
Abstract
Within the global carbon (C) cycle, there is still much debate as to the magnitude, location and turnover of the terrestrial C sinks (and sources). One of the major keys to closing this knowledge gap is that globally, the amount of C entering oceans maybe only ca. 33 % of the total C transported from terrestrial ecosystems to inland surface waters. Streams, lakes, rivers and transitional waters are areas for the active transformation and recycling of terrestrially-derived C indirectly back to the atmosphere (estimated range of 25–44 %). Understanding processes that control soil C losses to and its fate in surface waters is not only important in establishing accuracy of C fluxes, feedbacks and tradeoffs but also providing evidence to limit terrestrial ecosystem C contributions to atmospheric carbon dioxide (CO2).
The relationship between inland surface waters and C cycling are controlled by biogeochemical, physical and hydrometeorological metrics that integrate both lateral (soil to water) and longitudinal (along the riverine continuum) processes during C transport in its different forms, i.e., particulate, dissolved and gaseous C species. This chapter outlines processes affecting compositional “quality” of C within surface waters and in-stream physico-chemical and biotic mechanisms that are instrumental to understanding losses of C via the soil-surface water-atmosphere pathway.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Carbon
- Dissolved organic carbon
- Particulate organic carbon
- Organic matter
- Inland surface waters
- Carbon dioxide outgassing
- In-stream processes
- Biogeochemical quality
- Aquatic carbon cycle
9.1 Introduction
The global carbon (C) cycle is comprised of three main compartments: land (soil, vegetation and geological), ocean and atmospheric pools (Janzen 2004; Smith et al. 2008), each with their relative stocks of C that are constantly fluctuating. It is a complex system to understand and accurately estimate its C cycling processes, and there has been considerable disagreement over C stocks and the movement of C (i.e., C fluxes) between pools for disparate ecosystems (Dixon and Turner 1991; Malhi 2002). For example, total soil C content varies spatially within the same mapped soil unit and C stock estimates within a given area can vary depending on the scale of soil mapping (Dawson and Smith 2007). The myriad of spatio-temporal variations (e.g., soil characteristics, gaseous emissions, seasonality) combined with diverse land management strategies complicate derivation of C stocks, fluxes and processes in terrestrial (soil and vegetation) ecosystems (Chadwick et al. 1994; Cao et al. 2002). Furthermore, soil organic C stocks will be sensitive to changes in climate, but the magnitude and timescale of their response as well as potential feedbacks are not fully comprehended (Schmidt et al. 2011). Some discrepancies between literature estimates of the global C stocks and fluxes occur due to the different extrapolation approaches utilised. Limitation of sufficient representative data is one main source of uncertainty in estimates but further understanding of mechanisms and processes that underpin C cycling from microbial to global scales is also required to reduce the uncertainty that persists (Smith et al. 1993; Janzen 2004; Le Quéré et al. 2009).
It is important to note that the global C cycle does not operate independently but is linked to the nitrogen (N), phosphorus (P) and sulphur (S) cycles by biological processes such as mineralization and immobilisation (Richey 1983; Stevenson 1986). The ‘Anthropocene’ (Crutzen 2002) has been proposed as a name for the present epoch (commencing with the industrial revolution, ca 1750) in which consumption of fossil fuels releasing C, N and S to the atmosphere, together with N, P and ammonia fertilizer production and land-use changes (e.g., cultivation, clearing and harvesting of forests) have resulted in significant perturbations to these natural biogeochemical cycles (Bolin et al. 1983; Malhi 2002; Janzen 2004; Raupach and Canadell 2010). These perturbations have impacted climate, biodiversity and land cover resulting in multiple pressures on ecosystems that supply fundamental services for human well-being (Dawson and Smith 2010). One of the most striking examples of this ecosystem disequilibrium is the changing composition of the atmosphere as a result of anthropogenic consumption that has generated increasing amounts of greenhouse gases (GHGs, e.g., carbon dioxide [CO2] as well as methane [CH4] and nitrous oxide [N2O]) as end products (Janzen 2004).
The distribution of C within the land-atmosphere–ocean system for the past 10,000 years (Holocene), prior to the Anthropocene, had remained relatively stable. Atmospheric CO2 concentrations, determined from ice-cores, ranged between 260 and 280 ppmv (Barnola et al. 1987; Indermühle et al. 1999) and CH4 concentrations at 650–700 ppbv (Chappellaz et al. 1990). Between 1850 and 1950, the average annual C flux from fossil fuel combustion was 0.6 Pg C year−1 (1 Pg = 1015 g; Stuiver 1978). This flux had increased to 5.4 Pg C year−1 by 1990 (Tans et al. 1990; Dixon et al. 1994); the average emission of CO2 from fossil fuel combustion and cement manufacture for the 1990s was estimated at 6.3–6.5 Pg C year−1 (IGBP 1998; Schimel et al. 2001; Houghton 2003; Janzen 2004; Post et al. 2004). Since the turn of the century, rapid growth in these CO2 emissions has continued, reaching 9.1 ± 0.5 Pg C year−1 by 2010 (Peters et al. 2012). Changes in human land management strategies have also caused a net release of C to the atmosphere (Woodwell 1978; Houghton et al. 1983). Until ca. 1960 this was higher than the C emitted by fossil fuel combustion (Houghton et al. 1983; Malhi et al. 2002). By 1980 land-use changes emitted between 0.4 and 4.7 Pg C year−1 (Buringh 1984; Schlesinger 1984; Tans et al. 1990) and average annual emissions in the 1990s were between 1.7 and 2.2 Pg C year−1 (Houghton 2003; Janzen 2004). Net deforestation alone has been estimated to release 0.9–1.6 Pg C year−1 (IGBP 1998; Houghton 2000; Schlesinger and Andrews 2000; Raupach and Canadell 2010).
Although, the emission of CO2 by fossil fuel combustion and land-use changes since 1750 have been partly balanced by oceanic and terrestrial sinks (about 50–60 % of anthropogenic CO2 releases, Scholes and Noble 2001; Sabine et al. 2004; Le Quéré et al. 2009; Raupach and Canadell 2010), there was still a net increase of CO2 to the atmosphere of 3.2–3.4 Pg C year−1 during the 1990s (IGBP 1998; Schimel et al. 2001; Janzen 2004) resulting in atmospheric CO2 concentrations of 371 ppmv by 2001 (Malhi 2002; Janzen 2004; Post et al. 2004) and CH4 concentrations of 2,000 ppbv (Fowler et al. 1995). Recent estimates indicate that up to 5 Pg C year−1 maybe accumulating in the atmosphere (Le Quéré et al. 2009; Friedlingstein et al. 2010). Monthly mean CO2 concentrations, supplied by the US National Oceanic and Atmospheric Administration from the Mauna Loa Observatory in Hawaii, by April 2012 have reached 396 ppmv (National Oceanic and Atmospheric Administration 2012).
There is still much debate as to the magnitude, location and turnover of the terrestrial C sinks (and sources) (Dixon et al. 1994; Keeling et al. 1996; Houghton 2003; Aufdenkampe et al. 2011), particularly with changing climate and land use. As soil and vegetation combined (ca. 75 % of the terrestrial C stock occurs as organic matter [OM] in soils) contain three times as much C than in the atmosphere pool, small increases in net C losses from the terrestrial biosphere to the atmosphere could have significant impacts on atmospheric CO2 concentrations. Hence, the response of soils, particularly organic-rich soils (e.g., peatlands), to climate and land management future scenarios is crucial when assessing climate-C cycle feedbacks (Smith et al. 2008; Evans and Warburton 2010). The more C that can be retained resiliently via reduced turnover rates within the global terrestrial sink moderates that amount exported to the atmosphere as GHG. To achieve this, requires accurate assessment of terrestrial fluxes and processes. This has led to numerous studies with regards to determining net ecosystem exchange (NEE) and net ecosystem production (NEE incorporating lateral export, Lovett et al. 2006; Aufdenkampe et al. 2011) across different soil and vegetation classes. Others studied sensitivity to decomposition of soil organic matter (SOM) pools and land use/management strategies that can reduce losses of GHG from land to the atmosphere (e.g. Cao and Woodward 1998; Cannell 2003; Janssens et al. 2003; Janzen 2004; Dawson and Smith 2007; Smith et al. 2008; Le Quéré et al. 2009; Schulze et al. 2010).
However, to balance the C budget of anthropogenic emissions, land and ocean sinks and the measured atmospheric pool, it was proposed that the losses of C via inland surface waters equated to the amount of missing C adsorbed from the atmosphere but not sequestered in the terrestrial biosphere on land (Siemens 2003). Moreover, in contrast to the NEE dynamic equilibrium between CO2 inputs to land as gross primary production (GPP) and vegetation and soil biomass respiration losses of CO2 (Cao and Woodward 1998), C losses to surface waters are – redeposition on floodplains aside – uni-directional and are now considered integral to improving estimates of C budgets within terrestrial ecosystems from landscape to global scales (Billett et al. 2004, 2010; Cole et al. 2007; Dawson and Smith 2007; Battin et al. 2009; Aufdenkampe et al. 2011). Key to this is that the amount of C entering oceans is only a part of the total C transported from terrestrial ecosystems to surface waters. The remainder is cycled within the riverine continuum and returned back to the atmosphere as CO2 or buried in sediments as OM (Aufdenkampe et al. 2011).
Recent observations of increasing dissolved organic carbon (DOC) concentrations (Worrall et al. 2004; Evans et al. 2006a, b; Monteith et al. 2007; de Wit and Wright 2008) and increased erosion contributing particulate organic carbon (POC) (Evans et al. 2006a, b; Evans and Warburton 2010) within stream waters of upland (mainly organic-rich soils) ecosystems suggest that SOM stocks may be vulnerable and will potentially contribute to positive climate forcing. However, this assumes that the soil-derived OM entering surface waters is converted to CO2 with subsequent evasion from the water column to the atmosphere (Hope et al. 2001; Billett and Garnett 2010). Conversely, non-respired OM transported from the land to oceans, with burial and incorporation in marine sediments would mean that losses of SOM would not contribute to CO2 concentrations in the atmosphere. Therefore, understanding processes that control soil C losses to and its fate in surface waters is not only important in establishing accuracy of terrestrial and aquatic C fluxes, feedbacks and tradeoffs but also providing evidence for local, national and global land management and policy strategies that limit terrestrial ecosystem C contributions to atmospheric CO2.
