
Abstract
The application of layered materials as adsorbents for environmental issues was summarized. Various useful phenomena for environmental purification by removing toxic species from water and air have been reported. The concentration of noble species from environments has also been conducted. The impressive phenomena were derived during the careful and extended studies on the adsorption of various species onto layered solids from environments under different conditions. Due to the natural abundance, the materials’ variation, and useful adsorption behavior toward various cationic species, the application of bentonites and smectite group of clay minerals is promising for the cation fixation and removal. Some useful ion-exchange properties such as the heavy metal and the radioactive metal removal have been observed for various layered solids and the nature of the selectivity and efficiency has been investigated. The chemical modification of layered materials including pillaring and being organophilic is also useful to tune the adsorption behavior toward various nonionic organic species. Due to the increased demands for the efficient and selective concentration of target species from the environments, the materials’ design has been conducted through crystal engineering of layered materials as well as the chemical modification.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
Keywords
- Smectite
- Organophilic clay
- Pillared-layered structure
- Ion exchange
- Environmental purification
- Selectivity
1 Introduction
Concentration of noble elements and compounds from environments are topics of interests for environmental purification, pharmaceutical, and food sciences. For the concentration of a target-specific compound, adsorption is one of the most reliable and useful chemical phenomenon, therefore, the preparation of adsorbents and the studies on the adsorption phenomena to elucidate the adsorbent–adsorbate interactions have been investigated extensively. Extraction and storage, which are phenomena relevant to the adsorption, are topic of interests in materials chemistry because of the potential for the environments and future sustainable energy issues.
Layered materials, especially smectite group of clay minerals, have been used as adsorbents for a variety of species from metal ion, organic molecules, polymers, and colloidal particles. The adsorbent’s design toward specific targets has also been done based on the intercalation chemistry of smectites, chemical modification, and structural transformation into three-dimensional network (pillaring). Pillaring of various layered solids has been examined for the applications as adsorbents and catalysts [1]. As to the chemical modification, organophilic clays are the most famous example and there are several commercially available organophilic clays for industrial uses. The organophilic clays are developed in 1950s [2] and the successful industrial application and more recent advanced materials applications attract materials chemists for the organic modification of various layered solids.
Ion-exchanger application is also another useful example, in which noble (or toxic) metal ions in environments (sea water for example) have been collected by the ion exchangers. Materials variation of layered solids from cation exchangers with varied ion-exchange capacity and selectivity to anion exchangers, chemical stability superior to organic and polymeric ion exchangers, as well as possible morphosyntheses made layered materials more attractive as adsorbents of various ionic species for practical uses. In this chapter, we summarized the potential of layered materials as present and future adsorbents for practical uses. The attention is focused on the mechanism of the adsorption on layered solids and the chemical design of adsorbents from layered solids.
2 Adsorption of Metal Ions
2.1 Cation Exchange
Smectite is a group of layered clay mineral consisting of negatively charged silicate layers and readily exchangeable interlayer cations. Isomorphous substitution of metal cations with similar size and lower valency generates a net negative charge of layers. To compensate the negative charge, metal cations (generally Na+ and/or Ca2+) occupy the interlayer space as exchangeable cations [3]. Due to this structural feature, smectite has been investigated as a cation exchanger.
Thermodynamic data for alkali metal cation exchanges on smectite at 298 K (standard free energy) were summarized by Maes and Cremers [4a] based on the previous publications [4b–e], giving the following selectivity sequence for monovalent cations.
The selectivity is governed by the hydration energy; cations with smaller hydration energy are preferred.
It is known that K+ is fixed on weathered mica and vermiculite, which are 2:1 type layered silicates with the structures analogous to smectite [5]. K+ has lower hydration energy and the diameter of K+ (0.266 nm) is close to the size of the hexagonal hole of oxygen surface of the silicate layer. Therefore, K+ is stably (irreversibly) trapped in the hexagonal hole followed by the interlayer dehydration. K+ in mica is not exchangeable in general. There are some examples of the ion exchange of micas with some modification; the stable K+ in phlogopite, ideal formula of KMg3Si3AlO10(OH)2, was replaced with exchangeable Na+ using sodium tetraphenylboron [6]. The selective uptake of Cs+ has been shown on the Na+-exchanged phlogopite mica [7]. Na+–Cs+ exchange isotherm of the Na+-exchanged phlogopite mica (Fig. 9.1) laid above diagonal line, showing that Cs+ is highly preferred over Na+. The basal spacing (1.158 nm) of the phlogopite mica after the Cs+ uptake was slightly small compared to that (1.223 nm) of the Na+-exchanged phlogopite mica, while larger than that (1.003 nm) of K+-form. It was suggested that the diffusion of less hydrated Cs+ ions is possible. Taking the possibility of remediation of nuclear waste, the selective adsorption to concentrate metal ions from quite dilute solution has been investigated using various natural and synthetic micas [8].

Na/Cs exchange isotherm of K-depleted phlogopite mica, where diagonal line represents equal preference for both the cations. Reprinted from Ref. [7] with permission; copyright 1988, American Association for the Advancement of Science
Concentration of rare earth elements by the adsorption is an important topic from the viewpoints of environmental (collecting rare elements) geochemical interests. Competitive adsorption of a series of rare earth elements on a montmorillonite (Ceca bentonite) was examined [9] to show the adsorbed Na+ on the surface behaved as a barrier for the adsorption of rare earth ions at high ionic strength of 0.5 mol/L (background electrolytes are Na salts). Light rare earth elements showed this tendency due to their large ionic radius. At low ionic strength of 0.025 mol/L, the sorption coefficients were independent on the nature of the rare earths. Na-fluorotetrasilicic mica (abbreviated as Na-TSM) adsorbed Eu3+ effectively if compared with two smectites (Kunipia F, a natural montmorillonite and Sumecton SA, a synthetic saponite) as shown by the adsorption isotherms (Type-H for TSM, and type-L for smectites) [10]. The high affinity of Eu3+ to TSM is accounted for the structural in the octahedral sheet: TSM contains highly electronegative fluorine groups instead of the hydroxyls in smectites.
Layered alkali silicates such as octosilicate (SiO2/Na2O = 8), magadiite (SiO2/Na2O = 14–16) and kenyaite (SiO2/Na2O = 20–22) are known to possess cation exchange ability. Because protons of the silanol groups in the interlayer surface are exchanged with alkali ions, the cation-exchange capacity is strongly pH dependent [11]; protons compete with other cations at pH < 7, leading the silicates transform into the silicic acid [12]. In addition, sodium ions in the polysilicates cannot be exchanged with Fe3+ or Al3+, because these cations exist only in acidic aqueous solutions. On the other hand, exchange of sodium ions in a sodium silicate (SiO2/Na2O = 8) with various inorganic cations (e.g., lithium, potassium, ammonium, magnesium, copper) occurs [13]. Recently, concentration of Zn2+ on a layered alkali silicate, magadiite, from a seawater-mimicking aqueous solution was reported. Magadiite selectively and effectively adsorbed Zn2+ even when Cd2+ was present (Fig. 9.2a). The selectivity was not achieved for another sodium-type layered alkali silicate, octosilicate (Fig. 9.2b), and commercially available ion-exchange resin (Fig. 9.2c). Although the origin of the selectivity was not understood, ionic radius and hydration energy of cations are thought to be concerned for the affinity toward layer surface of magadiite [14].

