
Abstract
Acute Q fever is commonly resolved without an antibiotic regimen, but a primary infection may develop into a chronic infection in a minority of cases. Coxiella burnetii, the causative agent of Q fever, is known to infect macrophages both in vitro and in vivo. It has been observed that the intracellular survival of C. burnetii requires the subversion of the microbicidal properties of macrophages. Adaptive immunity is also essential to cure C. burnetii infection, as demonstrated by clinical studies and animal models. Indeed, the control of infection in patients with primary Q fever involves a systemic cell-mediated immune response and granuloma formation with an essential role for interferon-γ in the protection against C. burnetii. In contrast, chronic Q fever is characterized by defective cell-mediated immunity with the defective formation of granulomas and over-production of interleukin-10, an immunoregulatory cytokine. Finally, epidemiological data demonstrate that age and gender are risk factors for Q fever. The analysis of gene expression programs in mice reveals the importance of sex-related genes in C. burnetii infection because only 14% of the modulated genes are sex-independent, while the remaining 86% are differentially expressed in males and females. These results open a new field to understand how host metabolism controls C. burnetii infection in humans.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
14.1 The Natural History of Q Fever
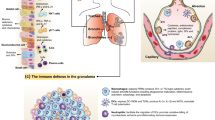
Q fever is caused by Coxiella burnetii, an obligate intracellular bacterium phylogenetically related to Legionellae species and Francisella tularensis (Weisburg et al. 1989). Q fever has a wide spectrum of clinical manifestations, including fever, pneumonia, hepatitis and, less frequently, neurological manifestations (Raoult et al. 2005). Naïve patients usually contract Q fever via aerosols and develop a primary infection (Fig. 14.1). More than half of patients will not exhibit any symptoms beyond a seroconversion, and only 2% will develop a severe disease that leads to their hospitalization. Acute Q fever almost always resolves without a specific antibiotic treatment. However, C. burnetii persists in patients who are apparently cured (Harris et al. 2000). In certain hosts, the primary infection may develop into a chronic infection (Raoult 1990). The latency between acute and chronic infection may last from months to years. Patients with valvular damage, patients with cancer (such as lymphomas) and pregnant women are at a high risk of a developing chronic infection (Fenollar et al. 2001). The main clinical manifestation of chronic Q fever is endocarditis (Brouqui et al. 1993), which represents 48% of the blood culture-negative cases of infective endocarditis (Houpikian and Raoult 2005). Q fever endocarditis, that may lead to death (Maurin and Raoult 1999), is characterized by fibrosis, calcification, slight inflammation and vascularization, small or absent vegetations, and a high level of antibodies specific for C. burnetii (Lepidi et al. 2003). Combination of doxycycline and chloroquine has changed the prognosis of the disease; now, less than 5% of patients experience a relapse after 18 months of treatment (Houpikian et al. 2002). There is a correlation between circulating concentrations of doxycycline and decreased titers of anti-C. burnetii antibodies (Rolain et al. 2003). Besides the two major clinical presentations of Q fever, two additional forms have recently been reported. The hyperinflammatory syndrome is associated with hepatitis and autoantibodies in middle-aged male patients; corticosteroids improve the patient outcome (Maurin and Raoult 1999). Persistent asthenia lasting for months or years has been reported in some patients with acute Q fever (Ayres et al. 1998). Finally, Q fever is deemed a category B biological terrorist agent (Madariaga et al. 2003). In this review, we will focus on the recent knowledge of innate and adaptive immune responses to C. burnetii infection.

The natural history of Q fever. The primary infection by C. burnetii is unapparent or symptomatic (acute Q fever). Months or years later, some individuals may develop a chronic infection that principally manifests as endocarditis
14.2 Innate Immunity
14.2.1 Monocytes and Macrophages
Monocytes and macrophages are the major targets of C. burnetii. The intracellular survival of C. burnetii organisms requires the subversion of the microbicidal properties of these cells through mechanisms that are described in the chapters on the bacterial persistence and intracellular trafficking of C. burnetii. Briefly, C. burnetii interacts with monocytes and macrophages via αvβ3 integrin, induces strong remodeling of the actin cytoskeleton, and activates protein tyrosine kinase pathways and cytokine production (Ghigo et al. 2009; Raoult et al. 2005). In addition to integrins, the Toll-like receptors (TLRs) play a role in macrophage infection because TLR4 is involved in the cytoskeleton remodeling induced by C. burnetii. Although TLR4 (which is known to recognize lipopolysaccharide (LPS)) controls the immune response against C. burnetii through granuloma formation and cytokine production, it is dispensable for bacterial clearancze in vivo (Honstettre et al. 2004). TLR2 (which is known to recognize peptidoglycan and lipopeptides) is also involved in C. burnetii infection. Indeed, Zamboni et al. showed that TLR2 is involved in TNF and IFN-γ production induced by avirulent variants of C. burnetii (phase II organisms) in macrophages from TLR2-deficient mice and that these macrophages are highly permissive for the intracellular growth of bacteria (Zamboni et al. 2004). In vivo experiments also indicated that TLR2 is involved in granuloma formation, as is TLR4 (Meghari et al. 2005).
