
Abstract
Atypical teratoid/rhabdoid tumor (AT/RT) is a highly malignant embryonal tumor most commonly occurring in young children, with the highest incidence in the first 2 years of life. The ratio of supratentorial to infratentorial localization of this tumor is reported to be 1.4:1. Supratentorial AT/RTs are located mostly in the cerebral hemispheres and affect more predominantly older children and adults, in contrast to AT/RTs occurring in the posterior fossa. AT/RTs of the pineal region are much rarer, and there have been several single case reports of both children and adults in the literature. There are no known symptoms or signs at presentation, or radiological features that are specific to AT/RTs of the pineal region, but a rapid progression of symptoms reflecting their aggressive biological behavior is common. The prognosis is dismal.
Histologically, AT/RTs are very cellular tumors and show marked regional heterogeneity, with primitive neuroectodermal tumor (PNET)-like, rhabdoid, epithelial, and mesenchymal components in variable proportions. Immunohistochemically, AT/RTs are polyphenotypic with constant expression of vimentin and variable expression of epithelial membrane antigen, smooth muscle actin, glial fibrillary acidic protein, cytokeratins, synaptophysin, and neurofilament protein. Loss of expression of INI1 is sensitive and specific marker for AT/RTs.
Histological differential diagnosis of AT/RTs of the pineal region includes so-called “malignant small blue cell tumors” (e.g., supratentorial PNETs, pineoblastoma), choroid plexus carcinoma, tumors with prominent rhabdoid features involving this region (e.g., rhabdoid meningioma, rhabdoid glioblastoma), and metastatic tumors. In addition to careful histological evaluation, a panel of immunohistochemical markers, including INI1, as well as detailed clinical information is crucial for this differential.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Glial Fibrillary Acidic Protein
- Epithelial Membrane Antigen
- Primitive Neuroectodermal Tumor
- Pineal Region
- Malignant Rhabdoid Tumor
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Atypical teratoid/rhabdoid tumor (AT/RT) is a rare, highly malignant central nervous system (CNS) neoplasm of unknown histogenesis with characteristic rapid progression and high potential for widespread dissemination throughout the CNS. It predominantly affects young children with the highest incidence in the first 2 years of life, while it rarely occurs in adults with less than 30 cases reported to date (Samaras et al. 2009; Takei et al. 2010). According to the current World Health Organization (WHO) 2007 classification of CNS tumors, this unique neoplasm constitutes one of three major embryonal tumor entities, all of which correspond to WHO grade IV (Judkins et al. 2007).
AT/RTs are immunophenotypically and genetically distinct from and are resistant to standard therapy for the other embryonal tumors; CNS primitive neuroectodermal tumors (PNETs) and medulloblastomas. Histologically, AT/RT is named for the presence of characteristic rhabdoid cells, which usually compose the tumor in part, with a variable combination of PNET-like, mesenchymal spindle-shaped and epithelial-type components. Although its unique similarity with the malignant rhabdoid tumor of the kidney is known, AT/RTs composed purely of rhabdoid cells are uncommon. Given the frequent overlapping histological features, the histological distinction from PNETs and medulloblastomas is sometimes very difficult, especially in small specimens. In keeping with their histologic diversity, they exhibit a broad spectrum of immunoreactivity (i.e., polyphenotypic), with frequent positivity for glial fibrillary acidic protein (GFAP), epithelial membrane antigen (EMA), cytokeratins, smooth muscle actin (SMA), and vimentin. The most distinctive and diagnostically almost pathognomonic feature of AT/RT is its association with the inactivation of the SMARCB1/hSNF5/INI1gene, located on chromosome 22q11.2, in the vast majority of cases. Thus, immunohistochemical study using an antibody against INI1 gene product or fluorescence in situ hybridization (FISH) to identify loss of the INI1 locus is the current routine work-up for diagnostic confirmation of AT/RT. Given its much worse prognosis, necessitating intensive therapy that differs markedly from the treatment of PNETs and medulloblastomas, this diagnostic confirmation is crucial.
In two large series of pediatric cases (a total of 73 cases) in the US, the ratio of supratentorial to infratentorial localization is 1.4:1 (Hilden et al. 2004; Tekautz et al. 2005). Supratentorial tumors are located mostly in the cerebral hemispheres and less frequently in the ventricular system and pineal region, and more predominantly affect older children as well as adults (Samaras et al. 2009). In contrast, posterior fossa, including cerebellum, cerebellopontine angle, and brainstem, is the most common primary tumor site among patients younger than 3 years.