Adsorption isotherms of (black circle) Zn2+ and (white square) Cd2+ on a magadiite, b octosilicate, and c ion exchange resin (IRC748, Organo Co.) from a seawater-mimicking solution containing Zn2+ and Cd2+. Horizontal line denotes the ion-exchange capacity for the dication of the silicates. Reprinted from Ref. [14]. Copyright 2011 Wiley VCH
Cation exchangeable-layered titanate and niobates are also available, and the selectivity ion exchange has been observed [15–18]. Komatsu and Fujiki [16] reported uptake of Cs+ on hydrous layered titanate, H2Ti4O9·nH2O, in the presence of alkali metal cations (Li+, Na+, H+, or Rb+). Even the concentration of Na+ or Li+ in solution increased, the uptake of Cs+ on H2Ti4O9·nH2O did not decrease. Rb0.14H7.9Nb22O59·8.2H2O; Na+ or K+ cations were selectively exchanged with protons of Rb0.14H7.9Nb22O59·8.2H2O in a concentrated LiCl aqueous solution [17]. HCa2Nb3O10·1.5H2O; Selective uptake of NH4 + from the mixture of Na+, K+, and NH4 + in solution on a protonated layered perovskite, HCa2Nb3O10·1.5H2O, has also been reported [18]. The uptake amount of NH4 + was 7- and 10-fold those of Na+ and K+, respectively, from mixed solution (NaCl, KCl, and NH4Cl) containing 1.0 mM of each ion at pH 2.2.
The cation exchange of K4Nb6O17·nH2O [19] shows a unique site selectivity. By the reactions using aqueous solutions of LiCl, NaCl, CaCl2, and NiCl2 at 90 °C, complete exchange K+ was attained only for Na+, while a half of K+ was exchanged with the divalent cations. The layered structures are composed of alternating two interlayers with different reactivities; interlayers I (hydrated) and II (not hydrated), which were determined from electron density projection along the b axis. It has been thought that selective exchange of K+ ions in the interlayer II with monovalent cations occurs, whereas both mono- and divalent cations can exchange with K+ in the interlayer I, from the observed changes in the basal spacings.
2.2 Anion Exchange
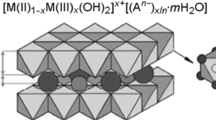
Layered double hydroxides (LDHs) have been investigated as anion exchangers. LDHs are a class of layered materials consisting of positively charged brucite like layer, where some M2+ cations are substituted with M3+ cations to give positive charge, and the charge compensating interlayer exchangeable anions. After the careful and extensive investigation on the anion-exchange properties of LDHs, the guest selectivity has been sequenced as the size and charge of anions [20–22]. Small and highly charged anions preferentially occupy the LDH interlayers with the order of preference
It is known that carbonate ion is the most stable as the interlayer anion and the replacement of the intercalated carbonate anion is difficult. Therefore, in order to incorporate various functional anions, nitrate or chloride forms of LDHs have been synthesized and employed as the intermediate for functional intercalation compounds. Experiments have been carefully done to find the experimental condition to overcome the anion-exchange sequence. In aqueous HCl solution, by the protonation of CO3 2− to form HCO3 −, Cl− was effectively intercalated into Zn/Al-LDH instead of CO3 2− [23a, b]. Iyi and Sasaki [23c] revealed that the deintercalation of CO3 2− from Mg/Al-LDHs with the Mg/Al ratios of 2 and 3 by using acetate-buffer (sodium acetate and acetic acid)/NaCl mixed solutions at 25 °C. The addition of NaCl to the acetate-buffer solution was shown to be important in the decarbonation without any morphological and weight changes. Even in the presence of acetate anion, only chloride ions were incorporated into the LDHs due to their extremely low acetate selectivity. Hayashi and Nakayama have reported the anion-exchange reactions of a CO3 2− form of Mg/Al-LDH with chloride, nitrate, and some organic anions in alcohols [24].
LDHs have been used as anion exchangers for environmental purifications. The removal of phosphates, arsenates, iodide, and chromate from water has been reported so forth [25, 26]. The removal of oxyanions such as phosphate from seawater has been investigated [26]. Dichromate anions (Cr2O7 2−) are reported to strongly interact with CO3 2− form of Mg/Al-LDH, which is an important advantage for the removal of monochromate (CrO4 2−) from the viewpoint of the efficient (two times) remediation of chromium contaminated water. The adsorption was possible under an acidic aqueous solution at pH 3 without dissolution of the LDHs, probably due to the large particle size of the used LDH [27]. In addition to anion exchange reactions, calcination-rehydration (reconstruction) is also useful for the uptake of anionic species [28–30]. The variation of removable anions is larger for the reconstruction method.
3 Adsorption of Molecules
3.1 Carbon Dioxides
Adsorption of CO2 has been a topic of interests in scientific and practical aspects, most notable example is global warming issue. Adsorption of CO2 onto layered solids also has several aspects. CO2 was used to determine specific surface areas of smectites [31, 32]. Thomas and Bohor [31] reported a larger specific surface area of the Cs+-exchanged from obtained from CO2 BET plot (315 m2/g at 195 K) than that (138 m2/g at 77 K) from N2 adsorption, even though the difference was negligible in kaolinite (a nonexpandable clay mineral) system. Because the adsorption temperature of CO2 was high, CO2 molecules are kinetically more energetic (with a larger diffusion coefficient), and thus the penetration into the interlayer space of the Cs+-smectite occurred.
Fripiat et al. [32] reported regarding the CO2 penetration from XRD analysis, where the basal spacing of dehydrated Na-montmorillonite (1.00 nm) increased to 1.23 nm. They also empirically predicted that the degree of hydration of the interlayer exchangeable cations (Li, Cs, K) should be a factor responsible for the greater amount of CO2 adsorption, because the amounts of adsorbed CO2 varied depending on the interlayer exchangeable cations. Although it was impossible to explicit about the detailed orientation of CO2 molecules in the interlayer space, a significant fraction is shown to be oriented with respect to the clay layer as revealed by infrared dichroic spectroscopy.
More recently, the interactions of CO2 with smectites have attracted another kind of attention as storage sites for anthropogenic CO2, which related to the CO2 sequestration, which is a global warming issue. Permeation ability of caprocks, which contain clay minerals such as smectites and kaolinite, for CO2 strongly correlates with the geological CO2 storage. Lower permeability is required for sealing CO2 by caprock for a long period. Exchange of water molecule in clay minerals with CO2 is negative phenomenon for sealing, therefore, it is important to understand the interactions of CO2 with water-bearing minerals (including intercalation behavior). Experiments [33–36] and simulations [37–41] have been conducted in order to elucidate the interactions between CO2 and smectites under supercritical conditions.
Michels et al. [34] reported the intercalation of gaseous CO2 into a synthetic fluorohectorite at ambient conditions. The retention ability of CO2 was strongly dependent on the nature of the interlayer cation (Li, Na, and Ni). For Li-exchanged fluorohectorite (LiFh), threshold temperature, which means deintercalation of CO2 from the interlayer space, was about 35 °C, whereas that was about −15 °C for Na-exchanged fluorohectorite (NaFh). Because of the difference in size between the Li+ and Na+, Li+ has a more concentrated charge than Na+ and can thus polarize CO2, forming a stronger bond. Similar discussion was reported [35] using such in situ techniques as XRD, 13C NMR, and ATR-IR. In Fig. 9.3, the CO2 adsorption isotherm on LiFh at room temperature is examined under a pressure range from 1 to 45 bar [34]. The initial part (0 to ca. 9 bar) of the adsorption isotherm represents diffusion of CO2 into LiFh. The swelling of LiFh occurred under the pressure above 9 bar, as confirmed by XRD. Further increase in CO2 pressure led the increased adsorption to be around 11 wt% at 38 bar. Above this pressure (approaching the critical pressure for CO2), the adsorbed amount started to decrease, probably due to the formation of an adsorbed layer with higher density and shrinking the volume of CO2 –LiFh complex to the pristine LiFh.

CO2 adsorption isotherm on Li-fluorohectorite at room temperature and pressure range 0–45 bar. Reprinted from Ref. [34]. Copyright 2015 Nature Publishing Group
Recently, in situ characterization (XRD and IR) on the intercalation behavior of CO2 using supercritical CO2 (scCO2) at 50 °C, 90 bar in the presence of water into Na-montmorillonite has been reported [36]. Molecular simulation studies on scCO2 intercalation into hydrated montmorillonite have been conducted using a Monte Carlo [38] and a DFT-based MD [38–41]. As an example of molecular simulations of CO2 intercalation into hydrated montmorillonite using Clayff force field [42], the change in the basal spacing upon increased water content and with addition of CO2 (two molecules per unit cell) is shown in Fig. 9.4 [37]. The basal spacing increased in the monolayer hydrate (arrow in Fig. 9.4) from 10.77 ± 0.04 to 12.22 ± 0.03 Å (12% swelling) after CO2 intercalation, and in the bi-layer hydrated system from 12.98 ± 0.09 to 14.78 ± 0.09 Å. Density profiles of atoms in the interlayer region showed separation of CO2 molecules into two distinct layers similar to that observed for a bi-layer water network.

Swelling behavior of Na-montmorillonite upon hydration (blue circles) and after additional intercalation of two molecules of CO2 per unit cell (red squares) based on NPT MD simulations at 310 K and 20 MPa. Arrows indicate swelling occurring after CO2 intercalation with approximately one and six water molecules per unit cell. Reprinted from Ref. [37]. Copyright 2012 American Chemical Society
Calcined LDHs has been recognized as CO2 adsorbents at high temperature [43–47]. While it has been widely understood that the pristine LDHs do not accommodate CO2 at ambient temperature, carbonate anion-intercalated LDH (hydrotalcite) underwent a carbon cycle with the uptake of atmospheric CO2 under ambient conditions [48]. As shown in Fig. 9.5, 13C-labeled CO2 (or CO3 2−) was used to monitor the dynamics of the exchange between initially intercalated 13C-labeled carbonate anions and carbonate anions derived from atmospheric CO2 by IR spectroscopy. Exchange was promoted at a lower humidity with a half-life of exchange of ∼24 h. The research group subsequently reported the interactions of CO2 with hydrotalcites of different Mg/Al ratios (Mg/Al = 2, 3, and 4, which are named as LDH2, LDH3, and LDH4, respectively) [49]. Adsorption/desorption isotherms of CO2 and N2 (the competitive adsorption test) have been done to evaluate how the Mg/Al ratio affects the adsorption capacities (Fig. 9.6); the LDHs selectively incorporated CO2 with the capacities sequence of LDH2 < LDH3 ≈ LDH4. They ascribed the adsorption site of CO2 to the basic Mg–OH based on the report that Mg-rich LDH promoted base catalyzed reactions [50].

a Dynamic exchange of carbonate anions of LDH with atmospheric CO2. b IR spectra of 13CO3 2−-LDH and CO3 2−-LDH. c Variation of IR frequency of 13CO 23 -LDH upon exposure to air at different relative humidity. Reprinted from Ref. [48]. Copyright 2013 American Chemical Society

N2 adsorption–desorption isotherms of a LDH2, b LDH3, and c LDH4 measured at 77.35 K. CO2 and N2 adsorption–desorption isotherms of d LDH2, e LDH3, and f LDH4 measured at 25 °C. Reprinted from Ref. [49]. Copyright 2014 American Chemical Society
3.2 Organic Molecules
Layered solids may adsorb polar organic molecules, and it is well known that smectites adsorb various polar molecules. Polar molecules such as alcohols, ketones, and amide are adsorbed by ion–dipole interactions with the interlayer cations and/or hydrogen bonding with the surface oxygen atom of the silicate layers. The intercalation of polar molecules usually occurs through solid–liquid or solid–gas reactions [1, 3, 51–54], while solid–solid reactions are also possible [55, 56].
In a smectite-polar molecule interactions in the solid-state, novel selectivity, which is not observed in the intercalation from solution [56], was observed. Maleic acid was intercalated into montmorillonite by solid–solid reactions, while its geometrical isomer, fumaric acid was not intercalated. From ethanolic solutions, both maleic and fumaric acids were intercalated. Such selectivity observed only in the solid–solid reaction was explained by the difference in the crystal structure of the molecules in addition to the ability of maleic acid to form a chelate like structure with the interlayer cations.
4 Adsorbents Design by the Modification of Layered Solids
4.1 Organic Modification with Ionic Species
Surface properties of layered solids have been modified to alter affinity toward target molecules. Cation exchange reactions of smectite [2, 57], layered titanate [58], layered niobate [59] with long-chain alkylammonium ions have been used to modify the surface property. The organophilic clays swell in aprotonic organic solvents for possible application as rheology controlling reagents [2, 60]. LDHs also became hydrophobic by incorporating long-chain alkyl carboxylates and sulfates [61, 62]. The adsorption characteristics of these ionic surfactants on layered solids via ion exchange reactions have been described separately in Chap. 6. Here, we will describe the adsorption of nonionic organic compounds from vapor and aqueous solution at lower concentration, as a way of the remediation of contaminated soil and water, which are important environmental problems.
The adsorption of nonionic organic compounds from water by soil depends on soil organic matter content [63]. Organic phase composed of hexadecyltrimethylammonium (HDTMA) behaved as partitioning medium that was more effective than natural soil organic matter for removing hydrocarbons (benzene and 1,2-dichlorobenzene) from water [64]. There are many fundamental studies on the adsorption behavior into hydrophobic clays with the structures varied by changing the surfactant amount and the alkyl-chain length [65].
Jaynes and Boyd [66] reported the sorption behavior of ethylbenzene in HDTMA-clays with different layer charge density from aqueous solution. Vermiculite (CEC = 80 meq/100 g clay), illite (CEC = 24 meq/100 g clay), kaolinite (CEC = 4 meq/100 g clay), and smectites (CECs = 87–130 meq/100 g clay) were used as host materials. Partition coefficients of ethylbenzene for HDTMA-clays varied depending upon the nature of smectites and HDTMA contents. Both the greater HDTMA contents and the larger basal spacings of high charged HDTMA-clays (vermiculite and smectites) led larger adsorption amounts of ethylbenzene.
Mortland et al. [67] have discussed hydrophobic interactions of nonionic organic compounds with HDTMA-montmorillonite (Wyoming bentonite). Whereas 3,4,5-trichlorophenol was adsorbed into HDTMA-montmorillonite from water, phenol was not adsorbed at all, as shown in the adsorption isotherms from water (Fig. 9.7 top). On the other hand, these two phenols were adsorbed into HDTMA-montmorillonite from hexane (Fig. 9.7 bottom). This difference has been ascribed to the relative importance of both adsorbate–solvent and adsorbate–adsorbent interactions on partitioning of adsorbates between adsorbent and solvent. In aqueous solution, phenol interacted strongly with water and was not attracted sufficiently to the hydrophobic surfaces of HDTMA-montmorillonite.

