
Abstract
Bile acids are the major driving force of bile excretion from hepatocytes; they are synthesized from cholesterol via at least 17 enzymatic reactions. They play a critical role in cholesterol disposal and the absorption of fat and fat-soluble vitamins. The concentration of intracellular bile acid is tightly regulated by modulating expression of bile acid transporters via nuclear receptors. This article provides a comprehensive overview of the characteristics and regulatory networks of hepatobiliary bile acid transporters.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
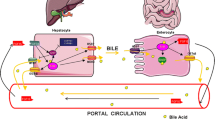
Bile acids are the major driving force of bile excretion from hepatocytes; they are synthesized from cholesterol via at least 17 enzymatic reactions. They play a critical role in cholesterol disposal and the absorption of fat and fat-soluble vitamins. After excretion from hepatocytes into the bile canaliculus, most bile acids (~95%) are reabsorbed in the terminal ileum and return to the liver via the portal vein (enterohepatic circulation). Influx and efflux of bile acids in organs is mediated by organ-specific transporters (Fig. 2.1). In hepatocytes, bile acids are absorbed from the sinusoid by the Na+-taurocholate cotransporting polypeptide (NTCP, SLC10A1) and secreted into the bile canaliculus by the bile salt export pump (BSEP, ABCB11). Other bile components are secreted by their corresponding transporters: phospholipids by multidrug resistance protein 3 (MDR3, ABCB4), organic anions by multidrug resistance-associated protein 2 (MRP2, ABCC2), and cholesterol by ABCG5/G8. In the terminal ileum, bile acids are absorbed from the intestinal lumen by the apical sodium-dependent bile acid transporter (ASBT, SLC10A2) and excreted into the portal vein by the organic solute transporters (OSTα/OSTβ, SLC51A/SLC51B) that facilitate bidirectional diffusion.

Bile acid transporters. In hepatocytes, bile acids are absorbed from the sinusoid by NTCP, and in part by OATPs, and are secreted into the bile canaliculus by BSEP. In the terminal ileum, they are absorbed from the intestinal lumen by ASBT and excreted into the portal vein by OSTα/OSTβ to return to the liver (enterohepatic circulation). Other components of bile are secreted into the bile canaliculus by MDR3 (phospholipids), MRP2 (organic anions), and ABCG5/G8 (cholesterol). OSTα/OSTβ, MRP3, and MRP4 are involved in retrograde bile acid elimination from the basolateral membrane to the sinusoid in cholestasis. In cholangiocytes, bile acids are absorbed by ASBT and secreted into the peribiliary plexus by OSTα/OSTβ, MRP3, and a truncated ASBT (t-ASBT) (cholehepatic shunting)
Bile acids are detergents and toxic to cells at high concentrations; therefore, their cellular concentration must be tightly regulated by refined feedback systems. Bile acids, now known as a signaling molecule, activate a nuclear receptor, farnesoid X receptor (FXR, NR1H4), and the G protein-coupled bile acid receptor, TGR5, thereby triggering a number of physiological reactions (see [Chap. 4] “Nuclear Receptor Regulation”). In hepatocytes, bile acids bind to FXR, which represses NTCP and prevents further bile acid uptake, downregulates cholesterol 7α-hydroxylase (CYP7A1) to inhibit further bile acid synthesis, and activates BSEP to induce bile acid secretion into the bile canaliculus. All of these events ultimately result in a reduction in the concentration of intracellular bile acids. This article provides a comprehensive overview of the characteristics and regulatory networks of hepatobiliary bile acid transporters.
2 BSEP
2.1 Characteristics
The human BSEP gene is located on chromosome 2 (2q24) and is translated into a protein comprising 1,321 amino acids, with a molecular mass of ~160 kDa [1]. BSEP belongs to the ABC subfamily B, harboring 12 potential transmembrane segments and two sets of Walker A and B motifs that bind to ATP [2–4]. BSEP is exclusively expressed in hepatocytes, where it resides along the canalicular membranes and exports bile acids into the bile canaliculus in an ATP-dependent fashion. The rat Bsep receives N-linked glycosylation at four asparagine residues in the first extracellular loop [5], sites that are also present in human BSEP. These glycans are required for correct trafficking to the canalicular membrane; loss of two or more glycans results in rapid degradation at the proteasome [5]. The intracellular distribution of Bsep was analyzed using pulse-chase studies in rats [6, 7]. Newly synthesized Bsep traffics directly from the Golgi to the canalicular membrane through a post-Golgi endosomal fraction. This is in contrast to other canalicular proteins, such as dipeptidyl peptidase IV and the canalicular cell adhesion molecule (cCAM105), which reach the basolateral membrane before arriving at the canalicular membrane (transcytosis) [6]. Bsep cycles between intracellular pools and the canalicular membrane, and taurocholic acid (TCA) and cAMP increase the amount of Bsep in the canalicular membrane [7]. Studies using WIFB9 cells, a stable hybrid of rat hepatoma and human fibroblasts with sealed bile canaliculi, revealed that Bsep constitutively cycles between the canalicular membrane and Rab11a-positive recycling endosomes [8]. HS-1-associated protein X-1 (HAX-1) [9] and non-muscle myosin II regulatory light chain 2a (MLC2a) [10] were identified as binding partners in a yeast two-hybrid screen. HAX-1 participates in clathrin-mediated endocytosis through interactions with cortactin [9]. MLC2a is involved in trafficking of newly synthesized Bsep to the canalicular membrane [10]. The AP2 adaptor complex is involved in clathrin-dependent endocytosis through interactions with a tyrosine motif at the carboxyl terminus of BSEP [11, 12].