The pineal gland is a small (5–8 mm) midline structure and embryologically develops during the second month of gestation as a diverticulum in the diencephalic roof of the third ventricle. It is located directly below the splenium of the corpus callosum and is flanked by the posterior and habenular commissures in the rostal portion of the midbrain. The “pineal region” includes a pineal gland, posterior wall of the third ventricle, tela choroidea, and velum interpositum. The primary pineal region tumors are derived from cells located in these anatomic structures, and have male and childhood predominance. The most common tumors are germ cell tumors and pineal parenchymal tumors. In older adults, metastatic tumors should be considered. It is known that AT/RTs can rarely arise in this region and that they are much rarer than those occurring in the cerebral hemispheres and posterior fossa. There have been several single case reports in the literature since the first case of “malignant rhabdoid tumor” of the pineal region was described in 1994 (Muller et al. 1994). In this chapter, we review those reported cases in the literature, including our case (Takei et al. 2010).
Clinical Features
Both pediatricas well as adult cases of AT/RT of the pineal region have been reported in the literature. However, of those reported, there are only three adult cases (Sugita et al. 1999; Ingold et al. 2006; Takei et al. 2010). No symptoms or signs at presentation specific to AT/RTs of the pineal region are known, but a rapid progression of symptoms reflecting their aggressive biological behavior is common. Tumors involving the pineal region that compress adjacent structures result in typical clinical symptoms, and one of the most common presentations includes headache, nausea, and vomiting, caused by aqueduct compression and resultant obstructive hydrocephalus, for which a shunt surgery should be considered. It is also commonly associated with bilateral papilloedema. Tumors compressing or directly infiltrating into the mesencephalic tectum, including the superior colliculus and adjacent oculomotor and Edinger-Westphal nuclei, result in dorsal midbrain syndrome (also known as Parinaud’s syndrome), characterized by upgaze paralysis and papillary and oculomotor nerve paresis.
Leptomeningeal dissemination is commonly seen in AT/RTs of the pineal region as is those occurring in other locations. Metastasis beyond the CNS from the pineal region has been reported to date in the following two patients. A hematogenous metastasis to lung, which was histologically diagnosed 18 months after the pineal tumor surgery, was described in a 27-year-old man (Sugita et al. 1999). A case of abdominal seeding along a ventriculoperitoneal shunt catheter, which was found at autopsy approximately 7 months after the initial diagnosis, was reported in a 45-year-old woman (Ingold et al. 2006).
The first-line treatment is surgery; however, deep localization and diffuse infiltration into the surrounding vital structures usually precludes total gross resection of this tumor. The prognosis of AT/RTs of the pineal region is very poor despite intensive adjuvant chemotherapy (and radiation therapy) after surgery. Adult patients appear to have longer survival than pediatric patients probably due to adjuvant aggressive chemoradiation therapy available in adults, which would not be tolerated by young children.
Radiological Features
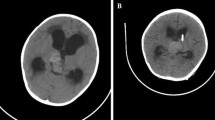
There are no specific imaging characteristics for intracranial AT/RTs (Parmar et al. 2006). No detailed magnetic resonance imaging (MRI) features of AT/RTs of the pineal region have been described in the literature. The MRI features of our case occurring in 33-year-old woman (Fig. 20.1a, b) are similar to those described in supratentorial AT/RTs, which are characterized by a large and lobulated lesion with heterogeneous signal intensity on T1 and T2-weighted images and heterogeneous enhancement, resulting from cystic change, necrosis, and/or hemorrhage (Cheng et al. 2005; Meyers et al. 2006; Parmar et al. 2006; Tez et al. 2008). Leptomeningeal disseminated foci can be detected.

MRI features, T1-weighted image with gadolinium contrast enhancement. (a) Sagittal section, (b) Transverse section. Heterogeneously enhancing, lobulated mass is present in the pineal region
Histopathological Features
No cytological or histological features unique to AT/RTs of the pineal region have been described. Cytomorphologically, the smears of AT/RTs stained with hematoxylin and eosin (H&E) are hypercellular with primitive-appearing neoplastic cells, which are characterized by a high nuclear/cytoplasmic (N/C) ratio, speckled chromatin, and small nucleoli, admixed with intermediate-sized, rhabdoid cells in varying proportions (Parwani et al. 2005). The rhabdoid cells are characterized by eccentrically placed nuclei, single prominent nucleoli, and granular-to-fibrillary, brightly eosinophilic cytoplasm with or without globoid “inclusions” (Fig. 20.2a). Apoptosis, mitosis and necrosis are commonly seen.