Top Adsorption isotherms (293 K) of phenols on the HDTMA-montmorillonite. A = 3,4,5-trichlorophenol; B = 3,5-dichlorophenol; C = 3-chlorophenol; D = phenol. Bottom Adsorption isotherms for 3,4,5-trichlorophenol from water (A) and hexane (A′), and phenol from water (B) and hexane (B′) onto HDTMA-smectite. Reprinted from Ref. [67]. Copyright 1986 The Clay Minerals Society
Modification of other layered solids as LDHs [68–70], layered hexaniobate [71, 72], and layered phosphate [73] provides hydrophobic intercalation compounds for uptake of chlorinated phenols from water. Competitive adsorption of three phenols (phenol, 4-chlorophenol and 2,4-dichlorophenol) from water on alkylammonium modified layered niobate was examined [72] to find preferential adsorption of 2,4-dichlorophenol among three phenols. The uptake of each solute was reduced by the presence of other solutes, and the reduction in the multi-solute systems depended on the hydrophobicity of the solutes coexisted in the solution.
In order to facilitate uptake of aromatic molecules, layered solids modified with various cationic species with aromatic nature have been reported. Cation exchange reactions of smectites with various aromatic ammonium ions have been reported [74]. The role of aromatic rings in the phenol adsorption was illustrated by Langmuir-type adsorption isotherm observed for the adsorption on tris(2,2′-bipyridine)nickel(II) modified Sumecton SA, while the adsorption of phenol on tris(ethylenediamine)nickel(II) modified Sumecton SA was less effective [75]. Charge-transfer interactions have been used for adsorption of phenols as reveal by using methylviologen (MV2+) modified smectites (Kunipia F and Sumecton SA) in the absence of solvent [76] and from aqueous solution [77]. Effective adsorption of 2,4-dichlorophenol was achieved through the charge-transfer interactions with MV2+ in the interlayer space of Sumecton SA. A possible reason for the effective adsorption was lower ionization potential of 2,4-dichlorophenol than phenol [77].
Adsorption selectivity of aromatic molecules from aqueous solutions has been emerged using aromatic functionality for the modification. Neostigmine, TMPA, and 3-(trifluoromethyl)phenyltrimethylammonium (Fig. 9.8) were used to find the effects of the surface modification on the adsorption capacity of 2-phenylphenol [78]. Among tested adsorbents, the neostigmanine-modified Sumecton SA was most effective to remove 2-phenylphenol. Even in the presence of sucrose, 2-phenylphenol was effectively adsorbed on neostigmine-modified smectites [79].

Molecular structures of a trimethylphenylammonium, b 3-(trifluoromethyl)phenyltrimethylammonium, c neostigmine, and d 2-phenylphenol
4.2 Pillared-Layered Materials
Layered solids have also been used as scaffolds to design nanospace by the intercalation of bulky organic species into the interlayer spaces as pillar. Specific surface area and micropore volume varied depending on the interlayer cations of a natural montmorillonite (SWy-1: Wyoming montmorillonite), and that increased with cationic radius in the order of TMA > Cs > K > Ca > Na, which correlates with the gallery height [80]. Intercalation of TMA ions produces significant volumes of micropore in the interlayer space, which are readily accessible to N2. The accessibility of the micropores to other gases would depend on the molecular dimensions (molecular sieving effects). This kind of materials has been named as “pillared clays.” Pillared clays have been studies for the application as hosts of photo- and electro-functional materials, catalyst supports and adsorbents [52–54, 81]. Pore size, porosity, and surface area vary depending on the molecular geometry of pillaring agents and the spatial distribution (density). Interactions of nonionic organic compounds with these pillared-layered solids are essentially different from those with the hydrophobic clays [52–54, 82, 83].
Detellier et al. reported a gas chromatographic separation of CH4/CO2 on montmorillonite (SWy-1 from Clay Source Repository, University of Missouri) modified with quaternarized polyammonium cations. A strong dependence of CO2 retention times upon the free surface area was observed and the CH4/CO2 separation was interpreted principally by interactions with the surface and not by interactions with the polar moieties of the organic cations [84].
The larger molecular size of tetraethylammonium (TEA) than TMA led to the smaller surface area of TEA-smectite, so that the adsorbed amounts of various molecules were relatively small [82]. We have reported the adsorption behavior of o-xylene and p-xylene vapor to a series of N,N′-hexamethylalkyldiammonium ions [(CH3)3N+(C n H2n+1)(CH3)3N+, where n = 2, 3, 6, 10, abbreviated as C 2+ n ]-exchanged montmorillonites (Kunipia F: CEC of 119 meq/100 g clay and BENGEL BLIGHT 11 (BB11) from Hojun, Japan: CEC of 78 meq/100 g clay) [85]. All the examined organo-montmorillonites adsorbed these gases to show their microporous nature. The adsorbed amounts of xylenes increased when the size of C 2+ n was smaller (Fig. 9.9), showing possible pore volume engineering.

Adsorption isotherms of p-xylene (298 K). (White circle) Adsorption isotherm for C2 2+-BB11, (white diamond) adsorption isotherm for C3 2+-BB11, (white up pointing triangle) adsorption isotherm for C6 2+-BB11, (white square) adsorption isotherm for C10 2+-BB11. Reprinted from Ref. [85]. Copyright 2009 Elsevier
Lee [86] reported the adsorption of benzene, toluene, and o-xylene on TMA-smectites with different layer charge density (SAz: Arizona montmorillonite: CEC = 120 meq/100 g clay and SAC: Wyoming montmorillonite: CEC = 90 meq/100 g clay) (Fig. 9.10). The adsorbed amount of toluene from vapor phase on the TMA-smectites varied depending upon the nature of the smectites. The closer packing of TMA in the TMA-SAz resulted in a higher degree of shape-selective adsorption (adsorbed amount decreased in the order of benzene > toluene > o-xylene). The adsorption capacities of aromatic molecules were significantly reduced in the presence of water (Fig. 9.10). The lower adsorption capacity of aromatic molecules accompanied the increased shape selectivity. The reduction in the capacity and the increased selectivity were pronounced for the water saturated, high-charge TMA-smectite than for the low charge TMA-smectite. Hydration of TMA ions or the siloxane surfaces apparently reduced the accessibility of the aromatic molecules to interlamellar regions. Phenol and 4-chlorophenol were adsorbed from aqueous solution on tetramethylphosphonium (TMP)-smectite (Wyoming montmorillonite, CEC = 90 meq/100 g), while a negligible adsorption of 2- and 3-chlorophenols was observed [87]. In addition, these phenols were not adsorbed on high CEC TMP-smectite (SAz, Arizona montmorillonite, CEC = 120 meq/100 g). These observations were interpreted from the difference in the size of the pore created by TMP. Phenols were not adsorbed on the TMA-smectite even when the host is lower layer charge density (CEC = 90 meq/100 g). It has been pointed out that higher degree of hydration of TMA compared with TMP resulted in the observed size exclusion.

Adsorption isotherms of toluene (left) from vapor phase and (right) from aqueous solution by high charge (triangles) and low charge (circles) TMA-smectite. Reprinted from Ref. [86]. Copyright 1990 The Clay Minerals Society
Adsorption behavior of phenols onto Kunipia F, Sumecton SA and a synthetic fluor-tetrasilicic mica (TSM) modified with [Ru(bpy)3]2+ is an example to show that the distance between adjacent cations plays a role in the adsorbent design [88]. Intercalation of [Ru(bpy)3]2+ into layered clay minerals has extensively been investigated for probing the surface of clays as well as for electrochemical applications [89–92]. A few examples on the adsorptive properties of the [Ru(bpy)3]2+ modified clays has been published [93, 94], where [Ru(bpy)3]2+ acts as pillar to create micropore in the interlayers. Sumecton SA produces relatively large pore volume in the interlayer space by [Ru(bpy)3]2+, resulting in larger adsorption capacities of phenols (phenol and chlorophenol). The [Ru(bpy)3]2+-Sumecton SA can be utilized as adsorbent for relatively bulkier compared with phenols used in this study to arise molecular sieving effects for such bulkier molecules. More defined porous structure, which is a 2-dimensional long range order for racemic and enantiomerically pure [Ru(bpy)3]2+, has been obtained by intercalating into synthetic fluorohectorite [95].
Thus, in smectite systems, the CEC is a key factor to determine the adsorptive properties for nonionic organic molecules [96]. However, some characteristics, which depend on the origin of smectites, as position of isomorphous substitution and particle size make it difficult to discuss the effects of CEC on the physicochemical properties of smectites and their intercalation compounds. It is possible to reduce the layer charge by thermal treatment of Li-exchanged forms (Hofmann-Klemen Effect) [97, 98]. Reduction of layer charge of montmorillonites [99] and a F-rich potassium hectorite (obtained by a high temperature melt synthesis) [100] by Hofmann–Klemen effect allows the tailoring of pore sizes of pillared clays.
Varied CEC by changing the amount of Mg2+/Li+ in hectorite-like layered silicates [101, 102] affected the size or volume of the interlayer pore created by MV2+ to vary adsorption capacity of organic molecules [102]. The intercalated N,N′-dimethylaniline polymerized to develop purple color when the layered silicates with the CECs of 74 and 42 meq/100 g were used. On the other hand, the intercalation compound with larger MV2+ content (CEC: 85 meq/100 g) suppressed the polymerization of N,N′-dimethylaniline due to the smaller pore. The smaller MV2+ content, the adsorption capacity of 2,4-dichlorophenol was larger.
Breu and co-workers [103] have (re-)established synthetic strategy of structurally well-ordered pillared clays (>100 μm) with homogeneous (ultra)micropore distribution. For this purpose, they have made efforts to make homogeneous charge distribution in synthetic fluorohectorite (Na0.5(Mg2.5Li0.5)Si4O10F2) [104] through very careful melt synthesis. There is no structural disorder in the synthetic fluorohectorite in contrast to naturally occurring swellable layered silicates. In order to avoid phase separation in the silicate melt, the crucible has been rotated during the synthesis. The homogeneous charge distribution has been confirmed by founding X-ray reflection due to the two-dimensional 3a × b super-cell (Fig. 9.11) in Me2DABCO2+(N,N-dimethyl-1,1-diazabicyclo[2.2.2]octane dication) [103a] and 2H-DABCO (diprotonated 1,4-diazabicyclo[2.2.2]octane) exchanged fluorohectorites [103b]. As a result, homogeneous micropore distribution and only ultramicropore with pore width of 0.4–0.6 nm were obtained. Charge homogeneity was also achieved for iron-containing Cs-taeniolite (Cs0.98Fe 2+1.93 Li1.01Si4O10F2) [103c]. They have provided preferred molecular structure of pillaring agents for generating homogeneous porous network by considering results of MD simulations. Figure 9.12 illustrates that the pillar (Me2DABCO2+) is oriented with its C 3 axis tilted by approximately 24° against the ab plane. While two of the C2H4 handles protrude into the silicate layers, the third one is in the hexagonal plane of the interlayer space [103d]. The sizes and volume of the micropores created by Me2DABCO2+ and [Rh(bpy)3]3+ were shown to be increased through layer charge reduction of the synthetic fluorohectorite using Hofmann–Klemen effect from CEC = 84 to 59 meq/100 g [100].

