
Abstract
Purpose
The aim of this study was to investigate and quantify drug movement to the brain via the neuro-olfactory system after intranasal dosing of four model drugs; three glycine receptor antagonists and one angiotensin antagonist.
Methods
The drugs were dosed to rats via intranasal or intravenous administration, after which a quantitative method for tissue distribution was utilised to determine drug distribution to the olfactory lobes, brain sections and the blood over 30 min. Autoradiography was used to visualise and quantify drug distribution throughout the brain and in the CSF. Micro-autoradiography was used to examine drug distribution throughout the olfactory nerve apparatus.
Results
The three glycine receptor antagonist compounds were transported to the CNS to differing degrees although they had similar molecular structures and similar physicochemical characteristics. All three compounds were shown to exploit a direct route of transport from nose to brain with Direct Transport Percentages (DTP) of 99.99%, 96.71% and 51.95%, respectively, although for the last molecule a major part of the brain content originated from systemic transport across the BBB. Intranasal administration of GR138950 resulted in over 3.5 times more drug in the olfactory lobes at 1 min post-dose compared to intravenous administration; and 5 times more drug was delivered to the olfactory lobes over 30 min. Micro-autoradiography showed that GR138950 could be found throughout the olfactory nerve apparatus. Autoradiography illustrated drug distribution throughout the brain and CSF, with drug concentrations in the CSF being equal or higher than in the brain tissue. It was determined that approximately 0.8% of the administered dose moved into the brain and CSF via the olfactory pathway over 30 min.
Conclusions
Intranasal administration resulted in greater delivery of the model drugs to the olfactory lobes and brain as compared to intravenous dosing. It is proposed that the drug moved through the neuro-olfactory system, primarily via paracellular pathways.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
For many drugs efficient delivery to the brain is problematic and has become a challenge for researchers in the pharmaceutical sector. Poor brain delivery is primarily due to the existence of the blood-brain barrier (BBB), which prevents many substances (e.g. polar drugs, peptides and proteins) from moving from the systemic circulation into the brain and cerebral spinal fluid (CSF) (1). Potentially debilitating conditions such as Alzheimer’s and Parkinson’s diseases involve the central nervous system (CNS), either partially or exclusively, and affect neurochemical levels and nerve pathways within the brain. In order to treat these conditions drugs need to gain access to the brain. Furthermore, for pain management, the drug needs to reach the brain where various receptors are stimulated by the therapeutic compound. Effective drug penetration into the brain may also be the key to successfully treatment of brain tumours. Undoubtedly many powerful therapeutic agents have been abandoned for the sole reason that effective delivery into the CNS was not achievable.
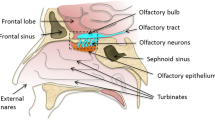
Hence, it would be beneficial to utilise an alternative non-invasive method of drug delivery to the brain that circumvents the BBB. The olfactory neuroepithelium in the nasal cavity is the only part of the CNS where dendritic processes of first order neurons are exposed directly to the environment in the upper nasal passage. Bundles of neuronal axons project through holes in the cribiform plate of the ethmoid bone to synaptic glomeruli in the olfactory bulb; therefore the nose has potential as a portal for direct delivery of drugs to the CNS (2). Considerable evidence now exists in the literature that drugs administered nasally in animals and in humans can be transported directly to the CNS (3).Various drug transport pathways have been described, such as the systemic pathway, in which the drug is absorbed across the nasal cavity into the systemic circulation and then across the BBB into the brain; the olfactory pathway, in which the drug passes through the olfactory epithelium (paracellularly or extracellularly) into the olfactory bulb and further into the brain tissue or into the cerebral spinal fluid (CSF); and the trigeminal pathway, in which the drug is transported via this nerve system (3,4). For example, it was shown in studies by Thorne et al. that 125I-IGF-1 was transported via the olfactory nerves mainly into the olfactory bulbs and the rostral (towards the nose) brain regions of rats, whereas the trigeminal pathway transferred the drug mainly to the brain stem and the spinal cord regions. However, it was not possible by the imaging methods used to clarify whether the drug was transported inside the neurons or extracellularly in the tissue surrounding the neural bundles.
Drug absorption (especially paracellular transport) via the olfactory system has been shown to be very fast, with drug being recorded in the CSF and brain tissue at 1 min post-dose (3). Hence a nasal application of selected drugs could also have an important impact on the treatment and management of crisis conditions such as migraine, panic attacks, nausea, breakthrough and acute pain, and other conditions that would benefit from very rapid therapeutic effect. However, it has been demonstrated in animal studies that the amount of drug that can be transported from the nasal cavity into the brain using simple formulations is minimal, typically in the order of less than 1% (3). Moreover, due to the anatomical differences between animals and humans, and the difficulty in reaching the olfactory region with normal nasal spray systems, it is likely that the amount of nose-to-brain transport of drugs would be lower in humans than in an animal model unless novel olfactory targeting delivery devices and nasal drug formulations specifically designed to remain on the olfactory region and promote the mucosal transport are used.
The aim of the present study was, in the first instance, to perform an initial evaluation in the rat model of nose-to-brain transport of three similar compounds (with slightly differing physicochemical characteristics) in the form of three glycine receptor antagonists. A more detailed evaluation of nose to brain transport was performed for one angiotensin antagonist and, furthermore, the pathway by which the drug moved through the neuro-olfactory system was investigated. GR138950 (an angiotensin antagonist) was the primary drug of interest. This compound is a trifluoromethanesulfonamide with high oral bioavailability (5). It has low BBB permeability, thus making it a good model for use in studies on trans-olfactory uptake because drug moving from the blood to the brain will not mask the extent of drug delivered through the olfactory system. In addition, three glycine receptor antagonist molecules (GW468816, GV196771, GV150526) were investigated. The target site for these molecules is within the brain, however, they exhibit a very high degree of binding to plasma proteins (unpublished data, GlaxoSmithKline), therefore restricting movement across the BBB.
Drug distribution in the blood and brain tissue was compared following intranasal or intravenous administration of the model drug molecules using a rat model. Quantitative tissue distribution determinations exploiting radioactive or analytical methods were used to measure drug concentration in the blood, olfactory lobes and the rest of the brain. Autoradiography was used to visualise drug distribution in the olfactory nerve tract, CSF and the brain.