9.2 The Soil-Surface Water-Atmosphere Pathway
9.2.1 The Role of Surface Waters in the Carbon Cycle
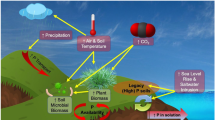
Within the global C cycle, inland surface waters (streams, lakes, rivers and transitional waters) act as a conduit for the transport of C from the land to the ocean (Fig. 9.1). The total global riverine organic C flux has been estimated at ca. 0.40 Pg C year−1 (Degens et al. 1991; Schlunz and Schneider 2000), of which particulate transport constitutes between 0.07 and 0.20 Pg C year−1 (Ittekkot and Laane 1991; Hedges et al. 1997).

Simplified global carbon cycle highlighting the uni-directional flux and aquatic cycling of carbon from the land to either atmosphere or ocean pools. See text for discussion of fluxes
Global inorganic C fluxes from rivers are of an equal magnitude to organic fluxes and have been estimated at between 0.26 and 0.53 Pg C year−1 (Meybeck 1993; Hope et al. 1994; Smith et al. 2008). Although these river fluxes are two orders of magnitude smaller than annual gross C fluxes between the atmosphere and land (ca. 120 Pg C year−1) and atmosphere and oceanic (ca. 90 Pg C year−1) pools of the global cycle (Janzen 2004; Smith et al. 2008), the global riverine flux of organic C to the oceans is comparable to the annual C sequestration in soil (0.4 Pg C year−1, Schlesinger 1990; Hope et al. 1994; Roulet and Moore 2006), suggesting that terrestrially-derived aquatic losses of organic C may contribute to regulating changes in SOC storage (Hope et al. 1997; Cole and Caraco 2001; Billett et al. 2006).
In surface waters, C is transported in particulate (non-solubilised), dissolved and gaseous forms (Dawson et al. 2002). OM covers a continuous size spectrum of compounds ranging from free monomers such as amino acids, carbohydrates and fatty acids, fulvic and humic acids, to macromolecules such as proteins and colloids, to aggregates and large particles including bacteria (Thurman 1985). POC and particulate inorganic carbon (PIC) are retained on a filter with pore sizes mainly between 0.45 and 0.7 μm. The upper limit is operationally defined by the sampling device used and is usually about a few mm (Ittekkot and Laane 1991). DOC is associated with the filtrate that passes through the filter paper (Thurman 1985; Hope et al. 1997a). Dissolved inorganic carbon (DIC) is also contained in the filtrate and in surface waters comprises H2CO3, HCO3 − and CO3 2−. These anions are associated with gaseous free CO2 via the carbonate equilibria. Their relative proportions dependent on pH and, to a lesser extent, on temperature (Stumm and Morgan 1981; Dawson et al. 1995). Although CH4 concentrations in surface waters are usually much lower than CO2, it is another important C-containing GHG associated with areas of anoxia within catchments (De Angelis and Scranton 1993; Jones and Mulholland 1998).
Continual C inputs to surface waters occur via tributaries, bank seepage and the hyporheic zone as headwaters – streams – rivers progress to estuaries, coastal environments and the deep ocean. However, surface waters are not inert channels but key hotspots for active transformation and recycling of C. Aquatic C has ecological significance as an energy source for both autotrophic and heterotrophic biota (Cole et al. 2007; Battin et al. 2008, 2009; Aufdenkampe et al. 2011). Inorganic and organic C, as a constituent of dissolved organic matter (DOM), influences the pH and ionic balance within surface waters (i.e., via organic acids and the carbonate equilibria). Both DOM and particulate organic matter (POM) are also involved in complexation mechanisms that affect solubility, transport and availability of ions, nutrients, heavy metals and organic pollutants (Hope et al. 1994; Dawson et al. 2002). Moreover, the quantity and quality of DOM can have economic consequences associated with treatment costs to remove colour for potable waters (Worrall et al. 2004; Dawson et al. 2009a).
9.2.2 Connectivity Between Soils and Surface Waters
In order to determine C losses to surface waters, most studies utilise the catchment as a functional unit. The catchment concept provides a natural environment that “links the atmosphere, plants, soils, groundwater, and streams through the convergence and interaction of material and energy flows” in an integrated manner (Lohse et al. 2009). Catchments provide a platform for maximising research within and between an infinite variety of different terrestrial systems (e.g., by area, characteristics or management), where hypotheses can be quantitatively assessed (Dawson and Smith 2010).
The climate, geology and land use/management of catchments and their interaction mainly determine soil type and vegetative communities and hence the terrestrial C stock. Topography, temperature, precipitation, hydrology and nutrients affect vegetative and soil-forming processes, such as primary production, respiration and biologically-mediated decomposition (Hope et al. 1994; Yoo et al. 2006), influencing the amount of potentially exportable C from the terrestrial pool. Decomposition and mineralization of litter and/or SOM converts primary production products to smaller organic components and inorganic forms, respectively, releasing C and nutrients for uptake by soil biota (plants, faunal and microbial communities). Factors, e.g., temperature, soil texture and moisture, and plant residue composition, that regulate OM stabilisation and decomposition rates in soils have been discussed extensively in the literature (e.g., Davidson and Janssens 2006; Von Lützow et al. 2006; Smith et al. 2008; Lohse et al. 2009; Schmidt et al. 2011). SOM decomposition differs considerably amongst terrestrial systems and results in losses of soil C as (i) mainly CO2 and CH4 to the atmosphere as part of NEE; (ii) particulate C and DOC by erosion processes and (iii) gaseous, dissolved and particulate C to surface waters (Dawson and Smith 2007).
Spatial and temporal (e.g., diurnal, seasonal) variations in concentrations and the type (particulate, dissolved or gaseous) of C exported in surface waters are determined by hydrological processes interacting with the biogeochemistry of the surrounding terrestrial environment (Hornberger et al. 1994; Lohse et al. 2009; Dawson et al. 2011). Within stream processes can constitute a secondary control on these inputs (Dawson et al. 2001a). Areas of SOM erosion, decomposition products within soil interstitial pore waters (e.g., temperature-related biological decomposition of available OM and solubility of DOC and gaseous components) and groundwater inputs contribute to C available for export from soils to surface waters (Dawson and Smith 2007). Connectivity of soils to the drainage network - controlled by soil distribution (Billett and Cresser 1992) and their hydrological properties (e.g., hydrology of soil types; Boorman et al. 1995) combined with hydrometeorological characteristics (e.g., antecedent conditions, rainfall-runoff ratios, mean transit times, discharge variability) are paramount in determining the extent and timing of C inputs (Brooks et al. 1999; Dawson et al. 2008, 2009b, 2011). However, the linkage between soil and surface waters is temporally dynamic and vulnerable to disturbance (Fraser et al. 2001; Billett et al. 2006).
9.2.3 Inputs of Particulate Carbon to Surface Waters
POC in surface waters originates from allochthonous (terrestrially-derived) sources such as soils and fragmented plant material (branches, cones, leaves, needles, twigs). It is also present in surface waters as microbial and algal biomass (biofilms) and from aggregation of DOM (Sollins et al. 1985; Ittekkot and Laane 1991; Hope et al. 1994). POC is less common, dependent on the nature of the underlying parent material within catchments, originating in areas containing carbonate minerals, such as limestone and dolomite (Meybeck 1982).
The main mechanism of POC inputs to surface waters is via erosion of topsoil aggregates containing SOM that are spatially close to receiving waters. Inputs of POC, as a variable component of suspended sediment (Dawson et al. 2012), tend to be episodic in nature. The majority of POC is transported via overland flow following intense rainfall events (and snowmelt), when runoff is greater than the infiltration capacity of the soil (Walling and Webb 1985, 1987; Reynolds 1986; Walling et al. 2002; Evans and Brazier 2005). In addition, sub-surface soil pipes can contribute substantially to POC content, particularly in peatlands that have been artificially drained (Warburton et al. 2004; Holden 2006). Bare soils, lacking in stability, are more liable to erosion than their equivalent soils that are naturally vegetated; e.g., under vegetation on arable land or have undergone peatland restoration. For riverbank soils, higher discharge and increased stream size enhances erosional processes, through weathering, fluid entrainment, “preparatory” bank weakening and eventual collapse of the bank (Lawler et al. 1997; Couper and Maddock 2001).
Soil erosion and production of suspended sediment is highly variable (e.g., Stott and Mount 2004; Dawson and Smith 2007). Altitude, slope and soil type have been correlated with extent of erosion. However, inadequate consideration of “best practice” protocols for land management influence losses such as tillage down slope by mechanised agriculture, burning and drainage of organic C-rich moorland, over-recreation, over-stocking and poaching by cattle are important contributors to erosional processes (McHugh et al. 2002; Dawson and Smith 2007). Temporally, the extent of erosion within a given area can also change markedly. Within the same locality, different stages of the forestry cycle comprising undisturbed peatland, ploughing and ditching, mature forests to harvesting have produced substantial changes to suspended sediment yields in adjacent surface waters (Stott and Mount 2004). The transport of eroded SOM also removes essential nutrients and minerals, which can affect the ability of the soil to support ecosystem functions (Neff and Asner 2001). Moreover, the mobilisation of soil to surface waters has other disadvantages as suspended sediment reduces light penetration, impacts on macro-invertebrate and fish communities, increases biochemical oxygen demand and contributes to diffuse pollution by controlling export of sediment-associated nutrients, pathogens and contaminants, such as trace metals and pesticides (Kronvang et al. 1997; Lawler et al. 2006; Collins and Anthony 2008).
9.2.4 Inputs of Dissolved Carbon to Surface Waters
DOC in surface waters originates from vegetation (leaching, rhizodeposition) and soil (leaching, OM decomposition and erosion) (Hope et al. 1994; Meyer et al. 1998). In-stream, processing of POM contributes to DOC concentrations (Dawson et al. 2004, 2012). DIC is derived from dissolution of carbonates and weathering of silicates in soils and bedrock. Speciation of DIC is also dependent on the carbonate equilibria (including its interaction with CO2 dynamics) within ground and soil water pools (Hope et al. 2004). Soil interstitial pore water is modified as it moves preferentially along flow paths through organic and mineral soil horizons to ground or surface waters (Grieve 1990; Hinton et al. 1998; Hagedorn et al. 2000). In upland peatland streams, isotopic studies have suggested that soil-derived DOC was commonly of recent (decadal) origin. However, ground water DOC was much older, ca. 8,500 years and associated with underlying mineral layers (Schiff et al. 1997; Palmer et al. 2001; Tipping et al. 2010). In larger rivers, a terrestrially-derived combination of young and old DOC and mainly old POC (>1,000 year) was noted. It has been suggested that the younger, more labile DOC has undergone preferential mineralization resulting in a residual older OM component (Raymond and Bauer 2001).
