
Abstract
The goal of this work was to design a sandwich assay with electrochemical transduction for the detection of a tumor marker, human epidermal growth factor receptor 2 (HER2), using a simple target capturing step by antibody-functionalised magnetic beads. The HER2 proto-oncogene is amplified and/or overexpressed in approximately 20–25% of invasive breast cancers. The assay is based on a sandwich format in which a primary monoclonal antibody anti-HER2 is coupled to protein A modified magnetic beads. The modified beads are then used to capture the protein from the sample solution and the sandwich assay is performed by adding a secondary monoclonal antibody anti-HER2 labelled with biotin. The enzyme alkaline phosphatase (AP) conjugated with streptavidin and its substrate (?-naphthyl-phosphate) are then used for the electrochemical detection by differential pulse voltammetry (DPV). The conditions for the antibody immobilization and for the protein binding have been first optimised. The performance of the assay in terms of sensitivity, reproducibility and selectivity has been studied in buffer.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
- Magnetic Bead
- Differential Pulse Voltammetry
- Sandwich Assay
- Epidermal Growth Factor Receptor Family
- Magnetic Block
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
HER2 (also known as neu, ErbB-2, ERBB2) stands for “Human Epidermal growth factor Receptor 2” and is a protein giving higher aggressiveness in breast cancers. It is a member of the ErbB protein family, more commonly known as the epidermal growth factor receptor family [1].
The HER2 oncogene is a tyrosine kinase receptor that is over-expressed in approximately 20–30% of high-grade invasive breast carcinomas and has been shown to be a valuable prognostic indicator [2]. HER2 positive tumors can be more aggressive and their status can predict response to targeting therapy such as chemotherapy. Knowing that a cancer is HER2 positive helps a medical team to select the appropriate treatment.
In this paper an electrochemical immunoassay is proposed as a screening device for the detection of HER2. The proposed approach uses disposable screen-printed electrodes as transducers and a simple target capturing step by antibody-functionalised magnetic beads [3].
2 Materials and Methods
All the reagents for the buffers and EDTA were from Merck (Milan, Italy). The protein HER2, the monoclonal antibody anti-HER2 and the biotinylated polyclonal antibody anti-HER2 (R&D Systems) were purchased from Space srl (Milan, Italy). Bovine serum albumin (BSA), streptavidin–alkaline phosphatase conjugated, ?-naphtyl phosphate, were from Sigma (Milan, Italy). Protein A-coated magnetic beads were purchased from Invitrogen (Milan, Italy).
Electrochemical measurements were performed using a ?Autolab type II PGSTAT with a GPES 4.9 software package (Metrohm, Italy). All measurements were carried out at room temperature by using Differential Pulse Voltammetry (DPV) with the following parameters: range potential 0/+600 mV, step potential 7 mV, modulation amplitude 70 mV, standby potential 200 mV, interval time 0.1 s. Carbon-based screen-printed electrochemical sensors were printed in house using a High Performance Multi Purpose Precision Screen Printer DEK 245 (DEK, Weymouth, UK), using different thermoplastic inks.
3 Procedure
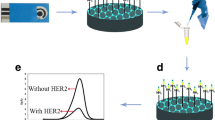
A sandwich assay for HER2 detection was developed, following the scheme reported in Fig. 23.1.

Scheme of the sandwich immunoassay onto screen printed electrochemical sensors
All the steps were performed at room temperature and the washing and coating phases were carried out under delicate stirring in the sample mixer.