MATERIALS AND METHODS
Materials and Equipment
14C-GR138950L (BN R2838/35/2, specific activity 3.01 MBq/mg), GW468816, GV196771, GV150526 were donated by GlaxoSmithKline (Stevenage, UK). The physicochemical characteristics of the drugs are given in Table I and the molecular structures in Fig. 1. Heparin sodium salt (porcine, 174 USP units/mg) was purchased from Sigma-Aldrich, Poole, Dorset, UK. Sodium chloride injection/infusion BP 0.9% w/v and water for injection (WFI) were purchased from Medical School Stores, QMC Hospital, Nottingham, UK. Hypnorm (Janssen-Cilag Ltd, High Wycombe, Bucks, UK) and Hypnovel (Roche Ltd, Welwyn Garden City, Herts, UK) were obtained from Biomedical Science Unit, QMC, Nottingham, UK. BD Microtainer plasma separator tubes with lithium heparin (Becton Dickson and Company, Franklin Lake, New Jersey, USA; purchased from NHS Supplies, UK) were used for blood collection and plasma isolation. Brain sections were prepared using a brain slicer (1 mm slice, for 200–300 g rats; Zivic Miller, supplied by Harvard Apparatus Ltd, Edenbridge, Kent, UK).

a Chemical structure of the three glycine receptor antagonists, GW468816, GV196771 and GV150526. b Chemical structure of the angiotensin antagonist GR138950.
OptiPhase ‘HiSafe’ 2 liquid scintillation cocktail and OptiSolv tissue solubiliser were purchased from Fisher Scientific UK, Loughborough, Leicestershire, UK, on behalf of EG & G Wallac, Milton Keynes, UK.
Monoclonal antibody to S-100 protein (BioGenex; Ab No. 058M, Clone 15E2E2) was purchased from A. Menarini Diagnostics, Wokingham, Berks, UK. The DAB substrate kit for peroxidase (SK-4100) was purchased from Vector Laboratories Ltd., Peterborough, UK. Ilford K5 and L4 emulsions, Phenisol™, Ilfostop Pro™ and Hypam™, produced by Ilford Imaging UK Ltd (Mobberley, Cheshire, UK), were used in the autoradiography imaging process.
Formulation Preparation
GW468816 was formulated by dissolving the drug in water for injection to produce a concentration of 2 mg/ml. GV196771 and GV150526 were formulated in DMSO and dextrose solution. 2 mg of drug was dissolved in 200 μl of neat DMSO; followed by dilution with 980 μl of 5% dextrose solution (pH 8.0) to produce a DMSO concentration of 2% and a final drug concentration of 2 mg/ml. The model drug 14C-GR138950L was dissolved in water for injection (WFI) to produce aqueous solutions of 2.5 mg/ml for intranasal testing; and 150 μg/ml for intravenous testing.
Concentrations of GW468816, GV196771 and GV150526 were quantified using an API 2000™ LC/MS/MS system and Analyst™ software (Applied Biosystems/MDS Sciex, Warrington, Cheshire, UK). Drug concentrations of 14C-GR138950L were determined from radioactivity as measured by liquid scintillation using a 1219 Rackbeta ‘Spectral’ Liquid Scintillation Counter (EG & G Wallac, Milton Keynes, UK).
Animals
Male Wistar rats weighing approximately 250 g were obtained from the Nottingham Biomedical Science Unit (Nottingham, UK) and Charles River Laboratories (Margate, UK). The rats were anesthetised intravenously with a solution of 2 ml Hypnorm (fentanyl/fluanisone) + 2 ml Hypnovel (midazolam) + 10 ml water for injection, via an indwelling needle implanted into the caudal (tail) vein. A dose of 0.2 ml was used to induce anaesthesia, followed by 0.1 ml doses as needed to maintain anaesthesia throughout the experiment. The rats were tracheotomised to maintain a patent airway and to prohibit airflow through the nasal cavity, and the oesophagus was occluded by ligation against the tracheal cannula to prevent drug absorption from the gastrointestinal tract. The carotid artery was cannulated to facilitate blood sampling. After cannulation and after blood sampling, heparinised saline (0.25 mg/ml heparin in 0.9% saline) was used to clear the cannula to prevent clot formation. This surgical methodology was taken from Hirai et al. (6) with the modifications described by Fisher et al. (7). The rats were laid on their backs on a heated table and body temperature was monitored throughout the study. All animal studies were carried out under a project license approved by the Home Office (UK), approved by the local ethics committee and conducted in accordance with the “Principles of Laboratory Animal Care” (NIH publication #85–23, revised in 1985).
Quantitative Tissue Distribution
The glycine receptor antagonists were dosed at 0.3 mg/kg (75 μg in 37.5 μl for a 250 g rat). Each intranasal dose was divided evenly between both nostrils and was administered using a flexible cannula (polyethylene, 0.28 mm I.D., 0.56 mm O.D.) that was inserted 7 mm into the nasal cavity. Each intravenous dose was administered as a single bolus injection in to the caudal (tail) vein. The rats (one for each time point) were sacrificed by cervical dislocation at the following terminal time points; 1, 3, 5, 20 and 60 min for GW468816 and 1, 5, 10, 20 and 60 min for GV196771 and GV150526. Blood samples (150–200 μl) were taken via cardiac puncture at the end-point and the olfactory lobes and brain were removed within 2 min following sacrifice. To excise the brain, an incision was first made in the skin to expose the skull. The occipital bone was removed using forceps, and then scissors were used to cut along the lateral cranial sutures of the parietal bone before using forceps to remove the parietal bone and reveal the brain. The brain was removed intact (excluding the olfactory lobes) and then the olfactory lobes were removed using spatulas.
For 14C-GR138950L, groups (n = 3) of rats for each time point were dosed at 0.3 mg/kg via intranasal (75 μg in 30 μl for a 250 g rat) or intravenous (75 μg in 0.5 ml for a 250 g rat) administration as described above. Each group was sacrificed by cervical dislocation at 1, 5, 10, 15 or 30 min post-dose, and then the olfactory lobes and brain were excised within 2 min following sacrifice. Dosing and subsequent sacrifice of each rat was staggered to allow prompt removal of the olfactory lobes and brain in a consistent manner. Blood samples (150–200 μl) were taken via the carotid cannula pre-dose and at 0 (immediately post-dose), 1, 2, 3, 5, 7, 8, 9, 10, 11, 15, 20, 25 and 30 min post-dose.