(a) Cytologic touch imprint: Rhabdoid cells with eccentrically placed nuclei and granular-to-fibrillary, brightly eosinophilic cytoplasm present (arrow) (H&E stain). Histological features (H&E stain): (b) A mixture of primitive neuroectodermal tumour (PNET)-like (malignant small blue cells), rhabdoid, epithelial-like components as well as cells with clear vacuolated cytoplasm. (c) Rhabdoid cells admixed with PNET-like cells. (d) Uncommon AT/RT composed exclusively of rhabdoid cells
Histologically, AT/RTs are very cellular tumors that show marked regional heterogeneity, with PNET-like, rhabdoid, epithelial, and mesenchymal components in variable proportions (Fig. 20.2b, c). There is often a fibrovascular stroma separating lobules and sheets of tumor cells. The cellular morphology varies from smaller PNET-like cells (so-called malignant small blue cells) indistinguishable from tumor cells of medulloblastomas/PNETs, characterized by hyperchromatic nuclei, scant cytoplasm, and a high N/C ratio, to large cells with eosinophilic, pale, or clear cytoplasm and large round to oval nuclei with or without prominent nucleoli. Tumor cells with clear cytoplasm are commonly seen. Characteristic rhabdoid cells with eccentric reniform nuclei, frequent prominent nucleoli, and eosinophilic discrete cytoplasm with or without intracytoplasmic hyaline globular inclusions, are usually present. Brisk mitotic activity and tumor necrosis is common. Spindle tumor cells having a sarcomatoid appearance are also seen, often as a minor component. In general, a PNET-like component is a more common feature than other components in AT/RTs (Ertan et al. 2009). AT/RTs composed exclusively of rhabdoid cells are uncommon (Fig. 20.2d). An unusual adult case of pineal AT/RT composed of rhabdoid tumor cells and chondroid formation without any other mesenchymal components was reported (Sugita et al. 1999). Given the pineal location, the distinction from very rare immature teratomas remains blurred in this particular case, although commonly used immunohistochemical markers for germ cell tumors were all negative.
Immunohistochemical Profile
The immunohistochemical profile of AT/RTs reflects their morphological heterogeneity. Immunoreactivity for a range of neuroectodermal markers, mesenchymal, and epithelial markers are known to be present, but of note is that the tumors are consistently negative for the germ cell markers. Vimentin is consistently expressed in this tumor, and labels hyaline globular intracytoplasmic inclusions of the rhabdoid cells. Expression of EMA and SMA is frequently observed. GFAP, cytokeratins, synaptophysin, and neurofilament protein are also known to be expressed, depending on the different cellular composition of this tumor (Fig. 20.3a–c). Loss of expression (absent nuclear staining in tumor cells) of INI1, the product of SMARCB1/hSNF5/INI1gene, is a sensitive and specific marker for AT/RTs (Fig. 20.3d). For the interpretation of INI1 immunohistochemistry results, given the negative nuclear staining, it is important to insure that normal components within the tumor such as endothelial cells preserve nuclear expression, as positive internal control. It has been recently reported that claudin-6, a key component of tight junctions, is expressed by most AT/RTs, but not by most other brain tumors, most importantly PNETs/medulloblastomas (Birks et al. 2010).

Immunohistochemical features: (a) Epithelial membrane antigen, (b) Glial fibrillary acidic protein, (c) Neurofilament protein, (d) INI1, The tumour nuclei are negative, while the internal controls (i.e., endothelial cells and inflammatory cells) are positive
In the diagnosis of AT/RTs, a panel of immunohistochemical markers, including vimentin, EMA, SMA, GFAP, cytokeratins, and synaptophysin, is likely to help confirm the diagnosis in the context of the appropriate morphological appearance. Claudin-6 may be a useful positive marker and can be included in the panel. INI1 immunostaining can be utilized in those cases having indeterminate histological features, or in small biopsies that may not be representative of the morphological heterogeneity typical of AT/RTs, since negative staining is intuitively not as desirable an end-result as compared with positive staining (Takei et al. 2007). The latter examples are very commonly encountered in pineal region tumors, in general, given the difficult location of surgical access.
