
Abstract
microRNAs are single-stranded noncoding RNA sequences of 18–24 nucleotide length. They play important role in post-transcriptional regulation of gene expression. Last decade witnessed immense research in microRNA identification, prediction, target identification, and disease associations. They are linked with up/down regulation of many diseases including cancer. The accurate identification of microRNAs is still complex and time-consuming process. Due to the unique structural and sequence similarities of microRNAs, many computational algorithms have been developed for prediction of microRNAs. According to the current status, 28645 microRNAs have computationally discovered from the genome sequences, and have reported 1961 human microRNAs (miRBase version 21, released on June 2014). There are several computational tools available for predicting the microRNA from the genome sequences. We have developed a support vector machine-based classifier for microRNA prediction. Top ranked 19 sequence, structural, and thermodynamic characteristics of validated microRNA sequence databases are employed for building the classifier. It shows an accuracy of 98.4 % which is higher than that of existing SVM-based classifiers such as Triplet-SVM, MiRFinder, and MiRPara.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
RNAs are single-stranded long sequences that are formed from the DNA sequences through transcription process. With the help of hydrogen bonding between the bases, a nucleotide sequence of RNA could form a nonlinear structure, called secondary structure [15, 16]. The components of a secondary structure can be classified as stem loop (hairpin loop), bulge loops, interior loops, and junctions (Multi-loops) [8]. Functionally, RNAs are responsible for protein synthesis and RNAs such as messenger RNA (mRNA), ribosomal RNA (rRNA), and transfer RNA (tRNA) have its own roles in this process [5]. A family of noncoding RNA, around 22 nt long, found in many eukaryotes including humans is called microRNA. The process of formation of microRNAs has many stages, initially longer primary transcript (pre-microRNA) is formed, which in turn converted into a pre-microRNA, and processes mandate presence of ribo-nucleolus Drosha, Exportion-5 [4, 9]. The pre-microRNAs are characterized by a hairpin-like structure. microRNAs play different roles in gene regulation by binding to specific sites in mRNA and causes translational repression or cleavage [22]. Due to the change in gene expression, microRNAs role as suppressor /oncogenes in different cancers such as colon, gastric, breast, and lung cancers are proved [3]. microRNA also helps for the proper functioning of brain and nervous system, and have regulatory roles in several other diseases like deafness, Alzheimers disease, Parkinson disease, Down’s syndrome, and Rheumatoid arthritis [1, 12]. microRNA-based cancer detection and therapy is underway [18]. As the in vivo identification of microRNAs is time consuming and complex, many computational tools had been developed to predict most provable microRNA sequences. The methods employed for computational prediction of microRNAs vary from search in conserved genomic regions, measuring structure, sequence, thermodynamic characteristics of RNA secondary structures, to properties of reads of next-generation sequencing data, together with advances in machine learning techniques [19].
Comparing DNA sequences of related species for conserved noncoding regions having regulatory functions were the initial approach employed for microRNA prediction. miRScan [11] and miRFinder [20] are examples of such tools. The sequence characteristics, especially the properties of blocks of three of consecutive nucleotides, namely triplet structure along with other parameters are used in Triplet-SVM [6], MiPred [17], and MiRank [25]. MiRank, developed by Yunpen et.al, works with a ranking algorithm based on random walks and reported prediction accuracy is 95 %. Peng et.al developed MiPred which classifies real and pseudo-microRNA precursors using random forest prediction model. MiPred has reported 88.21 % of total accuracy, and while combining the P-value randomization, the accuracy of prediction increased to 93.35 %. Mpred [18, 21] is a tool which uses artificial neural network for pre-microRNA validation and microRNA prediction by hidden Markov model. MiRPara [23], Triplet-SVM, and MiRFinder are the SVM-based classifier where reported accuracy of MiRPara is 80 % and that of Triplet-SVM is 90 %. MiRPara divides the input sequences into number of fragments of length around 60 nucleotides, filter out the fragments having an hairpin structure, extracts 77 different parameters from the sequence, and fed to SVM classifier. Triplet-SVM classifies the real and pseudo-microRNA precursor using structure and triplet sequence features. The positive training dataset collected miRNA registry database and the pseudo-miRNA datasets from the protein coding regions. MiRFinder tried to distinguish between microRNA and nonmicroRNA sequences using different representations of the sequence states such as paired, unpaired, insertion, deletion, and bulge with different symbols. They constructed the positive training data with the pre-miRNA sequences of human, mouse, pig, cattle, dog, and sheep collected from miRBase, and constructed the negative dataset with the sequences extracted from the UCSC genome pairwise alignments. MiRFinder used RNAfold [7] to predict the secondary structure of the sequences.