The interaction of monocytes/macrophages with C. burnetii leads to their functional polarization (Benoit et al. 2008). In resting monocytes in which C. burnetii survives but does not replicate, bacteria induce an M1-type program. This program, usually induced by interferon (IFN)-γ or bacterial products, contributes to the microbicidal activity of M1-polarized cells (Ghigo et al. 2004). In the macrophages in which C. burnetii replicates, bacteria induce the expression of M2 polarization-related genes, such as those encoding transforming growth factor β1, interleukin (IL)-1 receptor antagonist (IL-1ra), CCL18, mannose receptor and arginase 1, although C. burnetii organisms also induce the expression of the genes encoding IL-6 and CXCL8, which are two cytokines associated with an M1 program (Benoit et al. 2008). These findings indicate that the polarization of macrophages into M1- or M2-like phenotypes controls the intracellular life of C. burnetii.
14.2.2 Dendritic Cells
Dendritic cells (DCs) are immune sentinels that are necessary to orchestrate the immune response to infection. It has been reported that phase I C. burnetii (virulent bacteria) infects and replicates within human DCs without inducing maturation or inflammatory cytokine production. In contrast, phase II bacteria induce a dramatic maturation of DCs (Shannon et al. 2005). However, these findings raise several points. First, it is difficult to understand how Th1 polarization of T cells in acute Q fever is associated with a major defect of DC maturation. Second, we have found that DCs stimulated with phase I C. burnetii were able to induce T cell proliferation in an allogenic reaction. Third, the transcriptomic program of C. burnetii-stimulated DC clusters (as well as that of DCs stimulated with LPS from enterobacteria) is known to induce DC maturation (unpublished data). We believe that, compared to canonical activators, the maturation of DCs is only partial in response to phase I C. burnetii, and DCs retain the potential to activate T cells.
14.2.3 Others
The role of natural killer (NK) cells in C. burnetii infection remains poorly understood. Although histopathological lesions are more severe in SCIDβγ mice (which are devoid of T, B and NK cells) compared to SCID mice, the bacterial burden is similar in both types of mice (Andoh et al. 2007). It is likely that γδ T cells are not required for controlling primary pulmonary C. burnetii infection because mice depleted of both αβ CD4+ and CD8+ T cells are similarly susceptible to infection as SCID mice (Read et al. 2010).
14.3 Adaptive Immunity
It is well established that adaptive immunity is required to cure the infectious diseases that are caused by intracellular bacteria (Collins and Kaufmann 2002). Several lines of evidence (arising from both animal models and clinical studies) support the idea that adaptive immunity is essential to cure C. burnetii infection (Fig. 14.2). Hence, SCID mice that are devoid of T and B cells succumb to infection, demonstrating the major role of the adaptive immune response for the control of C. burnetii infection (Andoh et al. 2003). Patients that are experiencing immunosuppression (due to immunosuppressive treatment, pregnancy, and lymphomas) are at risk to develop chronic Q fever and to mount inefficient anti-C. burnetii immunity (Maurin and Raoult 1999).

