
Abstract
Bicuspid aortic valve (BAV) has a prevalence of 0.5–1.39% in the general population. The prevalence of BAV in aortic dissection is 3.5–11.8%. Unfortunately, the incidence of aortic dissection in BAV remains unknown. The etiology of BAV is polygenetic, where environmental factors and unknown genetic factors seem to interact to cause BAV. In some instances, chromosomal aberrations or defined gene defects cause BAV.
Congenital BAV must be distinguished from acquired BAV. Congenital aortic valve malformations differ by number of cusps, ranging from one to five. BAV cusps can be subclassified according to patterns of calcification, severity of calcification, presence of a raphe, and fusion of cusps. We tend to perceive BAV as an isolated congenital heart defect. However, we identified 20 well-defined syndromic, complex, or isolated congenital heart defects that are associated with BAV disease, some of which are apparently quite frequent.
BAV aortopathy can be classified according to presence and type of aortic valve dysfunction, shape of the proximal aorta, aortic arch involvement, and coexistence with coarctation of the aorta.
Factors that may increase the risk for aneurysmal formation, aortic rupture, or dissection in BAV comprise aortic valve characteristics, comorbidities of BAV, and behavioral factors. Candidates for biomarkers of BAV aortopathy comprise a family history with early dissection or death, increased aortic growth rates, proximal aortic shape, aortic stiffness and aortic elasticity markers, aortic wall shear stress, endothelial dysfunction, and serological biomarkers.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Bicuspid aortic valve (BAV) may simply be viewed as “an aortic valve that has two cusps instead of three,” and BAV aortopathy may simply be considered as “post-stenotic dilatation of the ascending aorta.”
More than 500 years ago, Leonardo da Vinci explained and depicted BAV for the first time. Paget recalled in 1844 the tendency of the BAV to develop disease, Peacock mentioned the tendency of these valves to develop obstructions or regurgitation early, Osler described the predilection of these valves to infective endocarditis, and Victor Babes (1891), in Germany, and 3 decades later Maude Abbott, in North America (1927), commented on an association between congenital BAV and aortic aneurysm, dissection, and rupture, and since Abbott described aortopathy in BAV [1]. Since then, the literature on BAV disease has grown into complex field on detailed and conflicting knowledge.
2 Frequency of BAV and BAV Aortopathy
Besides mitral valve prolapse, BAV is the most frequent congenital heart defect in the general population. Autopsy studies and echocardiographic screening studies yield strikingly similar results on the frequency of BAV in the general population. The prevalence ranges from 0.5 to 1.39% at autopsy and between 0.5 and 0.9% on echocardiographic screening. Autopsy studies and a clinical register of aortic dissection assess the prevalence of BAV in individuals with aortic dissection. Both types of studies report BAV in 7.5–11.2% and 3.5–11.8% of aortic dissections. Nistri et al. found aortic dilatation, which they defined with diameters >2 standard deviation above normal, in 68.9% of young Italian conscripts with BAV at echocardiographic screening [2]. The widely cited 2% prevalence of BAV appears to stem from a single autopsy study published in 1923 [3]. Cohort studies that register aortic events in BAV report on 0.5–0.8% incidence of aortic dissection with various difference of intervals of follow-up, treatment standards, reasons for inclusion, and ages at inclusion into the cohort. Hence, the accurate prevalence and natural history of BAV aortopathy are difficult to interpret in these studies (Table 15.1).
3 Classification of BAV: Etiology
BAV is a congenital heart anomaly, and to the best of our knowledge, the literature reports exclusively genetic causes of BAV. However, other causes of BAV such as intrauterine infection or intoxication appear to be a possible cause. Moreover, the BAV phenotype is highly variable, and epigenetic modifiers and environmental factors are likely to play an important role in BAV disease event when a distinct causative genetic defect can be identified [19].
We classify the genetic causes of BAV according to frequency and mechanisms (Table 15.2). First, male predominance and familial occurrence of BAV are found in BAV, which argue for a genetic mechanism in a majority of individuals with BAV. Second, only a small fraction of individuals with BAV have chromosomal disorders. However, some chromosomal disorders, such as Turner syndrome, have BAV in up to 30%. Third, BAV may be a monogenetic disease, where autosomal dominant traits are most frequent, but where other traits, such as autosomal recessive or X-linked traits, may occur. Monogenetic BAV can occur sporadically [20].
NOTCH1, TGFBR1, and FBN1 are examples for genes, where series of patients suggest a causative relationship between gene defect and BAV phenotype. Conversely, ACTA2 and SMAD6 are examples for genes, where studies of individual patients or studies of relatives suggest such a causative relationship. In most of these genes, the pathogenic mechanism is unclear, and the association of mutation with phenotype is not firmly established. The NOTCH1, however, is a good example for a gene, where the association with BAV is well established, whereas the FBN1 gene or the DMD gene is an example, where this relationship has been questioned. In all putatively causative genes however, the BAV phenotype and the associated cardiovascular and systemic phenotype are variable.
The etiology of BAV is polygenetic, where environmental factors and unknown genetic factors seem to interact. In some instances, chromosomal aberrations or defined gene defects cause BAV.
4 Classification of BAV: Valve Anatomy
An aortic valve may be considered “bicuspid” when we identify two cusps instead of three. However, numerous classification systems are available to further differentiate or classify BAV on the basis of anatomical criteria (Table 15.3).
First, anatomical classifications distinguish BAV from other anatomical variants of the aortic valve. These classifications include differentiation of congenital from acquired BAV and differentiation of BAV from unicuspid (UAV), quadricuspid (QAV), or pentacuspid (PAV) aortic valves according to the number of aortic valve cusps.
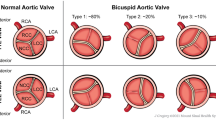
Second, anatomical classifications subclassify congenital BAV according to anatomical features of the aortic valve. Such classifications distinguish BAV according to patterns of valve calcification and the grade of valve calcification. However, most anatomical classifications focus on characterizing BAV according to which cusps are fused to one cusp and whether a raphe is present or absent. Unfortunately, there are many variants of such anatomical classifications, where some use the same expression to characterize different types of valves. We believe that a uniform classification system should be used. Buchner et al. distinguished BAV with raphe, where they classify BAV-RL, BAV-RN, and BAV-LN, depending on which aortic cusps, the right (R), left (L), or noncoronary (N), are fused, from BAV without raphe, where BAV-LA designated BAV with lateral orientation of the free edge of cusps and BAV-AP with anterior-posterior orientation. This classification covers all other classification systems and it is simple.
Third, we tabulate classification systems that combine the above described anatomical classification of BAV with other features of BAV disease, such as valvular function or aortic shape. However, such classifications may yield >20 subtypes, which are complicated to use without offering the reward of improving clinical management. Moreover, these combi-classifications are only in use to describe all possible combination of BAV anatomy with additional BAV disease features rather than that they establish new disease entities (such as a typical aortopathy in LR-BAV), and hence they do not provide additional insight into BAV disease.
Congenital BAV must be distinguished from acquired BAV. Congenital aortic valve malformations differ by number of cusps, ranging from one to five. BAV cusps can be subclassified according to patterns of calcification, severity of calcification, presence of a raphe, and fusion of cusps. Combi-classifications, where anatomical subtypes of BAV are combined with additional features of BAV disease, may be too complex for routine clinical use.