For all samples, blood was collected into Microtainers which were pre-coated with heparin and contained a polymer plug that allowed plasma separation by density-gradient centrifugation (13,000 rpm for 5 min). Following removal of the brain and olfactory lobes, as described above, the brain was divided into four sections, with the aid of a brain slicer as described previously by Charlton et al. (8). The first section was made across the transverse sinus, followed by two further cuts at 5 mm intervals towards the anterior of the cerebral hemispheres.
Drug concentration in the olfactory lobes and brain tissue samples were adjusted to compensate for drug carried within cerebral blood vessels, which is approximately 15 μl/g of tissue. This work is part of a series of experiments covering a range of doses, therefore all blood and tissue sample concentrations were normalised to a dose of 100 μg to simplify comparison. AUC values for the concentration-time curves (0–60 min) were calculated using GraphPad Prism version 4.03 for Windows (GraphPad Software, USA). All AUC values and AUCCNS/AUCplasma ratios for nasal and intravenous administrations were calculated per individual animal before determining the mean value. The AUCCNS/AUCplasma ratios were compared for intranasal delivery and intravenous delivery. As discussed by Van der Berg et al. (9), if the value of the ratio after intranasal delivery is smaller than after intravenous delivery this indicates a mainly systemic transport of the drug to the CNS. If the value of the ratio after intranasal delivery is larger than after intravenous delivery this indicates a mainly direct transport of the drug from the nose to the CNS.
In order to define the proportion of the drug being transported directly from the nose to the brain and not via the systemic circulation after nasal absorption, the direct transport percentage (DTP) was calculated (10).
where AUCX is the fraction of the CNS AUC contributed by drug in the systemic circulation passing the BBB.
Multiplying the individual AUC values by the total blood volume or weight of tissue gives the cumulative amount of drug delivered to the blood or brain tissue. This represents the ‘availability’ of drug. The total blood volume of a rat is 65 ml/kg (11). The average weight of each brain section was determined from the weights of the experimental samples.
LC/MS/MS
All glycine receptor antagonist drugs were expected to be >99% protein bound in the blood. Therefore, the drugs were extracted with the proteins from the plasma by precipitation using 3:1 acetonitrile. Drug was extracted from brain samples using methanol/ammonium acetate + 0.1% formic acid (80:20; 1 ml for olfactory lobes, and 3 ml for brain samples). The protein samples were then homogenised, followed by centrifugation. The supernatant, which contained the drug, was isolated and blown down to dryness. The sample was re-suspended in methanol/water 50:50 prior to analysis.
An API 2000™ LC/MS/MS triple quadrupole system (Applied Biosystems/MDS Sciex, Warrington, Cheshire, UK) was used to analyse the samples. The samples were first separated using liquid chromatography. The experimental setup at ambient temperature was:
The total run time was 5 min.
The samples were then introduced into the mass spectrometer at a flow rate of 0.25 ml/min. Multiple reaction monitoring (MRM) was used for quantification. All compounds were run in positive ion mode.
MRM transitions used for each drug were:
Samples of control plasma and homogenised control brain tissue were spiked with known quantities of drug to produce standards for calibration.
Drug concentration in olfactory lobe and brain tissue samples were adjusted to compensate for drug carried within cerebral blood vessels as above. The ‘availability’ of drug was calculated as described above. The average weight of each brain section was determined from the weights of the experimental samples.
Liquid Scintillation Counting
For 14C-GR138950L, drug concentration in blood or brain samples was determined by liquid scintillation using OptiPhase scintillation cocktail and a 1219 RackBeta ‘Spectral’ Liquid Scintillation Counter (EG & G Wallac, Milton Keynes, UK). Plasma analysis was performed using 80 μl of plasma with 10 ml of OptiPhase scintillation cocktail in a glass scintillation vial. Prior to analysis, tissue samples were treated using Optisolv (1 ml per 200 mg of tissue) and left to stand until fully solubilised (approximately 3 weeks) prior to analysis by liquid scintillation. The olfactory lobe samples were processed by analysing 200 μl of solubilised tissue/Optisolv with 10 ml of OptiPhase in a glass scintillation vial. For brain tissue samples, 1 ml of sample was added to 10 ml of OptiPhase in a glass scintillation vial. Before measuring radioactive content, samples were stored in the dark at 3–5°C for a minimum of 6 h to minimise chemiluminescence from the scintillation cocktail. Samples of control plasma and brain tissue were spiked with known quantities of drug to produce standards for calibration. Clean solubilised tissue samples and spiked controls were used as background and control samples.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism version 4.03 for Windows utilising two-way ANOVA followed by a Bonferroni multiple comparison test; the two factors being delivery route and time and the interaction component being used to determine a significant difference.
Autoradiography
14C-GR138950L was dissolved in water for injection at a concentration of 2.8 mg/ml. A larger dose was used for the autoradiography studies to aid visualisation of the drug in tissue sections. Five pairs of rats were administered intranasal doses of 0.16 ml/kg as described above, and were killed at 1, 5, 10, 30 or 60 min post-dose.
The carcasses were frozen in a heptane-dry ice bath and then embedded in 2% carboxymethylcellulose at −20°C. Coronal sections (40 μm thick) were taken through the olfactory apparatus, nasal cavity and brain using a cryomicrotome (Jung Cryomacrocut, Leica UK) and mounted on adhesive tape. The sections were allowed to dry by sublimation at −25°C for at least 96 h. When dry, the sections and standards were mounted onto carbon-14 sensitive imaging plates (Fuji Film Company, Japan). The imaging plates and sections were enclosed in exposure cassettes and allowed to expose in a lead safe at room temperature for up to 10 days. After exposure, the tape and sections were removed, and the plate was imaged using a BAS-1500 Bio-imaging analyser (Fuji Film Company, Japan). Images were read using BAS-reader version 2.9 and stored using TINA version 2.08e (Raytest Isotopenmebgerate GmbH).