The immune response to C. burnetii infection. In primary C. burnetii infection, the uptake of organisms by macrophages and DCs leads to the presentation of bacterial antigens to T lymphocytes. The production of IFN-γ by T lymphocytes (and NK cells) and that of TNF by immune cells including macrophages and T lymphocytes induce the apoptosis of infected macrophages and C. burnetii killing. In patients who are unable to mount an IFN-γ response (after defective presentation of antigen or other mechanisms), T lymphocytes are unable to produce IFN-γ. As a consequence, macrophages survive and their microbicidal activity is impaired
14.3.1 T Lymphocytes and Cytokines
The control of infection in patients with primary Q fever involves a systemic cell-mediated immune response and granuloma formation. A cell-mediated immune response, which manifests as a marked proliferative response to C. burnetii antigen, is observed in patients who have convalesced from acute Q fever and in patients with acute Q fever hepatitis (Koster et al. 1985a). Individuals vaccinated with formalin-inactivated C. burnetii exhibit specific lymphoproliferation and IFN-γ production in response to C. burnetii challenge (Izzo and Marmion 1993; Izzo et al. 1988). The combination of IFN-γ production and granuloma formation suggests a Th1-type polarization of the immune response. Nevertheless, immune control of Q fever does not lead to C. burnetii eradication because animals exhibit persistent shedding of C. burnetii (Maurin and Raoult 1999) and because C. burnetii DNA is found in circulating monocytes and bone marrow several months to years after recovery from acute Q fever (Harris et al. 2000). The role of T cells in the control of C. burnetii infection has been well documented in murine models. Nude and SCID mice are highly sensitive to C. burnetii infection, whereas wild type mice are resistant (Andoh et al. 2003; Kishimoto et al. 1978). The reconstitution of SCID mice with CD4+ T cells or CD8+ T cells was sufficient to control lung infection in a model of pneumonitis. Surprisingly, CD8+ T cells may play a more significant role in controlling splenomegaly, a marker of host response to the infection (Read et al. 2010). This finding may be related to the ability of CD8+ T cells to produce IFN-γ, which may also be produced by Th1 CD4+ T cells. It has been shown that inactivated phase I bacteria induce a strong Th1 response and protection, whereas inactivated phase II bacteria induce a weak Th1 response (Zhang et al. 2007). The essential role of IFN-γ in protection against C. burnetii is supported by the high mortality rate observed in IFN-γ−/− mice infected with C. burnetii (Andoh et al. 2007).
The microbicidal mechanism of IFN-γ directed against C. burnetii does not depend on the production of reactive oxygen intermediates (ROI). First, C. burnetii does not stimulate the release of superoxide anion and hydrogen peroxide by monocytes (Dellacasagrande et al. 1999), emphasizing previous results observed in neutrophils (Akporiaye et al. 1990). Second, the survival of C. burnetii is similar between control monocytes and monocytes from patients with chronic granulomatous disease (Akporiaye et al. 1990), in which the NADPH oxidase complex is not functional. The data on the role of reactive nitrogen intermediates are less clear. C. burnetii induces the production of nitric oxide (NO) by murine alveolar macrophages, but inhibitors of NO production do not modify the infection rate of macrophages (Yoshiie et al. 1999). IFN-γ controlled C. burnetii infection despite the absence of NO synthase in one report (Zamboni and Rabinovitch 2003) and less efficiently in another report (Brennan et al. 2004). In THP-1 macrophages, C. burnetii does not induce NO production, even in the presence of IFN-γ, and L-arginine inhibitors have no effect on the survival of C. burnetii (Dellacasagrande et al. 1999).
Experiments have indicated that IFN-γ stimulates the microbicidal program directed against C. burnetii (Fig. 14.2) through an oxygen-independent mechanism. Indeed, C. burnetii can survive in a phagosome that shares the properties of late endosomes but not those of lysosomes. IFN-γ restores phagosome-lysosome fusion and affects phagosomal pH (Ghigo et al. 2002). It is likely that this mechanism accounts for the restoration of phagosome-lysosome fusion in monocytes in patients with acute Q fever (Ghigo et al. 2004). In addition, changing phagosomal pH may have some therapeutic consequences, as demonstrated by the efficiency of the combination of doxycycline and chloroquine in the treatment of patients with chronic Q fever. This therapeutic regimen promotes vacuolar alkalization and restores the in vitro bactericidal activity of antibiotics against C. burnetii (Maurin and Raoult 1999).
While the survival of obligate intracellular organisms usually requires the prevention of host cell death (Clifton et al. 1998), IFN-γ is able to promote the apoptosis of C. burnetii-infected macrophages (Fig. 14.2). The apoptotic effect of IFN-γ on C. burnetii-infected cells is dependent on tumor necrosis factor (TNF). Indeed, IFN-γ up-regulates TNF production and induces the expression of membrane TNF. Neutralizing TNF with specific antibodies prevents macrophage apoptosis and the eradication of C. burnetii (Dellacasagrande et al. 1999). Concomitantly, IFN-γ induces homotypic adherence of C. burnetii-infected macrophages, which depends on β2 integrins and CD54. When adherence is disrupted by mechanical dissociation or by blocking the integrin receptors, cell apoptosis and the bacterial killing induced by IFN-γ are inhibited (Dellacasagrande et al. 2002). These findings may help to explain the mechanisms of granuloma formation in acute Q fever. In contrast, decreased IFN-γ production may impair the aggregation and the microbicidal activity of monocytes, as observed during chronic Q fever.