5 Classification of BAV: Associated Congenital Heart Defect (CHD)
We classify BAV into four categories according to the presence of associated congenital heart defects (CHDs):
-
1.
Smaller series report on the presence of BAV in syndromic or complex CHD, such as Ebstein’s anomaly, Shone’s complex, hypoplastic left heart syndrome, double-outlet right ventricle, tetralogy of Fallot, or complete transposition of the great arteries.
-
2.
BAV frequently associates with one typical additional CHD, where coarctation of the aorta (COA), patent ductus arteriosus (PDA), ventricular septal defect (VSD), and atrial septal defect (ASD) are the most common associates of BAV.
-
3.
Coronary arterial anomaly, bicuspid pulmonary valve (BPV), and mitral valve anomalies are rare in BAV, but their association with BAV is well established.
-
4.
There are only sparse or conflicting data on the potential association of BAV with CHD or vascular malformation such as myocardial abnormalities, familial aorto-cervicocephalic arterial dissections, intracranial aneurysms, and various arterial or venous vascular anomalies (Table 15.4).
Table 15.4 Classification of BAV according to associated congenital heart defect (CHD)
We tend to perceive BAV as an isolated CHD. However, we identified 20 well-defined syndromic, complex, or isolated congenital heart defects that are associated with BAV disease; some of them are apparently quite frequent.
6 Classification of BAV: Aortopathy
BAV may be associated with aortic dilatation or aneurysm of the proximal aorta, the aortic arch, the descending aorta, or the abdominal aorta. Some studies classified BAV into four groups by type of aortopathy. BAV aortopathy was classified:
-
1.
Type and presence of aortic valve dysfunction
-
2.
Geometrical configuration of the proximal part of the aorta
-
3.
Involvement of the aortic arch
-
4.
According to presence of coarctation of the aorta (COA)
None of these classifications have been applied prospectively in large cohorts of unselected individuals with BAV, and hence their overlap and comprehensiveness cannot be estimated properly. However, all classifications provide useful means to describe subtypes of BAV aortopathy for future assessment of prognosis (Table 15.5).
7 Classification of BAV Aortopathy: Risk Factors
Patients with BAV may exhibit additional factors that may increase diameter of the aorta and increase the risk of aortic aneurysm formation, aortic dissection, and rupture. We distinguish risk that may arise from three types of risk factors:
-
1.
From aortic valve characteristics, such as BAV morphotype, BAV stenosis, and BAV regurgitation
-
2.
From comorbidities of BAV, such as arterial hypertension (HTN), atherosclerosis, and coarctation of the aorta (BAV-COA), or from sleep apnea, comprising obstructive (OSA) and central sleep apnea (CSA)
-
3.
From behavioral factors including pregnancy, sports, high-performance aviation with G-force exposure, and drug abuse comprising cocaine, methamphetamine, and sildenafil
The evidence for increased risk for aortic complications is not equally strong for all factors in BAV (Table 15.6).
8 Classification of BAV Aortopathy: Candidate Biomarkers
Biomarkers should provide information of the development and evolution of BAV aortopathy. Aortic diameters clearly provide the single most important information on presence and risk of BAV aortopathy. Therefore, guidelines base their recommendations of timing for elective surgery of the aortic root predominantly on diameters [176–178]. Nonetheless, aortic size has to be judged differently depending on sex, body size, body surface, and the individual tissue stability of the aortic wall [179]. Additional biomarkers may be helpful to further stratify the risk of acute aortic events in BAV aortopathy.
Candidates for biomarkers of BAV aortopathy comprise a family history with dissection or death at younger age, increased aortic growth rates, proximal aortic shape, biomarkers of aortic stiffness and aortic elasticity markers, biomarkers of aortic wall shear stress (AWASS), biomarkers of endothelial dysfunction, and serological biomarkers (Table 15.7).
No single candidate biomarker of BAV aortopathy has currently accumulated evidence enough for introduction into clinical routine. However, many of the candidate biomarkers exhibit promising data. Some of these markers are likely to improve future management of BAV disease.
Abbreviations
- AOA:
-
Aortic arch
- ASC:
-
Ascending aorta
- AVA:
-
Aortic valve annulus (anatomical ventriculo-arterial junction)
- AVR:
-
Aortic valve replacement
- BAV:
-
Bicuspid aortic valve
- BAV-COA:
-
Coexistence of bicuspid aortic valve and coarctation of the aorta
- BAV-I:
-
BAV with predominant insufficiency
- BAV-LN:
-
BAV with fusion of the left and noncoronary cusp
- BAV-MO:
-
BAV morphotype
- BAV-RI:
-
BAV with balanced stenosis and insufficiency
- BAV-RL:
-
BAV with fusion of the right and left coronary cusp
- BAV-RN:
-
BAV with fusion of the right and noncoronary cusp
- BAV-S:
-
BAV with predominant stenosis
- CHD:
-
Congenital heart defect
- COA:
-
Coarctation of the aorta
- DESC:
-
Descending thoracic aorta
- HTN:
-
Arterial hypertension
- SOV:
-
Sinus of valsalva
- STJ:
-
Sinotubular junction
- TAV:
-
Tricuspid aortic valve
References
Braverman AC, Guven H, Beardslee MA, Makan M, Kates AM et al (2005) The bicuspid aortic valve. Curr Probl Cardiol 30:470–522
Nistri S, Basso C, Marzari C, Mormino P, Thiene G (2005) Frequency of bicuspid aortic valve in young male conscripts by echocardiogram. Am J Cardiol 96:718–721
Roberts WC (1970) The congenitally bicuspid aortic valve. A study of 85 autopsy cases. Am J Cardiol 26:72–83
Basso C, Boschello M, Perrone C, Mecenero A, Cera A et al (2004) An echocardiographic survey of primary school children for bicuspid aortic valve. Am J Cardiol 93:661–663
Edwards WD, Leaf DS, Edwards JE (1978) Dissecting aortic aneurysm associated with congenital bicuspid valve. Circulation 57:1022
Larson EW, Edwards WD (1984) Risk factors for aortic dissection: a necropsy study of 161 cases. Am J Cardiol 53:849–855
Roberts CS, Roberts WC (1991) Dissection of the aorta associated with congenital malformation of the aortic valve. J Am Coll Cardiol 17:712–716