Many studies in temperate and boreal catchments, but not all (e.g., Tao 1998; Dawson et al. 2008), indicate positive relationships between surface water concentrations of DOC (or POC) and hydrological discharge (Dawson and Smith 2007). However, seasonality and catchment-specific hydrological patterns (e.g., antecedent moisture levels, hysteresis related to rising and falling events on the hydrograph and OM source exhaustion following consecutive intense rainfall events) can affect DOC and POC concentrations for a particular discharge, increasing variation of individual concentration-discharge relationships (Walling 1977). Seasonal splitting of data can sometimes improve this relationship for DOC (Dawson et al. 2002, 2008, 2011). Surface water concentrations of DIC tend to show an inverse relationship with discharge as the contributions of ground water are reduced and the influence of sub-surface water derived from the upper organic soil horizons that contain less DIC increases at higher discharge (Hill and Neal 1997; Neal et al. 1997; Dawson et al. 2002, 2004). Understanding these relationships between discharge and concentrations is important for improving assessments of the total C lost from soils to surface waters.
9.2.5 Inputs of Gaseous Carbon to Surface Waters
Gaseous CO2 in surface waters is derived from the terrestrial environment, in-stream mineralization of OM and by exchange at the atmosphere/water interface (Dawson et al. 2009b). Microbial communities also convert OM to CH4 but only in areas of anoxia such as those in peat soils, riparian zones and bed sediments (Dahm et al. 1991; Jones and Mulholland 1998; Hope et al. 2004). In soils, gaseous components dissolve in soil water and are either trapped within soil pores, lost by diffusion to the soil surface, or hydrologically transported to ground or surface waters. The soil porosity and moisture levels control the proportion of gaseous C that is exported to surface waters (Skiba and Cresser 1991; Dawson et al. 2001a; Hope et al. 2004). In well-aerated, freely draining mineral soils, the rate of gaseous C efflux as CO2 to the atmosphere from the soil surface is dominant, and gaseous losses to the surface waters are a minor component. However, in typically saturated organic C-rich soils, gaseous C effluxes to the surface are reduced as CO2 and CH4 dissolves in soil pore waters more readily from where it is then transported laterally to surface waters increasing their losses via hydrological pathways (Clymo and Pearce 1995; Fowler et al. 1995; Billett et al. 2004).
9.2.6 Spatial Variability of Carbon Inputs to Surface Waters
As streams increase in size to rivers, C inputs from the surrounding terrestrial ecosystem become less significant as autochthonous (derived from up-stream contiguous inputs and in-stream processes) C content becomes more dominant. Anthropogenic inputs are also important in areas with a higher population density and pollution (Hope et al. 1994).
Studies indicate strong correlations between C in surface waters and soil type, e.g., % area of peat (Hope et al. 1997; Dawson et al. 2011) or the soil C pool and SOM C:N ratios (Aitkenhead et al. 1999; Billett et al. 2006; Aitkenhead-Peterson et al. 2007).
However, these often occur in smaller scale (<5 km2) catchments where soil-stream linkage is strongest (Aitkenhead et al. 1999; Dawson et al. 2001a; Hope et al. 2004; Billett et al. 2006). Figure 9.2 highlights an example of how changes in stream water DOC and CO2 concentrations link to spatial changes in the soil C pool along a headwater stream. Initially, at the source, shallow, immature soils (Leptosols) with little C content only supply small amounts of DOC or CO2 to the adjacent drainage network. As the stream flows through deep peat (>5 m in depth, Billett et al. 2006), increased DOC and CO2 are supplied directly to the drainage channel. These inputs then decrease in the lower part of the catchment as the stream flows through more freely drained organo-mineral soils. At this stage, the relationship between organic C in the soil and stream becomes weaker as complex interactions occur, e.g., continual inputs of detrital DOC from upstream, lower DOC and CO2 inputs from freely-drained minerals soils, continual outgassing of CO2 to the atmosphere and in-stream processing of DOC (Dawson et al. 2001a; Billett et al. 2006). This inter-relationship between allochthonous and autochthonous inputs and processes, affecting both organic and inorganic C concentrations and fluxes in surface waters, occurs continuously downstream to oceans with an increasing influence of autochthonous inputs as terrestrial connectivity with the surface water is often reduced in larger river systems (Minshall et al. 1985; Meyer and Edwards 1990; McTammany et al. 2003; Dawson et al. 2004, 2009b, 2011; Webster 2007).

Downstream spatial variability in DOC (▲) and CO2-C (◊) concentrations along the main stem of a head water peatland stream, indicating connectivity with soil type (■ = Leptosol ■ = Histosol (Peat) ■ = Histic Podzol) (Data obtained on 14/07/97, Adapted from Dawson et al. (2001a) and Billett et al. (2006)) (Color figure online)
9.2.7 In-Stream Processes Affecting Carbon Concentrations and Speciation
Many processes modify both organic and inorganic C concentrations within surface waters prior to entering the oceanic sinks (Fig. 9.3). It has been estimated that of the total amount of organic C entering global rivers, 50 % is transported to oceans, 25 % is oxidised and 25 % stored as sedimentary OM within impoundments, lakes, floodplains, and other wetlands (Hope et al. 1994). Recently, it has been suggested that ca. 33 % of global terrestrial C inputs to surface waters reach the oceanic sink and a minimum of 44 % enters the atmosphere as gaseous C at a rate of 1.2 Pg year−1 (range of 0.75–1.4 Pg year−1) with the remaining 22 % retained in sediments (Battin et al. 2009; Aufdenkampe et al. 2011). However, these rough estimates have an inherent uncertainty due to the substantial spatio-temporal variability within and between ecosystems.

Simplified diagram highlighting typical physical and biogeochemical processes affecting terrestrial inputs and in-stream carbon dynamics along an upland to lowland riverine continuum. The terrestrial input arrows give an indication of the relative magnitude of contribution for each carbon determinant; GPP Gross Primary Production, CR Community Respiration. Outputs of particulate organic carbon (POC), dissolved organic carbon (DOC), dissolved inorganic carbon (DIC) and gaseous components (e.g. CO2) to estuarine/ocean (and atmospheric) pools are dependent on the overall spatio-temporal dynamics within individual catchments
In organic C-rich uplands, POC is largely derived from low density eroded peat. Once this material enters a stream channel it is largely transported as washload which can be transported at very low discharge and is flushed downstream, only deposited once the flow weakens. Larger blocks of peat may be temporarily stored on the channel bed but these are soon broken down by in-stream microbial processes and rapid abrasion (Evans and Warburton 2001). OM deposited on the channel bed is re-suspended when discharge rises above a certain threshold. Sediment OM is liable to re-suspension (as POC) in an alternating sequence of deposition (with potential subsequent fates of burial or further decomposition) and re-suspension (Thurman 1985; Cushing et al. 1993; Evans and Warburton 2001, 2005) along the riverine continuum.
Whole stream metabolism of OM, related to the amount and type of biological activity within the stream ecosystem, exerts a significant control on C concentrations and is important for understanding differential speciation and fate of C within surface waters. Rates of in-stream GPP, community respiration (benthic and water column) and net ecosystem production rates are known to vary diurnally, seasonally and between different aquatic environments (Battin et al. 2008; Staehr et al. 2012). Increased input of nutrients in bioavailable forms to stream ecosystems may also stimulate primary productivity, increasing autotrophic organic biomass production such as phytoplankton and macrophytes, with concomitant increases in community respiration (Peterson et al. 1985; Neal et al. 2006; Stutter et al. 2008; Dawson et al. 2012). Physical aspects of the stream also play a role in determining OM processing (Fellows et al. 2001). This includes hydraulic parameters, such as water transient storage, hyporheic flow paths, pooling and flow characteristics, which can affect biotic establishment and stability, thus, influencing in-stream heterotrophic and autotrophic processes (Battin et al. 2008; Dawson et al. 2009b).
In detrital-based upland stream ecosystems and along organic C-rich rivers, heterotrophic decomposition of DOM often dominates (respiration > photosynthesis) acting as a source of CO2 (Meyer and Edwards 1990; Marzolf et al. 1994; Mulholland et al. 1997; Dawson et al. 2001b, 2004). This is due to large inputs of respiratory substrate (OM) from either upstream or adjacent terrestrial sources that constitute a major energy source for aquatic communities. Biota that utilise OM are likely to be acclimatised to specific DOC sources found within particular ecosystems (Kuserk et al. 1984). POM deposited as sediments can be broken down physically by macro-invertebrates and microbes to finer particles. Collectors and filter feeders (e.g., Diptera, Simulidae), which are known to process large amount of POC, break down these finer particles contributing DOC to the water column (Vannote et al. 1980; Meyer and Tate 1983; Malmqvist et al. 2001; Monaghan et al. 2001). Strong relationships between filter feeders and bioavailable-P within POM confirm the importance and reactivity of this substrate (Stutter et al. 2007). DOC inputs can be removed from the water column either by biotic or abiotic processes (Hope et al. 1994). Adsorption of DOC as well as via aggregation to form POC is usually controlled by its physical chemistry at the solid/liquid interface. OM can be adsorbed to inorganic particulates, undergo deposition and be retained on the stream bed by biofilm communities that act as major transformers of DOC in headwater streams (McDowell 1985; Fiebig and Lock 1991; Staehr et al. 2012). Dissolved humic substances (a component of the DOC pool), although often considered relatively recalcitrant, are also an important biodegradable source of C (Volk et al. 1997). In addition, there is evidence that photolytic degradation of DOM to more readily assimilated compounds occurs stimulating ecosystem productivity under suitable conditions, e.g., in lakes (Lindell et al. 1995; Granéli et al. 1996; De Lange et al. 2003). Particulate OM has been less studied as a source of C to the atmosphere compared to DOM as it is transported towards sedimentary pools in the estuarine and oceanic environments where it constitutes a major substrate for heterotrophic metabolism (Ittekkot and Larne 1991; Tappin et al. 2003; Cole et al. 2007). Consequently, “far less is known about potential transformations of POM, which govern POC fate and interaction within surface waters” (Dawson et al. 2012). The major OM microbial transformation processes associated with in-stream POM require initial colonisation on particulate material, respiration (either decomposition to DOC or mineralization to CO2) and nutrient recycling with formation of new biomass (Grossart and Ploug 2000; Zimmermann-Timm 2002; Battin et al. 2008).
