
Abstract
Cell-extrinsic signals and intrinsic cell cycle regulators strictly control proliferation. Cancers develop from a cell that escapes these tight controls and proliferates unrestrictedly. The primary cilium critically controls proliferation by mediating cell-extrinsic signals and regulating cell cycle entry. Accordingly, recent studies showed that defective cilia can either promote or suppress cancers, depending on the cancer-initiating mutation, and that presence or absence of primary cilia is associated with specific cancer types. These novel findings suggest that primary cilia play central but distinct roles in different cancer types, opening up a completely new avenue of research to understand the biology and treatment of cancers.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
FormalPara OutlineIn this chapter, we review evidence showing that primary cilia play important roles in cell cycle entry and multiple signaling pathways, and we discuss how these roles of primary cilia could contribute or inhibit brain cancer formation. First, we review the dual and opposing roles of primary cilia in medulloblastoma development, then discuss possible roles of primary cilia in other brain cancers focusing on diffuse intrinsic pontine glioma, glioblastoma multiforme, and cancer stem cell. Lastly, we discuss the potential of primary cilia in diagnosis and treatment for brain cancers.
8.1 Introduction
The primary cilium is at the crossroads of cell cycle progression and cellular signaling pathways. Cell cycle progression regulates assembly and disassembly of primary cilia, and primary cilia in turn regulate cell cycle entry (Rieder et al. 1979; Tucker et al. 1979; Kim et al. 2011b; Li et al. 2011). Primary cilia participate in multiple signaling pathways that control cell proliferation, differentiation, migration, polarity, and metabolism, deregulation of which are closely linked to oncogenesis (Huangfu et al. 2003; Schneider et al. 2005; Simons et al. 2005; Zhu et al. 2009; Boehlke et al. 2010; Berbari et al. 2011; Ezratty et al. 2011). Thus, it is reasonable to suspect that normal or abnormal functions of primary cilia may contribute to oncogenesis. Indeed, recent studies showed that primary cilia play fundamental roles in the development of basal cell carcinoma, the most common cancer in Caucasians, and medulloblastoma, the most common brain cancer in children (Han et al. 2009; Wong et al. 2009). Surprisingly, primary cilia are either required or suppressive for oncogenesis in mice, depending on the cancer-initiating mutation. Furthermore, the presence or absence of primary cilia is associated with specific cancer types in humans (Wheatley 1995; Han et al. 2009; Schraml et al. 2009; Seeley et al. 2009; Wong et al. 2009; Yuan et al. 2010; Kim et al. 2011a), suggesting that primary cilia can either promote or suppress human cancers as well. Therefore, understanding the mechanisms by which this intriguing organelle functions in normal and cancerous cells will reveal oncogenic mechanisms that were not apparent previously and will unmask new therapeutic approaches.
8.2 Primary Cilia and Cell Cycle Progression
The primary cilium contains a unique microtubule cytoskeleton called the axoneme, a ring of nine microtubule doublets (9 + 0) that runs longitudinally through the organelle. The axoneme grows from the basal body docked with the cell membrane. During cell division, the basal body detaches from membrane and transforms into the mother centriole to form the centrosome that organizes the mitotic spindle. Thus, ciliogenesis is tightly regulated throughout the cell cycle; proliferating cells disassemble cilia before mitosis to release the basal body and reassemble them after mitosis (Rieder et al. 1979; Tucker et al. 1979). Some cells also resorb cilia upon entry into the S phase (Tucker et al. 1979). Consistent with this regulation, Aurora kinase A, a centrosomal kinase that regulates mitotic entry, becomes activated at the basal body as cells enter the S phase and mitosis and triggers ciliary disassembly (Pugacheva et al. 2007). As a cell exits mitosis, CP110, a distal centriolar protein that inhibits ciliogenesis, disappears from the mother centriole, allowing ciliogenesis to occur (Spektor et al. 2007). Cyclin-dependent kinase inhibitors also influence ciliogenesis in cultured cells; knockdown of p16 INK4a positively influences ciliogenesis, and knockdown of p15 INK4b negatively regulates it (Bishop et al. 2010; Kim et al. 2010a). However, mutant mice defective for p16 INK4a and/or p15 INK4bdo not display developmental defects associated with defective cilia. Remarkably, recent studies showed that this cell-cycle-dependent organelle is not passively linked with the cell cycle but actively regulates cell cycle progression (Bielas et al. 2009; Jacoby et al. 2009; Kim et al. 2011b; Li et al. 2011).
One of the first pieces of evidence supporting the role of primary cilia in cell cycle progression came from studies on the inositol polyphosphate-5-phosphatase E (INPP5E) gene mutated in two cilia-associated diseases, Joubert and MORM syndromes (Bielas et al. 2009; Jacoby et al. 2009). INPP5E, which hydrolyzes the 5-phosphate of phosphatidylinositol 3,4,5-triphosphate and phosphatidylinositol 4,5-bisphosphate, localized exclusively at primary cilia; thus, mutation in INPP5E caused the ciliary membrane to accumulate these lipids, which play important roles in signaling pathways and membrane trafficking. Notably, INPP5E mutation did not affect assembly of cilia but accelerated both ciliary disassembly and S-phase entry after serum stimulation. Previous work showed that overexpression of INPP5E caused cell cycle arrest (Kisseleva et al. 2002). These studies suggest that ciliary disassembly can affect S-phase entry. More direct evidence supporting this notion came from the following two studies.
In one study, Sung and colleagues (Li et al. 2011) found that Tctex-1, a light chain subunit of cytoplasmic dynein, plays critical roles in ciliary disassembly and cell cycle progression. Knockdown of Tctex-1 inhibited both ciliary disassembly and S-phase entry that occur after serum addition to serum-starved NIH3T3 and RPE cells. Importantly, Tctex-1 knockdown did not affect S-phase entry in cells lacking primary cilia, indicating that failure to disassemble cilia underlies the blockage of S-phase entry. This inhibition was independent of the function of Tctex-1 as a cytoplasmic dynein component. Serum stimulation induced phosphorylation of Tctex-1 at threonine 94 (T94), leading to its dissociation from the dynein complex and accumulation at the transition zone between the basal body and the ciliary axoneme. The knockdown effect was rescued by wild-type and phosphorylation-mimicking Tctex-1 (Tctex-1T94E), but to a lesser extent by non-phosphorylatable Tctex-1T94A, which binds dynein. Furthermore, Tctex-1T94E accelerated ciliary disassembly and S-phase entry even in the absence of serum stimulation. In vivo, Tctex-1 is selectively enriched in proliferating neural progenitors. Remarkably, knockdown of Tctex-1 in radial glia, the neural progenitors in the embryonic brain, caused them to exit from the cell cycle and differentiate into neurons prematurely, whereas overexpression of Tctex-1T94E shortened the G1 phase, accelerated S-phase re-entry, and increased the proliferating progenitor cell population. These data indicate that the primary cilium blocks S-phase entry and cells use Tctex-1 to overcome this blockage.
In the other study, Tsiokas and colleagues (Kim et al. 2011b) found that Nde-1, a centrosomal protein, critically regulates cilia length and S-phase entry. Knockdown of Nde-1 in NIH3T3 and RPE cells lengthened cilia and delayed S-phase entry after serum stimulation. Importantly, similar to Tctex-1, the delay in S-phase entry upon Nde-1 knockdown was dependent on the presence of primary cilia; there was no delay in S-phase entry in the absence of primary cilia. Furthermore, expression of constitutively active form of Rab8a, which lengthens primary cilia independently of Nde-1, also inhibited S-phase entry, confirming that lengthened cilia delay S-phase entry. In vivo, knockdown of Nde-1 in zebrafish caused lengthening of primary cilia and defective proliferation in Kuffer’s vesicle. Interestingly, mutations in Nde-1 cause microcephaly in both humans and mice (Feng and Walsh 2004; Alkuraya et al. 2011; Bakircioglu et al. 2011), which may be partly due to premature cell cycle exit and differentiation of neural progenitors that have abnormally long cilia. Taken together, these studies revealed that primary cilia regulate cell division as a barrier blocking S-phase entry raising the possibility that primary cilia negatively regulate oncogenesis.