The tools discussed above uses different subsets of structural, sequence, and thermodynamic properties of secondary structure of microRNA sequence. Still there is relevance for a better tool with reduced feature set and higher level of accuracy. The main motivation of this work is to develop a classifier with high sensitivity (True Positive Rate), high specificity (True Negative Rate), low false positive rate, and an accuracy greater than 95 %. We have developed an SVM-based classifier and trained by the properties extracted from the experimentally validated database of human microRNAs.
2 SVM-Based Classifier Model
Figure 1 shows the system model. A trained and tested classifier could be able to predict whether a given input sequence is a probable microRNA or not. Figure 2 shows the preprocessing steps required for microRNA identification from an input gene sequence. The length of gene sequence vary from few hundreds to several thousands of nucleotides. A moving window divides input sequence into subsequence of length 100 with step size of 30. The candidate sequences with a lesser base pairing value than a threshold value can be discarded in the initial screening. The known microRNA sequences have at least 17 base pairs, and hence sequences having 17 or more pairs are only passed to feature extraction phase.

System model

Gene sequence preprocessing and feature extraction
2.1 Training Data Preparation
Sufficient quantities of positive and negative samples of data are required to train and test a classifier. The quality of the training dataset determines the accuracy of the classifier. miRBase is a primary microRNA sequence repository keeping identified pre-microRNA sequences, mature sequences, and gene coordinate information [10]. Presently, the database contains sequences from 223 species. 500 human microRNA sequences downloaded from miRBase database are used as positive dataset. The negative training dataset is prepared from the coding region of RNA, by filtering out sequences that contain a hairpin-like structure. The reason behind this selection is that the real microRNAs are characterized by their hairpin loop along with other properties. 500 sequences are selected for the negative dataset also.
Feature Extraction and Selection A major discriminating property of RNA secondary structure is free energy due to the hydrogen bonding between its bases, called minimum free energy (MFE). Several computational algorithms based on dynamic programming have been developed to find MFE. RNAfold [7] is one such algorithm. RNAfold generates the secondary structure in dot-bracket notation and predicts minimum free energy(MFE) of the structure.
A bracket represents a paired base with other end of sequence, while dot represents a unpaired base. Figure 3 shows secondary structure and its dot-bracket representation with respect to a given input RNA sequence. The dot-bracket representation obtained is the base for further computations in the development of this classifier. We have extracted 46 features which include 32 sequence-related features [6, 24], 9 structural features, and 5 thermodynamic features. When three adjacent nucleotides in a sequence are considered as a block, with brackets and dots as symbols, we have eight different combinations: ’(((’, ’((.’, ’(.(’, ’.((’, ’..(’, ’.(.’, ’(..’ and ’...’. For each block, there are four more possibilities when the middle nucleotide is fixed. For example, the consecutive paired bases can be of ’A(((’ ’C(((’, ’G(((’, ’U(((’, where the letter stands for nucleotide in the middle. The total possible combinations of triplets are \(8 \times 4 = 32\).
The following structural features were selected from the secondary structure of the sequence.
-
1.
Base Count: Total number of base pairs.
-
2.
Base Content: The ratio of total number of base pairs to the total number of nucleotides in that sequence.
-
3.
Lone loop 3: The count of lone loops that have 3 nts. (a lone loop is the one with first and last nucleotides of the loop as Watson Crick or wobble base pair).
-
4.
Lone loop 5: The count of lone loops that have 5 nts.
