
Abstract
Structural characterization of proteins by NMR spectroscopy begins with the process of sequence specific resonance assignments in which the 1H, 13C and 15N chemical shifts of all backbone and side-chain nuclei in the polypeptide are assigned. This process requires different isotope labeled forms of the protein together with specific experiments for establishing the sequential connectivity between the neighboring amino acid residues. In the case of spectral overlap, it is useful to identify spin systems corresponding to the different amino acid types selectively. With isotope labeling this can be achieved in two ways: (i) amino acid selective labeling or (ii) amino acid selective ‘unlabeling’. This chapter describes both these methods with more emphasis on selective unlabeling describing the various practical aspects. The recent developments involving combinatorial selective labeling and unlabeling are also discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
One of the first steps in elucidating the structure or dynamics of proteins is sequence specific resonance assignments. In this part of the structure determination process the identity of each resonance or peak observed in a given spectrum is established. In the case of small proteins (molecular mass <10 kDa) this can be accomplished using a set of two-dimensional (2D) homonuclear (1H-1H) correlation experiments [1, 2]. As the size of the protein increases beyond 10 kDa the number of observed resonances also increases. Consequently, given that chemical shift correlations of a given type are generally confined to the same region of the spectrum irrespective of the amino acid type or protein (e.g., amide 1H signals almost always occur in the range of 6–10 ppm or 13Cα-1Hα correlations occur in the range of 45–65 ppm etc.) the increase in the number of resonances results in their overlap with each other in the spectrum. Such spectral congestion can be alleviated by resorting to heteronuclear three or higher dimensional NMR experiments which provide resolution by correlating a larger number of different chemical shifts [2] using a protein sample enriched uniformly with 13C/15N/2H isotopes. However, in systems which are plagued with severe spectral overlap and/or chemical shift degeneracy such as proteins in solid state [3] (also discussed in Chap. 3 of this book), membrane [4, 5] or structurally disordered proteins [6] high dimensional shift correlations fail at times to provide sufficient resolution to accomplish unambiguous resonance assignments. Hence reduction of spectral overlap or spectral ‘simplification’ becomes pivotal. One solution is to reduce the number of peaks observed in the spectrum by detecting peaks selectively from a given amino acid type in the protein. This also helps in sequence specific resonance assignments if the type of residues around (i.e., the N-terminal or the C-terminal neighbor of) the selected residue can be identified and placed in the primary sequence. This chapter focuses on the different isotope labeling schemes to achieve the objectives of spectral simplification and sequence specific resonance assignments.
Systematic study of isotope labeling in proteins started in 1960s and one of the first amino acid type selective labeling method involved incorporation of specific protonated amino acids against a deuterated background [7]. Subsequently, selective incorporation of 13C/15N-labeled amino acids against an unlabeled (12C/14N) background was developed [8, 9]. In general, selective identification and assignment of different amino acid types can be accomplished in one of three ways: (i) using NMR experiments which detect a single or a set of amino acid-type(s) based on a specific magnetization transfer [10–18], (ii) selective 13C/15N labeling of amino acids using cell-based or cell-free methods [8, 19, 20] or/and (iii) selective unlabeling of amino acids using cell-based methods [21–25]. In the first approach, NMR experiments are implemented which exploit the topology of the side-chain of different amino acids. The magnetization transfer pathways in these experiments are designed to specifically detect a given amino acid type in the spectrum [10–18]. This approach suffers a loss in sensitivity with increase in size of proteins due to the long delay periods used in the pulse sequences for obtaining the desired magnetization transfer. Moreover, these experiments cannot be used for proteins which lose side-chain 1H upon deuteration.
The second approach involves selective labeling (13C/15N) of specific amino acid types in the protein while keeping the rest of the amino acids in the unlabeled form (i.e., 12C/14N). This is achieved either by cell-based [8, 9] or cell-free (in vitro) methods [20, 26, 27]. In the cell-based method the host organism is supplied with the desired isotopically labeled (13C/15N) amino acid, while supplying the rest of the amino acids in unlabeled form [8]. Isotope labeling of a specific site in the desired amino acid type can also be achieved using the appropriate site-selectively labeled amino acids or their precursors (e.g. methyl groups [28, 29]). In the cell-free based approach selective labelling is accomplished in vitro using the necessary components of the machinery used by the cells for protein biosynthesis [20, 26, 27, 30]. In recent years, this approach has emerged as a preferred alternative due to the following dis-advantages of the cell-based methods: (i) mis-incorporation of isotope labels at undesired sites (also known as ‘isotope scrambling’) and (ii) difficulty to produce certain types of proteins such as membrane proteins and/or those which are toxic to the cells expressing them. These are overcome to some extent in the cell-free methods. Cell free methods are also preferable in terms of the costs involved if site-selectively labeled amino acids need to be incorporated [31]. However, the cell-based methods have remained popular due to ease of expressing proteins with high yields especially in organisms such as E. Coli.
One of the drawbacks of the selective labeling approach described above is the requirement of expensive 13C/15N/2H (site selectively) enriched amino acids resulting in high costs especially if the protein yields are low, An inexpensive alternative is the method of amino acid selective “unlabeling” or ‘reverse’/‘inverse’ labeling [21–25]. This involves selective unlabeling of specific amino acid-types in the protein keeping the other amino acids uniformly 13C/15N labeled. This is accomplished by supplying the host microorganism with 15NH4Cl or/and 13C (1H/2H) -D-glucose as the sole source of nitrogen and carbon, respectively, along with the 12C/14N form of the desired amino acid(s) to be selectively unlabeled. As a result, resonances from residues that are unlabeled are not observed in the NMR spectra. A comparison of the spectrum is then made with a spectrum acquired on a control sample involving uniform 13C/15N labeling. This enables the assignment of chemical shifts of nuclei belonging to the unlabeled residue. This method requires relatively inexpensive sources of isotope labels (only unlabeled amino acids are used along with 15NH4Cl and/or 13C-D-glucose). It has been used in a large number of applications such as spectral simplification [22, 24, 32, 33], spin-system or amino acid-type identification [21, 23, 34], sequence specific resonance assignments [25], stereospecific assignment of the pro-chiral methyl groups of Val and Leu in large molecular weight proteins [35] and measurement of residual dipolar couplings (RDC) [36]. Its application has been proposed for structural studies of membrane and paramagnetic proteins [37, 38].
In addition to spectral simplification selective labeling or unlabeling is also useful for amino-acid type identification and sequence specific resonance assignments. This is achieved by using a specific combination of amino acid types for selective identification. Different samples of the protein are prepared in which different set of amino acid types are selectively labeled (sometime site-selectively) [39–53]. This approach is termed as “combinatorial selective labeling”. In a similar vein combinatorial amino acid selective unlabeling can be carried out in a combinatorial approach [21, 25]. In combinatorial selective labeling/unlabeling the combination of amino acid types are decided based on different critera such as the primary sequence of the protein of interest and the ease with which the set of amino acid types selected can be distinguished in the spectrum. Different algorithms have also been proposed for choosing an optimal combination of amino acid for combinatorial labeling [44, 50, 53, 54].
This chapter will focus primarily on two aspects: (1) amino acid selective unlabeling strategies for protein resonance assignment and structure determination and (2) combinatorial schemes involving selective labelling or unlabeling. The method for producing proteins with specific amino acid labeled selectively has been described/reviewed extensively in the past especially for proteins expressed in E. Coli [8, 9, 19, 55]. In Chaps. 10 and 11 of this book, different strategies for selective labeling of proteins expressed in higher organisms such as Yeast, insect cells and mammalian cells have been described. In Chap. 9 of this book amino acid selective labeling using the cell-free method is described. The procedure for producing selectively unlabeled protein samples with cell-based methods is described below.