Gray GW, Salisbury DA, Gulino AM (1995) Echocardiographic and color flow Doppler findings in military pilot applicants. Aviat Space Environ Med 66:32–34
Tutar E, Ekici F, Atalay S, Nacar N (2005) The prevalence of bicuspid aortic valve in newborns by echocardiographic screening. Am Heart J 150:513–515
Strader JR, Harrell TW, Adair A, Kruyer WB (2008) Efficacy of echocardiographic screening of pilot applicants. Aviat Space Environ Med 79:514–517
Januzzi JL, Isselbacher EM, Fattori R, Cooper JV, Smith DE et al (2004) Characterizing the young patient with aortic dissection: results from the International Registry of Aortic Dissection (IRAD). J Am Coll Cardiol 43:665–669
Di Eusanio M, Trimarchi S, Patel HJ, Hutchison S, Suzuki T et al (2013) Clinical presentation, management, and short-term outcome of patients with type A acute dissection complicated by mesenteric malperfusion: observations from the International Registry of Acute Aortic Dissection. J Thorac Cardiovasc Surg 145:385–390.e1
Michelena HI, Desjardins VA, Avierinos JF, Russo A, Nkomo VT et al (2008) Natural history of asymptomatic patients with normally functioning or minimally dysfunctional bicuspid aortic valve in the community. Circulation 117:2776–2784
McKellar SH, MacDonald RJ, Michelena HI, Connolly HM, Sundt TM 3rd (2011) Frequency of cardiovascular events in women with a congenitally bicuspid aortic valve in a single community and effect of pregnancy on events. Am J Cardiol 107:96–99
Tzemos N, Therrien J, Yip J, Thanassoulis G, Tremblay S et al (2008) Outcomes in adults with bicuspid aortic valves. JAMA 300:1317–1325
Davies RR, Kaple RK, Mandapati D, Gallo A, Botta DM Jr et al (2007) Natural history of ascending aortic aneurysms in the setting of an unreplaced bicuspid aortic valve. Ann Thorac Surg 83:1338–1344
La Canna G, Ficarra E, Tsagalau E, Nardi M, Morandini A et al (2006) Progression rate of ascending aortic dilation in patients with normally functioning bicuspid and tricuspid aortic valves. Am J Cardiol 98:249–253
Michelena HI, Khanna AD, Mahoney D, Margaryan E, Topilsky Y et al (2011) Incidence of aortic complications in patients with bicuspid aortic valves. JAMA 306:1104–1112
Prakash SK, Bossé Y, Muehlschlegel JD, Michelena HI, Limongelli G et al (2014) A roadmap to investigate the genetic basis of bicuspid aortic valve and its complications insights from the International BAVCon (Bicuspid Aortic Valve Consortium). J Am Coll Cardiol 64:832–839
Moran R, Robin NH (2013) Chapter 46: Congenital heart defects. In: Rimoin D, Korf RP (eds) Emery and Rimoin’s principles and practice of medical genetics, 6th edn. Academic, Oxford, pp 1–51
Hales AR, Mahle WT (2014) Echocardiography screening of siblings of children with bicuspid aortic valve. Pediatrics 133:e1212–e1217
Robledo-Carmona J, Rodríguez-Bailón I, Carrasco-Chinchilla F, Fernández B, Jiménez-Navarro M et al (2013) Hereditary patterns of bicuspid aortic valve in a hundred families. Int J Cardiol 168:3443–3449
Panayotova R, Macnab A, Waterworth PD (2013) A pilot project of familial screening in patients with bicuspid aortic valve disease. J Heart Valve Dis 22:150–155
Huntington K, Hunter AG, Chan KL (1997) A prospective study to assess the frequency of familial clustering of congenital bicuspid aortic valve. J Am Coll Cardiol 30:1809–1812
Carlson M, Silberbach M (2007) Dissection of the aorta in Turner syndrome: two cases and review of 85 cases in the literature. J Med Genet 44:745–749
Sybert VP, McCauley E (2004) Turner’s syndrome. N Engl J Med 351:1227–1238
Sybert VP (1998) Cardiovascular malformations and complications in Turner syndrome. Pediatrics 101:E11
Mazzanti L, Cacciari E (1998) Congenital heart disease in patients with Turner’s syndrome. Italian Study Group for Turner Syndrome (ISGTS). J Pediatr 133:688–692
Volkl TM, Degenhardt K, Koch A, Simm D, Dorr HG et al (2005) Cardiovascular anomalies in children and young adults with Ullrich-Turner syndrome the Erlangen experience. Clin Cardiol 28:88–92
Gotzsche CO, Krag-Olsen B, Nielsen J, Sorensen KE, Kristensen BO (1994) Prevalence of cardiovascular malformations and association with karyotypes in Turner’s syndrome. Arch Dis Child 71:433–436
Dawson-Falk KL, Wright AM, Bakker B, Pitlick PT, Wilson DM et al (1992) Cardiovascular evaluation in Turner syndrome: utility of MR imaging. Australas Radiol 36:204–209
Sachdev V, Matura LA, Sidenko S, Ho VB, Arai AE et al (2008) Aortic valve disease in Turner syndrome. J Am Coll Cardiol 51:1904–1909
Olivieri LJ, Baba RY, Arai AE, Bandettini WP, Rosing DR et al (2013) Spectrum of aortic valve abnormalities associated with aortic dilation across age groups in Turner syndrome. Circ Cardiovasc Imaging 6:1018–1023
Ben-Shachar S, Ou Z, Shaw CA, Belmont JW, Patel MS et al (2008) 22q11.2 distal deletion: a recurrent genomic disorder distinct from DiGeorge syndrome and velocardiofacial syndrome. Am J Hum Genet 82:214–221
Lopez-Rangel E, Maurice M, McGillivray B, Friedman JM (1992) Williams syndrome in adults. Am J Med Genet 44:720–729
De Rubens FJ, Rodriguez LM, Hach JL, Del Castillo RV, Martinez HO (2008) Cardiovascular spectrum in Williams-Beuren syndrome: the Mexican experience in 40 patients. Tex Heart Inst J 35:279–285
Brunetti-Pierri N, Berg JS, Scaglia F, Belmont J, Bacino CA et al (2008) Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat Genet 40:1466–1471
Tan TY, Aftimos S, Worgan L, Susman R, Wilson M et al (2009) Phenotypic expansion and further characterisation of the 17q21.31 microdeletion syndrome. J Med Genet 46:480–489
Kleefstra T, van Zelst-Stams WA, Nillesen WM, Cormier-Daire V, Houge G et al (2009) Further clinical and molecular delineation of the 9q subtelomeric deletion syndrome supports a major contribution of EHMT1 haploinsufficiency to the core phenotype. J Med Genet 46:598–606
Tandon R, Edwards JE (1973) Cardiac malformations associated with Down’s syndrome. Circulation 47:1349–1355
Mohamed SA, Aherrahrou Z, Liptau H, Erasmi AW, Hagemann C et al (2006) Novel missense mutations (p.T596 M and p.P1797H) in NOTCH1 in patients with bicuspid aortic valve. Biochem Biophys Res Commun 345:1460–1465
McKellar SH, Tester DJ, Yagubyan M, Majumdar R, Ackerman MJ et al (2007) Novel NOTCH1 mutations in patients with bicuspid aortic valve disease and thoracic aortic aneurysms. J Thorac Cardiovasc Surg 134:290–296