Micro-autoradiography
Three pairs of rats were prepared and dosed as per the method described above, and additionally the left and right common carotid arteries were cannulated to allow perfusion of formalin solution through the head and neck post-mortem. The animals were killed at 1, 5 and 10 min post-dose. After death, the cardiac ventricles were severed to allow ex-sanguination and normal saline was perfused into the coronary arteries at a rate of 6 ml.min−1. When the perfusate from the heart was clear of blood, formalin solution was perfused into the carotid arteries; the head was then removed and was immersed in formalin until further processing. The samples were submerged in EDTA solution for 4 weeks to decalcify and soften the bone prior to sectioning. Following this step, a 4–5 mm thick oblique lateral slice was selected that could be subsequently sectioned to show the nasal cavity, cribiform plate and the olfactory lobes. The selected tissue slices were processed to paraffin wax using a Tissue Tek® Vacuum Infiltration Processor (Bayer plc, UK) that exposed the selected tissues to a sequence of graded alcohols (70%, 90%, 3 × 100% IMS), followed by two changes of xylene and four changes of paraffin wax. Each reagent exposure was carried out using an alternating vacuum and pressure sequence. At the end of this process the tissues were orientated and embedded in molds of molten paraffin wax to provide subsequent support. When solidified, the blocks were cooled and sectioned at 5 μm using a rotary microtome (Leica, UK). The sections were floated onto a water bath at 46°C to flatten the sections prior to picking them up onto an adhesive coated glass microscope slide. The slides were dried overnight in an incubator at 37°C.
To achieve horseradish peroxidase (HRP) immunostaining of nerve cells, the sections were first soaked in xylene to remove the paraffin wax. This was followed by soaking the sections in IMS to remove the xylene, after which the sections were washed in distilled water. The sections were microwaved in pH 6 citrate buffer for 20 min and then cooled by adding cold distilled water to the heat induced epitope retrieval (HIER) buffer. The samples were treated with 3% H2O2 to block endogenous peroxidase activity, followed by immersion in 5% milk powder (diluted in PBS and 0.1% Triton X-100) to block non-specific proteins. The samples were incubated (room temperature, moist chamber, 60 min) with a primary monoclonal S-100 antibody that was diluted with 3% normal serum plus 0.1% Triton X100. The samples were washed in several changes of PBS and then incubated with biotinylated secondary anti-species (mouse) antibody (room temperature, moist chamber, 60 min). After binding the secondary antibody, the sections were washed in several changes of PBS, and then treated with streptavidin-HRP solution, which binds to the biotinylated secondary antibody. The sections were then incubated with diaminobenzidine (DAB), which forms a brown coloured insoluble precipitate, thus enabling visualisation of the bound antibody. Finally, the sections were dehydrated with IMS, cleared in xylene and mounted.
To achieve autoradiographic visualisation, the slides were dipped in Ilford K5 and L4 emulsions, which were maintained as a liquid at 40°C and to which was added 0.1% Triton X100 detergent to reduce surface tension and enable a more even film to be achieved. The back of each slide was wiped and the slides were then dried vertically in a damp atmosphere and in the dark for 2 h. They were then placed in light-tight boxes, sealed with tape and stored in a freezer at −15°C until developed. This entire stage was carried out under dark room conditions using a safe light with filter. The slides were developed in Ilford Phenisol™ (diluted 1 + 4 with distilled water) for 4 min at 18°C and then washed in Ilfostop Pro™ (diluted 1 + 19 with distilled water) for 1 min to stop the development process. The slides were then washed in distilled water for 1 min and fixed in Ilford Hypam™ (diluted 1 + 4 with distilled water) for 4 min to remove unexposed silver. They were then washed in tap water for 10 min, prior to counterstaining the nuclei with Mayer’s haemalum for 1–2 min. The sections were “blued” in tap water for 5 min, dehydrated in ascending concentrations of alcohol, cleared in xylene and mounted. The sections were examined using light microscopy.
RESULTS
Quantitative Tissue Distribution
Initial Experiments—Glycine Receptor Antagonists
The three glycine antagonists showed differences in absorption and distribution after nasal administration. GW468816 was rapidly absorbed into the blood from the nose, reaching a peak concentration at 5 min post-dose that was of a similar concentration to that following intravenous dosing at the same time point (Table II). Low drug levels were recorded in the olfactory lobes at 1 min after intranasal dosing, but no drug was detected at 3 and 5 min. Drug levels in the cerebral hemispheres and cerebellum were negligible throughout the study period. After intravenous administration, no drug was detected in the brain; indicating that the drug transported to the brain following intranasal administration was transported via the olfactory system. At 60 min post-dose, the bioavailability of the drug following nasal administration (AUC0–60) was 53.2% and 0.1% of the dose was transported into the brain. The ratio of drug absorbed into the plasma compared to the brain was 301:1. The ratio AUCCNS/AUCplasma following intranasal delivery was 0.67 compared to 0 (for calculation purposes set at 0.0001) for intravenous delivery and the DTP 99.9%, clearly showing that the drug was transported directly from the nose to the CNS.
Intranasal administration of GV196771 resulted in a plasma t max at 10 min (Table III). A steady increase in drug concentration was observed in the olfactory lobes over the duration of the study. No drug was observed in the rest of the brain. Bioavailability of the drug following nasal administration was 13.5% at 60 min post-dose and 0.05% of the dose was transported into the brain. The ratio of drug absorbed into the plasma compared to the brain was 238:1. The ratio AUCCNS/AUCplasma following intranasal delivery was 0.85 compared to 0.03 for intravenous delivery, and the DTP 96.7%, clearly showing that the drug was mainly transported directly from the nose to the CNS, with a slight contribution from drug transported to the brain from the systemic circulation.
Administration of GV150526 produced a peak concentration in the olfactory lobes at 1 min post-dose after intravenous and nasal administration, but the intravenous peak was sustained for twice as long, the peak durations being 5 and 10 min, respectively. Over 1 h following intravenous dosing, 26% more drug was delivered to the olfactory lobes compared to that after intranasal dosing (Table IV). Despite the relatively high drug uptake into the olfactory lobes after intravenous dosing, only a small amount of drug was detected at 60 min in the rest of the brain. At 60 min post-dose, the bioavailability following nasal administration was 38.0% and 0.05% of the dose was transported into the brain. The ratio of drug absorbed into the plasma compared to the brain was 349:1. The ratio AUCCNS/AUCplasma following intranasal delivery was 0.42 compared to 0.20 for intravenous delivery, indicating that, according to the criteria described above, the drug was mainly transported directly from the nose to the CNS. However, the DTP was 52.0% indicating that a major contribution to the brain content of drug after nasal administration was transported into the brain via the systemic circulation.