Finally, IFN-γ may control C. burnetii infection by different mechanisms such as cytokine production and/or regulation of the nutrient supply. C. burnetii-infected monocytes stimulated with IFN-γ release large amounts of TNF (Dellacasagrande et al. 1999). Besides its role in apoptosis, TNF may contribute to the microbicidal activity of macrophages. TNF affects phagocytosis but not the later steps of the microbicidal process. Indeed, neutralizing anti-TNF antibodies decreased C. burnetii internalization by monocytes from patients with Q fever endocarditis, but they have no effect on the long term survival of bacteria (Dellacasagrande et al. 2000a). In addition, IFN-γ controls iron metabolism in macrophages through the down-modulation of transferrin receptors, resulting in a decreased assimilation of iron. C. burnetii up-regulates the expression of transferrin receptors in murine macrophage cell lines, which results in an increased cell iron content and bacterial burden. An intracellular iron chelator, desferoxamine, suppresses the replication of C. burnetii (Howe and Mallavia 1999). It remains to be determined whether IFN-γ-mediated killing of C. burnetii involves a decrease in cellular iron content.
The major role of cell-mediated immunity in the protection against C. burnetii is emphasized by defective cell-mediated immunity during chronic Q fever (Fig. 14.2). Lymphocytes from patients with Q fever endocarditis do not proliferate in response to the C. burnetii antigen, in contrast to lymphocytes from patients with acute Q fever (Koster et al. 1985a). The mechanisms of this specific unresponsiveness may include alterations in T cell subsets, but CD4+ T cell lymphopenia was observed in patients with Q fever endocarditis and in cured patients who exhibited a normal immune response (Sabatier et al. 1997). This suppression is most likely mediated by immunoregulatory mediators such as the prostaglandin E2 (Koster et al. 1985b) or cytokines. IL-10, an immunoregulatory cytokine that is overproduced in chronic Q fever (Capo et al. 1996a; Honstettre et al. 2003), may be involved in Q fever-associated immunosuppression, perhaps via the induction of regulatory T cells. Finally, a defective imbalance of cytokines and chemokines may result in an impaired migration of immunocompetent cells to target organs (Meghari et al. 2006). Indeed, C. burnetii-infected monocytes exhibited defective transmigration through endothelium activated by TNF (Dellacasagrande et al. 2000b).
Immunosuppression in chronic Q fever is associated with an exacerbated inflammatory response. Severe inflammation is found in almost every patient with Q fever endocarditis and consists of up-regulation of the circulating levels of TNF, IL-6, two inflammatory cytokines, type II TNF receptors and IL-1ra (Capo et al. 1999). While IL-1ra levels are significantly higher in acute Q fever than in chronic Q fever, the levels of soluble CD23, a leukocyte activation marker also known as the low affinity receptor for immunoglobulin E, are specifically increased during chronic Q fever (Capo et al. 1999). In addition, TNF and IL-1β production is increased in monocytes from patients with Q fever endocarditis, whereas it remains low in patients with uncomplicated acute Q fever (Capo et al. 1996b; Dellacasagrande et al. 2000a). TNF production is related to the disease activity. First, it is higher in patients with Q fever recently diagnosed than both in those who have been monitored for more than 12 months and in cured patients. Second, there is a correlation between high levels of TNF production and titers of immunoglobulin G directed against C. burnetii (Capo et al. 1996b). Production of the chemokines CCL2 and CL5 is increased in monocytes from patients with Q fever endocarditis (Meghari et al. 2006). IL-6 production is high in both acute and chronic Q fever (Capo et al. 1999), which supports the overproduction of IL-6 that was reported in patients with post-Q fever fatigue syndrome (Penttila et al. 1998). Clearly, chronic Q fever is associated with immunosuppression and exacerbated inflammation.
14.3.2 Granulomas
The control of infection in patients with primary Q fever involves systemic cell-mediated immune responses and granuloma formation. The granulomatous lesions have a central open space and a fibrin ring, and they are referred to as doughnut granulomas. They consist of macrophages with epithelioid morphology and multinucleated giant cells, and they are paucibacillary in Q fever (Pellegrin et al. 1980; Srigley et al. 1985; Voigt et al. 1983). During chronic Q fever, granulomas are scarce and are replaced by lymphocyte infiltration and necrosis foci in the liver (Raoult et al. 1990). We recently adapted the in vitro method described by the Altare group to analyze mycobacterial granulomas (Puissegur et al. 2004) to that of C. burnetii granulomas (Delaby et al. 2010). The formation of C. burnetii granulomas is studied by incubating peripheral blood mononuclear cells with C. burnetii-coated sepharose beads. In the first step, monocytes induce granuloma formation, and in the second step, T lymphocytes are recruited by the nascent granuloma. The application of this method to mononuclear cells from patients with Q fever reveals that a large number of patients are unable to form granulomas due to a defective initial interaction of monocytes with bacterial antigens (Delaby et al. 2012).
