiation (IR), and cisplatin (CDDP) (Jin et al. 2008). The RTS cells also showed relative resistance to 4-NQO, unlike WS and BS cells which are hypersensitive to this drug (Jin et al. 2008). Mutant human cells lacking RECQL4 escaped from the S-phase arrest following UV or HU treatment, whereas BLM-defective cells exhibited a normal S-phase arrest following UV irradiation (Park et al. 2006). RECQL4 also formed discrete nuclear foci coincident with the nucleotide excision repair factor XPA, in response to UV irradiation and 4-NQO, suggesting that it could be involved in efficient removal of UV lesions (Fan and Luo 2008). However, the discrepancies among different reports might be due to different experimental approaches that have been employed for these studies. These results indicate functional differences among RecQ helicase family members in their possible involvement in various DNA repair and replication pathways.
The cellular functions of RECQL4 are largely unknown. However, data arising from its sensitivity towards different genotoxic agents are indicative of its involvement in distinct DNA metabolic and repair pathways. RECQL4 has been shown to interact with UBR1 and UBR2, members of a family of E3 ubiquitin ligase of the N-end rule pathway, which is a part of the ubiquitin-proteosome system (Yin et al. 2004). RECQL4 has been proposed to function in the initiation of DNA replication with its N terminus required for the recruitment of DNA polymerase α (Sangrithi et al. 2005; Matsuno et al. 2006). RECQL4 is also known to interact with Cut5 (a homologue of Dpb11 that is required for loading DNA polymerases onto chromatin (Hashimoto and Takisawa 2003). Further, RECQL4 interacts with poly (ADP-ribose) polymerase1 (PARP-1) which is involved in different pathways of DNA metabolism such as DNA recombination, repair, and transcriptional regulation (Woo et al. 2006). In response to the induction of DSBs by treatment with etoposide, a portion of RecQL4 and Rad51 nuclear foci colocalized, suggesting that RECQL4 plays a role in the repair of DSBs by homologous recombination (Petkovic et al. 2005). However, the mechanistic details of this interaction are unknown.
Roles of RECQL1 in genome stability
RECQL1 is found to be the most abundant of all five human RecQ helicases in resting B cells (Kawabe et al. 2000). Studies in chicken DT40 cells have shown that RECQL1 and RECQ5 have roles in cell viability under BLM-impaired conditions, indicating the redundant function of these helicases (Wang et al. 2003). Recent studies have shown that depletion of RECQL1 makes human cells sensitive to IR or camptothecin, and such cells show a high level of spontaneous γ-H2AX foci and elevated SCE, indicating an accumulation of double strand breaks. Further, its physical interaction with Rad51 suggests that RECQL1 may be involved in the repair of DSB by HR (Sharma and Brosh 2007). Consistent with a role of RECQL1 in HR, very recently, it has been shown that RECQL1 possesses ATPase-dependent DNA branch migration activity (Bugreev et al. 2008). A specific feature of RECQ1-catalysed branch migration is a strong preference towards the 3′ → 5′ polarity in both the three and four stranded reactions, which is very unique property of RECQ1. This specific 3′ → 5′ branch migration activity allows RECQL1 to disrupt recombination intermediates (D-loop) formed by invasion of tailed DNA with the 5′-protruding ends. These D-loops, in contrast to the D-loops formed by invasion of tailed DNA with the 3′-protruding ends, cannot be readily extended by DNA polymerase and therefore may represent unproductive recombination intermediates during DSB repair. Therefore, RECQL1 branch migration may prevent accumulation of these unconventional and potentially toxic intermediates in vivo.
Role of RecQ5 in maintaining genome stability
RecQ5 is one of the members of RecQ helicase family that has not been yet linked to any genetic disease. In both Drosophila and humans, RecQ5 exists in different isoforms generated by alternative splicing (Sekelsky et al. 1999). In humans there are three RecQ helicase isomers, RecQ5α, RecQ5β and RecQ5γ. Two of these isomers RecQ5α and RecQ5γ, are small and localized in the cytoplasm, while RecQ5β migrates into the nucleus and exists in the nucleoplasm, like other RecQ helicases (Shimamoto et al. 2000).
Recent studies in mouse models have shown that deletion of RecQ5 results in increased susceptibility to cancer. RecQ5-deleted cells exhibit elevated frequencies of spontaneous double stranded breaks (DSBs) and HR (Hu et al. 2007). Mechanistically, human RecQ5 binds to the Rad51 recombinase and inhibits Rad51 mediated D-loop formation by displacing Rad51 from ssDNA. These results suggest that RecQ5 may minimize gross chromosomal rearrangements (GCRs) and tumorigenesis by suppressing the accumulation of DSBs, and attenuate HR by disrupting the Rad51 pre-synaptic filament (Hu et al. 2007).