8.3 Primary Cilia and Signaling Pathways
Studies over the past decade have established primary cilia as a signaling hub for multiple signaling pathways, including Hedgehog (Goetz and Anderson 2010), Wnt (Wallingford and Mitchell 2011), receptor tyrosine kinases (RTKs) (Christensen et al. 2012), and Notch signaling (Ezratty et al. 2011). These signaling pathways control a myriad of cellular processes, including proliferation, differentiation, migration, polarity, and metabolism, all of which play critical roles in development, homeostasis, and oncogenesis. Here we briefly discuss the role of primary cilia in these signaling pathways.
8.3.1 Hedgehog Signaling
Forward genetic studies in mice have shown that Hedgehog signaling requires the primary cilium for both activation and repression of the pathway (Huangfu et al. 2003; Haycraft et al. 2005; Huangfu and Anderson 2005; Liu et al. 2005; May et al. 2005) (detailed in Chap. 2). Ciliary mutant mice have phenotypes similar to those of mutant mice defective in Hedgehog signaling. Epistasis studies placed primary cilia downstream of Hedgehog receptor Patched1 (Ptch1) and Smoothened (Smo), but upstream of the GLI-Kruppel family transcription factors (Gli1-3) and their binding protein Suppressor of Fused (Sufu) (Huangfu et al. 2003; Haycraft et al. 2005; Huangfu and Anderson 2005; Liu et al. 2005; May et al. 2005; Han et al. 2008; Chen et al. 2009; Jia et al. 2009). In the absence of Hedgehog, Ptch1 localizes to primary cilia and inhibits Hedgehog signaling by preventing Smo from entering primary cilia (Rohatgi et al. 2007). Upon binding of Hedgehog, Ptch1 moves out of the cilia, leading to accumulation and activation of Smo in the primary cilium (Corbit et al. 2005; Rohatgi et al. 2007). Activated Smo induces Sufu-Gli complex accumulation in primary cilia and dissociation of the complex, leading to formation of Gli transcriptional activators (Humke et al. 2010; Tukachinsky et al. 2010; Zeng et al. 2010). Without activated Smo in primary cilia, Gli2 and Gli3 are truncated by the proteasome to become transcriptional repressors and repress Hedgehog target gene expression. In the absence of primary cilia, the processing of Gli3 (and probably Gli2 also) repressor is greatly reduced, resulting in derepression of Hedgehog target genes (Haycraft et al. 2005; Huangfu and Anderson 2005; Liu et al. 2005; May et al. 2005).
In contrast to complete loss of primary cilia, which results in complete irresponsiveness to Hedgehog and inefficient processing of Gli repressors, distinctive structural abnormalities in primary cilia cause a range of Hedgehog signaling defects. Mutations in components of intraflagellar transport (IFT) complex A, which functions in retrograde transport of ciliary components from cilia to the cell body, cause abnormally short and swollen cilia and constitutive activation of Hedgehog signaling (Tran et al. 2008; Cortellino et al. 2009). Loss of Arl13b, a ciliary small GTPase, causes opening of axonemal microtubules and constitutive activation of Gli activators at low levels without affecting Gli3 repressor activity (Caspary et al. 2007). Mutations in Broad-minded, a Rab-GAP–like protein, cause detachment of ciliary membrane from the axoneme and selective loss of responsiveness to high Hedgehog levels (Ko et al. 2010). Thus, primary cilia play complex and active roles in Hedgehog signaling rather than merely concentrating signaling molecules to facilitate their interactions.
8.3.2 Wnt Signaling
Secreted protein Wnt binds to Frizzled receptors to trigger the signaling activity of cytoplasmic protein Dishevelled (Dvl), where the signaling diverges into canonical and non-canonical Wnt signaling pathways (Logan and Nusse 2004). In the canonical pathway, activated Dvl leads to accumulation and nuclear localization of β-catenin and subsequent activation of Wnt target genes. The non-canonical pathway is independent of β-catenin and primarily controls cytoskeletons involved in planar cell polarity (PCP) and cell migration. One of the first pieces of evidence linking primary cilia to Wnt signaling came from a study on Inversin, a ciliary protein whose mutation causes cystic kidney diseases and situs inversus (Simons et al. 2005). The study showed that, in fish and frogs, Inversin functions as a switch from the canonical to the non-canonical Wnt signaling pathway by targeting Dvl for destruction. Moreover, mutant mice defective in genes mutated in Bardet-Biedl syndrome, a disease linked to ciliary dysfunction, have phenotypes associated with PCP mutants (Ross et al. 2005). Other studies from kidney, pancreas, cochlea, fish embryos, and cultured cells also showed that defective primary cilia increase canonical Wnt signaling activity and disrupt non-canonical Wnt signaling (Lin et al. 2003; Cano et al. 2004; Gerdes et al. 2007; Corbit et al. 2008; Jonassen et al. 2008; Jones et al. 2008). Notably, however, mutant mice defective for primary cilia do not have obvious developmental phenotypes associated with defective Wnt signaling. Furthermore, two recent studies showed that defective primary cilia do not affect Wnt signaling in fish, mice, and cultured cells and suggested that Wnt signaling requires the basal body rather than primary cilia per se (Huang and Schier 2009; Ocbina et al. 2009). Thus, the primary cilium and the basal body appear to have a subtle and cell type–specific roles in Wnt signaling (Wallingford and Mitchell 2011).
8.3.3 RTK Signaling
RTKs are activated by growth factors and initiate a series of signaling cascades, including mitogen-activated protein kinase pathways, phosphatidylinositol 3-kinase pathways, and phospholipase C pathways. The first connection between RTKs and primary cilia came from evidence that platelet-derived growth factor receptor α (PDGFRα) signaling requires primary cilia in NIH3T3 cells and mouse embryonic fibroblasts (MEFs) (Schneider et al. 2005). Serum starvation of confluent cells induced PDGFRα expression and its localization to primary cilia. Subsequent stimulation of cells with PDGF-AA ligand induced phosphorylation of PDGFRα and downstream dual specificity mitogen-activated protein kinase kinase 1/2 (MEK1/2) inside primary cilia, leading to phosphorylation of retinoblastoma-associated (RB) protein, which marks S-phase entry. The activation of PDGFRα in primary cilia was also required for directional migration mediated by Na+/H + exchanger NHE1 (Schneider et al. 2009). Importantly, PDGF-AA failed to activate PDGFRα to induce S-phase entry and directional migration in MEFs isolated from hypomorphic IFT88 mutant mice (IFT88 Tg737Rpw) defective for ciliogenesis. These observations suggest that PDGF-AA and PDGFRα require primary cilia to transmit signals. Some of the observed defects, however, may be partly due to the low level of PDGFRα in IFT88 Tg737Rpw MEFs after serum starvation, which dramatically induced a higher PDGFRα level in wild-type but not in IFT88 Tg737Rpw MEFs (Schneider et al. 2005). Thus, it will be important to determine whether PDGF-AA can activate signaling in cells that lack primary cilia but have PDGFRα at a similar level as wild-type cells. It still remains to be determined whether primary cilia are required for PDGFRα signaling in vivo.
