
Abstract
It is a simple matter to conceptually classify trace metal fractions in soil in terms of their relative ‘availability’. However, the challenge has always been to translate this qualitative understanding into predictive models of metal solubility, ideally based on proven mechanistic processes of specific adsorption, ion exchange, precipitation, colloidal flocculation, reduction, intra-aggregate diffusion etc. This chapter starts with a brief examination of the surface properties of the main metal adsorbents in soil, including humus, and the colloidal minerals Fe/Mn oxides, alumino-silicate clays, zeolites and sparingly soluble Ca salts. The interaction of these phases with the transient soil variables (e.g., pH, redox potential, temperature) in controlling metal solubility is discussed alongside the overarching influence of time. The sheer complexity of these co-dependencies still confounds our efforts to resolve an exact description of metal solubility and speciation and the range of modelling approaches which has emerged from this ‘confrontation’ is outlined. Currently the literature still presents an uneasy co-existence of relatively simple descriptions of metal solid ↔ solution equilibrium along with thermodynamically consistent mechanistic models. To compensate for their limited predictive power in the face of soil heterogeneity, the empirical equations are often extended to incorporate key soil properties (soil organic carbon content, pH etc.) as determinants of the model parameters – with surprising success. The principal challenge for mechanistic models is to resolve a meaningful basis for their description of metal dynamics which can be confirmed experimentally. However, an equally important goal is to reconcile the demanding requirements for their operation with the paucity of such data in geochemical studies and datasets. With much wider use in recent years the remaining shortcomings of the mechanistic models are becoming more apparent and this has created new experimental imperatives. These include characterising the small proportions of high affinity sites on adsorbents and developing descriptions of mixed adsorbents beyond a purely additive approach. The future promises continued improvement of models describing trace metal dynamics in soils and their increasing incorporation into human and environmental risk assessment tools.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Adsorption
- Desorption
- Reaction kinetics
- Diffusion
- Labile metal
- Solubility
- Humus
- Hydrous oxides
- Phyllosilicates
- Zeolites
- Soil pH
- Redox potential
- Humic acid
- Freundlich
- Langmuir
- WHAM-VI
- NICA-Donnan
- Assemblage models
1 Introduction
This chapter outlines the nature of metal and metalloid interactions with soil and current approaches to modelling these interactions.
1.1 Background to the Study of Metal Interactions with Soil
Heavy metals and metalloids are adsorbed strongly by the organic, and inorganic, colloidal constituents of soil. The resulting moderation of mobility and bioavailability of metals in the environment must rank as one of the most important ‘ecosystem services’ attributable to soils with implications for both micronutrient availability and potential toxicity to terrestrial and aquatic organisms. The overall strength with which metals are retained in soils is a reflection of:
-
(i)
‘residual’ properties attributable to the metal source material (primary minerals within the soil parent material, smelter fallout, sewage sludge, mine spoil, urban anthropogenic artefacts etc.);
-
(ii)
the intrinsic affinity of individual metal ions for soil adsorption surfaces and for soluble soil ligands;
-
(iii)
the suite of adsorption surfaces present in soils (humus, metal oxides, alumina-silicate clays etc.);
-
(iv)
the more ‘transient’ properties of the soil, including pH, redox potential, water content, temperature, biological activity, salt concentration etc.
-
(v)
soil-metal contact time.
A complete understanding of the mechanisms through which intrinsic and transient soil properties influence metal solubility still eludes us, despite decades of investigative effort. Nevertheless, as in all applied sciences, the emergence of new analytical techniques has gradually improved our ability to predict the behaviour of metals in soils. For example, synchrotron-based spectroscopy (EXAFS; XANES) is able to elucidate the bonding environment and valence of sorbed metals, electron microprobe and laser-ablation-ICPMS reveal the elemental make up of individual particulates, isotopic dilution and diffuse-gradient technology (DGT) quantify the kinetics and reactivity of surface-adsorbed metal ions and ion chromatographic and Donnan Membrane techniques enable speciation of metals in the soil pore water. An increasing number of studies utilise these techniques in combination to reveal a fuller picture of the interactions between metals and soils. This new wave of analytical information feeds directly into the development of increasingly powerful geochemical modelling approaches. Such models fulfil a dual role in (i) providing predictive information on metal fractionation (in solids) and speciation (in solution) and (ii) revealing the research imperatives for those studying metal sorption reactions.
Chapter three deals primarily with adsorption and precipitation reactions as the two recognised mechanisms whereby metals and metalloids are retained in soil. The latter process involves added complexity in a soil environment where compounds may (i) precipitate on pre-existing surfaces, (ii) exist as heterogeneous solid-solutions rather than pure phases and (iii) will in turn be subject to surface ‘contamination’ through adsorption reactions. Thus the boundaries between the two processes are rather diffuse and they may eventually be described in a more unified manner. For trace metals the more important reaction is usually adsorption and this is reflected in the greater weight given to Sect. 3.2 which describes the main types of adsorption surfaces and the manner of their interaction with metals. The effects of transient soil conditions on adsorption strength are outlined in Sect. 3.3 and current approaches to modelling the interaction of trace metals with soils are described in Sect. 3.4.
1.2 Overview of Metal and Metalloid Interaction with Soils
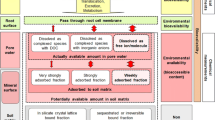
An emerging view of metal ‘fractionation’ in the solid phase, and ‘speciation’ in the solution phase, is illustrated in Fig. 3.1 below.

Schematic representation of three-phase divalent metal ion equilibria in soils. Broken and solid reversible arrows represent kinetically constrained and ‘instantaneous’ reactions respectively; ‘ML’ represents metal ions bound to soluble ligands
Minert is ‘inert’ metal, held in a form which only responds to changes in solution conditions over periods of years, possibly through mineral weathering, organic decomposition or changes in redox state. Examples include:
-
(i)
trace levels of metal ions buried within the structure of primary (or secondary) minerals by isomorphous substitution and released only by mineral weathering;
-
(ii)
resistant primary (e.g., sulphide) or secondary (e.g., phosphate, carbonate) metal compounds solubilised by slow oxidation reactions or dissolution over extended periods;
-
(iii)
recalcitrant organic compounds (containing metals);
-
(iv)
resistant anthropogenic contaminants such as metal fragments, vitrified particles etc.
Mnon-lab represents ‘non-labile’ metal that is not held reversibly and therefore shows apparent sorption hysteresis. It is held in forms which are ‘kinetically constrained’ and so responds to changing conditions slowly (days to months). However, desorption can occur without changes in redox status or decomposition of the absorbing substrate. Non-labile metal may exist within soil solids or even in solution/suspension linked to ligands (ML) such as humic/fulvic acid or contained within micron and sub-micron sized particulates [69]. Examples may include:
-
(i)
dehydrated metal ions which have diffused into the interior spaces or structural framework of minerals (e.g., metal hydrous oxides, collapsed planar alumino-silicates, selected voids within zeolites etc.) or become occluded through co-precipitation reactions;
-
(ii)
some surface-adsorbed metals held strongly, perhaps through multiple covalent linkages, on mineral or organic surfaces;
-
(iii)
an internal constituent of single or mixed metal compounds that are in solubility equilibrium with the soil solution;
-
(iv)
non-reversibly held metal complexes in solution and sub-micron particulates in suspension.
Mlabile represents ‘labile’ metal ions which respond reversibly (adsorption or desorption), and almost instantly, to changes in solution equilibrium and do not show ‘adsorption hysteresis’ [1]. These may exist in a wide range of chemical forms, including soluble ligand complexes (ML; inorganic and organic). Examples will include:
-
(i)
all exchangeably (electrostatically) held hydrated metal ions in clay interlayers and in the diffuse double layers of humus acids;
-
(ii)
some specifically-adsorbed metal ions on clay edges, Fe/Mn/Al hydrous oxide surfaces and humus;
-
(iii)
metal ions on the surface of metal compounds which are in solubility equilibrium with the soil solution;
-
(iv)
reversibly held metal complexes in solution including most inorganic species and a proportion of the metal bound to dissolved humic and fulvic acid.
Factors governing the distribution of inner-sphere surface complexes between ‘labile’ and ‘non-labile’ forms are still under investigation. However, the isotopically-exchangeable fraction of metals (Cd, Zn, Cu, Pb) always greatly exceeds the fraction exchangeable with salt cations (Ca2+, Mg2+) [82] implying that the ‘labile pool’ of metal must include (some) specifically-adsorbed metal. Furthermore, current models of metal binding to dissolved humic and fulvic acids are parameterised with datasets which assume complete reversibility of metal ion adsorption. This implies that even the most strongly bonded metal ions are nevertheless ‘labile’; of course this may not be the case.
M.(H2O)2+ represents free hydrated metal ions. These are often regarded as the ‘immediately’ bioavailable form of metal ions but there is good evidence that complexes in solution (MLlabile and possibly MLnon-labile) also contribute to apparent bioavailability. Obviously all forms in the soil solution (M.(H2O)2+, MLlabile, MLnon-labile) are subject to transportation in drainage water.
2 Adsorption of Metals on Soil Constituents
Trace metals are adsorbed on the surface of colloidal particles in soils, principally humus, hydrous oxides of Fe, Mn and Al, alumino-silicate clays and some sparingly soluble salts such as calcium carbonate.
2.1 Humus
2.1.1 Bulk Properties of Humus
Humus is the organic product of ongoing plant and animal decay in a soil environment. In contrast to the mineral constituents that adsorb metals, humus is markedly concentrated in the top 20 cm layer of most mineral soils, unless it has been redistributed by pedogenic processes, as in podzols. Soil organic matter content can vary from <1% (sandy subsoils) to over 90% (peats). The major elemental composition of humus includes carbon (C) (50–60%), oxygen (O) (30–40%) and both H and nitrogen (N)(c. 5%). Structurally, humic materials are around 20–30% aliphatic, 10–20% carbohydrate and 20–40% aromatic. About 20% of H is bound to O as carboxyl (Hu-COOH) and acidic (possibly phenolic) hydroxyl groups (Hu-OH), the rest is bound directly to C [113]. Humus is intensely heterogeneous and exists in a continuum of particulate, colloidal and molecular forms. These only broadly equate to the well known operational (extractable) fractions humin, humic acid (HA) and fulvic acid (FA) described in many texts [109, 113]. Comparatively little is known about the relatively hydrophobic ‘humin’ fraction, in particular. Furthermore, this simple fractionation scheme is complicated by the variable degree of molecular aggregation, and colloidal flocculation, to which humic substances are prone depending on soil pH, ionic strength, temperature and level of saturation with polyvalent cations.
2.1.2 Surface Chemistry of Humus Acids
The distribution and spacing of O-substituted acid groups are key determinants of the surface chemistry of colloidal and molecular HA and FA. Close juxtaposition of Hu-COOH and Hu-OH groups causes ‘mesomeric’ and ‘inductive’ electron-withdrawing effects, which create stronger acidity (lower pKa values). By contrast, electrostatic restriction from charged neighbouring groups, and hydrogen bonding, weaken the acidity of HA and FA (higher pKa values). In combination these effects extend the range of pH values over which surface charge is generated. Therefore, oxy-acid groups on humus dissociate across a wide pH range: pH 2–8 (Hu-COOH) and pH 7–11 (Hu-OH) generating an almost linear increase in negative charge from pH 3 to pH 10, often with the faint suggestion of an equivalence point (Hu-COO−) around pH 8. As a result, the strength of metal binding varies accordingly across the pH range (Sect. 3.3.1) with stronger adsorption at high pH.
2.1.3 Metal Binding by Humic and Fulvic Acids
Metals are thus bound to a range of sites on humus composed of a mix of oxygen, nitrogen and sulphur donor atoms. Potentiometric studies and a range of spectroscopic techniques (FTIR, EXAFS, XANES, ESR, NMR and others) have provided detailed, but not always consistent, information on the nature of metal binding. The negative charge on humus generates simple coulombic attraction of cationic metals, especially alkali and alkaline-earth cations. However, specific inner-sphere, complex formation of most heavy metals is likely and the presence of functional groups in close proximity suggests the formation of multidentate and multinuclear chelate sites. Binding of heavy metals is probably dominated by the formation of two or even three bonds to a mix of carboxyl and acidic hydroxyl groups. A prevailing idea for many years was that divalent trace metals were mainly bound in chelate structures on aromatic rings (essentially phthalic and salicylic acid units) although this has been difficult to confirm. The independence of binding parameters on humic acid concentration suggests that metals do not bridge between two humic molecules in solution but rather bond to single or multiple groups on one humic molecule [21].
There is also evidence for bonding to sites containing N (especially Cu) and sulphur (S) (especially Hg) but the relative importance of these ligands is unresolved [113]. Mercury (Hg) is thought to be associated with reduced S-containing ligands in humus [105]. Furthermore, Bernaus et al. [15] demonstrated, using XANES, that methylated forms of Hg (CH3HgCl) were covalently bound to humic acids, probably through thiol groups, into biologically unavailable forms. There is even evidence that (anionic) arsenate might be linked to DOC, possibly through bridge cations [20].
The relative importance of organic matter in metal binding has been demonstrated many times although this will clearly depend on the composition of the colloidal phases in individual soils. Spectroscopic techniques (e.g., EXAFS; [97]) show that even relatively weakly bound metals such as zinc (Zn) are predominantly held as organic complexes in soil. Removal of organic matter (e.g., by peroxide treatment) causes a profound reduction in metal adsorption capacity (e.g., for Cd; [87]). It is commonly found that strength of metal binding is affected by humus content and therefore by management practices which enhance humus [64].
2.1.4 ‘Active Organic Matter’
The organic fractions primarily responsible for metal binding, humic and fulvic acids, may be around 75% of the soil organic matter on average [109], but this varies between soils. Determining the proportion of what is termed the ‘active organic carbon’ (AOC) in soil solid and solution phases remains a challenge. To optimise their model of metal binding in a study of 98 upland soils from England and Wales, Tipping et al. [114] had to assume a range of AOC in the solid phase of 9–86%. Amery et al. [5] improved their prediction of metal (Cu) complex formation by equating the ‘active fulvic acid’ (AFA) fraction with ‘specific ultra-violet absorbance’ (SUVA, at 254 nm) as an index of ‘aromaticity’ in the dissolved organic carbon (DOC).