2 Amino Acid Selective Unlabeling
2.1 Sample Preparation
As of date, all selective unlabeling approaches have been cell-based. The procedure for preparing a selectively unlabeled protein sample using an E. coli expression system is similar to that employed in selective amino acid labeling [8, 9]. That is, the desired amino acid type is added in unlabeled form (i.e., 12C/14N) against a background of 13C and 15N labeling. The procedure typically employed when using an E. coli expression system is given below:
-
1.
Inoculate a 5 ml Luria Broth medium (LB/rich medium) with a single colony taken from a freshly transformed plate.
-
2.
After 8–10 h of growth, transfer it to 1 l of LB medium and continue to grow till the optical density (O. D.) reaches 0.6.
-
3.
Centrifuge the cells at 5,000 rpm for 10 min, re-suspend the cell pellet in 1 l of 1X minimal medium containing the desired amino acid to be selectively unlabeled at a concentration of 1.0 g/l. The minimal media consists of M9 salts containing 1 g/l 15NH4Cl supplemented with 0.250 g/l of MgSO4.2H2O, 0.015 g/l of CaCl2, 4.0 g/l [13C6] glucose. The minimal medium can be supplemented with vitamins/other salts (such as ZnSO4) if required.
-
4.
Allow the cells to grow for another 45 min and induce the desired protein expression suitably (e.g., using IPTG).
The procedure for harvesting/lysis of cells and protein purification remains the same as used for preparing a uniformly labeled sample. The amount of unlabeled amino acid to be used depends on the overall protein yield. For good protein expression up to 1 g/l of the desired amino acid to be selectively unlabeled can be used. In case of lower yields of the protein, the exogenously added amino acid can be reduced to 0.5 g/l. In case of amino acids which undergo isotope scrambling, larger amounts are required to be added, which is discussed below.
2.1.1 Identifying Peaks Corresponding to Unlabeled Amino Acids
There are two ways to identify peaks corresponding to the selectively unlabeled amino acid residue in the 2D [15N-1H] HSQC spectrum. The first method involves appropriate scaling and subtraction of the spectrum from that of the reference sample containing uniformly 15N labeled protein (which will be referred to as the ‘difference spectrum’). The peaks in the difference spectrum correspond to the specific amino acid residue being unlabeled. A drawback of this method is the imperfect cancellation of undesired peaks upon subtraction owing to the fact the two protein samples (reference and the selectively unlabeled) may have different concentrations and hence different signal-to-noise (S/N) ratios. In such cases a second method [25] can be used. In this method, first a reference or ‘control’ peak in the spectrum is chosen which is known to be un-affected by unlabeling (for instance a Gly residue). Next, the ratio of volume of each peak in the 2D HSQC spectrum of the uniformly labeled sample (also referred to as the reference sample) to the volume of the control peak in the same spectrum is taken. This ratio is called as IRref. In a similar manner, the ratio of volume of each peak in the 2D HSQC spectrum of the selectively unlabeled sample to the volume of the control residue in the same spectrum is taken. This ratio is called IRunlab. The ratio, IRunlab/IRref, is then independent of the differences in the signal-to-noise across the two spectra being compared (uniform and selectively unlabeled) and can be used in concert with the 2D difference spectrum. It also provides information on the extent to which the unlabeling of the particular amino acid has occurred. This is illustrated in Fig. 6.1b where IRunlab/IRref is plotted for each residue in a Lys selectively unlabeled sample of ubiquitin. A peak which is absent in the spectrum acquired for the selectively unlabeled sample will have IRunlab,i/IRref,i ∼0, whereas for any peak unaffected by selective unlabeling IRunlab,i /IRref,i with the ∼1 for any peak unaffected by selective unlabeling. In practice, a range of values from 0 to 1 are observed for the unlabeled residues reflecting the extent to which a given amino-acid type has been unlabeled. We consider IRunlab,i /IRref,i <0.5 to indicate that the residue i has undergone unlabeling or has been effected by unlabeling and IRunlab,i /IRref,i >0.5 is taken as an indication of no effect of unlabeling.

(a) An overlay of selected region of 2D [15N-1H] HSQC spectrum of uniformly 15N labeled (blue) and lysine selectively unlabeled (red) samples of ubiquitin. Assignments for lysine residues are indicated by the residue number. (b) A plot of IRunlab,i/IRref,i: IRunlab,i = I i unlab/I unlabcontrol and IRref, i = I i ref/I control ref where i denotes the residue number, I denotes volume of the peak and ‘control’ denotes a residue which does not undergo any effect of unlabeling in both selectively unlabeled and the reference sample. In this case the control residue chosen was G47. All residues that undergo the desired unlabeling are indicated as blue bars, also indicated with an arrow on the top. In the case of (K) sample almost complete unlabeling of Lys is observed (i.e., IRunlab,i/IRref ∼0). (c) Spectrum displaying scrambling of Glycine to Serine in the overlay of selected region of 2D [15N-1H] HSQC spectrum of uniformly 15N labeled (blue) and Glycine selectively unlabeled (red) samples of ubiquitin. (d) A plot similar to (b) for Glycine unlabeled ubiquitin where K6 was chosen as the control residue. Arrows in (d) point towards the residues having IRunlab,i/IRref ∼0. Note that E24 and G53 were not assigned and hence are absent along with Pro
2.1.2 Misincorporation of 14N at Undesired Sites (Isotope Scrambling)
The extent of misincorporation of 14N at the undesired sites (also referred to as isotope scrambling) is presented in Table 6.1. The overall nature of isotope scrambling is similar to that observed in the selective labeling approach [8], which is expected as the biosynthetic pathways involved are identical in the two methods. Figure 6.1c illustrates the istope scrambling for Gly. In a Gly selectively unlabeled spectrum of Ubiquitin (Fig. 6.1c), all Ser are unlabeled. Out of the 20 amino acid types, the best ones suited for selective unlabeling based on their abundance and cross labeling are: Ala, Arg, Asn, His, Lys, Pro. Unlabeling of Thr results in unlabeling of Gly and Ser but not vice-versa (i.e., Gly or Ser unlabel each other but not Thr; Fig. 6.1). Thr unlabeling also results in unlabeling of 13CO and 13Cδ1 of Ile. However, due to the ease with which Gly, Ser, and Thr can be distinguished from each other based on their 13Cα/β, misincorporation of 14N among these three amino acids is not harmful.
The amino acids Ile, Leu, Val inter-convert strongly to each other. For such amino acids which undergo isotope scrambling, larger amounts of the amino acid has to be added to the growth medium. This is illustrated in Fig. 6.2a, where IRunlab/IRref is plotted as a function of residue number for five samples prepared with different amounts of Ile added. The extent of unlabeling and isotope scrambling of Leu and Val (as seen in the ratio IRunlab/IRref) increases as the amount of Ile added is increased and reaches a stable value for 1.0 g/l of Ile. In contrast to this, for selective unlabeling of Lys which does not undergo isotope scrambling (Table 6.1), even 100 mg/l is sufficient for achieving complete unlabeling. This is evident from Fig. 6.2b, where the ratio IRunlab/IRref remains <0.5 even for small amounts of Lys added. Unlike in the selective labeling approach, unlabeling of glutamine is not detrimental. Its conversion to glutamic acid gets diluted by transamination of the latter to other amino acids. Only weak unlabeling of proline (via Glutamic acid) is observed in the samples prepared with Gln selectively unlabeled. On the other hand, glutamic acid or aspartic acid strongly unlabels many other amino acid types.

