
Abstract
Systemic fungal infections have increased over time due to the rise in the at-risk population, which includes immunocompromised patients, those submitted to organ transplantation or undergoing chemotherapy. Clinically available antifungals are limited since some of them have important side effects, being toxic to the host cells, and some can quell filamentous fungi, but their activity against pathogenic yeasts is not killing but controlling their multiplication. Antimicrobial peptides are multifunctional molecules expressed by several microorganisms or synthetized by different techniques. They can play a central role in infection and inflammation. Some of their other effects include chemotactic and immunomodulating activities and wound repair. Antimicrobial peptides (AMPs) can be isolated from a large variety of microorganisms, such as plants, vertebrates, insects, bacteria, and fungi. They are classified into categories according to their amino acid composition, size, and conformational structures, and naturally occurring peptides can be synthetized. Solid-phase peptide synthesis allows the use of nonproteinogenic amino acids and permits changes in structural and physicochemical properties. In these terms, peptide engineering is a useful tool to adjust features such as net charge, surface hydrophobicity, and polarity, and it may also optimize activity and overcome the limitations inherent to natural peptides. AMPs have potential applications in antifungal therapeutics in human health, and recent uses of synthetic AMPs against fungal infections are discussed in this article.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Antifungal peptides
- Antifungal therapy
- Antimicrobial peptides
- Synthetic peptides
- Peptide engineering
- Fungal infections
3.1 Introduction
Pathogenic fungal infections are the seventh most common cause of infection-related deaths in the United States, and the fourth cause of nosocomial infection is due to the fungal pathogen Candida albicans (McNeil et al. 2001; Fisher et al. 2012; Wisplinghoff et al. 2004). Systemic mycoses can be classified according to whether the causative agent is a systemic fungal pathogen (Coccidioides immitis, Histoplasma capsulatum, and Paracoccidioides brasiliensis) or one of the increasing number of opportunistic fungal pathogens, including C. albicans, Cryptococcus neoformans var. grubii (Maurya et al. 2011; Rodriguez-Cerdeira et al. 2014), and several other ones. Those infections have become more frequent among the increasingly large population of individuals with severe immune deficiencies, including those with human immunodeficiency virus (HIV ) and acquired immune deficiency syndrome (AIDS ) (Martinez and Temesgen 2006; Marukutira et al. 2014).
The treatments of those systemic fungal infections are primarily based on itraconazole, fluconazole, or amphotericin B (Rodriguez-Cerdeira et al. 2014; Kahn et al. 2014). The triazole group enhances the specificity for fungal cytochrome P450 target, and the extra methyl group in fluconazole enhances the hydrophobic interactions at the active site, but they present limitations of toxicity or bioavailability problems that affect their potential use as systemic agents (Fukuoka et al. 2003; Ostrosky-Zeichner et al. 2010).
Amphotericin B is used for the treatment of many types of invasive fungal infections and binds to ergosterol to form membrane pores, which leads to leakage of intracellular constituents (Gabrielska et al. 2006). Despite its undeniable antifungal activity, amphotericin B has many side effects, such as nephrotoxicity (Fanos and Cataldi 2001; Wong-Beringer et al. 1998). These side effects occur mainly because drug targets in fungi are homologues of some molecular sites in humans. In addition to this limitation, the development of antimicrobial resistance due to the use of broad-spectrum antifungal drugs and their limited number for clinical purposes is a concern for public health. Considering these facts, research into new molecules with future potential therapeutic application is extremely important. Antimicrobial peptides have potential use as antifungal agents, killing the microorganism directly, or immunomodulating the host immune system response (Maurya et al. 2011; Zhai et al. 2010; Lakshminarayanan et al. 2014; Wong et al. 2013; Steinstraesser et al. 2011; Lim et al. 2015).
3.2 Antimicrobial Peptides
Antimicrobial peptides (AMPs) are naturally occurring molecules that play an important role in the first line of defense against microbial threats. They can be isolated from organisms as diverse as humans, plants, insects, and even other microorganisms like bacteria (Zasloff 2002). They are produced due to an exposure to infecting microorganisms and act in order to kill or to slow the growth of invading microorganisms and to aid allied mechanisms of natural and adaptive immunity (Fox 2013; Brogden 2005).
AMPs have a broad spectrum of activity against bacteria, fungi, enveloped viruses, parasites, and even cancerous cells. They can act directly on microorganism membranes or other nonspecific cell targets, which is an advantage in avoiding the development of microbial resistance by gene mutation, as it might happen when drugs have specific proteins as targets (Peschel and Sahl 2006). Moreover, those peptides can be extremely variable in length, amino acid composition, and structure (Nguyen et al. 2011). According to their predominant secondary structure, AMPs are divided into four categories: (a) α-helical, (b) β-sheet, (c) mixed α-helix/β-sheet, and (d) extended. The net positive charge of cationic peptides (+2 or +9) mediates their selective activity against microorganisms’ cells that carry a negative net charge due to arginine and lysine residues. Those peptides also have approximately 50 % of hydrophobic amino acids that facilitate interactions with the fatty acyl chains (Steckbeck et al. 2014; Hancock and Patrzykat 2002; Garibotto et al. 2010; Hancock and Rozek 2002).
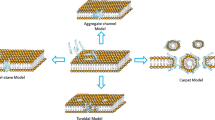
Some of the AMP mechanisms of action involve different membrane interactions. Membrane disruption can occur through the formation of toroidal pores, composed of loosely associated peptides with interdigitating phospholipid head groups among them. Those peptides are at a critical threshold concentration (Brogden 2005). Differently, in the “barrel-stave model,” the peptides are not associated with the lipid head groups, but their hydrophobic regions align with the lipid core region of the bilayer, and the hydrophilic peptide regions form the interior region of the pore (Yang et al. 2001). Another type of membrane interaction is through peptide accumulation on the bilayer surface, since they are electronically attracted to the anionic phospholipid head groups at numerous sites, thus covering the surface of the membrane in a carpet-like manner. At high peptide concentrations, these surface-oriented peptides may act like detergents, leading to the formation of micelles (Shai 1999; Ladokhin and White 2001).
The mechanism of action for the antifungal activity of peptides is generally more complex and often involves entry of the peptide into the cell. It occurs mainly because of the fungal cell architecture, briefly described since they are targets for antifungal drugs (Yu et al. 2014). The cell wall has been shown to be primarily composed of chitin, glucans, and glycoproteins, all of them being covalently cross-linked together. The glycoproteins presented in the cell wall are extensively modified with both N- and O-linked carbohydrates and, in many instances, contain a glycosylphosphatidylinositol (GPI ) anchor as well (Bowman and Free 2006). The β-glucan network consists largely of (1–3)-β-glucans with (1–6)-β-branches. In yeast (1–6)-β-glucans are also present. The cell wall is also composed of glycosylated proteins that can be decorated with mannose, galactose, glucose, and uronic acid residues. Chitin and (1-3)-β-glucan layer are a target for a wide range of antifungal molecules that can affect their synthesis and lead to a growth inhibitory effect (Fontaine et al. 2000; Schoffelmeer et al. 1999; Theis and Stahl 2004). Figure 3.1 is a schematic representation of some possible peptide membrane interactions and the basic fungi cell wall structure. Fungal plasma membranes are composed of three main lipids including phospholipids, sphingolipids, and sterols, mainly ergosterol (differently from mammalian cells, composed of cholesterol). The difference in sterol content has been exploited in the mechanism of antifungal drugs such as amphotericin B and azoles (van der Weerden et al. 2013; Kaminski 2014). The mechanisms through which AMPs have antimicrobial activity involve not only membrane physiology interference or disruptions (as described by the proposed models of barrel-stave, carpet, or toroidal pores). They might also interact with protein targets associated with the membrane or by intracellular targets, including DNA and protein synthesis, protein folding, enzymatic activity, and cell wall synthesis, which may confound the generation of resistance development (Lakshminarayanan et al. 2014; Brogden 2005; Jenssen et al. 2006; Hale and Hancock 2007; Hancock et al. 2012; Yount et al. 2006). As an example, histatins bind to a receptor in the fungal cell membrane, enter the cytoplasm, and induce the non-lytic loss of ATP from actively respiring cells. Their action can also disrupt the cell cycle and lead to the generation of reactive oxygen species (Kavanagh and Dowd 2004; De Smet and Contreras 2005) (Table 3.1).

Peptide interaction with the fungal membrane. Schematic representation of the fungal cell wall, composed of outer protein layer with carbohydrate residues (dark purple). Peptides with hydrophilic head group and hydrophobic acyl side chain regions (dark green, light green, and orange) interacting with fungal plasma membrane as barrel-stave pore (a), toroidal pore (b), or by membrane translocation (c) and membrane disruption in a carpet-like manner (d), as highlighted for LL-37. These interactions depend on the peptide, its concentration, and lipid composition of the membrane. Some peptides such as histatins, β-defensins, lactoferricins, RK-31, KS-30, and hLF (1-11) can reach internal targets such as the mitochondria (e). Histatin 5 also leads to the generation of (f) reactive oxygen species – ROS (black spindles). The non-lytic release of ATP (pink) (g) by HNP-1, HNP-2, HNP-3a, histatin-5, hLF(1–11), hLF(21–31), and B4010 might activate cell death pathway and (h) induce G1 phase arrest of the nucleus (i)
The isolation and characterization of a natural peptide is a long and laborious process that can hinder the clinical use of AMPs. A new approach is the design of synthetic sequences, which are the result of optimizing sequence and chemical characteristics that are common to many types of AMPs (pharmacophoric patron). Ideally, an antifungal peptide agent should be as short as possible, and therefore the de novo peptide design approaches help to minimize costs production and can help to overcome the low in vivo activity, the labile nature of peptides, and potential toxicity (Steckbeck et al. 2014; Garibotto et al. 2010).
De novopeptide design includes high-throughput combinatorial library screening, structure base modeling, predictive algorithms, and introduction of non-coded modifications to conventional peptide chemistry (Nguyen et al. 2011; Blondelle and Lohner 2010). The great importance of designed peptides is that many specific properties, such as hydrophobicity, hydrophobic length, or nature of flanking residues, can be systematically varied. Ideally, artificial transmembrane peptides should serve as mimics for transmembrane segments of membrane proteins (Holt and Killian 2010). It is possible to synthetize shorter peptides structurally related to another known peptide, exploring the influence of amino acid substitutions and deletions on its antifungal activity. Linear peptides are flexible, and their possible tridimensional conformations are therefore very complex to determine. It is necessary to use tools to perform conformational analysis for these structures (Garibotto et al. 2010). Figure 3.2 shows structures of some antifungal peptides deposited in RSCB Protein Data Bank website by their authors, including its PDB code (Barbault et al. 2003; Da Silva et al. 2003; Saravanan et al. 2013; Friedrich et al. 2001; Yang et al. 2009; Tack et al. 2002; Fahrner et al. 1996; Wang 2008; Zhao et al. 2013; Day et al. 1993).