2.1.5 Humus Mineral Interactions
Humic and fulvic acids are strongly adsorbed on mineral surfaces, mainly through chemi-sorption to hydrous oxide minerals (Fe, Al, Mn) and probably through co-flocculation with inorganic colloids. Mixed effects on metal and metalloid solubility arise as a result. Organic matter may interfere with the formation of ‘stable’ metal precipitates such as lead (Pb)-phosphates (chloropyromorphite; [65]). On the other hand, there is evidence for enhanced adsorption on mixed assemblages of fulvic acid and Fe oxide [122, 125]. Anionic metalloids (arsenate, selenate) and metals (chromate) must compete with humic and fulvic acids for their preferred adsorption sites on Fe or Mn hydrous oxides. A current priority in trace metal modelling is to provide a coherent description of mixed humus-mineral assemblages, such as humic acid adsorbed on the surface of Fe/Al/Mn hydrous oxides [126].
2.1.6 Dissolved Organic Carbon (DOC)
Soil colloids exist in a flocculated state; only small proportions (0.001–0.1%) of humic and fulvic acids are dissolved in solution at any given time. However, the presence of this dissolved organic carbon (DOC) has profound implications for transport of metals that strongly bind to humic and fulvic acids. In the case of Cu, for example, commonly up to 99% of Cu in the soil solution is complexed to fulvic acid [111]. In the context of risk to aquatic systems, strong bonding of metals to organic matter has always been recognised as a ‘double-edged sword’ in that the overall effect of strong bonding on metal mobility will depend on the solubility of the humus itself [63]. Metal transport in rivers and accumulation in floodplain soils are probably determined by the movement of humus acids and their subsequent deposition. Schroder et al. [99] found very strong correlations between humus content and metal loading in floodplain topsoils around the rivers Meuse and Rhine (Netherlands). Thus, predicting DOC solubility and the ratio of HA to FA in solution is another current challenge for heavy metal modelling in soils [86].
2.2 Hydrous Oxides and Hydroxides
2.2.1 Bulk Properties
2.2.1.1 Manganese Oxides
Manganese oxides in soil exist in a range of forms between MnIIO and MnIVO2, mainly in octahedral co-ordination. Phase changes are initiated by sufficient Mn atoms undergoing alteration of valence state (2, 3, or 4) to cause morphological transformation [77]. They are highly prone to dissolution, as Mn2+, under reducing conditions (Eh <200 mV), and re-precipitation within oxidising zones in the soil. Local accumulation of Mn (and Fe) may result from (re-) adsorption of reduced forms (Mn2+, Fe2+) from solution on to manganese and ferric oxide surfaces followed by re-oxidation and (thus) crystal growth of the original oxide surface. This leads to their characteristic appearance as coatings and nodules in subsoils subject to periodic water logging, often in association with Fe hydroxides. Much of the Mn hydrous oxide content of soil is amorphous although lithiophorite [Al2 Li Mn IV2 MnIIIO6(OH)6], birnessite [(Na, Ca)(MnIII, MnIV)7O14.2.8H2O], and others have been identified in contrasting soil environments. Lithiophorite is more common in older soils and birnessite in younger soils [33].
2.2.1.2 Iron Oxides
Iron oxides may be described as the inevitable end product of weathering in soils. Older, more heavily weathered soils often contain a greater free Fe oxide content than younger soils; the most obvious example is the tropical Ferralsol (Oxisol). Part of weathering, alongside dissolution and loss of silicic acid (H4SiO4), is the oxidation of ferrous iron (FeII) in alumino-silicates (e.g., the octahedral layer of biotite) to the ferric form (FeIII) and precipitation as a range of ferric oxide minerals. Thus Fe oxide is usually fairly evenly distributed through the profile of aerobic soils but is subject to the same re-distributive effects under fluctuating redox regimes as Mn oxides. In surface water and ground water gley soils Fe oxides may be concentrated on the outside of peds, and line the surface of major drainage pores and root channels, while in soils with an iron-pan, as the name suggests, they may exist as a consolidated layer determined partly by leaching with organic acids and partly by the prevailing redox state of the soil. Iron oxides often occur in poorly crystalline or amorphous forms but recognised crystal forms also exist. Goethite (α-FeOOH) is perhaps the only stable form in aerobic soils, Haematite (α-Fe2O3) is most common in hotter climates, Lepidocrocite (γ-FeOOH) is precipitated during the oxidative ‘phase’ of waterlogged soils and Ferrihydrite (Fe5(O4H3)3) is associated with re-precipitation in podzol Bf horizons and the bed of streams, ditches and ochre in field drains [101].
The bulk distribution of Fe and Mn oxides within soil profiles therefore contrasts markedly with the normal distribution of humus. However, local co-accumulation due to adsorption of humus on to oxide surfaces is also common.
2.2.2 Surface Chemistry
An electrical charge on Fe and Mn oxides arises firstly from adsorption and release of H+ ions from oxygen atoms at the interface between the mineral and the bathing solution (soil pore water). Hydrous oxides are amphoteric; they carry both positive and negative charges and the net surface charge is largely symmetrical around a zero point at a characteristic pH value [10]. This implies that three possible states apply to oxygen atoms on the surface of (e.g., Fe) oxides (Ox-Fe-OH):
The point of zero charge (pzc) for most Fe oxides is between pH 7 and pH 8.5 implying that they are positively charged in most soils. The pzc values of Mn hydrous oxides are lower than Fe oxides (pH 1.5–4.6) [77] which means they carry a net negative charge in most soils. Surface charge on oxides is also affected by salt concentration – i.e., the concentration of non-specifically adsorbed (electrostatically attracted) ‘counter-ions’ such as Na+ and Cl− – and temperature. Both these dependencies arise because the thermal diffusive forces resisting counter-ion accumulation close to charged surfaces have a greater influence at low salt concentration in the bulk solution and at higher temperature.
2.2.3 Metal Binding
Both Mn and Fe hydrous oxides have a high adsorption affinity for heavy metals which are bound as inner-sphere mono- and bi-dentate surface complexes. It is only recently that spectroscopic techniques have been able to determine the precise nature of metal bonding on Fe/Mn oxide surfaces. Arcon et al. [7] used EXAFS and XANES to show that arsenic (As) adsorbed in Cornish (UK) soils contaminated with mine spoil was primarily present as arsenate (AsV) bound to Fe oxides and Al hydroxides – despite the origins of the As, presumably as AsIII. Fendorf et al. [36] investigated arsenate and chromate bonding to goethite using EXAFS and suggested three types of bonding mechanism were apparent: monodentate, bidentate-mononuclear and bidentate-binuclear (Fig. 3.2). For both anions the monodentate form was most prevalent at low levels of surface coverage. Arai [6] employed EXAFS to distinguish between nickel (Ni) co-ordinated as face-, edge- and corner-sharing surface complexes on Goethite, Haematite and Ferrihydrite. They concluded that the majority of Ni was bound via edge-sharing, bidentate, inner-sphere Fe-O2-Ni units.

Arsenate bound to the surface of Goethite as monodentate, bidentate-mononuclear and bidentate-binuclear surface complexes (After Fendorf et al.[36])
The relative affinity of a metal for specific oxides is likely to be determined by oxide charge relations and the morphology and pore sizes within the oxide surface [42]. The affinity of different metal ions for a specific oxide will also be determined by the speciation of the metal ion in solution. Figure 3.3 below shows the effect of pH on adsorption of ten metals by Goethite [37]. All the metals show an increase in adsorption as pH rises, as a result of reduced competition with H+ ions and increasingly negative surface potential. The different positions of the curves reflect the intrinsic (chemical) affinity of the individual metals for the adsorption sites and their ability to overcome the positive surface charge on the mineral surface (pzc of Goethite is c. pH 8) and bond to surface oxygen atoms. However, note that the curves for Cr(III) (and Al) are particularly steep. Fischer et al. [37] suggest that, as pH rises, there is hydrolysis to CrOH2+ which may be the preferred adsorption species. This does not apply to the other ions which do not hydrolyse within their respective adsorption zones. Thus adsorption affinity follows the sequence: Hg > Cr > Al > Pb ≈ Cu > Zn> Co ≈ Ni > Cd > MnII. Relatively strong adsorption of trivalent ions, followed by Pb and Cu, and the similarity of Co and Ni behaviour, are features commonly seen in metal adsorption envelopes on other oxides.

Particularly strong associations have been noted between Mn/Fe oxides and Co, Ni, As, V, Pb, Cr and Zn [7, 63, 77]. In particular, correlations between Mn and Co in both solid and solution phases in soils are almost invariable high; it is likely that the majority of Co in soils is associated with Mn oxides. In addition to chemical bonding and electrostatic effects reduction-oxidation coupling may also be involved in some sorption reactions. For example, Co2+ is oxidised to CoIII on the surface of MnIVO2 and isomorphously incorporated into the oxide structure because CoIII is very similar in size to MnIV. A similar reaction (with Co) was suggested by Gerth [42] for ‘Cd substituted’ Goethite. It is recognised that Mn oxides also oxidise CrIII to highly toxic CrVI [63], however this effect is lessened if the oxide surface is highly substituted with adsorbed Co, Pb, Cu, Ni etc. [83]. Similarly, AsIII oxidation to AsV on oxide surfaces, which will enhance adsorption and reduce potential toxicity, may be possible [33].
There is clear evidence of time-dependent incorporation of metals into oxide structures [10], which can render the sorbed metal ions ‘non-labile’ (Fig. 3.1). This may occur by solid-phase diffusion or by occlusion through co-precipitation. The latter mechanism is particularly important under fluctuating redox regimes because both FeIII and MnIV hydrous oxides are subject to reductive dissolution and re-precipitation through oxidation over the redox potentials (Eh values) occurring in moist or saturated soils. These effects are discussed in more detail in Sects. 3.3.2 and 3.3.4; modelling metal adsorption is discussed in Sects. 3.4.3 and 3.4.4.
2.3 Planar Alumino-Silicate Clays (Phyllosilicates)
2.3.1 Bulk Properties
Planar alumino-silicate clays, or ‘phyllosilicates’, are composed of layers of silica tetrahedra and aluminium octahedra in ratios of 1:1 or 2:1. The types of clay minerals most commonly identified are Illite, Vermiculite, Smectite, Chlorite and Kaolinite; all are found in a wide range of soils. These may be very loosely associated with a ‘weathering sequence’ characterised by decreasing isomorphous substitution and variable accessibility of the interlayer space. Thus, Illite is the immediate alteration product of mica and most vermiculites are, in turn, derived from further weathering of Illite, or Mica. Smectites can occur through further alteration or through re-crystallization from solution. Pedogenic Chlorite forms through deposition of non-exchangeable Al-hydroxy polymers in the interlayer space and so is often present in acidic soils. The most common 1:1 clay is Kaolinite which is typically prominent as a result of geological inheritance or through prolonged and severe weathering, as found in tropical Ferralsols (Oxisols), for example [127].
2.3.2 Surface Chemistry
The surface area of phyllosilicates can be extremely large – up to 600 m2 g−1 for Smectites. Isomorphous substitution of the Al3+ or Si4+ in either the tetrahedral or octahedral layers creates a positive charge deficit which is balanced by cations in the ‘inter-layer space’. Thus, for example, the unit cell composition of a di-octahedral 2:1 clay is:
in which there is isomorphous substitution of nAl3+ for nSi4+ in the tetrahedral layer resulting in a positive charge deficit of ‘n’ and requiring n/v cations with valence v+ in the interlayer space of the clay to balance the negative charge from the structural oxygens and hydroxyls [O10(OH)2].
This gives rise to a ‘cation exchange capacity’ (CEC) which depends on both the negative charge of the layers and the degree of access to the interlayer space. Illite, derived from weathered mica, has the largest surface charge but provides only limited access to the interlayer space because it is mainly collapsed with dehydrated K+ ions balancing the isomorphous substitution (CEC = 10–40 cmolC kg−1). Vermiculite has less remaining isomorphous substitution but the interlayer space is accessible so its CEC is largest (CEC ≈ 100–160 cmolC kg−1). Smectite has a still lower remaining surface charge but again is wholly accessible (CEC ≈ 70–150 cmolC kg−1). Chlorite has a limited CEC because of non-exchangeable Al-hydroxy deposits in the interlayer space in acidic soils (CEC < 10 cmolC kg−1). Kaolinite is a very pure aluminosilicate with virtually no isomorphous substitution and a very limited ability to hold cations (CEC = 3–15 cmolC kg−1). Exchangeable cations are fully, or partially, hydrated and bound as outer-sphere complexes, primarily by electrostatic forces; specificity arises from ion size and ease of dehydration. Larger ions with weaker hydration shells are bound more strongly so that adsorption strength follows the sequence Cs+ > Rb+ > K+ > Na+ > Li+ and Ba2+ > Sr2+ > Ca2+ > Mg2+ for alkali and alkaline-earth cations respectively.
2.3.3 Metal Binding
Cationic heavy metals will be held in the interlayer space of phyllosilicates, as exchangeable cations, but they tend to be poorly represented because of the overwhelming presence of Ca, Mg and K in soils. However, trace levels of metal binding to clays can be considerably more complex. Furnare et al. [39] used EXAFS and XANES to study Cu2+ adsorption on Vermiculite and Smectite. They suggested that Cu was covalently bound through inner-sphere complexes to unbonded oxygens at clay edges and corners. They also identified occupancy of octahedral vacancies within the clay structure as well as simple outer-sphere electrostatic retention in the interlayers. The interlayer area can also support highly specific bonding: Ni, Co and Zn are thought to form multi-nuclear species which bind ‘irreversibly’.
There has been considerable interest in specific metal binding to clays with Al-hydroxide polymer species in the interlayer space (C_Al_HO systems). It may be that such systems bind some metals more strongly than simple oxides because of their greater surface area for chemi-sorption. Janssen et al. [56] found that Zn and Cu were specifically bound to AlHO in the clay interlayer space but Cd and Pb were only held exchangeably, through outer-sphere linkages, and were easily displaced by other cations. Sako et al. [96] showed specific adsorption of the platinum group elements, originating from catalytic convertors, in a kaolinitic soil (Pd > Rh > Pt). The trend in adsorption with pH was very similar to that expected for oxides (as in Fig. 3.3), with a steep rise in affinity over a single pH unit.