The effect of amount of unlabeled amino acid added to the growth medium on mis-incorporation of 14N label for isoleucine (a1–a5) and Lysine (b1–b5) in ubiquitin. Though Lysine residue gets completely unlabeled at almost all concentrations (blue bars), optimum unlabeling for Ile is achieved at an amount of 1 g (a4) of isoleucine (red bars) added per litre of culture. That is, beyond this amount any addition of Ile does not affect the extent of unlabeling. Plots of IRunlab,i/IRref,i: IRunlab,i = I i unlab/I unlabcontrol and IRref, i = I i ref/I control ref where i denotes the residue number (red-ile, blue-lys), I denotes volume of the peak and ‘control’ denotes a residue which does not undergo any effect of unlabeling in both selectively unlabeled and the reference sample. In this case the control residue chosen was G47. E24 and G53 were not assigned and hence are absent along with Pro
More than one amino acid type can be selectively unlabeled in a given sample. The particular combination of amino acids chosen depends on the relative ease with which the different amino acid types can be distinguished from one another in the spectrum. Such discrimination can be made on the basis of 13Cα and 13Cβ chemical shifts which are well-known indicators of amino acid-types [56, 57] and can be obtained from a 3D HNCACB spectrum acquired for the control sample. The 20 amino acids can be divided into nine groups (Table 6.2; labeled I–IX) with each group having a distinct range of 13Cα and 13Cβ chemical shifts. For instance, Arg and Ala can be chosen together for selective unlabeling as their 13Cβ shifts resonate in different spectral regions (Table 6.2). Similarly Ser and Gly which undergo intercoversion (Table 6.1) can be distinguished due to their distinct 13Cα and 13Cβ chemical shifts (Table 6.2). In the case of Ile/Leu/Val, their interconversion does not hamper analysis because these three amino acids belong to distinct categories (Table 6.2).
A method to reduce isotope scrambling was proposed recently by Rasia et al. for E. Coli based protein expression systems [58]. This is based on feeding the host organism with suitable metabolic precursors of specific amino acids such as Ile, Leu, Phe and Tyr. The metabolic precursors used are: α-ketobutyrate for Ile, α-ketoisovalerate for Leu and Val, phenylpyruvate for Phe and 4-hydroxy phenylpyruvate for Tyr. The precursors are directly converted to their respective amino acids without scrambling. An additional advantage of this method is that these amino acids are generated with a unique 13C/12C labeling pattern which can be exploited for their identification and assignment.
2.2 Applications of Selective Unlabeling
The application of selective unlabeling can be divided into two categories: (1) spectral simplification and (2) spin-system identification and resonance assignments.
2.2.1 Spectral Simplification
An application of selective unlabeling for spectral simplification is in stereospecific assignments of the prochiral methyl groups of valine and isoleucine [24, 35]. Among methyl groups there is a large overlap of 1H and 13C chemical shifts [24, 59, 60]. This hampers the unambiguous stereospecific assignments of Val and Leu. The overlap can be alleviated if resonances from residues overlapping with those of Val and Leu are suppressed. These residues typically are Ile, Leu, Lys and Thr [24]. Selective unlabeling of these specific amino acid residues along with fractional 13C labeling of other residues results in an enhanced spectral simplification. This methodology was demonstrated on proteins such as EhCaBP (15 kDa) [24, 61, 62] and MSG (80 kDa) [35]. Selective unlabeling of Ile and Leu was not found to adversely affect the 13C labeling of Val [24]. However, selective unlabeling of Leu resulted in unlabeling of Val (as discussed above in the case of 2D [15N-1H] HSQC).
2.2.2 Spectral Simplification by Isotope Filtering
Selective unlabeling is useful in NOESY based structure determination of proteins. In 3D 13C- or 15N-edited [1H-1H] NOESY a number of correlations between residues far in sequence but close in space are observed. Such ‘long range’ correlations are pivotal for high resolution structure determination [1]. Spectral overlaps hamper accurate analysis leading to incorrect or ambiguous assignments. One solution is to introduce 12C and 14N labeled amino acids in the protein via selective unlabeling and observe only those correlations which originate on 12C or 14N bound protons and get detected on 13C or 15N bound protons or vice-versa. This can be achieved by isotope filtered experiments or heteronuclear 14N/15N-half filters [63, 64] resulting in simplification of the NOESY spectrum thereby helping in assigning the cross peaks. Another advantage of the unlabeling method is that it removes the effects of 13C relaxation on proton linewidths thereby improving the sensitivity of the NOEs observed. Typically isotope filtered experiments are performed to select and assign intermolecular NOEs [64]. However, using selective unlabeling, these experiments can be applied to select NOEs within a given protein. Figure 6.3b shows a radio frequency (r.f.) pulse scheme of a 12C/14N (ω1) filtered-15N edited [1H-1H] NOESY which implements the magnetization transfer scheme shown in Fig. 6.3a. In this experiment, 12C and 14N bound protons are selected in the indirect dimension (ω1) and their NOE to 15N bound protons are detected in the direct (ω3) dimension. Using this pulse sequence a 3D spectrum was acquired for a Gln, Ile-selectively unlabeled- 13C, 15N labeled ubiquitin sample. A 2D {ω1 (1H), ω3 (1H)} strip plot from the 3D spectrum is shown in Fig. 6.3c wherein cross peaks from unlabeled Gln and Ile are observed in the spectrum.

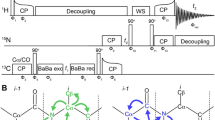
(a) R.f. pulse scheme of 3D 12C/14N (ω1) filtered-15N edited [1H-1H] NOESY. In this experiment, NOEs to all 15N bound protons originating from 12C/14N bound protons are detected as depicted in (b) Rectangular 90o and 180o pulses are indicated by thin and thick vertical bars, respectively, and phases are indicated above the pulses. Where no r.f. phase is marked, the pulse is applied along x. High-power 90o pulse lengths are: 9.4 ms for 1H, 31 ms for 15N and 11 ms for 13C. The 1H r.f. carrier is placed at the position of the solvent line at 4.7 ppm. The 13C and 15N carrier position is set to 38 ppm and 119 ppm, respectively, throughout the sequence. GARP [2] is employed to decouple 15N during acquisition. The shaped 180° pulses on 13C′ are of Gaussian cascade type with duration of 256 υs. All pulsed z-field gradients (PFGs) are sinc-shaped with gradient recovery delay of 200 ms. The duration and strengths of the PFGs are: G1 (1 ms, 26.8 G/cm); G2: (1 ms 16.08 G/cm); G3: (1 ms 42.88 G/ ms); G4: (1 ms 4.3 G/cm). The delays are: τ1 (≈1/2JNH) = 2.7 ms, τ2 (≈1/2JNH) = 2.5 ms, τ3 = 2.25 ms, δ = 0.5 ms and δ1 = 1.5 ms, δ2 = 1.2 ms, τ = 120 ms. Phase cycling: φ1 = x, x, x, x , −x,−x, −x, −x; φ2 = y, y, y, y, −y, −y, −y, −y; φ3 = x, −x; φ4 = x, x, −x, −x; φ5 = y, y, −y, −y; φrec = x, −x, −x, x, −x, x, x, −x. Quadrature detection along ω2(15N) is achieved using Echo-AntiEcho mode with sensitivity enhancement (G3 is inverted with a 180o shift for φ5). States-TPPI is used for quadrature of w1(1H) dimension. (b) Schematic depiction of the selection of NOE’s from 12C/14N bound protons to 15N bound protons. (c) and (d) A representative strip plot showing comparison of the normal 3D N-edited NOESY (b1) with the 3D 12C/14N (ω1) filtered-15N edited [1H-1H] NOESY (c) for residue 4F of Q & I unlabeled ubiquitin protein
An application for assignments of peaks in the NOESY spectrum was demonstrated by Vuister et al. [22] which was also one of the first applications of selective unlabeling. Subsequently, an application of the method to deuterated protein was proposed by Kelly et al. [33] in which five amino acids: Phe, Ile, Thr, Val and Tyr were selectively unlabeled against a background of 2H and 15N labeling in a 47 kDa homodimeric protein. Half-filter 2D and 3D 15N-edited [1H-1H] NOESY spectra were acquired for the sample which helped to selectively detect NOEs originating from the unlabeled amino acid residues to the rest of the residues in the protein [33].