Structures of antifungal peptides. Schematic representation of antifungal peptide based on nuclear magnetic resonance. Each PDB code is in parenthesis. Alo-3 (a), termicin (b), temporin-1 (c), indolicidin (d), BMAP-27(e), SMAP-29 (f), protegrin-1 (g), LL-37 (h), HNP1 (i), and (hLF) (j). All images were done using The Pymol molecular graphic system, v1.7.4
3.3 Insect Antifungal Peptides
Insect peptide Alo-3 was isolated from the coleopteran Acrocinus longimanus. This peptide contains six cysteine residues, forming three disulfide bridges and an antiparallel β-sheet with a long flexible loop connecting the first strand to the second strand and a series of turns. Alo-3 belongs to the knottin-type family of proteins with a cysteine-stabilized, “knotted” topology, defined by two parallel disulfide bonds, threaded by a third one. It has no negatively charged residues and displays a cationic pole on its surface that may contribute to its antifungal activity (van der Weerden et al. 2013). Barbault and coworkers (2003) tested two other homologous peptides, Alo-1 and Alo-2, with sequence identity above 80 %, but Alo-3 was the most effective against Candida glabrata and C. albicans, both tested not only against clinical isolates of those pathogens but also against ATCC strains (ATCC90030 and ATCC 36082, respectively). Another peptide derived from insects is termicin, isolated from the fungus-growing termite Pseudocanthotermes spiniger (heterometabole insect, Isoptera). Termicin is a cysteine-rich antifungal peptide with a α-helical segment and two antiparallel β-sheets forming a “cysteine αβ motif,” also found in antibacterial and antifungal defensins and from plants. Termicin showed activity against C. albicans and C. neoformans, but was inactive against C. glabrata (Da Silva et al. 2003; Lamberty et al. 2001).
Likewise, C. albicans growth was inhibited by holotricin-3 and tenecin-3 peptides, isolated from the hemolymph of the coleopteran insect Holotrichia diomphalia and from the larvae of Tenebrio molitor, respectively (Kim et al. 1998; Lee et al. 1995). Tenecin-3 had a better candidacidal effect, and its uptake and internalization by the cell are essential for its antimicrobial activity. This indicates an inner target involvement in the process of killing, since membrane permeabilization and calcein release were not observed. Uptake of tenecin-3 was inhibited at low temperature (0 °C) and by the presence of the oxidative phosphorylation inhibitor, sodium azide (Kim et al. 2001). Chae and coworkers (2012) isolated a new 14 kDa peptide, named tenecin-4, which was effective against Escherichia coli but not against Bacillus subtilis or C. albicans.
An interesting group of insect peptides is the cecropins, which were originally isolated from the cecropin moth (Hyalophora cecropia) and have been found in insects like Drosophila (van der Weerden et al. 2013). Cecropins are basic 35–39 amino acid residue peptides that can fold into two amphipathic α-helices, separated by a more flexible hinge. Their mode of action against bacteria is based on the formation of either voltage-dependent ion channels or general disruption of the membrane by a “carpet-like” mechanism (Steiner et al. 1988). Cecropin A, a 37 amino acid residue peptide, is complexed with lipopolysaccharide and in germinating cells of Aspergillus fumigatus induces death, whereas binding and cell death were not observed with non-germinating hyphae (De Lucca et al. 1997). De Lucca and coworkers (2000) have proposed the mode of action of this peptide as involving disruption of the plasma membrane. The ABP-dHC -cecropin A (antimicrobial peptide drury Hyphantria cunea), a highly cationic peptide isolated from the fat bodies of drury moths (H. cunea), has shown a strong antifungal activity against both C. albicans and Neurospora crassa as well as Rhyzopus, Fusarium, Alternaria, and Mucor species (Zhang et al. 2015).
Riciluca and coworkers (2012) have recently isolated a peptide named rondonin from the spider Acanthoscurria rondoniae. This peptide shows a molecular mass of 1236.77 Da and activity against C. albicans, C. krusei, C. glabrata, C. parapsilosis, C. tropicalis, and C. guilliermondii. Otherwise, no deleterious activities against human erythrocytes or Gram-positive and Gram-negative strains were observed.
3.4 Amphibian Antifungal Peptides
Amphibians inhabit environments that provide a great challenge for their immunity, providing valuable information about prospective functional molecules. In this context, peptides have been isolated and described. Some of them, like brevinins and temporins, were effective against human fungal pathogens (Xu and Lai 2015).
Brevinins consist of two families named brevinin-1 (24 residues) and brevinin-2 (33–34 residues), and they were first described in 1992 by Morikawa and coworkers (Morikawa et al. 1992), who isolated these peptides from the skin of the Japanese frog Rana brevipoda porsa, demonstrating microbicidal activity against a wide range of Gram-positive, Gram-negative bacteria and pathogenic fungi strains. Up to now about 350 types of brevinins have been discovered, sharing common features like linearity, amphipathicity, and cationicity, and some of them have a C-terminal disulfide-bridge cyclic heptapeptide, called a rana box (Novkovic et al. 2012; Savelyeva et al. 2014). Brevinin-1 exists as a random coil in aqueous solution, but adopts an amphipathic α-helical structure in a hydrophobic membrane-mimetic environment such as 50 % trifluoroethanol. The brevinin-1 peptides generally comprise an N-terminal hydrophobic region, a proline containing a hinge region in the central portion, and a C-terminal disulfide-bonded loop (Kwon et al. 1998). The α-helical structure leads to perturbation of the phospholipid bilayer of target membranes in the “barrel-stave” and “carpet-like” models (Savelyeva et al. 2014)
Conlon and coworkers (2003) isolated the peptide brevinin-1BYa (FLPILASLAAKFGPKLFCLVTKKC) from the norepinephrine-stimulated skin secretions from the foothill yellow-legged frog Rana boylii. This peptide was potent against C. albicans and Staphylococcus aureus, but its therapeutic potential is limited due to its strong hemolytic activity. The research group has also substituted amino acid in the original molecule, which leads to brevenin-1BYb and brevenin-1BYc peptides with fourfold and tenfold reduction against C. albicans, respectively. The change in the cationic residues can be the explanation of the observed result, since this global net charge reduction affects the initial binding to the negatively charged phospholipids in the microorganisms’ cell membranes (Yeaman and Yount 2003). Another study evaluated the antimicrobial activity of brevinin-1BYa and investigated the growth inhibitory activity of a synthetic replicate of this peptide: [Ser18, Ser24] brevinin-1BYa (FLPILASLAAKFGPKLFSLVTKKS). The group observed an eightfold reduced hemolytic activity compared to the native peptide and suggested that this reduction arises from destabilization of the α-helix in the C-terminal region of the peptide associated with replacement of the cysteine bridge. Antimicrobial activities against C. albicans and Gram-negative bacteria were reduced. In contrast, substituting the two cysteines for serines abolished the antifungal activity (Park et al. 2002a; Pal et al. 2006). Peptides isolated from Rana pipiens, such as brevinin-1Pa, brevinin-1Pb, and brevinin-1Pc, were also effective against C. albicans, S. aureus, and Escherichia coli (Goraya et al. 2000). In addition, brevinins are able to stimulate insulin release, which causes hypoglycemia in frogs attacking predators. This property can also be explored for the treatment of patients with type 2 diabetes (Marenah et al. 2004; Abdel-Wahab et al. 2010).
Another class of amphibian peptides is the temporins. They were initially identified in 1996 in skin secretion of the European red frog Rana temporaria (Simmaco et al. 1996), but they can be isolated from several other frog species as well as from wasp venom (Rollins-Smith et al. 2003). They are a group of linear short peptides (10–14 amino acid residues) with a net cationic charge and an amidated C-terminus. In an apolar environment, temporins showed a marked propensity to adopt an amphipathic α-helical structure. Temporins are not as basic as other cationic peptides, although in the most potent, temporin A, the one basic residue is essential for activity. Temporins A and B have been reported to be active against C. albicans and Gram-positive and Gram-negative bacteria. These peptides permeate both artificial and biological membranes, but they do not lyse human erythrocytes, which suggests there are additional factors involved in the mechanism of action on different cell types (van der Weerden et al. 2013; Mangoni et al. 2000; Wade et al. 2000; Hujakka et al. 2001; Carotenuto et al. 2008).
3.5 Mammalian Antifungal Cathelicidins
Cathelicidins are peptides of approximately 100 amino acid residues, and their sequences are related to cathelin, a cystatin-like protein. They are commonly found in humans and other species such as sheep, pigs, horses, cattle, chickens, rabbits, and some species of fishes, being usually stored in the secretory granules of neutrophils and macrophages. They can also be released extracellularly upon leukocyte activation (Zanetti 2005; Kosciuczuk et al. 2012). The term cathelicidins was first proposed in 1995 to acknowledge the evolutionary relationship of the novel protein family to cathelin, and it is used to denote holoproteins that contain a cathelin-like sequence and a cationic antimicrobial domain (Zanetti et al. 1995). The first is a conserved N-terminal sequence (“cathelin” domain, the cathepsin L inhibitor), and the second is a C-terminal antimicrobial domain of varied sequence and length (both interspecies and intraspecies), which express their activity after they have been cleaved from the holoprotein (Gennaro and Zanetti 2000). In mammals, cathelicidins were first identified in bone marrow myeloid cell, and therefore they are also named “myeloid antimicrobial peptides” (MAP) (Zanetti 2005).
Among the cathelicidin peptides, some present antifungal activity and this is stronger against yeast than against filamentous fungi (Benincasa et al. 2006). Indolicidin is a tryptophan-rich bovine cathelicidin peptide of 13 amino acid residues (ILPWKWPWWPWRR-NH2), purified from the cytoplasmic granules of neutrophils and found in bone marrow cells as a 144-long amino acid precursor (Selsted et al. 1992; Del Sal et al. 1992). This peptide showed activity against fungi C. albicans and C. neoformans, as well toward bacteria S. aureus and E. coli (Benincasa et al. 2006). The fungicidal activity involves membrane disruption, DNA binding and topoisomerase 1 inhibition. In the first situation, the peptide interacts with the lipid bilayers in a salt- and energy-dependent manner. The DNA interaction occurs by DNA synthesis inhibitors binding DNA or proteins involved in the process. Indolicidins may also interact in other biosynthesis pathways or in cell cycle signal transduction (Lee et al. 2003; Hsu et al. 2005).