Another RTK, insulin-like growth factor 1 receptor (IGF1R) appears to function preferentially in primary cilia in MEF-adipose-like 3T3-L1 cells (Zhu et al. 2009), which can be differentiated into adipocyte after growth arrest at confluence. IGF1R activation by insulin is essential to induce differentiation. Interestingly, in 3T3-L1 cells, insulin activated IGF1R in primary cilia faster than IGF1R outside primary cilia and induced accumulation of activated downstream signaling molecules, phosphorylated insulin receptor substrate 1 (IRS1) and protein kinase B (Akt), at the basal body. Remarkably, knockdown of IFT88 or Kif3a encoding a subunit of essential ciliogenic Kinesin-II motor disrupted IGF1R signaling and blocked differentiation of 3T3-L1, suggesting that IGF1R requires primary cilia to signal to induce differentiation. Like PDGFRα signaling, it is unknown whether primary cilia are required for IGF1R signaling in vivo.
In addition to PDGFRα and IGF1R, epidermal growth factor receptor (EGFR) and the angiopoietin receptors Tie-1 and Tie-2 localize to primary cilia (Ma et al. 2005; Teilmann and Christensen 2005; Danilov et al. 2009; Wu et al. 2009). Furthermore, recent studies showed that primary cilia negatively regulate the activity of mammalian target of rapamycin (mTOR) both in vitro and in vivo (DiBella et al. 2009; Boehlke et al. 2010; Berbari et al. 2011), whereas mTOR positively regulates the length of primary cilia (Yuan et al. 2012). mTOR is a key signaling molecule that integrates RTK signaling with cellular metabolism, a change in which is one of the hallmarks of cancers. Thus, primary cilia appear to participate in multiple RTK signaling pathways, providing them with a platform to crosstalk and integrate. Future work should investigate whether primary cilia participate in RTK signaling in vivo and in the crosstalk of multiple RTK signaling pathways.
8.3.4 Notch Signaling
A recent study showed that Notch signaling requires primary cilia during skin development (Ezratty et al. 2011). Notch is a transmembrane receptor protein that undergoes intramembrane proteolytic cleavage upon binding to its ligands, which are mainly transmembrane proteins as well (Kopan and Ilagan 2009). After cleavage, the Notch intracellular domain (NICD) enters the nucleus and activates transcription of target genes. Removing primary cilia in mouse embryonic skin cells by knockdown of IFT74 or conditional ablation of either IFT88 or Kif3a resulted in defective epidermal differentiation, a process dependent on Notch signaling. Consistently, expression of Notch responsive genes was disrupted in cells lacking primary cilia. Expression of NCID partially rescued expression of a Notch reporter gene and differentiation defects. The study also showed that Notch3 is selectively localized to primary cilia, and Presenilin-2, the catalytic subunit of γ-secretase that cleaves Notch receptor to generate NICD, is localized at the base of primary cilia in addition to intercellular membrane borders. These localizations were specific to suprabasal cells in the embryonic skin, where Notch signaling is active. Remarkably, nuclear NCID3, the processed Notch3, was observed only in ciliated suprabasal cells and not in Kif3a mutant cells. These findings raise interesting questions: Does Notch3 require primary cilia to signal in other tissues and animals? Does any other Notch require primary cilia to signal? Are membrane-bound ligands for Notch3 also exclusively localized to a specific domain of the signaling cell to be juxtaposed to the primary cilium of responding cells in developing skin? If so, what is the underlying mechanism?
Why do vertebrate cells require primary cilia for multiple signaling pathways? Simplistically, the concentration of signaling molecules in primary cilia whose large surface area relative to the small volume would provide high sensitivity for detection of low levels of extracellular signals. Subtle structural defects in primary cilia, however, cause unique Hh signaling defects, suggesting that the function of primary cilia are more than concentrating and sensitizing signaling molecules for extracellular signals. The primary cilium, although continuous from cytoplasm, is a distinct subcellular compartment, in which trafficking is restricted by IFT and the transition zone, a barrier at the base of cilia. This restriction may allow the coordinated spread or movement of second messengers or effecter molecules. In addition, the juxtaposition of the basal body and the Golgi complex at the base of cilia may facilitate rapid trafficking of molecules like Smo into the cilia upon receiving extracellular signals and could help coordinate the cell cycle.
8.4 Primary Cilia and Medulloblastoma
One of the first bits of direct evidence showing that primary cilia play salient roles in cancer came from a study on medulloblastoma (Han et al. 2009). Medulloblastoma is the most common malignant brain tumor in children, accounting for ∼20% of childhood brain tumors. Medulloblastomas mostly arise in the cerebellum, but a recent study showed that a subgroup of medulloblastoma arises in the dorsal brain stem (Gibson et al. 2010). Several transcriptional profiling studies revealed that medulloblastoma comprises four principal subgroups, which have distinct demographic, clinical, transcriptional, and mutational characteristics (Thompson et al. 2006; Kool et al. 2008; Cho et al. 2011; Northcott et al. 2011; Taylor et al. 2012). These subgroups include Sonic Hedgehog (SHH, one of three mammalian Hedgehog proteins), WNT, subgroup 3, and subgroup 4. The SHH and WNT subgroups are named after the signaling pathways thought to drive tumorigenesis of that subgroup. Subgroup 3 often shows amplification of MYC. Molecular mechanisms that drive subgroup 4 have not been identified.
SHH subgroup medulloblastoma is characterized by aberrant activation of SHH signaling and constitutes about 25% of medulloblastoma cases. The SHH subgroup arises from granule neuron precursors (GNPs) in the cerebellum (Schuller et al. 2008; Yang et al. 2008). GNPs are produced from radial glia in the anterior roof of the fourth ventricle, known as the upper rhombic lip, and migrate rostrally to form the external granular layer (EGL) on the surface of the developing cerebellum (Altman and Bayer 1997). In the EGL, GNPs proliferate extensively to produce cerebellar granule neurons, the most abundant neurons that constitute more than half of the neurons in the central nervous system. Immature granule neurons produced from the EGL migrate inward, passing Purkinje neurons and forming the internal granule neuron layer. SHH secreted from Purkinje neurons is an essential mitogen for GNPs in the EGL (Dahmane and Ruiz i Altaba 1999; Wallace 1999; Wechsler-Reya and Scott 1999). Consistent with the critical role of primary cilia in Hedgehog signaling, GNPs lacking primary cilia failed to proliferate, resulting in severe hypoplasia and underdevelopment of the cerebellum (Chizhikov et al. 2007; Spassky et al. 2008).
