
Abstract
Transcriptional initiation is arguably the most important control point for gene expression. It is regulated by a combination of factors, including DNA sequence and its three-dimensional topology, proteins and small molecules. In this chapter, we focus on the trans-acting factors of bacterial regulation. Initiation begins with the recruitment of the RNA polymerase holoenzyme to a specific locus upstream of the gene known as its promoter. The sigma factor, which is a component of the holoenzyme, provides the most fundamental mechanisms for orchestrating broad changes in gene expression state. It is responsible for promoter recognition as well as recruiting the holoenzyme to the promoter. Distinct sigma factors compete with for binding to a common pool of RNA polymerases, thus achieving condition-dependent differential expression. Another important class of bacterial regulators is transcription factors, which activate or repress transcription of target genes typically in response to an environmental or cellular trigger. These factors may be global or local depending on the number of genes and range of cellular functions that they target. The activities of both global and local transcription factors may be regulated either at a post-transcriptional level via signal-sensing protein domains or at the level of their own expression. In addition to modulating polymerase recruitment to promoters, several global factors are considered as “nucleoid-associated proteins” that impose structural constraints on the chromosome by altering the conformation of the bound DNA, thus influencing other processes involving DNA such as replication and recombination. This chapter concludes with a discussion of how regulatory interactions between transcription factors and their target genes can be represented as a network.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
2.1 Introduction: Regulation of Transcription Initiation in Bacteria
Flow of genetic information from DNA to proteins via transcription and translation is a tightly regulated process in bacteria, enabling optimal use of valuable nutritional resources and ensuring survival in rapidly changing environments. The initiation of transcription is arguably the most important control point for regulating gene expression. It is controlled by a wide range of molecule types: cis-acting DNA sequence and structural elements, and trans-acting proteins and small molecules.
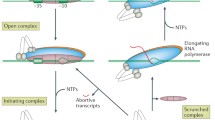
Transcription initiation begins with the recruitment of the RNA polymerase (RNAP) holoenzyme – a complex of the catalytically capable RNAP apoenzyme and a “σ-factor” – to a specific locus upstream of the gene known as its “promoter”. The σ-factor is responsible for promoter recognition as well as recruiting the holoenzyme to the promoter. The complex of RNAP holoenzyme and DNA (promoter) thus formed is called the “closed complex” [1]. In many cases, the σ-factor also facilitates the formation of the transcription bubble, i.e. the “open complex”, by stabilising the unwound DNA around 10 bp upstream of the transcription start site. Amidst extensive abortive initiation events, where the RNAP holoenzyme dissociates from the DNA after synthesising <15nt of RNA [2], processive elongation ensues followed by termination.
Successful transcription initiation requires several key components such as (a) DNA sequence and topology that permit promoter recognition, (b) σ-factors that can recognise promoters, (c) free RNAP for recruitment to the promoter concerned, and (d) trans-acting transcriptional regulators and their small molecule modulators, that enable condition-dependent differential gene expression.
In this chapter, we primarily discuss trans-acting protein factors that determine RNAP recruitment to promoters: namely σ-factors and transcription factors. The different categories of trans-acting protein factors are illustrated in Fig. 2.1. Other determinants like promoter architecture and the activity of RNAP apoenzyme have been extensively reviewed elsewhere, and will not be discussed here. First, we introduce the different families of σ-factors and highlight certain genome-scale investigations of their function. We then discuss transcription factors by focusing on their computational identification and occurrence in bacterial genomes. We will also discuss functional examples of transcription factors that regulate gene expression. Third, we highlight examples of functional interpretations derived from genome-scale analyses of transcriptional regulatory network structure. Fourth, we briefly discuss the architecture and evolution of transcription regulatory networks in Escherichia coli. Finally, we conclude the chapter with specific open questions that need to be addressed. Most of our discussion will pertain to the bacterium E. coli, for which there is extensive genomic scale experimental data.

Different groups of transcription factors based on their activity. Here we illustrate the different groups of transcription factors that are discussed in the text. Panel A depicts sigma factors that are an integral part of the transcription machinery (RNA polymerase holoenzyme). Panel B shows Nucleoid Associated Proteins (NAPs), which bind to chromosomal DNA and assist the formation of 3-dimensional nucleoid structure. Panel C depicts the classical TFs that aid the RNA polymerase holoenzyme in regulating transcription. These can be functionally divided into activators (panel D) or repressors (panel E) depending on their binding site relative to the transcriptional start site
2.2 Core Regulatory Members of the RNA Polymerase: The σ-Factors
σ-factors determine promoter specificity and are an integral part of the transcriptional machinery and the closed complex. These proteins provide most, if not all, of the determinants for promoter recognition and open complex formation, but only in complex with the rest of the RNAP [3].
There are two evolutionarily distinct families of σ-factors: σ70 and σ54. Typically, most transcription in rapidly growing cells is mediated by what is called the major σ-factor, which belongs to the σ70 family. Many bacterial genomes also code for several alternative σ-factors, which regulate specific sets of genes under different stresses and growth transformations, thus representing the most fundamental means of achieving major changes in transcription. Most alternative σ-factors also belong to different subgroups of the σ70 family. Whereas members of this family carry out open complex stabilisation on their own (as part of the RNAP holoenzyme), members of the second family, named σ54, require additional activators belonging to the AAA+ ATPase family to unwind the DNA. The σ70 family is almost ubiquitous in bacteria, and is mostly represented by multiple members. On the other hand, the σ54 family is found only in ~65% of sequenced bacterial genomes, and where present comprises a single member [3, 4]. For example, E. coli K12 encodes six members of the σ70 family (the major sigma factor RpoD, RpoH, RpoS, RpoE, FliA and FecI) but only one σ54 protein (RpoN).