2.2.3 Spin System Identification and Resonance Assignments
The identification of spin system forms an important step in the process of protein structure determination. It also helps in automated methods for resonance assignments [23, 56, 65]. Spin system identification by selective unlabeling is accomplished in a manner similar to that carried out in selective labeling. The objective here is to identify the resonances in a 2D [15N-1H] HSQC belonging to a specific amino acid-type. As discussed above, this is carried out using a difference spectrum (with respect to the 2D spectrum of a uniformly labeled sample) or using the ratios of intensities described above. One of the applications of unlabeling in spin system identification is the combinatorial labeling scheme proposed by Shortle [21]. Selective unlabeling can also be used for sequence specific resonance assignments [25]. This is explained in more detail below in the section on combinatorial labeling.
3 Combinatorial Amino Acid Selective Labeling
One useful starting point in sequence specific resonance assignments is the assignment of the resonances observed in the different 2D and 3D NMR spectra to the corresponding amino acid type (also referred to as ‘spin-system identification’) [1, 2]. In general this can be achieved by making 19 samples in each of which one out of the 19 amino acid types is selectively 15N-labeled (note that proline does not contain an amide proton and hence is not observed in the 2D [15N, 1H] HSQC spectrum). This is a tedious and time consuming approach. Hence it becomes important to develop a strategy to reduce the number of samples required by labeling more than one amino acid type in a sample. In selective labeling involving a combinatorial approach one or more protein samples are prepared with different combination of amino acids labeled selectively in a given sample [39–44, 46, 47, 50–53]. This requires proper selection of amino acids such that when taken together each amino acid has a unique labeling pattern across the different samples. This can be explained as follows.
Consider two labeling modes or states for a given amino acid (e.g., 15N-labeled or unlabeled). In N samples up to 2N − 1 amino acid types can be chosen such that each amino acid can have a unique labeling pattern across the N samples. This is explained in Table 6.3 for N = 4. For example, if the two labeling modes chosen are: 15N-labeling or no labeling, then in each sample a given amino acid will either be labeled or unlabeled. This gives rise to 2N amino acid types (indicated by numbering in the first column of Table 6.3) which can be distributed over N samples (Table 6.3a; left). It can be seen that no two amino acids have the same labeling pattern across the four samples shown in Table 6.3a. Thus based on a specific labeling pattern observed across the four samples (one spectrum is recorded per sample) the 16 amino acid types can be identified. Note that according to this calculation, one amino acid will remain unlabeled in all samples (the last amino acid shown in Table 6.3), which is of no use since its absence in all samples does not help in its assignment. Hence, (2N − 1) amino acids can be chosen with unique labeling pattern across the N samples. The number of amino acids assignable can be increased with lesser of number samples if more than two labeling modes are chosen. For instance, with three modes of labeling: 13C/15N, 15N-only labeling and no labeling (i.e., 12C/14N), up to 3N − 1 amino acid types can be chosen with unique labeling pattern across the N samples. For 20 amino acid types this results in requirement for just three samples (Table 6.3).
The early implementations of combinatorial labeling involved cell-based methods [39] whereas those developed recently are exclusively based on cell-free protein synthesis. In the cell-free method, site-specifically labeled amino acids (e.g., 1-13C labeled) are used during protein synthesis. The different combinatorial selective labeling approaches proposed till date can be divided into four types (Table 6.4; depending on the modes of labeling alluded to above): (1) use of 15N-only labeled amino acids in different combinations, (2) use of 13C, 15N and 15N-only labeled amino acids, (3) use of 1-13C and 15N-labeled amino acids and (4) use of 1-13C; 13C, 15N and 15N-only labeled amino acids. The first approach yields only amino-acid type identification whereas the remaining three approaches help in sequence specific resonance assignments. The different methods are summarized in Table 6.4. Each of the method is described below in detail.
3.1 Combinatorial Selective 15N-Labeling for Spin System Identification
A combinatorial scheme involving only selective 15N-labeling was proposed by Wu et al. [46]. The overall aim of the method is to achieve spin system identification by reducing the number of sample required (instead of 19) [45, 46, 48]. Five samples are made using cell-free protein synthesis because this enables the encoding of 20 amino acids in five different combinations such that each sample contains a unique combination of labeled amino acids (as shown in Table 6.3). The choice of amino acids in a given sample was based on their abundance in the protein and to minimize the overlap of peaks between different amino acid types. Those amino acid types which were present in higher numbers were kept single and not combined with other types in a sample. Each sample contained about 7–8 amino acids labeled selectively corresponding to ∼30% of the total number of residues in the protein. The 2D [15N-1H] HSQC spectrum was thus simplified by absence of ∼70% of resonances compared to complete labeling of the protein. A computational strategy was subsequently proposed to optimally select the combination of amino acid types in a given sample [54]. An analogous method involving selective unlabeling was proposed by Shortle [21]. As mentioned above, this approach helps only in assigning the resonances observed in the 2D [15N-1H] HSQC spectrum to the corresponding amino acid type. In order to obtain sequence specific resonance assignments, different labeling patterns such as the inclusion of 13C labels is required as described below.
3.2 Combinatorial Methods Using Two Selective Labeling Modes (Dual Selective Labeling)
In this approach the aim is to achieve both spin-system identification and sequence specific resonance assignments. The methods are based on the idea that if two contiguous amino acid residues: i, i + 1 (say X i and Y i+1) in the protein are labeled with 13C and 15N, respectively, then specific NMR experiments can be used which will selectively detect the di-peptide X i − Y i+1. There are two variants of this method (Table 6.4): (1) use of 1-13C and 15N-labeled amino acids [39, 40, 44, 53] and (2) use of 13C,15N and 15N- labeled amino acids [42, 43, 47, 52]. As mentioned above, the early implementations of combinatorial selective labeling involved cell-based methods [39] whereas those developed recently are exclusively based on cell-free protein synthesis.
3.2.1 Combinatorial Selective Labeling Using 1-13C and 15N Amino Acids
In this scheme two types of labeled amino acids are utilized: (i) 15N-labeled amino acids and (ii) those labeled in the carbonyl position (1-13C). If a residue i (X i ) gets labeled at the 1-13C position followed by a residue i + 1 (Y i ) which is 15N labeled, then 3D HNCO (or its 2D 15N-1HN projection) can be used to identify the di-peptide: X i − Y i+1. No other di-peptide will get detected in such a spectrum. If the dipeptide is unique in the protein primary sequence, it gets assigned sequence specifically. The combinations of amino acids with 1-13C or 15N-labeling can be chosen so as to maximize the uniqueness of the dipeptides. This methodology was demonstrated by Trbovic et al. for backbone assignments of membrane proteins [44]. The amino acid-types were selected using an algorithm which considered the protein primary sequence and the stretches of amino acids which could not be assigned using the conventional procedure involving triple resonance experiments [44]. Dipeptide segments: X i − Y i+1 with X i labeled at 1-13C and Y i labeled with 15N were identified using 2D [15N-1H] HSQC and 2D 15N-1HN projection of HNCO. The methodology was also used for resonance assignment of membrane domain structures and the combinations of amino acids were chosen using a Monte Carlo approach [53]. While the method may not result in 100% sequence specific resonance assignments, it helps in providing useful starting points which otherwise would render the resonance assignments ambiguous. Hence, it serves to supplement the conventional resonance assignment strategies.
3.2.2 Combinatorial Selective Labeling Using 15N and 13C,15N Amino Acids
In this scheme two types of labeled amino acids are utilized: (i) 15N-only labeled and (ii) 13C, 15N labeled. The strategy for assignment is same as mentioned above. That is, specific NMR experiments can be used to identify the di-peptide: X i − Y i+1 where the residue i is labeled with 13C and i + 1 is with 15N. While the NMR experiments employed are primarily 2D [15N-1H] HSQC and 2D [15N-1H] projection of 3D HNCO, different variants of this approach have been proposed depending on the choice of the amino acids for selection in a given sample.