Foffa I, Ait Ali L, Panesi P, Mariani M, Festa P et al (2013) Sequencing of NOTCH1, GATA5, TGFBR1 and TGFBR2 genes in familial cases of bicuspid aortic valve. BMC Med Genet 14:44
Hughes HE, Davies SJ (1994) Coarctation of the aorta in Kabuki syndrome. Arch Dis Child 70:512–514
Yoon JK, Ahn KJ, Kwon BS, Kim GB, Bae EJ et al (2015) The strong association of left-side heart anomalies with Kabuki syndrome. Korean J Pediatr 58:256–262
Padang R, Bagnall RD, Richmond DR, Bannon PG, Semsarian C (2012) Rare non-synonymous variations in the transcriptional activation domains of GATA5 in bicuspid aortic valve disease. J Mol Cell Cardiol 53:277–281
Fox JW, Lamperti ED, Eksioglu YZ, Hong SE, Feng Y et al (1998) Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron 21:1315–1325
Romano AA, Allanson JE, Dahlgren J, Gelb BD, Hall B et al (2010) Noonan syndrome: clinical features, diagnosis, and management guidelines. Pediatrics 126:746–759
Burch M, Sharland M, Shinebourne E, Smith G, Patton M et al (1993) Cardiologic abnormalities in Noonan syndrome: phenotypic diagnosis and echocardiographic assessment of 118 patients. J Am Coll Cardiol 22:1189–1192
Stevens CA, Bhakta MG (1995) Cardiac abnormalities in the Rubinstein-Taybi syndrome. Am J Med Genet 59:346–348
Girdauskas E, Schulz S, Borger MA, Mierzwa M, Kuntze T (2011) Transforming growth factor-beta receptor type II mutation in a patient with bicuspid aortic valve disease and intraoperative aortic dissection. Ann Thorac Surg 91:e70–e71
Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M et al (2005) A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet 37:275–281
Nistri S, Porciani MC, Attanasio M, Abbate R, Gensini GF et al (2012) Association of Marfan syndrome and bicuspid aortic valve: frequency and outcome. Int J Cardiol 155:324–325
Andelfinger G, Tapper AR, Welch RC, Vanoye CG, George AL Jr et al (2002) KCNJ2 mutation results in Andersen syndrome with sex-specific cardiac and skeletal muscle phenotypes. Am J Hum Genet 71:663–668
Guo DC, Pannu H, Tran-Fadulu V, Papke CL, Yu RK et al (2007) Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet 39:1488–1493
Guo DC, Papke CL, Tran-Fadulu V, Regalado ES, Avidan N et al (2009) Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am J Hum Genet 84:617–627
Liang CD, Hang CL (2001) Elongation of the aorta and multiple cardiovascular abnormalities associated with larsen syndrome. Pediatr Cardiol 22:245–246
Tan HL, Glen E, Töpf A, Hall D, O’Sullivan JJ et al (2012) Nonsynonymous variants in the SMAD6 gene predispose to congenital cardiovascular malformation. Hum Mutat 33:720–727
Lin X, Huo Z, Liu X, Zhang Y, Li L et al (2010) A novel GATA6 mutation in patients with tetralogy of Fallot or atrial septal defect. J Hum Genet 55:662–667
Gelb BD, Zhang J, Sommer RJ, Wasserman JM, Reitman MJ et al (1999) Familial patent ductus arteriosus and bicuspid aortic valve with hand anomalies: a novel heart-hand syndrome. Am J Med Genet 87:175–179
Feeley C, Rasbridge S (2006) Cardiomyopathy with a unique finding of bicuspid aortic valve in Becker’s muscular dystrophy. Cardiovasc Pathol Off J Soc Cardiovasc Pathol 15:347–351
Angelini A, Ho SY, Anderson RH, Devine WA, Zuberbuhler JR et al (1989) The morphology of the normal aortic valve as compared with the aortic valve having two leaflets. J Thorac Cardiovasc Surg 98:362–367
Mookadam F, Thota VR, Garcia-Lopez AM, Emani UR, Alharthi MS et al (2010) Unicuspid aortic valve in adults: a systematic review. J Heart Valve Dis 19:79–85
Mookadam F, Thota VR, Lopez AM, Emani UR, Tajik AJ (2010) Unicuspid aortic valve in children: a systematic review spanning four decades. J Heart Valve Dis 19:678–683
Novaro GM, Mishra M, Griffin BP (2003) Incidence and echocardiographic features of congenital unicuspid aortic valve in an adult population. J Heart Valve Dis 12:674–678
Thubrikar MJ, Aouad J, Nolan SP (1986) Patterns of calcific deposits in operatively excised stenotic or purely regurgitant aortic valves and their relation to mechanical stress. Am J Cardiol 58:304–308
Khan SK, Tamin SS, Araoz PA (2011) Quadricuspid aortic valve by cardiac magnetic resonance imaging: a case report and review of the literature. J Comput Assist Tomogr 35:637–641
Hurwitz LE, Roberts WC (1973) Quadricuspid semilunar valve. Am J Cardiol 31:623–626
Cemri M, Cengel A, Timurkaynak T (2000) Pentacuspid aortic valve diagnosed by transoesophageal echocardiography. Heart 84:e9
Kuroki H, Hirooka K, Ohnuki M (2012) Pentacuspid aortic valve causing severe aortic regurgitation. J Thorac Cardiovasc Surg 143:e11–e12
Beppu S, Suzuki S, Matsuda H, Ohmori F, Nagata S et al (1993) Rapidity of progression of aortic stenosis in patients with congenital bicuspid aortic valves. Am J Cardiol 71:322–327
Warren BA, Yong JL (1997) Calcification of the aortic valve: its progression and grading. Pathology 29:360–368
Brandenburg RO Jr, Tajik AJ, Edwards WD, Reeder GS, Shub C et al (1983) Accuracy of 2-dimensional echocardiographic diagnosis of congenitally bicuspid aortic valve: echocardiographic-anatomic correlation in 115 patients. Am J Cardiol 51:1469–1473
Yuan SM (2011) The anatomopathology of bicuspid aortic valve. Folia Morphol (Warsz) 70:217–227
Sadee AS, Becker AE, Verheul HA, Bouma B, Hoedemaker G (1992) Aortic valve regurgitation and the congenitally bicuspid aortic valve: a clinico-pathological correlation. Br Heart J 67:439–441
Tokunaga H, Koyanagi H, Hashimoto A, Nakano K, Hirayama T et al (1992) [Classification of congenitally bicuspid aortic valve and its angiographic and surgical significance]. [Zasshi] [Journal] Nihon Kyobu Geka Gakkai 40:467–71; discussion 71–72
Buchner S, Hulsmann M, Poschenrieder F, Hamer OW, Fellner C et al (2010) Variable phenotypes of bicuspid aortic valve disease: classification by cardiovascular magnetic resonance. Heart 96:1233–1240
Sonoda M, Takenaka K, Uno K, Ebihara A, Nagai R (2008) A larger aortic annulus causes aortic regurgitation and a smaller aortic annulus causes aortic stenosis in bicuspid aortic valve. Echocardiography 25:242–248
Sievers HH, Schmidtke C (2007) A classification system for the bicuspid aortic valve from 304 surgical specimens. J Thorac Cardiovasc Surg 133:1226–1233
Schaefer BM, Lewin MB, Stout KK, Gill E, Prueitt A et al (2008) The bicuspid aortic valve: an integrated phenotypic classification of leaflet morphology and aortic root shape. Heart 94:1634–1638