Proportional distribution of Cu, Cd and Pb between organic matter (solid line), Fe oxide (Δ), Mn oxide (●) and alumino-silicate clay (broken line) in the soil solid phase, as a function of pH, according to WHAM-VI. Hypothetical soil conditions included 1.2 kg suspended in 1 L solution; 5% humus with 30% HA and 20% FA (50% inert); 20% clay, 3.5% Fe oxide and 0.075% Mn oxide. The metals Cu, Cd and Pb were at the current UK ‘sludge limits’ (135, 3 and 300 mg kg−1). Solubility of Al3+ and Fe3+ were controlled by Al(OH)3(s) and Fe(OH)3(s). The solution phase contained 20 mg L−1 FA and 0.005 M Ca2+, 0.0005 M K+ balanced by 0.0095 M NO −3 and 0.001 M Cl−
2.4 Zeolites
2.4.1 Bulk Properties
Zeolites are ‘tectosilicates’, composed of silica tetrahedra which form a hydrated microporous three-dimensional framework, with intra-crystalline morphological features called ‘windows’, ‘cages’, ‘cavities’ and ‘channels’ [76]. The most common zeolites in nature are: analcime, chabazite, clinoptilolite, erionite, mordenite, phillipsite and ferrierite but they rarely comprise more than a few percent of the clay minerals in soils. Usually, zeolites in soils are inherited from parent materials derived from weathered volcanic deposits or from alluvial or eolian deposits [16]; the most commonly found mineral is clinoptilolite. Zeolites can also be ‘pedogenic’; these typically occur in saline-alkaline environments in which analcime is perhaps the most common example [16].
2.4.2 Surface Chemistry
Zeolites are negatively charged by isomorphous substitution of Al3+ for Si4+. Their generic composition is Cx/v[(AlO2)x (SiO2)y].mH2O, where C represents a charge-balancing cation of valence ‘v’. Their surface charge is therefore permanent and the resulting CEC can be extremely large, depending on the degree of Al substitution. Most zeolites have potential CEC values of 200–600 cmolC kg−1 which is considerably greater than the phyllosilicate clays (Sect. 3.2.3).
2.4.3 Metal Binding
A large proportion of the CEC of zeolites exhibits rapid and reversible cation exchange [22]. However, because of their complex 3-D structure zeolites also exhibit some specific adsorption capacity with respect to individual metal cations. The dimensions of the various ‘cages’, ‘channels’ etc. within the zeolite structure determine selectivity for different sizes of cation to a large degree. This form of selectivity has been called ‘ion sieving’ [16]. Fletcher and Townsend [38] demonstrated apparent irreversibility of trace metal sorption on Faujasite after only short contact times. Ören and Kaya [85] found evidence of surface precipitation of Zn in natural clinoptilolites above pH 6.0. By following changes in isotopic exchangeability with 109Cd2+, Ahmed et al. [1] demonstrated Cd adsorption hysteresis and prolonged time-dependent ‘fixation’ in a synthetic zeolite. The ‘fixation’ was well described as a reversible first order kinetic process and was thought to be associated with Cd ion dehydration and ingress to smaller pores within the zeolite structure.
The comparative rarity of zeolites in soils belies their potential importance in metal sorption and the application of this property to soil remediation [13] and water protection [58]. Natural zeolites have found widespread application in recovering trace metals (Pb, Cd, Zn, Ni, Co, CrIII, Cu and FeII) from wastewaters [90, 116] and the treatment of low-high level nuclear waste liquids [30, 51]. There have been several studies of the application of zeolites as amendments for metal-contaminated soils [25, 67].
2.5 Sparingly Soluble Calcium Salts and Metal Precipitation Reactions
2.5.1 Adsorption on Ca Salts
Sparingly soluble Ca salts are found in soils that are calcareous (CaCO3), gypsiferous (CaSO4.nH2O) or are heavily fertilized and contain Ca phosphates (e.g., hydroxyl apatite). Metals may bond to the surface of Ca salts either because of similarity to Ca (e.g., Cd) or because of a high affinity for the salt ligand (e.g., Pb for PO4). However, they may also precipitate on the Ca mineral surface as discrete compounds or form mixed solid-solutions.
There have been comparatively few studies of heavy metal adsorption on simple Ca compounds. Ahmed et al. [2] demonstrated that Cd is adsorbed in an isotopically exchangeable form on calcite but also undergoes time-dependent fixation within the calcite structure; they worked within the normal range of solution Cd concentrations found in soil pore water, below the ion activity product at which CdCO3 might precipitate. Papadopoulos and Rowell [88] suggested that a ‘mixed solid-solution’ of CdnCan−1(CO3) formed at higher Cd concentrations.
2.5.2 Discrete Metal Compounds in Soils
For metals to form discrete stable precipitates in soils the ion activity product (IAP) in the soil pore water must exceed the solubility product (Ks) of the compound (IAP > Ks). With the exception of the structural cations Fe, Mn and Al, this rarely occurs in uncontaminated soils because trace metals are present at comparatively small (total) concentrations and the free ion activity of trace metals in soil water is generally very small because of strong surface adsorption by soil constituents. Other factors may prevent formation of discrete metal compounds even if conditions are otherwise favourable for precipitation. Thus, Lang and Kaupenjohan [65] suggested that organic acids in soils could prevent crystal growth of the lead phosphate compound chloropyromorphite [Pb5(PO4)3Cl].
However, trace metals can exist as ‘guest elements’ within the matrices of other soil minerals. This is thought to occur through:
-
(i)
the process of isomorphous substitution, which occurs during the formation of the mineral;
-
(ii)
slow solid phase diffusion into pre-existing minerals following adsorption;
-
(iii)
occlusion within minerals during dissolution-precipitation cycles caused by fluctuating pH and/or Eh regimes.
It is not disputed that discrete metal compounds exist in soils,but their longevity, stability and the extent to which they can control metal solubility are poorly understood [32]. Soil processes act to dissolve introduced compounds which then become adsorbed on the soil colloidal phase even in quite heavily contaminated situations. Degryse et al. [31] showed that about 50% of Pb was isotopically exchangeable in soils heavily contaminated by smelter output and battery plants, despite having neutral pH values. Similarly, Sarret et al. [97] fractionated Zn in a soil contaminated by smelter output and showed by EXAFS that only 15% of the soil Zn was present as residual primary minerals while 54–92% was isotopically exchangeable and mainly bound to humus. Smolders and Degryse [106] showed that up to 40% of the Zn in tyres (mainly ZnO) becomes labile in soils within 1 year.
Nevertheless, unusual metal compounds have been identified in soils with extremely high residual contaminant loads or subject to contemporary inputs. For example, amorphous FeAsO4 was found in mine spoil contaminated soils with 8.8% As using EXAFS/XANES [7]. Sphalerite (ZnS) and Franklinite (ZnFe2O4) were identified in soils close to Zn smelters with >6,000 mg kg−1 using EXAFS and electron microprobe analysis [95]. Murakami et al. [80] also used electron probe microanalysis to identify particulates in roadside dust with >0.2% Cr and Pb, thought to originate from vehicle exhaust soot, brake lining and paint. Councell et al. [27] discuss the importance of vehicle tyre wear as a source of anthropogenic Zn in soils. Uzu et al. [118] found a range of Pb compounds (PbS, PbO.PbSO4, PbO, PbCO3) in PM10 and PM2.5 particles from a battery recycling plant and confirmed a link between particle size and bioavailability to plants.
Many studies have confirmed the presence of Pb compounds in particular. Some of these concern residual introduced material but many also focus on the pedogenic formation of discrete Pb phosphates. Chloropyromorphite formation has been encouraged by the introduction of soluble forms of phosphate to limit Pb solubility in urban brownfield soils [63] and land used as shooting ranges [49]. Other forms of Pb identified include: metallic Pb, PbO and PbCrO4 in industrially contaminated soils, identified by SEM-EDX [57]; Hydrocerrussite [Pb3(CO3)2(OH)2] resulting from weathering of shotgun pellets, identified by micro X-ray fluorescence [120]; Pb oxide in soils subject to smelter fallout [53].
2.6 Conclusions on Adsorption of Metals on Soil Constituents
As a very broad generalisation, the principal adsorption sites for trace metals may be on humus at low pH (<6) and on Fe oxides at high pH (>6). Thus, in a review of the behaviour of metals in tropical soils Rieuwerts [93] examined the perceived risk that (cationic) trace metals may be highly available in some oxisols and ultisols specifically because of the low pH and the relatively low content of humus. However, evidence for metal movement down the soil profile was inconclusive and it appeared that iron oxides provided effective adsorption sites despite low soil pH. Buekers et al. [18] examined fractionation using the geochemical model WHAM-VI (Sect. 3.4.3) to partition metals in the solid phase between humus, oxides and aluminosilicate clays. Similarly, Fig. 3.4 shows the fractionation of Cu, Pb and Cd, predicted by WHAM-VI, for a hypothetical soil (see Fig. 3.4 description) assumed to have metal concentrations at the UK sludge regulation limits. The model illustrates substantial differences between the three metals. Copper has the strongest affinity for humic and fulvic acids. Lead is bound almost exclusively by Mn oxide at low pH and Fe oxide at pH > 7. Cadmium is the most weakly bound metal, such that below pH 5 there is even substantial non-specific bonding to the alumina-silicate clay fraction (Sect. 3.2.3.3); however, above pH 7 Cd sorption is dominated by the Fe oxide fraction. The substantial differences that exist between metals and the dependence of fractionation on soil properties are clear; these are examined in more detail in the following section.
3 Effect of Soil Conditions on Metal and Metalloid Adsorption
3.1 Soil pH Value
Soil acidity is measured on the ‘pH’ scale, normally in a 1:2.5 (w/v) soil:solution suspension, using a combined H+-sensitive glass electrode and Ag-AgCl reference electrode, where:
Soil pH is both affected by, and indicative of, specific ecosystems or land use practices (Table 3.1).
3.1.1 Effect of Soil pH Value on Metal Solubility
Soil pH value has a greater influence on the solubility of heavy metals than any other factor. This would be predictable considering simple metal oxide, hydroxide and carbonate (MCO3) solubility reactions.
However, as discussed by Degryse et al. [32], the stoichiometry of M2+↔H+ exchange is usually less than the value of 2.0 required for many precipitation reactions which indicates the importance of specific adsorption in controlling metal ion solubility.
For cationic metals a change in soil pH has several important, and independent, effects which influence the apparent strength of adsorption by soil solids. Thus, as pH value rises, the following changes occur.
-
(i)
Surface charge, and electrical potential, will become more negative on the principal adsorption sites on Fe/Mn hydrous oxides, alumino-silicate clay edge sites and humus.
-
(ii)
Competition for adsorption sites from hydronium ions (H3O+) and from dissolved ‘structural’ cations (mainly Al3+ and AlOH2+) will decline as H3O+ ions are neutralised and soluble aluminium ions are (re-)precipitated as Al(OH)3(s).
-
(iii)
The speciation of the metal cation may change through formation of soluble hydroxy and bi-carbonate complexes, for example. If the principal adsorbing species is the divalent cation (e.g., Cu2+) then this effect will increase overall metal solubility. However, it has also been suggested that the first hydroxide complex of some metals (e.g., CrIIIOH2+) may be the more important adsorbing species and so changes in metal speciation may produce quite complex effects on adsorption.
-
(iv)
A related effect arises if the increase in soil pH dissolves metal-complexing anions. Again these will bring more metal into solution against the prevailing trend of greater sorption into the solid phase. The principal example here is the soluble components of humus: humic (HA) and fulvic (FA) acids. The solubility of HA and the strength of metal binding by both HA and FA increase as pH rises and the humus acids become more negatively charged and more hydrophilic. This effect is most pronounced for metals that bind most strongly to HA, such as Cu. It has been shown that the distribution coefficient (kd) of Cu in some soils is close to the kd value for dissolved organic carbon (DOC) [32, 111]. It may be that some sub-micron inorganic colloids, which both adsorb and contain trace metals, behave similarly although evidence for this is currently still emerging [69].
Notwithstanding some of the more subtle effects listed in (iii) and (iv) above, the dominant effect of an increase in soil pH is a decrease in cationic metal solubility. An example of this trend is shown in Fig. 3.5 below where samples of a soil used for sludge disposal for over a century were suspended in 0.01 M Ca salts and acidified to various levels. The solubility of Cd clearly follows a broadly exponential trend with pH within the range of the measurements.

Cadmium concentration in solution as a function of soil pH in batch equilibrations (1:10 w/v suspensions of 0.01 M Ca(NO3)2 or 0.01 M CaCl2) of an arable soil from a sewage disposal site. The soil was from one of the oldest and most heavily ‘sludged’ disposal areas (Cd = 59.3 mg kg−1; humus = 24.8%) solid lines are simple exponential fits
3.1.2 Liming Acid Soils to Lower Heavy Metal Solubility
Mineral soils and peats which have a pH (in water) below pH 6.5 or pH 5.8 respectively normally require liming with CaCO3 for arable agriculture, partly to avoid aluminium toxicity. Similarly, soil may be limed to around pH 7 to avoid toxicity from contaminant metals such as Pb, Ni, Cu, Cd, Zn, Hg, Sn. Examples include restoration of urban ‘brown-field’ sites, land affected by acid mine drainage, and management of arable land habitually used for sewage sludge disposal.
The alleviation of aluminium toxicity in acid soils can be shown by the simplified reaction illustrated below. Exchangeable aluminium hydroxide ions (Al(OH)2+), held in clay inter-layers and by humus functional groups (Humus-COO−), are precipitated as crystalline or amorphous forms of aluminium hydroxide (Al(OH)3). So the solubility and bio-availability of toxic aluminium is reduced.