Detailed Experiments—Angiotensin Antagonist
Intranasal administration resulted in rapid delivery of GR138950 to the olfactory lobes, with a mean concentration of 0.106 μg/g measured at 1 min post-dose (Fig. 2). A peak drug concentration (Cmax) of 0.15 μg/g was observed at 5 min post-dose (Table V); followed by a reduction in drug concentration up to 15 min, and then an increase at 30 min post-dose. The concentration profile for drug in the blood showed a C max of 0.13 μg/g at approximately 9–10 min post-dose, after which the concentration gradually decreased. At 1 min after intranasal dosing, drug concentration in the olfactory lobes was over five times higher than in the blood (0.11 μg/g vs 0.02 μg/g).

Concentration profiles of GR138950 in the olfactory lobes and plasma over 30 min following intranasal and intravenous dosing. Error bars represent the SEM, n = 3.
Intravenous dosing produced a typical drug-plasma concentration profile with first order elimination. Drug concentration in the olfactory lobes was relatively constant between 1 to 30 min post-dose —0.03 μg/g at 1 min and a C max of 0.04 μg/g at 5 min. The C max in plasma was 3.25 μg/g, measured immediately after dosing.
Intranasal administration resulted in consistently higher drug levels in the olfactory lobes compared to intravenous administration. Two-way ANOVA analysis indicated that the difference in concentration profiles was significant over the first 15 min (p = 0.0033) and at the 30 min time-point (p < 0.01).
The ratio of drug concentration in the olfactory lobes over the drug concentration in the plasma illustrates the difference in drug distribution between the two administration routes. The highest ratio after intranasal administration occurred at 1 min post-dose (5.01 μg.g−1/μg.ml−1), which highlights the rapid speed of drug movement from the nasal cavity to the brain. At this time the intranasal ratio was 400 times greater than the ratio associated with intravenous dosing (0.12 μg.g−1/μg.ml−1; p < 0.0001).
Analysis of distribution of GR138950 throughout the whole of the brain showed that intranasal administration resulted in a significantly higher drug concentration in the olfactory lobes at 1 min post-dose compared to the other parts of the brain (Fig. 3). At 5 min drug levels had increased in all regions of the brain, with the highest levels recorded in the anterior half of the brain, which is close to the olfactory system, thus supporting the theory that drug is entering the cranial cavity from the nasal cavity and then diffuses throughout the CNS. At this time (5 min) the mean drug concentrations in each of the brain sections were higher after intranasal administration than intravenous administration. Drug concentrations in the olfactory lobes and cerebral hemispheres were lower or relatively unchanged at later time points, whereas the average drug concentration measured in the cerebellum reached a maximum of 0.09 μg/g at 10 min before then decreasing (Table VI). Following intravenous dosing, GR138950 concentrations in the cerebral hemispheres and cerebellum at 1 min (t max) were comparable to that seen with intranasal dosing. A C max of 0.04 μg/g in the olfactory lobes was measured at 5 min. Drug concentrations throughout the olfactory lobes and brain sections were lower at 10 min, and were relatively unchanged at later time-points.

Concentration of GR138950 in sections of the brain at specific time points after intranasal or intravenous administration. Error bars represent the SEM, n = 3.
Based on the calculated availability data, intranasal dosing resulted in 159% more drug being delivered to the brain as a whole compared to intravenous dosing over the duration of the study (Table V, Fig. 4); whereas the bioavailability of the drug after intranasal dosing was 23%. 0.7% of the dose was delivered to the brain. Autoradiography indicated that drug availability in the CSF was approximately 0.23 μg min over 30 min, which is comparable to that determined for drug in the olfactory lobes. Thus, accounting for drug in brain tissue and CSF, approximately 0.8% of GR138950 dosed nasally was delivered to the brain. It should be noted that especially in the cerebellum the availability of the drug was significantly higher after nasal administration than after intravenous (Fig. 4). The ratio AUCCNS/AUCplasma following intranasal delivery was 1.28 compared to 0.07 for intravenous delivery, clearly showing that the drug was transported directly from the nose to the CNS. This was supported by the DTP of 94.6%, showing that for this drug applied nasally there was only a minor contribution from systemic transport to the brain.

Availability of GR138950 in brain regions over 30 min after intranasal and intravenous dosing. This value is calculated by multiplying AUC of drug concentration (μg/g) over time (min) by weight of brain section (g); n = 3.
Autoradiography
Autoradiographs of freeze-dried tissue sections presented in Fig. 5 show the distribution of GR138950 in the nasal cavity, brain and in the CSF around the brain over 60 min following intranasal dosing. Drug distribution was comparable for each of the two rats at any given time-point. The clearest image from either rat for each time-point is presented. All concentrations given are rounded to the nearest standard concentration, unless otherwise stated. In the images representing 1, 5, 10 and 60 min, the nasal cavity appears black due to the residence of the dose formulation. The section representing 30 min was cut through a plane above the nasal cavity.

Autoradiographs of freeze-dried tissue sections showing drug distribution in the nasal cavity and brain at 1, 5, 10, 30 and 60 min post-dose. The nasal cavity has a drug concentration of approximately 2.8 mg/g (based on dose concentration), and is seen as being very dark. The 30 min section was taken through a plane that did not pass through the nasal cavity. Drug concentration in the brain increased over time, with a concentration wave moving posteriorly from the olfactory lobes. A higher concentration of drug was also observed in the CSF compared to brain tissue.