Fernandes SM, Sanders SP, Khairy P, Jenkins KJ, Gauvreau K et al (2004) Morphology of bicuspid aortic valve in children and adolescents. J Am Coll Cardiol 44:1648–1651
Attenhofer Jost CH, Connolly HM, O’Leary PW, Warnes CA, Tajik AJ et al (2005) Left heart lesions in patients with Ebstein anomaly. Mayo Clin Proc 80:361–368
Barbara DW, Edwards WD, Connolly HM, Dearani JA (2008) Surgical pathology of 104 tricuspid valves (2000–2005) with classic right-sided Ebstein’s malformation. Cardiovasc Pathol Off J Soc Cardiovasc Pathol 17:166–171
Bolling SF, Iannettoni MD, Dick M 2nd, Rosenthal A, Bove EL (1990) Shone’s anomaly: operative results and late outcome. Ann Thorac Surg 49:887–893
Popescu BA, Jurcut R, Serban M, Parascan L, Ginghina C (2008) Shone’s syndrome diagnosed with echocardiography and confirmed at pathology. Eur J Echocardiogr 9:865–867
Brauner RA, Laks H, Drinkwater DC Jr, Scholl F, McCaffery S (1997) Multiple left heart obstructions (Shone’s anomaly) with mitral valve involvement: long-term surgical outcome. Ann Thorac Surg 64:721–729
Brown JW, Ruzmetov M, Vijay P, Hoyer MH, Girod D et al (2005) Operative results and outcomes in children with Shone’s anomaly. Ann Thorac Surg 79:1358–1365
Brenner JI, Berg KA, Schneider DS, Clark EB, Boughman JA (1989) Cardiac malformations in relatives of infants with hypoplastic left-heart syndrome. Am J Dis Child 143:1492–1494
Hinton RB Jr, Martin LJ, Tabangin ME, Mazwi ML, Cripe LH et al (2007) Hypoplastic left heart syndrome is heritable. J Am Coll Cardiol 50:1590–1595
Hinton RB, Martin LJ, Rame-Gowda S, Tabangin ME, Cripe LH et al (2009) Hypoplastic left heart syndrome links to chromosomes 10q and 6q and is genetically related to bicuspid aortic valve. J Am Coll Cardiol 53:1065–1071
Roberts WC, Morrow AG, Braunwald E (1962) Complete interruption of the aortic arch. Circulation 26:39–59
Duran AC, Frescura C, Sans-Coma V, Angelini A, Basso C et al (1995) Bicuspid aortic valves in hearts with other congenital heart disease. J Heart Valve Dis 4:581–590
Perloff JK (2010) The variant associations of aortic isthmic coarctation. Am J Cardiol 106:1038–1041
Thanassoulis G, Yip JW, Filion K, Jamorski M, Webb G et al (2008) Retrospective study to identify predictors of the presence and rapid progression of aortic dilatation in patients with bicuspid aortic valves. Nat Clin Pract Cardiovasc Med 5:821–828
Folger GM Jr, Stein PD (1984) Bicuspid aortic valve morphology when associated with coarctation of the aorta. Catheter Cardiovasc Diagn 10:17–25
Teo LL, Cannell T, Babu-Narayan SV, Hughes M, Mohiaddin RH (2011) Prevalence of associated cardiovascular abnormalities in 500 patients with aortic coarctation referred for cardiovascular magnetic resonance imaging to a tertiary center. Pediatr Cardiol 32:1120–1127
Deshpande J, Kinare SG (1991) The bicuspid aortic valve – an autopsy study. Indian J Pathol Microbiol 34:112–118
Suzuki T, Nagai R, Kurihara H, Kurihara Y, Yamazaki T et al (1994) Stenotic bicuspid aortic valve associated with a ventricular septal defect in an adult presenting with congestive heart failure: a rare observation. Eur Heart J 15:402–403
Hutchins GM (1971) Coarctation of the aorta explained as a branch-point of the ductus arteriosus. Am J Pathol 63:203–209
Siu SC, Silversides CK (2010) Bicuspid aortic valve disease. J Am Coll Cardiol 55:2789–2800
Eichhorn P, Vogt P, Ritter M, Widmer V, Jenni R (1995) Abnormalities of the atrial septum in adults: kind, prevalence and clinical relevance. Schweiz Med Wochenschr 125:1336–1341
Lerer PK, Edwards WD (1981) Coronary arterial anatomy in bicuspid aortic valve. Necropsy study of 100 hearts. Br Heart J 45:142–147
Cho S, Jeon KN, Bae K (2015) Anomalous origin and aneurysm of the right coronary artery associated with congenital bicuspid aortic valve: MDCT findings. SpringerPlus 4:426
Ayusawa M, Sato Y, Kanamaru H, Kunimasa T, Sumitomo N et al (2010) MDCT of the anomalous origin of the right coronary artery from the left sinus of Valsalva associated with bicuspid aortic valve. Int J Cardiol 143:e45–e47
Aoyagi S, Suzuki S, Kosuga K, Ohishi K (1991) Anomalous origin of the right coronary artery associated with congenital bicuspid aortic valve. Kurume Med J 38:199–202
Palomo AR, Schrager BR, Chahine RA (1985) Anomalous origin of the right coronary artery from the ascending aorta high above the left posterior sinus of Valsalva of a bicuspid aortic valve. Am Heart J 109:902–904
Kang N, Tan SY, Ding ZP, Chua YL (2013) Abnormal origin of right coronary artery from left ventricle with bicuspid aortic stenosis. Ann Thorac Surg 96:e43–e45
Bossert T, Walther T, Doll N, Gummert JF, Kostelka M et al (2005) Anomalous origin of the right coronary artery from the pulmonary artery combined with aortic valve stenosis. Ann Thorac Surg 79:347–348
Tejada JG, Albarran A, Hernandez F, Jimenez S, Tascon JC (2001) Anomalous coronary artery origin associated with bicuspid aortic valve in a patient with rheumatic mitral stenosis: a case report. Angiology 52:649–652
Schang SJ, Pepine CJ, Bemiller CR (1975) Anomalous coronary artery origin and bicuspid aortic valve. Vasc Surg 9:67–72
Ishida N, Shimabukuro K, Matsuno Y, Ogura H, Takemura H (2014) Single coronary artery with bicuspid aortic valve stenosis and aneurysm of the ascending aorta: report of a case. Surg Today 44:550–552
Stajic Z, Mijailovic Z (2013) Single coronary artery associated with severe bicuspid aortic valve stenosis. J Saudi Heart Assoc 25:277–278
Rashid A, Saucedo JF, Hennebry TA (2005) Association of single coronary artery and congenital bicuspid aortic valve with review of literature. J Interv Cardiol 18:389–391
Morimoto K, Taniguchi I, Miyasaka S, Marumoto A (2005) Bicuspid aortic valve stenosis with single coronary artery. Ann Thorac Cardiovasc Surg 11:267–269
Kemaloglu Oz T, Karadeniz FO, Gundlapalli H, Erer B, Sharma RK et al (2014) Coexisting bicuspid aortic and pulmonary valves with normally related great vessels diagnosed by live/real time three-dimensional transesophageal echocardiography. Echocardiography 31:218–221
Ghez OY, Chetaille PM, Campbell BJ, Van Praagh R, Metras D (2002) Tetralogy of Fallot with aortic valvular stenosis: surgical correction in one case. Ann Thorac Surg 73:967–969