14.3.3 B Lymphocytes and Antibodies
Antibodies are considered dispensable in Q fever, according to the paradigm of protection that is mediated by T cells in chronic infection. It is likely that the reality is less simple. First, large amounts of antibodies are produced in humans and animals infected with C. burnetii. Antibodies develop within 3–4 weeks of the onset of symptoms of primary infection: the majority of the antibodies are directed against phase II antigens and a minority are directed against phase I antigens. A similar model accounts for the response to the Q fever vaccine. Nevertheless, past studies have reported that not all vaccinated or convalescent patients have detectable levels of antibodies (Marmion et al. 1990). Increased levels of antibodies directed against phase I antigens are related to the chronic development of Q fever, with a diagnostic value for titers higher than 800 (Fournier et al. 1998). We have shown that mice that overexpress the human IL-10 transgene in macrophages produce high amounts of antibodies directed against phase I and II bacteria, similar to chronic human infection (Meghari et al. 2008). Second, initial descriptions of C. burnetii infection have indicated that anti-C. burnetii serum confers passive protection in mice and guinea pigs (Shannon and Heinzen 2009). Vaccination with phase I cellular vaccine induces both an antibody response and protection (Waag et al. 2008). Third, wild type mice and mice deficient in the receptors for the Fc fragment of IgG (FcγR) are equally protected by passive immunization, suggesting that antibodies and their receptors are not essential in C. burnetii clearance (Shannon and Heinzen 2009). This conclusion is strengthened by the finding that athymic mice and SCID mice are not protected by passive immunization (Humphres and Hinrichs 1981; Zhang et al. 2007). However, B cell-deficient mice are more severely affected than are wild type mice after C. burnetii infection, indicating that antibodies play a regulatory role in the infection even if bacterial clearance is not affected (Andoh et al. 2007). One can hypothesize that immunoregulatory cytokines are produced by macrophages only when FcγRs are engaged in the presence of an infectious trigger, as documented in leishmaniosis (Humphres and Hinrichs 1981; Zhang et al. 2007). We showed that macrophages incubated with C. burnetii that has been opsonized with specific IgG release higher amounts of IL-10 than those incubated with unopsonized bacteria (Desnues et al. 2009). The fate of microorganisms in this environment is a source of debate. In our experimental conditions, C. burnetii opsonization favors the formation of multibacillary vacuoles and increased bacterial replication (Desnues et al. 2009). The Heinzen group confirmed the opsonizing activity of anti-C. burnetii antibodies but did not find any effect of these antibodies on the growth rate of bacteria (Shannon and Heinzen 2009). Taken together, these results indicate that anti-C. burnetii antibodies may have a dual role in Q fever: they confer protection in synergy with T cells in acute Q fever and exacerbate immune depression in chronic Q fever through the production of IL-10.
14.4 Host Metabolism and Control of C. burnetii Infection
It is well known that age is a risk factor for Q fever. Indeed, symptomatic Q fever occurs more frequently in people who are over 15 years of age than in people who are under 15 years. The prevalence of clinical cases in children significantly increases with age (Maltezou and Raoult 2002). We have recently found that the bacterial burden and the number of granuloma are increased in the tissues of 14-month-old mice compared to 1-month-old mice (Leone et al. 2007).
Gender also affects C. burnetii infection. Men are more frequently symptomatic than are women (with a man:woman ratio of 2.5), despite comparable exposure and seroprevalence (Maltezou and Raoult 2002; Tissot-Dupont et al. 1992). The predisposition for infection in men may be explained by differences in sex hormones such as 17β-oestradiol. Indeed, female C56BL/6 mice exhibit fewer granulomas and a lower bacterial burden than do males, and ovariectomized mice exhibit disease rates that are comparable to those of males. The administration of 17β-oestradiol prevents the effect of ovariectomy on host response and tissue burden (Leone et al. 2004). The study of gene expression programs in mice reveals the importance of sex-related genes during C. burnetii infection. Multiclass analysis has identified 2,777 probes for which expression is specifically modulated by C. burnetii infection. Only 14% of the modulated genes are sex-independent, and the remaining 86% are differentially expressed in males and females. Castration of males and females has indicated that sex hormones are responsible for more than 60% of the observed gene modulation, and this reduction is most pronounced in males. Using functional annotation of modulated genes, we have identified four clusters that are enriched in males and are related to cell-cell adhesion, signal transduction, defensins and cytokine/Jak-Stat pathways. Up-regulation of the IL-10 and Stat-3 genes may account for the high susceptibility of men with Q fever to C. burnetii infection and to autoantibody production. Two clusters have been identified in females, including the circadian rhythm pathway, which consists of a feedback loop with positive (Clock, Arntl) and negative (Per) limbs. We have found that Clock and Arntl are down-regulated, whereas Per is up-regulated. These changes may be associated with efficient bacterial elimination in females but not in males, in which an exacerbated host response would be prominent (Textoris et al. 2010). Preliminary results in humans suffering from Q fever indicate that these gene pathways are differentially modulated in men and women.