a Powder XRD pattern of 2H-DABCO-hectorite. Asterisks (*) mark reflections due to the two-dimensional 3a × b super-cell; b Scheme of the a × b super-cell of pillars in 2H-DABCO-hectorite. The unit cell of the parent hectorite is shown as dotted lines. Reprinted from Ref. [103a]. Copyright 2008 Royal Society of Chemistry

Results of the X-ray single-crystal refinement (tetrahedral layer of the layered silicate and Me2DABCO2+ in the interlayer space). Reprinted from Ref. [103d]. Copyright 2008 Wiley VCH
4.3 Grafting
Immobilization of organic functionality into the interlayer space through covalent bonding is a way of the surface modification, and this is available for layered solids with interlayer hydroxyl groups. Reactions between layered silicates and silane coupling agents became possible after the pioneering work by Ruiz-Hitzky and Rojo [105], and studies on this topic have been summarized in a recent review [106]. Introduction of trimethylsilyl groups into a layered silicate (kanemite) led to effective benzene adsorption by weakening the interaction between the interlayer hydrogen bonds of the surface hydroxyl groups [107]. Distance between adjacent organic groups has been controlled by covalent attachment of octylsilyl groups to layered silicate, magadiite [108–111]. Alkyl alcohols were intercalated into the organic derivatives modified with controlled amounts of octylsilyl groups, while alkanes were not adsorbed [108]. Larger amounts of alcohols were intercalated into the derivatives with lower surface coverage. These results have been explained as the cooperative effect of the geometry and the chemical nature of the surface covered with octyl groups and silanol groups.
Selective adsorption of 4-nonylphenol from the mixtures of n-nonane, phenol and 4-nonylphenol in water was observed by attaching both functionalities of octadecyl and phenyl groups when layered lithium potassium titanate (K0.8Ti1.73Li0.27O4) [112] and layered alkali silicate (octosilicate) [113] were modified through silylation. These phenomena were explained as the result of the cooperative effect of chemical interactions and geometrical matching of the modified titanate and silicates, where two functional units (octadecyl and phenyl groups) were arranged to give the selective binding of 4-nonylphenol.
4.4 Inorganic Modification
Deposition/immobilization of nanoparticles of metals and their oxides onto inorganic layered solids have been widely investigated for applications as developing molecular-sieve-like catalysts. The reduction of Cu2+-exchanged montmorillonite (Kunipia F) with ethylene glycol resulted in the formation of Cu nanoparticle with the diameter less than 0.5 nm on montmorillonite [114]. We have investigated the immobilization of gold nanoparticles into the interlayer space of a layered alkali silicate and titanate utilizing a thiol-bearing organoammonium and silane coupling reagent [115, 116]. Disk-like gold nanoparticles formed in the interlayer space of a layered titanate modified with 3-(mercaptopropyl)trimethoxysilane, and the hybrid showed efficient and selective photocatalytic oxidation of aqueous benzene to phenol under visible light irradiation (λ > 420 nm) when the reaction was conducted in the presence of aqueous phenol [117]. Choy, Hwang and co-workers have reported pillaring layered titanate with NiO by the reaction between the aqueous (exfoliated) H0.67Ti1.83□0.17O4 (□ = vacancy) suspension and the precursor of NiO and the application for selective cyclohexane epoxidation [118].
Smectites pillared with metal oxides, represented by Al2O3- or ZrO2-pillared clays, have been studied as useful adsorbents and catalyst supports because of their thermal stability [119–123]. An alumina pillared montmorillonite (Wyoming bentonite) fabricated by using [AlO4Al12(OH)24(H2O)12]7+ polyoxycations as the pillaring agent was proposed as a recyclable surfactant support for the adsorption of organic toxicants in water and subsequent combustion [124]. Besides smectites, layered alkali silicates [125] and layered titanates [125, 126] have also been used as scaffolds for designing such pillared materials.
Controlled hydration of layered titanates (M x Ti2−x/3Li x/3O4; M = K, Li, Na; x = 0.61–0.76) in water affected adsorption of organic molecules and photocatalytic activity [127]. The interlayer cation and layer charge density of the titanates determined the water vapor adsorption and hydration in liquid water, and the Na-form with the lowest layer charge density (Na x Ti2−x/3Li x/3O4; x = 0.61) showed a larger amount of water vapor uptake and the expansion of the interlayer space (Fig. 9.13). The greater water uptake played an important role in photocatalytic selective benzene oxidation on the titanate from aqueous mixture; benzene was preferentially decomposed in the aqueous mixture with phenol and 4-butylphenol by UV irradiation (Fig. 9.14). The selectivity was thought to be due to the access into the interlayer space; size of benzene was smaller than the gallery height (~0.7 nm) of the Na-form for effective access of benzene to the interlayer surface to be decomposed. On the other hand, benzene was hardly intercalated into the K-titanate, which does not swell, and into the Li-titanate with lower swelling ability than that of the Na-form (Fig. 9.14).

Water adsorption (filled symbol)/desorption (open symbol) isotherms of a K x Ti2−x/3Li x/3O4 [x = (red filled/open circle) 0.61, (blue filled/open circle) 0.67, (green filled/open circle) 0.74], b Li x Ti2−x/3Li x/3O4 [x = (red filled/open square) 0.61, (blue filled/open square) 0.67, (green filled/open square) 0.76], and c Na x Ti2−x/3Li x/3O4 [x = (red filled/open triangle) 0.61, (blue filled/open triangle) 0.67, (green filled/open triangle) 0.76]. Reprinted from Ref. [127]. Copyright 2010 American Chemical Society

Photocatalytic decomposition of (white circle) benzene, (white square) phenol, and (cross sign) 4-butylphenol in water by a K x Ti2−x/3Li x/3O4 (x = 0.74), b Li x Ti2−x/3Li x/3O4 (x = 0.76), c Na x Ti2−x/3Li x/3O4 (x = 0.61), and d titanium dioxide (P25). Vertical lines indicate the beginning of ultraviolet light irradiation. Reprinted from Ref. [127]. Copyright 2010 American Chemical Society
4.5 Characterization
Understanding the interlayer hydrated structures of smectites (the orientation and the dynamics of the intercalated water) has been recognized as prime importance for assessing chemical and geological phenomena. The phenomena include barriers for underground storage of radioactive waste and rheological behavior of soil, which contributes to slipping processes in plate-boundary faults. Intercalation of water molecules is characterized by the stepwise expansion of the interlayer space with increasing water amount, which was controlled by the atmospheric humidity [128, 129]. The interlayer hydrated structures have been assessed using in situ XRD along water adsorption/desorption isotherms [130], 1H and 2H NMR [131], and neutron scattering [132]. A recent synchrotron-radiation X-ray diffraction study by Sasai et al. [133] showed large thermal vibration of hydrated Cl ions in the interlayer space because of higher mobility compared with the dehydrated form (Fig. 9.15). Difference in the vibration in the presence of water depending on the nature of anion should be important in anion-exchange properties of LDHs. The structure and dynamics of interlayer water molecules in smectites and LDHs can be revealed by computer simulations including Monte Carlo and molecular dynamics (MD). The structure and swelling behavior of smectites and LDHs have been more precisely understood at a molecular level [42, 134–139], although the nano-sized nature and lack of suitable large single crystals for accurate structure determinations except for a recent example on Na-fluorohectorite with quite large aspect ratio [140a]. Hydrated interlayer structure of well-ordered crystalline Na-fluorohectorite have been evaluated at 2 GPa in an excess of liquid water, resulting in successful detection of the phase transition from 2 to 3 water layers as a stepwise fashion [140b].

Fine crystal structure and electron density distribution of Cl–Mg/Al-LDH (Mg/Al = 2) derived by the MEM and Rietveld method from SXRD data measured at 300 and 523 K. Isosurface level of the charge density is 0.5eÅ−3. Reprinted from Ref. [133]. Copyright 2013 The Chemical Society of Japan
A transmission X-ray diffraction technique has been adopted to pursue the interlayer structural change upon the intercalation of caffeine from aqueous solution [141]; benzylammonium-exchanged smectites with the basal spacing of 1.3–1.4 nm swelled in liquid water to be 1.60 nm, and further expanded to 1.70 nm in an aqueous caffeine solution (Fig. 9.16). It has been assumed that the expansion by water molecules facilitates caffeine adsorption (intercalation) into the interlayer space.

Change in the XRD pattern of caffeine-adsorbing benzylammonium-Kunipia F. The patterns of the aqueous dispersions were measured by a transmission technique in the presence and absence of caffeine (2.1 mM). The powder diffractions were recorded using a conventional method at room temperature with relative humidity ca. 50%
4.6 Stimuli Responsive Adsorbents
Stimuli responsive properties have been investigated for the creation of smart materials. Intercalation of photo-responsive functional groups into layered solids has been examined. In some cases, reversible change in the basal spacing has been observed in response to alteration of molecular configuration triggered by photoirradiation. Azobenzene is a photochromic dye, which shows reversible trans-to-cis photoisomerization by UV irradiation and subsequent visible light irradiation or thermal treatment (Fig. 9.17a) [142]. When a cationic azo dye, p-[2-(2-hydroxyethyldimethylammonio)ethoxy]azobenzene (Fig. 9.17b, abbreviated as AZ(OH)+) was intercalated in magadiite with the adsorbed amount of 1.90 meq/g silicate, the basal spacing changed after UV irradiation from 2.69 to 2.75 nm and the value came back to 2.69 nm upon visible light irradiation [143]. Intercalated AZ(OH)+ as J-aggregate in montmorillonite (CEC: 1.19 meq/g) did not exhibit the photoresponses of the basal spacing during the photoisomerization [144]. Even careful consideration of controlled orientation using amphiphilic cationic azobenzenes in layered silicates (smectites and magadiite), photoresponse of the basal spacing has hardly been observed [145–148]. A molecular dynamics simulation [149] indicates an importance of the flexibility of the functional groups attached to azobenzene in inducing such XRD detectable structural change. The AZ(OH)+ dye does not contain flexible long alkylchains, therefore, the photoisomerization induced the change in the microstructure detectable by XRD. At the photostationary state, a densely packed aggregate of AZ(OH)+ is possibly difficult to form in magadiite due to the geometric difference of the two isomers (approximate molar trans:cis ratio of 1:1), and this causes the change in the basal spacing.