Dissolved CO2 content in surface waters is determined as a concentration (mg L−1) or partial pressure (pCO2) in the gas phase. The ‘excess partial pressure of CO2’ (epCO2) has been defined as ‘the ratio of the pCO2 in solution to the atmospheric pCO2 value’ (Neal 1988; Dawson et al. 2009b). Water in equilibrium with the atmosphere at current values has a partial pressure of dissolved CO2 (pCO2) of ca. 390 ppmv. A recent collation of estimates has stated that nearly all fresh waters contain CO2 in concentrations that are supersaturated with respect to that of the atmosphere (epCO2 values > 1). Measured pCO2 values range from 1,000 to 12,000 ppmv in rivers and from 350 to 10,000 ppmv in lakes and reservoirs (Aufdenkampe et al. 2011). This supersaturated state is due to the allochthonous inputs of CO2 from ground and soil pore waters, and because aquatic community respiration rates from OM mineralization exceed photosynthetic uptake (Cole and Caraco 2001; Dawson et al. 2001b, 2009b; Jones et al. 2003; Griffiths et al. 2007; Billett and Moore 2008; Teodoru et al. 2009).
The loss of supersaturated CO2 in streams as it equilibrates with the atmospheric CO2 concentration is termed the “outgassing effect” (Skiba and Cresser 1991). Like CO2, CH4 can also be supersaturated in streams (Jones and Mulholland 1998), where it undergoes oxidation to CO2 and outgassing once it enters surface waters (Hope et al. 2001). Historically, losses of CO2 via this soil-surface water-atmosphere pathway have tended to be unaccounted for in most inland surface water C flux estimations and ecosystem C budgets but it is now considered an important indirect transfer mechanism to the atmosphere from the terrestrial C pool (Kling et al. 1991; Cole et al. 1994; Cole and Caraco 2001; Richey et al. 2002). Considerable differences exist between studies that have investigated evasion rates of CO2 and CH4 from streams, rivers, lakes and wetland ponds due to spatio-temporal differences in catchment soils and their moisture levels affecting mineralization rates and hydrological flow paths e.g., lateral transport, as well as in-stream processes (Dawson and Smith 2007). Figure 9.2 illustrates how CO2 concentrations in a headwater stream can vary spatially within the same network (and hence outgassing flux) along short distances in relation to changes in source areas (e.g., peat). Once the elevated levels of CO2 have been lost from the stream by outgassing, further inputs are reduced as the stream enters areas adjacent to more freely drained peaty podzols. Often, significant outgassing ‘hotspots’ occur along a riverine continuum, such as organic C-rich soil pools, where soil C losses to the atmosphere are reduced and the soil-stream linkage is strong (Dawson et al. 2001a, 2004; Hope et al. 2001). In-stream physico-chemical factors controlling CO2 losses to the atmosphere include temperature-dependent solubility, volume, depth, velocity, gradient, turbulence and wind speed, as well as the initial concentration of CO2 in the stream water (Neal 1988; Rebsdorf et al. 1991; Hope et al. 2001; Dawson et al. 2001a, 2009b).
Hope et al. (2001) showed that gaseous C evasion fluxes from a peatland stream to the atmosphere were similar to fluxes of the total C (particulate, dissolved and gaseous) transported within the stream itself. Other studies have indicated its importance as part of an overall total C balance (sink/source status) in landscapes (Billet et al. 2004; Dinsmore et al. 2009, 2010). Moreover, processes influencing CO2 in surface waters, along with other chemical inputs, alter pH that determine speciation/calcite precipitation of the ground water and terrestrially-derived DIC (i.e. HCO3 −/CO3 2−) via the carbonate equilibria (Stumm and Morgan 1981). Isotopic studies of outgassed CO2 from carbonate free waters suggest that a “small, rapidly cycling pool (2–5 year) of organic C is responsible for the large C fluxes from land to water to atmosphere”. However, some systems show greater contributions from ground water sources which can increase the mean CO2 age to several decades (Mayorga et al. 2005).
Even in highly heterotrophic streams and rivers, autotrophic processes continue, but their dominance is restricted to occasional periods of the day or year when conditions for photosynthetic processes are optimised. During these periods when epCO2 is <1, influx of CO2 from the atmosphere can occur (Rosenfeld and Roff 1991; Dawson et al. 2001b, 2009b; Battin et al. 2008). Further downstream from organic C-rich headwaters, heterotrophic communities can potentially utilise C derived from “upstream processing inefficiencies” and autotrophic communities utilise CO2 derived from continual ground and soil pore-water inputs as well as in-stream OM mineralization and solubility dependent gas exchange with the atmosphere (Vannote et al. 1980; Griffiths et al. 2007; Dawson et al. 2009b). There is a direct correlation between in-stream primary production and stream order (Minshall et al. 1985). The deposition of C increases as streams and rivers meander and hydraulic energy is dispersed to levels suitable for increased sedimentation. These physical changes that lead to increases in sedimentation, encourages colonisation and establishment of benthic autotrophic communities that sequester CO2 from the water column. Along with the high input of allochthonous respiratory C substrate in organic C-rich upland systems, this produces a U-shaped curve of energy between contiguous headwaters, tributaries and main rivers ecosystems (Fiebig and Lock 1991; Webster 2007; Dawson et al. 2009b).
However, the dynamic equilibrium between efflux and sequestration of CO2 within surface waters is spatially dependent as the dynamics of physical (outgassing/gas exchange) and biogeochemical processes vary (Dawson et al. 2009b). Moreover, different processes also dominate depending on the time-scale of the analysis (Hanson et al. 2006). For example, (i) atmospheric deposition, climate and land use changes influence DOC dynamics in soil and stream waters at different spatio-temporal scales which make it difficult to discern the overriding control on widespread long-term trends of DOC in surface water (Clark et al. 2010) and, (ii) photosynthetic consumption of CO2 is light dependent but in-stream community respiration and CO2 terrestrial inputs continue throughout periods of darkness and diurnal variations in concentrations of CO2 concentrations can be significant (Guasch et al. 1998; Dawson et al. 2001b; Neal et al. 2004; Griffiths et al. 2007). Maximum diurnal amplitude of CO2 concentrations tend to occur during summer when an increased prevalence of lower discharges and higher temperatures encourages biotic activity (Dawson et al. 2001b; Griffiths et al. 2007; Battin et al. 2008).
9.2.8 Biogeochemical Quality of Organic Matter
In the past, the majority of studies on organic C distribution within surface waters have concentrated on quantifying concentrations or fluxes and discerning processes that influence the main organic components, i.e., DOC and POC, and production of GHG. However, the biogeochemical composition and degradability of OM will affect its transformation dynamics and hence distribution within surface waters. This has implications for selective processing of OM and hence its fate within the aquatic environment (McCallister et al. 2006; Dawson et al. 2012).
Moreover, as OM mineralization and decomposition rates can depend on the physico-chemical characteristics of the OM itself, soil management and land use practices that affect spatio-temporal variability of the quantity and type of OM entering watercourses will influence whether terrestrially-derived C eventually resides in atmospheric or oceanic pools. Therefore, there is a need to understand SOM quality inputs and in-stream processes that control the proportions and kinetics of aquatic (POC and/or DOC) and benthic sediment OM transformations to GHG across a diverse range of environmental scenarios. Furthermore, nutrient enrichment from e.g., agricultural diffuse such as N and P pollutants (Stutter et al. 2008), influences the “biogeochemical environment” of aquatic biotic activity (both photosynthetic and respiratory processes) changing nutrient and C flows and ecological stoichiometry within food webs (Cross et al. 2005; Penton and Newman 2007; Manzoni and Poporato 2011).
Different functional forms of aquatic OM influence biological and physico-chemical processes, such as its biological decomposition potential e.g., labile to recalcitrant compounds; ecosystem energy, hydrophobicity, pH buffering and photochemical fading (Baker et al. 2008). The degradability of OM may also be limited by the availability of other elements such as N and P (Taylor and Townsend 2010). Stoichiometric ratios (C:N:P) of OM are generally a simple yet robust indicator of OM quality (Elser et al. 2000; Cross et al. 2003).
In order to monitor and assess the extreme spatio-temporal variability of OM quality, standardised biogeochemical techniques and assays are required that can be performed relatively easily. These assays should also relate to functional properties of OM that impact on their fate within the aquatic environment. In terms of DOC, The relationship between ultra-violet (UV) absorbance and DOC concentrations has been used to infer changes in the proportion of hydrophobic (aromatic, recalcitrant) C and hence potential biodegradability of DOC (Marschner and Kalbitz 2003; Chen et al. 2002; Weisharr et al. 2003; Dawson et al. 2009a). Ågren et al. (2008) have suggested that the bioavailability of the DOC maybe also be related to molecular weight (indicted by Abs254/Abs365 ratios), encompassing both aliphatic and aromatic components. More complex characterisation of DOC can be achieved by fluorescence spectroscopy, which include determination of humic-like, fulvic-like, tryptophan or tyrosine (protein) like groups, chlorophyll as well as linking to functional characteristics of DOC (Baker and Spencer 2004; Baker et al. 2008). This technique has also been utilised to assess biodegradability of stream water DOC (Fellman et al. 2009). Furthermore, the use of Fourier Transform Infrared (FT-IR) spectroscopy has been utilised to assess chemical functional groups of humic substances (Lumsdon and Fraser 2005).
The inherent biodegradability of OM derived from surface waters and benthic sediment can be assessed by an integrated measurement of its respiration kinetics. Benthic sediment samples can be collected relatively easily and measured under defined conditions. However, respiration rates have also been measured from suspended sediments collected on filter papers using the MicroRespTM technique (Dawson et al. 2012). The principal of the MicroRespTM system is that a colour change forms in an indicator gel during incubation due to the CO2 evolved from microbial-induced substrate decomposition (Campbell et al. 2003). Other measurements of “biogeochemical reactivity” of OM include stoichiometric (C, N, P) assessments of both DOM and POM as well as chlorophyll-α concentrations (Sobczak et al. 2002; Neal et al. 2006) that assess autotrophic OM contributions (autochthonous apportionment, Dawson et al. 2012). The amount of respired CO2 produced as a proportion of the total C associated with POM on the suspended sediment and POM autotrophic activity could be related to contributory land use pressures as well as the biogeochemical water environment (Dawson et al. 2012).