14.5 Conclusions: New Basis for Vaccine Development
The immune response to C. burnetii is organized within granulomas. A better understanding of this response is becoming possible through the development of new approaches, including live imaging. However, this response does not protect the patients from the risk of relapse. The innate immune response is sufficient to control the infection, and the adaptive immune response allows for bacterial clearance. Both responses are also involved in features of the disease such as the uncontrolled inflammatory response in some patients with acute Q fever and in patients with chronic Q fever. The fact that a minority of exposed patients develop chronic Q fever suggests that certain genetic factors related to host immune response are critical. The relationship between the clinical expression of Q fever and gender is an excellent illustration of this interplay. The next step in understanding the immune response to C. burnetii will consist of the evaluation of individual host responses by systemic approaches to relate each clinical expression of Q fever with a specific genetic signature. These relations will be essential to analyze treatment failures.
References
Akporiaye ET, Stefanovich D, Tsosie V et al (1990) Coxiella burnetii fails to stimulate human neutrophil superoxide anion production. Acta Virol 34:64–70
Andoh M, Naganawa T, Hotta A et al (2003) SCID mouse model for lethal Q fever. Infect Immun 71:4717–4723
Andoh M, Zhang G, Russell-Lodrigue KE et al (2007) T cells are essential for bacterial clearance, and γ interferon, tumor necrosis factor α, and B cells are crucial for disease development in Coxiella burnetii infection in mice. Infect Immun 75:3245–3255
Ayres JG, Flint N, Smith EG et al (1998) Post-infection fatigue syndrome following Q fever. QJM 91:105–123
Benoit M, Barbarat B, Bernard A et al (2008) Coxiella burnetii, the agent of Q fever, stimulates an atypical M2 activation program in human macrophages. Eur J Immunol 38:1065–1070
Brennan RE, Russell K, Zhang G et al (2004) Both inducible nitric oxide synthase and NADPH oxidase contribute to the control of virulent phase I Coxiella burnetii infections. Infect Immun 72:6666–6675
Brouqui P, Tissot-Dupont H, Drancourt M et al (1993) Chronic Q fever. Ninety-two cases from France, including 27 cases without endocarditis. Arch Intern Med 153:642–648
Capo C, Zaffran Y, Zugun F et al (1996a) Production of interleukin-10 and transforming growth factor beta by peripheral blood mononuclear cells in Q fever endocarditis. Infect Immun 64:4143–4147
Capo C, Zugun F, Stein A et al (1996b) Upregulation of tumor necrosis factor-α and interleukin-1β in Q fever endocarditis. Infect Immun 64:1638–1642
Capo C, Amirayan N, Ghigo E et al (1999) Circulating cytokine balance and activation markers of leucocytes in Q fever. Clin Exp Immunol 115:120–123
Clifton DR, Goss RA, Sahni SK et al (1998) NF-κB-dependent inhibition of apoptosis is essential for host cell survival during Rickettsia rickettsii infection. Proc Natl Acad Sci U S A 95:4646–4651
Collins H, Kaufmann SHE (2002) Acquired immunity against bacteria. In: Kaufmann SHE, Sher A, Ahmed R (eds) Immunology of infectious diseases. ASM Press, Washington, pp 207–221
Delaby A, Espinosa L, Lepolard C et al (2010) 3D reconstruction of granulomas from transmitted light images implemented for long-time microscope applications. J Immunol Methods 360:10–19
Delaby A, Gorvel L, Espinosa L et al (2012) Defective monocyte dynamics in Q fever granuloma deficiency. J Infect Dis 205:1086–1094
Dellacasagrande J, Capo C, Raoult D et al (1999) IFN-γ-mediated control of Coxiella burnetii survival in monocytes: the role of cell apoptosis and TNF. J Immunol 162:2259–2265
Dellacasagrande J, Ghigo E, Capo C et al (2000a) Coxiella burnetii survives in monocytes from patients with Q fever endocarditis: involvement of tumor necrosis factor. Infect Immun 68:160–164
Dellacasagrande J, Moulin PA, Guilianelli C et al (2000b) Reduced transendothelial migration of monocytes infected by Coxiella burnetii. Infect Immun 68:3784–3786