Kutty S, Kaul S, Danford CJ, Danford DA (2010) Main pulmonary artery dilation in association with congenital bicuspid aortic valve in the absence of pulmonary valve abnormality. Heart 96:1756–1761
Chisholm JC (1981) Mitral valve prolapse syndrome associated with congenital bicuspid aortic valve. J Natl Med Assoc 73:921–923
Charitos EI, Hanke T, Karluss A, Hilker L, Stierle U et al (2013) New insights into bicuspid aortic valve disease: the elongated anterior mitral leaflet. Eur J Cardiothorac Surg 43:367–370
Roberts WC, Janning KG, Vowels TJ, Ko JM, Hamman BL et al (2012) Presence of a congenitally bicuspid aortic valve among patients having combined mitral and aortic valve replacement. Am J Cardiol 109:263–271
Wrigley BJ, Rosin M, Banerjee P (2009) Replacement of a congenital bicuspid aortic valve in a patient with left ventricular noncompaction. Tex Heart Inst J 36:241–243
Krishnan U, Kilner PJ, Money-Kyrle A, Ramrakha P (2006) Two diverticula of the left ventricular outflow tract adjacent to the commissures of a bicuspid aortic valve. Congenit Heart Dis 1:332–334
Yildirir A, Batur MK, Kabakci G (2001) Left ventricular septal aneurysm in association with bicuspid aortic valve – a case report. Angiology 52:73–76
Vanelli P, Carro C, Scrofani R, Turiel M, Antona C et al (2002) Subannular left ventricular aneurysm in a patient with bicuspid aortic valve stenosis. Italian Heart J Off J Ital Fed Cardiol 3:608–610
Feizi O, Farrer Brown G, Emanuel R (1978) Familial study of hypertrophic cardiomyopathy and congenital aortic valve disease. Am J Cardiol 41:956–964
Schievink WI, Mokri B (1995) Familial aorto-cervicocephalic arterial dissections and congenitally bicuspid aortic valve. Stroke 26:1935–1940
Schievink WI, Raissi SS, Maya MM, Velebir A (2010) Screening for intracranial aneurysms in patients with bicuspid aortic valve. Neurology 74:1430–1433
Shaulov A, Leibowitz D, Rott D (2012) Prevalence of bicuspid aortic valve in patients presenting with subarachnoid hemorrhage related to an intracerebral aneurysm. Int J Cardiol 157:142–143
Goyal MS, Gottumukkala R, Bhalla S, Kates A, Zipfel GJ et al (2015) Bicuspid aortic valves and thoracic aortic aneurysms in patients with intracranial aneurysms. Neurology 84:46–49
Gupta P, Theut SB, Joshi A, Aggarwal S (2011) Circumaortic innominate vein in a patient with bicuspid aortic valve and coarctation. Ann Thorac Surg 92:e37
Zeina AR, Nachtigal A, Troitsa A, Admon G, Avshovich N (2010) Isolated spontaneous dissection of the celiac trunk in a patient with bicuspid aortic valve. Vasc Health Risk Manag 6:383–386
Westaby S, Bradlow WM, Newton JD, Mahesh B, Jin XY et al (2011) Rupture of an aneurysmal aortic diverticulum associated with coarctation and bicuspid aortic valve. Circulation 123:102–103
Di Martino L, Correale M, Cocco D, Di Biase M, Brunetti ND (2013) Bicuspid aortic valve and lusory artery: an unusual association. Intern Emerg Med 8:539–540
Kono T, Kitahara H, Watanabe T, Takano T, Sakaguchi M et al (2004) Successful repair of aberrant right subclavian artery aneurysm combined with bicuspid aortic valve through a median sternotomy. Ann Thorac Surg 77:2196–2197
Mandila C, Papanikolaou J, Saranteas T, Dounis G, Kostopanagiotou G et al (2009) Bicuspid aortic valve associated with persistent left and absent right superior vena cava. J Cardiothorac Vasc Anesth 23:579–580
Aydin A, Desai N, Bernhardt AM, Treede H, Detter C et al (2013) Ascending aortic aneurysm and aortic valve dysfunction in bicuspid aortic valve disease. Int J Cardiol 164:301–305
Hahn RT, Roman MJ, Mogtader AH, Devereux RB (1992) Association of aortic dilation with regurgitant, stenotic and functionally normal bicuspid aortic valves. J Am Coll Cardiol 19:283–288
Bauer M, Gliech V, Siniawski H, Hetzer R (2006) Configuration of the ascending aorta in patients with bicuspid and tricuspid aortic valve disease undergoing aortic valve replacement with or without reduction aortoplasty. J Heart Valve Dis 15:594–600
Fazel SS, Mallidi HR, Lee RS, Sheehan MP, Liang D et al (2008) The aortopathy of bicuspid aortic valve disease has distinctive patterns and usually involves the transverse aortic arch. J Thorac Cardiovasc Surg 135:901–907, 907.e1–907.e2
Preventza O, Livesay JJ, Cooley DA, Krajcer Z, Cheong BY et al (2013) Coarctation-associated aneurysms: a localized disease or diffuse aortopathy. Ann Thorac Surg 95:1961–1967; discussion 7
von Kodolitsch Y, Aydin AM, Bernhardt AM, Habermann C, Treede H et al (2010) Aortic aneurysms after correction of aortic coarctation: a systematic review. VASA 39:3–16
von Kodolitsch Y, Aydin AM, Koschyk DH et al (2002) Predictors of aneurysm formation after surgical correction of aortic coarctation. J Am Coll Cardiol 39:617–624
Della Corte A, Bancone C, Dialetto G, Covino FE, Manduca S et al (2014) Towards an individualized approach to bicuspid aortopathy: different valve types have unique determinants of aortic dilatation. Eur J Cardiothorac Surg 45:e118–e124
Schaefer BM, Lewin MB, Stout KK, Byers PH, Otto CM (2007) Usefulness of bicuspid aortic valve phenotype to predict elastic properties of the ascending aorta. Am J Cardiol 99:686–690
Russo CF, Cannata A, Lanfranconi M, Vitali E, Garatti A et al (2008) Is aortic wall degeneration related to bicuspid aortic valve anatomy in patients with valvular disease? J Thorac Cardiovasc Surg 136:937–942
Holmes KW, Lehmann CU, Dalal D, Nasir K, Dietz HC et al (2007) Progressive dilation of the ascending aorta in children with isolated bicuspid aortic valve. Am J Cardiol 99:978–983
Della Corte A, Bancone C, Quarto C, Dialetto G, Covino FE et al (2007) Predictors of ascending aortic dilatation with bicuspid aortic valve: a wide spectrum of disease expression. Eur J Cardiothorac Surg 31:397–404; discussion −5
Kang JW, Song HG, Yang DH, Baek S, Kim DH et al (2013) Association between bicuspid aortic valve phenotype and patterns of valvular dysfunction and bicuspid aortopathy: comprehensive evaluation using MDCT and echocardiography. JACC Cardiovasc Imaging 6:150–161
Kim YG, Sun BJ, Park GM, Han S, Kim DH et al (2012) Aortopathy and bicuspid aortic valve: haemodynamic burden is main contributor to aortic dilatation. Heart 98:1822–1827
Westhoff-Bleck M, Meyer GP, Lotz J, Tutarel O, Weiss T et al (2005) Dilatation of the entire thoracic aorta in patients with bicuspid aortic valve: a magnetic resonance angiography study. VASA 34:181–185