The results discussed above suggest that higher organisms have multiple pathways to regulate HR and that members of the RecQ helicase family might have overlapping functions in modulating HR. Studies in chicken B-lymphocyte line DT40 cells showed that both RecQ1−/−/BLM−/− and RecQ5−/−/BLM−/− cells grew much more slowly than BLM−/− cells, indicating that RecQ1 and RecQ5 are involved in cell viability when BLM function is impaired. Moreover, RecQ5−/−/BLM−/− cells also showed a higher frequency of SCE than BLM−/− cells, indicating that RecQ5 suppresses SCE under the BLM function-impaired conditions. These results suggest that RecQ1 and RecQ5 in combination with TOP3α partially substitute the function of BLM under BLM function-impaired conditions, indicating the existence of functional redundancy between different RecQ helicase members (Otsuki et al. 2008; Wang et al. 2003). However, a contradicting result has been observed in mouse embryonic stem (ES) cells, in which disruption of either Blm or the Recq5 gene resulted in a significant increase in the frequency of sister chromatid exchange (SCE) compared to wild type (wt), whereas deleting both Blm and Recq5 leads to an even higher frequency of SCE (Hu et al. 2005). Further, these authors also show that embryonic fibroblasts derived from Recq5 knockout mice also exhibit a significantly increased frequency of SCE compared to corresponding wild type controls. These results indicate that BLM and Recq5 have non-redundant functions in suppressing crossovers in mouse ES cells (Hu et al. 2005).
Conclusions and future perspectives
The RecQ helicases are essential parts of cellular machinery engaged in DNA metabolism and genome stability. The presence of five different types of RecQ helicases in human and mouse is an adaptive feature towards complexity in the genome among higher eukaryotes. RecQ helicases are involved in various DNA metabolic pathways and ensure error-free DNA transactions in each generation. Therefore, defective RecQ helicases lead to diverse chromosomal abnormalities, genomic instability and premature aging. Existing literature suggests that different RecQ helicases have overlapping functions in the DNA metabolic pathways. There is likely to be interplay among RecQ helicases in cellular pathways, with one helicase complementing the function of other in a particular type of stress response; some might have non-redundant functions. In the future it would be very interesting to gain insight into how different RecQ helicases function together to ensure genome stability. Among the RecQ helicases only WRN and BLM have been studied in detail, however, the evidence discussed here suggests that other members are equally important in maintaining genome stability. A major focus in the future would be to characterize the cellular and biological functions of other RecQ helicases in response to various stresses.
References
Adams MD, McVey M, Sekelsky JJ (2003) Drosophila BLM in double-strand break repair by synthesis-dependent strand annealing. Science 299:265–267
Akkari YM, Bateman RL, Reifsteck CA, Olson SB, Grompe M (2000) DNA replication is required to elicit cellular responses to psoralen-induced DNA interstrand cross-links. Mol Cell Biol 20:8283–8289. doi:10.1128/MCB.20.21.8283-8289.2000
Anbari KK, Ierardi-Curto LA, Silber JS, Asada N, Spinner N, Zackai EH, Belasco J, Morrissette JD, Dormans JP (2000) Two primary osteosarcomas in a patient with Rothmund–Thomson syndrome. Clin Orthop Relat Res 378:213–223. doi:10.1097/00003086-200009000-00032
Bachrati CZ, Hickson ID (2003) RecQ helicases: suppressors of tumorigenesis and premature aging. Biochem J 374:577–606. doi:10.1042/BJ20030491
Bachrati CZ, Hickson ID (2008) RecQ helicases: guardian angels of the DNA replication fork. Chromosoma 117:219–233. doi:10.1007/s00412-007-0142-4
Baird DM, Davis T, Rowson J, Jones CJ, Kipling D (2004) Normal telomere erosion rates at the single cell level in Werner syndrome fibroblast cells. Hum Mol Genet 13:1515–1524. doi:10.1093/hmg/ddh159
Baynton K, Otterlei M, Bjoras M, von Kobbe C, Bohr VA, Seeberg E (2003) WRN interacts physically and functionally with the recombination mediator protein RAD52. J Biol Chem 278:36476–36486. doi:10.1074/jbc.M303885200
Beamish H, Kedar P, Kaneko H, Chen P, Fukao T, Peng C, Beresten S, Gueven N, Purdie D, Lees-Miller S, Ellis N, Kondo N, Lavin MF (2002) Functional link between BLM defective in Bloom’s syndrome and the ataxia-telangiectasia-mutated protein, ATM. J Biol Chem 277:30515–30523. doi:10.1074/jbc.M203801200