Other bovine cathelicidins with fungicidal activity are bovine myeloid antimicrobial peptides (BMAP ) of 27 and 38 amino acid residues, BMAP-27 and BMAP-28, respectively. BMAP-28 is toxic for mammalian tumor cells, inducing their apoptosis, and it was also demonstrated that it induces mitochondrial permeability, forming transition pores (MPTP ), resulting in the release of cytochrome c. The cytotoxic activity has been related to the structural features of the peptide, which consists of a cationic N-terminal sequence predicted to assume an amphipathic α-helical conformation (residues 1–18) and a C-terminal hydrophobic tail (residues 19–27). This hydrophobic tail is responsible for the peptide activity, since its analogue, BMAP28 (1–18), which comprises the 18 N-terminal residues, showed a reduction in MPTP effect. BMAP28 cytotoxicity requires an active metabolism of the target cells (Risso et al. 2002).
SMAP -29 is cathelicidin-like peptide derived from myeloid sheep with α-helical structure in a hydrophobic environment, and its C-terminal hydrophobic domain has a strong membrane permeability (Chen et al. 2011; Skerlavaj et al. 1999). This peptide concentrates on the plasma membrane of treated cells and causes propidium iodide (PI ) uptake, provided by the cells that are metabolically active. Lee and coworkers (2002a) suggest that membrane disruption by SMAP-29 occurs via pore formation, due to a direct interaction with the lipid bilayers and irregularly disrupted fungal membranes in an energy- and salt-dependent manner. SMAP-29, however, is strongly hemolytic against human erythrocytes. A variant of SMAP-29, [K22,25,27]-SMAP-29, is effective against bacterial and fungal cells in physiological salt concentrations and was not injurious to eukaryotic cells, such as human erythrocytes (Shin et al. 2001; Dawson and Liu 2011).
Isolated from porcine myeloid, peptide PMAP -23 was identified by cDNA cloning and is 23 residues long, cationic (+7), and amphipathic. In a hydrophobic environment, it forms two α-helices joined by a flexible region when membrane-bound (Roversi et al. 2014; Park et al. 2002b). This peptide is capable of binding to plasma membrane of C. albicans protoplasts, indicating that an interaction with the cell wall is not a requirement for the inhibitory activity of this peptide, which also did not show hemolytic activity (Park et al. 2002b; Lee et al. 2001). Lee and coworkers (2002b) designed several analogs of PMAP-23, with amino acid substitutions in order to increase the net hydrophobicity by Trp (W)-substitution at positions 10, 13, or 14 on the hydrophilic face of the peptide. In C. albicans the P6 analog peptide exerted its fungicidal effect on the blastoconidia by disrupting the mycelial forms, causing significant morphological changes. Meanwhile, P6 also displayed about fourfold greater antitumor activity than the parent PMAP-23.
Porcine cathelicidins, such as protegrin-1 (PG-1), showed activity against clinical isolates of fungi, including those resistant to conventional medicines used in human therapy (Benincasa et al. 2006). These peptides and their variants have a rather rigid antiparallel β-sheet (β-hairpin) structure that is stabilized by two intramolecular disulfide bonds. The linear peptide forms have been reported to be considerably less active than the native form, being sensitive to physiological salt concentrations (Dawson and Liu 2010). PG-1, BMAP-27, BMAP-28, SMAP-29, and indolicidin showed deleterious activity against a number of nosocomial yeast strains, mainly Candida spp. and C. neoformans (Benincasa et al. 2006).
The most famous prototype of cathelicidin group is human cathelicidin LL-37, which attains a helical structure when bounded to cell wall and plasma membrane of treated cells and has the protein hCAP-18 as its precursor. LL-37 is secreted in human sweat and further processed into RK-31 and KS-30, more active peptides that retain their activity even in high salt conditions. Moreover, they are able to enter the cytoplasm of C. albicans cells, suggesting that their increased activity may result from interaction with intracellular targets. In contrast, LL-37 could not be detected in the cytoplasm (den Hertog et al. 2005, 2006; Lopez-Garcia et al. 2005). LL-37 showed a pH-dependent activity against C. albicans, disrupting its membrane and allowing leakage of proteins of up to 40 kDa into the medium (den Hertog et al. 2005). This peptide discriminates against phospholipid monolayers containing negatively charged lipids, as SMPA-29 does. However, experiments comparing these two peptides demonstrated that the LL-37 peptide had a more potent effect than the SMAP-29, suggesting that its interaction with monolayers involves other factors, such as hydrophobicity, size, and charge distribution, although SMAP-29 has a higher net positive charge (+10), and it would be expected to be more attracted to the negatively charged lipid monolayers (Neville et al. 2010). When compared to histatin-5, LL-37 induced higher morphological defects, but the efflux of nucleotides is similar in comparable candidacidal concentrations, suggesting that the loss of nucleotides plays an important role in the killing process (den Hertog et al. 2005). LL-37 has been found to have additional activities, such as regulating the inflammatory response and chemo-attracting cells of the adaptive immune system to wound or infection sites, helping to neutralize the microorganism and promoting re-epithelization and wound closure (Durr et al. 2006; Oudhoff et al. 2010).
3.6 Mammalian Antifungal Defensins
Mammalian defensins are a large group of peptides with an important role in the host’s immune system. They can be divided into the α- and β-defensins based on their structural characteristics and cysteine spacing pattern (van der Weerden et al. 2013). Both defensins were first identified as antimicrobial compounds involved in innate immunity. In humans, α- and β-defensins are expressed mainly in different sites: the α-defensins are mostly expressed in neutrophils (known as human neutrophil peptides (HNP) , or human defensins (HP) when expressed in natural killer cells) and the β-defensins are secreted by the epithelial cells of the skin and mucosae and known as HβD (Suarez-Carmona et al. 2014). Apart from the antimicrobial activities, the defensins appeared as modulators of the adaptive immune system and angiogenesis, key mediators of wound healing and determinant players in male fertility (Ganz 2003; Oppenheim et al. 2003). The α-defensins comprise 29–35 amino acid peptides that share six conserved cysteine residues with three disulfide bonds. Their structure is formed by an amphipathic and antiparallel β-sheet. They also have a β-hairpin loop containing cationic charged molecules. The β-defensins are longer than their α-counterparts (34–42 residues in length) and are triple-stranded with an antiparallel β-sheet as well as a short α-helix (De Smet and Contreras 2005; Selsted and Ouellette 2005). The human defensins present activity against a wide range of microorganisms, including fungal pathogens. HNP 1–3 are identical apart from one N-terminal amino acid, which makes HNP 3 completely inactive against C. albicans, while HNP 1–2 is candidacidal. HNP 4 is also toxic to C. albicans cells, and the mechanism of action of those peptides on fungal cells has been proposed to be by membrane permeabilization (Ganz and Lehrer 1995; Ganz 2005). NP 1 seems to work in the same way, but differently from HNP, and it was shown to be dependent on the metabolic activity of the target cells. HNP-1 causes C. albicans to release ATP, just as histatin 5 does, but in contrast, it did not seem to lyse cells. NP-1, NP2, and NP3a were highly effective against C. albicans, and NP-1 was effective against C. neoformans, Coccidioides immitis, and hyphae and germinating conidia of Rhizopus oryzae and Aspergillus fumigatus (van der Weerden et al. 2013; Raj and Dentino 2002; Lehrer et al. 1988).
Moreover, β-defensins, HβD 2 and HβD 3, are potent inhibitors of C. albicans. Exposure to this microorganism and to Trichophyton rubrum and A. fumigatus stimulates HβD 2 expression. A. fumigatus also induces the expression of HβD 9. The mechanisms of action of these peptides are not well known, but some requirements seem to be necessary for their activities, such as metabolically active cells and a low concentration, since they have a strong positive net charge, which in high concentrations of cations may decrease efficacy (Dhople et al. 2006; Joly et al. 2004; Liu et al. 2002; Alekseeva et al. 2009). Another antifungal peptide is Novexatin, a cyclic and highly cationic peptide (1,093 daltons), arginine rich, based on the human α and β defensins. It is currently in phase 1/2 of clinical trials for use against fungal infections of the toenails; NovaBiotics (Aberdeen, UK) is the company responsible for its development (Fox 2013).
3.7 Mammalian Antifungal Histatins
The histatin family consists of 12 members of histidine-rich peptides from which histatins 1 and 3 (the full-length proteins and gene products) and histatin 5 (a cleavage product of histatin 3) are the main ones and constitute 70–80 % of the total amount (Xu et al. 1999). The other nine members are proteolytic cleavage products of these peptides (Fitzgerald et al. 2003). Although they are named due to a high number of histidine residues, other amino acids including lysines (Lys5 and Lys13) rather than histidines have key importance for fungicidal activity (Kumar et al. 2011; Rothstein et al. 2001). Studies demonstrate that histatins have a number of biological activities in vitro, such as the maintenance of tooth surface integrity, histamine release induction, and potentiation of rabbit chondrocyte growth (Hay 1975; Oppenheim et al. 1986; Sugiyama et al. 1985; Murakami et al. 1994). The histatins are encoded by two closely related genes (HIS1 and HIS2), with histatin 1 and histatin 3 as primary products of HISI and HIS2, respectively (Sabatini and Azen 1989).
Histatin 5 was obtained from histatin 3 (Raj et al. 1998) and has the strongest antimicrobial activity against pathogenic fungi C. albicans, C. neoformans, and A. fumigatus (Kavanagh and Dowd 2004). This histatin is 24-amino acid residues long with seven histidines, four arginines, and three lysines, and its fungicidal activity resides in a region of 11–24 residues at the C-terminal, referred to as the functional domain or dh-5 (Driscoll et al. 1995). In a nonaqueous environment, the peptide adopts a α-helical conformation, and, like histatin 3, in an aqueous environment it adopts a random coil structure (van der Weerden et al. 2013; Helmerhorst et al. 1999a; Tsai and Bobek 1997). The ability to form a α-helix was thought to be important for the mode of action of histatin 5, but Situ and coworkers (2000) demonstrated that one variant of histatin 5 (3P) with reduced ability to form α-helix had an antifungal ability comparable to that of histatin 5. Its antifungal mechanisms against C. albicans involve binding to a specific receptor, translocation across the membrane, and interaction with internal targets such as mitochondria and non-lytic release of cellular ATP (Fitzgerald et al. 2003; Helmerhorst et al. 1999b; Koshlukova et al. 1999, 2000). Histatins do not display lytic activities to lipid membranes, measured by release and dequenching of the fluorescent dye calcein (Edgerton et al. 1998).
Histatin interaction with the cell wall and its uptake by the cell are two independent events, since the fungal cell wall binding itself does not result in uptake of histidines. Li and coworkers (2006) demonstrated that the heat shock protein Ssa2p is the binding site for histatin 5 measured by yeast two-hybrid analyses, whereas Ssa1p appears to have a lesser role in histatin 5 toxicity. This heat shock protein 70 (Ssap 1/2) is located in the fungal cell envelope. These interactions, however, can be prevented in the presence of Ca+, which presumably disrupts the interaction between histatin 5 and Ssa2p (Li et al. 2006). Internalization of histatin 5 occurs by translocation, and its uptake is dependent on two polyamine transporters, Dur3 and Dur31 (which usually function in spermidine uptake), since C. albicans showed a reduced intracellular transport of histatin 5 upon growth in a medium rich in spermidine, implicating polyamine transporters in uptake of this peptide (van der Weerden et al. 2013; Kumar et al. 2011).