While SHH signaling is essential for the proliferation of GNPs, abnormal activation of SHH signaling leads to uncontrolled expansion of GNPs, resulting in medulloblastoma (Hatten and Roussel 2011). A recent study revealed surprising dual roles of primary cilia in medulloblastoma development driven by abnormal activation of SHH signaling (Han et al. 2009). Mice expressing a constitutively active form of Smo (SmoM2), which was identified in medulloblastoma and basal cell carcinoma (Lam et al. 1999), in GNPs develop medulloblastoma (Hallahan et al. 2004; Mao et al. 2006; Schuller et al. 2008; Han et al. 2009). In these mice, SmoM2 concentrated in the primary cilia of tumor cells and required this organelle to induce tumors; concomitant removal of primary cilia in SmoM2-expressing cells completely blocked medulloblastoma development (Han et al. 2009). Unlike SmoM2, GNPs expressing a constitutively active form of GLI2, a downstream transcription factor, that lacks an N-terminal repressor domain (GLI2ΔN) did not form medulloblastoma (Roessler et al. 2005; Pasca di Magliano et al. 2006; Han et al. 2009). Surprisingly, however, concomitant removal of primary cilia in GNPs expressing GLI2ΔN resulted in 100% medulloblastoma development, suggesting that primary cilia suppress medulloblastoma development when the oncogenic mutation is in the GLI2 transcription factor. Thus, the primary cilium plays opposing dual roles in medulloblastoma: it is required for SmoM2 but suppressive for GLI2ΔN to induce medulloblastoma (Fig. 8.1). The molecular mechanism by which primary cilia suppress GLI2ΔN-driven medulloblastoma development remains to be determined. In the presence of primary cilia, Gli3 repressors may counteract GLI2ΔN and inhibit medulloblastoma development, whereas in the absence of primary cilia, Gli3 repressors do not form (Haycraft et al. 2005; Huangfu and Anderson 2005; Liu et al. 2005; May et al. 2005), which may allow GLI2ΔN to induce medulloblastoma. Alternatively, primary cilia may be required for another signaling pathway to suppress tumorigenesis. Primary cilia may also function as a general barrier for cell cycle entry, as discussed above. Similar opposing dual functions of primary cilia were observed in basal cell carcinoma driven by SmoM2 and GLI2ΔN (Wong et al. 2009). Taken together, these suggest that, to induce cancer, some oncogenic mutations may require intact primary cilia but others may require losing them. Notably, in support of this hypothesis, the presence or absence of primary cilia is tightly associated with specific subgroups of medulloblastoma (Fig. 8.2). In humans, primary cilia are almost exclusively present in the SHH and WNT subgroups of medulloblastoma but absent in subgroups 3 and 4 (Han et al. 2009). Thus, primary cilia may be required for the SHH and WNT subgroups but suppressive for subgroups 3 and 4. The presence of primary cilia in the WNT subgroup is somewhat contradictory to several studies showing that primary cilia constrain canonical WNT signaling; however, as discussed above, the role of primary cilia in WNT signaling is still controversial and specific to cell type. Recently developed mouse models each representing WNT-subgroup medulloblastoma (Gibson et al. 2010) and subgroup 3 (Kawauchi et al. 2012; Pei et al. 2012) will provide an excellent opportunity to investigate the role of primary cilia in these tumors. These investigations will also provide clues to whether the dual roles of primary cilia are generally applicable to cancers in addition to those driven by Hedgehog signaling.

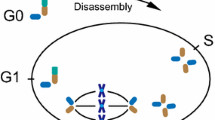
Dual and opposing roles of primary cilia in medulloblastoma formation. (a) SmoM2 is insensitive to inhibition by Ptch1 and constitutively localizes to primary cilia, where it inhibits production of repressor forms of Gli2 and Gli3 and induces production of activator forms. Uncontrolled activation of the signaling leads to medulloblastoma formation in hGFAP::Cre; SmoM2 fl/+ mice. (b) Without primary cilia, SmoM2 cannot activate downstream signaling, thus loss of primary cilia in hGFAP::Cre; SmoM2 fl/+; Kif3a fl/fl mice completely blocks medulloblastoma formation. (c, d) Constitutively active form of Gli2 (Gli2ΔN) is not sufficient to induce medulloblastoma in hGFAP::Cre; CLEG2 fl/+ mice. Loss of primary cilia in hGFAP::Cre; CLEG2 fl/+; Kif3a fl/fl mice allows Gli2ΔN to induce medulloblastoma. Repressor forms of Gli2 and/or Gli3, whose formation requires primary cilia, may inhibits the tumorigenic activity of Gli2ΔN. Alternatively, unknown mechanism through primary cilia may inhibit the tumorigenic activity of Gli2ΔN

Primary cilia are present in human medulloblastomas showing oncogenic activation of SHH or WNT signaling (a), but mostly absent in other molecular subgroups of medulloblastomas (b). Arrows indicate basal bodies stained with an antibody against pericentrin and arrowheads indicate primary cilia stained with an antibody against acetylated tubulin
Currently, SMO inhibitors are under clinical trials to treat SHH subgroup medulloblastoma and basal cell carcinoma. Although these clinical trials show promise for SMO inhibitors for treating medulloblastoma, resistance to a SMO inhibitor was observed in a patient who initially showed a dramatic response (Yauch et al. 2009). Subsequently, a point mutation in SMO conferring resistance to the SMO inhibitor was found in the medulloblastoma of this patient. Furthermore, in vivo and in vitro studies using the same SMO inhibitor revealed frequent appearance of resistance (Dijkgraaf et al. 2011). Currently, SMO is the only molecular target under clinical trials to treat medulloblastoma. Thus, it is necessary to develop strategies to overcome the resistance. Since Smo requires primary cilia to function and ciliogenesis requires a number of cellular processes involving a large number of proteins, primary cilia may provide multiple novel targets that can overcome resistance to SMO inhibitors.
8.5 Primary Cilia and Other Brain Cancers
Given that primary cilia play important roles in cell cycle progression and signaling pathways frequently involved in oncogenesis, they are also likely to play important roles in other brain cancers in addition to medulloblastoma. Here we will discuss two types of brain cancers, the deadliest brain cancers of children and adults, in which primary cilia may have important roles.
Diffuse intrinsic pontine gliomas (DIPGs) are diffusely infiltrative high-grade gliomas in the ventral pons. DIPGs affect mostly children, peaking at age 6–7 years, constituting 10–15% of pediatric brain cancer (Hawkins et al. 2011). DIPGs are extremely aggressive cancers that are almost universally fatal in less than a year. Yet, little is known about the biology of this tumor for which no effective therapy exists. The age- and region-specific natures of DIPGs suggest that these tumors arise from deregulation of a specific postnatal development process occurring in the ventral pons in children. Indeed, a recent study identified a putative neural precursor cell population positive for neural precursor markers, Nestin, Vimentin, and Olig2 in the ventral pons of both humans and mice, whose spatiotemporal distribution matches very closely to that of DIPGs (Monje et al. 2011). Interestingly, the Hedgehog signaling pathway was active in this precursor population in mice. Expression of SmoM2 increased proliferation of these cells in mice, leading to hypertrophy of the ventral pons. Furthermore, blocking Hedgehog signaling reduced self-renewal of neurospheres generated form human DIPGs, whereas addition of SHH increased self-renewal. Thus, aberrant activation of Hedgehog signaling may contribute to DIPG formation by driving proliferation of these precursor cells. Unlike medulloblastoma, however, expression of SmoM2 was not sufficient to induce DIPGs, suggesting that another hit is necessary for DIPGs to form. This is consistent with the fact that people having germline mutations of PTCH1 are predisposed to medulloblastoma but not DIPG (Johnson et al. 1996). Notably, recent genome-wide analyses identified frequent amplification or overexpression of PDGFRα, PDGF-A and IGF1R, all of which encode proteins that may signal through primary cilia (Zarghooni et al. 2010; Paugh et al. 2011). Cancer cells that have amplification or overexpression of these oncogenes may rely on normal signaling mechanisms through primary cilia. Thus, primary cilia may play important roles in the development of this devastating disease. It would be interesting to find whether DIPGs are ciliated and co-activation of SHH and PDGFRα can cause DIPGs in mouse models, as well as to test the role of primary cilia in such mouse models. If primary cilia play a significant role, targeting ciliogenesis would be a valid treatment option.