a cis–trans Photoisomerization of azobenzene and b molecular structure of the cationic azobenzene derivatives used in our studies
Inoue and his co-workers have reported a densely packed intercalation of a polyfluorinate cationic azo dye ([2-(2,2,3,3,4,4,4-heptafluorobutylamino)ethyl]-{2-[4-(4-hexyphenylazo)-phenoxy]-ethyl}dimethylammonium: C3F-Azo) into a layered niobate [150] and a layered titanoniobate [151]. Reversible trans-to-cis photoisomerization of the intercalated azobenzene moiety accompanied the repeated changes in the basal spacings. The direction of the interlayer distance upon each isomerization was opposite to that observed in the magadiite system as described above. Morphological change was detected by AFM when the C3F-Azo-layered niobate hybrid film was used [152]. After the irradiation of ca. 370 nm light, the bottom edge of the film slid out (the sliding distance of ca. 1500 nm), while the height of the top edge maintained. Subsequent higher wavelength light (ca. 460 nm) irradiation resulted in sliding back to the original position before the photoirradiation. Photoresponsive morphological changes have also been reported in a hybrid film composed of an azobenzene containing polymer and a LDH obtained by using a layer-by-layer self-assembly technique [153]. In this case, AFM observations of the resulting film exhibited changes in the surface roughness with responses to UV and visible light irradiation.
These phenomena demonstrated a type of photomechanical effect and larger volume change is expected. In order to amplify the change in the basal spacing upon the photoirradiation, we examined photoresponse of phenol intercalation in smectites modified with AZ(OH)+ [154]. Phenol was intercalated into AZ(OH)+-Kunipia F to expand the interlayer space from 1.8 to 2.6 nm by mechanical mixing without solvent. As shown in the XRD patterns (Fig. 9.18), the basal spacing increased to 3.2 nm by the UV light irradiation, indicating phenol intercalation. Subsequent visible light irradiation resulted in the decrease in the basal spacing to the value before the UV irradiation, suggesting phenol deintercalation. It was assumed to the intercalation and deintercalation of phenol induced by reversible trans-to-cis isomerization of the intercalated AZ(OH)+. More polar nature of cis-AZ(OH)+ compared to the trans-form possibly caused the intercalation of phenol. We have defined this kind of materials as photoresponsive adsorbents.

Change in the XRD patterns of AZ(OH)+-Kunipia F by the reaction with phenol and by photoirradiations. Reprinted from Ref. [154]. Copyright 2004 Royal Society of Chemistry
To investigate the effects of the nano-structure on the photoinduced intercalation behavior, layered silicates with different layer charge density (Kunipia F, Sumecton SA and TSM) have been used [155]. While intercalation and photoinduced intercalation of phenol into AZ(OH)+-TSM and AZ(OH)+-Kunipia F were observed, those phenomena were not seen for AZ(OH)+-Sumecton SA. The structure of the AZ(OH)+-clays before the intercalation is different; an interdigitated monolayer of AZ(OH)+ with the longer molecular axis inclined to the silicate layer in Kunipia and TSM, and a monolayer coverage of AZ(OH)+ with their molecular long axis parallel to the silicate layer in Sumecton SA. The dye orientation correlates the intercalation and photoinduced intercalation of phenol.
Another cationic azo dye, AZ(CH3)+, in which hydroxyl group is absent (Fig. 9.17b), was synthesized to compare the adsorptive properties of AZ(OH)+-Kunipia F [156] (Fig. 9.19). Although the gallery height of AZ(CH3)+-Kunipia F also changed by the phenol intercalation as also seen in the AZ(OH)+-Kunipia F system, the amount of the intercalated phenol into AZ(CH3)+-Kunipia F was relatively large compared with that in AZ(OH)+-Kunipia F. Due to the absence of the hydroxyl group, the hydrophobic nature enhanced to lead larger amount of the intercalated phenol before irradiation. On the contrary, the amount of the intercalated phenol after the UV irradiation for AZ(CH3)+-Kunipia F was relatively small. No intercalation at ground state, larger amount of intercalation by irradiation, complete deintercalation by subsequent irradiation and smooth response will be required to find practical application of these phenomena as advanced smart adsorbents.

The change in the gallery heights of AZ(CH3)+- and AZ(OH)+-Kunipia F; a before the intercalation of phenol, b after phenol intercalation, c after UV irradiation and d after subsequent visible light irradiation. Reprinted from Ref. [156]. Copyright 2008 Elsevier
5 Adsorption of Polymers
Layered solids dispersed in water often adsorb polymers including polyelectrolytes and nonionic polymers from aqueous solution. The adsorption of water-soluble polymers onto clay minerals (montmorillonite, in particular) have long been studied, because of many useful applications as flocculation aids, soil conditioners, drilling mud-extenders, and protection of salt–water drilling fluids [157]. The sol of clay minerals (suspension) in water readily flocculated by adding only small amount of positively charged polyelectrolyte due to the electrostatic interactions. Crystal edges of montmorillonite are thought to be a possible adsorption sites for a negatively charged polyelectrolyte. Although the polyanions (e.g., Na-polymethacrylate) alone do not flocculate the clay sol, addition of salt to the suspension causes the flocculation [158, 159]. Small amount of polyanion is thought to be adsorbed on several clay sol particles, followed by forming a bridge between them. The salt played a role to allow the sol particles to come closer together by the reduction of the range of double-layer repulsion and thus to promote bridging of the particles. The sequence of the addition of the polyanion and the salt is important in order to prevent re-dispersion (repeptized) by washing with water (or in order to act as flocculation aids effectively); salt should be added to the sol before the polyelectrolyte addition. Otherwise, the suspension is repeptized during the washing with water, because the clay surfaces are not covered completely with the polyions due to less coagulated clay particles in the absence of salt (Fig. 9.20) [157, 158].

Schematic representation of the interaction of clay particles with polyanions and salt as a function of the sequence of addition. a Salt added before adding polyanions. b Salt added after the addition of polyanions. Reprinted from Ref. [157]
Adsorption of enzymes (urease and glucose oxidase) on smectites [160, 161] was also reported in order to apply smectites as a support to reproduce enzymatic function. While the adsorption of enzyme on smectites is pH dependent, the enzyme is strongly bound by alkylammonium-smectites through hydrophobic interactions. Such interactions may involve hydrophobic portion of the enzymes interacting with the hydrophobic alkyl groups on the surface of clays.
Nonionic polymers (for examples of polysaccharides and polyoxyethylenes) were also adsorbed on smectites. At higher concentrations, the polymers usually have a protective action on clay sols (behaving as protective colloids), while at lower concentrations, they sometimes flocculate a clay sol (the flocculation does not occur for low-molecular weight polymers) [157]. Adsorption of a water-soluble polymer has been used to control the state of organic cations as well as the spatial distribution on smectites [162–164]. The isolation of [Ru(bpy)3]2+ has been achieved by the co-intercalation of poly(vinyl pyrrolidone) (abbreviated as PVP) [162, 163]. The controlled expansion of the interlayer space (design the size of two-dimensional nanospace) was achieved by changing PVP amount on synthetic saponite (Sumecton SA) [164].
6 Adsorption of Nanoparticles
Adsorption of colloidal nanoparticles including silica [165, 166], titania [167, 168], and CdS [169, 170] into montmorillonite was reported. Material’s applications of these resulting solids have been investigated in fields of photo- and electrochemistry. The adsorption often leads to provide intercalation compounds (to form pillared-layered solids). TiO2 sol (1.5 nm in diameter) pillared montmorillonite was prepared for the photocatalytic decomposition of n-carboxylic acids with up to 8 carbons, while the catalytic activity for capric acid was lower than that over bare TiO2. The adsorption, in some cases, gives homogeneous gels in which the order in the layer stacking is partially or completely lost (hetero-coagulation) [166, 168].
Besides the interest in advanced materials applications, the immobilization of nanoparticles on solids is a concern involving general environmental issues [171–173]. Nanoparticles in nature (especially those in air and water) are often regarded as contaminants and reservoir of toxic compounds, and their removal is required for environmental remediation. Depending on the nature of environmental nanoparticles (clays, pollen, etc.), various methods (filtration, precipitation, chemically induced aggregation, and so forth) have been used for their removal from the environment. Electrostatic interactions between negatively charged colloidal particles (β-FeOOH and a synthetic smectite) and positively charged LDH (hydrotalcite) have been used as a driving force for the nanoparticle immobilization [174]. β-FeOOH, which dispersed stably in water, precipitated after the addition of LDH as shown in the photograph (Fig. 9.21). The relationship of the β-FeOOH amounts between remaining and removed in/from the aqueous phase was similar to the shape of the Langmuir-type adsorption isotherm, suggesting strong interactions between β-FeOOH and LDH. A synthetic saponite (Sumecton SA) also precipitated after the LDH addition. The concept was adopted to cause precipitation of a positively charged colloidal particle (fine LDH particle) by micrometer-sized platy particles (octosilicate: a layered sodium silicate).

β-FeOOH precipitation after addition of LDH. The photograph was taken after the sample stood for 7 h. Reprinted from Ref. [174]. Copyright 2013 American Chemical Society
7 Morphosynthesis of Layered Solids for System Design
In order to regulate macroscopic shapes required for some uses and sensing and electronics, bottom-up self-assembly of layered solid nanosheets using the electrostatic interactions has been developed by depositing exfoliated nanosheets on a substrate and by molding to useful shapes using spray-drying [175] and freeze-drying techniques [176–178]. Optically transparent films [179–181] have been obtained by means of various fabrication techniques, including simple casting, Langmuir–Blodgett method [182], and layer-by-layer deposition technique [183–186].
Thanks to the optically transparent films, sensors with optical detection are possible. When the adsorption of a certain molecular species causes color change of the transparent films, it can be applied to colorimetric sensing for the environmental pollutants detection. Takagi et al. have reported solvatochromism of porphyrin-Sumecton SA complex membrane prepared by filtering the aqueous dye-clay suspension with PTFE membrane filter before transferring the residual membrane on the PTFE to a cover glass. The orientation of the porphyrin in the clay interlayer space changes by organic solvents to cause the color change [187].
Structural color (interference) is also applicable for the detection. The films coated on a high reflective index substrate (titania or Si) leads to the structural color as a result of the controlled thickness, which is a possible way to detect the adsorption without chromophores [185, 186]. By the adsorption (intercalation) of organic molecules or cations, an interference color changes, according to the optical Bragg low as the following equation:
(where n eff and Λ are the refractive index of a substrate and its period, respectively). This equation can be met by modulation of n eff and by changing the clay layer dimension (Λ) through swelling by the intercalation of organic substances. Since the observed interference color (derived from λ B) is a function of clay film thickness and refractive index, changes in the latter parameters directly translate into optically perceptible color change. A color change, corresponding to a red-shift of stop-band (which does not allow visible light to pass), was able to be visually observed, when exposing the film to aliphatic quaternary organoammoniums ions, cationic polyelectrolytes and a nonionic surfactant. Bulky surfactants and their higher loading gave rise to the increase in the basal spacing of Laponite, thus leading to a larger red-shift of the stop-band (Fig. 9.22). On the contrary, only a slight change in the stop-band was observed when immersing in a solution of neutral aromatic compound probably due to small amount of the intercalate.