Girdauskas E, Rouman M, Disha K, Espinoza A, Misfeld M et al (2015) Aortic dissection after previous aortic valve replacement for bicuspid aortic valve disease. J Am Coll Cardiol 66:1409–1411
Milan A, Avenatti E, Tosello F, Iannaccone A, Leone D et al (2013) Aortic root dilatation in essential hypertension: prevalence according to new reference values. J Hypertens 31:1189–1195
Agmon Y, Khandheria BK, Meissner I, Schwartz GL, Sicks JD et al (2003) Is aortic dilatation an atherosclerosis-related process? Clinical, laboratory, and transesophageal echocardiographic correlates of thoracic aortic dimensions in the population with implications for thoracic aortic aneurysm formation. J Am Coll Cardiol 42:1076–1083
Oliver JM, Alonso-Gonzalez R, Gonzalez AE, Gallego P, Sanchez-Recalde A et al (2009) Risk of aortic root or ascending aorta complications in patients with bicuspid aortic valve with and without coarctation of the aorta. Am J Cardiol 104:1001–1006
Bauer M, Bauer U, Siniawski H, Hetzer R (2007) Differences in clinical manifestations in patients with bicuspid and tricuspid aortic valves undergoing surgery of the aortic valve and/or ascending aorta. Thorac Cardiovasc Surg 55:485–490
Quenot JP, Boichot C, Petit A, Falcon-Eicher S, d’Athis P et al (2005) Usefulness of MRI in the follow-up of patients with repaired aortic coarctation and bicuspid aortic valve. Int J Cardiol 103:312–316
Keshavarz-Motamed Z, Garcia J, Kadem L (2013) Fluid dynamics of coarctation of the aorta and effect of bicuspid aortic valve. PLoS One 8:e72394
Kohler M, Pitcher A, Blair E, Risby P, Senn O et al (2013) The impact of obstructive sleep apnea on aortic disease in Marfan’s syndrome. Respiration 86:39–44
Hata M, Yoshitake I, Wakui S, Unosawa S, Takahashi K et al (2012) Sleep disorders and aortic dissection in a working population. Surg Today 42:403–405
Rybczynski M, Koschyk D, Karmeier A, Gessler N, Sheikhzadeh S et al (2010) Frequency of sleep apnea in adults with the Marfan syndrome. Am J Cardiol 105:1836–1841
Keles T, Durmaz T, Bayram NA, Ciftci B, Yeter E et al (2009) Effect of continuous positive airway pressure therapy on aortic stiffness in patients with obstructive sleep apnea syndrome. Echocardiography 26:1217–1224
Serizawa N, Yumino D, Takagi A, Gomita K, Kajimoto K et al (2008) Obstructive sleep apnea is associated with greater thoracic aortic size. J Am Coll Cardiol 52:885–886
Sampol G, Romero O, Salas A, Tovar JL, Lloberes P et al (2003) Obstructive sleep apnea and thoracic aorta dissection. Am J Respir Crit Care Med 168:1528–1531
Immer FF, Bansi AG, Immer-Bansi AS, McDougall J, Zehr KJ et al (2003) Aortic dissection in pregnancy: analysis of risk factors and outcome. Ann Thorac Surg 76:309–314
Yuan SM (2013) Aortic dissection during pregnancy: a difficult clinical scenario. Clin Cardiol 36:576–584
Maron BJ, Shirani J, Poliac LC, Mathenge R, Roberts WC, Mueller FO (1996) Sudden death in young competitive athletics. Clinical, and pathologic profiles. JAMA 276:199–204
Galanti G, Stefani L, Toncelli L, Vono MC, Mercuri R et al (2010) Effects of sports activity in athletes with bicuspid aortic valve and mild aortic regurgitation. Br J Sports Med 44:275–279
Carter D, Pokroy R, Azaria B, Matetzky S, Prokopetz A et al (2007) Effect of G-force on bicuspid aortic valve in aviators. Cardiology 108:124–127
Hsue PY, Salinas CL, Bolger AF, Benowitz NL, Waters DD (2002) Acute aortic dissection related to crack cocaine. Circulation 105:1592–1595
Eagle KA, Isselbacher EM, DeSanctis RW (2002) Cocaine-related aortic dissection in perspective. Circulation 105:1529–1530
Palmiere C, Burkhardt S, Staub C, Hallenbarter M, Paolo Pizzolato G et al (2004) Thoracic aortic dissection associated with cocaine abuse. Forensic Sci Int 141:137–142
Singh S, Trivedi A, Adhikari T, Molnar J, Arora R et al (2007) Cocaine-related acute aortic dissection: patient demographics and clinical outcomes. Can J Cardiol 23:1131–1134
Wako E, LeDoux D, Mitsumori L, Aldea GS (2007) The emerging epidemic of methamphetamine-induced aortic dissections. J Card Surg 22:390–393
Westover AN, Nakonezny PA (2010) Aortic dissection in young adults who abuse amphetamines. Am Heart J 160:315–321
Tiryakioglu SK, Tiryakioglu O, Turan T, Kumbay E (2009) Aortic dissection due to sildenafil abuse. Interact Cardiovasc Thorac Surg 9:141–143
Boodhwani M, Andelfinger G, Leipsic J, Lindsay T, McMurtry MS et al (2014) Canadian Cardiovascular Society position statement on the management of thoracic aortic disease. Can J Cardiol 30:577–589
Erbel R, Aboyans V, Boileau C, Bossone E, Di Bartolomeo R et al (2014) ESC Guidelines on the diagnosis and treatment of aortic diseases: document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (ESC). Eur Heart J 2014
Hiratzka LF, Bakris GL, Beckman JA, Bersin RM, Carr VF et al (2010) ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with thoracic aortic disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation 121:e266–e369
von Kodolitsch Y, Robinson P, Berger J (2014) When should surgery be performed in Marfan syndrome and other connective tissue disorders to protect against type a dissection? In: Bonser RS, Pagano D, Haverich A, Mascaro J (eds) Controversies in aortic dissection and aneurysmal disease. Springer, London, pp 17–47
Biner S, Rafique AM, Ray I, Cuk O, Siegel RJ et al (2009) Aortopathy is prevalent in relatives of bicuspid aortic valve patients. J Am Coll Cardiol 53:2288–2295
Martin LJ, Hinton RB, Zhang X, Cripe LH, Benson DW (2011) Aorta measurements are heritable and influenced by bicuspid aortic valve. Front Genet 2:61
Biddinger A, Rocklin M, Coselli J, Milewicz DM (1997) Familial thoracic aortic dilatations and dissections: a case control study. J Vasc Surg 25:506–511
Ferencik M, Pape LA (2003) Changes in size of ascending aorta and aortic valve function with time in patients with congenitally bicuspid aortic valves. Am J Cardiol 92:43–46
Novaro GM, Griffin BP (2004) Congenital bicuspid aortic valve and rate of ascending aortic dilatation. Am J Cardiol 93:525–526
Etz CD, Zoli S, Brenner R, Roder F, Bischoff M et al (2010) When to operate on the bicuspid valve patient with a modestly dilated ascending aorta. Ann Thorac Surg 90:1884–1890; discussion 91–92.