-
5.
AU content: The ratio of number of AU base pairs to the total number of base pairs.
-
6.
GC content: The ratio of number of GC base pairs to the total number of base pairs.
-
7.
GU content: The ratio of number of GU base pairs to the total number of base pairs.
-
8.
Hairpin length: The number of nucleotides in the hairpin loop.
-
9.
Number of Bulges: Total number of bulges.

Secondary structure and dot-bracket representation corresponding to a typical RNA sequence
The features related with the structural stability in terms of energy value due to the bonding of bases are known as thermodynamic features [14].
-
1.
Minimum Free Energy: Minimum free energy of the structure.
-
2.
MFE content: The ratio of MFE to the number of nucleotides in the sequence [24].
-
3.
GC/Fe: The ratio of number of GC pairs to the MFE.
-
4.
AU/Fe: The ratio of number of AU pairs to the MFE.
-
5.
GU/Fe: The ratio of number of GU pairs to the MFE.
This is quite large number of parameters, and dimensionality reduction is applied based on principle component analysis (PCA) [2]. PCA is a mathematical method for dimensionality reduction. This can be viewed as rotation of axes of original variable coordinate system to new orthogonal axes called principal axes, which coincide with the direction maximum variation of original observations. Thus, principal components represent a reduced set of uncorrelated variables corresponding to the original set of correlated variables. We used WEKA [13] to build the classifier. Based on the value of variance specified, WEKA chooses sufficient number of Eigen vectors to account original data. Ranking of attribute can be performed with WEKA by selecting an option to transform back to original space. The top ranked 19 features are only used for final classification, as there is very little improvement in accuracy when others are also considered. The selected features and their rank are shown in Table 1. This includes seven features from sequence-related features such as ’A(((’, ’C(((’, ’G(((’, ’A((.’, ’G((.’, ’A.((’ and ’G.((’; and eight features from structure-related group; and four from thermodynamic group. Although many subsets of these features are used by other computational tools for microRNA prediction, we uniquely identified three new features. They are ratio of GC and free energy (GC/Fe), ratio of AU and free energy (AU/Fe), and ratio of GU and free energy (GU/Fe). It is evident that they have decisive role as they have ranked 1st, 11th, and 13th in the select list of attributes.
Machine Learning Support vector machines(SVM) are supervised learning model with associated learning algorithms [6, 23]. Given a set of training examples, each marked as belonging to one of two classes, an SVM training algorithm builds a model that assigns new examples into the appropriate class, making it a non-probabilistic binary classifier. SVMs effectively do this classification by a technique called kernel trick, implicitly mapping their inputs into high-dimensional feature space. A linear classifier is based on discriminant function of the form \(f(x)=\omega ^{T} \cdot {x~+~b}\), where \(\omega \) is the weight vector, and b is the bias. The set of points \(\omega ^{T} \cdot x~=~0\) define a hyperplane, and b translates hyperplane away from the origin. A nonlinear classifier is based on discriminant function of form \(f(x)=\omega ^T\phi (x)~+~b\), where \(\phi \) is a nonlinear function. Performance of the SVM classifier with a linear kernel, and two nonlinear kernels, namely radial basis function kernel (RBF) and Pearson VII kernel (PUK), are analyzed. The RBF kernel is defined by
and Pearson VII kernel is defined by
where \(\omega \) and \(\sigma \) control half width and trailing factor of peak, respectively.
3 Performance Analysis of the Classifier
The performance of the classifier with linear and nonlinear kernel, with complete and reduced feature set, is evaluated. Table 2 shows the confusion matrix in SVM with PUK kernel function when 10-fold cross validation is employed. A classifier gives best result when it reaches high TP and TN rates. The efficiency and quality of a tool depend upon a number of factors such as sensitivity (TP rate), specificity (TN rate), and accuracy. The accuracy of the classifier can be calculated using the following equations [23, 24]:
When all the 46 features are used with PUK kernel and 10-fold cross validation, the sensitivity, specificity, and accuracy reached 98.6 %, and this recorded as the best result from the classifier. However, if top ranked 19 features are used, the classifier provides sensitivity, specificity, and accuracy as 98.4 % (same value for all the parameters). When compared with the performance with whole feature set, variation is insignificant, but computational cost will be definitely higher in the former case. The classifier performance under different conditions is shown in Table 3. It is also evident from the data in the table, when RBF kernel is used, that change in value of parameter \(\gamma \) from 0.01 to 1 makes considerable increase in accuracy.