The mitochondrion’s primeval functionality is as an energy-generating organelle, although the molecular machinery in charge of its role shows a broad divergence among different phyla (Tielens et al. 2002). It has been proposed that a negatively charged phospholipid in the mitochondrion membrane, called cardiolipin, attracts histatin 5 toward the mitochondrion. This interaction causes ATP release into the cytoplasm, and it was shown by the evidence of unchanged levels of ATP when cells were treated with respiration uncouplers, such as azide or cyanide, and then exposed to histatin 5, indicating that respiration is essential for histatin activity to occur (Helmerhorst et al. 1999a; Koshlukova et al. 1999). Gyurko and coworkers (2000) also demonstrated that fungal cells incapable of respiration (respiratory-deficient or petite mutants) are resistant to the action of histatins. The ATP efflux occurs via ATP-binding cassette (ABC) proteins, and extracellular ATP activates a purigenic-like receptor that triggers cell death (Koshlukova et al. 2000). Another ATP release consequence is the effect on the regulation of cell volume homeostasis, which can halt cells at the G1 phase, disrupting the cell cycle and leading to cell death that is not in a programmed pathway (Wunder et al. 2004). Histatin 5 is responsible for the generation of reactive oxygen species (ROS), which is measured using an oxygen radical sensitive probe (dihydroethidium), and it is one of the components responsible for the disruption of cell organelle structure or function (Baev et al. 2001). Another important factor of histatin-5 is that it retards the transition of C. albicans from the blastopore to the hyphal stage of growth, a process that may assist in arresting tissue penetration by the fungus (Helmerhorst et al. 1997).
3.8 Mammalian Lactoferrin-Derived Antifungal Peptides
Lactoferrin (Lf) is a multifunctional protein (80 kDa), a member of the transferrin family of non-heme iron-binding glycoproteins that is expressed and secreted by granular epithelial cells and secreted into mucosal fluids that bathe the body surface; it is found in the secondary granules of neutrophils during the myelocyte stage of maturation (Ward et al. 2005; Levay and Viljoen 1995). It was first isolated from bovine milk and later identified in mice, pigs, and humans (van der Weerden et al. 2013). On the mucosal surface, this peptide works as a component of the first line of host defense, being released from neutrophils during infection, inflammation, tumor development, and iron overload (Levay and Viljoen 1995; Legrand et al. 2008). It also acts in the regulation of iron homeostasis, cellular growth, and differentiation and protection against cancer development and metastasis (Ward et al. 2005; Shimamura et al. 2004).
Proteolytic cleavages of lactoferrin revealed some peptides (lactoferricins) with better antifungal activity than that of the whole protein, such as the first and second cationic domains derived from human lactoferrin (hLF ) hLF (1–11) and hLF (21–31), respectively (Lupetti et al. 2000). A study using those synthetic peptides revealed a dose-dependent release of ATP by C. albicans upon exposure to hLF (1–11). The same study demonstrated that a metabolic active cell is necessary for the hLF (1–11) mode of action, since cells incubated with sodium azide had a reduced candidacidal activity and a lower PI uptake. The use of the fluorescent dye rhodamine 123 showed an accumulation inside the mitochondria and later was released into the cytoplasm when cells were treated with hLF (1–11), which indicates that the peptide triggers the energized mitochondrion (Lupetti et al. 2000). This ATP efflux was also observed in a short synthetic peptide following the N-terminal amino sequence of bovine lactoferrin (peptide 2 or Pep2). Pep 2 activated pertussis toxin-insensitive and cholera toxin-sensitive G-protein and activated signals downstream through phosphatidylinositol 3-kinase to protein kinase C. This indicates that Pep 2 induced ATP efflux mediated by G-protein activation (Tanida et al. 2006; Helmerhorst et al. 2002). Another study showed that cell wall interaction and therefore membrane binding with hLF are not the major mode of action, thus demonstrating a slight efflux of K+ from C. albicans cells, but this did not allow Na+ release or membrane disruption (Viejo-Diaz et al. 2004). Some other peptides, derived both from bovines (LfcinB), which comprise the region spanning 17–40 residues, and from humans (LfcinH), dissipated the proton gradient across the plasma membrane. However, they did not seem to act by nonspecific permeabilization of the membrane as they did not cause calcein release from artificial liposomes (Nguyen et al. 2005; Viejo-Diaz et al. 2003).
3.9 Synthetic Antifungal Peptides
Natural peptides can have their activity enhanced by peptide engineering, which can help to develop novel peptides with desirable biological properties (Ryu et al. 2014). The synthetic peptide B4010 originated from a 10-residue peptide (RGRKVVRRKK), which in turn is a synthetic analogue of human β-defensin-3 (HβD-3). Its properties have been previously reported by Lakshminarayanan and coworkers (2014) as showing potent activity against Pseudomonas aeruginosa, but poor activity against fungi (Bai et al. 2009).
That research group also extended the previous analysis to a higher order of covalently linked peptides. B4010 is a tetravalent synthetic peptide, which carries four copies of the sequence RGRKVVRR through a branched lysine core. This strategy of developing multivalent peptides by assembling multiple copies of monomeric peptides around a core molecule is an alternative to circumventing the drawbacks of antimicrobial peptides, such as the loss of antimicrobial properties in a physiological concentration of salts and polyanionic polymers (Knappe et al. 2010; Eckert 2011).
B4010 presented deleterious activity against C. albicans strains (Lakshminarayanan et al. 2014). The researchers also tested another peptide by linking two copies of the sequence through a branched lysine, B2088, and observed a substantial decrease in MIC (minimal inhibitory concentration) values when compared to the monomer or linear retrodimer peptide (RGRKVVRRKKRRVVVKRGR). The MIC values of B4010 (0.37 μM) for two clinical isolates of C. albicans were also lower when compared to the MIC values for amphotericin B (1.4 μM) and natamycin (15 μM). This last one is the only US FDA-approved antifungal for ophthalmic applications (Lakshminarayanan et al. 2014; Arora et al. 2011), which is also nonhemolytic and nontoxic to mice when administrated by intravenous (100 mg.kg−1) or intraperitoneal (200 mg.kg−1) routes and had no affinity for cell wall polysaccharides. It was proposed that its mode of action includes a rapid dissipation of membrane potential and release of vital ions and ATP when challenged by C. albicans, and some studies suggest that the first arginine is important for mediating peptide-bilayer interactions (Lakshminarayanan et al. 2014; Nguyen et al. 2010).
Cell-penetrating peptides (CPPs) are part of a group of synthetic peptides with up to 30 peptides and are able to enter cells in an energy-independent manner, translocating across the membrane (Milletti 2012). Penetratin 1 is a 16 amino acid long CPP from the third helix of the Antennapedia homeodomain of Drosophila. It can be classified as a cationic amphipathic peptide, and its proposed mechanism of action is by “inverted micelle” pathway. This peptide was synthetized and had its fungicidal activity evaluated by Masman and coworkers (2009). This research group tested not only penetratin 1 against C. albicans and C. neoformans but also tested other peptide sequences, among them a trapeptide (RQKK-NH2), identified as 8 in their study. Both peptides, 1 and 8, displayed the most potent inhibitory effect against those pathogenic fungi (Garibotto et al. 2010; Masman et al. 2009).
3.10 Prospects for Clinical Use of Antimicrobial Peptides
The development of new drugs is a remarkable challenge. The last new class of antibiotics, the lipopeptide daptomycin, was introduced in 2003, more than 40 years after the introduction of fluoroquinolones, the last antibiotics used to treat multidrug-resistant (MDR ) organisms such as Klebsiella and Acinetobacter species. Peptides are a class of molecules with potential for therapeutic use against fungal infections (Steckbeck et al. 2014), but their use as novel drugs has to overcome some therapeutic difficulties, such as their poor chemical and physical stability and short circulating plasma half-life. Additionally, those molecules are antagonized by physiological concentrations of salt and polyanionic polymers (mucins, DNA, and glycosaminoglycans) and by the action of proteolytic enzymes, thus limiting their therapeutic potential (Lakshminarayanan et al. 2014; Knappe et al. 2010; Otvos and Wade 2014). Another difficulty is the expensive manufacturing of peptides when compared to small-molecule drugs. Companies are scrambling for financial support both from federal programs and from corporate partners through the later and most expensive stages of clinical evaluations (Fox 2013).
AMPs have only been tested in clinical trials relatively recently, and to date, with the exception of gramicidin for topical administrations, none has received US Food and Drug Administration (FDA ) approval. Kaspar and Reichert (Kaspar and Reichert 2013) highlight that in 2012 the first marketing approvals were made for six peptides (lucinactant, peginesatide, pasireotide, carfilzomib, linaclotide, and teduglutide) in the United States and European Union, which only disapproved peginesatide due to safety issues. Another example of a peptide that was undergoing clinical trials is pexiganan, a synthetic analog of the AMP magainin. Pexiganan had impressive results in early phase I and II clinical trials to treat diabetic ulcers, but its performance was not superior when compared to traditional antibiotics used in treating feet ulcers (Moore 2003). At present, approximately 140 peptide therapeutics are being evaluated in clinical trials (Fosgerau and Hoffmann 2015).
In spite of all the challenges, antimicrobial peptides have the potential for development as novel antifungal agents, because they have rapid action and a broad spectrum, being active against species that are multiple resistant to currently used antimycotics. Moreover, peptides are selective and efficacious signaling molecules with specific targets, properties that are important features for drug safety, and they show diminished side effects for the patient (Matsuzaki 2009; Wang et al. 2010; Takahashi et al. 2010). Natural or designed peptides can avoid cell toxicity and hemolytic activity and other undesirable features (Fjell et al. 2012). Some chemical strategies that have been used are sugar molecules incorporated in the peptide N-terminus, which may improve tissue penetration, and the conjugation to passive and active transport enhancers in order to increase oral bioavailability (Charlton et al. 2008; Gupta et al. 2013; Sachdeva et al. 2013). In addition, peptides play important roles in innate immune response and wound healing, additional features for their antimicrobial activity (Pena et al. 2013; Steinstraesser et al. 2012).
In summary, peptides are promising molecules with potential as future therapeutics for fungal diseases. Research is imperative to understand their mode of action and specific targets against fungi and to improve their pharmacological properties to meet clinical therapeutics and drug manufacturing needs.