This involves a series of individual reaction steps, including:
-
(i)
dissolution of CaCO3(s) to release Ca2+ and CO 2−3 into solution
-
(ii)
cation exchange between Al(OH)2+ and Ca2+ on clay and humus;
-
(iii)
hydrolysis of \( {\hbox{CO}}_3^{{2 - }} \) to produce OH− ions and CO2;
-
(iv)
precipitation of solid aluminium hydroxide, Al(OH)3(s).
By contrast, when a soil contaminated with divalent heavy metals is limed it is unlikely that precipitation of the (divalent) Pb, Ni, Cu, Cd hydroxides, carbonates or oxides will occur (IAP < Ks). Therefore, it might be expected that Ca2+ from liming agents would simply liberate adsorbed metal ions into solution, which would increase the potential toxic hazard. Of course the solubility of the contaminant trace metals almost always decreases due to the rise in pH caused by the addition of lime. Fundamental to the successful use of lime to reduce the solubility and bioavailability of potentially toxic metals is the difference in bonding to soil of alkali/alkaline-earth cations (e.g., Ca2+) and most heavy metals. The former are largely held weakly as exchangeable hydrated ions, by electrostatic forces, within alumino-silicate clay interlayers or on charged humus carboxyl groups. Heavy metals, by contrast, are strongly (specifically) bonded to exposed oxy-acid groups on humus and hydrous oxides as inner-sphere (partially dehydrated) complexes. Therefore, addition of lime creates two opposing reactions affecting metal solubility. Calcium ions may release into solution the limited amount of ‘exchangeable’ trace metals that are held in clay interlayer sites and humus. However this effect is completely negated by the titrating effect of the carbonate ions which neutralise H+ ions from organic carboxyl and surface hydroxyl groups on Fe/Mn hydrous oxides and thereby free sites for occupancy by strongly binding divalent heavy metals. The latter occurs as a combination of reduced competition from H+ ions (and AlOH2+ ions) and increased electrostatic attraction between negatively charged humus or hydrous oxide sites and metal ions, as discussed above.
The example below represents liming an acidic soil contaminated with lead initially present in an available, in this case exchangeable, form. Calcium ions displace exchangeable forms of Pb into solution but carbonate ions neutralise specifically adsorbed H+ ions on humus carboxyl groups (Hu-COOH), increasing (negative) surface charge and encouraging greater specific adsorption of Pb2+ ions.
Liming acid soils to pH 7–8 remains the most expedient way of reducing the solubility of heavy metals, in most situations.
3.2 Soil Redox Potential (Eh)
Redox potential (Eh, mV) is an intensive thermodynamic property related to the hypothetical activity of electrons (e−) through the well known Nernst equation and, in practice, is normally measured with a platinum electrode with Ag/AgCl chloride electrode as reference (pe = −log10(e−)).
Although the derived value of (e−) can be used in equilibrium calculations, as if it were a discrete chemical species, it is accepted that Eh values are more safely regarded as an ‘index’ of redox status when measured in soils. This is partly due to difficulties in measuring Eh [100] and partly because there is always doubt whether redox equilibrium has truly been established [61].
A decrease in soil redox potential (Eh value becomes less positive) typically occurs when soils become wet. The resulting impediment to the diffusion of oxygen into the soil causes the utilization of alternative electron acceptors as organic matter decomposition continues under progressively more anaerobic conditions. The general reduction sequence followed in soils is well established [91]. Table 3.2 shows the broad range of Eh values in which specific redox reactions are ‘poised’ (buffered) as soils become progressively more anaerobic.
The rate at which the redox potential falls is mainly determined by the nature and availability of the carbon substrate, the degree of restriction on oxygen ingress and the availability of the alternative electron acceptors. It is recognised that some heavy metals become more soluble under anaerobic conditions. Thus Pareuil et al.[89] have suggested that it may be useful to characterise the risk of metal mobilisation in contaminated soils by equilibrating with a range of concentrations of reducing agents, such as ascorbate, to generate a ‘redox mobilization edge’ (metal solubility vs Eh). However, as redox potential falls naturally, due to microbial activity in saturated soils, several changes occur which may affect metal and metalloid solubility in contradictory ways. These are discussed below.
3.2.1 Change in pH
Most redox reactions in soils involve transfer of electrons and protons. This dependence can be described by Eq. 3.4.
Eo is the standard redox potential for the reaction and the ratio nH/ne denotes the stoichiometric ratio for electron and proton transfer. In practice it is unlikely that Eq. 3.4 will be followed exactly because (i) there are widely varying concentrations of specific redox couples and (ii) soil pH is strongly buffered by other proton adsorption reactions which are not directly involved in redox reactions [78]. Nevertheless, redox reactions do have a marked effect on soil pH. In fact, soil pH values gravitate to a very restricted circumneutral range under anaerobic conditions (c. pH 6.5–7.5) [91]. This is thought to arise from the conflicting effects of (i) a build up of CO2 partial pressure which lowers pH in alkaline soils by hydrolysis to carbonic acid and (ii) the consumption of H+ ions by most reduction reactions which raises pH in acid soils (Table 3.2) [94]. Thus for soils which have pH values <6.5 under aerobic conditions, the net result of anaerobism may be an increase in the strength of cationic metal adsorption by humus and metal oxides and an increased possibility of (e.g.) metal carbonate precipitation (Sect. 3.3.1).
3.2.2 Dissolution of Fe/Mn Hydrous Oxides
At pH > 6 Fe and Mn oxides are likely to be significant adsorbers of cationic metals and anionic oxyacids (Sect. 3.2.2). If the oxides are reduced and solubilized then two significant effects on metal solubility should occur. Firstly, metals and metalloids adsorbed on the surface will be released as the underlying substrate dissolves. This can be represented as a surface reduction-dissolution reaction for (e.g.,) Pb sorbed on ferric oxide (balancing ions omitted):
The heavy metals may be re-adsorbed on the freshly exposed oxide surface but this must depend on the extent of oxide dissolution and the thickness of the oxide coating. Secondly, the release of large concentrations of Fe2+ and Mn2+ into solution will present intense competition for metal adsorption sites on humus, clay edges and, of course, on the newly created FeIII/MnIV oxide surfaces. The overall result of reduction may therefore be increased solubility of trace metals.
When a flooded soil drains, or dries through evaporation, redox potential rises and Fe/Mn oxides re-precipitate. This will cause re-adsorption of liberated metals and if co-precipitation occurs, may even result in ‘fixation’ of metals within oxide particles. Contin et al. [23, 24] suggested that deliberately implementing redox cycles in contaminated soils could be used to lower metal bioaccessibility in some soils. Metal fixation followed the qualitative order Cu > Cd > Zn> > Pb> > Ni using three cycles with anaerobic periods of 40 days each, interspersed with short drying periods. Lee [66] suggested that a similar fixation process may have maintained low metal availability in a kaolinitic paddy soil contaminated by mine tailings.
3.2.3 Changes in Metal and Metalloid Valence
Some metals and metalloids undergo valence changes within the range of redox potentials experienced in anaerobic soils – arsenic is a prime example. The stable form of As in aerobic soils is arsenate (mainly H2AsO −4 and HAsO 2−4 in the pH range 6–7) which behaves very similarly to phosphate (pKa2 = 7.2) and is adsorbed strongly by Fe oxides. As Eh values fall below 250 mV AsV is reduced mainly to AsIII (arsenite) and possibly methylated forms such as dimethylarsinic acid (DMA; (CH3)2HAsO2) and monomethylarsonic acid (MMA; CH3H2AsO3). The closest analogue to arsenite is perhaps silicic acid (pKa1 = 9.8), and so AsIII forms a neutral molecule (H3AsO o3 ) that is very poorly adsorbed. This may be the prime mechanism whereby As is released into groundwater in aquifers which become even mildly anaerobic, as experienced in West Bengal and Bangladesh [60].
Figure 3.6 shows that transformations between arsenate and arsenite are predicted to occur within the pH-Eh ranges often encountered in wet topsoils, or aquifers with sufficient organic matter to undergo reduction.

Predominance diagram for arsenic species as a function of pH and Eh (Produced using The Geochemist’s Workbench® program)
Chromium is also subject to changes in valence within the range of conditions found in soils. Reduction of CrVI to CrIII increases adsorption (as Cr3+ or CrOH2+) and renders carcinogenic Cr VI less harmful. Thus Banks et al. [8] found that adding organic manure detoxified CrVI and promoted greater adsorption. Cobalt is strongly adsorbed within Mn oxides as CoIII [42] (see Sect. 3.2.2) and, correspondingly, is usually strongly correlated with Mn2+ release under anaerobic conditions [47], partly through reduction of CoIII to Co2+. Selenate reduction to selenite should increase adsorption as selenite is a specifically adsorbed anion while selenate is a strong acid anion which is indifferently adsorbed and, at pH 6–7, will remain almost entirely in solution.
3.2.4 Release of Complex-Forming Anions and Dissolved Organic Carbon (DOC)
Under anaerobic conditions, anions (phosphate, bicarbonate, fluoride, molydate, humic and fulvic acids etc.), normally adsorbed on Fe/Mn oxides, come into solution as pH rises and the oxides are partially solubilised. In addition, in flooded soils, DOC concentration normally increases as a result of decomposition reactions under oxygen-free conditions. This may have two conflicting effects on cationic metal solubility. Complex formation in solution, especially with HA, FA and other DOC compounds such as propionic and butyric acids [91] will clearly increase the solubility of strongly bound metals such as Cu, Pb and rare earth elements [47]. On the other hand, the formation of new mixed solid phase assemblages, such as humic acid with Fe oxides, may actually increase adsorption of metals [122].
3.2.5 Precipitation of Metal Sulphides and Carbonates
Increased soil pH and partial pressure of trapped CO2 following soil flooding may cause precipitation of metal carbonates [70]; this has been reported for Cd, Zn and Pb [78]. However, under strongly anaerobic conditions (Eh < −100 mV), especially in soils with a significant (reducible) sulphate content, it may be possible for metal sulphide precipitation to occur [34]. From solubility modelling, Schroder et al. [100] suggested there was strong evidence for Pb, Zn, Cd and Cu sulphide precipitation in floodplain soils under such conditions. Cornu et al. [26] incubated an industrially contaminated arable soil (Cd = 3.8 mg kg−1) under saturated conditions and observed an initial increase in Cd solubility followed by a decline as pe fell below 3. It was thought possible that CdS (Greenockite) may have formed but this could not be confirmed thermodynamically. Theoretically, the trace metals Cd, Zn and Pb should all precipitate before ferrous sulphide formation ‘poises’ the system and thereby prevents further decline in Eh. However, this depends on the free ion activity of the trace metals supported which is difficult to predict when progressive anaerobism usually coincides with changes in soil pH and complexing ligand concentration.
3.2.6 Biomethylation
Some metals and metalloids are biologically transformed to methylated forms under anaerobic conditions. These may be more (Hg) or less (As) hazardous than inorganic forms. The range and significance of methylated and organic forms of As are relatively unknown. Many studies have focussed only on arsenite (AsIII) and arsenate (AsV) in studies of As transformations in soils. However, Huang and Matzner [54] identified seasonally dependent microbial methylation of As (MMA, DMA, TMAO) in pore water from wetland soils in Germany, along with several unknown organic forms of As.
3.3 Soluble Ligands
In most soils, metal solubility is significantly affected by the presence of complex-forming ligands. The most common organic ligands are fulvic and humic acids which are present in all soil pore solutions; these are discussed in Sects. 3.2.1 and 3.4.3. However inorganic ligands can also increase metal solubility. These typically include SO 2−4 , F−, NO −3 , Cl− with HCO −3 and CO 2−3 at high pH. Chloro-complexation in particular has received considerable attention because Cd has a moderately high affinity for Cl-, forming soluble CdCl+ and CdCl 02 complexes with overall stability constants: logβ(CdCl+) = 1.98 and logβ(CdCl 02 ) = 2.6. In fact, Ahmed et al. [1] demonstrated that Cd adsorption (on zeolites) was controlled exclusively by Cd2+ and chloro-complexation increased Cd solubility quantitatively. Thus Cd mobilization in soils subject to elevated chloride has been the subject of some concern [107]. Millward and Liu [79] found the mobilization of metals from sediments suspended in seawater followed the sequence Ni > Cd > Zn > Cu. Rasa et al. [92] suggested that Cd may be more soluble in roadside soils due to NaCl use for de-icing. However, the net effect on metal solubility in salt-affected environments will be a combination of several independent processes in addition to complex formation, including cation exchange with Na+, weakening of adsorption by increased ionic strength, possible flocculation or dispersion of colloidal particles and effects on pH and Eh (Sects. 3.3.1 and 3.3.2). Thus Du Laing et al. [34] reviewed the factors affecting metal behaviour in (saline) estuarine soils and noted, for Cd in particular, that mobilization by chloro-complexation is offset by precipitation with sulphide in anoxic saline environments.
Non-humic organic ligands which can mobilize metals may arise from natural root exudates (citrate, malate, oxalate) [131] and from the application of certain pesticides, such as Glyphosate [9]. However, the effect of these ligands on metal solubility will partly depend on the solubility and longevity of the organic acid in soils.
3.4 Time, Temperature and ‘The Slow Reaction’
3.4.1 Time
It is generally recognised that metal ions added to soils in solution will rapidly attain an apparent equilibrium with adsorption surfaces over a period of several hours. However, most metals and metalloids then exhibit further (slower) sorption, over an extended period (months), in which metal ions are transferred from a ‘labile’ to a ‘non-labile’ state [32, 117]. This is generally referred to as ‘the slow reaction’, ‘ageing’ or ‘fixation’ [123] and may be partly (or wholly) responsible for the phenomenon of ‘desorption hysteresis’ commonly observed for metal ions adsorbed from solution on to soil minerals such as calcite, zeolites and goethite [1, 2, 11]. However, it is important to note that ‘fixation’ is not always a time-dependent process and may also be caused by very rapid reactions such as surface precipitation or possibly strong inner-sphere adsorption. Thus it is often found that a proportion of metal ions, added to soil in solution, become fixed almost instantly – as best judged by isotopic exchangeability. Nakhone and Young [82] noted this effect for Cd fixation in calcareous soils while Ma et al. [71–73] observed a similar process for Cu adsorbed by soils high in organic matter.