Blasco MA (2005) Telomeres and human disease: ageing, cancer and beyond. Nat Rev Genet 6:611–622
Bohr VA, Souza Pinto N, Nyaga SG, Dianov G, Kraemer K, Seidman MM, Brosh RM Jr (2001) DNA repair and mutagenesis in Werner syndrome. Environ Mol Mutagen 38:227–234. doi:10.1002/em.1076
Braybrooke JP, Li JL, Wu L, Caple F, Benson FE, Hickson ID (2003) Functional interaction between the Bloom’s syndrome helicase and the RAD51 paralog, RAD51L3 (RAD51D). J Biol Chem 278:48357–48366. doi:10.1074/jbc.M308838200
Brosh RM Jr, Bohr VA (2007) Human premature aging, DNA repair and RecQ helicases. Nucleic Acids Res 35:7527–7544. doi:10.1093/nar/gkm1008
Bugreev DV, Yu X, Egelman EH, Mazin AV (2007) Novel pro- and anti-recombination activities of the Bloom’s syndrome helicase. Genes Dev 21:3085–3094. doi:10.1101/gad.1609007
Bugreev DV, Brosh RM Jr, Mazin AV (2008) RECQ1 possesses DNA branch migration activity. J Biol Chem 283:20231–20242. doi:10.1074/jbc.M801582200
Bussen W, Raynard S, Busygina V, Singh AK, Sung P (2007) Holliday junction processing activity of the BLM-topo IIIalpha-BLAP75 complex. J Biol Chem 282:31484–31492. doi:10.1074/jbc.M706116200
Cabral RE, Queille S, Bodemer C, de Prost Y, Neto JB, Sarasin A, Daya-Grosjean L (2008) Identification of new RECQL4 mutations in Caucasian Rothmund–Thomson patients and analysis of sensitivity to a wide range of genotoxic agents. Mutat Res 643:41–47. doi:10.1016/j.mrfmmm.2008.06.002
Celli GB, Denchi EL, de Lange T (2006) Ku70 stimulates fusion of dysfunctional telomeres yet protects chromosome ends from homologous recombination. Nat Cell Biol 8:885–890. doi:10.1038/ncb1444
Chaganti RS, Schonberg S, German J (1974) A manyfold increase in sister chromatid exchanges in Bloom’s syndrome lymphocytes. Proc Natl Acad Sci USA 71:4508–4512
Chakraverty RK, Hickson ID (1999) Defending genome integrity during DNA replication: a proposed role for RecQ family helicases. Bioessays 21:286–294. doi:10.1002/(SICI)1521-1878(199904)21:4<286::AID-BIES4>3.0.CO;2-Z
Cheng WH, von Kobbe C, Opresko PL, Arthur LM, Komatsu K, Seidman MM, Carney JP, Bohr VA (2004) Linkage between Werner syndrome protein and the Mre11 complex via Nbs1. J Biol Chem 279:21169–21176. doi:10.1074/jbc.M312770200
Cheng WH, Kusumoto R, Opresko PL, Sui X, Huang S, Nicolette ML, Paull TT, Campisi J, Seidman M, Bohr VA (2006) Collaboration of Werner syndrome protein and BRCA1 in cellular responses to DNA interstrand cross-links. Nucleic Acids Res 34:2751–2760. doi:10.1093/nar/gkl362
Cheng WH, Muftic D, Muftuoglu M, Dawut L, Morris C, Helleday T, Shiloh Y, Bohr VA (2008) WRN is required for ATM Activation and the S-phase checkpoint in response to interstrand crosslink-induced DNA double strand breaks. Molecular biology of the cell. Mol Biol Cell 19:3923–3933
Choi D, Whittier PS, Oshima J, Funk WD (2001) Telomerase expression prevents replicative senescence but does not fully reset mRNA expression patterns in Werner syndrome cell strains. FASEB J 15:1014–1020. doi:10.1096/fj.00-0104com
Constantinou A, Tarsounas M, Karow JK, Brosh RM, Bohr VA, Hickson ID, West SC (2000) Werner’s syndrome protein (WRN) migrates Holliday junctions and co-localizes with RPA upon replication arrest. EMBO Rep 1:80–84. doi:10.1093/embo-reports/kvd004
Cooper MP, Machwe A, Orren DK, Brosh RM, Ramsden D, Bohr VA (2000) Ku complex interacts with and stimulates the Werner protein. Genes Dev 14:907–912
Cox LS, Faragher RG (2007) From old organisms to new molecules: integrative biology and therapeutic targets in accelerated human ageing. Cell Mol Life Sci 64:2620–2641. doi:10.1007/s00018-007-7123-x
Davalos AR, Kaminker P, Hansen RK, Campisi J (2004) ATR and ATM-dependent movement of BLM helicase during replication stress ensures optimal ATM activation and 53BP1 focus formation. Cell Cycle 3:1579–1586
Davies SL, North PS, Dart A, Lakin ND, Hickson ID (2004) Phosphorylation of the Bloom’s syndrome helicase and its role in recovery from S-phase arrest. Mol Cell Biol 24:1279–1291. doi:10.1128/MCB.24.3.1279-1291.2004
de Lange T (2005) Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev 19:2100–2110. doi:10.1101/gad.1346005
De Silva IU, McHugh PJ, Clingen PH, Hartley JA (2000) Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross-links in mammalian cells. Mol Cell Biol 20:7980–7990. doi:10.1128/MCB.20.21.7980-7990.2000
Der Kaloustian VM, McGill JJ, Vekemans M, Kopelman HR (1990) Clonal lines of aneuploid cells in Rothmund–Thomson syndrome. Am J Med Genet 37:336–339. doi:10.1002/ajmg.1320370308