Molecular Pathology
Most AT/RTs exhibit monosomy 22 or deletions of the chromosome 22q11 locus containing the SMARCB1/hSNF5/INI1gene, and less commonly show mutations of the SMARCB1/hSNF5/INI1 gene. This gene encodes a component of the SWI/SNF chromatin remodeling complex that has an important role in transcriptional regulation (Imbalzano and Jones 2005). Immunohistochemical staining with anti-INI1 antibody (i.e., nuclear immunonegativity) correlates with these molecular alterations.
Differential Diagnosis
The differential diagnosis of AT/RTs of the pineal region includes so-called “malignant small blue cell tumors”, choroid plexus carcinoma (CPC), tumors with prominent rhabdoid features involving this region, and metastatic tumors.
Malignant small blue cell tumors as a differential diagnosis include pineoblastomas and supratentorial PNETs involving the pineal region. Pineoblastoma is a highly malignant primitive embryonal tumour of the pineal gland, preferentially affecting young children, and is composed of primitive cells resembling PNETs/medulloblastomas. This tumor is distinct from PNETs in other locations in its photosensory differentiation, characterized by Flexner-Wintersteiner rosettes and fleurettes. Since AT/RTs usually contain PNET-like tumor cells as a major component, these tumors are considered to be the most important differential diagnoses, especially in children. In addition, this histological differential is particularly difficult or sometimes impossible in small biopsies, unless typical rosette formation is found. Rare pineoblastomas contain pigmented epithelium and mesenchymal components such as skeletal muscle and cartilage and are referred to as ‘pineal anlage tumors’ (Nakazato et al. 2007), which can further complicate the differential diagnosis. Immunohistochemically, pineoblastomas express not only neuronal, but also photosensory markers (e.g., retinal S-antigen), as do some PNETs/medulloblastomas (Janss et al. 1996; Kramm et al. 1991). No studies to test photosensory marker immunoreactivity on AT/RTs have been reported to date. As mentioned above, INI-1 immunohistochemical staining (i.e., nuclear immunonegativity) is of great help for this differential diagnosis.
CPCs arising in the posterior third ventricle can involve the pineal region. CPCs and AT/RTs are known to share numerous clinical, radiological, and pathological (i.e., histological and immunohistochemical) features. Prominence of rhabdoid tumor cells favors AT/RT over CPC. However, histopathologic distinction between these tumors using routine immunohistochemical panels is often very difficult or may be impossible, especially if the AT/RT shows predominant epithelial differentiation, or if the CPC exhibits minimal epithelial differentiation. In general, significant immunoreactivity for SMA and EMA is a feature of AT/RT, while transthyretin (prealbumin) labeling favors CPC, although the expression is variable in this malignant choroid plexus tumor. Given that INI1expression is preserved in the majority of CPCs and is lost in AT/RTs, this immunohistochemical marker is particularly important for this differential diagnosis.
Tumors with prominent rhabdoid features that can involve the pineal region include rhabdoid meningiomas and rhabdoid glioblastomas, although they are much less commonly encountered as a differential diagnosis. Given the age incidence, this differential diagnosis is probably more important in older children and adults. Histologically, it is very important to find a subpopulation of an otherwise classic meningioma or glioblastoma, respectively. Moreover, a PNET-like component is not a feature of these tumors. Immunohistochemically, glial and neuronal markers are usually negative in rhabdoid meningiomas. Of note is that cytogenetic evidence of monosomy 22 cannot exclude a rhabdoid meningioma, since this is the most common cytogenetic alteration in meningiomas. INI1 nuclear expression is preserved in reported cases of rhabdoid meningioma (Wakabayashi et al. 2005; Buccoliero et al. 2010). In addition, cases of rhabdoid glioblastoma with cytogenetical evidence of monosomy 22 have been reported (Wyatt-Ashmead et al. 2001; Kleinschmidt-DeMasters et al. 2010). Interestingly, it is recently reported that two cases of rhabdoid glioblastoma show focal loss of INI1 protein nuclear immunoexpression and no expression of claudin-6, in contrast to AT/RTs, and it is suggested that claudin-6 immunohistochemical staining may be a good discriminator of AT/RTs from rhabdoid glioblastomas (Kleinschmidt-DeMasters et al. 2010).