Dellacasagrande J, Ghigo E, Raoult D et al (2002) IFN-γ-induced apoptosis and microbicidal activity in monocytes harboring the intracellular bacterium Coxiella burnetii require membrane TNF and homotypic cell adherence. J Immunol 169:6309–6315
Desnues B, Imbert G, Raoult D et al (2009) Role of specific antibodies in Coxiella burnetii infection of macrophages. Clin Microbiol Infect 15(Suppl 2):161–162
Fenollar F, Fournier PE, Carrieri P et al (2001) Risks factors and prevention of Q fever endocarditis. Clin Infect Dis 33:312–316
Fournier PE, Marrie TJ, Raoult D (1998) Diagnosis of Q fever. J Clin Microbiol 36:1823–1834
Ghigo E, Capo C, Tung CH et al (2002) Coxiella burnetii survival in THP-1 monocytes involves the impairment of phagosome maturation: IFN-γ mediates its restoration and bacterial killing. J Immunol 169:4488–4495
Ghigo E, Honstettre A, Capo C et al (2004) Link between impaired maturation of phagosomes and defective Coxiella burnetii killing in patients with chronic Q fever. J Infect Dis 190:1767–1772
Ghigo E, Pretat L, Desnues B et al (2009) Intracellular life of Coxiella burnetii in macrophages. Ann N Y Acad Sci 1166:55–66
Harris RJ, Storm PA, Lloyd A et al (2000) Long-term persistence of Coxiella burnetii in the host after primary Q fever. Epidemiol Infect 124:543–549
Honstettre A, Imbert G, Ghigo E et al (2003) Dysregulation of cytokines in acute Q fever: role of interleukin-10 and tumor necrosis factor in chronic evolution of Q fever. J Infect Dis 187:956–962
Honstettre A, Ghigo E, Moynault A et al (2004) Lipopolysaccharide from Coxiella burnetii is involved in bacterial phagocytosis, filamentous actin reorganization, and inflammatory responses through Toll-like receptor 4. J Immunol 172:3695–3703
Houpikian P, Raoult D (2005) Blood culture-negative endocarditis in a reference center: etiologic diagnosis of 348 cases. Medicine (Baltimore) 84:162–173
Houpikian P, Habib G, Mesana T et al (2002) Changing clinical presentation of Q fever endocarditis. Clin Infect Dis 34:E28–E31
Howe D, Mallavia LP (1999) Coxiella burnetii infection increases transferrin receptors on J774A. 1 cells. Infect Immun 67:3236–3241
Humphres RC, Hinrichs DJ (1981) Role of antibody in Coxiella burnetii infection. Infect Immun 31:641–645
Izzo AA, Marmion BP (1993) Variation in interferon-γ responses to Coxiella burnetii antigens with lymphocytes from vaccinated or naturally infected subjects. Clin Exp Immunol 94:507–515
Izzo AA, Marmion BP, Worswick DA (1988) Markers of cell-mediated immunity after vaccination with an inactivated, whole-cell Q fever vaccine. J Infect Dis 157:781–789
Kishimoto RA, Rozmiarek H, Larson EW (1978) Experimental Q fever infection in congenitally athymic nude mice. Infect Immun 22:69–71
Koster FT, Williams JC, Goodwin JS (1985a) Cellular immunity in Q fever: specific lymphocyte unresponsiveness in Q fever endocarditis. J Infect Dis 152:1283–1289
Koster FT, Williams JC, Goodwin JS (1985b) Cellular immunity in Q fever: modulation of responsiveness by a suppressor T cell-monocyte circuit. J Immunol 135:1067–1072
Leone M, Honstettre A, Lepidi H et al (2004) Effect of sex on Coxiella burnetii infection: protective role of 17β-estradiol. J Infect Dis 189:339–345
Leone M, Bechah Y, Meghari S et al (2007) Coxiella burnetii infection in C57BL/6 mice aged 1 or 14 months. FEMS Immunol Med Microbiol 50:396–400
Lepidi H, Houpikian P, Liang Z et al (2003) Cardiac valves in patients with Q fever endocarditis: microbiological, molecular, and histologic studies. J Infect Dis 187:1097–1106
Madariaga MG, Rezai K, Trenholme GM et al (2003) Q fever: a biological weapon in your backyard. Lancet Infect Dis 3:709–721
Maltezou HC, Raoult D (2002) Q fever in children. Lancet Infect Dis 2:686–691
Marmion BP, Ormsbee RA, Kyrkou M et al (1990) Vaccine prophylaxis of abattoir-associated Q fever: eight years’ experience in Australian abattoirs. Epidemiol Infect 104:275–287
Maurin M, Raoult D (1999) Q fever. Clin Microbiol Rev 12:518–553
Meghari S, Honstettre A, Lepidi H et al (2005) TLR2 is necessary to inflammatory response in Coxiella burnetii infection. Ann N Y Acad Sci 1063:161–166
Meghari S, Desnues B, Capo C et al (2006) Coxiella burnetii stimulates production of RANTES and MCP-1 by mononuclear cells: modulation by adhesion to endothelial cells and its implication in Q fever. Eur Cytokine Netw 17:253–259