Du X, Shen J, Kugan N, Furth EE, Lombard DB, Cheung C, Pak S, Luo G, Pignolo RJ, DePinho RA, Guarente L, Johnson FB (2004) Telomere shortening exposes functions for the mouse Werner and Bloom syndrome genes. Mol Cell Biol 24:8437–8446. doi:10.1128/MCB.24.19.8437-8446.2004
Durand F, Castorina P, Morant C, Delobel B, Barouk E, Modiano P (2002) Rothmund–Thomson syndrome, trisomy 8 mosaicism and RECQ4 gene mutation. Ann Dermatol Venereol 129:892–895
Eller MS, Liao X, Liu S, Hanna K, Backvall H, Opresko PL, Bohr VA, Gilchrest BA (2006) A role for WRN in telomere-based DNA damage responses. Proc Natl Acad Sci USA 103:15073–15078. doi:10.1073/pnas.0607332103
Epstein CJ, Martin GM, Motulsky AG (1965) Werner’s syndrome; caricature of aging. A genetic model for the study of degenerative diseases. Trans Assoc Am Physicians 78:73–81
Fan W, Luo J (2008) RecQ4 facilitates UV-induced DNA damage repair through interaction with nucleotide excision repair factor XPA. J Biol Chem 283:29037–29044
Faragher RG, Kill IR, Hunter JA, Pope FM, Tannock C, Shall S (1993) The gene responsible for Werner syndrome may be a cell division “counting” gene. Proc Natl Acad Sci USA 90:12030–12034. doi:10.1073/pnas.90.24.12030
Franchitto A, Pichierri P (2002) Protecting genomic integrity during DNA replication: correlation between Werner’s and Bloom’s syndrome gene products and the MRE11 complex. Hum Mol Genet 11:2447–2453. doi:10.1093/hmg/11.20.2447
Franchitto A, Pichierri P (2004) Werner syndrome protein and the MRE11 complex are involved in a common pathway of replication fork recovery. Cell Cycle 3:1331–1339
Fukuchi K, Martin GM, Monnat RJ Jr (1989) Mutator phenotype of Werner syndrome is characterized by extensive deletions. Proc Natl Acad Sci USA 86:5893–5897. doi:10.1073/pnas.86.15.5893
Fukuchi K, Tanaka K, Kumahara Y, Marumo K, Pride MB, Martin GM, Monnat RJ Jr (1990) Increased frequency of 6-thioguanine-resistant peripheral blood lymphocytes in Werner syndrome patients. Hum Genet 84:249–252. doi:10.1007/BF00200569
German J (1995) Bloom’s syndrome. Dermatol Clin 13:7–18
Griffith JD, Comeau L, Rosenfield S, Stansel RM, Bianchi A, Moss H, de Lange T (1999) Mammalian telomeres end in a large duplex loop. Cell 97:503–514. doi:10.1016/S0092-8674(00)80760-6
Hanada K, Hickson ID (2007) Molecular genetics of RecQ helicase disorders. Cell Mol Life Sci 64:2306–2322. doi:10.1007/s00018-007-7121-z
Harley CB, Futcher AB, Greider CW (1990) Telomeres shorten during ageing of human fibroblasts. Nature 345:458–460. doi:10.1038/345458a0
Hashimoto Y, Takisawa H (2003) Xenopus Cut5 is essential for a CDK-dependent process in the initiation of DNA replication. EMBO J 22:2526–2535. doi:10.1093/emboj/cdg238
Hickson ID (2003) RecQ helicases: caretakers of the genome. Nat Rev Cancer 3:169–178. doi:10.1038/nrc1012
Hoehn H, Bryant EM, Au K, Norwood TH, Boman H, Martin GM (1975) Variegated translocation mosaicism in human skin fibroblast cultures. Cytogenet Cell Genet 15:282–298. doi:10.1159/000130526
Hoeijmakers JH (2001) Genome maintenance mechanisms for preventing cancer. Nature 411:366–374. doi:10.1038/35077232
Hu P, Beresten SF, van Brabant AJ, Ye TZ, Pandolfi PP, Johnson FB, Guarente L, Ellis NA (2001) Evidence for BLM and topoisomerase IIIalpha interaction in genomic stability. Hum Mol Genet 10:1287–1298. doi:10.1093/hmg/10.12.1287
Hu Y, Lu X, Barnes E, Yan M, Lou H, Luo G (2005) Recql5 and Blm RecQ DNA helicases have nonredundant roles in suppressing crossovers. Mol Cell Biol 25:3431–3442. doi:10.1128/MCB.25.9.3431-3442.2005
Hu Y, Raynard S, Sehorn MG, Lu X, Bussen W, Zheng L, Stark JM, Barnes EL, Chi P, Janscak P, Jasin M, Vogel H, Sung P, Luo G (2007) RECQL5/Recql5 helicase regulates homologous recombination and suppresses tumor formation via disruption of Rad51 presynaptic filaments. Genes Dev 21:3073–3084. doi:10.1101/gad.1609107
Huang P, Pryde FE, Lester D, Maddison RL, Borts RH, Hickson ID, Louis EJ (2001) SGS1 is required for telomere elongation in the absence of telomerase. Curr Biol 11:125–129. doi:10.1016/S0960-9822(01)00021-5
James SE, Faragher RG, Burke JF, Shall S, Mayne LV (2000) Werner’s syndrome T lymphocytes display a normal in vitro life-span. Mech Ageing Dev 121:139–149. doi:10.1016/S0047-6374(00)00205-0
Jin W, Liu H, Zhang Y, Otta SK, Plon SE, Wang LL (2008) Sensitivity of RECQL4-deficient fibroblasts from Rothmund–Thomson syndrome patients to genotoxic agents. Hum Genet 123:643–653. doi:10.1007/s00439-008-0518-4
Johnson RD, Jasin M (2000) Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. EMBO J 19:3398–3407. doi:10.1093/emboj/19.13.3398