Abbreviations
- ABP-dHC:
-
Antimicrobial peptide drury Hyphantria cunea
- AIDS:
-
Acquired immune deficiency syndrome
- AMPs:
-
Antimicrobial peptides
- ATP:
-
Adenosine triphosphate
- BMAP:
-
Bovine myeloid antimicrobial peptides
- DNA:
-
Desoxyribonucleic acid
- EDMC:
-
Electrostatically driven Monte Carlo
- FDA:
-
US Food and Drug Administration
- GPI:
-
Glycosylphosphatidylinositol
- HIV:
-
Human immunodeficiency virus
- hLF:
-
Human lactoferrin
- HNP:
-
Human neutrophils peptides
- HP:
-
Human defensins
- MDR:
-
Multidrug-resistant
- MPTP:
-
Mitochondrial permeability forming transition pores
- PI:
-
Propidium iodate
- PMAP:
-
Porcine myeloid antimicrobial peptide
- SMAP:
-
Sheep myeloid antimicrobial peptide
References
Abdel-Wahab YH, Patterson S, Flatt PR, Conlon JM (2010) Brevinin-2-related peptide and its [D4K] analogue stimulate insulin release in vitro and improve glucose tolerance in mice fed a high fat diet. Hormone Metabol Res Horm Stoffwechselforschung Horm Metab 42(9):652–656. doi:10.1055/s-0030-1254126
Alekseeva L, Huet D, Femenia F, Mouyna I, Abdelouahab M, Cagna A et al (2009) Inducible expression of beta defensins by human respiratory epithelial cells exposed to Aspergillus fumigatus organisms. BMC Microbiol 9:33. doi:10.1186/1471-2180-9-33
Arora R, Gupta D, Goyal J, Kaur R (2011) Voriconazole versus natamycin as primary treatment in fungal corneal ulcers. Clin Experiment Ophthalmol 39(5):434–440. doi:10.1111/j.1442-9071.2010.02473.x
Baev D, Li X, Edgerton M (2001) Genetically engineered human salivary histatin genes are functional in Candida albicans: development of a new system for studying histatin candidacidal activity. Microbiology 147(Pt 12):3323–3334
Bai Y, Liu S, Jiang P, Zhou L, Li J, Tang C et al (2009) Structure-dependent charge density as a determinant of antimicrobial activity of peptide analogues of defensin. Biochemistry 48(30):7229–7239. doi:10.1021/bi900670d
Barbault F, Landon C, Guenneugues M, Meyer JP, Schott V, Dimarcq JL et al (2003) Solution structure of Alo-3: a new knottin-type antifungal peptide from the insect Acrocinus longimanus. Biochemistry 42(49):14434–14442. doi:10.1021/bi035400o
Benincasa M, Scocchi M, Pacor S, Tossi A, Nobili D, Basaglia G et al (2006) Fungicidal activity of five cathelicidin peptides against clinically isolated yeasts. J Antimicrob Chemother 58(5):950–959. doi:10.1093/jac/dkl382
Blondelle SE, Lohner K (2010) Optimization and high-throughput screening of antimicrobial peptides. Curr Pharm Des 16(28):3204–3211
Bowman SM, Free SJ (2006) The structure and synthesis of the fungal cell wall. BioEssays: News Rev Mol Cell Dev Biol 28(8):799–808. doi:10.1002/bies.20441
Brogden KA (2005) Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol 3(3):238–250. doi:10.1038/nrmicro1098
Carotenuto A, Malfi S, Saviello MR, Campiglia P, Gomez-Monterrey I, Mangoni ML et al (2008) A different molecular mechanism underlying antimicrobial and hemolytic actions of temporins A and L. J Med Chem 51(8):2354–2362. doi:10.1021/jm701604t
Chae JH, Kurokawa K, So YI, Hwang HO, Kim MS, Park JW et al (2012) Purification and characterization of tenecin 4, a new anti-Gram-negative bacterial peptide, from the beetle Tenebrio molitor. Dev Comp Immunol 36(3):540–546. doi:10.1016/j.dci.2011.09.010
Charlton ST, Whetstone J, Fayinka ST, Read KD, Illum L, Davis SS (2008) Evaluation of direct transport pathways of glycine receptor antagonists and an angiotensin antagonist from the nasal cavity to the central nervous system in the rat model. Pharm Res 25(7):1531–1543. doi:10.1007/s11095-008-9550-2
Chen C, Wu S, Li X, Zhang X, Yan M (2011) [Structure, function and molecular design strategies of antibacterial peptide SMAP-29: a review]. Sheng Wu Gong Cheng Xue Bao Chin J Biotechnol 27(6):846–859
Conlon JM, Sonnevend A, Patel M, Davidson C, Nielsen PF, Pal T et al (2003) Isolation of peptides of the brevinin-1 family with potent candidacidal activity from the skin secretions of the frog Rana boylii. J Pept Res 62(5):207–213
Da Silva P, Jouvensal L, Lamberty M, Bulet P, Caille A, Vovelle F (2003) Solution structure of termicin, an antimicrobial peptide from the termite Pseudacanthotermes spiniger. Protein Sci: Publ Protein Soc 12(3):438–446. doi:10.1110/ps.0228303
Dawson RM, Liu CQ (2010) Disulphide bonds of the peptide protegrin-1 are not essential for antimicrobial activity and haemolytic activity. Int J Antimicrob Agents 36(6):579–580. doi:10.1016/j.ijantimicag.2010.08.011
Dawson RM, Liu CQ (2011) Analogues of peptide SMAP-29 with comparable antimicrobial potency and reduced cytotoxicity. Int J Antimicrob Agents 37(5):432–437. doi:10.1016/j.ijantimicag.2011.01.007
Day CL, Anderson BF, Tweedie JW, Baker EN (1993) Structure of the recombinant N-terminal lobe of human lactoferrin at 2.0 A resolution. J Mol Biol 232(4):1084–1100. doi:10.1006/jmbi.1993.1462
De Lucca AJ, Bland JM, Jacks TJ, Grimm C, Cleveland TE, Walsh TJ (1997) Fungicidal activity of cecropin A. Antimicrob Agents Chemother 41(2):481–483
De Lucca AJ, Bland JM, Vigo CB, Jacks TJ, Peter J, Walsh TJ (2000) D-cecropin B: proteolytic resistance, lethality for pathogenic fungi and binding properties. Med Mycol 38(4):301–308
De Smet K, Contreras R (2005) Human antimicrobial peptides: defensins, cathelicidins and histatins. Biotechnol Lett 27(18):1337–1347. doi:10.1007/s10529-005-0936-5
Del Sal G, Storici P, Schneider C, Romeo D, Zanetti M (1992) cDNA cloning of the neutrophil bactericidal peptide indolicidin. Biochem Biophys Res Commun 187(1):467–472
den Hertog AL, van Marle J, van Veen HA, Van’t Hof W, Bolscher JG, Veerman EC et al (2005) Candidacidal effects of two antimicrobial peptides: histatin 5 causes small membrane defects, but LL-37 causes massive disruption of the cell membrane. Biochem J 388(Pt 2):689–695. doi:10.1042/BJ20042099
den Hertog AL, van Marle J, Veerman EC, Valentijn-Benz M, Nazmi K, Kalay H et al (2006) The human cathelicidin peptide LL-37 and truncated variants induce segregation of lipids and proteins in the plasma membrane of Candida albicans. Biol Chem 387(10–11):1495–1502. doi:10.1515/BC.2006.187
Dhople V, Krukemeyer A, Ramamoorthy A (2006) The human beta-defensin-3, an antibacterial peptide with multiple biological functions. Biochim Biophys Acta 1758(9):1499–1512. doi:10.1016/j.bbamem.2006.07.007
Driscoll J, Zuo Y, Xu T, Choi JR, Troxler RF, Oppenheim FG (1995) Functional comparison of native and recombinant human salivary histatin 1. J Dent Res 74(12):1837–1844
Durr UH, Sudheendra US, Ramamoorthy A (2006) LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim Biophys Acta 1758(9):1408–1425. doi:10.1016/j.bbamem.2006.03.030
Eckert R (2011) Road to clinical efficacy: challenges and novel strategies for antimicrobial peptide development. Future Microbiol 6(6):635–651. doi:10.2217/fmb.11.27
Edgerton M, Koshlukova SE, Lo TE, Chrzan BG, Straubinger RM, Raj PA (1998) Candidacidal activity of salivary histatins. Identification of a histatin 5-binding protein on Candida albicans. J Biol Chem 273(32):20438–20447
Fahrner RL, Dieckmann T, Harwig SS, Lehrer RI, Eisenberg D, Feigon J (1996) Solution structure of protegrin-1, a broad-spectrum antimicrobial peptide from porcine leukocytes. Chem Biol 3(7):543–550
Fanos V, Cataldi L (2001) Renal transport of antibiotics and nephrotoxicity: a review. J Chemother 13(5):461–472. doi:10.1179/joc.2001.13.5.461
Fisher MC, Henk DA, Briggs CJ, Brownstein JS, Madoff LC, McCraw SL et al (2012) Emerging fungal threats to animal, plant and ecosystem health. Nature 484(7393):186–194. doi:10.1038/nature10947
Fitzgerald DH, Coleman DC, O’Connell BC (2003) Binding, internalisation and degradation of histatin 3 in histatin-resistant derivatives of Candida albicans. FEMS Microbiol Lett 220(2):247–253
Fjell CD, Hiss JA, Hancock RE, Schneider G (2012) Designing antimicrobial peptides: form follows function. Nat Rev Drug Discov 11(1):37–51. doi:10.1038/nrd3591
Fontaine T, Simenel C, Dubreucq G, Adam O, Delepierre M, Lemoine J et al (2000) Molecular organization of the alkali-insoluble fraction of Aspergillus fumigatus cell wall. J Biol Chem 275(36):27594–27607. doi:10.1074/jbc.M909975199
Fosgerau K, Hoffmann T (2015) Peptide therapeutics: current status and future directions. Drug Discov Today 20(1):122–128. doi:10.1016/j.drudis.2014.10.003
Fox JL (2013) Antimicrobial peptides stage a comeback. Nat Biotechnol 31(5):379–382. doi:10.1038/nbt.2572
Friedrich CL, Rozek A, Patrzykat A, Hancock RE (2001) Structure and mechanism of action of an indolicidin peptide derivative with improved activity against gram-positive bacteria. J Biol Chem 276(26):24015–24022. doi:10.1074/jbc.M009691200
Fukuoka T, Johnston DA, Winslow CA, de Groot MJ, Burt C, Hitchcock CA et al (2003) Genetic basis for differential activities of fluconazole and voriconazole against Candida krusei. Antimicrob Agents Chemother 47(4):1213–1219
Gabrielska J, Gagos M, Gubernator J, Gruszecki WI (2006) Binding of antibiotic amphotericin B to lipid membranes: a 1H NMR study. FEBS Lett 580(11):2677–2685. doi:10.1016/j.febslet.2006.04.021