In the method proposed by Shi et al. [43] four samples were prepared. In each sample two amino acid-types were chosen with 13C, 15N labeling (double labeled) and one with only 15N labeling. The double labeled amino acid types were chosen each from two different groups (Group I and II) while the 15N labeled amino acid were selected from a third group (Group III). Groups I and II contained amino acids such that those belonging to one group could be distinguished from the other based on the 13Cα and 13Cβ chemical shifts. Group I contained amino acids Arg, Asp, Glu, Gln, Leu and Lys and Group II consisted of Ala, Ile, Ser, Thr and Val. In Group III less abundant amino acid types were chosen such as Cys, Gly, His, Met, Phe, Tyr, Trp. For each specifically labeled sample, different NMR spectra were acquired [43]. In addition to specific labeling of different amino acids in a sample, differential amount of labeling was also tried. In this approach both unlabeled and labeled forms of different amino acid types are added in different pre-determined ratios. The intensities of the peaks corresponding to the amino acids follow these ratios and thus help in their identification and assignments. A similar method was proposed by Staunton et al. [52] using the two labeling modes similar to that of Shi et al. [43].
A combinatorial method based on differential amount of labeling was proposed by Parker et al. [42, 47]. It is a dual labeling scheme with two modes of labeling for each amino acid type: 13C, 15N labeling (double labeled) or 50% 15N/50% 14N labeling. Five samples are prepared. One uniform 13C, 15N labeled sample serves as a control and in four other samples different amino acids are labeled in one of the two labeling modes (Table 6.5). If I i is the intensity of a peak in the 2D [15N-1H] HSQC spectrum of one of the four selectively labeled samples, its corresponding intensity in the HSQC spectra of the other samples will either be I i or I i /2 depending on whether it is 13C, 15N or 50% 15N/50% 14N labeled. Based on this, the comparison of relative peak intensity pattern in the 2D [15N-1H] HSQC spectra acquired on the four selectively labeled samples helps in identifying a given amino acid type. Since four samples are prepared, this approach allows the assignment of 16 amino acid types (see Table 6.3). For sequence specific resonance assignments, a 2D HNCO spectrum is acquired on the four selectively labeled samples and instead of a relative peak intensity pattern as observed in the HSQC spectrum, the absence or presence of a peak in the spectrum across the four samples becomes unique for a given di-peptide segment. Taken together, the two spectra 2D HSQC and 2D HNCO are sufficient to identify all 16*16 amino acid pairs.
3.3 Combinatorial Methods Using Three Selective Labeling Modes
In a recent work by Lohr et al. [51] a combinatorial approach using three types of labeled amino acids: (i) 1-13C, (ii) 13C,15N and (iii) 15N was proposed. In this method, in addition to combinatorial selective labeling using 1-13C and 15N amino acids, a 13C,15N labeled amino acid was included. This increased the number of amino acids which can be identified while reducing the number of samples. A dipeptide segment in the protein: X i − Y i+1 with a specific labeling pattern on X or Y was selected using a set of six 2D NMR experiments. The amino acids used for the different samples were based on the amino acid composition of the protein being studied.
4 Combinatorial Methods Based on Amino Acid Selective Unlabeling
In combinatorial methods based on amino acid selective unlabeling, a set of samples are prepared in each of which specific amino acids are selectively unlabeled (i.e., 12C/14N). As a result the peaks due to these amino acids are absent in the spectrum acquired. Similar to methods based on selective labeling, amino acid types in selective unlabeling are chosen based on the abundance of the amino acids in the protein and the ease with which they can be distinguished from each other. As of date, all amino acid selective unlabeling schemes proposed have been cell-based. Two of the combinatorial methods involving selective unlabeling are described below.
4.1 Combinatorial Selective Unlabeling for Spin System Identification
In combinatorial selective unlabeling approach proposed by Shortle [21], four proteins samples containing a combination of different 14N labeled amino acids (or unlabeled amino acids) against a background of 13C and 15N labeling was proposed. The protein was expressed using an E. Coli based expression system. The amino acids were identified based on the intensity pattern (i.e., absence of the peaks) in different samples. Since the number of samples were four, up to 16 amino acids (24) could be assigned. Amino acids which show extensive isotope scrambling such as Glutamic acid and Aspartic acid (Table 6.1) were left out. The method was one of the earliest combinatorial methods proposed and is similar to that proposed later by Wu et al. [46].
4.2 Combinatorial Selective Unlabeling for Sequence Specific Resonance Assignments
A method for sequence specific resonance assignments based on selective unlabeling was recently proposed by Krishnarjuna et al. [25]. The method involves unlabeling selectively a set of amino acid types in different samples. Identification and assignment of resonances in a 2D [15N, 1H] HSQC spectrum corresponding to the selectively unlabeled amino acids is done by comparing a HSQC spectrum of the selectively unlabeled sample with that of a reference sample of the same protein which is uniformly 13C/15N labeled. The particular combination of amino acids chosen depends on their relative abundance in the protein and the relative ease with the different amino acid types can be distinguished from one another in the spectrum. Such discrimination can be made on the basis of 13Cα and 13Cβ chemical shifts which are well-known indicators of amino acid-types [56, 57]. The 20 amino acids can be divided into nine groups (Table 6.2; labeled I–IX) with each group having a distinct range of 13Cα and 13Cβ chemical shifts. Based on these considerations four samples each containing two selectively unlabeled amino acid types were prepared [25]: (i) Gln, Ile (Q,I), (ii) Arg, Asn (R,N) (iii) Phe, Val (F, V) and (iv) Lys, Leu (K, L). A uniformly 13C or/and 15N labelled sample was additionally prepared as a reference. These amino acids were chosen on the basis of their high abundance in proteins (80% together with Ala, Gly, Ser and Thr). With an appropriate choice of the amino acid type, isotope scrambling (Table 6.1) does not hamper the analysis. For instance, selective unlabeling of Ile results in significant unlabeling of Leu and Val (Table 6.1; Fig. 6.2). However, the 13Cα and 13Cβ chemical shifts of these amino acids occur in different spectral regions (Table 6.2) enabling their distinction. Thus, by avoiding selective unlabeling of amino-acid types belonging to the same group, the effect of isotope scrambling can be ignored.
In addition to spin-system identification, sequence specific resonance assignments can be carried out with the help of a 2D {12CO i -15N i+1}-filtered HSQC spectrum, which aids in linking the 1HN/15N resonances of a selectively unlabeled residue i and i + 1. The radio-frequency (r.f.) pulse sequence and the di-peptide selection scheme for 2D {12CO i -15N i+1}-filtered HSQC is shown in Fig. 6.4. The experiment works by selecting those residues (i + 1) which are 15N labeled and have an unlabeled residue as their N-terminal neighbor (i.e., residue i). This is achieved by tuning the delay periods in the pulse sequence to generate anti-phase 15N i+1-13C′ i magnetization which is not detectable in the 2D HSQC spectrum. If the residue i is an unlabeled amino acid, no antiphase 15N i+1-13C′ i magnetization is generated and 15N i+1 is observed as in a regular 2D HSQC spectrum. Thus, the resulting 2D spectrum consists of peaks corresponding only to those residues which have selectively unlabeled N-terminal neighbors.