9.3 Conclusion
The understanding and quantification of in-stream processes, including in-stream cycling of C derived from non-terrestrial sources, provides increased certainty of the proportions of terrestrially-derived aquatic OM transformed to GHG and that is eventually exported from streams, lakes and rivers to estuarine and marine environments.
The relationships between inland surface waters and C cycling can be dependent on the integration of many different processes that requires understanding of both lateral (soil to water) and longitudinal (along the riverine continuum) transport of C. This also requires an integrated understanding and assessment that incorporates particulate, dissolved and gaseous C species. These processes need to be explored to evaluate the ultimate fate of terrestrially-derived C in surface waters. Many surface waters are dominated by in-stream biological systems which may ultimately control the decomposition of complex natural OM. This is where an integrative quantitative assessment of these processes that link the compositional ‘quality’ of OM forms to biological accessibility for respiration under ‘realistic’ scenarios is required. There are, however, interconnected physical processes of stream morphology, deposition and re-suspension of particulates, photo-oxidation of DOM and outgassing of gaseous C. Hydrometeorological metrics affect connectivity of soil sources controlling the amount and nature of the material initially reaching the surface waters and the stability and hence processing capacity of in-stream biological communities. Moreover, climate change as well as progressive anthropogenic influences along the river continuum, such as bank disturbance, nutrient/pollutant inputs and water abstraction also influences OM degradation rates both in the water column and benthic sediments.
It must be noted that OM losses to inland surface waters are an important component of its natural ecosystem functioning. In-stream C provides energy for processing of other terrestrially-derived materials, e.g., processing and reduction of nitrate relies on metabolically available DOC. However, perturbation of biogeochemical cycles has meant that although C performs this important role, consequently being respired and lost to the atmosphere, it is appropriate to devise strategies that also reduce excessive point/diffuse pollutant terrestrial inputs to surface waters, resulting in a more balanced equilibrium between cycles.
Therefore, to reduce GHG production via the soil-stream-atmosphere pathway requires reduction in the first stage of this process, i.e., unnecessary losses of C (and nutrients) to waters, via soil erosion and hydrologically-mediated transport of SOM decomposition products. Strategies increasing SOM stabilization and reducing the rate of decomposition and mineralization to DOC and CO2, respectively, are instrumental to this hydrological export. Therefore, land use and best management practices that mitigate against C losses from terrestrial environments (both directly to the atmosphere and via surface waters) are important as to which strategies have the ability to store the most C.
Abbreviations
- C:
-
Carbon
- CH4 :
-
Methane
- CO2 :
-
Carbon Dioxide
- DIC:
-
Dissolved Inorganic Carbon
- DOC:
-
Dissolved Organic Carbon
- DOM:
-
Dissolved Organic Matter
- FT-IR:
-
Fourier Transform Infrared
- GHG:
-
Greenhouse Gas
- GPP:
-
Gross Primary Production
- OM:
-
Organic Matter
- N2O:
-
Nitrous Oxide
- NEE:
-
Net Ecosystem Exchange
- PIC:
-
Particulate Inorganic Carbon
- POC:
-
Particulate Organic Carbon
- POM:
-
Particulate Organic Matter
- SOC:
-
Soil Organic Carbon
- SOM:
-
Soil Organic Matter
- UV:
-
Ultra-Violet
References
Ågren A, Berggren M, Laudon H, Jansson M (2008) Terrestrial export of highly bioavailable carbon from small boreal catchments in spring floods. Freshw Biol 53:964–972
Aitkenhead JA, Hope D, Billett MF (1999) The relationship between dissolved organic carbon in streamwater and soil organic carbon pools at different spatial scales. Hydrol Proc 13:1289–1302
Aitkenhead-Peterson JA, Smart RP, Aitkenhead MJ, Cresser MS, McDowell WH (2007) Spatial and temporal variation of dissolved organic carbon export from gauged and ungauged watersheds of Dee Valley, Scotland: effect of land cover and C:N. Water Resour Res 43:W05442
Aufdenkampe AK, Mayorga E, Raymond PA, Melack JM, Doney SC, Alin SR, Aalto RE, Yoo K (2011) Riverine coupling of biogeochemical cycles between land, oceans, and atmosphere. Front Ecol Environ 9:53–60
Baker A, Spencer RGM (2004) Characterization of dissolved organic matter from source to sea using fluorescence and absorbance spectroscopy. Sci Total Environ 333:217–232
Baker A, Tipping E, Thacker SA, Gondar D (2008) Relating dissolved organic matter fluorescence and functional properties. Chemosphere 73:1765–1772
Barnola JM, Raynaud D, Korotkevich YS, Lorius C (1987) Vostok ice core provides 160,000 year record of atmospheric CO2. Nature 329:408–414
Battin TJ, Kaplan LA, Findlay S, Hopkinson CS, Marti E, Packman AI, Newbold JD, Sabater F (2008) Biophysical controls on organic carbon fluxes in fluvial networks. Nat Geosci 1:95–100
Battin TJ, Luyssaert S, Kaplan LA, Aufdenkampe AK, Richter A, Tranvik LJ (2009) The boundless carbon cycle. Nat Geosci 2:598–600
Billett MF, Cresser MS (1992) Predicting stream-water quality using catchment and soil chemical characteristics. Environ Pollut 77:263–268
Billett MF, Garnett MH (2010) Isotopic composition of carbon dioxide lost by evasion from surface water to the atmosphere: methodological comparison of a direct and indirect approach. Limnol Oceanogr Method 8:45–53
Billett MF, Moore TR (2008) Supersaturation and evasion of CO2 and CH4 in surface waters at Mer Bleue peatland, Canada. Hydrol Proc 22:2044–2054
Billett MF, Palmer SM, Hope D, Deacon C, Storeton-West R, Hargreaves KJ, Flechard C, Fowler D (2004) Linking land-atmosphere-stream carbon fluxes in a lowland peatland system. Glob Biogeochem Cyc 18:GB1024
Billett MF, Deacon CM Palmer SM, Dawson JJC, Hope D (2006) Connecting organic carbon in streamwater and soils in a peatland catchment. J Geophys Res Biosci 111:G02010
Billett MF, Charman DJ, Clark JM, Evans CD, Evans MG, Ostle NJ, Worrall F, Burden A, Dinsmore KJ, Jones T, McNamara NP, Parry L, Rowson JG, Rose R (2010) Carbon balance of UK peatlands: current state of knowledge and future research challenges. Clim Res 45(Sp Iss 24):13–29
Bolin B, Crutzen PJ, Vitousek PM, Woodmansee RG, Goldberg ED, Cook RB (1983) Interactions of biogeochemical cycles. In: Bolin B, Cook RB (eds) The major biogeochemical cycles and their interactions – SCOPE 21. Wiley, Chichester, pp 1–39
Boorman DB, Hollis JM, Lilly A (1995) Hydrology of soil types: a hydrological classification of the soils of the United Kingdom. Institute of Hydrology Report 126, Institute of Hydrology, Wallingford
Brooks PD, McKnight DM, Bencala KE (1999) The relationship between soil heterotrophic activity, soil dissolved organic carbon (DOC) leachate, and catchment-scale DOC export in headwater catchments. Water Resour Res 35:1895–1902
Buringh P (1984) Organic carbon in soils of the world. In: Woodwell GM (ed) The role of terrestrial vegetation in the global carbon cycle: measurement by remote sensing – SCOPE 23. Wiley, Chichester, pp 91–109
Campbell CD, Chapman SJ, Cameron CM, Davidson MS, Potts JM (2003) A rapid microtiter plate method to measure carbon dioxide evolved from carbon amendments so as to determine the physiological profiles of soil microbial communities by using whole soil. Appl Environ Microbiol 69:3593–3599
Cannell MGR (2003) Carbon sequestration and biomass energy offset: theoretical, potential and achievable capacities globally, in Europe and the UK. Biomass Bioenergy 24:97–116
Cao M, Woodward FI (1998) Dynamic responses of terrestrial ecosystem carbon cycling to global climate change. Nature 393:249–252
Cao M, Prince SD, Shugart HH (2002) Increasing terrestrial carbon uptake from the 1980s to the 1990s with changes in climate and atmospheric CO2. Glob Biogeochem Cycle 16:1069
Chadwick OA, Kelly EF, Merritts DM, Amundson RG (1994) Carbon dioxide consumption during soil development. Biogeochemistry 24:115–127
Chappellaz J, Barnola JM, Raynaud D, Korotkevich YS, Lorius C (1990) Ice-core record of atmospheric methane over the past 160,000 years. Nature 345:127–131
Chen J, Gu B, LeBoeuf EJ, Pan H, Dai S (2002) Spectroscopic characterization of the structure and functional properties of natural organic matter fractions. Chemosphere 48:59–68
Clark JM, Bottrell SH, Evans CD, Monteith DT, Bartlett R, Rose R, Newton RJ, Chapman PJ (2010) The importance of the relationship between scale and process in understanding long-term DOC dynamics. Sci Total Environ 408:2768–2775
Clymo RS, Pearce DME (1995) Methane and carbon dioxide production in, transport through and efflux from a peatland. Philos Trans R Soc Lond (A) 350:249–259
Cole JJ, Caraco NF (2001) Carbon in catchments: connecting terrestrial carbon losses with aquatic metabolism. Mar Freshw Res 52:101–110
Cole JJ, Caraco NF, Kling GW, Kratz TK (1994) Carbon dioxide supersaturation in the surface waters of lakes. Science 265:1568–1570