Different σ-factors in bacterial cell compete for a limited number of RNAP apoenzyme molecules, and the outcome of this competition determines the cellular gene expression state. The dynamics of this competition depend on (i) relative concentrations of various sigma factors [5–8], (ii) presence of σ-factor sequestering anti-σ-factor proteins (Rsd in E. coli) [9], (iii) presence of modulating small molecule second-messengers such as (p)ppGpp [10], (iv) small non-coding RNA such as 6S RNA [11], (v) presence of other players such as H-NS [12], and (vi) finally, the ability of the sigma factor to recognize evolutionarily divergent promoter sites [13].
All these factors play a role in determining the outcome of stress σ-factor (RpoS) regulation in E. coli. During the stationary phase, RpoS is highly expressed, albeit at a third of the RpoD expression levels. However, the major σ-factor itself is sequestered by its anti-σ-factor Rsd. Also, the presence of (p)ppGpp during starvation conditions reduces transcription from the RpoD promoters, as does the 6S RNA. The presence of H-NS on chromosomal DNA also negatively impacts transcriptional initiation by RpoD. Also, molecular level studies have shown that RpoS is more tolerant to mutations in its promoters, and hence is more robust at initiating transcription from mutant promoters. All these factors facilitate transcription by RpoS at the promoters. Further, the activity of RpoS is also enhanced by the presence of A/T rich tracts upstream, and sometimes downstream, of the promoter [14].
Thus, a combination of dynamic (small molecules/proteins) and static properties (promoter sequence/architecture) determines the condition specific dominance of various sigma factors. However, it is not known how much the target gene repertoires (regulons) of different σ-factors in an organism overlap with one another. Even though a recent study reports a significant overlap between the regulons of two distinct σ-factors (RpoD and RpoH) in E. coli [15], these conclusions are controversial and await further clarification [16].
The role of σ-factors in initiating transcription, coupled with results of earlier molecular studies [17–19], suggested that the σ-factor dissociates after a successful initiation. However, later studies have shown that as much as 90% of early elongation complexes contain the σ-factor [20, 21] and provide evidence for some σ-factor retention well inside gene bodies [22]. These studies, in concert with earlier results, suggest that σ-factors play a complex role by regulating expression during initiation and controlling RNAP pausing in the elongation phases [23, 24].
2.3 Transcription Factors
Transcription factors (TFs) are proteins that bind to specific sequences on the DNA near their target genes, thus modulating transcription initiation. TFs can activate or repress transcription depending where they bind relative to the transcription start site of the target gene [1]. Each TF regulates a set of genes, in response to specific environmental and/or intracellular triggers. A complete transcriptional regulatory interaction between a TF and its target gene-(s) encompasses (1) signal sensing, (2) signal transduction, (3) the TF; and (4) the target gene-(s) [25]. In the following sections, we will focus primarily on identification of the TFs and transcriptional regulation by these TFs.
2.3.1 Identification and Genomic Distribution of Transcription Factors
Both prokaryotic and eukaryotic TFs are generally identified by the presence of a DNA-binding domain using sequence searches against protein family databases such as PFAM [26], and by BLAST-based [27] detection of homologs of experimentally-verified TFs. Several databases of computationally identified transcription factors are publicly available; most are specific to certain phylogenetic groups such as the FlyTF [28], and RegulonDB [29]. On the other hand, DBD (which in this chapter refers to “DNA-Binding Domain Database”) includes many completely sequenced genomes [30]. This database contains TF predictions for about 480 of >1,000 bacterial genomes that have been completely sequenced.
Transcription factors in the above-mentioned DBD contain one of 131 distinct protein families or domains, of which 61 are found in bacteria. Such studies showed that the number of TFs scales in a nearly quadratic fashion with genome size [31–33]. For bacteria with comparatively large genomes such as E. coli and Bacillus subtilis, TFs account for ~6% of their total gene count. These organisms may require a large proportion of transcription factors in order to regulate functionally specialised groups of genes or they might make use of more complex, and longer cascades of regulatory interactions [34]. On the other hand, organisms in host-associated symbiosis or parasitism have an extremely poor TF gene content consistent with their lack of need for sensing and responding to changing environments. Examples include Mycobacterium leprae [35] which encodes only 42 TFs (2.4% of gene count), and Rickettsia prowazekii [36] which has only nine TFs (<1%).
The E. coli genome is predicted to code for around 270 TFs, which accounts for 6% of protein-coding genes in this organism [33]. Based on the hierarchical classification of protein structures in the SCOP database, it was found that these TFs all belong to one of 11 different families, of which 10 contain the helix-turn-helix structural motif. Over 75% of all predicted TFs in E. coli contain an additional domain, belonging to a wider range of 46 different protein families. These domains are largely involved in sensing signals. Significantly, 40–50% of all TFs contain a second domain that can potentially bind to small-molecules [33, 37] and more than a third of these have been experimentally verified according to the Ecocyc database [38]. Such a high percentage of TFs with small-molecule-binding capability is not known in eukaryotes [39]. Another 10% of TFs are part of two-component signalling cascades where they are phosphorylated by an upstream histidine kinase, which in almost every case is the top-level signal sensor. Overall, these patterns of domain coupling suggest extensive and immediate interactions between signals and the transcriptional machinery, which in eukaryotes takes place through longer cascades of signal-transduction events.
2.3.2 Classification of Transcription Factors Based on Their Regulatory Scope: Global and Local Regulators
TFs in bacteria can have either a broad or a narrow regulatory scope. The scope of regulation of various TFs can be studied for the E. coli genome using the RegulonDB database. This is a collection of experimentally validated and computationally predicted TF–target interactions for majority of TFs in E. coli genome. Despite not representing many TFs, this database is useful for analyzing trends of TF–target interactions in the genome.