p38MAPK is involved in BSEP trafficking from the Golgi to the canalicular membrane, and tauroursodeoxycholic acid (TUDCA)-induced choleretic action is dependent on p38MAPK activation [13]. Short-chain ubiquitination is associated with BSEP degradation and is modulated by 4-phenylbutyrate (4PBA) [14].
Human BSEP transports glycine and taurine conjugates of the two primary bile acids, cholic acid (CA) and chenodeoxycholic acid (CDCA), with high affinity and selectivity [2, 15–17].
2.2 Gene Regulation
BSEP expression is tightly regulated by the nuclear receptor, FXR. When bile acids bind to FXR, FXR forms a heterodimer with retinoid X receptor (RXR) [18] and induces BSEP upregulation via binding to the FXR-responsive element (FXRE) in the promoter region [19, 20]. Besides endogenous FXR agonists, such as CDCA, deoxycholic acid (DCA), and CA [18], more potent synthetic FXR agonists such as 6α-ethyl-CDCA (obeticholic acid, INT-747) [21], 6α-ethyl-3α,7α,23-trihydroxy-24-nor-5β-cholan-23-sulfate (INT-767) [22], and GW4064 [23] upregulate BSEP expression in various cell lines and animal models. As for obeticholic acid, clinical trials for primary biliary cholangitis (PBC), primary sclerosing cholangitis (PSC), and nonalcoholic steatohepatitis (NASH) are ongoing. Ursodeoxycholic acid (UDCA), which is often used to treat cholestasis, is not an FXR ligand. Notably, muricholic acid, one of the major bile acid species present in rodents but not in humans, is antagonistic to FXR [24]. In Fxr −/− mice, Bsep expression levels are markedly reduced at baseline and are not induced further by bile acid feeding [25], suggesting a critical role for FXR in the regulation of BSEP expression. Furthermore, a recent report documented four patients from two families with a homozygous loss of FXR exhibiting severe neonatal cholestasis [26].
Besides FXR, several other transcriptional factors regulate BSEP gene expression. Liver receptor homolog 1 (LRH-1, NR5A2), a transcriptional regulator for the biosynthesis and transport of cholesterol and bile acids, activates the BSEP promoter [27], and Bsep expression is reduced in LRH-1 knockout mice [28].
Nuclear factor erythroid 2-related factor 2 (Nrf2) is another transcriptional regulator for BSEP [29]. Nrf2 maintains redox homeostasis by regulating many phase I and II drug-metabolizing and detoxification enzymes. Nrf2 upregulates BSEP by binding to musculoaponeurotic fibrosarcoma recognition element (MARE) 1 in the BSEP promoter. Upregulation of Bsep expression by alpha-naphthyl isothiocyanate (ANIT) is abrogated in Nrf2-null mice [30].
Recently, Song et al. reported that 17β-estradiol (E2) repressed BSEP expression through direct interaction with estrogen receptor α (ERα) and FXR in the late stage of pregnancy [31], implicating a mechanistic role for the E2/ERα/FXR pathway in intrahepatic cholestasis of pregnancy (ICP).
2.3 BSEP-Associated Diseases
There are two types of hereditary intrahepatic cholestatic disease: progressive familial intrahepatic cholestasis (PFIC) and benign recurrent intrahepatic cholestasis (BRIC). PFIC patients progress to liver failure and require liver transplantation in childhood, whereas BRIC patients display intermittent and usually nonprogressive jaundice (reviewed in [32]). PFIC1 and BRIC1 are caused by mutations in the FIC1 (ATB8B1) gene, which encodes a P-type ATPase functioning as a flippase for phosphatidylserine, whereas PFIC3 is caused by mutations in the MDR3 gene. PFIC2 and BRIC2 are caused by mutations in BSEP, and more than 150 genetic abnormalities, including missense, nonsense, deletions, insertions, and splice-site mutations, have been identified [3, 33–36]. Some missense mutations and single nucleotide polymorphisms (SNPs) can cause aberrant pre-mRNA splicing, resulting in impaired BSEP function [35]. PFIC2 is characterized by absent or much reduced canalicular BSEP expression [34, 37] as well as a markedly diminished concentration of biliary bile acid [37].