At 1 min the olfactory lobes have a dark outline around the anterior surface and the sides, which can be attributed to drug in the CSF. The drug concentration at the anterior edge of the olfactory lobes was 0.8 μg/g, and was approximately 0.3 μg/g at the sides. The drug concentration within the olfactory lobes ranged from 0.84 μg/g at the anterior to 0.11 μg/g at the posterior edge. The cerebral hemispheres appear to contain GR138950 in the anterior quarter only, and the concentration was less than 0.01 μg/g. At 5 min, the drug concentration in the CSF around the olfactory lobes had increased to 3.2 μg/g; and extended back to the mid-line of the cerebral hemispheres at a concentration of 0.1 μg/g. The drug concentration in the anterior half of the olfactory lobes was 0.84 μg/g, decreasing to 0.2 μg/g at the posterior. The anterior section of the cerebral hemispheres contained drug at a concentration of 0.05 μg/g. At 10 min, GR138950 was detectable in the CSF surrounding the brain; the concentrations ranging from 3.2 μg/g around the olfactory lobes to 0.3 μg/g over the paraflocculus (a part of the cerebellum). Drug concentrations were 0.3–3.2 μg/g in the olfactory lobes, 0.1–0.3 μg/g in the cerebral hemispheres and 0.1 μg/g in the cerebellum. The section used to show drug distribution at 30 min was cut through a plane above the nasal cavity, at the top of the olfactory lobes and cerebellum. This enabled visualisation of CSF around the midline between the cerebral hemispheres and above the cerebellum. The drug concentration over the cerebral hemispheres was 0.06 μg/g, with concentrations rising to 0.8 μg/g at areas of CSF pooling, such as between the olfactory lobes and cerebral hemispheres, and over the pineal body and flocculus. The distribution and concentrations of drug at 60 min were very similar to those recorded at 10 min post-dose.
By averaging the concentration of the drug in the CSF between the olfactory lobes and the cerebellum at each time-point, and then plotting it against time and calculating the AUC gave a value of approximately 1.56 min μg/g. The CSF volume in the rat is approximately 150 μl (12); therefore the calculated availability was approximately 0.23 μg min over 30 min.
Micro-Autoradiography
HRP immunostaining and DAB have been used to visualise nerve tissue (coloured brown) in the olfactory apparatus (Fig. 6a). Autoradiographic visualisation was used to illustrate distribution of GR138950. Figure 6a shows a section that details the lumen of the nasal cavity, the olfactory epithelium lining the nasal cavity, olfactory nerves and bone comprising the cribiform plate. A gap in the cribiform plate is clearly visible, permitting passage of the olfactory nerve bundles from the nasal area to the olfactory lobes. The olfactory epithelium was found to contain a high concentration of GR138950. This was expected due to its intimate contact with the formulation. Drug was shown to be distributed throughout the olfactory nerves and the olfactory lobes; whereas the cribiform plate, which is bone, had a negligible drug content.

a Micro-autoradiography section showing the physiological positioning of the olfactory nerve bundle (leading to the olfactory lobes), cribiform plate and nasal lumen. The section was immunostained with HRP and incubated with DAB to achieve visualisation (brown colouration) of nerve tissue; magnification ×50. b Autoradiographic visualisation revealed the presence of drug, represented by dark spots. There was significantly more drug visualised in the olfactory nerve region compared to the cribiform plate. Magnification ×1000.
At higher magnification (Fig. 6b), autoradiographic visualisation shows that a significant amount of drug was in the olfactory nerve bundle, confirming that GR138950 exploits this route of transport between the nasal cavity and the brain. Drug distribution was uniform throughout the olfactory nerve bundles at 1 min post-dose, and measured levels of GR138950 were comparable at 1, 5 and 10 min. Consequently, it was not possible to follow a concentration front along the olfactory nerve tract and thereby attempt to calculate the rate of drug movement. The relative drug level in the olfactory nerve bundles compared to the cribiform plate was quantified by determining the density of autoradiograph ‘spots’; however, actual drug concentration could not be determined using this method. On average, the distribution of GR138950 in the olfactory nerve tissue was more than eight times greater than in the cribiform plate (p < 0.0001, t test).
DISCUSSION
The initial experiments on the glycine receptor antagonists showed that although all of the compounds were of similar size and lipophilicity, and were administered at the same dose, they showed variation in absorption profiles. The main difference between the compounds was the pKa values, with the GW468816 and GV150526 being zwitterionic (pKa 1.90, 4.22 and 3.77, 13.22, respectively) and GV196771 a weak acid (pKa 3.65) (pKa values were calculated using ACD/pKa DB software (Advanced Chemistry Development Inc. (ACD/Labs), Toronto, Canada). This means that at pH 6 (water for injection) the GW468816 and the GV196771 are negatively charged whereas GV150526 has both negative and positive charges (i.e. it exists as a zwitterion) resulting in partial intramolecular charge neutralisation. GW468816 and GV196771 showed the highest AUCCNS/AUCplasma ratio and the highest DTP, indicating clear direct nose to brain transport and only minimal contribution from the nasal-systemic-brain pathway. GV150526 (zwitterionic at pH 6.0) was the only one of the three molecules that resulted in significant drug distribution in the cerebral hemispheres and cerebellum in addition to the olfactory lobes; however, intravenous administration resulted in greater delivery to the olfactory lobes. This was supported by only a small difference in AUCCNS/AUCplasma ratios and a DTP of 52.0%. It was to be expected that charged molecules would show a lower nasal absorption in to the systemic circulation as compared to “neutral” molecules and hence a higher transport to the brain. This has previously been suggested by Sakane et al. (13). It should be noted, however, that replicates were not performed when testing these molecules, so the possibility of experimental and biological variation cannot be dismissed. These results suggest that drug delivery to the CNS via intranasal administration may be prone to significant variation between different molecules due to their molecular characteristics. The use of 2% DMSO as solvents for GV196771 and GV150526 may have had a small effect on the cilia beat frequency (CBF) of the nasal epithelial cells and hence on the nasal absorption of the drugs (14,15). However, it has been shown that DMSO showed no damaging effect of cell DNA in cultures of human nasal epithelial cells (16). In the present studies it is suggested that the effects of DMSO would have been negligible since very different absorption patterns were found for the two drugs dissolved in 2% DMSO.
The quantitative determination of tissue distribution demonstrated that intranasal dosing resulted in delivery of the angiotensin antagonist GR138950 to the olfactory lobes within 1 min, with over 3.5 times more drug in the olfactory lobes after intranasal dosing compared to intravenous dosing, despite only 4% of the intranasal dose having been absorbed from the nasal cavity at this time point. Over 30 min, five times more drug was delivered to the olfactory lobes after intranasal dosing compared to intravenous, and the C max was four times greater. Furthermore, the AUCCNS/AUCplasma ratio was significantly higher (1.28) for the intranasal dosing than for the intravenous dosing (0.07) and the DTP 94.6% showed direct nose to brain transport with only a small contribution from systemically absorbed drug. Hence, this drug behaved very similarly to the compounds GW468816 and GV196771, and at pH 6.0 would be considered to have a similar net negative charge.