A cursory analysis of RegulonDB reveals that ten TFs in E. coli are responsible for more than 61% of regulatory interactions in this bacterium. Thus, a small proportion of TFs in E. coli have a global scope (global TFs), while most others target specific gene (s) and/or operon (s) (local TFs). This leaves an open question of classifying a TF as “global” or “local”, which was addressed by Martinez-Antonio and Collado-Vides [40].
Martinez-Antonio and Collado-Vides have defined a set of characteristics that distinguish global TFs from “local” players that go beyond the number of genes it regulates [40]. These characteristics include (1) number and nature of co-regulating TFs, (2) ability to regulate genes which belong to target-groups of different σ-factors, (3) capacity to regulate genes belonging to diverse functional categories, and (4) potential to respond to a wide range of environmental conditions. Besides these characteristics, global TFs have been recently shown to bind extensively to the chromosomal DNA, not necessarily causing expression changes in proximal genes [41]. Only seven TFs in E. coli satisfy all the above criteria to be a global TF: the catabolite-responsive CRP, anaerobiosis regulators FNR and ArcA, the feast or famine LRP, and three other DNA structuring proteins FIS, IHF and H-NS. Based on an analysis of target genes involved in small molecule metabolism, we have shown that six of the seven above TFs regulate multiple functional categories, but show a statistical enrichment for targeting a single function. On the other hand, most of the remaining TFs regulate genes from a single metabolic pathway or a broader functional grouping of pathways [42].
Moreover, at least five of the above seven global TFs have been classified as “nucleoid-associated proteins” (NAP) (Fig. 2.1b), primarily based on their ability to bind extensively to the DNA and to alter the topology of the bound DNA by bending, bridging or wrapping it. However, such classification is unlikely to be definite in the absence of further data; for example, there is evidence that one of the global TFs not usually considered as a NAP – FNR – can bend DNA. Finally, some global TFs have signal sensing or phosphorylation-receiving domains, which regulate their DNA binding activity; the activities of other global TFs may be regulated primarily at the level of their expression levels and/or competition or interaction with other proteins. Different NAPs show distinct patterns of gene expression during batch growth and also differ from each other in the degree of sequence specificity (see below); for instance H-NS displays preferential binding to A/T-rich sequences, and the [A/G]ATA[A/T][T/A] motif in particular, whereas others such as Hu have not been associated with any motifs so far. The properties of global TFs are illustrated with examples below.
2.3.3 Signal Dependent Activity of Global Regulators: CRP and LRP
2.3.3.1 Lrp: The Feast or Famine Global Transcription Factor
Lrp was first identified as a regulator of branched amino acid transport [43]. It was also observed in many cases that in turn its own activity is modulated by the amino acid leucine, which acts as a nutritional indicator [44, 45]. In E. coli, the TF regulates genes involved in amino acid metabolism and transport, and non-metabolic functions such as pili biosynthesis. A recent study interrogating the genome-wide binding of Lrp to the DNA identified sequence-specific interactions with ~140 chromosomal sites with an identifiable sequence motif, thus expanding the catalogue of known Lrp targets by a factor of five [46, 47]. The authors showed that absence of leucine and stationary phase increase the number of Lrp-binding regions by 3 to 4-fold, the latter effect in agreement with the inverse relationship between Lrp expression and growth rate.
Lrp and its signal, leucine, can interact in three distinct ways: (a) independent response where leucine has no effect on Lrp action; (b) concerted response in which leucine enhances the effect of Lrp; and (c) reciprocal response in which leucine antagonises the effect of Lrp. Lrp exists largely in two forms: octameric (Lrp8) and hexadecameric (Lrp16). Leucine binding favours the dissociation of Lrp to the octameric form (Lrp8-leu) [48]. Differences among promoters in their affinities to the different oligomeric forms of Lrp might explain the manner in which they are regulated by leucine [48].
Lrp can also bend and wrap the DNA [49], and its ortholog in Bacillus subtilis can, in addition, help form DNA bridges [50, 51]. These results, combined with its global scope of binding, imply that Lrp can influence the 3D topology of the chromosome. For these reasons, Lrp is considered as a NAP.
2.3.3.2 Crp and Transcriptional Responses to Carbon-Source Nutrition
Crp is the most prolific global transcription factor in E. coli, based on the information available in RegulonDB [29]. It is activated by the binding of the second messenger cyclic-AMP (cAMP) in response to glucose starvation and other stresses. Though commonly described in the context of catabolite repression (utilization of an alternative carbon source in the absence of glucose), a microarray study investigating gene expression changes in a Δcrp strain revealed a much broader regulatory scope for CRP [52], including regulation of motility in E. coli [53]. Another study investigating differential expression of genes following a change of carbon source from glucose to another (of poorer quality) highlighted that most targets of CRP are likely to be regulated indirectly [54]. Genome-wide binding studies on Crp in E. coli revealed fewer strong binding sites (~70) than expected, with a relative high background generated by many weak binding events at low-affinity sites [55]. The study also noted that only a minority of binding events directly affected target gene transcription. Based on these results and the ability of CRP to bend DNA [56, 57], the authors of this study [55] propose that CRP is too a NAP.