There are several mechanisms which may explain the slow reaction, including diffusive limitations, adsorption energy barriers and surface precipitation; in practice these are not entirely independent.
3.4.2 Diffusive Limitations
It is recognised that diffusive limitations exist in solution as ions slowly penetrate the microporous structure of soil aggregates. This may be especially true for anionic species, such as HAsO 2−4 and CrO 2−4 , due to repulsion (anion exclusion) from micro-pores lined with negatively charged surfaces [84]. Immediately following adsorption there is likely to be progressive ‘surface diffusion’ as metal ions re-distribute themselves between adsorption sites to achieve lower free energy states. However, it is ‘solid phase diffusion’, following initial surface adsorption [11], which is usually most closely associated with the extended slow reaction. This involves penetration of the solid phase matrix by replacement of the native cation (or anion) and may occur by true solid phase diffusion but could also occur by occlusion resulting from the continuous process of dissolution and re-precipitation. Diffusive processes may be particularly important at pH values below those required for precipitation reactions; Ma et al. [73] suggested that a diffusive process was solely responsible for slow fixation of Cu in soils below pH 5.
3.4.3 Adsorption Energy Barriers to Specific Reactions
Energy barriers to adsorption may occur where there is a specific part of the adsorption mechanism that is rate-limited, such as dehydration, the formation of multiple bonds or slow exchange reactions. Ahmed et al. [1] found that both slow adsorption and desorption of Cd on zeolites was better explained as a first-order kinetic process rather than by continuous diffusion into the zeolite particles. From EXAFS data they provisionally associated the Cd fixation process with retention of dehydrated Cd2+ in a specific cavity in the zeolite structure [3].
3.4.4 Discrete and Surface Precipitation
Precipitation of metals may occur on existing mineral surfaces or as discrete compounds. In both cases there may be slow reactions involving (i) ‘Ostwald ripening’ to reduce the surface : volume ratio, (ii) formation of compounds with more stable stoichiometries and (iii) continued penetration of mineral surfaces to form solid solutions. Ma et al. [73] suggested that the slow reaction between Cu and calcareous soils signified Malachite (Cu2(OH)2CO3) and Cu(OH)2 formation and ‘precipitation/nucleation’ reactions generally occurring above pH 6.
3.4.5 Effect of Soil Properties on the Slow Reaction
It seems likely that soil components which have little scope for (internal) absorption because of the lack of a porous structure or no clear means of occluding metals will show little if any slow reactions. Thus Covelo et al. [28] found that metals (Cd, Cr, Cu, Ni, Zn, Pb) were most reversibly adsorbed on 2:1 alumino-silicate clays and least reversibly adsorbed on Mn oxides. Palagyi et al. [87] investigated sorption of 109Cd by forest and arable topsoils and subsoils, with peroxide treatment to remove humus. They showed that Cd is reversibly held by humus but undergoes time-dependent fixation in mineral constituents. Shirvani et al. [104] demonstrated Cd sorption hysteresis in the Mg-silicates Palygorskite and Sepiolite and in Calcite. Ahmed et al. [2] measured and modelled time-dependent fixation and adsorption hysteresis of Cd on Calcite. Nevertheless, it may also be possible for organic matter to ‘fix’ metals. Vega et al. [121] expressed irreversible Cd and Cu sorption in soils as a ‘hysteresis index’ (HI) and showed that although HI was greatest for Cd at high pH and MnO2 content, it was greatest for Cu at high humus content. Young et al. [130] reported isotopically non-exchangeable Hg in filtered solutions of 203Hg spiked soils which they tentatively interpreted as fixation in DOC. Thus, fixation of metals may also occur simply through strong surface complexation, without occlusion within solids.
Slow fixation reactions have been demonstrated most successfully using isotopic exchange to discriminate between ‘labile’ and ‘non-labile’ forms [1, 19, 29]. Figure 3.7 shows the reduction in isotopic exchangeability of Zn, initially added to soils in solution, as a function of pH. As expected, fixation was greater at high pH. However, a crucial point is that the slow reaction had to be modelled as a reversible first-order kinetic reaction [1, 2, 29]; the decline in metal lability does not tend to zero but to an apparent steady state in which non-labile metal ions re-enter the labile pool. This also supports the universal finding that historically contaminated soils typically have substantial proportions of isotopically exchangeable metal. Therefore metal ions determined as non-labile should be regarded as ‘kinetically restricted’ in their interaction with the soil solution phase rather than permanently ‘fixed’. This reinforces the distinction between ‘non-labile’ and ‘inert’ in Fig. 3.1; Degryse et al. [32] discuss this issue in more detail.

Change in isotopic exchangeability of Zn initially added in solution to 23 soils categorised by pH range. The slow reaction is modelled as a reversible first-order kinetic process (solid lines)
3.4.6 Temperature
A higher temperature increases the proportion of the metal ions which have sufficient energy to overcome energy barriers to adsorption or desorption and therefore movement in general. This effect can be described in terms of an activation energy (Ea, kJ mol−1). The rate constant for adsorption or desorption (k) is related to temperature through the well known Arrhenius equation:
A is the ‘pre-exponential factor’, with the same units as the rate constant (k). Temperature will therefore also affect the final position of the equilibrium if the value of Ea for adsorption differs from that for desorption. Equation 3.5 translates directly into describing the temperature dependence of diffusion (Eq. 3.6) where Dapp is the apparent diffusion coefficient for the solid and Do is Dapp when the activation energy is zero [108].
Thus, fixation by diffusion into spherical solid particles can be described by equations of the form:
Where Pfo and Pft are the proportions of added metal ions fixed at time zero and time ‘t’.
The linear dependence of metal fixation on √t is often presented as strong evidence for a diffusion-controlled process and most studies appear to conclude that a higher temperature will increase the rate of fixation in soils. Thus, Ma et al. [72] used a combination of Eqs. 3.6 and 3.7 to model the decline in isotopic exchangeability of Cu added to soils and incubated at three temperatures for 30 days. A similar response has been observed for Cd adsorption on goethite [81], Pb reaction with carbonate hydroxyl apatite [128] and several other studies. In addition, the relative affinity of different metals may change with temperature and differences in activation energy may indicate the underlying fixation process, such as diffusion [43] or reflect the nature (e.g., porosity) of the soil adsorption surfaces [11].
4 Modelling Metal and Metalloid Adsorption
In the soil chemistry literature many of the early empirical approaches to modelling trace metal equilibria, intended mainly for predictive purposes, can still be found published alongside the very latest developments in ‘mechanistic’ models of metal adsorption.
Adsorption of metals by soil, and soil constituents, is often expressed as the mathematical relationship between the concentrations of metal in the solid phase (Mads) and in an equilibrated solution (Msoln). However, both Mads and Msoln may represent several alternative variables. The variable Mads may be the total concentration of metal in the soil solid phase, or it may be a specific fraction such as ‘extractable’ or ‘isotopically exchangeable’ metal [40]. Similarly, Msoln can represent either the ‘total’ or the ‘non-complexed’ concentrations of metal ions in solution, or their free ion activity [117].
The two most common objectives of characterising the solid-solution equilibration of metals are:
-
(i)
to predict concentration (or activity) of metal species in solution, perhaps under changing circumstances, to address questions of plant availability, ecotoxicological risk to water systems etc.;
-
(ii)
to assess alternative adsorption mechanisms and (ideally) gain greater understanding of the adsorption process itself.
4.1 Simple Adsorption Isotherm Equations
The simplest expression of the strength of metal adsorption by soil is the ‘distribution coefficient’ (kd): the ratio of Mads to Msoln (Eq. 3.8).
The most obvious shortcomings of this approach to describing metal equilibria are the implied assumptions of (i) infinite adsorption capacity, (ii) homogeneity of adsorption site type, (iii) no effect of site occupancy on the free energy of (further) adsorption and (iv) no effect of competitor ions (e.g., H+ or other metals). Clearly for sorption of metal ions on to charged humus and oxide surfaces, in the chemical cocktail of a soil environment, all of these assumptions are invalid. Nevertheless, the simple distribution coefficient has found widespread application in risk assessment approaches which characterise sorption of radionuclides and pesticides. For metals also, if we assume that the characteristics (pH, humus content, texture, metal content) of individual soils are fairly conservative, then metal kd values could provide a reasonable assessment of adsorption strength for comparative purposes. Sheppard et al. [102] measured kd values for 54 elements in Canadian soils intended as an aid to critical load assessment. Covelo et al. [28] suggested a standard comparative index (kd100) which is the value of kd measured following adsorption by 12 g soil from 200 mL of an initial solution concentration of 100 mg L−1 after 24 h equilibration. They also proposed extending this to a general index of metal adsorption strength (kdΣ100) for a suite of different metals.
Nevertheless, in most studies of metal adsorption, the non-linearity of the relationship between Mads and Msoln is acknowledged and a wide range of equations have been deployed to describe metal adsorption isotherms in Soil Science. Giles et al. [44] classified adsorption isotherms on the basis of their ‘shape’ (initial slope and curvature). Their basic classification includes constant partition (C), high affinity (H), Langmuir (L) and sigmoidal (S) forms. The choice of which adsorption equation to use is discussed in several publications [50, 59]. Traditionally, however, the two most widely used functions are the well known Langmuir and Freundlich adsorption equations.
The Langmuir equation (LE, Eq. 3.9) is attractive because it is asymptotic [50] and therefore ‘provides’ an important soil characteristic – the maximum adsorption capacity or ‘number of sites’ (Ns). Its failings include an assumption that the standard free energy of adsorption is constant, regardless of ‘site coverage’ which, for ion adsorption on to charged surfaces, is incorrect. Although the single LE also ignores competition from other ions and assumes a single type of adsorption site, these can easily be included in extended forms of the equation [129].
The Freundlich equation (FE, Eq. 3.10) is an empirical relation that is consistent with an exponential distribution of binding energies [12]. Strobel et al. [110] demonstrated that the kd values for Cd and Cu in arable soils increased with depletion of the metal; therefore a measured kd value may overestimate the solubility of the majority of the soil metal content. Thus the FE addresses the issue of site heterogeneity, to some degree, and it is probably fair to say that it is considerably more useful than the LE for trace metal studies [12]. Although the FE ignores the notion of an adsorption maximum it could be argued that this is seldom an issue because the concentration of adsorbed metal present is normally much less than the metal-adsorption capacity of the soil.
Both equations are still used to describe metal adsorption for comparative purposes. This approach is perhaps most valid when the soils being studied have a restricted range of conditions (especially pH) or when single soils or pure mineral substrates are being assessed. Sako et al. [96] used the FE to compare adsorption of platinum group elements (Pd, Rh and Pt) on a single kaolinitic soil. Jalali and Moharrami [55] tested both the FE and LE as descriptions of metal adsorption in calcareous soils from Western Iran with a narrow pH range (c. pH 7.0–7.5). Markiewicz-Patkowska et al. [75] used the simple LE to estimate the adsorption capacities of (urban) brownfield soil material at just two (controlled) pH values (pH 2 and pH 7).
The simple LE and FE models are often applied when the adsorption of metals is only a part of the overall reaction process being studied and it is convenient to adopt a simple description of metal equilibrium. Thus, Őren and Kaya [85] applied the FE to Zn adsorption on zeolite. Shirvani et al. [104] described adsorption of Cd on pure clays (Sepiolite and Palygorskite) and calcite using the FE. Ahmed et al. [2] used the FE to describe Cd adsorption on zeolite. In all these studies the primary objective was not to derive a definitive description of adsorption per se but to integrate a convenient adsorption equation into a wider study of adsorption kinetics, desorption hysteresis and/or reaction mechanisms.
There have been several attempts to develop more flexible forms of the FE and LE which address the problems such as site heterogeneity and competition from other ions. Temminghoff et al. [111] tested a ‘two species Freundlich’ (TSF) model alongside the more mechanistic NICA model (Sect. 3.4.3) to describe Cu2+-H+ competition for adsorption on humus in a sandy soil. Barrow et al. [12] used a modification of the FE, in which the exponent (n) varied with solution concentration (Msoln), to explain competition between selenite and phosphate in a Chilean Andosol. Xue et al. [129] adapted the LE to include competition and non-integer reaction stoichiometry to describe Cd and Zn adsorption on a yellow cinnamon soil from China.
4.2 Relating Adsorption Equations to Soil Properties
It is inevitable that ‘single component’ (i.e., Msoln) forms of the LE and FE provide only ‘soil-specific’ parameters at best and, for the same reasons, they will fail if the characteristics (such as pH) of a single soil are changed. The recognition of correlations between soil properties (e.g., pH) and metal adsorption strength has therefore encouraged the development of regression approaches so that metal adsorption may be empirically predicted for soils with differing characteristics. The soil properties chosen for regression are usually those known to have direct, or indirect, effects on metal binding strength and are normally fairly ‘accessible’ soil variables. Typically they include various combinations of pH, cation exchange capacity (CEC), humus content (%C or %LOI), Fe or Mn oxide content, clay content, concentration of the major cations or competing trace metals etc.
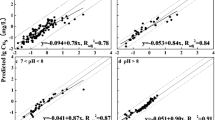
In fact, such regression equations are essentially an extended form of the FE if it is assumed that logkF is a linear function of multiple soil properties (Pi), while the Freundlich exponent (n) remains constant across all soils, (Eqs. 3.11 and 3.12):
This has been called the ‘general purpose Freundlich’ model (GPF) [35]. Sauve et al. [98] reviewed over 70 studies and optimised GPF expressions for Cd, Cu, Pb, Ni and Zn with pH and SOM content as the predictive soil properties (Pi, Eq. 3.12). This approach explained 61 – 88% of the variation in dissolved metal concentration; for Pb the fit was considerably poorer. Gray and McLaren [45] included the predictive properties pH, %C and amorphous Fe/Al oxide content in a study of 200 New Zealand soils contaminated from a wide range of sources. Their optimised equations were able to predict between 50% and 85% of the variation in Msoln for six metals across the range of soils studied. Horn et al. [52] parameterised GPF equations for Cd with a dataset of nearly 500 soils from Germany; they included pH, clay, soil organic carbon, CEC and Fe/Mn oxides as predictive soil properties. Similar dependencies on soil properties were found by Lair et al. [64] for long term field experiment soils from Austria. Tipping et al. [114] included just pH and %LOI as predictive properties in a study of metal solubility in 98 upland soils from the UK and achieved results which were only slightly inferior to a more complex mechanistic model (Sect. 3.4.3). Gandois et al. [41] found that the solubility of eight trace metals, in a range of soils, could be predicted from soil organic carbon content, pH value, total Fe content and DOC.