AT/RTs with predominant epithelioid/rhabdoid components can be mistaken as metastatic carcinoma or melanoma, especially in older adults. Carcinomas with rhabdoid features have been reported in many anatomic sites, including stomach (Ueyama et al. 1993) and lung (Shimazaki et al. 2001; Yilmazbayhan et al. 2005). Some pulmonary cases were reported to show neuroendocrine differentiation immunohistochemically (Shimazaki et al. 2001). Malignant melanomas are known to show plasmacytoid features, and rare cases of rhabdoid phenotype have been reported (Abbott et al. 2004; Gavino and Gillies 2007). Characteristic polyphenotypic immunohistochemical profile described above supports the diagnosis of AT/RT rather than metastatic tumors. Interestingly, complete loss of S-100 protein and/or HMB-45 expression in some cases of rhabdoid melanoma has been reported (Gavino and Gillies 2007), while a single case of AT/RT with few HMB-45 immunoreactive melanin-containing tumors cells in an 8-year-old girl was reported (Tekkok and Sav 2005). Although younger age favors AT/RT rather than metastasis, clinical and radiological information are crucial for this distinction.
References
Abbott JJ, Amirkhan RH, Hoang MP (2004) Malignant melanoma with a rhabdoid phenotype: histologic, immunohistochemical, and ultrastructural study of a case and review of the literature. Arch Pathol Lab Med 128:686–388
Birks DK, Kleinschmidt-DeMasters BK, Donson AM, Barton VN, McNatt SA, Foreman NK, Handler MH (2010) Claudin 6 is a positive marker for atypical teratoid/rhabdoid tumors. Brain Pathol 20:140–150
Buccoliero AM, Castiglione F, Degl’innocenti DR, Franchi A, Sanzo M, Cetica V, Giunti L, Sardi I, Mussa F, Giordano F, Genitori L, Taddei GL (2010) Pediatric rhabdoid meningioma: a morphological, immunohistochemical, ultrastructural and molecular case study. Neuropathology 31:59–65
Cheng YC, Lirng JF, Chang FC, Guo WY, Teng MM, Chang CY, Wong TT, Ho DM (2005) Neuroradiological findings in atypical teratoid/rhabdoid tumor of the central nervous system. Acta Radiol 46:89–96
Ertan Y, Sezak M, Turhan T, Kantar M, Ersahin Y, Mutluer S, Vergin C, Oniz H, Akalin T (2009) Atypical teratoid/rhabdoid tumor of the central nervous system: clinicopathologic and immunohistochemical features of four cases. Childs Nerv Syst 25:707–711
Gavino ACP, Gillies EM (2007) Metastatic rhabdoid melanoma: report of a case with a comparative review of the literature. J Cutan Pathol 35:337–342
Hilden JM, Meerbaum S, Burger P, Finlay J, Janss A, Scheithauer BW, Andrew WW, Rorke LB, Biegel JA (2004) Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry. J Clin Oncol 22:2877–2884
Imbalzano AN, Jones SN (2005) Snf5 tumor suppressor couples chromatin remodeling, checkpoint control, and chromosomal stability. Cancer Cell 7:294–295
Ingold B, Moschopulos M, Hutter G, Seeger H, Rothlisberger B, Landolt H, Yonekawa Y, Jochum W, Heppner FL (2006) Abdominal seeding of an atypical teratoid/rhabdoid tumor of the pineal gland along a ventriculoperitoneal shunt catheter. Acta Neuropathol 111:56–59
Janss AJ, Yachnis AT, Silber JH, Trojanowski JQ, Lee VM, Sutton LN, Perilongo G, Rorke LB, Phillips PC (1996) Glial differentiation predicts poor clinical outcome in primitive neuroectodermal brain tumors. Ann Neurol 39:481–489
Judkins AR, Eberhart CG, Wasseling P (2007) Atypical teratoid/rhabdoid tumour. In: Louis DN, Ohgaki H, Wiestler OD (eds) World Health Organization classification of tumours of the central nervous system. IARC Press, Lyon, pp 147–149
Kleinschmidt-DeMasters BK, Alassiri AH, Birks DK, Newell KL, Moore W, Lillehei KO (2010) Epithelioid versus rhabdoid glioblastomas are distinguished by monosomy 22 and immunohistochemical expression of INI-1 by not Claudin 6. Am J Surg Pathol 34:341–354