2.3.4 Expression and Protein–Protein Interaction Dependent Activity of Global Regulators: FIS and H-NS
2.3.4.1 Fis: An Enigmatic Transcriptional Regulator
Fis is a versatile DNA binding protein that can affect multiple processes including transcription. In E. coli, it is thought to be a major regulator of growth transitions [58]. Fis is expressed in a growth phase dependent fashion, showing high expression during logarithmic growth [59]. It activates more genes than it represses [41], though it represses several non-essential genes during exponential growth [60–62]. At least two independent genomic studies in E. coli have demonstrated that Fis mediates global changes in gene expression with over 20% of all genes being affected by Fis [41, 63, 64]. Δfis mutants of E. coli show unnaturally high negative supercoiling during stationary phase growth [58], which might lead to a general increase in transcription during this phase of growth.
Though certain FIS-binding characteristics such as localisation to gene-upstream regions may be associated with gene expression, it is being realised that, as with CRP [55], a majority of Fis binding events do not lead to proximal gene expression changes [41]. This might be because Fis has complex effects on the 3D topology of chromosomal DNA [65, 66] that go beyond just proximity binding effects.
2.3.4.2 H-NS: “The Genome Sentinel”
H-NS is a global repressor of gene expression in enterobacteria and is one of the best-studied NAPs. It is expressed throughout all the growth phases in E. coli and simultaneously affects DNA structure and transcription by forming DNA–H-NS–DNA bridges and reinforcing plectonemically supercoiled structures [67–71]. Genome-scale analysis [41, 72] showed that H-NS binds to tracts of DNA [72] and it spreads linearly from high affinity sites to flanking lower affinity regions [41]. This analysis further provided genome-scale evidence for the existence of two modes of H-NS-mediated gene regulation. Short binding regions provide mild modulation, typically repression, of the expression of proximal genes whereas long binding tracts lead to total transcriptional silencing [41].
Genome-scale investigations of H-NS-binding in Salmonella revealed a surprising mechanism for bacterial defence against foreign DNA: the protein selectively silences the transcription of large numbers of horizontally acquired genes, including those within its major pathogenicity islands [73, 74]. This arises because the protein preferentially binds A/T-rich DNA, and these acquired genomic regions tend to display high AT-content. Removal of H-NS leads to uncontrolled expression of several pathogenicity islands, which has deleterious consequences for bacterial fitness. The mechanism appears to be general for other enterobacteria, since introduction of non-native plasmids into Δhns cells can cause severe growth and infectivity defects [74–76]. Although the acquired genes are silenced during log growth, the combination of H-NS interactions with other regulatory factors and promoter-binding by the stress-associated RpoS σ-factor enables expression under stress conditions [77–79]. Thus, H-NS enables DNA to be acquired from exogenous sources, while avoiding their unregulated expression.
Thus, global regulators such as Lrp, CRP, Fis and H-NS modulate gene expression on a genome wide scale, in response to various stresses. Their responses are characterized by a global scope combined with a specific focus, such as repression of horizontally acquired genes by H-NS.
2.3.5 Local Transcription Factors and Specific Responses
The global TFs set the generic response mode such as stress, starvation and utilization of alternative carbon sources. However, in many cases, they are aided by many other TFs that make up the bulk of TF repertoire in the bacterial genome. These specific TFs, also known as local TFs, usually have a restricted regulatory scope comprising a few genes or operons. These are nonetheless responsible and necessary for regulation of their respective targets. In many known cases these TFs also act as signal sensing modules by sensing the environmental concentration of their small molecule “trigger”. We will discuss two specific examples of local TFs, both of which bind to a small molecule metabolite that modulates their activity.
LacI is a canonical local TF, which regulates the expression of the lac operon, in response to a combination of glucose starvation (CRP/c-AMP) and presence of allolactose inside the cell (LacI). The regulation of the lac operon also presents a classic case of combinatorial regulation by CRP. When the cell senses the absence of glucose, and the presence of alternative carbon source in the form of lactose/allolactose, the lac operon is activated and lactose catabolism ensues. So far, the only known target of LacI in E. coli is the lac operon.
Another example of specific local regulation involves the tryptophan synthesis operon (trp), which is regulated by the TrpR (trp Repressor). TrpR senses the levels of free tryptophan, which is the end-product of the trp operon, inside the cell by binding it. When levels of tryptophan increase inside the cell, the repressor binds to the amino acid, which stabilizes its active conformation [80], allowing it to bind upstream of the trp operon. Upon depletion of intracellular tryptophan, this process is reversed and the repression is relieved.
There are many such examples of specific repression/activation of genes and pathways by local TFs in bacteria.
2.4 Structure and Evolution of Bacterial Transcriptional Regulatory Networks
The ensemble of TF-target gene interactions in a bacterium determines its gene expression profile, and subsequently, its temporary phenotype. Such interactions can be analyzed in the form of networks, in order to gain a deeper understanding of bacterial biology. In this section, we will introduce bacterial gene regulatory networks and discuss their implications.
2.4.1 Modular Architecture of the Transcriptional Regulatory Network
A functional module is defined as a discrete entity whose function is separable from those of other modules [81]. Although there are numerous algorithms for identifying modules based on network topologies [82–85], perhaps the best characterised types of modules are network motifs that were originally described by Alon and colleagues [86]. Network motifs can be thought of as recurring circuits of regulatory interactions between TFs and target genes. Such motifs were originally defined in E. coli, in which they were detected as patterns of connections that occurred in the transcriptional network more often than would be expected in random networks.
One of the most important motifs is called the Feed Forward Loop (FFL), in which TF A regulates TF B and both A and B regulate a target gene C (Fig. 2.2a). The top-level TF in many FFLs is a global regulator: this is particularly exemplified by the classical catabolite repression which involves CRP as the top-level regulator and one of various sugar-responsive local TFs as the second regulator. Removal of global TFs from the dataset led to loss of many FFLs within the network [82, 84, 86], highlighting their importance in establishing this motif.