Glioma is the most frequent brain tumor in adults, and malignant glioma (glioblastoma multiforme, GBM) comprises 80% of malignant tumors in the central nervous system (Chen et al. 2012). Currently, GBM patients’ 5 year survival rate is less than 5%, and median survival is about 1 year. Two recent genome-wide studies including gene expression profiling, DNA copy number variation, protein-encoding gene sequencing, and DNA methylation status revealed three core pathways that commonly mutated in GBM: the p53 pathway, the RB pathway, and the RTK pathway (Parsons et al. 2008; TCGA 2008). The majority (74%) of GBMs had alterations in all three pathways, which enables cancer cells to proliferate unrestrictedly, escaping from cell-cycle checkpoints, senescence, and apoptosis. On the other hand, alterations affecting components in the same core pathway were mutually exclusive. Among RTKs, frequent aberrations were found in EGFR, ERBB2, PDGFRα, and MET. Similar to what was seen in medulloblastoma, several gene expression profiling studies identified distinct molecular subtypes in GBM (Vitucci et al. 2011). A recent study grouped GBM into four subtypes: proneural, neural, classical, and mesenchymal (Verhaak et al. 2010). By integrating gene expression profiles with the previous genome-wide analysis of GBM, this study showed that aberrant status of EGFR, NF1, and PDGFRα/Isocitrate dehydrogenase 1 define the classic, mesenchymal, and proneural subtypes, respectively. Notably, the classic subtype expressed high levels of Hedgehog (SMO, GLI2, and GAS1) and NOTCH (NOTCH3, JAG1, and LFNG) signaling components. Given the important role of primary cilia in Hedgehog, Notch, and PDGFRα signaling pathways, the classic and proneural subtypes may require primary cilia for their growth. Thus, it would be interesting to determine whether the presence or absence of primary cilia is associated with specific GBM subtypes. Such associations may indicate distinct roles of primary cilia in different GBM subtypes. Primary cilia are absent in several GBM cell lines (Moser et al. 2009); however, the molecular subtypes to which the cell lines belong are unknown. If the presence or absence of primary cilia is associated with specific GBM subgroups and such an association is important for oncogenesis, primary cilia will be an important diagnostic tool and a treatment target for GBM.
Although the cancer stem cell (CSC) theory is controversial, it suggests that primary cilia may have important roles in GBM and possibly in other brain cancers. CSCs are a subpopulation of cells in a cancer that can self-renew and give rise to highly heterogeneous cancer cells that make up the bulk of cancer. GBM CSCs were one of the first CSCs isolated from solid cancers (Singh et al. 2004). Resistance to radiation and chemotherapies is thought to be partly due to CSCs, which have preferentially active DNA repair pathways (Bao et al. 2006) and high levels of ATP-binding cassette transporters to export chemotherapy agents (Bleau et al. 2009); thus, CSCs have important implications for cancer targeting strategy. CSCs are thought to have properties similar to those of somatic stem cells. A number of studies have shown that signaling pathways that critically regulate the behavior of normal somatic stem cells also regulate that of GBM CSCs (Clark et al. 2007; Takebe et al. 2011). These pathways include Hedgehog, Wnt, and Notch signaling pathways, for which primary cilia play important roles. Consistently, recent studies showed that expression of Gli1 and β-catenin are associated with recurrence after therapy and poor prognosis in GBM patients (Rossi et al. 2011; Kim et al. 2012). Therefore, targeting these pathways is a vital therapeutic approach to increase the efficacy of radiation and chemotherapies. Understanding the mechanism by which primary cilia function in these signaling pathways in normal stem cells and CSCs will be important for developing such a therapeutic intervention.
8.6 Conclusion
Brain cancer is a complex and heterogeneous disease. Its treatments, however, are largely similar, including surgical resection, radiation, and chemotherapy, thus resulting in individually different outcomes. Recent advances in genome-wide studies on large cohorts of brain cancer patients elucidated that cancers that otherwise appear identical are highly heterogeneous at the molecular level, with distinctive oncogenic mutations and gene expression profiles. These recent advances call for new treatment paradigms building on better understandings of the molecular and cellular processes involved in initiation and progression of particular brain cancer types. We envision that investigating the function of primary cilia together with oncogenic mutations specific to distinct cancer types will reveal oncogenic mechanisms that were not appreciated previously. We also envision that primary cilia hold a great therapeutic potential for treatment for brain cancer patients. Some cancers have primary cilia, but others do not (Wheatley 1995; Han et al. 2009; Schraml et al. 2009; Seeley et al. 2009; Wong et al. 2009; Yuan et al. 2010; Kim et al. 2011a). Most cells in our body have the primary cilium; thus, some cancers may have it as a default. Building and maintaining primary cilia requires complex processes involving a wide variety of proteins that function in cell cycle progression (Pugacheva et al. 2007; Spektor et al. 2007; Kim et al. 2011b; Li et al. 2011), cytoskeletal dynamics (Kim et al. 2010a), apicobasal polarity (Fan et al. 2004), planar cell polarity (Kim et al. 2010b; Wallingford 2010), intraflagellar transport (Rosenbaum and Witman 2002), vesicle trafficking (Nachury et al. 2007; Zuo et al. 2009; Knodler et al. 2010), and transcriptional regulation (Thomas et al. 2010). Thus, some cancers may have lost primary cilia secondarily as they progress and accumulate mutations. In cancers driven by Hedgehog signaling, however, the presence or absence of primary cilia directly controls oncogenesis (Han et al. 2009; Wong et al. 2009). Furthermore, the presence or absence of primary cilia is associated with specific cancer types. Thus, the status of primary cilia in a particular cancer may reflect the role of primary cilia in that cancer; some cancers may keep primary cilia and others may eliminate them for growth and progression. Primary cilia will be important targets for such cancers. Since many proteins are involved in ciliogenesis, inhibiting ciliogenesis or ciliary function may be a plausible strategy to treat cancers that require primary cilia for their growth. Indeed, a high-throughput screening for inhibitors of Hedgehog signaling discovered a small molecule that inhibits cytoplasmic dynein and ciliogenesis (Firestone et al. 2012). It will be challenging to restore primary cilia in cancers that have eliminated them for growth. Yet, recent studies showed that small molecules targeting signaling molecules or fatty acid synthesis can restore primary cilia even in cancer cells (Wang et al. 2009; Willemarck et al. 2010). Therefore, research in primary cilia will open up a completely new avenue of research to understand the biology and treatment of cancers.