Cole JJ, Prairie YT, Caraco NF, McDowell WH, Tranvik LJ, Striegl RG, Duarte CM, Kortelainen P, Downing JA, Middelburg JJ, Melack J (2007) Plumbing the global carbon cycle: integrating inland waters into the terrestrial carbon budget. Ecosystem 10:171–184
Collins AL, Anthony SG (2008) Predicting sediment inputs to aquatic ecosystems across England and Wales under current environmental conditions. Appl Geogr 28:281–294
Couper PR, Maddock IP (2001) Subaerial river bank erosion processes and their interaction with other bank erosion mechanisms on the River Arrow, Warwickshire, UK. Earth Surf Proc Land 26:631–646
Cross WF, Benstead JP, Rosemond AD, Wallace JB (2003) Consumer-resource stoichiometry in detritus-based streams. Ecol Lett 6:721–732
Cross WF, Benstead JP, Frost PC, Thomas SA (2005) Ecological stoichiometry in freshwater benthic systems: recent progress and perspectives. Freshw Biol 50:1895–1912
Crutzen PJ (2002) The “anthropocene”. J de Physique Iv 12:1–5
Cushing CE, Minshall GW, Newbold JD (1993) Transport dynamics of fine particulate organic matter in two Idaho streams. Limnol Oceanogr 38:1101–1115
Dahm CN, Carr DL, Coleman RL (1991) Anaerobic carbon cycling in a stream ecosystem. Verh Internat Verein Limnol 24:1600–1604
Davidson EA, Janssens IA (2006) Temperature sensitivity of soil carbon decomposition and feedbacks to climate change. Nature 440:165–173
Dawson JJC, Smith P (2007) Carbon losses from soil and its consequences for land-use management. Sci Total Environ 382:165–190
Dawson JJC, Smith P (2010) Integrative management to mitigate diffuse pollution in multi-functional landscapes. Curr Opin Environ Sustain 2:375–382
Dawson JJC, Hope D, Cresser MS, Billett MF (1995) Downstream changes in free CO2 in an upland catchment in northeastern Scotland. J Environ Qual 24:699–706
Dawson JJC, Bakewell C, Billett MF (2001a) Is within stream processing an important control on spatial changes in headwater carbon fluxes? Sci Total Environ 265:153–167
Dawson JJC, Billett MF, Hope D (2001b) Diurnal variations in the carbon chemistry of two acidic peatland streams in north-east Scotland. Freshw Biol 46:1309–1322
Dawson JJC, Billett MF, Neal C, Hill S (2002) A comparison of particulate, dissolved and gaseous carbon in two contrasting upland streams in the UK. J Hydrol 257:226–246
Dawson JJC, Billett MF, Hope D, Palmer SM, Deacon CM (2004) Sources and sinks of aquatic carbon in a peatland stream continuum. Biogeochemistry 70:71–92
Dawson JJC, Soulsby C, Tetzlaff D, Hrachowitz M, Dunn SM, Malcolm IA (2008) Influence of hydrology and seasonality on DOC exports from three contrasting upland catchments. Biogeochemistry 90:93–113
Dawson JJC, Malcolm IA, Middlemas S, Tetzlaff D, Soulsby C (2009a) Is the composition of dissolved organic carbon changing in upland acidic streams? Environ Sci Technol 43:7748–7753
Dawson JJC, Soulsby C, Hrachowitz M, Speed M, Tetzlaff D (2009b) Seasonality of epCO2 atdifferent scales along an integrated river continuum within the Dee basin, NE Scotland. Hydrol Proc 23:2929–2942
Dawson JJC, Tetzlaff D, Speed M, Hrachowitz M, Soulsby C (2011) Seasonal controls on DOC dynamics in nested upland catchments in NE Scotland. Hydrol Proc 25:1647–1658
Dawson JJC, Adhikari YR, Soulsby C, Stutter MI (2012) The biogeochemical reactivity of suspended particulate matter at nested sites in the Dee basin, NE Scotland. Sci Total Environ 434:159–170
De Angelis MA, Scranton MI (1993) Fate of methane in the Hudson River and estuary. Glob Biogeochem Cycle 7:509–523
De Lange HJ, Morris DP, Williamson CE (2003) Solar ultraviolet photodegradation of DOC may stimulate freshwater food webs. J Plankton Res 25:111–117
De Wit HA, Wright RF (2008) Projected stream water fluxes of NO3 and total organic carbon from the Storgama headwater catchment, Norway, under climate change and reduced acid deposition. Ambio 37:56–63
Degens ET, Kempe S, Richey JE (1991) Biogeochemistry of major world rivers – SCOPE 42. Wiley, Chichester, 380 pp
Dinsmore KJ, Billett MF, Moore TR (2009) Transfer of carbon dioxide and methane through the soil-water-atmosphere system at Mer Bleue peatland, Canada. Hydrol Proc 23:330–341
Dinsmore KJ, Billett MF, Skiba UM, Rees RM, Drewer J, Helfter C (2010) Role of the aquatic pathway in the carbon and greenhouse gas budgets of a peatland catchment. Glob Change Biol 16:2750–2762
Dixon RK, Turner DP (1991) The global carbon cycle and climate change: responses and feedbacks from below-ground systems. Environ Pollut 73:245–262
Dixon RK, Brown S, Houghton RA, Solomon AM, Trexler MC, Wisniewski J (1994) Carbon pools and flux of forest global ecosystems. Science 263:185–190
Elser JJ, Fagan WF, Denno RF, Dobberfuhl DR, Folarin A, Huberty A, Interlandi S, Kilham SS, McCauley E, Schulz KL, Siemann EH, Sterner RW (2000) Nutritional constraints in terrestrial and freshwater food webs. Nature 408:578–580
Evans R, Brazier R (2005) Evaluation of modelled spatially distributed predictions of soil erosion by water versus field-based assessments. Environ Sci Policy 8:493–501
Evans MG, Warburton J (2001) Transport and dispersal of organic debris (peat blocks) in upland fluvial systems. Earth Surf Proc Land 26:1087–1102
Evans MG, Warburton J (2005) Sediment budget for an eroding peat-moorland catchment in northern England. Earth Surf Proc Land 30:557–577
Evans MG, Warburton J (2010) Peatland geomorphology and carbon cycling. Geogr Compass 4:1513–1531
Evans CD, Chapman PJ, Clark JM, Monteith DT, Cresser MS (2006a) Alternative explanations for rising dissolved organic carbon export from organic soils. Glob Change Biol 12:2044–2053
Evans MG, Warburton J, Yang J (2006b) Eroding blanket peat catchments: global and local implications of upland organic sediment budgets. Geomorphology 79:45–57
Fellman JB, Hood E, D’Amore DV, Edwards RT, White D (2009) Seasonal changes in the chemical quality and biodegradability of dissolved organic matter exported from soils to streams in coastal temperate rainforest watersheds. Biogeochemistry 95:277–293
Fellows CS, Valett HM, Dahm CN (2001) Whole-stream metabolism in two montane streams: contribution of the hyporheic zone. Limnol Oceanogr 46:523–531
Fiebig DM, Lock MA (1991) Immobilization of dissolved organic matter from groundwater discharging through the streambed. Freshw Biol 26:45–55
Fowler D, Hargreaves KJ, Skiba U, Milne R, Zahniser MS, Moncrieff JB, Beverland IJ, Gallagher MW (1995) Measurements of CH4 and N2O fluxes at the landscape scale using micrometeorological methods. Philos Trans R Soc Lond (A) 351:339–356
Fraser CJD, Roulet NT, Moore TR (2001) Hydrology and dissolved organic carbon biogeochemistry in an ombrotrophic bog. Hydrol Proc 15:3151–3166
Friedlingstein P, Houghton RA, Marland G, Hackler JL, Boden TA, Conway TJ, Canadell JG, Raupach MR, Ciais P, Le Quéré C (2010) Update on CO2 emissions. Nat Geosci 3:811–812
Granéli W, Lindell M, Tranvik L (1996) Photo-oxidative production of dissolved inorganic carbon in lakes of different humic content. Limnol Oceanogr 41:698–706
Grieve IC (1990) Seasonal, hydrological and land management factors controlling dissolved organic carbon concentrations in the Loch Fleet catchments, Southwest Scotland. Hydrol Proc 4:231–239
Griffiths J, Nutter J, Binley A, Crook N, Young A, Pates J (2007) Variability of dissolved CO2 in the Pang and Lambourn chalk rivers. Hydrol Earth Syst Sci 11:328–339
Grossart HP, Ploug H (2000) Bacterial production and growth efficiencies: direct measurements on riverine aggregates. Limnol Oceanogr 45:436–445
Guasch H, Armengol J, Marti E, Sabater S (1998) Diurnal variation in dissolved oxygen and carbon dioxide in two low-order streams. Water Res 32:1067–1074
Hagedorn F, Schleppi P, Waldner P, Fluhler H (2000) Export of dissolved organic carbon and nitrogen from Gleysol dominated catchments – the significance of water flow paths. Biogeochemistry 50:137–161
Hanson PC, Carpenter SR, Armstrong DE, Stanley EH, Kratz TK (2006) Lake dissolved inorganic carbon and dissolved oxygen: changing drivers from days to decades. Ecol Monogr 76:343–363
Hedges JI, Keil RG, Benner R (1997) What happens to terrestrial organic matter in the ocean? Org Geochem 27:195–212
Hill T, Neal C (1997) Spatial and temporal variation in pH, alkalinity and conductivity in surface runoff and groundwater for the Upper River Severn catchment. Hydrol Earth Syst Sci 1:697–715
Hinton MJ, Schiff SL, English MC (1998) Sources and flowpaths of dissolved organic carbon during storms in two forested watersheds of the Precambrian Shield. Biogeochemistry 41:175–197
Holden J (2006) Sediment and particulate carbon removal by pipe erosion increase over time in blanket peatlands as a consequence of land drainage. J Geophys Res Earth Surf 111:F02010
Hope D, Billett MF, Cresser MS (1994) A review of the export of carbon in river water: fluxes and processes. Environ Pollut 84:301–324
Hope D, Billett MF, Cresser MS (1997) Exports of organic carbon in two river systems in NE Scotland. J Hydrol 193:61–82
Hope D, Palmer S, Billett MF, Dawson JJC (2001) Carbon dioxide and methane evasion from a temperate peatland stream. Limnol Oceanogr 46:847–857
Hope D, Palmer SM, Billett MF, Dawson JJC (2004) Variations in dissolved CO2 and CH4 in a headwater catchment: an investigation of soil-stream linkages. Hydrol Proc 18:3255–3275