Johnson FB, Lombard DB, Neff NF, Mastrangelo MA, Dewolf W, Ellis NA, Marciniak RA, Yin Y, Jaenisch R, Guarente L (2000) Association of the Bloom syndrome protein with topoisomerase IIIalpha in somatic and meiotic cells. Cancer Res 60:1162–1167
Johnson FB, Marciniak RA, McVey M, Stewart SA, Hahn WC, Guarente L (2001) The Saccharomyces cerevisiae WRN homolog Sgs1p participates in telomere maintenance in cells lacking telomerase. EMBO J 20:905–913. doi:10.1093/emboj/20.4.905
Karow JK, Constantinou A, Li JL, West SC, Hickson ID (2000) The Bloom’s syndrome gene product promotes branch migration of holliday junctions. Proc Natl Acad Sci USA 97:6504–6508. doi:10.1073/pnas.100448097
Kawabe T, Tsuyama N, Kitao S, Nishikawa K, Shimamoto A, Shiratori M, Matsumoto T, Anno K, Sato T, Mitsui Y, Seki M, Enomoto T, Goto M, Ellis NA, Ide T, Furuichi Y, Sugimoto M (2000) Differential regulation of human RecQ family helicases in cell transformation and cell cycle. Oncogene 19:4764–4772. doi:10.1038/sj.onc.1203841
Khakhar RR, Cobb JA, Bjergbaek L, Hickson ID, Gasser SM (2003) RecQ helicases: multiple roles in genome maintenance. Trends Cell Biol 13:493–501. doi:10.1016/S0962-8924(03)00171-5
Kitao S, Shimamoto A, Goto M, Miller RW, Smithson WA, Lindor NM, Furuichi Y (1999) Mutations in RECQL4 cause a subset of cases of Rothmund–Thomson syndrome. Nat Genet 22:82–84. doi:10.1038/8788
Krogh BO, Symington LS (2004) Recombination proteins in yeast. Annu Rev Genet 38:233–271. doi:10.1146/annurev.genet.38.072902.091500
Kusumoto R, Dawut L, Marchetti C, Wan Lee J, Vindigni A, Ramsden D, Bohr VA (2008) Werner protein cooperates with the XRCC4-DNA ligase IV complex in end-processing. Biochemistry 47:7548–7556. doi:10.1021/bi702325t
Laud PR, Multani AS, Bailey SM, Wu L, Ma J, Kingsley C, Lebel M, Pathak S, DePinho RA, Chang S (2005) Elevated telomere-telomere recombination in WRN-deficient, telomere dysfunctional cells promotes escape from senescence and engagement of the ALT pathway. Genes Dev 19:2560–2570. doi:10.1101/gad.1321305
Lee JW, Harrigan J, Opresko PL, Bohr VA (2005) Pathways and functions of the Werner syndrome protein. Mech Ageing Dev 126:79–86. doi:10.1016/j.mad.2004.09.011
Lee JY, Kozak M, Martin JD, Pennock E, Johnson FB (2007) Evidence that a RecQ helicase slows senescence by resolving recombining telomeres. PLoS Biol 5:e160. doi:10.1371/journal.pbio.0050160
Li X, Heyer WD (2008) Homologous recombination in DNA repair and DNA damage tolerance. Cell Res 18:99–113. doi:10.1038/cr.2008.1
Liang F, Han M, Romanienko PJ, Jasin M (1998) Homology-directed repair is a major double-strand break repair pathway in mammalian cells. Proc Natl Acad Sci USA 95:5172–5177. doi:10.1073/pnas.95.9.5172
Liberi G, Maffioletti G, Lucca C, Chiolo I, Baryshnikova A, Cotta-Ramusino C, Lopes M, Pellicioli A, Haber JE, Foiani M (2005) Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev 19:339–350. doi:10.1101/gad.322605
Lillard-Wetherell K, Machwe A, Langland GT, Combs KA, Behbehani GK, Schonberg SA, German J, Turchi JJ, Orren DK, Groden J (2004) Association and regulation of the BLM helicase by the telomere proteins TRF1 and TRF2. Hum Mol Genet 13:1919–1932. doi:10.1093/hmg/ddh193
Lindor NM, Furuichi Y, Kitao S, Shimamoto A, Arndt C, Jalal S (2000) Rothmund–Thomson syndrome due to RECQ4 helicase mutations: report and clinical and molecular comparisons with Bloom syndrome and Werner syndrome. Am J Med Genet 90:223–228. doi:10.1002/(SICI)1096-8628(20000131)90:3<223::AID-AJMG7>3.0.CO;2-Z
Lonn U, Lonn S, Nylen U, Winblad G, German J (1990) An abnormal profile of DNA replication intermediates in Bloom’s syndrome. Cancer Res 50:3141–3145
Luo G, Santoro IM, McDaniel LD, Nishijima I, Mills M, Youssoufian H, Vogel H, Schultz RA, Bradley A (2000) Cancer predisposition caused by elevated mitotic recombination in Bloom mice. Nat Genet 26:424–429. doi:10.1038/82548
Machwe A, Xiao L, Orren DK (2004) TRF2 recruits the Werner syndrome (WRN) exonuclease for processing of telomeric DNA. Oncogene 23:149–156. doi:10.1038/sj.onc.1206906
Machwe A, Xiao L, Lloyd RG, Bolt E, Orren DK (2007) Replication fork regression in vitro by the Werner syndrome protein (WRN): Holliday junction formation, the effect of leading arm structure and a potential role for WRN exonuclease activity. Nucleic Acids Res 35:5729–5747. doi:10.1093/nar/gkm561
Mann MB, Hodges CA, Barnes E, Vogel H, Hassold TJ, Luo G (2005) Defective sister-chromatid cohesion, aneuploidy and cancer predisposition in a mouse model of type II Rothmund–Thomson syndrome. Hum Mol Genet 14:813–825. doi:10.1093/hmg/ddi075