Two prominent architectures found in E. coli gene regulatory networks. Two sub-network architectures are prominently found in E. coli gene regulatory networks. They are the feed forward loop (FFL – Panel A), and the cascade (Panel B). In a FFL, a primary TF (tfA in figure) regulates a secondary TF (tfB) and both tfA and tfB regulate the expression of the target gene (TG in figure). Such combinatorial regulation is observed in the regulation of catabolic genes by the global regulator CRP. The second architecture, the cascade, occurs in regulation of developmental processes such as flagellar biosynthesis. In this mode of regulation, a primary TF (tfA) regulates a secondary TF (tfB), which in turn regulates the target genes
In addition to describing topological relationships between TFs and targets, different types of network motifs have been shown to carry out specific information-processing functions that are particularly suited to the biological requirements of the involved genes. For instance, FFLs filter out transient or rapidly varying input signals, thus enforcing the requirement of persistent signals for activation [86]. Thus an interesting question that can be addressed using network-based approaches is whether different types of cellular functions are regulated by distinct network architectures. For instance, the use of FFLs in controlling sugar metabolism ensures that catabolic enzymes are not expressed unless there are steady levels of the correct nutrients in the environment.
2.4.2 Subnetwork Architectures for Different Gene Functions
An important question is how these network motifs combine to form the whole regulatory system. Using symbols for different types of motifs can help depict an entire regulatory system in a compact way. In E. coli, it becomes immediately clear that FFLs feed into a layer of densely interconnected TFs, an arrangement commonly known as multi-input motifs (MIMs). Here, each TF regulates many target genes, and in turn each target is controlled by many TFs; thus a MIM can be conceptualised as a gate-array that translates multiple inputs into multiple outputs. E. coli has several discrete MIMs with hundreds of output genes, each responsible for a broad biological function, such as anaerobic growth and stress response.
Long regulatory cascades are rare in E. coli: thus most FFLs connect directly into a MIM, and in most cases, each MIM produces a final output. A possible reason for this shallow architecture is that single-celled organisms need to respond rapidly to changing environmental conditions. An exception is the relatively long cascade controlling flagella assembly: the temporal ordering afforded by multiple TFs is thought to be useful in processes requiring several stages to complete. This type of mechanism also helps explain the experimentally observed temporal programme in the expression of flagella biosynthesis genes [87].
Despite the discrete network organisation of different cellular functions (such as sugar metabolism and flagella assembly above), there is also a great deal of interconnection between them. In particular, glucose is a positive regulator of biofilm formation [88], thus linking sugar metabolism/carbon nutrition with long-term cellular decisions. This is potentially due to CRP, which is indirectly controlled by glucose availability and is a top-level regulator of both sugar metabolism and these developmental processes. A second control point integrating these two functions operates at a post-transcriptional level [89].
Architectural features of regulatory sub-networks can vary even within a single functional group. For instance, the three broad functions within metabolism, viz. catabolism, anabolism and central metabolism, differ from each other in the number and types of their regulators [42]. The genes involved in catabolism undergo combinatorial regulation, with a global regulator such as CRP and a local TF. On the other hand, anabolic pathways are often regulated by a single specific TF, and the central metabolism is regulated by multiple global TFs [42]. Further, despite the similarity in network architectures of catabolic genes, different sugar operons display distinct output patterns in response to input signals [90].
2.4.3 Evolution of Transcription Networks: Implications for Regulatory Networks
TFs and their networks are dynamic evolving entities. In fact, TFs are less conserved that other protein types such as enzymes [34, 91]. Such evolution is often directed by the environment of the bacterium and, in some cases, its interaction with a higher eukaryotic host. Interaction of bacteria with higher eukaryotes, often as pathogens, means that certain transcriptional response networks in phylogenetically distinct organisms may undergo convergent evolution. The outcome of such evolution is that phylogenetically unrelated networks might assume similar functional architectures, where related ones will differ. The evolution of transcriptional regulatory networks between phylogenetically related organisms, and its driving forces, pose some of the important questions to be addressed in this field.
2.5 Conclusions
Transcriptional regulation is essential for ensuring that the correct genes are expressed at the right amounts at the appropriate time. It is controlled by a combination of cis-effects such as DNA sequence and topology, and trans-acting factors, the focus of this chapter. Sigma factors, a component of the RNA polymerase holoenzyme, are responsible for promoter-recognition and recruitment of the holoenzyme to specific promoters; therefore they provide the most fundamental level of control for the expression of large numbers of genes. Among DNA-binding TFs, global regulators target a disproportionately large numbers of genes, and exert their control over diverse functional categories. In E. coli, five out of seven global TFs are also nucleoid-associated proteins, “histone-like” proteins that bind extensively to the genome, and alter the topology of the bound DNA. The role of such proteins appear to extend well beyond the traditional confines of transcriptional regulation, since a large proportion of binding sites do not appear to cause expression changes in proximal genes. Finally, local TFs comprise most of the regulatory repertoire in bacterial genomes, and usually have a narrow regulatory scope restricted to specific gene functions.
A crucial point to consider in bacterial gene regulation is that RNA polymerase is in very short supply: in E. coli there are estimated ~1,500 to ~11,500 polymerase molecules per cell depending on growth condition. In combination, the above factors ensure that the RNA polymerase holoenzyme is correctly distributed among the 2,000 or so competing promoters in the genome. Molecular and biophysical studies over the past 50 years have elucidated distinct mechanisms for modulating the expression of individual genes: some mechanisms allow for fine tuning of expression levels, whereas others define much sharper transitions between active and inactive transcriptional states. In contrast, genome-scale studies during the last decade have generated unprecedented quantities of information describing the location of binding sites; however, our understanding of how all these binding events lead to transcriptional regulation is still very preliminary. A major challenge over the next decade will be to bridge the gap between the detailed molecular descriptions and genome-scale overviews so that we can understand how every gene in a bacterial genome is transcriptionally regulated.