Kramm CM, Korf HW, Czerwionka M, Schachenmayr W, de Grip WJ (1991) Photoreceptor differentiation in cerebellar medulloblastoma: evidence for a functional photopigment and authentic S-antigen (arrestin). Acta Neuropathol 81:296–302
Meyers SP, Khademian ZP, Biegel JA, Chuang SH, Korones DN, Zimmerman RA (2006) Primary intracranial atypical teratoid/rhabdoid tumors of infancy and childhood: MRI features and patient outcomes. AJNR Am J Neuroradiol 27:962–971
Muller M, Hubbard SL, Provias J, Greenberg M, Becker LE, Rutka JT (1994) Malignant rhabdoid tumour of the pineal region. Can J Neurol Sci 21:273–277
Nakazato Y, Jouvet A, Scheithauer BW (2007) Pineoblastoma. In: Louis DN, Ohgaki H, Wiestler OD (eds) World Health Organization classification of tumours of the central nervous system. IARC Press, Lyon, pp 126–129
Parmar H, Hawkins C, Bouffet E, Rutka J, Shroff M (2006) Imaging findings in primary intracranial atypical teratoid/rhabdoid tumors. Pediatr Radiol 36:126–132
Parwani AV, Stelow EB, Pambuccian SE, Burger PC, Ali SZ (2005) Atypical teratoid/rhabdoid tumor of the brain. Cancer 105:65–70
Samaras V, Stamatelli A, Samaras E, Stergiou I, Konstantopoulou P, Varsos V, Judkins AR, Biegel JA, Barbatis C (2009) Atypical teratoid/rhabdoid tumor of the central nervous system in an 18-year-old patient. Clin Neuropathol 28:1–10
Shimazaki H, Aida S, Sato M, Deguchi H, Ozeki Y, Tamai S (2001) Lung carcinoma with rhabdoid cells: a clinicopathological study and survival analysis of 14 cases. Histopathology 38:425–434
Sugita Y, Takahashi Y, Hayashi I, Morimatsu M, Okamoto K, Shigemori M (1999) Pineal malignant rhabdoid tumor with chondroid formation in an adult. Pathol Int 49:1114–1118
Takei H, Bhattacharjee MB, Rivera A, Dancer Y, Powell SZ (2007) New immunohistochemical markers in the evaluation of central nervous system tumors: a review of 7 selected adult and pediatric brain tumors. Arch Pathol Lab Med 131:234–241
Takei H, Adesina AM, Mehta V, Powell SZ, Langford LA (2010) Atypical teratoid/rhabdoid tumor of the pineal region in an adult. J Neurosurg 113:374–379
Tekautz TM, Fuller CE, Blaney S, Fouladi M, Broniscer A, Merchant TE, Krasin M, Dalton J, Hale G, Kun LE, Wallace D, Gilbertson RJ, Gajjar A (2005) Atypical teratoid/rhabdoid tumors (ATRT): improved survival in children 3 years of age and older with radiation therapy and high-dose alkylator-based chemotherapy. J Clin Oncol 23:1491–1499
Tekkok IH, Sav A (2005) Primary malignant rhabdoid tumor of the central nervous system- a comprehensive review. J Neuro-Oncol 73:241–252
Tez S, Koktener A, Guler G, Ozisik P (2008) Atypical teratoid/rhabdoid tumors: imaging findings of two cases and review of the literature. Turk Neurosurg 18:30–34
Ueyama T, Nagai E, Yao T, Tsuneyoshi M (1993) Vimentin-positive gastric carcinomas with rhabdoid features. A clinicopathologic and immunohistochemical study. Am J Surg Pathol 17:813–819
Wakabayashi K, Suzuki N, Mori F, Kamada M, Hatanaka M (2005) Rhabdoid cystic papillary meningioma with diffuse subarachnoid dissemination. Acta Neuropathol 110:196–198
Wyatt-Ashmead J, Kleinschmidt-DeMasters BK, Hill DA, Mierau GW, McGavran L, Thompson SJ, Foreman NK (2001) Rhabdoid glioblastoma. Clin Neuropathol 20:248–255
Yilmazbayhan D, Ates LE, Dilege S, Gulluoglu M, Tanju S, Kalayci G (2005) Pulmonary large cell carcinoma with rhabdoid features. Ann Diagn Pathol 9:223–226
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Takei, H., Langford, L.A. (2013). Atypical Teratoid/Rhabdoid Tumor of the Pineal Region: Diagnosis. In: Hayat, M. (eds) Pediatric Cancer, Volume 4. Pediatric Cancer, vol 4. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-6591-7_20
Download citation
DOI: https://doi.org/10.1007/978-94-007-6591-7_20
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-6590-0
Online ISBN: 978-94-007-6591-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)