R.f. pulse scheme of 2D {12CO i 15N i+1}-filtered HSQC [25]. Rectangular 90o and 180o pulses are indicated by thin and thick vertical bars, respectively, and phases are indicated above the pulses. Where no r.f. phase is marked, the pulse is applied along x. High-power 90o pulse lengths are: 8.7 ms for 1H, 37 ms for 15N and 15.8 ms for 13C. The 1H r.f. carrier is placed at the position of the solvent line at 4.7 ppm. The 13C and 15N carrier position is set to 176 ppm and 118.5 ppm, respectively, throughout the sequence. GARP [2] is employed to decouple 15N during acquisition. The shaped 180° pulses on 13C′ are of Gaussian cascade type [2] with duration of 256 μs. All pulsed z-field gradients (PFGs) are sinc-shaped with gradient recovery delay of 200 ms. The duration and strengths of the PFGs are: G1–G2 (1 ms, 26.8 G/cm); G3: (1 ms 42.9 G/cm) G4: (1 ms 4.3 G/cm). The delays are: τ1 (≈1/2JNH) = 2.3 ms, τ2 (≈1/2JNC′) = 15.0 ms, τ3 (≈1/2JNH) = 2.3 ms, δ = 1.2 ms and δ1 = 1.2 ms. Phase cycling: φ1 = x,−x; φ2 = f 3 = x, x, –x, –x; φrec = x, −x. Quadrature detection along ω1(15N) is achieved with sensitivity enhancement (G3 is inverted with a 180o shift for hφ4)
The 2D {12CO i -15N i+1}-filtered HSQC is combined with 3D HNCACB and 3D CBCA(CO)NH spectrum acquired for the control sample for sequence specific resonance assignments as follows. First resonances in the 2D HSQC spectrum belonging to the selectively unlabeled amino acid residues are identified. This is done by generating a difference spectrum and/or using the method of ratios described above (Fig. 6.1). Such peaks serve as starting points. The next step is to map or establish a link between each of the 1HN/15N resonances observed in the difference spectrum with the corresponding resonances of their C-terminal neighbors (i + 1) observed in the 2D {12CO i -15N i+1}-filtered HSQC. This is accomplished by an inspection of the 3D HNCACB and 3D CBCA(CO)NH spectra acquired for the reference sample as illustrated in Fig. 6.5. For a given spin-system i in the 2D difference spectrum, the spin system corresponding to i + 1 in 2D {12CO i -15N i+1}-filtered HSQC is the one for which the 13Cα and 13Cβ chemical shifts observed in 3D CBCA(CO)NH spectrum matches that of the residue i in HNCACB (Fig. 6.5). Further, using 3D CBCA(CO)NH for a given spin-system i in the 2D difference spectrum the spin system corresponding to i − 1 can be identified based on the 13Cα and 13Cβ chemical shifts (Table 6.2). This results in the identification of a tri-peptide segment from the knowledge of the amino acid types of residues: i − 1, i and i + 1. An alternative method to obtain the amino acid types of i − 1 and i + 1 is to acquire the 3D HN(CA)NH experiment [66] for the control sample. This experiment directly correlates the chemical shifts of 15N i and 1H i with those of residues i − 1 and i + 1 [66]. Once 15N i − 1/i+1 and 1H i − 1/i+1 are identified using 3D HN(CA)NH, the amino acid-types corresponding to i − 1/i + 1 can be identified based on their respective 13Cα and 13Cβ chemical shifts observed in 3D HNCACB. The tri-peptide thus identified using one of the above methods can then be mapped on to the protein primary sequence for sequence specific resonance assignments. The mapping is carried out as depicted in Fig. 6.6. A unique number code is assigned to amino acids in each of the nine categories shown in Table 6.2 except for the residue being selectively unlabeled, which is assigned a separate code ‘0’ (given that its type is known exactly). Thus, all amino acid types in a given category get the same code. The different codes assigned are: Ala-1; Gly-2; Ser-3; Thr-4; Lys, Arg, Gln, Glu, His, Trp, Cysred, Met-5; Asp, Asn, Phe, Tyr, Cysoxd and Leu-6; Ile-7; Val-8; Pro-9. Next the protein primary sequence is converted into a new sequence of codes using the same codes as assigned to the amino acids in the different groups (Fig. 6.6). The tri-peptide segment (containing three codes) is then mapped on to the primary sequence of codes to identify its location for sequence specific resonance assignments. This is illustrated in Fig. 6.7 for a particular tripeptide in ubiquitin. Since the method relies on the presence of 15N, 1H on the residue i + 1 following a residue i which is unlabeled, the following types of tri-peptide segments cannot be assigned: (i) the segment in which the immediate C-terminal neighbor of the central (unlabeled) residue is a Pro (i.e., i+1ºPro), (ii) the segment which contains both residues i and i + 1 as selectively unlabeled and (iii) the residue is the last amino acid in the polypeptide chain (i.e., located at the C-terminal end). Compared to the other methods which assign di-peptides, the combinatorial selective unlabeling method described above helps in assigning tri-peptide segments which are more uniquely mapped on the sequence. Once the tri-peptides are assigned, the remaining residues in the protein can be assigned in di-peptide pairs, which earlier could not be assigned due to degeneracy.

Schematic illustration of the sequential assignment strategy used for selective unlabeling

Schematic illustration of mapping the tri-peptide segment identified using the methodology depicted in Fig. 6.5 onto the protein primary sequence. First, based on the 2D difference spectrum and 2D {12CO i -15N i+1}-filtered HSQC spectrum in concert with CBCA(CO)NH and HNCACB, the amino acid type corresponding to the tri-peptide segment (i − 1, i, i + 1) is identified (see Fig. 6.5). Each residue of the tripeptide segment is assigned a code based on the group to which it belongs (Table 6.2). The selectively unlabeled residue is assigned an unique code ‘0’ because its amino acid type is known exactly. Next, the protein primary sequence is converted into an array of codes for each amino acid based again on the group to which each amino acid belongs in Table 6.2. The tripeptide segment identified above is then mapped on to the primary sequence wherever the tri-peptide code matches that in the primary sequence

Illustration of sequence specific resonance assignment of a tri-peptide segment in Ubiquitin. Given a peak in the 2D difference spectrum of (R, N) selectively unlabeled sample, (e.g., R54), its C-terminal neighbour (i + 1) is identified using the procedure shown in Fig. 6.5 and its amino acid type is assigned using 13Cα and 13Cβ chemical shift in 3D HNCACB. The amino acid type of residue i−1 is assigned like-wise from 3D CBCA(CO)NH and the tri-peptide segment (containing the amino acid type information) is then mapped on to the protein sequence shown for sequence specific assignments
We have carried out a statistical analysis of ∼160,000 non-homologues primary sequences to estimate the extent to which such tri-peptide segments can be placed uniquely in a protein sequence (Fig. 6.8). In many proteins the uniqueness of the tri-peptide segment is high if the central amino acid type in the tripeptide segment is known by selective unlabeling. In a given protein the information of amino acid type significantly reduces the multiple occurrences of tri-peptide segments. For instance, on an average 65% of tri-peptide segments containing lysine as the central residue were unique given the knowledge of its amino acid type (Fig. 6.8). On the contrary, in the absence of identification of lysine only ∼5% of the tripeptide sequences are unique. Note that the uniqueness is based on the code system described above based on categorizing the amino acids into different groups (Table 6.2). Taken together, the combinatorial selective unlabeling approach not only helps in reducing the search space for sequential assignments (with 2D {12CO i -15N i+1}-filtered HSQC), but also increases the uniqueness of the tri-peptide segment identified, resulting in unambiguous assignments. The advantage with selective unlabeling is more in the case of Group V and Group VI amino acid residues (Table 6.2), which are composed of a large number of amino acid types.

Statistical analysis of the uniqueness of tri-peptide segments in proteins rendered with selective unlabeling. 161792 proteins from UniRef50 were considered for analysis. (a) The bars show the percentage of proteins which have unique tri-peptide sequences containing a given amino acid residue in the central position. To calculate the uniqueness of such tri-peptide segments, the residue preceding and following the central residue is given a number code according to the group to which it belongs (Table 6.2) and the central residue is given a unique code. The entire polypeptide chain (also converted to a sequence of codes) is then searched for multiple occurrences of such tri-peptide segments. (b) The average percentage of unique tri-peptide segments in a protein centred on a given amino acid type. The average and standard deviation was measured considering all the proteins (161,792). When considering the selective unlabeling approach (shown in dark red), the central residue in a tri-peptide segment is given a unique code whereas the two residues around it are given codes according to the respective groups (Table 6.2) to which they belong. While considering uniform labelling (or non-selective labelling) as shown in blue, the central residue in a tri-peptide is given the code according to the group to which it belongs. Thus the increase in uniqueness due to selective unlabeling arises from the identification of the amino acid being selectively unlabeled (the central residue)
The number of assignable residues can be increased and the number of samples reduced if more than two amino acids are selectively unlabeled in the same sample. However, no two amino acids from the same group should be selectively unlabeled in one sample owing to the fact that their identity is purely based on the 13Cα and 13Cβ chemical shifts. Thus, for example two samples can be prepared: (1) one containing the amino acids R, N, G, S, T and A and (2) containing K. The combination of multiple samples also helps to verify the overlapping assignments.