Meghari S, Bechah Y, Capo C et al (2008) Persistent Coxiella burnetii infection in mice overexpressing IL-10: an efficient model for chronic Q fever pathogenesis. PLoS Pathog 4:e23
Pellegrin M, Delsol G, Auvergnat JC et al (1980) Granulomatous hepatitis in Q fever. Hum Pathol 11:51–57
Penttila IA, Harris RJ, Storm P et al (1998) Cytokine dysregulation in the post-Q-fever fatigue syndrome. QJM 91:549–560
Puissegur MP, Botanch C, Duteyrat JL et al (2004) An in vitro dual model of mycobacterial granulomas to investigate the molecular interactions between mycobacteria and human host cells. Cell Microbiol 6:423–433
Raoult D (1990) Host factors in the severity of Q fever. Ann N Y Acad Sci 590:33–38
Raoult D, Raza A, Marrie TJ (1990) Q fever endocarditis and other forms of chronic Q fever. In: Marrie TJ (ed) Q fever. The disease. CRC Press, Boca Raton, pp 3784–3786
Raoult D, Marrie TJ, Mege JL (2005) Natural history and pathophysiology of Q fever. Lancet Infect Dis 5:219–226
Read AJ, Erickson S, Harmsen AG (2010) Role of CD4+ and CD8+ T cells in clearance of primary pulmonary infection with Coxiella burnetii. Infect Immun 78:3019–3026
Rolain JM, Mallet MN, Raoult D (2003) Correlation between serum doxycycline concentrations and serologic evolution in patients with Coxiella burnetii endocarditis. J Infect Dis 188:1322–1325
Sabatier F, Dignat-George F, Mege JL et al (1997) CD4+ T-cell lymphopenia in Q fever endocarditis. Clin Diagn Lab Immunol 4:89–92
Shannon JG, Heinzen RA (2009) Adaptive immunity to the obligate intracellular pathogen Coxiella burnetii. Immunol Res 43:138–148
Shannon JG, Howe D, Heinzen RA (2005) Virulent Coxiella burnetii does not activate human dendritic cells: role of lipopolysaccharide as a shielding molecule. Proc Natl Acad Sci U S A 102:8722–8727
Srigley JR, Vellend H, Palmer N et al (1985) Q-fever. The liver and bone marrow pathology. Am J Surg Pathol 9:752–758
Textoris J, Ban LH, Capo C et al (2010) Sex-related differences in gene expression following Coxiella burnetii infection in mice: potential role of circadian rhythm. PLoS One 5:e12190
Tissot-Dupont H, Raoult D, Brouqui P et al (1992) Epidemiologic features and clinical presentation of acute Q fever in hospitalized patients: 323 French cases. Am J Med 93:427–434
Voigt JJ, Delsol G, Fabre J (1983) Liver and bone marrow granulomas in Q fever. Gastroenterology 84:887–888
Waag DM, England MJ, Bolt CR et al (2008) Low-dose priming before vaccination with the phase I chloroform-methanol residue vaccine against Q fever enhances humoral and cellular immune responses to Coxiella burnetii. Clin Vaccine Immunol 15:1505–1512
Weisburg WG, Dobson ME, Samuel JE et al (1989) Phylogenetic diversity of the Rickettsiae. J Bacteriol 171:4202–4206
Yoshiie K, Matayoshi S, Fujimura T et al (1999) Induced production of nitric oxide and sensitivity of alveolar macrophages derived from mice with different sensitivity to Coxiella burnetii. Acta Virol 43:273–278
Zamboni DS, Rabinovitch M (2003) Nitric oxide partially controls Coxiella burnetii phase II infection in mouse primary macrophages. Infect Immun 71:1225–1233
Zamboni DS, Campos MA, Torrecilhas AC et al (2004) Stimulation of toll-like receptor 2 by Coxiella burnetii is required for macrophage production of pro-inflammatory cytokines and resistance to infection. J Biol Chem 279:54405–54415
Zhang G, Russell-Lodrigue KE, Andoh M et al (2007) Mechanisms of vaccine-induced protective immunity against Coxiella burnetii infection in BALB/c mice. J Immunol 179:8372–8380
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Capo, C., Mege, JL. (2012). Role of Innate and Adaptive Immunity in the Control of Q Fever. In: Toman, R., Heinzen, R., Samuel, J., Mege, JL. (eds) Coxiella burnetii: Recent Advances and New Perspectives in Research of the Q Fever Bacterium. Advances in Experimental Medicine and Biology, vol 984. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-4315-1_14
Download citation
DOI: https://doi.org/10.1007/978-94-007-4315-1_14
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-4314-4
Online ISBN: 978-94-007-4315-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)