Hornberger GM, Bencala KE, McKnight DM (1994) Hydrological controls on dissolved organic carbon during snowmelt in the Snake River near Montezuma, Colorado. Biogeochemistry 25:147–165
Houghton RA (2000) A new estimate of global sources and sinks of carbon from land-use change. Eos 81(Suppl):S281
Houghton RA (2003) Why are estimates of the terrestrial carbon balance so different? Glob Change Biol 9:500–509
Houghton RA, Hobbie JE, Melillo JM, Moore B, Peterson BJ, Shaver GR, Woodwell GM (1983) Changes in the carbon content of terrestrial biota and soils between 1860 and 1980: a net release of CO2 to the atmosphere. Ecol Monogr 53:235–262
IGBP (International Geosphere-Biosphere Programme) Terrestrial Carbon Working Group (1998) The terrestrial carbon cycle: implications for the Kyoto Protocol. Science 2801:393–1394
Indermühle A, Stocker TF, Joos F, Fischer H, Smith HJ, Wahlen M, Deck B, Mastroianni D, Tschumi J, Blunier T, Meyer R, Stauffer B (1999) Holocene carbon-cycle dynamics based on CO2 trapped in ice at Talyer Dome, Antarctica. Nature 398:121–126
Ittekkot V, Laane RWPM (1991) Fate of riverine particulate organic matter. In: Degens ET, Kempe S, Richey JE (eds) Biogeochemistry of major world rivers – SCOPE 42. Wiley, Chichester, pp 233–243
Janssens IA, Freibauer A, Ciais P, Smith P, Nabuurs GJ, Folberth G, Schlamadinger B, Hutjes RWA, Ceulemans R, Schulze ED, Valentini R, Dolman AJ (2003) Europe’s terrestrial biosphere absorbs 7 to 12 % of European anthropogenic CO2 emissions. Science 300:1538–1542
Janzen HH (2004) Carbon cycling in earth systems – a soil science perspective. Agric Ecosyst Environ 104:399–417
Jones JB, Mulholland PJ (1998) Methane input and evasion in a hardwood forest stream: effect of subsurface flow from shallow and deep pathways. Limnol Oceanogr 43:1243–1250
Jones JB Jr, Stanley EH, Mulholland PJ (2003) Long-term decline in carbon dioxide supersaturation in rivers across the contiguous United States. Geophys Res Lett 30:1495
Keeling RF, Piper SC, Heimann M (1996) Global and hemispheric CO2 sinks deduced from changes in atmospheric O2 concentration. Nature 381:218–221
Kling GW, Kipphut GW, Miller MC (1991) Arctic lakes and streams as gas conduits to the atmosphere: implications for tundra carbon budgets. Science 25:298–301
Kronvang B, Laubel A, Grant R (1997) Suspended Sediment and particulate phosphorus transport and delivery pathways in an arable catchment, Gelbæk Stream. Hydrol Proc 11:627–642
Kuserk FT, Kaplan LA, Bott T (1984) In situ measures of dissolved organic carbon flux in a rural stream. Can J Fish Aquat Sci 41:964–973
Lawler DM, Couperthwaite J, Bull LJ, Harris NM (1997) Bank erosion events and processes in the Upper Severn basin. Hydrol Earth Syst Sci 1:523–534
Lawler DM, Petts GE, Foster IDL, Harper S (2006) Turbidity dynamics during spring storm events in an urban headwater river system: the Upper Tame, West Midlands, UK. Sci Total Environ 360:109–126
Le Quéré C, Raupach MR, Canadell JG, Marland G, Bopp L, Ciais P, Conway TJ, Doney SC, Feely RA, Foster P, Friedlingstein P, Gurney KR, Houghton RA, House JI, Huntingford C, Levy PE, Lomas MR, Majkut J, Metzl N, Ometto J, Peters GP, Prentice IC, Randerson JT, Running SW, Sarmiento JL, Schuster U, Sitch S, Takahashi T, Viovy N, van der Werf GR, Woodward FI (2009) Trends in the sources and sinks of carbon dioxide. Nat Geosci 2:831–836
Lindell MJ, Granéli W, Tranvik L (1995) Enhanced bacterial growth in response to photochemical transformation of dissolved organic matter. Limnol Oceanogr 40:195–199
Lohse KA, Brooks PD, McIntosh JC, Meixner T, Huxman TE (2009) Interactions between biogeochemistry and hydrologic systems. Ann Rev Environ Resour 34:65–96
Lovett G, Cole J, Pace M (2006) Is net ecosystem production equal to ecosystem carbon accumulation? Ecosystem 9:152–155
Lumsdon DG, Fraser AR (2005) Infrared spectroscopic evidence supporting heterogeneous site binding models for humic substances. Environ Sci Technol 39:6624–6631
Malhi Y (2002) Carbon in the atmosphere and terrestrial biosphere in the 21st century. Philos Trans R Soc Lond (A) 360:2925–2945
Malhi Y, Meir P, Brown S (2002) Forests, carbon and global climate. Philos Trans R Soc Lond (A) 360:1567–1591
Malmqvist B, Wotton RS, Zhang YX (2001) Suspension feeders transform massive amounts of seston in large northern rivers. Oikos 92:35–43
Manzoni S, Poporato A (2011) Common hydrologic and biogeochemical controls along soil–stream continuum. Hydrol Proc 25:1355–1360
Marschner B, Kalbitz K (2003) Controls of bioavailability and biodegradability of dissolved organic matter in soils. Geoderma 113:211–235
Marzolf ER, Mulholland PJ, Steinman AD (1994) Improvements to the diurnal upstream-downstream dissolved oxygen change technique for determining whole-stream metabolism in small streams. Can J Fish Aquat Sci 51:1591–1599
Mayorga E, Aufdenkampe AK, Masiello CA, Krusche AV, Hedges JI, Quay PD, Richey JE, Brown TA (2005) Young organic matter as a source of carbon dioxide outgassing from Amazonian rivers. Nature 436:538–541
McCallister SL, Bauer JE, Ducklow HW, Canuel EA (2006) Sources of estuarine dissolved and particulate organic matter: a multi-tracer approach. Org Geochem 37:454–468
McDowell WH (1985) Kinetics and mechanisms of dissolved organic carbon retention in a headwater stream. Biogeochemistry 1:329–352
McHugh M, Harrod T, Morgan R (2002) The extent of soil erosion in upland England and Wales. Earth Surf Proc Land 27:99–107
McTammany ME, Webster E, Benfield EF, Neatrour MA (2003) Longitudinal patterns of metabolism in a southern Appalachian river. J North Am Benthol Soc 22:359–370
Meybeck M (1982) Carbon, nitrogen and phosphorus transport by World Rivers. Am J Sci 282:401–450
Meybeck M (1993) Riverine transport of atmospheric carbon: sources, global typology and budget. Water Air Soil Pollut 70:443–464
Meyer JL, Edwards RT (1990) Ecosystem metabolism and turnover of organic carbon along a blackwater river continuum. Ecology 71:668–677
Meyer JL, Tate CM (1983) The effects of watershed disturbance on dissolved organic carbon dynamics of a stream. Ecology 64:33–44
Meyer JL, Wallace JB, Eggert SL (1998) Leaf litter as a source of dissolved organic carbon in streams. Ecosystem 1:240–249
Minshall GW, Cummins KW, Petersen RC, Cushing CE, Bruns DA, Sedell JR, Vannote RL (1985) Developments in stream ecosystem theory. Can J Fish Aquat Sci 42:1045–1055
Monaghan MT, Thomas SA, Minshall GW, Newbold JD, Cushing CE (2001) The influence of filter-feeding benthic macroinvertebrates on the transport and deposition of particulate organic matter and diatoms in two streams. Limnol Oceanogr 46:1091–1099
Monteith DT, Stoddard JL, Evans CD, de Wit HA, Forsius M, Hogasen T, Wilander A, Skjelkvale BL, Jeffries DS, Vuorenmaa J, Keller B, Kopacek J, Vesely J (2007) Dissolved organic carbon trends resulting from changes in atmospheric deposition chemistry. Nature 450:537–540
Mulholland PJ, Marzolf ER, Webster JR, Hart DR, Hendricks SP (1997) Evidence that hyporheic zones increase heterotrophic metabolism and phosphorus uptake in forest streams. Limnol Oceanogr 42:443–451
National Oceanic and Atmospheric Administration (2012) Trends in atmospheric carbon dioxide – Recent monthly mean CO2 atMauna Loa. http://www.esrl.noaa.gov/gmd/ccgg/trends/mlo.html. Accessed 20 May 2012
Neal C (1988) Determination of dissolved CO2 in upland streamwater. J Hydrol 99:127–142
Neal C, Wilkinson J, Neal M, Harrow M, Wickham H, Hill L, Morfitt C (1997) The hydrochemistry of the headwaters of the River Severn, Plynlimon. Hydrol Earth Syst Sci 1:583–617
Neal C, Jarvie HP, Wade AJ, Neal M, Wyatt R, Wickham H, Hill L, Hewitt N (2004) The water quality of the LOCAR Pang and Lambourn catchments. Hydrol Earth Syst Sci 8:614–635
Neal C, Hilton J, Wade AJ, Neal M, Wickham H (2006) Chlorophyll-α in the rivers of Eastern England. Sci Total Environ 365:84–104
Neff JC, Asner GP (2001) Dissolved organic carbon in terrestrial ecosystems: synthesis and a model. Ecosystem 4:29–48
Palmer SM, Hope D, Billett MF, Dawson JJC, Bryant CL (2001) Sources of aquatic carbon in a headwater stream: evidence from carbon isotope studies. Biogeochemistry 52:321–338
Penton CR, Newman S (2007) Enzyme activity responses to nutrient loading in subtropical wetlands. Biogeochemistry 84:83–98
Peters GP, Marland G, Le Quéré C, Boden T, Canadell JG, Raupach MR (2012) Rapid growth in CO2 emissions after the 2008–2009 global financial crisis. Nat Clim Change 2:2–4
Peterson BJ, Hobbie JE, Hershey AE, Lock MA, Ford TE, Vestal JR, Mckinley VL, Hullar MAJ, Miller MC, Ventullo RM, Volk GS (1985) Transformation of a tundra river from heterotrophy to autotrophy by addition of phosphorus. Science 229:1383–1386
Post WM, Izaurralde RC, Jastrow JD, McCarl BA, Amonette JE, Bailey VL, Jardine PM, West TO, Zhou JZ (2004) Enhancement of carbon sequestration in US soils. BioScience 54:895–908
Raupach MR, Canadell JG (2010) Carbon and the Anthropocene. Curr Opin Environ Sustain 2:210–218
Raymond PA, Bauer JE (2001) Riverine export of aged terrestrial organic matter to the North Atlantic Ocean. Nature 409:497–500
Rebsdorf A, Thyssen N, Erlandsen M (1991) Regional and temporal variation in pH, alkalinity and carbon dioxide in Danish streams, related to soil type and land use. Freshw Biol 25:419–435