a Relative optical shifts of Laponite thin films after exposure to 0.03 M surfactant solutions in ethanol. Increasing bulkiness translates into an enhanced optical response. b Position of the stop-band of a TiO2 hybridized film before (black) and after (gray) exposure of DDTMABr (top) and PDDA (bottom). c Relative optical shifts of three films with respect to different analytes. Legend of abbreviations: CTAB cetyltrimethylammonium bromide; DDDMABr didodecyldimethylammonium bromide; DDTMABr dodecyltrimethylammonium bromide; NaDOSS sodium dioctyl sulfosuccinate; PDDA poly(diallyldimethylammonium chloride); POE-10 polyoxyethylene-10-laurylether; SDS sodium dodecylsulfate; TEABr tetraethylammonium bromide; TBABr tetrabutylammonium bromide; TOABr tetraoctylammonium bromide. Reprinted from Ref. [185]. Copyright 2008 Wiley VCH
The film of layered solids was also applied for electrochemical detection of organic contaminants. Redox reactions in response to selective adsorption of analytes on electrodes modified with layered solids (e.g., smectites and LDHs) have been widely investigated as electrochemical sensors and biosensors [188]. Piezoelectronic sensors have also been developed; Organoammonium-hectorites (i.e., ODTMA-, TMA-hectorite) were coated on the gold electrodes of the QCM by dipping the crystal in their suspensions [189]. Water, acetonitrile/water or chloroform was used as solvents depending upon the dispersion properties of the organoammonium-hectorites. The ODTMA-hectorite film on the electrode adsorbed dichloromethane and toluene. On the contrary, the adsorption of n-hexane and n-octane on the ODTMA-hectorite did not occur even when the interlayer microenvironment was hydrophobic (Fig. 9.23). They deduced that the molecular size of sorbates as well as hydrophobic interactions affected the unique sorption behavior.

Transient sorption of vapor pulses at 100 °C on 6.0 MHz QCMs coated with ODTMA-hectorite film (181 μg/cm2). Reprinted from Ref. [189]. Copyright 1993 American Chemical Society
In addition to the macroscopic organization of layered materials, controlled particle morphology has been a topic of interests. Application as chromatography stationary phase was achieved by the processing of layered materials into spherical particle. Spherical particles of a synthetic hectorite (average radius 5 μm) were obtained by spray-drying and has been used as a HPLC column packing material. The ion exchange of the spherical particles of hectorite (RU-1 from Shiseido Co. Ltd) with enantiomers has been reported for chiral discrimination [175, 190–192]; an ion-exchanged adduct of a clay and enantiomeric tris(1,10-phenanthroline)ruthenium(II) ion (Δ- or Λ-[Ru(phen)3]2+) was used as a packing material [193–197], where a vacant space surrounded by enantio pure [Ru(phen)3]2+ molecules provides an adsorption site with high chiral discrimination [198].
Rectangular single crystals such as layered niobate with mm-scale [199–201], silicate (octosilicate) with μm-scale [13, 202, 203] are known examples of controlled and defined morphology. Crystal (particle) size and morphology of LDHs are also controllable [204]. Monodisperse particles of LDHs have been obtained [205–210]. To obtain high quality-homogeneous crystals, homogeneous precipitation using urea hydrolysis has been adopted because pH rises homogeneously in the solution to avoid heterogeneity of mixing (or pH change); successful preparation of LDHs with huge hexagonal platy hydrotalcite particles (25 μm) [207] and associated to a relatively narrow particle size distribution [208–210]. Nanometer-sized LDH particles with narrow particle size distribution have been obtained using an anion-exchange resin to adjust the pH for the precipitation [211]. Using a tripodal ligand of tris(hydroxymethyl)aminomethane is an example on producing smaller LDH particles (10 nm) [212].
The direct (in situ) crystallization of layered solids on bulk solid substrates has been reported as an adsorbent design [213–223], which is a way to produce mechanically improved supporting materials (avoid exfoliation). When other chemical sources are supplied from the solution, the solid substrate has been used as a source of layered materials. As a result of the reactions at the interface (heterogeneous nucleation reactions), fine crystals form only on the substrates (maintaining the shapes of the substrate). The choice of solid substrate chemicals in a solution leads to forming various fine crystals at the interfaces such as M(=Ni, Zn)/Al layered double hydroxides (LDHs)–Al2O3 plates (Fig. 9.24) [213, 214], M(=Mg, Zn)/Al-LDH–Al2O3 microfibers [215, 216], hectorite–silica microspheres [217–219], saponite–silica microspheres [220], hectorite–silica microfiber filter paper (Fig. 9.25) [221], and titanosilicate–silica microfibers (Fig. 9.26) [222].

SEM images of Ni/Al-LDH films obtained after 36 h at 120 °C on porous alumina/aluminum substrates: a top view of the Ni/Al-LDH film, b a high-magnification SEM image of a, and c cross-section of the Ni/Al-LDH film and the corresponding EDX line scan. Reprinted from Ref. [213]. Copyright 2006 Wiley-VCH

SEM images of a the pristine silica fiber and b hectorite–silica microfiber. c XRD pattern. d A cross-sectional TEM image of the hectorite–silica microfiber. Reprinted from Ref. [221]. Copyright 2016 Royal Society of Chemistry