ROC is plot of fraction of true positives out of the total actual positives (TPR = true positive rate) versus the fraction of false positives out of the total actual negatives (FPR = false positive rate), at various threshold settings. TPR is also known as sensitivity or recall in machine learning. The FPR is also known as the fall-out and can be calculated as one minus specificity. The ROC curve is then the sensitivity as a function of fall-out. In general, if both of the probability distributions for detection and false alarm are known, the ROC curve can be generated by plotting the cumulative distribution function (area under the probability distribution from \(-\)inf to \(+\)inf) of the detection probability in the y-axis versus the cumulative distribution function of the false alarm probability in x-axis. Figure 4 A shows the ROC curve of the classifier, with RBF and PUF kernel functions. Area under the ROC curve should be high for an excellent classifier. In our classifier, the area under the ROC curve is 0.984, which indicates TP rate attains its highest values when FP rate is as low as 0.016.
Figure 4 A shows the ROC curve of the classifier with RBF, and
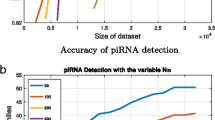
MiRPara, Triplet-SVM, MiRFinder, etc. are the main examples of SVM-based classifiers. We tried to compare performance of our SVM classifier with that of above three tools. Figure 5 B shows the comparison of their accuracy.

ROC curves of SVM with RBF kernel and PUK kernel

Comparison of accuracy of different microRNA prediction tools
4 Conclusion
The classifier that we developed distinguishes microRNAs and nonmicroRNAs very accurately. When compared with other tools that employ SVM as the classifier, our results sense better possibly due to the use of hybrid future set, precise feature selection, and selection of best classifier algorithm. The accuracy of our tool is 98.4 % which is higher than that of existing SVM-based classifier such as MiRFinder, Triplet-SVM, and MirPara. The classifier sensitivity is 98.4 % and specificity is 98.4 % which is also higher than that of existing classifiers.
References
Anastasis, O., Martin, R., Panayiota, P.: Image encryption based on chaotic modulation of wavelet coefficients. IEEE Trans. Inf. Technol. Biomed. 13(1), (2009)
Arnaz, M., Robert, X.G.: Pca-based feature selection scheme for machine defect classification. IEEE Trans. Instrum. Measur. 53(6), (2004)
Aurora, E.K., Frank, J.S.: Oncomirs micrornas with a role in cancer. Nat. Rev. Cancer 6, 259–270 (2006)
Bartel, D.: Micrornas: genomics, biogenesis, mechanism, and function. Cell 116, 281–297 (2004)
Bruce, A., Alexander, J., Julian, L., Martin, R., Keith, R., Peter, W.: Molecular biology of the cell. Garland Sci. (2002)
Chenghai, X., Fei, L., Tao, H., Guo-Ping, L., Yanda, L., Xuegong, Z.: Classification of real and pseudo microrna precursors using local structure-sequence features and support vector machine. BMC Bioinform (2005)
Dianwei, H., Jun, Z., Guiliang, T.: MicroRNAfold: microRNA secondary structure prediction based on modified NCM model with thermodynamics-based scoring strategy. University of Kentucky, Department of Computer Science, Lexington (2008)
Giulio, P., Giancarlo, M., Graziano, P.: Predicting conserved hairpin motifs in unaligned rna sequences. In: Proceedings of the 15th IEEE International Conference on Tools with Artificial Intelligence (ICTAI03) (2003)