Reynolds B (1986) A comparison of element outputs in solution, suspended sediments and bedload for a small upland catchment. Earth Surf Proc Land 11:217–221
Richey JE (1983) Interactions of C, N, P and S in river systems: a biogeochemical model. In: Bolin B, Cook RB (eds) The major biogeochemical cycles and their interactions – SCOPE 21. Wiley, Chichester, pp 365–383
Richey JE, Melack JM, Aufdenkampe AK, Ballester VM, Hess LL (2002) Outgassing from Amazonian rivers and wetlands as a large tropical source of atmospheric CO2. Nature 416:617–620
Rosenfeld J, Roff JC (1991) Primary production and potential availability of autochthonous carbon in southern Ontario streams. Hydrobiologia 224:99–109
Roulet N, Moore TR (2006) Browning the waters. Nature 444:283–284
Sabine CL, Feely RA, Gruber N, Key RM, Lee K, Bullister JL, Wanninkhof R, Wong CS, Wallace DWR, Tilbrook B, Millero FJ, Peng T-H, Kozyr A, Ono T, Rios AF (2004) The oceanic sink for anthropogenic CO2. Science 305:367–371
Schiff S, Aravena R, Trumbore SE, Hinton MJ, Elgood R, Dillon PJ (1997) Export of DOC from forested catchments on the Precambrian shield of Central Ontario: clues from 13C and 14C. Biogeochemistry 36:43–65
Schimel DS, House JI, Hibbard KA, Bousquet P, Ciais P, Peylin P, Braswell BH, Apps MJ, Baker D, Bondeau A, Canadell J, Churkina G, Cramer W, Denning AS, Field CB, Friedlingstein P, Goodale C, Heimann M, Houghton RA, Melillo JM, Moore B III, Murdiyarso D, Noble I, Pacala SW, Prentice IC, Raupach MR, Rayner PJ, Scholes RJ, Steffen WL, Wirth C (2001) Recent patterns and mechanisms of carbon exchange by terrestrial ecosystems. Nature 414:169–172
Schlesinger WH (1984) Soil organic matter: a source of atmospheric CO2. In: Woodwell GM (ed) The role of terrestrial vegetation in the global carbon cycle: measurement by remote sensing – SCOPE 23. Wiley, Chichester, pp 111–127
Schlesinger WH (1990) Evidence from chronosequence studies for a low carbon-storage potential of soils. Nature 348:232–234
Schlesinger WH, Andrews JA (2000) Soil respiration and the global carbon cycle. Biogeochemistry 48:7–20
Schlunz B, Schneider RR (2000) Transport of terrestrial organic carbon to the oceans by rivers: re-estimating flux- and burial rates. Int J Earth Sci 88:599–606
Schmidt MWI, Torn MS, Abiven S, Dittmar T, Guggenberger G, Janssens IA, Kleber M, Kögel-Knabner I, Lehmann J, Manning DAC, Nannipieri P, Rasse DP, Weiner S, Trumbore SE (2011) Persistence of soil organic matter as an ecosystem property. Nature 478:49–56
Scholes RJ, Noble IR (2001) Storing carbon on land. Science 294:1012–1013
Schulze ED, Ciais P, Luyssaert S, Schrumpf M, Janssens IA, Thiiruchittampalam B, Theloke J, Saurat M, Bringezu S, Lelieveld J, Lohila A, Rebmann C, Jung M, Bastviken D, Abril G, Grassi G, Leip A, Freibauer A, Kutsch W, Don A, Nieschulze J, Borner A, Gash JH, Dolman AJ (2010) The European carbon balance. Part 4. Integration of carbon and other trace-gas fluxes. Glob Change Biol 16:1451–1469
Siemens J (2003) The European carbon budget: a gap. Science 302:1681
Skiba U, Cresser MS (1991) Seasonal changes in soil atmospheric CO2 concentrations in two upland catchments and associated changes in river water chemistry. Chem Ecol 7:217–225
Smith TM, Cramer WP, Dixon RK, Leemans R, Neilson RP, Solomon AM (1993) The global terrestrial carbon-cycle. Water Air Soil Pollut 70:19–37
Smith P, Fang C, Dawson JJC, Moncrieff JB (2008) Impact of global warming on soil organic carbon. Adv Agron 97:1–43
Sobczak WV, Cloern JE, Jassby AD, Muller-Solger AB (2002) Bioavailability of organic matter in a highly disturbed estuary: the role of detrital and algal resources. Proc Natl Acad Sci U S A 99:8101–8105
Sollins P, Glassman CA, Dahm CN (1985) Composition and possible origin of detrital material in streams. Ecology 66:297–299
Staehr PA, Testa JM, Kemp WM, Cole JJ, Sand-Jensen K, Smith SV (2012) The metabolism of aquatic ecosystems: history, applications, and future challenges. Aquat Sci 74:15–29
Stevenson FJ (1986) The carbon cycle. In: Stevenson FJ, Cole MA (eds) Cycles of soil – carbon, nitrogen, phosphorus, sulfur, micronutrients, 2nd edn. Wiley, New York, pp 1–44
Stott T, Mount N (2004) Plantation forestry impacts on sediment yields and downstream channel dynamics in the UK: a review. Prog Phys Geogr 28:197–240
Stuiver M (1978) Atmospheric carbon dioxide and carbon reservoir changes. Science 199:253–258
Stumm W, Morgan JJ (1981) Aquatic chemistry – an introduction emphasizing chemical equilibria in natural waters. Wiley, New York
Stutter MI, Langan SJ, Demars BOL (2007) River sediments provide a link between catchment pressures and ecological status in a mixed land use Scottish River system. Water Res 41:2803–2815
Stutter MI, Langan SJ, Cooper RJ (2008) Spatial and temporal dynamics of streamwater particulate and dissolved N, P and C forms along a catchment transect, NE Scotland. J Hydrol 350:187–202
Tans PP, Fung IY, Takahashi T (1990) Observational constraints on the global atmospheric CO2 budget. Science 247:1431–1438
Tao S (1998) Spatial and temporal variation in DOC in the Yichun River, China. Water Res 32:2205–2210
Tappin AD, Harris JRW, Uncles RJ (2003) The fluxes and transformations of suspended particles, carbon and nitrogen in the Humber estuarine system (UK) from 1994 to 1996: results from an integrated observation and modelling study. Sci Total Environ 314:665–713
Taylor PG, Townsend AR (2010) Stoichiometric control of organic carbon-nitrate relationships from soils to the sea. Nature 464:178–1181
Teodoru CR, del Giorgio PA, Prairie YT, Camire M (2009) Patterns in pCO2 in boreal streams and rivers of northern Quebec, Canada. Glob Biogeochem Cycle 23:GB2012
Thurman EM (1985) Organic geochemistry of natural waters. Nijhoff/Junk Publishers, Dordrecht/Holland
Tipping E, Billett MF, Bryant CL, Buckingham S, Thacker SA (2010) Sources and ages of dissolved organic matter in peatland streams: evidence from chemistry mixture modelling and radiocarbon data. Biogeochemistry 100:121–137
Vannote RL, Minshall GW, Cummins KW, Sedell JR, Cushing CE (1980) The river continuum concept. Can J Fish Aquat Sci 37:130–137
Volk CJ, Volk CB, Kaplan LA (1997) Chemical composition of biodegradable dissolved organic matter in streamwater. Limnol Oceanogr 42:39–44
Von Lützow M, Kögel-Knabner I, Ekschmitt K, Matzner E, Guggenberger G, Marschner B, Flessa H (2006) Stabilization of organic matter in temperate soils: mechanisms and their relevance under different soil conditions – a review. Eur J Soil Sci 57:426–445
Walling DE (1977) Assessing the accuracy of suspended sediment rating curves for a small basin. Water Resour Res 13:531–538
Walling DE, Webb BW (1985) Estimating the discharge of contaminants to coastal waters and rivers: some cautionary comments. Mar Pollut Bull 16:488–492
Walling DE, Webb BW (1987) Suspended load in gravel bed rivers: UK experience. In: Thorne CR, Bathurst JC, Hey RD (eds) Sediment transport in gravel-bed rivers. Wiley, Chichester, pp 767–782
Walling DE, Russell MA, Hodgkinson RA, Zhang Y (2002) Establishing sediment budgets for two small lowland agricultural catchments in the UK. Catena 47:323–353
Warburton J, Holden J, Mills AJ (2004) Hydrological controls of surficial mass movements in peat. Earth Sci Rev 67:139–156
Webster JR (2007) Spiraling down the river continuum: stream ecology and the U-shaped curve. J North Am Benthol Soc 26:375–389
Weisharr JL, Aiken GR, Bergamaschi BA, Fram MS, Fujii R, Mopper K (2003) Evaluation of specific ultraviolet absorbance as an indicator of the chemical composition and reactivity of dissolved organic carbon. Environ Sci Technol 37:4702–4708
Woodwell GM (1978) The carbon dioxide question. Sci Am 238:34–43
Worrall F, Harriman R, Evans CD, Watts CD, Adamson J, Neal C, Tipping E, Burt T, Grieve I, Monteith D, Naden PS, Nisbet T, Reynolds B, Stevens P (2004) Trends in dissolved organic carbon in UK rivers and lakes. Biogeochemistry 70:369–402
Yoo K, Amundson R, Heimsath AM, Dietrich WE (2006) Spatial patterns of soil organic carbon on hillslopes: integrating geomorphic processes and the biological C cycle. Geoderma 130:47–65
Zimmermann-Timm H (2002) Characteristics, dynamics and importance of aggregates in rivers – an invited review. Int Rev Hydrobiol 87:197–240
Acknowledgements
The author would like to thank the Scottish Government’s Rural and Environment Science and Analytical Services (RESAS) Division for funding. Thanks to Pete Smith, Chris Soulsby at the University of Aberdeen and Mike Billett, CEH Edinburgh for helpful advice in relation to studies undertaken by the author contributing to this chapter. Finally, thanks to Marc Stutter (James Hutton Institute) for constructive comments.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Dawson, J.J.C. (2013). Loss of Soil Carbon to the Atmosphere via Inland Surface Waters. In: Lal, R., Lorenz, K., Hüttl, R., Schneider, B., von Braun, J. (eds) Ecosystem Services and Carbon Sequestration in the Biosphere. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-6455-2_9
Download citation
DOI: https://doi.org/10.1007/978-94-007-6455-2_9
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-6454-5
Online ISBN: 978-94-007-6455-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)