References
Alkuraya FS, Cai X, Emery C, Mochida GH, Al-Dosari MS, Felie JM, Hill RS, Barry BJ, Partlow JN, Gascon GG, Kentab A, Jan M, Shaheen R, Feng Y, Walsh CA (2011) Human mutations in NDE1 cause extreme microcephaly with lissencephaly [corrected]. Am J Hum Genet 88:536–547
Altman J, Bayer SA (1997) Development of the cerebellar system: in relation to its evolution, structure, and functions. CRC Press, Boca Raton
Bakircioglu M et al (2011) The essential role of centrosomal NDE1 in human cerebral cortex neurogenesis. Am J Hum Genet 88:523–535
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444:756–760
Berbari NF, Kin NW, Sharma N, Michaud EJ, Kesterson RA, Yoder BK (2011) Mutations in Traf3ip1 reveal defects in ciliogenesis, embryonic development, and altered cell size regulation. Dev Biol 360:66–76
Bielas SL et al (2009) Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat Genet 41:1032–1036
Bishop CL, Bergin AM, Fessart D, Borgdorff V, Hatzimasoura E, Garbe JC, Stampfer MR, Koh J, Beach DH (2010) Primary cilium-dependent and -independent Hedgehog signaling inhibits p16(INK4A). Mol Cell 40:533–547
Bleau AM, Hambardzumyan D, Ozawa T, Fomchenko EI, Huse JT, Brennan CW, Holland EC (2009) PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell 4:226–235
Boehlke C, Kotsis F, Patel V, Braeg S, Voelker H, Bredt S, Beyer T, Janusch H, Hamann C, Godel M, Muller K, Herbst M, Hornung M, Doerken M, Kottgen M, Nitschke R, Igarashi P, Walz G, Kuehn EW (2010) Primary cilia regulate mTORC1 activity and cell size through Lkb1. Nat Cell Biol 12:1115–1122
Cano DA, Murcia NS, Pazour GJ, Hebrok M (2004) Orpk mouse model of polycystic kidney disease reveals essential role of primary cilia in pancreatic tissue organization. Development 131:3457–3467
Caspary T, Larkins CE, Anderson KV (2007) The graded response to Sonic Hedgehog depends on cilia architecture. Dev Cell 12:767–778
Chen MH, Wilson CW, Li YJ, Law KK, Lu CS, Gacayan R, Zhang X, Hui CC, Chuang PT (2009) Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev 23:1910–1928
Chen J, McKay RM, Parada LF (2012) Malignant glioma: lessons from genomics, mouse models, and stem cells. Cell 149:36–47
Chizhikov VV, Davenport J, Zhang Q, Shih EK, Cabello OA, Fuchs JL, Yoder BK, Millen KJ (2007) Cilia proteins control cerebellar morphogenesis by promoting expansion of the granule progenitor pool. J Neurosci 27:9780–9789
Cho YJ, Tsherniak A, Tamayo P, Santagata S, Ligon A, Greulich H, Berhoukim R, Amani V, Goumnerova L, Eberhart CG, Lau CC, Olson JM, Gilbertson RJ, Gajjar A, Delattre O, Kool M, Ligon K, Meyerson M, Mesirov JP, Pomeroy SL (2011) Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J Clin Oncol 29:1424–1430
Christensen ST, Clement CA, Satir P, Pedersen LB (2012) Primary cilia and coordination of receptor tyrosine kinase (RTK) signalling. J Pathol 226:172–184
Clark PA, Treisman DM, Ebben J, Kuo JS (2007) Developmental signaling pathways in brain tumor-derived stem-like cells. Dev Dyn 236:3297–3308
Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF (2005) Vertebrate Smoothened functions at the primary cilium. Nature 437:1018–1021
Corbit KC, Shyer AE, Dowdle WE, Gaulden J, Singla V, Chen MH, Chuang PT, Reiter JF (2008) Kif3a constrains beta-catenin-dependent Wnt signalling through dual ciliary and non-ciliary mechanisms. Nat Cell Biol 10:70–76
Cortellino S, Wang C, Wang B, Bassi MR, Caretti E, Champeval D, Calmont A, Jarnik M, Burch J, Zaret KS, Larue L, Bellacosa A (2009) Defective ciliogenesis, embryonic lethality and severe impairment of the Sonic Hedgehog pathway caused by inactivation of the mouse complex A intraflagellar transport gene Ift122/Wdr10, partially overlapping with the DNA repair gene Med1/Mbd4. Dev Biol 325:225–237
Dahmane N, Ruiz i Altaba A (1999) Sonic hedgehog regulates the growth and patterning of the cerebellum. Development 126:3089–3100
Danilov AI, Gomes-Leal W, Ahlenius H, Kokaia Z, Carlemalm E, Lindvall O (2009) Ultrastructural and antigenic properties of neural stem cells and their progeny in adult rat subventricular zone. Glia 57:136–152
DiBella LM, Park A, Sun Z (2009) Zebrafish Tsc1 reveals functional interactions between the cilium and the TOR pathway. Hum Mol Genet 18:595–606
Dijkgraaf GJ, Alicke B, Weinmann L, Januario T, West K, Modrusan Z, Burdick D, Goldsmith R, Robarge K, Sutherlin D, Scales SJ, Gould SE, Yauch RL, de Sauvage FJ (2011) Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer Res 71:435–444
Ezratty EJ, Stokes N, Chai S, Shah AS, Williams SE, Fuchs E (2011) A role for the primary cilium in Notch signaling and epidermal differentiation during skin development. Cell 145:1129–1141
Fan S, Hurd TW, Liu CJ, Straight SW, Weimbs T, Hurd EA, Domino SE, Margolis B (2004) Polarity proteins control ciliogenesis via kinesin motor interactions. Curr Biol 14:1451–1461
Feng Y, Walsh CA (2004) Mitotic spindle regulation by Nde1 controls cerebral cortical size. Neuron 44:279–293
Firestone AJ, Weinger JS, Maldonado M, Barlan K, Langston LD, O’Donnell M, Gelfand VI, Kapoor TM, Chen JK (2012) Small-molecule inhibitors of the AAA + ATPase motor cytoplasmic dynein. Nature 484:125–129
Gerdes JM, Liu Y, Zaghloul NA, Leitch CC, Lawson SS, Kato M, Beachy PA, Beales PL, Demartino GN, Fisher S, Badano JL, Katsanis N (2007) Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat Genet 39(11):1350–1360
Gibson P et al (2010) Subtypes of medulloblastoma have distinct developmental origins. Nature 468:1095–1099
Goetz SC, Anderson KV (2010) The primary cilium: a signalling centre during vertebrate development. Nat Rev 11:331–344
Hallahan AR, Pritchard JI, Hansen S, Benson M, Stoeck J, Hatton BA, Russell TL, Ellenbogen RG, Bernstein ID, Beachy PA, Olson JM (2004) The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Cancer Res 64:7794–7800
Han YG, Spassky N, Romaguera-Ros M, Garcia-Verdugo JM, Aguilar A, Schneider-Maunoury S, Alvarez-Buylla A (2008) Hedgehog signaling and primary cilia are required for the formation of adult neural stem cells. Nat Neurosci 11:277–284
Han YG, Kim HJ, Dlugosz AA, Ellison DW, Gilbertson RJ, Alvarez-Buylla A (2009) Dual and opposing roles of primary cilia in medulloblastoma development. Nat Med 15:1062–1065
Hatten ME, Roussel MF (2011) Development and cancer of the cerebellum. Trends Neurosci 34:134–142
Hawkins CE, Bartels U, Bouffet E (2011) Molecular genetic approaches and potential new therapeutic strategies for pediatric diffuse intrinsic pontine glioma. J Clin Oncol 29:3956–3957
Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK (2005) Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet 1:e53
Huang P, Schier AF (2009) Dampened Hedgehog signaling but normal Wnt signaling in zebrafish without cilia. Development 136:3089–3098
Huangfu D, Anderson KV (2005) Cilia and Hedgehog responsiveness in the mouse. Proc Natl Acad Sci USA 102:11325–11330
Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV (2003) Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 426:83–87
Humke EW, Dorn KV, Milenkovic L, Scott MP, Rohatgi R (2010) The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev 24:670–682
Jacoby M, Cox JJ, Gayral S, Hampshire DJ, Ayub M, Blockmans M, Pernot E, Kisseleva MV, Compere P, Schiffmann SN, Gergely F, Riley JH, Perez-Morga D, Woods CG, Schurmans S (2009) INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat Genet 41:1027–1031