Attempts have also been made to improve the basis of expression for the two metal variables (Mads and Msoln) in order to describe metal equilibria on a more mechanistic basis. Thus, Elzinga et al. [35] used 92 published sources to collect 2,000, 700 and 1,500 data for Cd, Cu and Zn adsorption respectively. They tested for improvements in the fit of a GPF model by varying the definition of Msoln as total concentration or corrected for ionic strength, inorganic complexation and organic complexation. Overall these refinements to Msoln produced fairly small improvements. Model predictions of Mads from field datasets under-estimated by a factor of 5–10. This was thought to be due to overestimation of ‘adsorbed reactive’ metal by acid digestion of field soils and partly because of metal competition effects which are not included in the adsorption studies but limit metal adsorption in field soils. Trancoso et al. [115] tried to improve the fit of a GPF description of Pb equilibria in a study of Portuguese soils (Pi = pH, Fe/Al oxides) by measuring labile forms of Pbsoln using differential pulse polarography. Tye et al. [117] used what was effectively a GPF approach, applied to UK soils incubated with added Cd (Pi = %LOI, pH, Znsoln, ionic strength). They defined Cdsoln as the free ion activity (Cd2+), calculated from a solution speciation model (WHAM-VI; Sect. 3.4.3) and Cdads was measured as the isotopically exchangeable (labile) soil Cd concentration. Groenenberg et al. [46] used a similar approach, but with nitric acid extractable metal as a proxy for the ‘reactive’ metal pool (Mads). They confirmed the controlling influence of humus content and pH value on free ion activity of Cd, Cu, Pb, Ni and Zn in over 200 soil solutions from the Netherlands and UK. Broadly speaking, in most applications of the GPF to large multi-soil datasets, the resulting ‘models’ are able to predict Msoln from soil properties (Pi) to within approximately 0.5–1.0 log10 units.
4.3 Mechanistic Models of Metal Adsorption
A wholly rigorous treatment of adsorption reactions is simply not possible because of the inherent complexity of soils. Thus, when models of metal adsorption are described as ‘mechanistic’ this normally means that (i) defined adsorption mechanisms form the operational basis of the model and (ii) the model is thermodynamically consistent in that the masses of all defined species and their associated charges are conserved and balanced respectively.
Several geochemical models describing metal bonding to organic matter, Fe/Mn/Al oxides and alumina-silicate clays have been combined to predict equilibrium speciation and fractionation in ‘whole soils’ (see for example the Visual MINTEQ site managed by Jon Petter Gustafson [48]). Perhaps the two models which have been most widely applied are the ‘extended versions’ of the Windermere Humic Acid Model (WHAM-VI) and the Non-Ideal Competitive Adsorption model (NICA-Donnan). Both these models started life as descriptions of metal binding to humic and fulvic acid in solution and have been extended to include surface complexation models dealing with exchange reactions on clays and specific adsorption on Fe/Mn/Al oxides to form the ‘assemblage’ models ‘SCAMP’ (e.g., [68]) and ‘ECOSAT’ (e.g., [124]) respectively. Crucially, this progression enables a complete audit of metal speciation in solution and fractionation in the solid phase. However, the description of metal binding to humus acids probably remains the most important component of both extended models and, in this function, they have elements in common. They both recognise that: (i) HA and FA have two types of group; (ii) there is heterogeneity within each type of group; (iii) metals may bond to HA and FA through multiple linkages; (iv) HA and FA are charged molecules, partitioned between solid and solution phases, with non-specifically adsorbed counter ions. The basic approach of both models is briefly outlined below.
4.3.1 WHAM-VI Model
WHAM-VI [112, 113] has its origins in ‘conventional’ descriptions of equilibria between metals and anionic molecules in solution but extended to allow for build up of electrostatic charge on macromolecules with multiple functional groups. In WHAM-VI, humic and fulvic acids are assumed to be impenetrable spheres (radii 1.72 and 0.8 nm) with the functional groups (Hu-COOH and Hu-OH; Sect. 3.2.1) confined to the surface of an otherwise hydrophobic molecule.
The existence of negative surface charge (Z) on humic acid molecules (Hu-COO−) weakens proton dissociation constants and so the apparent acidity constant at a given humic charge (KZ) is calculated from an ‘intrinsic’ dissociation constant (Ki), which would apply if the humic molecule carried zero surface charge, modified by an electrostatic interaction factor (w) and the surface charge (Z). The electrostatic interaction factor (w) is an empirical function (P) of ionic strength to reflect suppression of the electrostatic field around the charged humic molecule or colloid at high salt concentrations.
Group heterogeneity is incorporated by assuming two types of group (A and B) which probably, but not explicitly, represent carboxyl and phenolic-hydroxyl groups. Each group type is subdivided into four and it is assumed that (i) the ratio of Type A: Type B groups is 2:1 and (ii) each of the four subgroups are equally represented on the humic molecule. The resulting eight subgroups all have individual ‘intrinsic’ pKi values. These are calculated from the average intrinsic pK (pKA or pKB) and a ‘spreading factor’ to provide ‘group heterogeneity’ within type A and B groups, For example, for carboxyl (A) groups:
Typical parameter values for humic acid are pKA = 4.0 and ΔpKA = 1.8 which would give four carboxyl groups with intrinsic pKi values of 3.1, 3.7, 4.3, 4.9.
Specific sorption of metals is described with analogous intrinsic equilibrium constants and allowance for electrostatic effects. Metal-specific spreading factors provide heterogeneity and there is also allowance for mono-, bi- and tri-dentate bonding between metals and functional groups. Values of intrinsic stability constants for bidentate (log(Ki,j) and tridentate (logKi,j,k) bonding are expressed as the sum of the component monodentate constants but with additional terms (x ΔLK2 and y ΔLK2) which extend the heterogeneity caused by multi-site bonding to allow for high affinity chelate sites. Thus, for tridentate bonding, y is set to 0, 1 or 2 for 90.1%, 9% and 0.9% of the adsorption sites. Considering the large number of possible sites arising from combinations of groups (e.g., 120 tridentate sites), the number of sites used in the model is rationalised by restricting bi- and tri-dentate sites to combinations of dissimilar groups types and utilizing a representative sub-set (80 sites in total, of which 8 are monodentate).
Finally, metal ions are also bound (weakly) as non-specific counter-ions within the diffuse double layer around the charged humic molecule. In the WHAM model this zone is simply described as a discrete Donnan layer, calculated from ionic strength, and the amount of electrostatically bound trace metals reflects their relative abundance in the bulk solution and their valence. Normally the ratio of chemically bound to electrostatically bound trace metals would be high.
4.3.2 NICA-Donnan Model
The origins of the NICA-Donnan model are closer to the mathematical descriptions of adsorption isotherms; the model was derived originally from the Langmuir-Freundlich equation (LFE). The NICA model also recognises (two) discrete classes of group on humic acid but within each class assumes a continuous ‘affinity distribution’ of functional groups rather than trying to subdivide into individually identified groups. The LFE includes provision for site heterogeneity and can be extended to describe adsorption of multiple species (protons and metal ions). However, in order to describe the specific adsorption affinity shown by individual metal ions, a further term was introduced. The equation below describes adsorption of a divalent metal ion (Mads) by humic acid, again with two types of group (A and B) in which T represents the total concentration of group A or B. The exponent (n) primarily reflects metal-specific stoichiometry; the ratio nM/nH may be seen as the average binding stoichiometry of metals to individual groups and so should be <1 although this is not always true for optimised values. The parameter ‘P’ (0–1) describes the heterogeneity of ion affinity and is a characteristic of the molecule rather than individual metal ions; (M2+) is the free ion activity of the adsorbing metal species and K is the average affinity constant. The compound term Tj(nM,j)/(nH,j) reflects the different binding maximum, to group A or B, for individual ions.
The NICA-Donnan model again includes provision for non-specific metal adsorption within a diffuse double layer of the charged humic acid molecule. More detailed descriptions of this widely used model, its applications, and its extension to (whole soil) assemblage models incorporating the metal oxide binding model ‘CD-MUSIC’ can be found in many publications, e.g., [62, 119].
4.3.3 Applications of WHAM and NICA-Donnan Models
An obvious, and probably unfair, criticism of these models is that they are ‘data-hungry’. To work effectively, as a means of predicting speciation in solution and fractionation in the solid phase, they should ideally be useable in ‘default mode’ with input of data from straightforward soil chemical analysis. If users have to measure or infer a large number of unique soil parameters (specific surface areas, active humus fractions, intrinsic stability constants etc.) for each application then their value diminishes. However, when such models are applied to whole soils it is often found that some ‘fine tuning’ of model parameters is required. Furthermore, in comparison with empirical approaches, such as the GPF or regression equations, mechanistic models may sometimes provide only marginal advantages.
Thus Tipping et al. [114] applied WHAM-VI to 98 upland soils from the UK and improved the fit by using (optimised) soil-specific values for the active organic matter (AOM) fraction. Shi et al. [103] also had to optimise the proportion of AOM when using WHAM-VI to predict Cd adsorption by 14 non-calcareous soils from the USA. Amery et al. [5] used WHAM as an assemblage model to predict metal speciation in an arable field site in Belgium and tried to improve estimates of ‘active fulvic acid’ in solution by UV-spectroscopic measurement. Ålmas et al. [4] applied WHAM to Norwegian arable soils contaminated with smelter output and had to optimise the active fraction of metals in the soil; for arable soils they found that the results were sensitive to the ‘active’ Ca concentration in particular. Buekers et al. [18, 19] applied WHAM-VI to 28 soils with widely varying characteristics. Their prediction of solution speciation was good and the distribution of Cd, Ni, Zn and Cu in the solid phase showed the expected pH-trend across the range of soils with Fe oxides deposing organic matter as the principal metal adsorbent at pH > 6. However, they identified several complications to the application of the model including possible irreversible ‘fixation’ of Cu within organic matter, the need to refine the estimate of ‘reactive’ metal (using isotopic exchange) and some uncertainty in the specific surface area used for Fe oxides.
Benedetti [14] applied the NICA-Donnan (with CD-MUSIC) model to a woodland soil from France affected by Pb/Zn smelting activity and found good agreement with measured solubility of Cd, Zn, Cu and Pb, following minor changes to humic-Pb binding parameters. MacDonald and Hendershot [74] applied the NICA-Donnan model to a study of Canadian forest soils contaminated with smelter output and had to optimise the ‘active organic matter’ fraction and the ratio of HA to FA for each soil. Bonten et al. [17] applied the NICA-Donnan model to a large dataset from the Netherlands and found disparities in Zn-contaminated soils where the simple measure of ‘active Zn’ (0.43 M HNO3 extraction) failed. They also identified possible problems with the description of Zn binding to metal oxides, Cd binding constants to humus acids and overestimation of reactive Pb. They suggested that there may be a possible problem with the ‘additivity’ principle of assemblage models where adsorption surfaces (humus, oxides, clays) are considered only as discrete entities and interactions between them are not considered.
4.4 Conclusions
Ultimately, mechanistic models of metal adsorption will probably supersede the simpler approaches outlined in Sects. 3.4.1 and 3.4.2. This will arise partly because of an increasing awareness of the importance of both fractionation in the solid phase and speciation in solution to accurately assess mobility, bioavailability and ecotoxicological risk of heavy metals in soil. In future, it will not be sufficient to predict Msoln to within an order of magnitude from broad empirical relationships, nor, as at present, to rely on measurement of total soil metal content in risk assessments. However, key to providing more effective mechanistic ‘assemblage’ models will be resolving some important underpinning processes.
4.4.1 Single Adsorbents
Aspects still requiring improvement include:
-
(i)
better characterisation of adsorption surface capacity – predicting or measuring the reactive surface area of Fe/Mn/Al oxides and the proportion of ‘active organic matter’ in both the solid and solution phases;
-
(ii)
developing better descriptions of adsorption on alumina-silicate clay edges and within ‘pillared’ clays;
-
(iii)
allowing for more complex ion-surface associations within existing surface-complexation models, such as ternary complexes in which specifically adsorbed anions bridge heavy metal associations with (Fe)oxides.
4.4.2 Multi-Phase Adsorbents
There is also a need for greater understanding of colloidal-molecular interactions between the individual metal adsorbents (FA/HA, Fe/Mn/Al oxides and silicate clays). There is evidence that the aggregate behaviour of organic-mineral assemblages is not simply additive, due partly to interacting electric field effects [122]; modelling the resulting effects on metal adsorption is an active research area [125, 126].
4.4.3 Reduction and Oxidation Processes
The integration of changing redox conditions into adsorption models has been neglected in the past, despite a clear qualitative understanding of its importance, and is likely to feature in future developments [100].
4.4.4 The ‘Slow Reaction’
Following early work integrating solid phase diffusion processes with surface complexation models [10] there is now the emergence of coupled ‘kinetic-adsorption’ models [18, 19, 37, 123].
4.4.5 Solution Speciation
Finally, in the solution phase, a greater appreciation of more complex metal speciation is emerging with evidence of slowly reactive metal in sub-micron sized particulates [69, 71] which might require a re-assessment of consequences for mobility and ecotoxicology.
References
Ahmed, I. A. M., Young, S. D., & Crout, N. M. J. (2006). Mechanisms of cadmium sorption and fixation on CaX-Zeolite. Geochimica et Cosmochimica Acta, 70, 4850–4861.
Ahmed, I. A. M., Crout, N. M. J., & Young, S. D. (2008). Kinetics of Cd sorption, desorption and fixation by calcite: A long term radiotracer study. Geochimica et Cosmochimica Acta, 72, 1498–1512.
Ahmed, I. A. M., Young, S. D., Crout, N. M. J., Mosselmans, J. F. W., & Bailey, E. H. (2009). Coordination of Cd2+ ions in the internal pore system of zeolite-X: A combined EXAFS and isotopic exchange study. Geochimica et Cosmochimica Acta, 73, 1577–1587.