Matsuno K, Kumano M, Kubota Y, Hashimoto Y, Takisawa H (2006) The N-terminal noncatalytic region of Xenopus RecQ4 is required for chromatin binding of DNA polymerase alpha in the initiation of DNA replication. Mol Cell Biol 26:4843–4852. doi:10.1128/MCB.02267-05
Neumann AA, Reddel RR (2002) Telomere maintenance and cancer–look, no telomerase. Nat Rev Cancer 2:879–884. doi:10.1038/nrc929
Ogburn CE, Oshima J, Poot M, Chen R, Hunt KE, Gollahon KA, Rabinovitch PS, Martin GM (1997) An apoptosis-inducing genotoxin differentiates heterozygotic carriers for Werner helicase mutations from wild-type and homozygous mutants. Hum Genet 101:121–125. doi:10.1007/s004390050599
Okada M, Goto M, Furuichi Y, Sugimoto M (1998) Differential effects of cytotoxic drugs on mortal and immortalized B-lymphoblastoid cell lines from normal and Werner’s syndrome patients. Biol Pharm Bull 21:235–239
Opresko PL (2008) Telomere ResQue and preservation–roles for the Werner syndrome protein and other RecQ helicases. Mech Ageing Dev 129:79–90. doi:10.1016/j.mad.2007.10.007
Opresko PL, von Kobbe C, Laine JP, Harrigan J, Hickson ID, Bohr VA (2002) Telomere-binding protein TRF2 binds to and stimulates the Werner and Bloom syndrome helicases. J Biol Chem 277:41110–41119. doi:10.1074/jbc.M205396200
Opresko PL, Otterlei M, Graakjaer J, Bruheim P, Dawut L, Kolvraa S, May A, Seidman MM, Bohr VA (2004) The Werner syndrome helicase and exonuclease cooperate to resolve telomeric D loops in a manner regulated by TRF1 and TRF2. Mol Cell 14:763–774. doi:10.1016/j.molcel.2004.05.023
Opresko PL, Mason PA, Podell ER, Lei M, Hickson ID, Cech TR, Bohr VA (2005) POT1 stimulates RecQ helicases WRN and BLM to unwind telomeric DNA substrates. J Biol Chem 280:32069–32080. doi:10.1074/jbc.M505211200
Orstavik KH, McFadden N, Hagelsteen J, Ormerod E, van der Hagen CB (1994) Instability of lymphocyte chromosomes in a girl with Rothmund–Thomson syndrome. J Med Genet 31:570–572
Oshima J, Huang S, Pae C, Campisi J, Schiestl RH (2002) Lack of WRN results in extensive deletion at nonhomologous joining ends. Cancer Res 62:547–551
Otsuki M, Seki M, Inoue E, Abe T, Narita Y, Yoshimura A, Tada S, Ishii Y, Enomoto T (2008) Analyses of functional interaction between RECQL1, RECQL5, and BLM which physically interact with DNA topoisomerase IIIalpha. Biochim Biophys Acta 1782:75–81
Otterlei M, Bruheim P, Ahn B, Bussen W, Karmakar P, Baynton K, Bohr VA (2006) Werner syndrome protein participates in a complex with RAD51, RAD54, RAD54B and ATR in response to ICL-induced replication arrest. J Cell Sci 119:5137–5146. doi:10.1242/jcs.03291
Ouyang KJ, Woo LL, Ellis NA (2008) Homologous recombination and maintenance of genome integrity: cancer and aging through the prism of human RecQ helicases. Mech Ageing Dev 129:425–440. doi:10.1016/j.mad.2008.03.003
Park SJ, Lee YJ, Beck BD, Lee SH (2006) A positive involvement of RecQL4 in UV-induced S-phase arrest. DNA Cell Biol 25:696–703. doi:10.1089/dna.2006.25.696
Pedrazzi G, Bachrati CZ, Selak N, Studer I, Petkovic M, Hickson ID, Jiricny J, Stagljar I (2003) The Bloom’s syndrome helicase interacts directly with the human DNA mismatch repair protein hMSH6. Biol Chem 384:1155–1164. doi:10.1515/BC.2003.128
Petkovic M, Dietschy T, Freire R, Jiao R, Stagljar I (2005) The human Rothmund-Thomson syndrome gene product, RECQL4, localizes to distinct nuclear foci that coincide with proteins involved in the maintenance of genome stability. J Cell Sci 118:4261–4269. doi:10.1242/jcs.02556
Pichierri P, Rosselli F (2004) The DNA crosslink-induced S-phase checkpoint depends on ATR-CHK1 and ATR-NBS1-FANCD2 pathways. EMBO J 23:1178–1187. doi:10.1038/sj.emboj.7600113
Pichierri P, Franchitto A, Mosesso P, Palitti F (2000) Werner’s syndrome cell lines are hypersensitive to camptothecin-induced chromosomal damage. Mutat Res 456:45–57. doi:10.1016/S0027-5107(00)00109-3
Pichierri P, Franchitto A, Mosesso P, Palitti F (2001) Werner’s syndrome protein is required for correct recovery after replication arrest and DNA damage induced in S-phase of cell cycle. Mol Biol Cell 12:2412–2421
Pichierri P, Rosselli F, Franchitto A (2003) Werner’s syndrome protein is phosphorylated in an ATR/ATM-dependent manner following replication arrest and DNA damage induced during the S phase of the cell cycle. Oncogene 22:1491–1500. doi:10.1038/sj.onc.1206169
Poot M, Gollahon KA, Rabinovitch PS (1999) Werner syndrome lymphoblastoid cells are sensitive to camptothecin-induced apoptosis in S-phase. Hum Genet 104:10–14. doi:10.1007/s004390050903