Jia J, Kolterud A, Zeng H, Hoover A, Teglund S, Toftgard R, Liu A (2009) Suppressor of Fused inhibits mammalian Hedgehog signaling in the absence of cilia. Dev Biol 330:452–460
Johnson RL, Rothman AL, Xie J, Goodrich LV, Bare JW, Bonifas JM, Quinn AG, Myers RM, Cox DR, Epstein EH Jr, Scott MP (1996) Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science (NY) 272:1668–1671
Jonassen JA, San Agustin J, Follit JA, Pazour GJ (2008) Deletion of IFT20 in the mouse kidney causes misorientation of the mitotic spindle and cystic kidney disease. J Cell Biol 183:377–384
Jones C, Roper VC, Foucher I, Qian D, Banizs B, Petit C, Yoder BK, Chen P (2008) Ciliary proteins link basal body polarization to planar cell polarity regulation. Nat Genet 40:69–77
Kawauchi D, Robinson G, Uziel T, Gibson P, Rehg J, Gao C, Finkelstein D, Qu C, Pounds S, Ellison DW, Gilbertson RJ, Roussel MF (2012) A mouse model of the most aggressive subgroup of human medulloblastoma. Cancer Cell 21:168–180
Kim J, Lee JE, Heynen-Genel S, Suyama E, Ono K, Lee K, Ideker T, Aza-Blanc P, Gleeson JG (2010a) Functional genomic screen for modulators of ciliogenesis and cilium length. Nature 464:1048–1051
Kim SK, Shindo A, Park TJ, Oh EC, Ghosh S, Gray RS, Lewis RA, Johnson CA, Attie-Bittach T, Katsanis N, Wallingford JB (2010b) Planar cell polarity acts through septins to control collective cell movement and ciliogenesis. Science (NY) 329:1337–1340
Kim J, Dabiri S, Seeley ES (2011a) Primary cilium depletion typifies cutaneous melanoma in situ and malignant melanoma. PLoS One 6:e27410
Kim S, Zaghloul NA, Bubenshchikova E, Oh EC, Rankin S, Katsanis N, Obara T, Tsiokas L (2011b) Nde1-mediated inhibition of ciliogenesis affects cell cycle re-entry. Nat Cell Biol 13:351–360
Kim Y, Kim KH, Lee J, Lee YA, Kim M, Lee SJ, Park K, Yang H, Jin J, Joo KM, Nam DH (2012) Wnt activation is implicated in glioblastoma radioresistance. Lab Invest 92:466–473
Kisseleva MV, Cao L, Majerus PW (2002) Phosphoinositide-specific inositol polyphosphate 5-phosphatase IV inhibits Akt/protein kinase B phosphorylation and leads to apoptotic cell death. J Biol Chem 277:6266–6272
Knodler A, Feng S, Zhang J, Zhang X, Das A, Peranen J, Guo W (2010) Coordination of Rab8 and Rab11 in primary ciliogenesis. Proc Natl Acad Sci USA 107:6346–6351
Ko HW, Norman RX, Tran J, Fuller KP, Fukuda M, Eggenschwiler JT (2010) Broad-minded links cell cycle-related kinase to cilia assembly and hedgehog signal transduction. Dev Cell 18:237–247
Kool M, Koster J, Bunt J, Hasselt NE, Lakeman A, van Sluis P, Troost D, Meeteren NS, Caron HN, Cloos J, Mrsic A, Ylstra B, Grajkowska W, Hartmann W, Pietsch T, Ellison D, Clifford SC, Versteeg R (2008) Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One 3:e3088
Kopan R, Ilagan MX (2009) The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137:216–233
Lam CW, Xie J, To KF, Ng HK, Lee KC, Yuen NW, Lim PL, Chan LY, Tong SF, McCormick F (1999) A frequent activated smoothened mutation in sporadic basal cell carcinomas. Oncogene 18:833–836
Li A, Saito M, Chuang JZ, Tseng YY, Dedesma C, Tomizawa K, Kaitsuka T, Sung CH (2011) Ciliary transition zone activation of phosphorylated Tctex-1 controls ciliary resorption, S-phase entry and fate of neural progenitors. Nat Cell Biol 13:402–411
Lin F, Hiesberger T, Cordes K, Sinclair AM, Goldstein LS, Somlo S, Igarashi P (2003) Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc Natl Acad Sci USA 100:5286–5291
Liu A, Wang B, Niswander LA (2005) Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development 132:3103–3111
Logan CY, Nusse R (2004) The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 20:781–810
Ma R, Li WP, Rundle D, Kong J, Akbarali HI, Tsiokas L (2005) PKD2 functions as an epidermal growth factor-activated plasma membrane channel. Mol Cell Biol 25:8285–8298
Mao J, Ligon KL, Rakhlin EY, Thayer SP, Bronson RT, Rowitch D, McMahon AP (2006) A novel somatic mouse model to survey tumorigenic potential applied to the Hedgehog pathway. Cancer Res 66:10171–10178
May SR, Ashique AM, Karlen M, Wang B, Shen Y, Zarbalis K, Reiter J, Ericson J, Peterson AS (2005) Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev Biol 287:378–389
Monje M, Mitra SS, Freret ME, Raveh TB, Kim J, Masek M, Attema JL, Li G, Haddix T, Edwards MS, Fisher PG, Weissman IL, Rowitch DH, Vogel H, Wong AJ, Beachy PA (2011) Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proc Natl Acad Sci USA 108:4453–4458
Moser JJ, Fritzler MJ, Rattner JB (2009) Primary ciliogenesis defects are associated with human astrocytoma/glioblastoma cells. BMC Cancer 9:448
Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peranen J, Merdes A, Slusarski DC, Scheller RH, Bazan JF, Sheffield VC, Jackson PK (2007) A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 129:1201–1213
Northcott PA, Korshunov A, Witt H, Hielscher T, Eberhart CG, Mack S, Bouffet E, Clifford SC, Hawkins CE, French P, Rutka JT, Pfister S, Taylor MD (2011) Medulloblastoma comprises four distinct molecular variants. J Clin Oncol 29:1408–1414
Ocbina PJ, Tuson M, Anderson KV (2009) Primary cilia are not required for normal canonical Wnt signaling in the mouse embryo. PLoS One 4:e6839
Parsons DW et al (2008) An integrated genomic analysis of human glioblastoma multiforme. Science (NY) 321:1807–1812
Pasca di Magliano M, Sekine S, Ermilov A, Ferris J, Dlugosz AA, Hebrok M (2006) Hedgehog/Ras interactions regulate early stages of pancreatic cancer. Genes Dev 20:3161–3173
Paugh BS, Broniscer A, Qu C, Miller CP, Zhang J, Tatevossian RG, Olson JM, Geyer JR, Chi SN, da Silva NS, Onar-Thomas A, Baker JN, Gajjar A, Ellison DW, Baker SJ (2011) Genome-wide analyses identify recurrent amplifications of receptor tyrosine kinases and cell-cycle regulatory genes in diffuse intrinsic pontine glioma. J Clin Oncol 29:3999–4006
Pei Y, Moore CE, Wang J, Tewari AK, Eroshkin A, Cho YJ, Witt H, Korshunov A, Read TA, Sun JL, Schmitt EM, Miller CR, Buckley AF, McLendon RE, Westbrook TF, Northcott PA, Taylor MD, Pfister SM, Febbo PG, Wechsler-Reya RJ (2012) An animal model of MYC-driven medulloblastoma. Cancer Cell 21:155–167
Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA (2007) HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell 129:1351–1363
Rieder CL, Jensen CG, Jensen LC (1979) The resorption of primary cilia during mitosis in a vertebrate (PtK1) cell line. J Ultrastruct Res 68:173–185
Roessler E, Ermilov AN, Grange DK, Wang A, Grachtchouk M, Dlugosz AA, Muenke M (2005) A previously unidentified amino-terminal domain regulates transcriptional activity of wild-type and disease-associated human GLI2. Hum Mol Genet 14:2181–2188
Rohatgi R, Milenkovic L, Scott MP (2007) Patched1 regulates hedgehog signaling at the primary cilium. Science (NY) 317:372–376
Rosenbaum JL, Witman GB (2002) Intraflagellar transport. Nat Rev Mol Cell Biol 3:813–825
Ross AJ et al (2005) Disruption of Bardet-Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat Genet 37:1135–1140
Rossi M, Magnoni L, Miracco C, Mori E, Tosi P, Pirtoli L, Tini P, Oliveri G, Cosci E, Bakker A (2011) Beta-catenin and Gli1 are prognostic markers in glioblastoma. Cancer Biol Ther 11:753–761
Schneider L, Clement CA, Teilmann SC, Pazour GJ, Hoffmann EK, Satir P, Christensen ST (2005) PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr Biol 15:1861–1866