References
Browning DF, Busby SJ (2004) The regulation of bacterial transcription initiation. Nat Rev Microbiol 2:57–65
Goldman SR, Ebright RH, Nickels BE (2009) Direct detection of abortive RNA transcripts in vivo. Science 324:927–928
Gruber TM, Gross CA (2003) Multiple sigma subunits and the partitioning of bacterial transcription space. Annu Rev Microbiol 57:441–466
Pérez-Rueda E, Janga SC, Martínez-Antonio A (2009) Scaling relationship in the gene content of transcriptional machinery in bacteria. Mol Biosyst 5:1494–1501
Holland AM, Rather PN (2008) Evidence for extracellular control of RpoS proteolysis in Escherichia coli. FEMS Microbiol Lett 286:50–59
Balandina A, Claret L, Hengge-Aronis R, et al (2001) The Escherichia coli histone-like protein HU regulates rpoS translation. Mol Microbiol 39:1069–1079
Zhou Y, Gottesman S, Hoskins JR, et al (2001) The RssB response regulator directly targets sigma (S) for degradation by ClpXP. Genes Dev 15:627–637
Yamashino T, Ueguchi C, Mizuno T (1995) Quantitative control of the stationary phase-specific sigma factor, sigma S, in Escherichia coli: involvement of the nucleoid protein H-NS. EMBO J 14:594–602
Jishage M, Ishihama A (1998) A stationary phase protein in Escherichia coli with binding activity to the major sigma subunit of RNA polymerase. Proc Natl Acad Sci U S A 95:4953–4958
Jishage M, Kvint K, Shingler V, et al (2002) Regulation of sigma factor competition by the alarmone ppGpp. Genes Dev 16:1260–1270
Wassarman KM, Storz G (2000) 6S RNA regulates E. coli RNA polymerase activity. Cell 101:613–623
Shin M, Song M, Rhee JH, et al (2005) DNA looping-mediated repression by histone-like protein H-NS: specific requirement of Esigma70 as a cofactor for looping. Genes Dev 19:2388–2398
Typas A, Hengge R (2006) Role of the spacer between the -35 and -10 regions in sigmas promoter selectivity in Escherichia coli. Mol Microbiol 59:1037–1051
Typas A, Becker G, Hengge R (2007) The molecular basis of selective promoter activation by the sigmaS subunit of RNA polymerase. Mol Microbiol 63:1296–1306
Wade JT, Roa DC, Grainger DC, et al (2006) Extensive functional overlap between sigma factors in Escherichia coli. Nat Struct Mol Biol 13:806–814
Waldminghaus T, Skarstad K (2010) ChIP on Chip: surprising results are often artifacts. BMC Genomics 11:414
Hansen UM, McClure WR (1980) Role of the sigma subunit of Escherichia coli RNA polymerase in initiation. II. Release of sigma from ternary complexes. J Biol Chem 255:9564–9570
Travers AA, Burgess RR (1969) Cyclic re-use of the RNA polymerase sigma factor. Nature 222:537–540
Straney DC, Crothers DM (1985) Intermediates in transcription initiation from the E. coli lac UV5 promoter. Cell 43:449–459
Kapanidis AN, Margeat E, Laurence TA, et al (2005) Retention of transcription initiation factor sigma70 in transcription elongation: single-molecule analysis. Mol Cell 20:347–356
Mukhopadhyay J, Kapanidis AN, Mekler V, et al (2001) Translocation of sigma (70) with RNA polymerase during transcription: fluorescence resonance energy transfer assay for movement relative to DNA. Cell 106:453–463
Reppas NB, Wade JT, Church GM, et al (2006) The transition between transcriptional initiation and elongation in E. coli is highly variable and often rate limiting. Mol Cell 24:747–757
Mooney RA, Landick R (2003) Tethering sigma70 to RNA polymerase reveals high in vivo activity of sigma factors and sigma70-dependent pausing at promoter-distal locations. Genes Dev 17:2839–2851
Ring BZ, Yarnell WS, Roberts JW (1996) Function of E. coli RNA polymerase sigma factor sigma 70 in promoter-proximal pausing. Cell 86:485–493
Salgado H, Martínez-Antonio A, Janga SC (2007) Conservation of transcriptional sensing systems in prokaryotes: a perspective from Escherichia coli. FEBS Lett 581:3499–3506
Finn RD, Mistry J, Tate J, et al (2010) The Pfam protein families database. Nucleic Acids Res 38:D211–D222
Altschul SF, Madden TL, Schäffer AA, et al (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Pfreundt U, James DP, Tweedie S, et al (2010) FlyTF: improved annotation and enhanced functionality of the Drosophila transcription factor database. Nucleic Acids Res 38:D443–D447
Gama-Castro S, Jiménez-Jacinto V, Peralta-Gil M, et al (2008) RegulonDB (version 6.0): gene regulation model of Escherichia coli K-12 beyond transcription, active (experimental) annotated promoters and Textpresso navigation. Nucleic Acids Res 36:D120–D124
Charoensawan V, Wilson D, Teichmann SA (2010) Genomic repertoires of DNA-binding transcription factors across the tree of life. Nucleic Acids Res 38:7364–7377
van Nimwegen E (2003) Scaling laws in the functional content of genomes. Trends Genet 19:479–484
Ranea JA, Grant A, Thornton JM, et al (2005) Microeconomic principles explain an optimal genome size in bacteria. Trends Genet 21:21–25
Madan Babu M, Teichmann SA (2003) Evolution of transcription factors and the gene regulatory network in Escherichia coli. Nucleic Acids Res 31:1234–1244