FE-SEM images of a a cross-section of a titanosilicate (JDF-L1)-glass fiber hybrid where they observed the growth of the titanosilicate crystals radial to the glass fibers, b as-made JDF-L1, c naked glass fiber, and d the hybrid obtained after 4 days of reaction. Reprinted from Ref. [222]. Copyright 2013 American Chemical Society
Because the direct crystallization technique leads thin layer coating of layered solids on a substrate, the adsorption–desorption equilibrium for such core-shell particle is likely accomplished within the thin shell layer. For the application to a chiral HPLC application, elution would proceed rapidly, maintaining high degree of chiral discrimination. Motivated by this expectation, a clay column based on a hectorite-coated silica gel was prepared and its chromatographic behavior has been investigated [223]. A number of racemic mixtures including chiral neutral metal complexes and 1,1′-binaphthol were optically resolved. In case of tris(acetylacetonato)ruthenium(III), for example, the total elution volume was 100 mL when the racemic mixture was eluted with methanol on a 4 mm (i.d.) × 25 cm column in which Δ-[Ru(phen)3]2+ was mounted on the commercial RU-1, indicating that the clay column adsorbed molecules so strongly that a large volume of solvent was required for elution. On the other hand, the silica gel with a thin layer (ca. 0.1 μm thickness) elution volume was reduced to 10% when it was compared with RU-1.
8 Summary and Future Perspectives
Studies on layered materials as advanced adsorbents were summarized. Various useful phenomena for environmental purification or concentration of noble species have been found during the careful and extended studies on the adsorption of various species onto layered solids. Due to the natural abundance, the materials’ variation and useful adsorption behavior toward various cationic species, the application of bentonites and smectite group of clay minerals is promising. In addition, some useful ion exchange properties such as the heavy metal concentration and the radioactive metal removal have been observed and the nature of the selectivity and efficiency has been investigated. The chemical modification of layered materials is also quite useful to tune the adsorption behavior. Due to the increased demands for the efficient and selective concentration of a target species from the environments, the materials’ design will be conducted further along the line of careful syntheses of layered materials and their chemical modification. Toward more specific targets (like huge amount of waste from industry and atomic power plant, very low concentration of biohazard etc.) the optimization of the materials design including process and production cost should be done.
References
Barrer RM (1978) Zeolites and clay minerals as sorbents and molecular sieves. Academic Press, London
Jordan JW (1950) J Phys Colloid Chem 54:294
Bergaya F, Theng BKG, Lagaly G (eds) (2006) Handbook of clay science. Elsevier Science, Amsterdam
(a) Maes A, Cremers A (1978) J Chem Soc Faraday Trans 174:1234: (b) Cremers A, Thomas HC (1968) Israel J Chem 6:949; c Martin H, Laudelout H (1963) J Chim Phys 60:1086; (d) Gast RG (1969) Soil Sci Soc Amer Proc 33:661; (e) Gast RG (1972) Soil Sci Soc Amer Proc 36:14
Sawhney L (1972) Clays Clay Miner 20:93
Scott AD, Smith SJ (1966) Clays Clay Miner 14:69
Komarneni S, Roy R (1988) Science 239:1286
Shimizu K, Hasegawa K, Nakamuro Y, Kodama T, Komarneni S (2004) J Mater Chem 14:1031
Coppin F, Berger G, Bauer A, Castet S, Loubet M (2002) Chem Geol 182:57
Okada T, Ehara Y, Ogawa M (2007) Clays Clay Miner 55:348
Lagaly G (1979) Adv Colloid Interface Sci 11:105
Lagaly G, Beneke K, Weiss A (1975) Am Mineral 60:642
Iler RK (1964) J Colloid Sci 19:648
Ide Y, Ochi N, Ogawa M (2011) Angew Chem Int Ed 50:654
Sasaki T, Komatsu Y, Fujiki Y (1986) J Radioanal Nucl 107:111
Komatsu Y, Fujiki Y (1980) Chem Lett 9:1525
Yang X, Makita Y, Hosokawa J, Sakane K, Ooi K (2005) Chem Mater 17:5420
Chitrakar R, Tezuka S, Sonoda A, Kakita H, Sakane K, Ooi K, Hirotsu T (2008) Ind Eng Chem Res 47:176
Kinomura N, Kumada N, Muto F (1985) J Chem Soc Dalton Trans 2349
Reichle WT (1986) Solid States Ionics 22:135
Miyata S (1983) Clays Clay Miner 31:305
Yamaoka T, Abe M, Tsuji M (1989) Mater Res Bull 24:1183
(a) Hines DR, Solin SA, Costantino U, Nocchetti M (2000) Phys Rev B 61:11348; (b) Iyi N, Matsumoto T, Kaneko Y, Kitamura K (2004) Chem Mater 16:2926; (c) Iyi N, Sasaki T (2008) J Colloid Interface Sci 322:237
Hayashi A, Nakayama H (2011) Chem Lett 40:276
Tezuka S, Chitrakar R, Sonoda A, Ooi K, Tomida T (2004) Green Chem 6:104
Chitrakar R, Tezuka S, Hosokawa J, Makita Y, Sonoda A, Ooi K, Hirotsu T (2010) J Colloid Interface Sci 349:314
Kaneko S, Ogawa M (2013) Appl Clay Sci 75–76:109
Ogawa M, Asai S (2000) Chem Mater 12:3253
Ogawa M, Inomata K (2005) Chem Lett 34:810
Aisawa S, Higashiyama N, Takahashi S, Hirahara H, Ikematsu D, Kondo H, Nakatama H, Narita E (2007) Appl Clay Sci 35:146
Thomas J, Bohor BF (1968) Clays Clay Miner 16:83
Fripiat JJ, Cruz MI, Bohor BF, Thomas J Jr (1974) Clays Clay Miner 22:23
Hemmen H, Rolseth EG, Fonseca DM, Hansen EL, Fossum JO, Plivelic TS (2012) Langmuir 28:1678
Michels L, Fossum JO, Rozynek Z, Hemmen H, Rustenberg K, Sobas PA, Kalantzopoulos GN, Knudsen KD, Janek M, Plivelic TS, de Sliva GJ (2015) Sci Rep 5:8775
Loring JS, Schaef HT, Turcu RVF, Thompson CJ, Miller QRS, Martin PF, Hu J, Hoyt DW, Qafoku O, Ilton ES, Felmy AR, Rosso KM (2012) Langmuir 28:7125
Loring JS, Ilton ES, Chen J, Thompson CJ, Martin PF, Benezeth P, Rosso KM, Felmy AR, Schaef HT (2014) Langmuir 30:6120
Cygan RT, Romanov VN, Myshakin EM (2012) J Phys Chem C 116:13079
Botan A, Rotemberg B, Marry V, Turq P, Noetinger B (2010) J Phys Chem C 114:14962
Myshakin EM, Saidi WA, Romanov VN, Cygan RT, Jordan KD (2013) J Phys Chem C 117:11028
Lee MS, McGrail BP, Glezakou V-M (2014) Environ Sci Technol 48:8612
Sena MM, Morrow CP, Kirkpatrick RJ, Krishnan M (2015) Chem Mater 27:6946
Cygan RT, Liang J-J, Kalinichev AG (2004) J Phys Chem B 108:1255
Miyata S, Hirose T (1978) Clays Clay Miner 26:441
Hutson ND, Speakman SA, Payzant EA (2004) Chem Mater 16:4135
Reddy MK, Xu ZP, Lu GQ, Diniz JC (2006) Ind Eng Chem Res 45:7504
Wang Q, Gao Y, Luo J, Zheng Z, Borgna A, Guo Z, O’Hare D (2013) RSC Adv 3:3414
Gao Y, Zhang Z, Wu J, Yi X, Zheng A, Umar A, O’Hare D, Wang Q (2013) J Mater Chem A 1:12782
Ishihara S, Sahoo P, Deguchi K, Ohki S, Tansho M, Shimizu T, Labuta J, Hill JP, Ariga K, Watanabe K, Yamauchi Y, Suehara S, Iyi N (2013) J Am Chem Soc 135:18040
Sahoo P, Ishihara S, Yamada K, Deguchi K, Ohki S, Tansho M, Shimizu T, Eisaku N, Sasai R, Labuta J, Ishikawa D, Hill JP, Ariga K, Prasad B, Bastakoti P, Yamauchi Y, Iyi N, Appl ACS (2014) Mater Interfaces 6:18352
Constantino VRL, Pinnavaia TJ (1995) Inorg Chem 34:883
Theng BKG (1974) The chemistry of clay-organic reactions. Adam Hilger, London
Ogawa M, Kuroda K (1997) Bull Chem Soc Jpn 70:2593
Okada T, Ide Y, Ogawa M (2012) Chem–Asian J 7:1980
Okada T, Seki Y, Ogawa M (2014) J Nanosci Nanotech 14:2121
Ogawa M, Kuroda K, Kato C (1989) Chem Lett 18:1659
Ogawa M, Hirata K, Kuroda K, Kato C (1992) Chem Lett 21:365
Lagaly G (1981) Clay Miner 16:1
Izawa H, Kikkawa S, Koizumi M (1983) Polyhedron 2:741
Lagaly G, Beneke K (1976) J Inorg Nucl Chem 38:1513
Jones TR (1983) Clay Miner 18:399
Meyn M, Beneke K, Lagaly G (1990) Inorg Chem 29:5201
Morioka S, Tagaya H, Karasu M, Kadokawa J, Chiba K (1995) J Solid State Chem 117:337
Chiou CT, Peters LJ, Freed VH (1979) Science 207:831
Boyd SA, Lee JF, Mortland MM (1988) Nature 333:345
Xu S, Sheng G, Boyd SA (1997) Adv Agron 59:25
Jaynes WF, Boyd SA (1991) Soil Sci Soc Am J 55:43
Mortland MM, Shaobai S, Boyd SA (1986) Clays Clay Miner 34:581
Barriga C, Gaitan M, Pavlovic I, Ulbarri MA, Hermonsin MC, Cornejo J (2002) J Mater Chem 12:1027
Klumpp E, Ortegac CC, Klahrea P, Tinoa FJ, Yapard S, Prtilloc C, Stegenc S, Queiroloc F, Schwuger MJ (2004) Colloids Surf A 230:111
Chuang YH, Tzou YM, Wang MK, Liu CH, Chiang PN (2008) Ind Eng Chem Res 47:3813
Nakato T, Miyashita H, Yakabe S (2003) Chem Lett 32:72
Wei Q, Nakato T (2006) Microporous Mesoporous Mater 96:84
Hayashi A, Nakayama H (2003) Bull Chem Soc Jpn 76:2315
Jaynes WF, Vance GF (1999) Clays Clay Miner 47:358
Okada T, Morita T, Ogawa M (2004) Clay Sci 12:277
Okada T, Ogawa M (2002) Chem Lett 812
Okada T, Ogawa M (2003) Chem Commun 1378
Seki Y, Ogawa M (2010) Bull Chem Soc Jpn 83:712
Seki Y, Ide Y, Okada T, Ogawa M (2015) Appl Clay Sci 109–110:64
Rutherford D, Chiou CT, Eberl DD (1997) Clays Clay Miner 45:534
Ogawa M (2004) Photoprocesses in clay-organic complexes. In: Auerbach SM, Carrado KA, Dutta PK (eds) Handbook of layered materials handbook of layered materials. Marcel Dekker, New York, pp 191–259
Barrer RM (1989) Clays Clay Miner 37:385
Smith JA, Jaffe PR (1991) Environ Sci Technol 25:2054
Lao H, Latieule S, Detellier C (1991) Chem Mater 3:1009
Seki Y, Okada T, Ogawa M (2009) Microporous Mesoporous Mater 129:30
Lee JF, Mortland MM, Chiou CT, Kile DE, Boyd SA (1990) Clays Clay Miner 38:113
Lawrence MAM, Kukkadapu RK, Boyd SA (1998) Appl Clay Sci 13:13
Okada T, Morita T, Ogawa M (2005) Appl Clay Sci 29:45
Ogawa M, Kuroda K (1995) Chem Rev 95:399
Ogawa M (1998) Ann Rep (Sec. C) 94:209
Thomas JK (1988) Acc Chem Res 21:275
Yamagishi A (1987) J Coord Chem 16:131
Traynor MF, Mortland MM, Pinnavaia TJ (1978) Clays Clay Miner 26:318
(a) Yamagishi A, Taniguchi M, Imamura Y, Sato H (1996) Appl Clay Sci 11:1; (b) Fujita S, Sato H, Kakegawa N, Yamagishi A (2006) J Phys Chem B 110:2533