The olfactory lobes have a very close proximity to the point of drug entry into the cranial space, so it is not surprising that there was a high drug concentration in this area of the brain at all time-points. The cerebellum is farthest from the olfactory system, hence the later t max; however, it is believed (due to the rapid appearance in the CNS) that GR138950 was primarily absorbed via a paracellular route and was then distributed in the CSF—the volume of CSF is greater around the cerebellum than the cerebral hemispheres, therefore it is likely that there was a significant local pool of available drug that could penetrate into the cerebellum, resulting in higher drug levels compared to that in the cerebral hemispheres.
The autoradiography data indicated that GR138950 was in the CSF at 1 min post-dose, and had distributed past the rear edge of the olfactory lobes. Drug concentration in the CSF at 5 min post-dose was nearly four times higher than the concentration recorded in the olfactory lobe tissue. At 10 min the concentration in the olfactory lobes was comparable to the concentration in the surrounding CSF; but the concentration in the CSF around the cerebellum was three times higher than in the contiguous tissue. This data suggests that GR138950 was primarily distributed in the CSF. Autoradiography data shows higher drug concentrations in the olfactory lobes compared to those determined by quantitative tissue distribution, but levels in the cerebral hemispheres and cerebellum were comparable.
GR138950 concentrations in the olfactory lobes and CSF were constantly higher than in the blood over the first 5 min after intranasal administration, and the C max was earlier and larger than the peak in plasma, indicating that drug was moving into the brain from the nasal cavity via a pathway that did not involve the drug being carried by the blood. It is suggested that GR138950 moved through the olfactory pathway. The arrangement of olfactory nerve bundles generally channels drug towards the olfactory lobes, which explains why higher drug concentrations were recorded in the olfactory lobes compared to other sections of the brain; however, the perineural sheaths around the cells are integrated with the subarachnoid and thus drugs moving via a paracellular route pass into the subarachnoid space which is filled with CSF (17). The drug is then distributed throughout the CSF, which circulates around the brain. Autoradiography data showed that GR138950 was in the CSF and olfactory lobes at 1 min post-dose, and the drug concentration in the CSF was higher than in the olfactory lobes. This pattern of distribution indicates that GR138950 moved via the paracellular pathway. FITC-dextran molecules of M w 18.9 kDa have been shown to pass along paracellular pathways to the brain, therefore it is possible that GR138950 (Mw 611) was absorbed via the same route (18).
However, the rise in drug concentration in the olfactory lobes at 30 min post-dose may suggest that GR138950 is also transported by the slower transcellular route. Intranasal administration of dopamine to mice has resulted in a peak concentration in the olfactory lobes at 30 min, and a second peak at 4 h post-dose (19). The authors suggested that the first peak was due to dopamine transport via the paracellular pathway and the second peak to transaxonal transport. It is reasonable to assume that molecules are able to utilise both transport pathways simultaneously. Eriksson et al. have investigated the distribution of four carboxylic acids following intranasal administration (20). Autoradiography showed that the test substances were distributed throughout the olfactory nerve apparatus and the glomerular layer (outer layer) of the olfactory lobes at 0.5–4 h post-dose. There also appeared to be a high concentration of the test molecules in the CSF surrounding the olfactory lobes. The authors commented that the data did not allow determination of paracellular or transcellular transport.
Movement of GR138950 through the olfactory nerve system is supported by the micro-autoradiography data, which illustrates the direct pathway between the nasal cavity and the olfactory lobes, and showed a significant amount of drug distributed throughout the olfactory nerve apparatus. However, due to the two dimensional nature of the sections it is difficult to determine if the drug molecules are distributed in the nerve cells or around the exterior of the cells, which relates to paracellular or intracellular movement of the drug. Drug distribution appears to be uniform throughout the nerve bundles, therefore it is reasonable to assume that GR138950 travels via paracellular routes in addition to penetrating into the olfactory nerve cells. Thorne et al. (4) showed in an investigation of the nasal distribution of IGF-1 by high resolution phosphor imaging of tissue sections that the drug was transported to the CNS via two routes, one associated with the peripheral olfactory system connecting the nasal passages with the olfactory bulb and also the peripheral trigeminal system connecting the nasal passages with the brainstem and spinal cord region. They also found that the distribution within the olfactory bulb displayed a decreasing gradient with distance from the nasal passages. The techniques used did not allow for identification of the exact pathway through the neuronal pathways but due to the rapid appearance in the olfactory bulb it was suggested that the route was extracellular.
Based on the quantitative tissue distribution data, the proportion of the dose delivered to the brain was a relatively small amount, however, the small size of the brain compartment means that a small amount of drug can produce a relatively high drug concentration. Intranasal drug delivery may allow the use of lower doses that will achieve therapeutic levels in the CNS with lower systemic levels compared to traditional dosing routes. This may be of great benefit for drugs that target the brain but cause systemic and peripheral toxicity. However, the relatively low levels of drug observed in the brain and CNS indicate that intranasal delivery is more suited to potent molecules to ensure a therapeutic response unless means are taken to improve delivery across the olfactory epithelium into the CNS (8).
CONCLUSIONS
Studies with rats have demonstrated that intranasal administration of all of the model drugs, GR138950, GW468816, GV196771 and GV150526, resulted in greater delivery to the brain, and particularly the olfactory lobes, compared to intravenous administration. Intranasal administration of GR138950 resulted in over 3.5 times more drug in the olfactory lobes at 1 min post-dose compared to intravenous administration; and 5 times more drug was delivered to the olfactory lobes over 30 min. Micro-autoradiography has shown GR138950 throughout the olfactory nerve apparatus. Autoradiography has illustrated drug distribution throughout the brain and CSF, with drug concentrations in the CSF being equal or higher than in the brain tissue over 30 min post-dose.
The appearance of GR138950 in the olfactory nerves, olfactory lobes and CSF at 1 min post-dose leads us to conclude that the drug is primarily moving via paracellular pathways, however, it is reasonable to assume that transcellular transport also occurs. The rapid appearance of drug in the CNS after intranasal administration could be very important for the treatment of crisis conditions, such as pain and panic attacks, which would benefit from a fast acting treatment.