Madan Babu M, Teichmann SA, Aravind L (2006) Evolutionary dynamics of prokaryotic transcriptional regulatory networks. J Mol Biol 358:614–633
Cole ST, Eiglmeier K, Parkhill J, et al (2001) Massive gene decay in the leprosy bacillus. Nature 409:1007–1011
Andersson SG, Zomorodipour A, Andersson JO, et al (1998) The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature 396:133–140
Anantharaman V, Koonin EV, Aravind L (2001) Regulatory potential, phyletic distribution and evolution of ancient, intracellular small-molecule-binding domains. J Mol Biol 307:1271–1292
Keseler IM, Bonavides-Martínez C, Collado-Vides J, et al (2009) EcoCyc: a comprehensive view of Escherichia coli biology. Nucleic Acids Res 37:D464–D470
Sellick CA, Reece RJ (2005) Eukaryotic transcription factors as direct nutrient sensors. Trends Biochem Sci 30:405–412
Martínez-Antonio A, Collado-Vides J (2003) Identifying global regulators in transcriptional regulatory networks in bacteria. Curr Opin Microbiol 6:482–489
Kahramanoglou C, Seshasayee ASN, Prieto AI, et al (2011) Direct and indirect effects of H-NS and Fis on global gene expression control in Escherichia coli. Nucleic Acids Res 39:2073–2091
Seshasayee AS, Fraser GM, Babu MM, et al (2009) Principles of transcriptional regulation and evolution of the metabolic system in E. coli. Genome Res 19:79–91
Anderson JJ, Quay SC, Oxender DL (1976) Mapping of two loci affecting the regulation of branched-chain amino acid transport in Escherichia coli K-12. J Bacteriol 126:80–90
Lin R, D’Ari R, Newman EB (1992) Lambda placMu insertions in genes of the leucine regulon: extension of the regulon to genes not regulated by leucine. J Bacteriol 174:1948–1955
Chen S, Hao Z, Bieniek E, et al (2001) Modulation of Lrp action in Escherichia coli by leucine: effects on non-specific binding of Lrp to DNA. J Mol Biol 314:1067–1075
Cho BK, Barrett CL, Knight EM, et al (2008) Genome-scale reconstruction of the Lrp regulatory network in Escherichia coli. Proc Natl Acad Sci U S A 105:19462–19467
Calvo JM, Matthews RG (1994) The leucine-responsive regulatory protein, a global regulator of metabolism in Escherichia coli. Microbiol Rev 58:466–490
Chen S, Rosner MH, Calvo JM (2001) Leucine-regulated self-association of leucine-responsive regulatory protein (Lrp) from Escherichia coli. J Mol Biol 312:625–635
McFarland KA, Lucchini S, Hinton JC, et al (2008) The leucine-responsive regulatory protein, Lrp, activates transcription of the fim operon in Salmonella enterica serovar typhimurium via the fimZ regulatory gene. J Bacteriol 190:602–612
Tapias A, López G, Ayora S (2000) Bacillus subtilis LrpC is a sequence-independent DNA-binding and DNA-bending protein which bridges DNA. Nucleic Acids Res 28:552–559
Beloin C, Jeusset J, Revet B, et al (2003) Contribution of DNA conformation and topology in right-handed DNA wrapping by the Bacillus subtilis LrpC protein. J Biol Chem 278:5333–5342
Zheng D, Constantinidou C, Hobman JL, et al (2004) Identification of the CRP regulon using in vitro and in vivo transcriptional profiling. Nucleic Acids Res 32:5874–5893
Soutourina O, Kolb A, Krin E, et al (1999) Multiple control of flagellum biosynthesis in Escherichia coli: role of H-NS protein and the cyclic AMP-catabolite activator protein complex in transcription of the flhDC master operon. J Bacteriol 181:7500–7508
Liu M, Durfee T, Cabrera JE, et al (2005) Global transcriptional programs reveal a carbon source foraging strategy by Escherichia coli. J Biol Chem 280:15921–15927
Grainger DC, Hurd D, Harrison M, et al (2005) Studies of the distribution of Escherichia coli cAMP-receptor protein and RNA polymerase along the E. coli chromosome. Proc Natl Acad Sci U S A 102:17693–17698
Lin SH, Lee JC (2003) Determinants of DNA bending in the DNA-cyclic AMP receptor protein complexes in Escherichia coli. Biochemistry 42:4809–4818
Napoli AA, Lawson CL, Ebright RH, et al (2006) Indirect readout of DNA sequence at the primary-kink site in the CAP-DNA complex: recognition of pyrimidine-purine and purine-purine steps. J Mol Biol 357:173–183
Schneider R, Travers A, Muskhelishvili G (1997) FIS modulates growth phase-dependent topological transitions of DNA in Escherichia coli. Mol Microbiol 26:519–530
Ali Azam T, Iwata A, Nishimura A, et al (1999) Growth phase-dependent variation in protein composition of the Escherichia coli nucleoid. J Bacteriol 181:6361–6370
Browning DF, Cole JA, Busby SJ (2008) Regulation by nucleoid-associated proteins at the Escherichia coli nir operon promoter. J Bacteriol 190:7258–7267
Grainger DC, Goldberg MD, Lee DJ, et al (2008) Selective repression by Fis and H-NS at the Escherichia coli dps promoter. Mol Microbiol 68:1366–1377
Squire DJ, Xu M, Cole JA, et al (2009) Competition between NarL-dependent activation and Fis-dependent repression controls expression from the Escherichia coli yeaR and ogt promoters. Biochem J 420:249–257
Bradley MD, Beach MB, de Koning AP, et al (2007) Effects of Fis on Escherichia coli gene expression during different growth stages. Microbiology 153:2922–2940