Ganz T (2003) Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol 3(9):710–720. doi:10.1038/nri1180
Ganz T (2005) Defensins and other antimicrobial peptides: a historical perspective and an update. Comb Chem High Throughput Screen 8(3):209–217
Ganz T, Lehrer RI (1995) Defensins. Pharmacol Ther 66(2):191–205
Garibotto FM, Garro AD, Masman MF, Rodriguez AM, Luiten PG, Raimondi M et al (2010) New small-size peptides possessing antifungal activity. Bioorg Med Chem 18(1):158–167. doi:10.1016/j.bmc.2009.11.009
Gennaro R, Zanetti M (2000) Structural features and biological activities of the cathelicidin-derived antimicrobial peptides. Biopolymers 55(1):31–49. doi:10.1002/1097-0282(2000)55:1<31::AID-BIP40>3.0.CO;2-9
Goraya J, Wang Y, Li Z, O’Flaherty M, Knoop FC, Platz JE et al (2000) Peptides with antimicrobial activity from four different families isolated from the skins of the North American frogs Rana luteiventris, Rana berlandieri and Rana pipiens. Eur J Biochem/FEBS 267(3):894–900
Gupta V, Hwang BH, Lee J, Anselmo AC, Doshi N, Mitragotri S (2013) Mucoadhesive intestinal devices for oral delivery of salmon calcitonin. J Control Release: Off J Control Release Soc 172(3):753–762. doi:10.1016/j.jconrel.2013.09.004
Gyurko C, Lendenmann U, Troxler RF, Oppenheim FG (2000) Candida albicans mutants deficient in respiration are resistant to the small cationic salivary antimicrobial peptide histatin 5. Antimicrob Agents Chemother 44(2):348–354
Hale JD, Hancock RE (2007) Alternative mechanisms of action of cationic antimicrobial peptides on bacteria. Expert Rev Anti Infect Ther 5(6):951–959. doi:10.1586/14787210.5.6.951
Hancock RE, Patrzykat A (2002) Clinical development of cationic antimicrobial peptides: from natural to novel antibiotics. Curr Drug Targets Infect Disord 2(1):79–83
Hancock RE, Rozek A (2002) Role of membranes in the activities of antimicrobial cationic peptides. FEMS Microbiol Lett 206(2):143–149
Hancock RE, Nijnik A, Philpott DJ (2012) Modulating immunity as a therapy for bacterial infections. Nat Rev Microbiol 10(4):243–254. doi:10.1038/nrmicro2745
Hay DI (1975) Fractionation of human parotid salivary proteins and the isolation of an histidine-rich acidic peptide which shows high affinity for hydroxyapatite surfaces. Arch Oral Biol 20(9):553–558
Helmerhorst EJ, Van’t Hof W, Veerman EC, Simoons-Smit I, Nieuw Amerongen AV (1997) Synthetic histatin analogues with broad-spectrum antimicrobial activity. Biochem J 326(Pt 1):39–45
Helmerhorst EJ, Reijnders IM, van’t Hof W, Simoons-Smit I, Veerman EC, Amerongen AV (1999a) Amphotericin B- and fluconazole-resistant Candida spp., Aspergillus fumigatus, and other newly emerging pathogenic fungi are susceptible to basic antifungal peptides. Antimicrob Agents Chemother 43(3):702–704
Helmerhorst EJ, Breeuwer P, van’t Hof W, Walgreen-Weterings E, Oomen LC, Veerman EC et al (1999b) The cellular target of histatin 5 on Candida albicans is the energized mitochondrion. J Biol Chem 274(11):7286–7291
Helmerhorst EJ, Troxler RF, Oppenheim FG (2001) The human salivary peptide histatin 5 exerts its antifungal activity through the formation of reactive oxygen species. Proc Natl Acad Sci U S A 98(25):14637–14642. doi:10.1073/pnas.141366998
Helmerhorst EJ, Murphy MP, Troxler RF, Oppenheim FG (2002) Characterization of the mitochondrial respiratory pathways in Candida albicans. Biochim Biophys Acta 1556(1):73–80
Holt A, Killian JA (2010) Orientation and dynamics of transmembrane peptides: the power of simple models. Eur Biophys J: EBJ 39(4):609–621. doi:10.1007/s00249-009-0567-1
Hsu CH, Chen C, Jou ML, Lee AY, Lin YC, Yu YP et al (2005) Structural and DNA-binding studies on the bovine antimicrobial peptide, indolicidin: evidence for multiple conformations involved in binding to membranes and DNA. Nucleic Acids Res 33(13):4053–4064. doi:10.1093/nar/gki725
Hujakka H, Ratilainen J, Korjamo T, Lankinen H, Kuusela P, Santa H et al (2001) Synthesis and antimicrobial activity of the symmetric dimeric form of Temporin A based on 3-N, N-di(3-aminopropyl)amino propanoic acid as the branching unit. Bioorg Med Chem 9(6):1601–1607
Jenssen H, Hamill P, Hancock RE (2006) Peptide antimicrobial agents. Clin Microbiol Rev 19(3):491–511. doi:10.1128/CMR.00056-05
Joly S, Maze C, McCray PB Jr, Guthmiller JM (2004) Human beta-defensins 2 and 3 demonstrate strain-selective activity against oral microorganisms. J Clin Microbiol 42(3):1024–1029
Kahn A, Carey EJ, Blair JE (2014) Universal fungal prophylaxis and risk of coccidioidomycosis in liver transplant recipients living in an endemic area. Liver Transplant: Off Publ Am Assoc Study Liver Dis Int Liver Transplant Soc. doi:10.1002/lt.24055
Kaminski DM (2014) Recent progress in the study of the interactions of amphotericin B with cholesterol and ergosterol in lipid environments. Eur Biophys J: EBJ 43(10–11):453–467. doi:10.1007/s00249-014-0983-8
Kaspar AA, Reichert JM (2013) Future directions for peptide therapeutics development. Drug Discov Today 18(17–18):807–817. doi:10.1016/j.drudis.2013.05.011
Kavanagh K, Dowd S (2004) Histatins: antimicrobial peptides with therapeutic potential. J Pharm Pharmacol 56(3):285–289. doi:10.1211/0022357022971
Kim DH, Lee YT, Lee YJ, Chung JH, Lee BL, Choi BS et al (1998) Bacterial expression of tenecin 3, an insect antifungal protein isolated from Tenebrio molitor, and its efficient purification. Mol Cells 8(6):786–789
Kim DH, Lee DG, Kim KL, Lee Y (2001) Internalization of tenecin 3 by a fungal cellular process is essential for its fungicidal effect on Candida albicans. Eur J Biochem/FEBS 268(16):4449–4458
Knappe D, Henklein P, Hoffmann R, Hilpert K (2010) Easy strategy to protect antimicrobial peptides from fast degradation in serum. Antimicrob Agents Chemother 54(9):4003–4005. doi:10.1128/AAC.00300-10
Kosciuczuk EM, Lisowski P, Jarczak J, Strzalkowska N, Jozwik A, Horbanczuk J et al (2012) Cathelicidins: family of antimicrobial peptides. A review. Mol Biol Rep 39(12):10957–10970. doi:10.1007/s11033-012-1997-x
Koshlukova SE, Lloyd TL, Araujo MW, Edgerton M (1999) Salivary histatin 5 induces non-lytic release of ATP from Candida albicans leading to cell death. J Biol Chem 274(27):18872–18879
Koshlukova SE, Araujo MW, Baev D, Edgerton M (2000) Released ATP is an extracellular cytotoxic mediator in salivary histatin 5-induced killing of Candida albicans. Infect Immun 68(12):6848–6856
Kumar R, Chadha S, Saraswat D, Bajwa JS, Li RA, Conti HR et al (2011) Histatin 5 uptake by Candida albicans utilizes polyamine transporters Dur3 and Dur31 proteins. J Biol Chem 286(51):43748–43758. doi:10.1074/jbc.M111.311175
Kwon MY, Hong SY, Lee KH (1998) Structure-activity analysis of brevinin 1E amide, an antimicrobial peptide from Rana esculenta. Biochim Biophys Acta 1387(1–2):239–248
Ladokhin AS, White SH (2001) ‘Detergent-like’ permeabilization of anionic lipid vesicles by melittin. Biochim Biophys Acta 1514(2):253–260
Lakshminarayanan R, Liu S, Li J, Nandhakumar M, Aung TT, Goh E et al (2014) Synthetic multivalent antifungal peptides effective against fungi. PLoS One 9(2):e87730. doi:10.1371/journal.pone.0087730
Lamberty M, Zachary D, Lanot R, Bordereau C, Robert A, Hoffmann JA et al (2001) Insect immunity. Constitutive expression of a cysteine-rich antifungal and a linear antibacterial peptide in a termite insect. J Biol Chem 276(6):4085–4092. doi:10.1074/jbc.M002998200
Lee SY, Moon HJ, Kurata S, Natori S, Lee BL (1995) Purification and cDNA cloning of an antifungal protein from the hemolymph of Holotrichia diomphalia larvae. Biol Pharm Bull 18(8):1049–1052
Lee DG, Kim DH, Park Y, Kim HK, Kim HN, Shin YK et al (2001) Fungicidal effect of antimicrobial peptide, PMAP-23, isolated from porcine myeloid against Candida albicans. Biochem Biophys Res Commun 282(2):570–574. doi:10.1006/bbrc.2001.4602
Lee DG, Kim PI, Park Y, Park SC, Woo ER, Hahm KS (2002a) Antifungal mechanism of SMAP-29 (1–18) isolated from sheep myeloid mRNA against Trichosporon beigelii. Biochem Biophys Res Commun 295(3):591–596
Lee DG, Kim PI, Park Y, Woo ER, Choi JS, Choi CH et al (2002b) Design of novel peptide analogs with potent fungicidal activity, based on PMAP-23 antimicrobial peptide isolated from porcine myeloid. Biochem Biophys Res Commun 293(1):231–238. doi:10.1016/S0006-291X(02)00222-X
Lee DG, Kim HK, Kim SA, Park Y, Park SC, Jang SH et al (2003) Fungicidal effect of indolicidin and its interaction with phospholipid membranes. Biochem Biophys Res Commun 305(2):305–310
Legrand D, Pierce A, Elass E, Carpentier M, Mariller C, Mazurier J (2008) Lactoferrin structure and functions. Adv Exp Med Biol 606:163–194. doi:10.1007/978-0-387-74087-4_6
Lehrer RI, Ganz T, Szklarek D, Selsted ME (1988) Modulation of the in vitro candidacidal activity of human neutrophil defensins by target cell metabolism and divalent cations. J Clin Invest 81(6):1829–1835. doi:10.1172/JCI113527
Levay PF, Viljoen M (1995) Lactoferrin: a general review. Haematologica 80(3):252–267