It has been determined that, over the 30 min period after nasal administration of GR138950, approximately 0.8% of the dose moved into the brain and CSF via the olfactory pathway, with 22.8% being absorbed into the blood. The proportion of dose delivered to the CNS is relatively small, though it is significantly higher compared to intravenous delivery. Hence, it is suggested that this delivery route is better suited for more potent therapeutic molecules.
References
W. M. Pardridge. Non-invasive drug delivery to the human brain using endogenous blood-brain barrier transport systems. Pharm. Sci. Technol. To. 2:49–59 (1999).
P. A. Hilger. Applied anatomy and physiology of the nose. In: Otolaryngology. A textbook of ear, nose and throat diseases. W. B. Saunders, Philadelphia, pp177–195 (1989).
L. Illum. Is nose-to-brain transport of drugs in man a reality. J. Pharm. Pharmacol. 56:3–17 (2004).
R. G. Thorne, G. J. Pronk, V. Padmanabhan, and W. H. Frey II. Delivery of insulin-like growth factor-1 top the rat brain and spinal cord along olfactory and trigeminal pathways following intranasal administration. Neuroscience 127:481–496 (2004).
D. B. Judd, M. D. Dowle, D. Middlemiss, D. I. Scopes, B. C. Ross, T. I. Jack, M. Pass, E. Tranquillini, J. E. Hobson, T. A. Panchal, P. Stuart, J. M. S. Paton, T. Hubbard, A. Hilditch, G. M. Drew, M. J. Robertson, K. L. Clark, A. Travers, A. A. E. Hunt, J. Polley, P. J. Eddershaw, M. K. Bayliss, G. R. Manches, M. D. Donelly, D. G. Walker, and S. A. Richards. Bromobenzofuran-based non-peptide antagonists of angiotensin LL: GR138950, a potent antihypertensive agent with high oral bioavailability. J. M. Chem. 16:3108–3120 (1994).
S. Hirai, T. Yashiki, T. Matsuzawa, and H. Mima. Absorption of drugs from the nasal mucosa of rat. Int. J. Pharm. 7:317–325 (1981).
A. N. Fisher, K. Brown, S. S. Davis, G. D. Parr, and D. A. Smith. The nasal absorption of sodium cromoglycate in the albino-rat. J. Pharm. Pharmacol. 37:38–41 (1985).
S. T. Charlton, S. S. Davis, and L. Illum. Nasal administration of an angiotensin antagonist in the rat model: Effect of bioadhesive formulations on the distribution of drugs to the systemic and central nervous systems. Int. J. Pharm. 338:94–103 (2007).
M. P. Van der Berg, P. Merkus, S. G. Romeijn, J. Coos Verhoef, and F. W. H. M. Merkus. Hydroxycobalamin uptake into the cerebrospinal fluid after nasal and intravenous delivery in rats and humans. J. Drug Target. 11:325–331 (2003).
Q. Zhang, X. Jiang, W. Jiang, W. Lu, L. Su, and Z. Shi. Prepration of nimodine-loaded microsemulsion for intranasal delivery and evaluation of the targeting efficiency to brain. Int. J. Pharm. 275:85–96 (2004).
Laboratory Animal Science Association. Collection of Blood Samples. LASA, Tamworth, UK, 1998.
H. Davson, K. Welch, and M. B. Segal. Secretion of cerebrospinal fluid. In: The Physiology and Pathophysiology of the Cerebrospinal Fluid, Churchill Livingstone, London (1987).
T. Sakane, M. Akizuki, S. Yamashita, H. Sezaki, and T. Nagai. Direct drug transport from the rat nasal cavity to the cerebrospinal fluid: the relation to the dissociation of the drug. J. Pharm. Pharmacol. 46:378–379 (1994).
W. M. Boek, K. Graamans, and E. M. Huizing. Ciliary beat frequency of human SPHENOID SINUS MUCOSA AFTER CRYPRESERVATION. Eur. Arch. Otorhinolaryngol. 255:135–137 (1998).
R. U. Agu, M. Jorissen, T. Willems, R. Kinget, and N. Verbeke. Mechanistic appraisal of the effects of some protease inhibitors on ciliary beat frequency in a sequential cell culture system of human nasal epithelium. Eur. J. Pharm. Biopharm. 55:283–289 (2003).
N. H. Kleinsasser, J. Juchhoff, B. C. Wallner, A. Bergner, U. A. Harreus, F. Gamarra, M. Buhrlen, R. M. Huber, and A. W. Rettenmeiser. The use of mini-organ cultures of human upper aerodigestive tract epithelia in ecogenotoxicology. Mut. Res. 561:63–73 (2004).
W. M. Faber. The nasal mucosa and the subarachnoid space. Am.J.Anat. 62:121–148 (1937).
T. Sakane, M. Akizuki, Y. Taki, S. Yamashita, H. Sezaki, and T. Nadai. Direct drug transport from the rat nasal cavity to the cerebrospinal fluid: The relation to the molecular weight of drugs. J. Pharm. Pharmacol. 47:379–381 (1995).
M. Dahlin, U. Bergman, B. Jansson, E. Bjork, and E. Brittebo. Transfer of dopamine in the olfactory pathway following nasal administration in mice. Pharm.Res. 17:737–742 (2000).
C. Eriksson, U. Bergman, A. Franzen, M. Sjoblom, and E. B. Brittebo. Transfer of some carboxylic acids in the olfactory system following intranasal administration. J. Drug Target. 7:131–142 (1999).
Acknowledgements
Thanks are extended to GlaxoSmithKline for the provision of a Case award to support this work.
Author information
Authors and Affiliations
Corresponding author
Additional information
To be submitted to: Pharmaceutical Research
Rights and permissions
About this article
Cite this article
Charlton, S.T., Whetstone, J., Fayinka, S.T. et al. Evaluation of Direct Transport Pathways of Glycine Receptor Antagonists and an Angiotensin Antagonist from the Nasal Cavity to the Central Nervous System in the Rat Model. Pharm Res 25, 1531–1543 (2008). https://doi.org/10.1007/s11095-008-9550-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-008-9550-2