Breu J, Stoll A, Lange KG, Probst T (2001) Phys Chem Chem Phys 3:1232
Mermut AR (ed) (1994) Layer charge characteristics of 2:1 silicate clay minerals. CMS workshop lectures, vol 6. Clay Minerals Society
Hofmann U, Klemen RZ (1950) Anorg Allg Chem 262:95
Jaynes WF, Traina SJ, Bigham JM, Johnston CT (1992) Clays Clay Miner 40:397
Jaynes WF, Boyd SA (1991) Clays Clay Miner 39:428
Herling MM, Kalo H, Seibt S, Schobert R, Breu J (2012) Langmuir 28:14713
Ogawa M, Matsutomo T, Okada T (2008) J Ceram Soc Jpn 116:1309
Okada T, Matsutomo T, Ogawa M (2010) J Phys Chem C 114:539
(a) Stöcker M, Seidl W, Seyfarth L, Senker J, Breu J (2008) Chem Commun 629; (b) Stöcker M, Seyfarth L, Hirsemann D, Senker J, Breu J (2010) Appl Clay Sci 48:146; (c) Mariychuk R, Baumgartner A, Wagner FE, Lerf A, Dubbe A, Moos R, Breu J (2007) Chem Mater 19:5377; (d) Baumgartner A, Sattler K, Thun J, Breu J (2008) Angew Chem Int Ed 47:1640
Breu J, Seidl W, Stoll AJ, Lange KG, Probst TU (2001) Chem Mater 13:4213
Ruiz-Hitzky E, Rojo JM (1980) Nature 287:28
Takahashi N, Kuroda K (2011) J Mater Chem 21:14336
Toriya S, Kobayashi S, Takei T, Fuji M, Watanabe T, Chikazawa M (2003) Colloid Polym Sci 281:1121
Ogawa M, Okutomo S, Kuroda K (1998) J Am Chem Soc 120:7361
Okutomo S, Kuroda K, Ogawa N (1999) Appl Clay Sci 15:253
Fujita I, Kuroda K, Ogawa M (2003) Chem Mater 15:3134
Fujita I, Kuroda K, Ogawa M (2005) Chem Mater 17:3717
Ide Y, Ogawa M (2007) Angew Chem Int Ed 46:8449
Ide Y, Iwasaki S, Ogawa M (2011) Langmuir 27:2522
Malla PB, Ravindranathanm P, Komarneni S, Roy R (1991) Nature 351:555
Ide Y, Fukuoka A, Ogawa M (2007) Chem Mater 19:964
Ide Y, Nakasato Y, Ogawa M (2008) Bull Chem Soc Jpn 81:757
Ide Y, Matsuoka M, Ogawa M (2010) J Am Chem Soc 132:16762
Kim TW, Hwang S-J, Jhung SH, Chang J-S, Park H, Choi W, Choy J-H (2008) Adv Mater 20:539
(a) Pinnavaia TJ (1983) Science 220:365; (b) Ohtsuka K (1997) Chem Mater 9:2039
Michot LJ, Barres O, Hegg EL, Pinnavaia TJ (1993) Langmuir 9:1794
Galarneau A, Barodawalla A, Pinnavaia TJ (1995) Nature 374:529
Nakatsuji M, Ishii R, Wang Z-M, Ooi K (2004) J Colloid Interface Sci 272:158
Németh J, Rodríguez-Gattorno G, Díaz D, Vázquez-Olmos AR, Décany I (2004) Langmuir 20:2855
Michot LJ, Pinnavaia TJ (1991) Clays Clay Miner 39:634
Landis ME, Aufdembrink BA, Chu P, Johnson ID, Kirker GW, Rubin MK (1991) J Am Chem Soc 113:3189
Anderson MW, Klinowski J (1990) Inorg Chem 29:3260
Ide Y, Nakasato Y, Ogawa M (2010) J Am Chem Soc 132:3601
Norrish K (1954) Discuss Faraday Soc 18:120
Mooney RW, Keenan AG, Wood LA (1952) J Am Chem Soc 74:1371
Ferrage E, Lanson B, Michot LJ, Robert J-L (2010) J Phys Chem C 114:4515
Tenorio RP, Alme LR, Engelsberg M, Fossum JO, Hallwass F (2008) J Phys Chem C 112:575
Malikova N, Cadene A, Dubois E, Marry V, Durand-Vidal S, Turq P, Breu J, Longeville S, Zanotti J-M (2007) J Phys Chem C 111:17603
Sasai R, Matsuoka Y, Sato H, Moriyoshi C, Kuroiwa Y (2013) Chem Lett 42:1285
Emiel EJM, Hensen B (2002) J Phys Chem B 106:12664
Young DA, Smith DE (2000) J Phys Chem B 104:9163
Boek ES, Coveney PV, Skipper NT (1995) J Am Chem Soc 117:12608
Morrow CP, Yazaydin AO, Krishnan M, Bowers GM, Kalinichev AG, Kirkpatrick RJ (2013) J Phys Chem C 117:5172
Dazas B, Lanson B, Delville A, Robert J-L, Komarneni S, Michot LJ, Ferrage E (2015) J Phys Chem C 119:4158
Wang J, Kalinichev AG, Kirkpatrick RJ, Hou X (2001) Chem Mater 13:145
(a) Stöcker M, Kunz D, Schmidt M, Hirsemann D, Kalo H, Putz B, Senker J, Breu J (2013) Langmuir 29:1280; (b) You S, Kunz D, Stöcker M, Kalo H, Putz B, Breu J, Talyzin AV (2013) Angew Chem Int Ed 52:3891
Okada T, Oguchi J, Yamamoto K, Shiono T, Fujita M, Iiyama T (2015) Langmuir 31:180
Rau H (1990) In: Dürr H, Bouas-Laurent H (eds) Studies in organic chemistry: photochromism, molecules and systems, vol. 40. Elsevier, Amsterdam, pp 165–192
Ogawa M, Ishii T, Miyamoto N, Kuroda K (2001) Adv Mater 13:1107
Ogawa M, Ishii T, Miyamoto N, Kuroda K (2003) Appl Clay Sci 22:179
Ogawa M, Ishikawa A (1998) J Mater Chem 8:463
Ogawa M, Goto R, Kakegawa N (2000) Clay Sci 11:231
Ogawa M (1996) Chem Mater 8:1347
Ogawa M, Yamamoto M, Kuroda K (2001) Clay Miner 36:263
Heinz H, Vaia RA, Koerner H, Farmer BL (2008) Chem Mater 20:6444
Tong ZW, Takagi S, Shimada T, Tachibana H (2006) Inoue J Am Chem Soc 128:684
Tong ZW, Sasamoto S, Shimada T, Takagi S, Tachibana H, Zhang XB, Tryk DA, Inoue H (2008) J Mater Chem 18:4641
Nabetani Y, Takamura H, Hayasaka Y, Shimada T, Takagi S, Tachibana H, Masui D, Tong Z, Inoue H (2011) J Am Chem Soc 133:17130
Han J, Yan D, Shi W, Ma J, Yan H, Wei M, Evans DG, Duan X (2010) J Phys Chem B 114:5678
Okada T, Watanabe Y, Ogawa M (2004) Chem Commun 320
Okada T, Watanabe Y, Ogawa M (2005) J Mater Chem 15:987
Okada T, Sakai H, Ogawa M (2008) Appl Clay Sci 40:187
Van Olphen H (1977) An introduction to clay colloid chemistry, 2nd edn. Wiley-Interscience, New York, pp 174–181
Ruehrwein RA, Ward DW (1952) Soil Sci 73:485
Warkentin BP, Miller RD (1958) Soil Sci 85:14
Garwood GA, Mortland MM, Pinnavaia TJ (1983) J Mol Catal 22:153
Boyd SA, Mortland MM (1986) J Mol Catal 34:1
Ogawa M, Tsujimura M, Kuroda K (2000) Langmuir 16:4202
Kakegawa N, Ogawa M (2004) Langmuir 20:7004
Sohmiya M, Omata S, Ogawa M (2012) Polym Chem 3:1069
Letaïef S, Ruiz-Hitzky E (2003) Chem Commun 24:2996
Letaïef S, Martín-Luengo MA, Aranda P, Ruiz-Hitzky E (2006) Adv Funct Mater 16:401
Yoneyama H, Haga S, Yamanaka S (1989) J Phys Chem 93:4833
Mogyorósi K, Dékány I, Fendler JH (2003) Langmuir 19:2938
Enea O, Bard AJ (1986) J Phys Chem 90:301
Stramel RD, Nakamura T, Thomas JK (1986) Chem Phys Lett 130:423
Bradford S, Simunek J, Bettahar M, Van Genuchten MT, Yates SR (2003) Environ Sci Technol 37:2242
Sauer JE, Davis EJ (1994) Environ Sci Technol 28:737
Zevi Y, Dathe A, McCarthy JF, Richards BK, Steenhuis TS (2005) Environ Sci Technol 39:7055
Ogawa M, Kaneko S (2013) Langmuir 29:14469
Nakamura Y, Yamagishi A, Matsumoto S, Tohkubo K, Ohtsu Y, Yamaguchi M (1989) J Chromatogr 482:165
Call F (1953) Nature 172:126
Nakazawa H, Yamada H, Fujita T, Ito Y (1987) Clay Sci 6:269
Okada T, Kato T, Yamaguchi T, Sakai T, Mishima S (2013) Ind Eng Chem Res 52:12018
Ogawa M, Takahashi M, Kato C, Kuroda K (1994) J Mater Chem 4:519
Sasai R, Iyi N, Fujita T, Arbeloa LF, Martinez MV, Takagi K, Itoh H (2004) Langmuir 20:4715
Ishida Y, Kulasekharan R, Shimada T, Takagi S, Ramamurthy V (2013) Langmuir 29:1748
Inukai K, Hotta Y, Taniguchi M, Tomura S, Yamagishi A (1994) J Chem Soc Chem Commun 959
Kleinfeld ER, Ferguson GS (1994) Science 265:370
Lvov Y, Ariga K, Ichinose I, Kunitake T (1996) Langmuir 12:3038
Lotsch BV, Ozin GA (2008) Adv Mater 20:4079
Lotsch BV, Ozin GA (2008) ACS Nano 10:2065
Takagi S, Shimada T, Masui D, Tachibana H, Ishida Y, Tryk DA, Inoue H (2010) Langmuir 26:4639
Mously C (2004) Appl Clay Sci 27:159
Yan Y, Bein T (1993) Chem Mater 5:905
Ogawa T, Ohtsu T, Yamaguchi M (1992) Kuromatogurafi (Japanese) 13:315
Hirose K, Asada K, Darwish I, Akizawa T, Yoshioka M (1997) J Pharam Biomed Anal 1241
Hirose K, Akizawa T, Yoshioka M (1998) Anal Chim Acta 129
Yamagishi A, Soma M (1981) J Am Chem Soc 103:4640
Yamagishi A (1985) J Am Chem Soc 107:732
Yamagishi A, Taniguchi M, Imamura Y, Sato H (1996) Appl Clay Sci 11:1
Yamagishi A (1983) J Chromatogr A 262:41
Yamagishi A (1985) J Chromatogr A 319:299
Sato H, Yamagishi A, Kato S (1992) J Am Chem Soc 114:10933
Miyamoto N, Kuroda K, Ogawa M (2001) J Am Chem Soc 123:6949
Gasperin M, Bihan MTL (1982) J Solid State Chem 43:346
Nassau K, Shiever JW, Bernstein JL (1969) J Electrochem Soc 116:348
Ogawa M, Iwata D (2010) Cryst Growth Des 10:2068
Endo K, Sugawara Y, Kuroda K (1994) Bull Chem Soc Jpn 67:3352
Ogawa M, Inomata K (2011) Clay Sci 15:131
Li L, Ma R, Ebina Y, Iyi N, Sasaki T (2005) Chem Mater 17:4386
Liu Z, Ma R, Osada M, Iyi N, Ebina Y, Takada K, Sasaki T (2006) J Am Chem Soc 128:4872
Ogawa M, Kaiho H (2002) Langmuir 18:4240
Kayano M, Ogawa M (2006) Clays Clay Miner 54:382
Kayano M, Ogawa M (2006) Bull Chem Soc Jpn 79:1988
Arai Y, Ogawa M (2009) Appl Clay Sci 42:601
(a) Nitoh K, Ayral A, Ogawa M (2010) Chem Lett 39:1018; (b) Naito S, Nitoh K, Ayral A, Ogawa M (2012) Ind Eng Chem Res 51:14414
Kuroda Y, Miyamoto Y, Hibino M, Yamaguchi K, Mizuno N (2013) Chem Mater 25:2291
Chen H, Zhang F, Fu S, Duan X (2006) Adv Mater 18:3089
Zhang F, Zhan L, Chen H, Xu S, Evans DG, Duan X (2008) Angew Chem Int Ed 47:2466
Zhang T, Zhou Y, He M, Zhu Y, Bu X, Wang Y (2013) Chem Eng J 219:278
Zhang T, Zhou Y, Bu X, Xue J, Hu J, Wang Y, Zhang M (2014) Microporous Mesoporous Mater 188:37
Okada T, Yoshido S, Miura H, Yamakami T, Sakai T, Mishima S (2012) J Phys Chem C 116:21864
Okada T, Suzuki A, Yoshido S, Minamisawa MH (2015) Microporous Mesoporous Mater 215:168
Okada T, Suzuki A (2015) Clay Sci 19:45
Okada T, Sueyoshi M, Minamisawa HM (2015) Langmuir 31:13842
Okada T, Shimizu K, Yamakami T (2016) RSC Adv 6:26130
Pérez-Carvajal J, Aranda P, Berenguer-Murcia A, Cazorla-Amorós D, Coronas J, Ruiz-Hitzky E (2013) Langmuir 29:7449
Okada T, Kumasaki A, Shimizu K, Yamagishi A, Sato H (2016) J Chromatogr Sci 54:1238
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Japan KK
About this chapter
Cite this chapter
Okada, T., Ogawa, M. (2017). Adsorbents Derived from Layered Solids. In: Nakato, T., Kawamata, J., Takagi, S. (eds) Inorganic Nanosheets and Nanosheet-Based Materials. Nanostructure Science and Technology. Springer, Tokyo. https://doi.org/10.1007/978-4-431-56496-6_9
Download citation
DOI: https://doi.org/10.1007/978-4-431-56496-6_9
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-56494-2
Online ISBN: 978-4-431-56496-6
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)