Beroukhim RS, Kruzick TL, Taylor AL, Gao D, Yetman AT (2006) Progression of aortic dilation in children with a functionally normal bicuspid aortic valve. Am J Cardiol 98:828–830
Roman MJ, Rosen SE, Kramer-Fox R, Devereux RB (1993) Prognostic significance of the pattern of aortic root dilation in the Marfan syndrome. J Am Coll Cardiol 22:1470–1476
Cotrufo M, Della Corte A (2009) The association of bicuspid aortic valve disease with asymmetric dilatation of the tubular ascending aorta: identification of a definite syndrome. J Cardiovasc Med (Hagerstown) 10:291–297
Robicsek F, Thubrikar MJ (1994) Hemodynamic considerations regarding the mechanism and prevention of aortic dissection. Ann Thorac Surg 58:1247–1253
Warner PJ, Al-Quthami A, Brooks EL, Kelley-Hedgepeth A, Patvardhan E et al (2013) Augmentation index and aortic stiffness in bicuspid aortic valve patients with non-dilated proximal aortas. BMC Cardiovasc Disord 13:19
Tzemos N, Lyseggen E, Silversides C, Jamorski M, Tong JH et al (2010) Endothelial function, carotid-femoral stiffness, and plasma matrix metalloproteinase-2 in men with bicuspid aortic valve and dilated aorta. J Am Coll Cardiol 55:660–668
Grotenhuis HB, Ottenkamp J, Westenberg JJ, Bax JJ, Kroft LJ et al (2007) Reduced aortic elasticity and dilatation are associated with aortic regurgitation and left ventricular hypertrophy in nonstenotic bicuspid aortic valve patients. J Am Coll Cardiol 49:1660–1665
Aydin A, Mortensen K, Rybczynski M, Sheikhzadeh S, Willmann S et al (2011) Central pulse pressure and augmentation index in asymptomatic bicuspid aortic valve disease. Int J Cardiol 147:466–468
Nistri S, Sorbo MD, Basso C, Thiene G (2002) Bicuspid aortic valve: abnormal aortic elastic properties. J Heart Valve Dis 11:369–373; discussion 73–74
Shim CY, Cho IJ, Yang WI, Kang MK, Park S et al (2011) Central aortic stiffness and its association with ascending aorta dilation in subjects with a bicuspid aortic valve. J Am Soc Echocardiogr 24:847–852
Jackson V, Petrini J, Caidahl K, Eriksson MJ, Liska J et al (2011) Bicuspid aortic valve leaflet morphology in relation to aortic root morphology: a study of 300 patients undergoing open-heart surgery. Eur J Cardiothorac Surg 40:e118–e124
Nistri S, Grande-Allen J, Noale M, Basso C, Siviero P et al (2008) Aortic elasticity and size in bicuspid aortic valve syndrome. Eur Heart J 29:472–479
Pees C, Michel-Behnke I (2012) Morphology of the bicuspid aortic valve and elasticity of the adjacent aorta in children. Am J Cardiol 110:1354–1360
Donato Aquaro G, Ait-Ali L, Basso ML, Lombardi M, Pingitore A et al (2011) Elastic properties of aortic wall in patients with bicuspid aortic valve by magnetic resonance imaging. Am J Cardiol 108:81–87
Oulego-Erroz I, Alonso-Quintela P, Mora-Matilla M, Gautreaux Minaya S, de Armentia SLL (2012) Ascending aorta elasticity in children with isolated bicuspid aortic valve. Int J Cardiol 168(2):1143–1146
Hope MD, Hope TA (2013) Functional and molecular imaging techniques in aortic aneurysm disease. Curr Opin Cardiol 28:609–618
Hope MD, Hope TA, Meadows AK, Ordovas KG, Urbania TH et al (2010) Bicuspid aortic valve: four-dimensional MR evaluation of ascending aortic systolic flow patterns. Radiology 255:53–61
Burris NS, Sigovan M, Knauer HA, Tseng EE, Saloner D et al (2014) Systolic flow displacement correlates with future ascending aortic growth in patients with bicuspid aortic valves undergoing magnetic resonance surveillance. Investig Radiol 49:635–639
Hope MD, Hope TA, Crook SE, Ordovas KG, Urbania TH et al (2011) 4D flow CMR in assessment of valve-related ascending aortic disease. JACC Cardiovasc Imaging 4:781–787
Hope MD, Wrenn J, Sigovan M, Foster E, Tseng EE et al (2012) Imaging biomarkers of aortic disease: increased growth rates with eccentric systolic flow. J Am Coll Cardiol 60:356–357
Nathan DP, Xu C, Plappert T, Desjardins B, Gorman JH 3rd et al (2011) Increased ascending aortic wall stress in patients with bicuspid aortic valves. Ann Thorac Surg 92:1384–1389
Ali OA, Chapman M, Nguyen TH, Chirkov YY, Heresztyn T et al (2014) Interactions between inflammatory activation and endothelial dysfunction selectively modulate valve disease progression in patients with bicuspid aortic valve. Heart 100:800–805
Foffa I, Murzi M, Mariani M, Mazzone AM, Glauber M et al (2012) Angiotensin-converting enzyme insertion/deletion polymorphism is a risk factor for thoracic aortic aneurysm in patients with bicuspid or tricuspid aortic valves. J Thorac Cardiovasc Surg 144:390–395
Kilickesmez KO, Abaci O, Kocas C, Yildiz A, Kaya A et al (2012) Dilatation of the ascending aorta and serum alpha 1-antitrypsin level in patients with bicuspid aortic valve. Heart Vessel 27:391–397
Ikonomidis JS, Ivey CR, Wheeler JB, Akerman AW, Rice A et al (2013) Plasma biomarkers for distinguishing etiologic subtypes of thoracic aortic aneurysm disease. J Thorac Cardiovasc Surg 145:1326–1333
Boon RA, Seeger T, Heydt S, Fischer A, Hergenreider E et al (2011) MicroRNA-29 in aortic dilation: implications for aneurysm formation. Circ Res 109:1115–1119
Schmoker JD, McPartland KJ, Fellinger EK, Boyum J, Trombley L et al (2007) Matrix metalloproteinase and tissue inhibitor expression in atherosclerotic and nonatherosclerotic thoracic aortic aneurysms. J Thorac Cardiovasc Surg 133:155–161
Boyum J, Fellinger EK, Schmoker JD, Trombley L, McPartland K et al (2004) Matrix metalloproteinase activity in thoracic aortic aneurysms associated with bicuspid and tricuspid aortic valves. J Thorac Cardiovasc Surg 127:686–691
Wang Y, Wu B, Dong L, Wang C, Wang X et al (2014) Circulating matrix metalloproteinase patterns in association with aortic dilatation in bicuspid aortic valve patients with isolated severe aortic stenosis. Heart Vessels 31:189–197
Irtiuga OB, Zhiduleva EV, Dubrovskaia OB, Moiseeva OM (2014) Concentration of osteoprotegerin and RANKL in blood serum of patients with aortic stenosis. Kardiologiia 54:44–48
Branchetti E, Bavaria JE, Grau JB, Shaw RE, Poggio P et al (2014) Circulating soluble receptor for advanced glycation end product identifies patients with bicuspid aortic valve and associated aortopathies. Arterioscler Thromb Vasc Biol 34:2349–2357
Hillebrand M, Millot N, Sheikhzadeh S, Rybczynski M, Gerth S et al (2014) Total serum transforming growth factor-beta1 is elevated in the entire spectrum of genetic aortic syndromes. Clin Cardiol 37:672–679
Black KM, Masuzawa A, Hagberg RC, Khabbaz KR, Trovato ME et al (2013) Preliminary biomarkers for identification of human ascending thoracic aortic aneurysm. J Am Heart Assoc 2:e000138
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Japan KK
About this chapter
Cite this chapter
von Kodolitsch, Y., Kaemmerer, H. (2017). Bicuspid Aortic Valve. In: Niwa, K., Kaemmerer, H. (eds) Aortopathy. Springer, Tokyo. https://doi.org/10.1007/978-4-431-56071-5_15
Download citation
DOI: https://doi.org/10.1007/978-4-431-56071-5_15
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-56069-2
Online ISBN: 978-4-431-56071-5
eBook Packages: MedicineMedicine (R0)