Li XS, Sun JN, Okamoto-Shibayama K, Edgerton M (2006) Candida albicans cell wall ssa proteins bind and facilitate import of salivary histatin 5 required for toxicity. J Biol Chem 281(32):22453–22463. doi:10.1074/jbc.M604064200
Lim K, Chua RR, Ho B, Tambyah PA, Hadinoto K, Leong SS (2015) Development of a catheter functionalized by a polydopamine peptide coating with antimicrobial and antibiofilm properties. Acta Biomater 15:127–138. doi:10.1016/j.actbio.2014.12.015
Liu AY, Destoumieux D, Wong AV, Park CH, Valore EV, Liu L et al (2002) Human beta-defensin-2 production in keratinocytes is regulated by interleukin-1, bacteria, and the state of differentiation. J Invest Dermatol 118(2):275–281. doi:10.1046/j.0022-202x.2001.01651.x
Lopez-Garcia B, Lee PH, Yamasaki K, Gallo RL (2005) Anti-fungal activity of cathelicidins and their potential role in Candida albicans skin infection. J Invest Dermatol 125(1):108–115. doi:10.1111/j.0022-202X.2005.23713.x
Lupetti A, Paulusma-Annema A, Welling MM, Senesi S, van Dissel JT, Nibbering PH (2000) Candidacidal activities of human lactoferrin peptides derived from the N terminus. Antimicrob Agents Chemother 44(12):3257–3263
Mangoni ML, Rinaldi AC, Di Giulio A, Mignogna G, Bozzi A, Barra D et al (2000) Structure-function relationships of temporins, small antimicrobial peptides from amphibian skin. Eur J Biochem/FEBS 267(5):1447–1454
Marenah L, Flatt PR, Orr DF, McClean S, Shaw C, Abdel-Wahab YH (2004) Brevinin-1 and multiple insulin-releasing peptides in the skin of the frog Rana palustris. J Endocrinol 181(2):347–354
Martinez J, Temesgen Z (2006) Opportunistic infections in patients with HIV and AIDS. Fungal and parasitic infections. J Med Liban Liban Med J 54(2):84–90
Marukutira T, Huprikar S, Azie N, Quan SP, Meier-Kriesche HU, Horn DL (2014) Clinical characteristics and outcomes in 303 HIV-infected patients with invasive fungal infections: data from the Prospective Antifungal Therapy Alliance registry, a multicenter, observational study. Hiv/Aids 6:39–47. doi:10.2147/HIV.S53910
Masman MF, Rodriguez AM, Raimondi M, Zacchino SA, Luiten PG, Somlai C et al (2009) Penetratin and derivatives acting as antifungal agents. Eur J Med Chem 44(1):212–228. doi:10.1016/j.ejmech.2008.02.019
Matsuzaki K (2009) Control of cell selectivity of antimicrobial peptides. Biochim Biophys Acta 1788(8):1687–1692. doi:10.1016/j.bbamem.2008.09.013
Maurya IK, Pathak S, Sharma M, Sanwal H, Chaudhary P, Tupe S et al (2011) Antifungal activity of novel synthetic peptides by accumulation of reactive oxygen species (ROS) and disruption of cell wall against Candida albicans. Peptides 32(8):1732–1740. doi:10.1016/j.peptides.2011.06.003
McNeil MM, Nash SL, Hajjeh RA, Phelan MA, Conn LA, Plikaytis BD et al (2001) Trends in mortality due to invasive mycotic diseases in the United States, 1980–1997. Clin Infect Dis: Off Publ Infect Dis Soc Am 33(5):641–647. doi:10.1086/322606
Milletti F (2012) Cell-penetrating peptides: classes, origin, and current landscape. Drug Discov Today 17(15–16):850–860, doi:http://dx.doi.org/10.1016/j.drudis.2012.03.002
Moore A (2003) The big and small of drug discovery. EMBO Rep 4(2):114–117. doi:10.1038/sj.embor.embor748
Morikawa N, Hagiwara K, Nakajima T (1992) Brevinin-1 and -2, unique antimicrobial peptides from the skin of the frog, Rana brevipoda porsa. Biochem Biophys Res Commun 189(1):184–190
Murakami Y, Nagata H, Shizukuishi S, Nakashima K, Okawa T, Takigawa M et al (1994) Histatin as a synergistic stimulator with epidermal growth factor of rabbit chondrocyte proliferation. Biochem Biophys Res Commun 198(1):274–280. doi:10.1006/bbrc.1994.1038
Neville F, Ivankin A, Konovalov O, Gidalevitz D (2010) A comparative study on the interactions of SMAP-29 with lipid monolayers. Biochim Biophys Acta 1798(5):851–860. doi:10.1016/j.bbamem.2009.09.017
Nguyen LT, Schibli DJ, Vogel HJ (2005) Structural studies and model membrane interactions of two peptides derived from bovine lactoferricin. J Pept Sci: Off Publ Eur Pept Soc 11(7):379–389. doi:10.1002/psc.629
Nguyen LT, Chau JK, Perry NA, de Boer L, Zaat SA, Vogel HJ (2010) Serum stabilities of short tryptophan- and arginine-rich antimicrobial peptide analogs. PLoS One 5(9):e12684. doi:10.1371/journal.pone.0012684
Nguyen LT, Haney EF, Vogel HJ (2011) The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol 29(9):464–472, doi:http://dx.doi.org/10.1016/j.tibtech.2011.05.001
Novkovic M, Simunic J, Bojovic V, Tossi A, Juretic D (2012) DADP: the database of anuran defense peptides. Bioinformatics 28(10):1406–1407. doi:10.1093/bioinformatics/bts141
Oppenheim FG, Yang YC, Diamond RD, Hyslop D, Offner GD, Troxler RF (1986) The primary structure and functional characterization of the neutral histidine-rich polypeptide from human parotid secretion. J Biol Chem 261(3):1177–1182
Oppenheim JJ, Biragyn A, Kwak LW, Yang D (2003) Roles of antimicrobial peptides such as defensins in innate and adaptive immunity. Ann Rheum Dis 62(Suppl 2):ii17–ii21
Ostrosky-Zeichner L, Casadevall A, Galgiani JN, Odds FC, Rex JH (2010) An insight into the antifungal pipeline: selected new molecules and beyond. Nat Rev Drug Discov 9(9):719–727. doi:10.1038/nrd3074
Otvos L Jr, Wade JD (2014) Current challenges in peptide-based drug discovery. Front Chem 2:62. doi:10.3389/fchem.2014.00062
Oudhoff MJ, Blaauboer ME, Nazmi K, Scheres N, Bolscher JG, Veerman EC (2010) The role of salivary histatin and the human cathelicidin LL-37 in wound healing and innate immunity. Biol Chem 391(5):541–548. doi:10.1515/BC.2010.057
Pal T, Abraham B, Sonnevend A, Jumaa P, Conlon JM (2006) Brevinin-1BYa: a naturally occurring peptide from frog skin with broad-spectrum antibacterial and antifungal properties. Int J Antimicrob Agents 27(6):525–529. doi:10.1016/j.ijantimicag.2006.01.010
Park SH, Kim HE, Kim CM, Yun HJ, Choi EC, Lee BJ (2002a) Role of proline, cysteine and a disulphide bridge in the structure and activity of the anti-microbial peptide gaegurin 5. Biochem J 368(Pt 1):171–182. doi:10.1042/BJ20020385
Park K, Oh D, Shin SY, Hahm KS, Kim Y (2002b) Structural studies of porcine myeloid antibacterial peptide PMAP-23 and its analogues in DPC micelles by NMR spectroscopy. Biochem Biophys Res Commun 290(1):204–212. doi:10.1006/bbrc.2001.6173
Pena OM, Afacan N, Pistolic J, Chen C, Madera L, Falsafi R et al (2013) Synthetic cationic peptide IDR-1018 modulates human macrophage differentiation. PLoS One 8(1):e52449. doi:10.1371/journal.pone.0052449
Peschel A, Sahl HG (2006) The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nat Rev Microbiol 4(7):529–536. doi:10.1038/nrmicro1441
Raj PA, Dentino AR (2002) Current status of defensins and their role in innate and adaptive immunity. FEMS Microbiol Lett 206(1):9–18
Raj PA, Marcus E, Sukumaran DK (1998) Structure of human salivary histatin 5 in aqueous and nonaqueous solutions. Biopolymers 45(1):51–67. doi:10.1002/(sici)1097-0282(199801)45:1<51::aid-bip5>3.0.co;2-y
Riciluca KC, Sayegh RS, Melo RL, Silva PI Jr (2012) Rondonin an antifungal peptide from spider (Acanthoscurria rondoniae) haemolymph. Results Immunol 2:66–71. doi:10.1016/j.rinim.2012.03.001
Risso A, Braidot E, Sordano MC, Vianello A, Macri F, Skerlavaj B et al (2002) BMAP-28, an antibiotic peptide of innate immunity, induces cell death through opening of the mitochondrial permeability transition pore. Mol Cell Biol 22(6):1926–1935
Rodriguez-Cerdeira C, Arenas R, Moreno-Coutino G, Vasquez E, Fernandez R, Chang P (2014) Systemic fungal infections in patients with human immunodeficiency virus. Actas Dermosifiliogr 105(1):5–17. doi:10.1016/j.ad.2012.06.017
Rollins-Smith LA, Carey C, Conlon JM, Reinert LK, Doersam JK, Bergman T et al (2003) Activities of temporin family peptides against the chytrid fungus (Batrachochytrium dendrobatidis) associated with global amphibian declines. Antimicrob Agents Chemother 47(3):1157–1160
Rothstein DM, Spacciapoli P, Tran LT, Xu T, Roberts FD, Dalla Serra M et al (2001) Anticandida activity is retained in P-113, a 12-amino-acid fragment of histatin 5. Antimicrob Agents Chemother 45(5):1367–1373. doi:10.1128/aac.45.5.1367-1373.2001
Roversi D, Luca V, Aureli S, Park Y, Mangoni ML, Stella L (2014) How many antimicrobial peptide molecules kill a bacterium? The case of PMAP-23. ACS Chem Biol 9(9):2003–2007. doi:10.1021/cb500426r
Ryu JS, Cho AY, Seo SW, Min H (2014) Engineering bioactive peptide-based therapeutic molecules. Methods Mol Biol 1088:35–50. doi:10.1007/978-1-62703-673-3_3
Sabatini LM, Azen EA (1989) Histatins, a family of salivary histidine-rich proteins, are encoded by at least two loci (HIS1 and HIS2). Biochem Biophys Res Commun 160(2):495–502
Sachdeva V, Zhou Y, Banga AK (2013) In vivo transdermal delivery of leuprolide using microneedles and iontophoresis. Curr Pharm Biotechnol 14(2):180–193
Saravanan R, Joshi M, Mohanram H, Bhunia A, Mangoni ML, Bhattacharjya S (2013) NMR structure of temporin-1 ta in lipopolysaccharide micelles: mechanistic insight into inactivation by outer membrane. PLoS One 8(9):e72718. doi:10.1371/journal.pone.0072718