5 Conclusions
Amino acid selective labeling and unlabeling are very important tools in protein structure determination. In the case of large proteins and/or proteins with severe chemical shift overlap such as intrinsically disordered/membrane proteins the process of sequence specific resonance assignments becomes a tedious and time consuming task. In such cases selective identification of amino acid residue by selective labeling or unlabeling improves the efficiency of assignments. Selective identification of amino acids helps not only in obtaining starting points for carrying out assignments, they also help directly in sequence specific resonance assignment. This is achieved either using single or a group of amino acid types labeled/unlabeled selectively in a given sample. The method of selective unlabeling is an inexpensive approach and has the advantage of providing assignments for residues preceding and following the amino acid type being unlabeled. It is possible to devise new experiments which can extend this methodology to identify residues further away (i.e., i + 2, i − 2) from the unlabeled site thereby providing larger segments of sequentially connected residues which can map uniquely onto the protein primary sequence.
References
Wüthrich K (1986) NMR of proteins and nucleic acids. Wiley, New York
Cavanagh J, Fairbrother WJ, Palmer AG, Skelton NJ (1996) Protein NMR spectroscopy. Academic, San Diego
McDermott AE (2004) Structural and dynamic studies of proteins by solid-state NMR spectroscopy: rapid movement forward. Curr Opin Struct Biol 14:554–561
Marassi F, Opella SJ (1998) NMR structural studies of membrane proteins. Curr Opin Struct Biol 8:640–648
Sanders CR, Sonnichsen FD (2006) Solution NMR of membrane proteins: practice and challenges. Magn Reson Chem 44:S24–S40
Dyson HJ, Wright PE (1998) Equilibrium NMR studies of unfolded and partially folded proteins. Nat Struct Biol 5:499–503
Markley JL, Putter I, Jardetzky O (1968) High resolution nuclear magnetic resonance spectra of selectively deuterated staphylococcal nuclease. Science 161:1249–1251
Muchmore DD, McIntosh LP, Russell CB, Anderson DE, Dahlquist FW (1989) Expression and nitrogen-15 labelling of proteins for proton and nitrogen-15 nuclear magnetic resonance. Methods Enzymol 177:44–73
McIntosh LP, Dahlquist FW (1990) Biosynthetic incorporation of 15N and 13C for assignment and interpretation of nuclear magnetic resonance spectra of proteins. Q Rev Biophys 23:1–38
Dötsch V, Matsuo H, Wagner G (1996) Amino-acid type identification for deuterated proteins with a b-carbon edited HNCOCACB experiment. J Magn Reson B 112:95–100
Dötsch V, Oswald RE, Wagner G (1996) Amino acid type selective triple resonance experiments. J Magn Reson B 110:107–111
Dötsch V, Wagner G (1996) Editing of amino acid type in CBCA(CO)NH experiments based on the 13Cb -13Cg coupling. J Magn Reson B 111:310–313
Schubert M, Schmalla M, Schmeider P, Oschkinat H (1999) MUSIC in triple-resonance experiments: amino acid type-selective 15N/1H correlations. J Magn Reson 141:34–43
Schubert M, Oschkinat H, Schmeider P (2001) MUSIC and aromatic residues: amino acid type-selective backbone 15N/1H correlations, part III. J Magn Reson 153:186–192
Schubert M, Oschkinat H, Schmeider P (2001) Amino acid type-selective backbone 15N/1H correlations for Arg and Lys. J Biomol NMR 20:379–384
Schubert M, Oschkinat H, Schmeider P (2001) MUSIC, selective pulses and tuned amino acid type-selective 15N/1H correlations, part II. J Magn Reson 148:61–72
Schubert M, Labudde D, Leitner D, Oschkinat H, Schmeider P (2005) A modified strategy for sequence specific assignment of protein NMR spectra based on amino acid type selective experiments. J Biomol NMR 31:115–128
Barnwal RP, Rout AK, Atreya HS, Chary KVR (2008) Identification of C-terminal neighbours of amino acid residues without an aliphatic C-13(gamma) as an aid to NMR assignments in proteins. J Biomol NMR 41:191–197
Goto NK, Kay LE (2000) New developments in isotope labeling strategies for protein solution NMR spectroscopy. Curr Opin Struct Biol 10:585–592
Ohki S, Kainosho M (2008) Stable isotope labeling methods for protein NMR. Prog NMR Spectrosc 53:208–226
Shortle D (1994) Assignment of amino acid type in 1H-15N correlation spectra by labeling with 14N-amino acids. J Magn Reson B105:88–90
Vuister GW, Kim SJ, Wu C, Bax A (1994) 2D and 3D NMR-study of phenylalanine residues in proteins by reverse isotopic labeling. J Am Chem Soc 116:9206–9210
Atreya HS, Chary KVR (2000) Amino acid selective ‘unlabelling’ for residue-specific NMR assignments in proteins. Curr Sci 79:504–507
Atreya HS, Chary KVR (2001) Selective ‘unlabeling’ of amino acids in fractionally C-13 labeled proteins: an approach for stereospecific NMR assignments of CH3 groups in Val and Leu residues. J Biomol NMR 19:267–272
Krishnarjuna B, Jaipuria G, Thakur A, D’Silva P, Atreya HS (2011) Amino acid selective unlabeling for sequence specific resonance assignments in proteins. J Biomol NMR 49:39–51
Vinarov DA, Lytle BL, Peterson FC, Tyler EM, Volkman BF, Markley JL (2004) Cell-free protein production and labeling protocol for NMR-based structural proteomics. Nat Methods 1:149–153
Apponyi MA, Ozawa K, Dixon NE, Otting G (2008) Cell-free protein synthesis for analysis by NMR spectroscopy. Methods Mol Biol 426:257–268
Tugarinov V, Hwang PM, Kay LE (2004) Nuclear magnetic resonance spectroscopy of high-molecular-weight proteins. Annu Rev Biochem 73:107–146
Tugarinov V, Kay LE (2004) An isotope labeling strategy for methyl TROSY spectroscopy. J Biomol NMR 28:165–172
Katzen F, Chang G, Kudlicki W (2005) The past, present and future of cell-free protein synthesis. Trends Biotechnol 23:150–156
Kainosho M, Torizawa T, Iwashita Y, Terauchi T, Ono AM, Güntert P (2006) Optimal isotope labelling for NMR protein structure determinations. Nature 440:52–57
Hong M, Jakes K (1999) Selective and extensive 13C labeling of a membrane protein for solid-state NMR investigation. J Biomol NMR 14:71–74
Kelly MJS, Krieger C, Ball LJ, Yu Y, Richter G, Schmieder P, Bacher A, Oschkinat H (1999) Application of amino acid type-specific 1H and 14N labeling in a 2H-, 15N-labeled background to a 47 kDa homodimer: potential for NMR structure determination of large proteins. J Biomol NMR 14:79–83
Mohan PMK, Barve M, Chatteljee A, Ghosh-Roy A, Hosur RV (2008) NMR comparison of the native energy landscapes of DLC8 dimer and monomer. Biophys Chem 134:10–19
Tugarinov V, Kay LE (2004) Stereospecific NMR assignments of prochiral methyls, rotameric states and dynamics of valine residues in malate synthase G. J Am Chem Soc 126:9827–9836
Mukherjee S, Mustafi SM, Atreya HS, Chary KVR (2005) Measurement of (1)J(N-i, C-i(alpha)), (1)J(N-i, C ′(i-1)), (2)J(N-i, C-alpha (i-1)), (2)J (H-N (i), C ′ (i-1)) and (2)J(H-N (i), C-alpha (i)) values in C-13/N-15-labelled proteins. Magn Reson Chem 43:326–329