Schneider L, Stock CM, Dieterich P, Jensen BH, Pedersen LB, Satir P, Schwab A, Christensen ST, Pedersen SF (2009) The Na+/H+ exchanger NHE1 is required for directional migration stimulated via PDGFR-alpha in the primary cilium. J Cell Biol 185:163–176
Schraml P, Frew IJ, Thoma CR, Boysen G, Struckmann K, Krek W, Moch H (2009) Sporadic clear cell renal cell carcinoma but not the papillary type is characterized by severely reduced frequency of primary cilia. Mod Pathol 22:31–36
Schuller U, Heine VM, Mao J, Kho AT, Dillon AK, Han YG, Huillard E, Sun T, Ligon AH, Qian Y, Ma Q, Alvarez-Buylla A, McMahon AP, Rowitch DH, Ligon KL (2008) Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell 14:123–134
Seeley ES, Carriere C, Goetze T, Longnecker DS, Korc M (2009) Pancreatic cancer and precursor pancreatic intraepithelial neoplasia lesions are devoid of primary cilia. Cancer Res 69:422–430
Simons M, Gloy J, Ganner A, Bullerkotte A, Bashkurov M, Kronig C, Schermer B, Benzing T, Cabello OA, Jenny A, Mlodzik M, Polok B, Driever W, Obara T, Walz G (2005) Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat Genet 37:537–543
Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB (2004) Identification of human brain tumour initiating cells. Nature 432:396–401
Spassky N, Han YG, Aguilar A, Strehl L, Besse L, Laclef C, Ros MR, Garcia-Verdugo JM, Alvarez-Buylla A (2008) Primary cilia are required for cerebellar development and Shh-dependent expansion of progenitor pool. Dev Biol 317:246–259
Spektor A, Tsang WY, Khoo D, Dynlacht BD (2007) Cep97 and CP110 suppress a cilia assembly program. Cell 130:678–690
Takebe N, Harris PJ, Warren RQ, Ivy SP (2011) Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol 8:97–106
Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, Eberhart CG, Parsons DW, Rutkowski S, Gajjar A, Ellison DW, Lichter P, Gilbertson RJ, Pomeroy SL, Kool M, Pfister SM (2012) Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 123(4):465–472
TCGA (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455:1061–1068
Teilmann SC, Christensen ST (2005) Localization of the angiopoietin receptors Tie-1 and Tie-2 on the primary cilia in the female reproductive organs. Cell Biol Int 29:340–346
Thomas J, Morle L, Soulavie F, Laurencon A, Sagnol S, Durand B (2010) Transcriptional control of genes involved in ciliogenesis: a first step in making cilia. Biol Cell 102:499–513
Thompson MC, Fuller C, Hogg TL, Dalton J, Finkelstein D, Lau CC, Chintagumpala M, Adesina A, Ashley DM, Kellie SJ, Taylor MD, Curran T, Gajjar A, Gilbertson RJ (2006) Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol 24:1924–1931
Tran PV, Haycraft CJ, Besschetnova TY, Turbe-Doan A, Stottmann RW, Herron BJ, Chesebro AL, Qiu H, Scherz PJ, Shah JV, Yoder BK, Beier DR (2008) THM1 negatively modulates mouse sonic hedgehog signal transduction and affects retrograde intraflagellar transport in cilia. Nat Genet 40:403–410
Tucker RW, Pardee AB, Fujiwara K (1979) Centriole ciliation is related to quiescence and DNA synthesis in 3 T3 cells. Cell 17:527–535
Tukachinsky H, Lopez LV, Salic A (2010) A mechanism for vertebrate Hedgehog signaling: recruitment to cilia and dissociation of SuFu-Gli protein complexes. J Cell Biol 191:415–428
Verhaak RG et al (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17:98–110
Vitucci M, Hayes DN, Miller CR (2011) Gene expression profiling of gliomas: merging genomic and histopathological classification for personalised therapy. Br J Cancer 104:545–553
Wallace VA (1999) Purkinje-cell-derived Sonic Hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr Biol 9:445–448
Wallingford JB (2010) Planar cell polarity signaling, cilia and polarized ciliary beating. Curr Opin Cell Biol 22:597–604
Wallingford JB, Mitchell B (2011) Strange as it may seem: the many links between Wnt signaling, planar cell polarity, and cilia. Genes Dev 25:201–213
Wang G, Krishnamurthy K, Bieberich E (2009) Regulation of primary cilia formation by ceramide. J Lipid Res 50:2103–2110
Wechsler-Reya RJ, Scott MP (1999) Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron 22:103–114
Wheatley DN (1995) Primary cilia in normal and pathological tissues. Pathobiology: J Immunopathol Molecul Cell Biol 63:222–238
Willemarck N, Rysman E, Brusselmans K, Van Imschoot G, Vanderhoydonc F, Moerloose K, Lerut E, Verhoeven G, van Roy F, Vleminckx K, Swinnen JV (2010) Aberrant activation of fatty acid synthesis suppresses primary cilium formation and distorts tissue development. Cancer Res 70:9453–9462
Wong SY, Seol AD, So PL, Ermilov AN, Bichakjian CK, Epstein EH Jr, Dlugosz AA, Reiter JF (2009) Primary cilia can both mediate and suppress Hedgehog pathway-dependent tumorigenesis. Nat Med 15:1055–1061
Wu J, Du H, Wang X, Mei C, Sieck GC, Qian Q (2009) Characterization of primary cilia in human airway smooth muscle cells. Chest 136:561–570
Yang ZJ, Ellis T, Markant SL, Read TA, Kessler JD, Bourboulas M, Schuller U, Machold R, Fishell G, Rowitch DH, Wainwright BJ, Wechsler-Reya RJ (2008) Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer Cell 14:135–145
Yauch RL, Dijkgraaf GJ, Alicke B, Januario T, Ahn CP, Holcomb T, Pujara K, Stinson J, Callahan CA, Tang T, Bazan JF, Kan Z, Seshagiri S, Hann CL, Gould SE, Low JA, Rudin CM, de Sauvage FJ (2009) Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science (NY 326:572–574
Yuan K, Frolova N, Xie Y, Wang D, Cook L, Kwon YJ, Steg AD, Serra R, Frost AR (2010) Primary cilia are decreased in breast cancer: analysis of a collection of human breast cancer cell lines and tissues. J Histochem Cytochem 58:857–870
Yuan S, Li J, Diener DR, Choma MA, Rosenbaum JL, Sun Z (2012) Target-of-rapamycin complex 1 (Torc1) signaling modulates cilia size and function through protein synthesis regulation. Proc Natl Acad Sci USA 109:2021–2026
Zarghooni M, Bartels U, Lee E, Buczkowicz P, Morrison A, Huang A, Bouffet E, Hawkins C (2010) Whole-genome profiling of pediatric diffuse intrinsic pontine gliomas highlights platelet-derived growth factor receptor alpha and poly (ADP-ribose) polymerase as potential therapeutic targets. J Clin Oncol 28:1337–1344
Zeng H, Jia J, Liu A (2010) Coordinated translocation of mammalian Gli proteins and suppressor of fused to the primary cilium. PLoS One 5:e15900
Zhu D, Shi S, Wang H, Liao K (2009) Growth arrest induces primary-cilium formation and sensitizes IGF-1-receptor signaling during differentiation induction of 3 T3-L1 preadipocytes. J Cell Sci 122:2760–2768
Zuo X, Guo W, Lipschutz JH (2009) The exocyst protein Sec10 is necessary for primary ciliogenesis and cystogenesis in vitro. Mol Biol Cell 20:2522–2529
Acknowledgments
American Lebanese Syrian Associated Charities (Y.-G.H.) and the Cancer Center Support Grant CA021765 from the National Cancer Institute (Y.-G.H.) supported this work.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Hou, S., Han, YG. (2013). Primary Cilia and Brain Cancer. In: Tucker, K., Caspary, T. (eds) Cilia and Nervous System Development and Function. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-5808-7_8
Download citation
DOI: https://doi.org/10.1007/978-94-007-5808-7_8
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-5807-0
Online ISBN: 978-94-007-5808-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)