Poot M, Yom JS, Whang SH, Kato JT, Gollahon KA, Rabinovitch PS (2001) Werner syndrome cells are sensitive to DNA cross-linking drugs. FASEB J 15:1224–1226
Poot M, Gollahon KA, Emond MJ, Silber JR, Rabinovitch PS (2002) Werner syndrome diploid fibroblasts are sensitive to 4-nitroquinoline-N-oxide and 8-methoxypsoralen: implications for the disease phenotype. FASEB J 16:757–758
Prince PR, Emond MJ, Monnat RJ Jr (2001) Loss of Werner syndrome protein function promotes aberrant mitotic recombination. Genes Dev 15:933–938. doi:10.1101/gad.877001
Ralf C, Hickson ID, Wu L (2006) The Bloom’s syndrome helicase can promote the regression of a model replication fork. J Biol Chem 281:22839–22846. doi:10.1074/jbc.M604268200
Raynard S, Bussen W, Sung P (2006) A double Holliday junction dissolvasome comprising BLM, topoisomerase IIIalpha, and BLAP75. J Biol Chem 281:13861–13864. doi:10.1074/jbc.C600051200
Rodriguez-Lopez AM, Jackson DA, Iborra F, Cox LS (2002) Asymmetry of DNA replication fork progression in Werner’s syndrome. Aging Cell 1:30–39. doi:10.1046/j.1474-9728.2002.00002.x
Rodriguez-Lopez AM, Whitby MC, Borer CM, Bachler MA, Cox LS (2007) Correction of proliferation and drug sensitivity defects in the progeroid Werner’s syndrome by Holliday junction resolution. Rejuvenation Res 10:27–40. doi:10.1089/rej.2006.0503
Saintigny Y, Makienko K, Swanson C, Emond MJ, Monnat RJ Jr (2002) Homologous recombination resolution defect in werner syndrome. Mol Cell Biol 22:6971–6978. doi:10.1128/MCB.22.20.6971-6978.2002
Salk D, Au K, Hoehn H, Martin GM (1985) Cytogenetic aspects of Werner syndrome. Adv Exp Med Biol 190:541–546
Sangrithi MN, Bernal JA, Madine M, Philpott A, Lee J, Dunphy WG, Venkitaraman AR (2005) Initiation of DNA replication requires the RECQL4 protein mutated in Rothmund–Thomson syndrome. Cell 121:887–898. doi:10.1016/j.cell.2005.05.015
Sekelsky JJ, Brodsky MH, Rubin GM, Hawley RS (1999) Drosophila and human RecQ5 exist in different isoforms generated by alternative splicing. Nucleic Acids Res 27:3762–3769. doi:10.1093/nar/27.18.3762
Sharma S, Brosh RM Jr (2007) Human RECQ1 is a DNA damage responsive protein required for genotoxic stress resistance and suppression of sister chromatid exchanges. PLOS One 2:e1297. doi:10.1371/journal.pone.0001297
Sharma S, Doherty KM, Brosh RM Jr (2006) Mechanisms of RecQ helicases in pathways of DNA metabolism and maintenance of genomic stability. Biochem J 398:319–337. doi:10.1042/BJ20060450
Shen JC, Loeb LA (2000) The Werner syndrome gene: the molecular basis of RecQ helicase-deficiency diseases. Trends Genet 16:213–220. doi:10.1016/S0168-9525(99)01970-8
Shen J, Loeb LA (2001) Unwinding the molecular basis of the Werner syndrome. Mech Ageing Dev 122:921–944. doi:10.1016/S0047-6374(01)00248-2
Shimamoto A, Nishikawa K, Kitao S, Furuichi Y (2000) Human RecQ5beta, a large isomer of RecQ5 DNA helicase, localizes in the nucleoplasm and interacts with topoisomerases 3alpha and 3beta. Nucleic Acids Res 28:1647–1655. doi:10.1093/nar/28.7.1647
Sidorova JM, Nianzhen L, Folch A, Monnat RJ (2008) The RecQ helicase WRN is required for normal replication fork progression after DNA damage or replication fork arrest. Cell Cycle 7:796–807
Siitonen HA, Kopra O, Kaariainen H, Haravuori H, Winter RM, Saamanen AM, Peltonen L, Kestila M (2003) Molecular defect of RAPADILINO syndrome expands the phenotype spectrum of RECQL diseases. Hum Mol Genet 12:2837–2844. doi:10.1093/hmg/ddg306
Sowd G, Lei M, Opresko PL (2008) Mechanism and substrate specificity of telomeric protein POT1 stimulation of the Werner syndrome helicase. Nucleic Acids Res 36:4242–4256. doi:10.1093/nar/gkn385
Stavropoulos DJ, Bradshaw PS, Li X, Pasic I, Truong K, Ikura M, Ungrin M, Meyn MS (2002) The Bloom syndrome helicase BLM interacts with TRF2 in ALT cells and promotes telomeric DNA synthesis. Hum Mol Genet 11:3135–3144. doi:10.1093/hmg/11.25.3135
Stinco G, Governatori G, Mattighello P, Patrone P (2008) Multiple cutaneous neoplasms in a patient with Rothmund–Thomson syndrome: case report and published work review. J Dermatol 35:154–161. doi:10.1111/j.1346-8138.2008.00436.x
Sung P, Krejci L, Van Komen S, Sehorn MG (2003) Rad51 recombinase and recombination mediators. J Biol Chem 278:42729–42732. doi:10.1074/jbc.R300027200
Takata M, Sasaki MS, Sonoda E, Morrison C, Hashimoto M, Utsumi H, Yamaguchi-Iwai Y, Shinohara A, Takeda S (1998) Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J 17:5497–5508. doi:10.1093/emboj/17.18.5497