Ålmas, Å. R., Lofts, S., Mulder, J., & Tipping, E. (2007). Solubility of major cations and Cu, Zn and Cd in soil extracts of some contaminated agricultural soils near a zinc smelter in Norway: Modelling with a multisurface extension of WHAM. European Journal of Soil Science, 58, 1074–1086.
Amery, F., Degryse, F., Cheyns, K., De Troyer, I., Mertens, J., Merckx, R., & Smolders, E. (2008). The UV-absorbance of dissolved organic matter predicts the fivefold variation in its affinity for mobilizing Cu in an agricultural soil horizon. European Journal of Soil Science, 59, 1087–1095.
Arai, Y. (2008). Spectroscopic evidence for Ni(II) surface speciation at the iron oxyhydroxides-water interface. Environmental Science and Technology, 42, 1151–1156.
Arcon, I., van Elteren, J. T., Glass, H. J., Kodre, A., & Slejkovec, Z. (2005). EXAFS and XANES study of arsenic in contaminated soil. X-Ray Spectrometry, 34, 435–438.
Banks, M. K., Schwab, A. P., & Henderson, C. (2006). Leaching and reduction of chromium in soil as effected by soil organic content and plants. Chemosphere, 62, 255–264.
Barrett, K. A., & McBride, M. B. (2006). Trace element mobilization in soils by glyphospate. Soil Science Society of America Journal, 70, 1882–1888.
Barrow, N. J. (1987). Reactions with variable-charge soils. Dordrecht: Martinus Nijhoff Publishers.
Barrow, N. J., Gerth, J., & Brümmer, G. W. (1989). Reaction kinetics of the adsorption and desorption of nickel, zinc and cadmium by goethite II. Modelling the extent and rate of reaction. European Journal of Soil Science, 40, 437–450.
Barrow, N. J., Cartes, P., & Mora, M. L. (2005). Modifications to the Freundlich equation to describe anion sorption over a large range and to describe competition between pairs of ions. European Journal of Soil Science, 56, 601–606.
Baydina, N. L. (1996). Inactivation of heavy metals by humus and zeolites in industrially contaminated soil. European Journal of Soil Science, 28, 96–105.
Benedetti, M. F. (2006). Metal ion binding to colloids from database to field systems. Journal of Geochemical Exploration, 88, 81–85.
Bernaus, A., Gaona, X., Ivask, A., Kahru, A., & Valiente, M. (2005). Analysis of sorption and bioavailability of different species of Mercury on model soil components using XAS techniques and sensor bacteria. Analytical and Bioanalytical Chemistry, 382, 1541–1548.
Boettinger, J. L., & Ming, D. W. (2002). Zeolites. In J. B. Dixon & D. G. Schulze (Eds.), Soil mineralogy with environmental applications (Soil science society of America book series, Vol. 7). Madison: SSSA Inc.
Bonten, L. T. C., Groenenberg, J. E., Weng, L., & van Riemsdijk, W. H. (2008). Use of speciation and complexation models to estimate heavy metal sorption in soils. Geoderma, 146, 303–310.
Buekers, J., Amery, F., Maes, A., & Smolders, E. (2008). Long term reactions of Ni, Zn and Cd with iron oxyhydroxides depend on crystallinity and structure and on metal concentrations. European Journal of Soil Science, 59, 706–715.
Buekers, J., Degryse, F., Maes, A., & Smolders, E. (2008). Modelling the effects of ageing on Cd, Zn, Ni and Cu solubility in soils using an assemblage model. European Journal of Soil Science, 59, 1160–1170.
Chen, Z., Cai, Y., Solo-Gabrielle, H., Snyder, G. H., & Cisar, J. L. (2006). Interactions of arsenic and the dissolved substances derived from turf soils. Environmental Science and Technology, 40, 4659–4665.
Christl, I., Metzger, A., Heidmann, I., & Kretzschmar, R. (2005). Effect of humic and fulvic acid concentrations and ionic strength on copper and lead binding. Environmental Science and Technology, 39, 5319–5326.
Colella, C. (1996). Ion exchange equilibrium in zeolite minerals. Mineralium Deposita, 31, 554–562.
Contin, M., Mondini, C., Leita, L., & De Nobili, M. (2007). Enhanced soil toxic metal fixation in iron (hydr)oxides by redox cycles. Geoderma, 140, 164–175.
Contin, M., Mondini, C., Leita, L., Zaccheo, P., Crippa, L., & De Nobili, M. (2008). Immobilisation of soil toxic metals by repeated additions of Fe(II) sulphate solution. Geoderma, 147, 133–140.
Coppola, E. I., Battaglia, G., Bucci, M., Ceglie, D., Colella, A., Langella, A., Buondonno, A., & Colella, C. (2003). Remediation of Cd- and Pb- polluted soil by treatment with organo-zeolite conditioner. Clays and Clay Minerals, 51, 609–615.
Cornu, J. Y., Denaix, L., Schneider, A., & Pellerin, S. (2007). Temporal evolution of redox processes and free Cd dynamics in a metal-contaminated soil after rewetting. Chemosphere, 70, 306–314.
Councell, T. B., Duckenfield, K. U., Landa, E. R., & Callender, E. (2004). Tire wear as a source of zinc to the environment. Environmental Science and Technology, 38, 4206–4214.
Covelo, E. F., Vega, F. A., & Andrade, M. L. (2007). Competitive sorption and desorption of heavy metals by individual soil components. Journal of Hazardous Materials, 140, 308–315.
Crout, N. M. J., Tye, A. M., Zhang, H., Mcgrath, S., & Young, S. D. (2006). Kinetics of metal fixation in soils: Measurement and modelling by isotopic dilution. Environmental Toxicology and Chemistry, 25, 659–663.
De Gennaro, B., Collela, A., Aprea, P., & Colella, C. (2003). Evaluation of an intermediate-silica sedimentary chabazite as exchanger for potentially radioactive cations. Microporous and Mesoporous Materials, 61, 159–165.
Degryse, F., Waegeneers, N., & Smolders, E. (2007). Labile lead in polluted soils measured by stable isotope dilution. European Journal of Soil Science, 58, 1–7.
Degryse, F., Smolders, E., & Parker, D. R. (2009). Partitioning of metals (Cd, Co, Cu, Ni, Pb, Zn) in soils: Concepts, methodologies, prediction and applications – a review. European Journal of Soil Science, 60, 590–612.
Dixon, J. B., & White, G. N. (2002). Manganese oxides. In J. B. Dixon & D. G. Schulze (Eds.), Soil mineralogy with environmental applications. Madison: Soil Science Society of America.
Du Laing, G., Rinklebe, J., Vandecasteele, B., Meers, E., & Tack, F. M. G. (2009). Trace metal behaviour in estuarine and riverine floodplain soils and sediments: A review. Science of the Total Environment, 407, 3972–3985.
Elzinga, E. L., van Grinsven, J. J. M., & Swartjes, F. A. (1999). General purpose Freundlich isotherms for cadmium, copper and zinc in soils. European Journal of Soil Science, 50, 139–149.
Fendorf, S., Eick, M. J., Grossl, P., & Sparks, D. L. (1997). Arsenate and chromate retention mechanisms on Goethite. 1. Surface structure. Environmental Science and Technology, 31, 315–320.
Fischer, L., Brummer, G. W., & Barrow, N. J. (2007). Observations and modelling of the reactions of 10 metals with goethite: Adsorption and diffusion processes. European Journal of Soil Science, 58, 1304–1315.
Fletcher, P., & Townsend, R. P. (1982). Transition metal ion exchange in mixed ammonium sodium X and Y zeolites. Journal of Chromatography. A, 238, 59–68.
Furnare, L. J., Vailionis, A., & Strawn, D. G. (2005). Polarized XANES and EXAFS spectroscopic investigation into copper(II) complexes on vermiculite. Geochimica et Cosmochimica Acta, 69, 5219–5231.
Gabler, H. E., Bahr, A., Heidkamp, A., & Utermann, J. (2007). Enriched stable isotopes for determining the isotopically exchangeable element content in soils. European Journal of Soil Science, 58, 746–757.
Gandois, L., Probst, A., & Dumat, C. (2010). Modelling trace metal extractability and solubility in French forest soils by using soil properties. European Journal of Soil Science, 61, 271–286.
Gerth, J. (2005). Effects of crystal modification on the binding of trace metals and arsenate by Goethite. Journal of Soils and Sediments, 5, 30–36.
Gerth, J., Brummer, G. W., & Tiller, K. G. (1993). Retention of Ni, Zn and Cd by Si-associated goethite. Zeitschrift für Pflanzenernährung und Bodenkunde, 156, 123–129.
Giles, C. H., Smith, D., & Huitson, A. (1974). A general treatment and classification of the solute adsorption isotherm I. Theoretical. Journal of Colloid and Interface Science, 47, 755–765.
Gray, C. W., & McLaren, R. G. (2006). Soil factors affecting heavy metal solubility in some New Zealand soils. Water, Air, and Soil Pollution, 175, 3–14.
Groenenberg, J. E., Römkens, P. F. A. M., Comans, R. N. J., Luster, J., Pampura, T., Shotbolt, L., Tipping, E., & De Vries, W. (2010). Transfer functions for solid-solution partitioning of cadmium, copper, nickel, lead and zinc in soils: Derivation of relationships for free metal ion activities and validation with independent data. European Journal of Soil Science, 61, 58–73.
Grybos, M., Davranche, M., Gruau, G., & Petitjean, P. (2007). Is trace metal release in wetland soils controlled by organic matter mobility or Fe-oxyhydroxides reduction? Journal of Colloid and Interface Science, 314, 490–501.
Gustafsson, J. P. (2010). http://www2.lwr.kth.se/English/OurSoftware/vminteq/
Hashimoto, Y., Matsufuru, H., & Sato, T. (2008). Attenuation of lead leachability in shooting range soils using poultry waste amendments in combination with indigenous plant species. Chemosphere, 73, 643–649.
Hinz, C. (2001). Description of sorption data with isotherm equations. Geoderma, 99, 225–243.
Hofstetter, K. J., & Hitz, C. G. (1983). The use of the submerged demineralizer system at three-mile-island. Separation Science and Technology, 18, 1747–1764.
Horn, A. L., Reiher, W., Düring, R.-A., & Gäth, S. (2005). Efficiency of pedotransfer functions describing cadmium sorption in soils. Water, Air, and Soil Pollution, 170, 229–247.
Hrsak, J., Fugass, M., & Vadjic, V. (2000). Soil contamination by Pb, Zn and Cd from a lead smelter. Environmental Monitoring and Assessment, 60, 359–366.
Huang, J.-H., & Matzner, E. (2006). Dynamics of organic and inorganic arsenic in the solution phase of an acidic fen in Germany. Geochimica et Cosmochimica Acta, 70, 2023–2033.
Jalali, M., & Moharrami, S. (2007). Competitive adsorption of trace elements in calcareous soils of western Iran. Geoderma, 140, 156–163.
Janssen, R. P. T., Bruggenwert, M. G. M., van Dijk, G., & van Riemsdijk, W. H. (2007). Lead ion adsorption on montmorillonite-Al hydroxide polymer systems. European Journal of Soil Science, 58, 1136–1144.
Jensen, P. E., Ottosen, L. M., & Pederson, A. J. (2006). Speciation of Pb in industrially polluted soils. Water, Air, and Soil Pollution, 170, 359–382.
Kesraoul-Ouki, S., Cheeseman, C., & Perry, R. (1994). Natural zeolite utilization in pollution control – a review of applications to metal effluents. Journal of Chemical Technology & Biotechnology, 59, 121–126.
Kinniburgh, D. G. (1986). General purpose adsorption isotherms. Environmental Science and Technology, 20, 895–904.
Kinniburgh, D. G., & Smedley, P. L. (2001). Arsenic contamination of groundwater in Bangladesh (Summary of BGS Technical Report WC/00/19, Volume 1). Keyworth: British Geological Survey.
Kirk, G. J. D. (2004). The biogeochemistry of submerged soils. Chichester: Wiley.
Koopal, L. K., Saito, T., Pinheiro, J. P., & van Riemsdijk, W. H. (2005). Ion binding to natural organic matter: General considerations and the NICA-Donnan model. Colloids and Surfaces A, 265, 40–54.
Kumpiene, J., Lagerkvist, A., & Maurice, C. (2008). Stabilization of As, Cr, Cu, Pb and Zn in soil using amendments – a review. Waste Management, 28, 215–225.
Lair, G. J., Gerzabek, M. H., & Haberhauer, G. (2007). Retention of copper, cadmium and zinc in soil and its textural fractions influenced by long term field management. European Journal of Soil Science, 58, 1145–1154.
Lang, F., & Kaupenjohann, M. (2003). Effect of dissolved organic matter on the precipitation and mobility of the lead compound chloropyromorphite in solution. European Journal of Soil Science, 54, 139–147.
Lee, S. (2006). Geochemistry and partitioning of trace metals in paddy soils affected by metal mine tailings in Korea. Geoderma, 135, 26–37.
Leggo, P. J., Ledésert, B., & Christie, G. (2006). The role of clinoptilolite in organo-zeolitic-soil systems used for phytoremediation. Science of the Total Environment, 363, 1–10.
Lofts, S., & Tipping, E. (1999). Modelling the solid-solution partitioning of metals in environmental systems. Environmental Geochemistry and Health, 21, 299–304.
Lombi, E., Hamon, R. E., McGrath, S. P., & McLaughlin, M. J. (2003). Lability of Cd, Cu, and Zn in polluted soils treated with lime, beringite, and red Mud and identification of a non-labile colloidal fraction of metals using isotopic techniques. Environmental Science and Technology, 37, 979–984.
Ma, Q. Y., & Lindsay, W. L. (1993). Measurement of free Zn2+ activity in uncontaminated and contaminated soils using chelation. Soil Science Society of America Journal, 57, 963–967.
Ma, Y. B., Lombi, E., Nolan, A. L., & McLaughlin, M. J. (2006). Determination of labile Cu in soils and isotopic exchangeability of colloidal Cu complexes. European Journal of Soil Science, 57, 147–153.