Cho BK, Knight EM, Barrett CL, et al (2008) Genome-wide analysis of Fis binding in Escherichia coli indicates a causative role for A-/AT-tracts. Genome Res 18:900–910
Maurer S, Fritz J, Muskhelishvili G (2009) A systematic in vitro study of nucleoprotein complexes formed by bacterial nucleoid-associated proteins revealing novel types of DNA organization. J Mol Biol 387:1261–1276
Schneider R, Lurz R, Lüder G, et al (2001) An architectural role of the Escherichia coli chromatin protein FIS in organising DNA. Nucleic Acids Res 29:5107–5114
Dorman CJ (2004) H-NS: a universal regulator for a dynamic genome. Nat Rev Microbiol 2:391–400
Dame RT, Luijsterburg MS, Krin E, et al (2005) DNA bridging: a property shared among H-NS-like proteins. J Bacteriol 187:1845–1848
Dame RT, Noom MC, Wuite GJ (2006) Bacterial chromatin organization by H-NS protein unravelled using dual DNA manipulation. Nature 444:387–390
Dorman CJ (2007) Probing bacterial nucleoid structure with optical tweezers. Bioessays 29:212–216
Noom MC, Navarre WW, Oshima T, Wuite GJ, Dame RT (2007) H-NS promotes looped domain formation in the bacterial chromosome. Curr Biol 17:R913–R914
Oshima T, Ishikawa S, Kurokawa K, et al (2006) Escherichia coli histone-like protein H-NS preferentially binds to horizontally acquired DNA in association with RNA polymerase. DNA Res 13:141–153
Doyle M, Fookes M, Ivens A, et al (2007) An H-NS-like stealth protein aids horizontal DNA transmission in bacteria. Science 315:251–252
Lucchini S, Rowley G, Goldberg MD, et al (2006) H-NS mediates the silencing of laterally acquired genes in bacteria. PLoS Pathog 2:e81
Schechter LM, Jain S, Akbar S, et al (2003) The small nucleoid-binding proteins H-NS, HU, and Fis affect hilA expression in Salmonella enterica serovar Typhimurium. Infect Immun 71:5432–5435
Hinton JC, Santos DS, Seirafi A, et al (1992) Expression and mutational analysis of the nucleoid-associated protein H-NS of Salmonella typhimurium. Mol Microbiol 6:2327–2337
Baños RC, Vivero A, Aznar S, et al (2009) Differential regulation of horizontally acquired and core genome genes by the bacterial modulator H-NS. PLoS Genet 5:e1000513
Barth M, Marschall C, Muffler A, et al (1995) Role for the histone-like protein H-NS in growth phase-dependent and osmotic regulation of sigma S and many sigma S-dependent genes in Escherichia coli. J Bacteriol 177:3455–3464
Stoebel DM, Free A, Dorman CJ (2008) Anti-silencing: overcoming H-NS-mediated repression of transcription in Gram-negative enteric bacteria. Microbiology 154:2533–2545
Grillo AO, Brown MP, Royer CA (1999) Probing the physical basis for trp repressor-operator recognition. J Mol Biol 287:539–554
Hartwell LH, Hopfield JJ, Leibler S, et al (1999) From molecular to modular cell biology. Nature 402:C47–C52
Ma HW, Kumar B, Ditges U, et al (2004) An extended transcriptional regulatory network of Escherichia coli and analysis of its hierarchical structure and network motifs. Nucleic Acids Res 32:6643–6649
Balázsi G, Barabási AL, Oltvai ZN (2005) Topological units of environmental signal processing in the transcriptional regulatory network of Escherichia coli. Proc Natl Acad Sci U S A 102:7841–7846
Freyre-González JA, Alonso-Pavón JA, Treviño-Quintanilla LG, et al (2008) Functional architecture of Escherichia coli: new insights provided by a natural decomposition approach. Genome Biol 9:R154
Resendis-Antonio O, Freyre-González JA, Menchaca-Méndez R, et al (2005) Modular analysis of the transcriptional regulatory network of E. coli. Trends Genet 21:16–20
Shen-Orr SS, Milo R, Mangan S, et al (2002) Network motifs in the transcriptional regulation network of Escherichia coli. Nat Genet 31:64–68
Martínez-Antonio A, Janga SC, Thieffry D (2008) Functional organisation of Escherichia coli transcriptional regulatory network. J Mol Biol 381:238–247
Cerca N, Jefferson KK (2008) Effect of growth conditions on poly-N-acetylglucosamine expression and biofilm formation in Escherichia coli. FEMS Microbiol Lett 283:36–41
Romeo T (1998) Global regulation by the small RNA-binding protein CsrA and the non-coding RNA molecule CsrB. Mol Microbiol 29:1321–1330
Kaplan S, Bren A, Zaslaver A, et al (2008) Diverse two-dimensional input functions control bacterial sugar genes. Mol Cell 29:786–792
Lozada-Chávez I, Janga SC, Collado-Vides J (2006) Bacterial regulatory networks are extremely flexible in evolution. Nucleic Acids Res 34:3434–3445
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2011 Springer Science+Business Media B.V.
About this chapter
Cite this chapter
Seshasayee, A.S.N., Sivaraman, K., Luscombe, N.M. (2011). An Overview of Prokaryotic Transcription Factors. In: Hughes, T. (eds) A Handbook of Transcription Factors. Subcellular Biochemistry, vol 52. Springer, Dordrecht. https://doi.org/10.1007/978-90-481-9069-0_2
Download citation
DOI: https://doi.org/10.1007/978-90-481-9069-0_2
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-90-481-9068-3
Online ISBN: 978-90-481-9069-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)