Kim, V.: Small RNAS: classification, biogenesis mechanism and function. Mol. Cell 19, 1–15 (2005)
Kozomara, A., Griffiths-Jones, S.: Mirbase: annotating high confidence micrornas using deep sequencing data. Nucl. Acids Res. 42, D68–D73 (2014)
Lim, L., Lau, N., Weinstein, E., Abdelhakim, A., Yekta, S., Rhoades, M., Burge, C., Bartel, D.: The micrornas of caenorhabditis elegans. Genes Dev. 17, 991–1008 (2003)
Manel, E.: Non-coding rnas in human disease. Nat. Rev., Genet. (2011)
Mark, H., Eibe, F., Geoffrey, H., Bernhard, P., Peter, R., Ian H., W.: The weka data mining software: an update. SIGKDD Explorations, pp. 10–18 (2009)
Markus, E., Nebel, Anika, S.: Analysis of the free energy in a stochastic rna secondary structure model. IEEE/ACM Trans. Comput. Biol. Bioinform. 8(6) (2011)
Michael, A., Andy, M., Tyrrell: Regulatory motif discovery using a population clustering evolutionary algorithm. IEEE/ACM Trans. Comput. Biol. Bioinform. 4(3) (2007)
Modan, K.D., Ho-Kwok, D.: A survey of dna motif finding algorithms. BMC Bioinfor. 8(doi:10.1186/1471-2105-8-S7-S21) (2007)
Peng, J., Haonan, W., Wenkai, W., Wei, M., Xiao, S., Zuhong, L.: Mipred: classification of real and pseudo microrna precursors using random forest prediction model with combined features. Nucleic Acids Res. 35(1) (2007)
Reshmi, G.: Vinod Chandra, S., Janki, M., Saneesh, B., Santhi, W., Surya, R., Lakshmi, S., Achuthsankar, S.N., Radhakrishna, P.: Identification and analysis of novel micrornas from fragile sites of human cervical cancer: computational and experimental approach. Genomics 97(6), 333–340 (2011)
Salim, A., Vinod Chandra, S.: Computational prediction of micrornas and their targets. J. Proteomics Bioinform. 7:7, 193–202 (2014)
Ting-Hua, H., Bin, F., Max, F.R., Zhi-Liang, H., Kui, L., Shu-Hong, Z.: Mirfinder: an improved approach and software implementation for genome-wide fast microrna precursor scans. BMC Bioinform. (2007)
Vinod Chandra, S., Reshmi, G.: A pre-microrna classifier by structural and thermodynamic motifs (2009)
Vinod Chandra, S., Reshmi, G., Achuthsankar, S.N., Sreenathan, S., Radhakrishna, P.: MTAR: a computational microrna target prediction architecture for human transcriptome. BMC Bioinform. 10(S1), 1–9 (2010)
Wu, Y., Wei, B., Liu, H., Li, T., Rayner, S.: Mirpara: a SVM-based software tool for prediction of most probable microrna coding regions in genome scale sequences. BMC Bioinform. 12(107) (2011)
Ying-Jie, Z., Qing-Shan, N., Zheng-Zhi, W.: Identification of microrna precursors with new sequence-structure features. J. Biomed. Sci. Eng. 2, 626–631 (2009)
Yunpen, X., Xuefeng, Z., Weixiong, Z.: Microrna prediction with a novel ranking algorithm based on random walks. Bioinformatics 24 (2008)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer India
About this paper
Cite this paper
Sumaira, K.A., Salim, A., Vinod Chandra, S.S. (2016). SVM-Based Pre-microRNA Classifier Using Sequence, Structural, and Thermodynamic Parameters. In: Das, S., Pal, T., Kar, S., Satapathy, S., Mandal, J. (eds) Proceedings of the 4th International Conference on Frontiers in Intelligent Computing: Theory and Applications (FICTA) 2015. Advances in Intelligent Systems and Computing, vol 404. Springer, New Delhi. https://doi.org/10.1007/978-81-322-2695-6_6
Download citation
DOI: https://doi.org/10.1007/978-81-322-2695-6_6
Published:
Publisher Name: Springer, New Delhi
Print ISBN: 978-81-322-2693-2
Online ISBN: 978-81-322-2695-6
eBook Packages: EngineeringEngineering (R0)