Ma, Y. B., Lombi, E., Nolan, A. L., & McLaughlin, M. J. (2006). Short-term natural attenuation of copper in soils: Effects of time, temperature and soil characteristics. Environmental Toxicology and Chemistry, 25, 652–658.
Ma, Y. B., Lombi, E., Oliver, I. W., Nolan, A. L., & McLaughlin, M. J. (2006). Long term aging of copper added to soils. Environmental Science and Technology, 40, 6310–6317.
MacDonald, J. D., & Hendershot, W. H. (2006). Modelling trace metal partitioning in forest floors of northern soils near metal smelters. Environmental Pollution, 143, 228–240.
Markiewicz-Patowska, J., Hursthouse, A., & Przybyla-Kij, H. (2005). The interaction of heavy metals with urban soils: Sorption behaviour of Cd, Cu, Cr, Pb and Zn with a typical mixed brownfield deposit. Environment International, 31, 513–521.
McCusker, L. B., Liebau, F., & Engelhardt, G. (2001). Nomenclature of structural and compositional characteristics of ordered microporous and mesoporous materials with inorganic hosts: IUPAC recommendations 2001. Pure and Applied Chemistry, 73, 381–394.
McKenzie, R. M. (1977). Manganese oxides and hydroxides. In J. B. Dixon & S. B. Weed (Eds.), Minerals in soil environments. Madison: Soil Science Society of America.
Menzies, N. (2007). Redox processes and attenuation of metal availability in soils. In R. Hamon, M. McLaughlin, & E. Lombi (Eds.), Natural attentuation of trace element availability in soils. Pensacola: Society of Environmental Toxicology and Chemistry (SETAC).
Millward, G. E., & Liu, Y.-P. (2003). Modelling metal desorption kinetics in estuaries. Science of the Total Environment, 314/316, 613–623.
Murakami, M., Nakajima, F., Furumai, H., Tomiyasu, B., & Owari, M. (2007). Identification of particles containing chromium and lead in road dust and soakaway sediment by electron probe microanalyser. Chemosphere, 67, 2000–2010.
Mustafa, G., Kookana, R. S., & Singh, B. (2006). Desorption of cadmium from goethite: Effects of pH, temperature and aging. Chemosphere, 64, 856–865.
Nakhone, L. N., & Young, S. D. (1993). The significance of (radio-) labile cadmium pools in soil. Environmental Pollution, 82, 73–77.
Negra, C., Ross, D. S., & Lanzirotti, A. (2005). Soil manganese oxides and trace metals: Competitive sorption and microfocused synchrotron X-ray fluorescence mapping. Soil Science Society of America Journal, 69, 353–361.
Nye, P. H., & Tinker, P. B. (1977). Solute movement in the soil-root system (Studies in ecology, Vol. 4). Oxford: Blackwell Publishing.
Őren, A. H., & Kaya, A. (2006). Factors affecting adsorption characteristics of Zn2+ on two natural zeolites. Journal of Hazardous Materials, 131, 59–65.
Oste, L. A., Temminghoff, E. J. M., & van Riemsdijk, W. H. (2002). Solid-solution partitioning of organic matter in soils as influenced by an increase in pH or Ca concentration. Environmental Science and Technology, 36, 208–214.
Palágyi, Š., Salzer, P., & Mitro, A. (2006). Sorption desorption and extraction of cadmium from some arable and forest soils. Journal of Radioanalytical and Nuclear Chemistry, 269, 103–113.
Papadopoulos, P., & Rowell, D. L. (1988). The reactions of cadmium with calcium carbonate surfaces. Journal of Soil Science, 39, 23–36.
Pareuil, P., Penilla, S., Ozkan, N., Bordas, F., & Bollinger, J.-C. (2008). Influence of reducing conditions on metallic elements released from various contaminated soil samples. Environmental Science and Technology, 42, 7615–7621.
Peric, J., Trgo, M., & Medvidovic, N. V. (2004). Removal of zinc, copper and lead by natural zeolite – a comparison of adsorption isotherms. Water Research, 38, 1893–1899.
Ponnamperuma, F. N. (1972). The chemistry of submerged soils. Advances in Agronomy, 24, 29–96.
Rasa, K., Peltovuori, T., & Hartikainen, H. (2006). Effects of de-icing chemicals sodium chloride and potassium formate on cadmium solubility in a coarse mineral soil. Science of the Total Environment, 366, 819–825.
Rieuwerts, J. S. (2007). The mobility and bioavailability of trace metals in tropical soils: A review. Chemical Speciation and Bioavailability, 19, 75–85.
Ritvo, G., Avnimelech, Y., & Kochba, M. (2003). Empirical relationship between conventionally determined pH and in situ values in waterlogged soils. Aquacultural Engineering, 27, 1–8.
Roberts, D. R., Scheinost, A. C., & Sparks, D. L. (2002). Zinc speciation in a smelter-contaminated soil profile using bulk and microspectroscopic techniques. Environmental Science and Technology, 36, 1742–1750.
Sako, A., Lopes, L., & Roychoudhury, N. (2009). Adsorption and surface complexation modelling of palladium, rhodium and platinum in surficial semi-arid soils and sediment. Applied Geochemistry, 24, 86–95.
Sarret, G., Balesdent, J., Bouziri, L., Garnier, J.-M., Marcus, M. A., Geoffroy, N., Panfili, F., & Manceau, A. (2004). Zn speciation in the organic horizon of a contaminated soil by micro-X-ray fluorescence, micro- and powder-EXAFS spectroscopy, and isotopic dilution. Environmental Science and Technology, 38, 2792–2801.
Sauvé, S., Hendershot, W., & Allen, H. E. (2000). Solid-solution partitioning of metals in contaminated soils: Dependence on pH, total metal burden and organic matter. Environmental Science and Technology, 34, 1125–1131.
Schröder, T. J., Hiemstra, T., Vink, J. P. M., & van der Zee, S. E. A. T. M. (2005). Modeling of the solid-solution partitioning of heavy metals and arsenic in embanked flood plain soils of the rivers Rhine and Meuse. Environmental Science and Technology, 39, 7176–7184.
Schröder, T. J., van Riemsdijk, W. H., van der Zee, S. E. A. T. M., & Vink, J. P. M. (2008). Monitoring and modelling of the solid-solution partitioning of metals and as in a river floodplain redox sequence. Applied Geochemistry, 23, 2350–2363.
Schwertmann, U., & Taylor, R. M. (1982). Iron oxides. In J. B. Dixon & S. B. Weed (Eds.), Minerals in the soil environment. Madison: Soil Science Society of America.
Sheppard, M. I., Sheppard, S. C., & Grant, C. A. (2007). Solid/liquid partition coefficients to model trace element critical loads for agricultural soils in Canada. Canadian Journal of Soil Science, 87, 189–201.
Shi, Z., Allen, H. E., Di Toro, D. M., Lee, S.-Z., Flores Meza, D. M., & Lofts, S. (2007). Predicting cadmium adsorption on soils using WHAM-VI. Chemosphere, 69, 605–612.
Shirvani, M., Kalbasi, M., Shariatmadari, H., Nourbakhsh, F., & Najafi, B. (2006). Sorption–desorption of cadmium in aqueous palygorskite, sepiolite and calcite suspensions: Isotherm hysteresis. Chemosphere, 65, 2178–2184.
Skyllberg, U., Bloom, P. R., Qian, J., Lin, C.-M., & Bleam, W. F. (2006). Complexation of mercury(II) in soil organic matter: EXAFS evidence for linear two-coordination with reduced sulfur groups. Environmental Science and Technology, 40, 4174–4180.
Smolders, E., & Degryse, F. (2002). Fate and effect of zinc from tire debris in soil. Environmental Science and Technology, 36, 3706–3710.
Smolders, E., Lambregts, R. M., McLaughlin, M. J., & Tiller, K. G. (1998). Effect of soil solution chloride on cadmium availability to Swiss chard. Journal of Environmental Quality, 27, 426–431.
Sparks, D. L. (2003). Kinetics of soil chemical processes. In D. L. Sparks (Ed.), Environmental soil chemistry (2nd ed.). London: Academic.
Stevenson, F. J. (1994). Humus chemistry: Genesis, composition, reactions (2nd ed.). New York: Wiley.
Strobel, B. W., Borggaard, O. K., Hansen, H. C. B., Andersen, M. K., & Raulund-Rasmussen, K. (2005). Dissolved organic carbon and decreasing pH mobilize cadmium and copper in soil. European Journal of Soil Science, 56, 189–196.
Temminghoff, E. J. M., van der Zee, S. E. A. T. M., & de Haan, F. A. M. (1997). Copper mobility in a copper-contaminated sandy soil as affected by pH and solid and dissolved organic matter. Environmental Science and Technology, 31, 1109–1115.
Tipping, E. (1998). Humic ion binding model VI: An improved description of the interactions of protons and metal ions with humic substances. Aquatic Geochemistry, 4, 3–48.
Tipping, E. (2002). Cation binding by humic substances (Cambridge environmental chemistry series, Vol. 12). New York: Cambridge University Press.
Tipping, E., Rieuwerts, J., Pan, G., Ashmore, M. R., Lofts, S., Hill, M. T. R., Farago, M. E., & Thornton, I. (2003). The solid–solution partitioning of heavy metals (Cu, Zn, Cd, Pb) in upland soils of England and Wales. Environmental Pollution, 125, 213–225.
Trancoso, M. A., Correia dos Santos, M. M., & Gonçalves, M. L. S. (2007). Lead sorption to selected Portuguese soils. European Journal of Soil Science, 58, 854–863.
Turkman, A. E., Aslan, S., & Ege, I. (2004). Treatment of metal-containing wastewaters by natural zeolites. Fresenius Environmental Bulletin, 13, 574–580.
Tye, A. M., Young, S. D., Crout, N. M. J., Zhang, H., Preston, S., Barbosa-Jefferson, V. L., Davison, W., McGrath, S. P., Paton, G. I., Kilham, K., & Resende, L. (2003). Predicting the activity of Cd2+ and Zn2+ in soil pore water from the radio-labile metal fraction. Geochimica et Cosmochimica Acta, 67, 375–385.
Uzu, G., Sobanska, S., Aliouane, Y., Pradere, P., & Dumat, C. (2009). Study of lead phytoavailability for atmospheric industrial micronic and sub-micronic particles in relation with lead speciation. Environmental Pollution, 157, 1178–1185.
Van Riemsdijk, W. H., Koopal, L. K., Kinniburgh, D. G., Benedetti, M. F., & Weng, L. (2006). Modeling the interactions between humics, ions, and mineral surfaces. Environmental Science and Technology, 40, 7473–7480.
Vantelon, D., Lanzirotti, A., Scheinost, A. C., & Kretzschmar, R. (2005). Spatial distribution and speciation of lead around corroding bullets in a shooting range soil studied by micro-X-ray fluorescence and absorption spectroscopy. Environmental Science and Technology, 39, 4808–4815.
Vega, F. A., Covelo, E. F., & Andrade, M. L. (2009). Hysteresis in the individual and competitive sorption of cadmium, copper and lead by various soil horizons. Journal of Colloid and Interface Science, 331, 312–317.
Vermeer, A. W. P., McCulloch, J. K., van Riemsdijk, W. H., & Koopal, L. K. (1999). Metal ion adsorption to complexes of humic acid and metal oxides: Deviations from the additivity rule. Environmental Science and Technology, 33, 3892–3897.
Wendling, L. A., Ma, Y., Kirby, J. K., & McLaughlin, M. J. (2009). A predictive model of the effects of aging on cobalt fate and behaviour in soil. Environmental Science and Technology, 43, 135–141.
Weng, L., Temminghoff, E. J. M., Lofts, S., Tipping, E., & van Riemsdijk, W. H. (2002). Complexation with dissolved organic matter and solubility control of heavy metals in a sandy soil. Environmental Science and Technology, 36, 4804–4810.
Weng, L., van Riemsdijk, W. H., Koopal, L. K., & Hiemstra, T. (2006). Ligand and charge distribution (LCD) model for the description of fulvic acid adsorption to goethite. Journal of Colloid and Interface Science, 302, 442–457.
Weng, L., van Riemsdijk, W. H., & Hiemstra, T. (2008). Cu2+ and Ca2+ adsorption to goethite in the presence of fulvic acids. Geochimica et Cosmochimica Acta, 72, 5857–5870.
White, G. N., & Dixon, J. B. (2002). Kaolin-serpentine minerals. In J. B. Dixon & D. G. Schulze (Eds.), Soil mineralogy with environmental applications. Madison: Soil Science Society of America.
Xu, H.-Y., Yang, L., Wang, P., Liu, Y., & Peng, M.-S. (2008). Kinetic research on the sorption of aqueous lead by synthetic carbonate hydroxyapatite. Journal of Environmental Management, 86, 319–328.
Xue, W.-B., Yi, A.-H., Zhang, Z.-Q., Tang, C.-L., Zhang, X.-C., & Gao, J.-M. (2009). A New competitive adsorption isothermal model of heavy metals in soils. Pedosphere, 19, 251–257.
Young, S. D., Crout, N. M. J., Hutchinson, J., Tye, A., Tandy, S., & Nakhone, L. (2007). Techniques for measuring attenuation: Isotopic dilution methods. In R. Hamon, M. McLaughlin, & E. Lombi (Eds.), Natural attenuation of trace element availability in soils. Pensacola: Society of Environmental Toxicology and Chemistry (SETAC).
Zhang, S., Li, W., Shan, X.-Q., Lu, A., & Zhou, P. (2005). Effects of low molecular weight organic anions on the release of arsenite and arsenate from a contaminated soil. Water, Air, and Soil Pollution, 167, 111–122.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Young, S.D. (2013). Chemistry of Heavy Metals and Metalloids in Soils. In: Alloway, B. (eds) Heavy Metals in Soils. Environmental Pollution, vol 22. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-4470-7_3
Download citation
DOI: https://doi.org/10.1007/978-94-007-4470-7_3
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-4469-1
Online ISBN: 978-94-007-4470-7
eBook Packages: Earth and Environmental ScienceEarth and Environmental Science (R0)