Hilty C, Fernandez C, Wider G, Wüthrich K (2002) Side chain NMR assignments in the membrane protein OmpX reconstituted in DHPC micelles. J Biomol NMR 23:289–301
Turano P, Lalli D, Felli IC, Theil EC, Bertini I (2010) NMR reveals pathway for ferric mineral precursors to the central cavity of ferritin. Proc Natl Acad Sci USA 107:545–550
Kainosho M, Tsuji T (1982) Assignment of the three methionyl carbonyl carbon resonances in streptomyces subtilisin inhibitor by a carbon-13 and nitrogen-15 double-labeling technique. A new strategy for structural studies of proteins in solution. Biochemistry 21:6273–6279
Yabuki T, Kigawa T, Dohmae N, Takio K, Terada T, Ito Y, Laue ED, Cooper JA, Kainosho M, Yokoyama S (1998) Dual amino acid-selective and site-directed stable isotope-labeling of the human c-Ha-Ras protein by cell free synthesis. J Biomol NMR 11:295–306
Weigelt J, van Dongen M, Uppenberg J, Schultz J, Wikstrom M (2002) Site-selective screening by NMR spectroscopy with labeled amino acid pairs. J Am Chem Soc 124:2446–2447
Parker MJ, Aulton-Jones M, Hounslow AM, Craven CJ (2004) A combinatorial selective labeling method for assignment of backbone amide resonances. J Am Chem Soc 126:5020–5021
Shi J, Pelton JG, Cho HS, Wemmer DE (2004) Protein signal assignments using specific labeling and cell-free synthesis. J Biomol NMR 28:235–247
Trbovic N, Klammt C, Koglin A, Lohr F, Bernhard F, Dotsch V (2005) Efficient strategy for the rapid backbone assignment of membrane proteins. J Am Chem Soc 127:13504–13505
Ozawa K, Wu PSC, Dixon NE, Otting G (2006) 15N-labelled proteins by cell-free protein synthesis: strategies for high-throughput NMR studies of proteins and protein-ligand complexes. FEBS J 273:4154–4159
Wu PSC, Ozawa K, Jergic S, Su XC, Dixon NE, Otting G (2006) Amino-acid type identification in 15N-HSQC spectra by combinatorial selective 15N-labeling. J Biomol NMR 34:13–21
Craven CJ, Al-Owais M, Parker MJ (2007) A systematic analysis of backbone amide assignments achieved via combinatorial selective labelling of amino acids. J Biomol NMR 38:151–159
Xun Y, Tremouilhac P, Carraher C, Gelhaus C, Ozawa K, Otting G, Dixon NE, Leippe M, Grotzinger J, Dingley AJ, Kralicek AV (2009) Cell-free synthesis and combinatorial selective 15N-labeling of the cytotoxic protein amoebapore A from Entamoeba histolytica. Protein Expr Purif 68:22–27
Sobhanifar S, Reckel S, Junge F, Schwarz D, Kai L, Karbyshev M, Lohr F, Bernhard F, Dotsch V (2010) Cell-free expression and stable isotope labelling strategies for membrane proteins. J Biomol NMR 46:33–43
Hefke F, Bagaria A, Reckel S, Ullrich SJ, Dotsch V, Glaubitz C, Guntert P (2011) Optimization of amino acid type-specific 13C and 15N labeling for the backbone assignment of membrane proteins by solution- and solid state NMR with the UPLABEL algorithm. J Biomol NMR 49:75–84
Lohr F, Reckel S, Karbyshev M, Connolly PJ, Abdul-Manan N, Bernhard F, Moore JM, Dotsch V (2012) Combinatorial triple-selective labeling as a tool to assist membrane protein backbone resonance assignment. J Biomol NMR 52:197–210
Staunton D, Schlinkert R, Zanetti G, Colebrook SA, Campbell ID (2006) Cell-free expression and selective isotope labelling in protein NMR. Magn Reson Chem 44:S2–S9
Maslennikov I, Klammt C, Hwang E, Kefala G, Okamura M, Esquivies L, Mors K, Glaubitz C, Kwiatkowski W, Jeon YH, Choe S (2010) Membrane protein structure of three classes of histidine kinase receptors by cell free expression and rapid NMR analysis. Proc Natl Acad Sci USA 107:10902–10907
Sweredoski MJ, Donovan KJ, Nguyen BD, Shaka AJ, Baldi P (2007) Minimizing the overlap problem in protein NMR: a computational framework for precision amino acid labeling. Bioinformatics 23:2829–2835
Cai ML, Huang Y, Sakaguchi K, Clore GM, Gronenborn AM, Craigie R (1998) An efficient and cost-effective isotope labeling protocol for proteins expressed in Escherichia coli. J Biomol NMR 11:97–102
Atreya HS, Sahu SC, Chary KVR, Govil G (2000) A tracked approach for automated NMR assignments in proteins (TATAPRO). J Biomol NMR 17:125–136
Atreya HS, Chary KVR, Govil G (2002) Automated NMR assignments of proteins for high throughput structure determination: TATAPRO II. Curr Sci 83:1372–1376
Rasia RM, Brutscher B, Plevin MJ (2012) Selective isotopic unlabeling of proteins using metabolic precursors: application to NMR assignment of intrinsically disordered proteins. Chembiochem 13:732–739
Barnwal RP, Atreya HS, Chary KVR (2008) Chemical shift based editing of CH3 groups in fractionally C-13-labelled proteins using GFT (3,2)D CT-HCCH-COSY: stereospecific assignments of CH3 groups of Val and Leu residues. J Biomol NMR 42:149–154
Jaipuria G, Thakur A, D’Silva P, Atreya HS (2010) High-resolution methyl edited GFT NMR experiments for protein resonance assignments and structure determination. J Biomol NMR 48:137–145
Sahu SC, Atreya HS, Chauhan S, Bhattacharya A, Chary KVR, Govil G (1999) Letter to the editor: sequence-specific 1H, 13C and 15N assignments of a calcium binding protein from Entamoeba histolytica. J Biomol NMR 14:93–94
Atreya HS, Sahu SC, Bhattacharya A, Chary KVR, Govil G (2001) NMR derived solution structure of an EF-hand calcium-binding protein from Entamoeba histolytica. Biochemistry 40:14392–14403
Otting G, Wüthrich K (1990) Heteronuclear filters in two-dimensional [1H,1H]-NMR spectroscopy: combined use with isotope labelling for studies of macromolecular conformation and intermolecular interactions. Q Rev Biophys 23:39–96
Breeze AL (2000) Isotope-filtered NMR methds for the study of biomolecular structure and interactions. Prog NMR Spectrosc 36:323–372
Baran MC, Huang YJ, Moseley HNB, Montelione GT (2004) Automated analysis of protein NMR assignments and structures. Chem Rev 104:3541–3555
Chandra K, Jaipuria G, Shet D, Atreya HS (2011) Efficient sequential assignments in proteins with reduced dimensionality 3D HN(CA)NH. J Biomol NMR 52:115–126
Acknowledgments
The facilities provided by NMR Research Centre at IISc supported by Department of Science and Technology (DST), India is gratefully acknowledged. HSA acknowledges support from DST-SERC and DAE-BRNS research awards. GJ acknowledges fellowship from Council of Scientific and Industrial Research (CSIR). We thank Dr. John Cort, Pacific Northwest National Laboratory, for providing the Ubiquitin plasmid.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Jaipuria, G., Krishnarjuna, B., Mondal, S., Dubey, A., Atreya, H.S. (2012). Amino Acid Selective Labeling and Unlabeling for Protein Resonance Assignments. In: Atreya, H. (eds) Isotope labeling in Biomolecular NMR. Advances in Experimental Medicine and Biology, vol 992. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-4954-2_6
Download citation
DOI: https://doi.org/10.1007/978-94-007-4954-2_6
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-4953-5
Online ISBN: 978-94-007-4954-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)