Magnetic beads coated with protein A were completely re-suspended and a suspension of 100 ?L of the beads was introduced in a tube containing 400 ?L of primary antibody solution. After 30 min of incubation, the tube was positioned on a magnetic block to allow the precipitation of the beads on the bottom of the test tube; the supernatant was then removed and the beads were washed twice with PBS pH 7.4 containing 0.02% Tween 20. The antibody-coated beads could also be prepared in advance and kept at +4°C for several days. 50 ?L of suspension containing the primary antibody-coated beads were mixed with 450 ?L of solution containing HER2 at different concentrations in 20 mM Tris pH 7.3, 150 mM NaCl. After 30 min or 1 h of incubation time, the beads were magnetically separated to remove the supernatant and then washed twice using Tris buffer. After the binding of HER2, the beads were re-suspended in 500 ?L of secondary antibody solution in Tris buffer for 30 min or 1 h and then separated and washed twice using Tris. The beads carrying the affinity complex (primary antibody-analyte-secondary antibody) were re-suspended in 500 ?L of streptavidin–alkaline phosphatase conjugated (2.23 U/mL) in Tris buffer for 10 min. After separation and washing, the beads were re-suspended in 50 ?L of Tris buffer. For the electrochemical detection, 10 ?L of the beads suspension were transferred onto the surface of the working electrode of a screen-printed electrochemical sensor. To better localise the beads, the magnetic block was placed on the bottom of the electrode. Then 60 ?L of a solution containing the enzymatic substrate (?-naphtyl phosphate) 1 mg/mL in detection buffer (0.1 M diethanolamine buffer containing 1 mM MgCl2, 100 mM KCl and 0.1% pH 9.6) were deposited on the screen-printed strip. After 5 min, the enzymatic product was determined by DPV.
4 Results and Discussion
The sandwich assay was optimised with respect to several parameters such as concentration of primary and secondary antibodies, incubation time of antigen and secondary antibody, blocking agent. In Table 23.1 we report the optimization of the concentration of the secondary antibody. The optimization was performed by testing the blank and two concentrations of HER2 (10 and 50 ng/mL) with two different concentrations of secondary antibody (0.5 and 1 mg/L). Better performances both in terms of reproducibility and response linearity were obtained with a 1 mg/L of secondary antibody concentration.
The final conditions for the assay were: 100 mg/L primary antibody, 1 mg/L secondary antibody, 1 h antigen incubation time, 0.5% BSA as blocking agent. The sandwich assay was performed using HER2 standard solutions in the concentration range 2.5–25 ng/mL (Fig. 23.2). The assay was repeated in order to evaluate the reproducibility; at this purpose, three repetitions of each standard solution were carried out. The average coefficient of variation was 7%, calculated as mean of all the concentrations considered. The limit of detection (LOD) of the assay was evaluated as minimum detectable concentration and was calculated by the evaluation of the average response of the blank plus three times the standard deviation; in this case the recorded blank signal was 1.59 ± 0.19 ?A, leading to a LOD of 1.7 ng/mL.

Calibration curve for HER2. The points correspond to the current ± SD calculated for n = 3 repetitions
Preliminary experiments were conducted on commercial normal human serum diluted 1:5 and spiked with two concentrations of HER2 (15 and 25 ng/mL). The results evidenced the possibility to distinguish between non-spiked serum and serum spiked with HER2 and to distinguish between the two concentrations of HER2 added to serum.
5 Conclusions
The different parameters of the assay were optimised and a first calibration was produced in buffer with good sensitivity (1.7 ng/mL) and reproducibility (average CV% = 7%). The reached detection limit is adequate for the analysis of serum samples since 15 ng/mL was defined as the appropriate cut-off between normal and elevated serum HER2 protein levels. Further experiments will be conducted to optimise the measurements in serum and the assay will be then applied to the analysis of serum samples from hospital patients.
References
Cussens L et al (1985) Tyrosine kinase receptor with extensive homology to EGF receptor shares chromosomal location with neu oncogene. Science 230:1132–1139
Kushlinskii NE, Shirokii VP, Gershtein ES, Yermilova VD, Chemeris GY, Letyagin VP (2007) Soluble fragment of HER2/neu receptor in the serum of patients with breast cancer with different levels of this protein expression in the tumor. Bull Exp Biol Med 143:449–451
Centi S, Laschi S, Mascini M (2007) Improvement of analytical performances of a disposable electrochemical immunosensor by using magnetic beads. Talanta 73:394–399
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2011 Springer Science+Business Media B.V.
About this paper
Cite this paper
Al-Khafaji, Q.A.M., Tombelli, S., Laschi, S., Marrazza, G., Mascini, M., Mohammed, N.A.M. (2011). Detection of a Tumor Marker in Serum by an Electrochemical Assay Coupled to Magnetic Beads. In: Neri, G., Donato, N., d'Amico, A., Di Natale, C. (eds) Sensors and Microsystems. Lecture Notes in Electrical Engineering, vol 91. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-1324-6_23
Download citation
DOI: https://doi.org/10.1007/978-94-007-1324-6_23
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-1323-9
Online ISBN: 978-94-007-1324-6
eBook Packages: EngineeringEngineering (R0)