Savelyeva A, Ghavami S, Davoodpour P, Asoodeh A, Los MJ (2014) An overview of Brevinin superfamily: structure, function and clinical perspectives. Adv Exp Med Biol 818:197–212. doi:10.1007/978-1-4471-6458-6_10
Schoffelmeer EA, Klis FM, Sietsma JH, Cornelissen BJ (1999) The cell wall of Fusarium oxysporum. Fungal Genet Biol: FG & B 27(2–3):275–282. doi:10.1006/fgbi.1999.1153
Selsted ME, Ouellette AJ (2005) Mammalian defensins in the antimicrobial immune response. Nat Immunol 6(6):551–557. doi:10.1038/ni1206
Selsted ME, Novotny MJ, Morris WL, Tang YQ, Smith W, Cullor JS (1992) Indolicidin, a novel bactericidal tridecapeptide amide from neutrophils. J Biol Chem 267(7):4292–4295
Shai Y (1999) Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by alpha-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim Biophys Acta 1462(1–2):55–70
Shimamura M, Yamamoto Y, Ashino H, Oikawa T, Hazato T, Tsuda H et al (2004) Bovine lactoferrin inhibits tumor-induced angiogenesis. Int J Cancer J Int Cancer 111(1):111–116. doi:10.1002/ijc.20187
Shin SY, Park EJ, Yang ST, Jung HJ, Eom SH, Song WK et al (2001) Structure-activity analysis of SMAP-29, a sheep leukocytes-derived antimicrobial peptide. Biochem Biophys Res Commun 285(4):1046–1051. doi:10.1006/bbrc.2001.5280
Simmaco M, Mignogna G, Canofeni S, Miele R, Mangoni ML, Barra D (1996) Temporins, antimicrobial peptides from the European red frog Rana temporaria. Eur J Biochem/FEBS 242(3):788–792
Situ H, Balasubramanian SV, Bobek LA (2000) Role of alpha-helical conformation of histatin-5 in candidacidal activity examined by proline variants. Biochim Biophys Acta 1475(3):377–382
Skerlavaj B, Benincasa M, Risso A, Zanetti M, Gennaro R (1999) SMAP-29: a potent antibacterial and antifungal peptide from sheep leukocytes. FEBS Lett 463(1–2):58–62
Steckbeck JD, Deslouches B, Montelaro RC (2014) Antimicrobial peptides: new drugs for bad bugs? Expert Opin Biol Ther 14(1):11–14. doi:10.1517/14712598.2013.844227
Steiner H, Andreu D, Merrifield RB (1988) Binding and action of cecropin and cecropin analogues: antibacterial peptides from insects. Biochim Biophys Acta 939(2):260–266
Steinstraesser L, Kraneburg U, Jacobsen F, Al-Benna S (2011) Host defense peptides and their antimicrobial-immunomodulatory duality. Immunobiology 216(3):322–333, doi:http://dx.doi.org/10.1016/j.imbio.2010.07.003
Steinstraesser L, Hirsch T, Schulte M, Kueckelhaus M, Jacobsen F, Mersch EA et al (2012) Innate defense regulator peptide 1018 in wound healing and wound infection. PLoS One 7(8):e39373. doi:10.1371/journal.pone.0039373
Suarez-Carmona M, Hubert P, Delvenne P, Herfs M (2014) Defensins: “Simple” antimicrobial peptides or broad-spectrum molecules? Cytokine Growth Factor Rev. doi:10.1016/j.cytogfr.2014.12.005
Sugiyama K, Suzuki Y, Furuta H (1985) Isolation and characterization of histamine-releasing peptides from human parotid saliva. Life Sci 37(5):475–480
Tack BF, Sawai MV, Kearney WR, Robertson AD, Sherman MA, Wang W et al (2002) SMAP-29 has two LPS-binding sites and a central hinge. Eur J Biochem/FEBS 269(4):1181–1189
Takahashi D, Shukla SK, Prakash O, Zhang G (2010) Structural determinants of host defense peptides for antimicrobial activity and target cell selectivity. Biochimie 92(9):1236–1241, doi:http://dx.doi.org/10.1016/j.biochi.2010.02.023
Tanida T, Okamoto T, Ueta E, Yamamoto T, Osaki T (2006) Antimicrobial peptides enhance the candidacidal activity of antifungal drugs by promoting the efflux of ATP from Candida cells. J Antimicrob Chemother 57(1):94–103. doi:10.1093/jac/dki402
Theis T, Stahl U (2004) Antifungal proteins: targets, mechanisms and prospective applications. Cell Mol Life Sci: CMLS 61(4):437–455. doi:10.1007/s00018-003-3231-4
Tielens AG, Rotte C, van Hellemond JJ, Martin W (2002) Mitochondria as we don’t know them. Trends Biochem Sci 27(11):564–572
Tsai H, Bobek LA (1997) Human salivary histatin-5 exerts potent fungicidal activity against Cryptococcus neoformans. Biochim Biophys Acta (BBA) Gen Subj 1336(3):367–369, doi:http://dx.doi.org/10.1016/S0304-4165(97)00076-7
van der Weerden NL, Bleackley MR, Anderson MA (2013) Properties and mechanisms of action of naturally occurring antifungal peptides. Cell Mol Life Sci: CMLS 70(19):3545–3570. doi:10.1007/s00018-013-1260-1
Viejo-Diaz M, Andres MT, Perez-Gil J, Sanchez M, Fierro JF (2003) Potassium efflux induced by a new lactoferrin-derived peptide mimicking the effect of native human lactoferrin on the bacterial cytoplasmic membrane. Biochem Biokhim 68(2):217–227
Viejo-Diaz M, Andres MT, Fierro JF (2004) Effects of human lactoferrin on the cytoplasmic membrane of Candida albicans cells related with its candidacidal activity. FEMS Immunol Med Microbiol 42(2):181–185. doi:10.1016/j.femsim.2004.04.005
Wade D, Silberring J, Soliymani R, Heikkinen S, Kilpelainen I, Lankinen H et al (2000) Antibacterial activities of temporin A analogs. FEBS Lett 479(1–2):6–9
Wang G (2008) Structures of human host defense cathelicidin LL-37 and its smallest antimicrobial peptide KR-12 in lipid micelles. J Biol Chem 283(47):32637–32643. doi:10.1074/jbc.M805533200
Wang P, Nan YH, Yang ST, Kang SW, Kim Y, Park IS et al (2010) Cell selectivity and anti-inflammatory activity of a Leu/Lys-rich alpha-helical model antimicrobial peptide and its diastereomeric peptides. Peptides 31(7):1251–1261. doi:10.1016/j.peptides.2010.03.032
Ward PP, Paz E, Conneely OM (2005) Multifunctional roles of lactoferrin: a critical overview. Cell Mol Life Sci: CMLS 62(22):2540–2548. doi:10.1007/s00018-005-5369-8
Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB (2004) Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis: Off Publ Infect Dis Soc Am 39(3):309–317. doi:10.1086/421946
Wong JH, Ye XJ, Ng TB (2013) Cathelicidins: peptides with antimicrobial, immunomodulatory, anti-inflammatory, angiogenic, anticancer and procancer activities. Curr Protein Pept Sci 14(6):504–514
Wong-Beringer A, Jacobs RA, Guglielmo BJ (1998) Lipid formulations of amphotericin B: clinical efficacy and toxicities. Clin Infect Dis: Off Publ Infect Dis Soc Am 27(3):603–618
Wunder D, Dong J, Baev D, Edgerton M (2004) Human salivary histatin 5 fungicidal action does not induce programmed cell death pathways in Candida albicans. Antimicrob Agents Chemother 48(1):110–115
Xu X, Lai R (2015) The chemistry and biological activities of peptides from amphibian skin secretions. Chem Rev 115(4):1760–1846. doi:10.1021/cr4006704
Xu Y, Ambudkar I, Yamagishi H, Swaim W, Walsh TJ, O’Connell BC (1999) Histatin 3-mediated killing of Candida albicans: effect of extracellular salt concentration on binding and internalization. Antimicrob Agents Chemother 43(9):2256–2262
Yang S, Jung, H., Kim J (2009) Solution structure of BMAP-27. doi:10.2210/pdb2ket/pdb
Yang L, Harroun TA, Weiss TM, Ding L, Huang HW (2001) Barrel-stave model or toroidal model? A case study on melittin pores. Biophys J 81(3):1475–1485. doi:10.1016/S0006-3495(01)75802-X
Yeaman MR, Yount NY (2003) Mechanisms of antimicrobial peptide action and resistance. Pharmacol Rev 55(1):27–55. doi:10.1124/pr.55.1.2
Yount NY, Bayer AS, Xiong YQ, Yeaman MR (2006) Advances in antimicrobial peptide immunobiology. Biopolymers 84(5):435–458. doi:10.1002/bip.20543
Yu S, Chai X, Wang Y, Cao Y, Zhang J, Wu Q et al (2014) Triazole derivatives with improved in vitro antifungal activity over azole drugs. Drug Des Devel Ther 8:383–390. doi:10.2147/DDDT.S58680
Zanetti M (2005) The role of cathelicidins in the innate host defenses of mammals. Curr Issues Mol Biol 7(2):179–196
Zanetti M, Gennaro R, Romeo D (1995) Cathelicidins: a novel protein family with a common proregion and a variable C-terminal antimicrobial domain. FEBS Lett 374(1):1–5
Zasloff M (2002) Antimicrobial peptides of multicellular organisms. Nature 415(6870):389–395. doi:10.1038/415389a
Zhai B, Zhou H, Yang L, Zhang J, Jung K, Giam CZ et al (2010) Polymyxin B, in combination with fluconazole, exerts a potent fungicidal effect. J Antimicrob Chemother 65(5):931–938. doi:10.1093/jac/dkq046
Zhang J, Movahedi A, Xu J, Wang M, Wu X, Xu C et al (2015) In vitro production and antifungal activity of peptide ABP-dHC-cecropin A. J Biotechnol 199C:47–54. doi:10.1016/j.jbiotec.2015.02.018
Zhao L, Tolbert WD, Ericksen B, Zhan C, Wu X, Yuan W et al (2013) Single, double and quadruple alanine substitutions at oligomeric interfaces identify hydrophobicity as the key determinant of human neutrophil alpha defensin HNP1 function. PLoS One 8(11):e78937. doi:10.1371/journal.pone.0078937
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer India
About this chapter
Cite this chapter
Freitas, C.G., Franco, O.L. (2016). Antifungal Peptides with Potential Against Pathogenic Fungi. In: Basak, A., Chakraborty, R., Mandal, S. (eds) Recent Trends in Antifungal Agents and Antifungal Therapy. Springer, New Delhi. https://doi.org/10.1007/978-81-322-2782-3_3
Download citation
DOI: https://doi.org/10.1007/978-81-322-2782-3_3
Published:
Publisher Name: Springer, New Delhi
Print ISBN: 978-81-322-2780-9
Online ISBN: 978-81-322-2782-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)