Thompson LH, Schild D (2002) Recombinational DNA repair and human disease. Mutat Res 509:49–78. doi:10.1016/S0027-5107(02)00224-5
van Brabant AJ, Stan R, Ellis NA (2000) DNA helicases, genomic instability, and human genetic disease. Annu Rev Genomics Hum Genet 1:409–459. doi:10.1146/annurev.genom.1.1.409
Van Maldergem L, Siitonen HA, Jalkh N, Chouery E, De Roy M, Delague V, Muenke M, Jabs EW, Cai J, Wang LL, Plon SE, Fourneau C, Kestila M, Gillerot Y, Megarbane A, Verloes A (2006) Revisiting the craniosynostosis-radial ray hypoplasia association: Baller–Gerold syndrome caused by mutations in the RECQL4 gene. J Med Genet 43:148–152. doi:10.1136/jmg.2005.031781
Vennos EM, James WD (1995) Rothmund–Thomson syndrome. Dermatol Clin 13:143–150
Vennos EM, Collins M, James WD (1992) Rothmund–Thomson syndrome: review of the world literature. J Am Acad Dermatol 27:750–762. doi:10.1016/0190-9622(92)70249-F
Wang W, Seki M, Narita Y, Nakagawa T, Yoshimura A, Otsuki M, Kawabe Y, Tada S, Yagi H, Ishii Y, Enomoto T (2003) Functional relation among RecQ family helicases RecQL1, RecQL5, and BLM in cell growth and sister chromatid exchange formation. Mol Cell Biol 23:3527–3535. doi:10.1128/MCB.23.10.3527-3535.2003
Wang Y, Erdmann N, Giannone RJ, Wu J, Gomez M, Liu Y (2005) An increase in telomere sister chromatid exchange in murine embryonic stem cells possessing critically shortened telomeres. Proc Natl Acad Sci USA 102:10256–10260. doi:10.1073/pnas.0504635102
Werner SR, Prahalad AK, Yang J, Hock JM (2006) RECQL4-deficient cells are hypersensitive to oxidative stress/damage: insights for osteosarcoma prevalence and heterogeneity in Rothmund–Thomson syndrome. Biochem Biophys Res Commun 345:403–409. doi:10.1016/j.bbrc.2006.04.093
West SC (2003) Molecular views of recombination proteins and their control. Nat Rev Mol Cell Biol 4:435–445
Woo LL, Futami K, Shimamoto A, Furuichi Y, Frank KM (2006) The Rothmund–Thomson gene product RECQL4 localizes to the nucleolus in response to oxidative stress. Exp Cell Res 312:3443–3457. doi:10.1016/j.yexcr.2006.07.023
Wu L, Hickson ID (2003) The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature 426:870–874. doi:10.1038/nature02253
Wu L, Hickson ID (2006) DNA helicases required for homologous recombination and repair of damaged replication forks. Annu Rev Genet 40:279–306. doi:10.1146/annurev.genet.40.110405.090636
Wu L, Davies SL, Levitt NC, Hickson ID (2001) Potential role for the BLM helicase in recombinational repair via a conserved interaction with RAD51. J Biol Chem 276:19375–19381. doi:10.1074/jbc.M009471200
Wu L, Bachrati CZ, Ou J, Xu C, Yin J, Chang M, Wang W, Li L, Brown GW, Hickson ID (2006) BLAP75/RMI1 promotes the BLM-dependent dissolution of homologous recombination intermediates. Proc Natl Acad Sci USA 103:4068–4073. doi:10.1073/pnas.0508295103
Wyllie FS, Jones CJ, Skinner JW, Haughton MF, Wallis C, Wynford-Thomas D, Faragher RG, Kipling D (2000) Telomerase prevents the accelerated cell ageing of Werner syndrome fibroblasts. Nat Genet 24:16–17. doi:10.1038/71630
Yeager TR, Neumann AA, Englezou A, Huschtscha LI, Noble JR, Reddel RR (1999) Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res 59:4175–4179
Yin J, Kwon YT, Varshavsky A, Wang W (2004) RECQL4, mutated in the Rothmund–Thomson and RAPADILINO syndromes, interacts with ubiquitin ligases UBR1 and UBR2 of the N-end rule pathway. Hum Mol Genet 13:2421–2430. doi:10.1093/hmg/ddh269
Yin J, Sobeck A, Xu C, Meetei AR, Hoatlin M, Li L, Wang W (2005) BLAP75, an essential component of Bloom’s syndrome protein complexes that maintain genome integrity. EMBO J 24:1465–1476. doi:10.1038/sj.emboj.7600622
Zheng H, Wang X, Warren AJ, Legerski RJ, Nairn RS, Hamilton JW, Li L (2003) Nucleotide excision repair- and polymerase eta-mediated error-prone removal of mitomycin C interstrand cross-links. Mol Cell Biol 23:754–761. doi:10.1128/MCB.23.2.754-761.2003
Acknowledgments
We would like to thank Drs. Jian Lu and Avik K. Ghosh for critical reading of the manuscript. This work was in part supported by funds from the Intramural Program of the National Institute on Aging, NIH. This work was also in part supported by funds from the BK 21 Project in 2008 and KRF-2008-521-C00211 from KRF.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Singh, D.K., Ahn, B. & Bohr, V.A. Roles of RECQ helicases in recombination based DNA repair, genomic stability and aging. Biogerontology 10, 235–252 (2009). https://doi.org/10.1007/s10522-008-9205-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10522-008-9205-z