To elucidate the effects of these mutations and SNPs on BSEP function, a number of studies have been performed using cell lines expressing a mutated form of BSEP [38–42]. The BSEP protein harboring a disease-associated missense mutation is unstable and is degraded in the proteasome [41, 42]. TCA transport activity of BSEP was analyzed in BSEP-expressing MDCKII cells. The activity of PFIC2 mutants (D482G, E297G, K461E, G982R, R1153C, R1268Q, and 3767–3768insC) was 0–30% that of wild type, and BRIC2 mutants (A570T and R1050C) exhibited 50–60% wild-type activity [42]. The reduced activity corresponded to the stability of synthesized BSEP protein. Thus, the difference in the severity of the clinical phenotype between PFIC2 and BRIC2 may be explained by the differences in transport activity of BSEP harboring corresponding mutations [41, 42]. Several patients who had clinical and histopathological characteristics of BRIC progressed to PFIC [33, 43], suggesting a possible phenotypic progression between BRIC2 and PFIC2. Furthermore, the E297G mutation, which is responsible for PFIC2, is also found in BRIC2 patients [33]. Therefore, although the BSEP genotype appears to play an important role in determining clinical severity, other precipitating factors, including viral infection and pregnancy, may also participate [33].
Impaired BSEP function as a cause of cholestasis has been suggested for other congenital diseases. Although the exact molecular mechanism underlying cholestasis in PFIC1 is not fully understood, FIC1 deficiency may lead to a loss of asymmetric phospholipid distribution in the canalicular membrane, decreasing membrane stability, thereby disturbing the function of transporters including BSEP [44, 45]. Microvillus inclusion disease (MVID), a hereditary disorder manifesting intractable diarrhea associated with mutations in the MYO5B gene, occasionally accompanies PFIC-like cholestasis. Reduced BSEP expression in the canalicular membrane due to disturbed MYO5B/RAB11A apical recycling endosome pathway has been proposed as a molecular mechanism for cholestasis in this disease [46].
An association between BSEP SNP and acquired intrahepatic cholestasis has been reported. The C-allele frequency of BSEP c.1331T>C (p. V444A) (rs2287622) SNP was higher among patients with ICP (67% in patients versus 54% in controls, P < 0.001) [47]. In a recent comprehensive study, two intronic SNPs (rs7577650 and rs3815676) were identified as significant risk alleles associated with ICP. The V444A SNP remained associated with the disease, but the association was driven by rs7577650 [48], suggesting that the V444A SNP is not causative. The effect of an amino acid substitution at position 444 on BSEP function is controversial. Western blot analysis on normal liver tissues from patients undergoing liver resection revealed that canalicular BSEP expression was slightly, but not significantly, reduced in individuals carrying the 444A polymorphism [49]. In another study that utilized a bank of human liver samples, BSEP mRNA, but not protein, expression was significantly attenuated in individuals with the 444A polymorphism [50]. The bile acid transport activity of 444A BSEP was not reduced when expressed in Sf9 [51] and HeLa [50] cells and was slightly reduced by up to 20% in MDCKII cells [52].
Impaired BSEP function is also involved in drug-induced liver injury; its severity is associated with dual inhibition of BSEP and mitochondrial function [53]. The association of BSEP V444A SNP in drug-induced cholestasis has been reported in European populations (76% in patients versus 57% in controls) [51]. However, this association was not reproducible among Japanese patients with drug-induced cholestasis (66% in patients versus 78% in controls) [52]. Further investigation is necessary to identify underlying causative risk alleles in the different populations.
2.4 Choleretic Agents
Given that bile acids are the major driving force for bile excretion, drugs that upregulate or activate BSEP are good candidates to treat intrahepatic cholestasis. UDCA is one of the drugs most commonly used for hepatobiliary diseases including PBC, PSC, cholestasis, and cholelithiasis. UDCA exerts a choleretic effect by targeting BSEP to the canalicular membrane [13, 54–56] via activation of p38MAPK and a Ca2+-independent protein kinase C (PKC) isoform [13, 55]. In fact, UDCA administration induced remission at least transiently in children with PFIC2 by retargeting BSEP to the canalicular membrane [57].
4PBA enhanced the cell surface expression and transport capacity of wild-type BSEP and BSEP carrying a PFIC2 mutation (E297G and D482G) in MDCKII cells [58]. Administration of 4PBA also induced canalicular Bsep expression, accompanied by an increase in biliary excretion of TCA in rats [58]. These effects may be achieved by decreasing short-chain ubiquitination-mediated Bsep degradation [14] and by reducing AP2 adaptor complex-mediated clathrin-dependent endocytosis [11]. In the clinical setting, 4PBA therapy improved liver function tests, liver histology, and itching in patients with PFIC2 [59, 60] and BRIC2 [61].
2.5 Antibody-Induced BSEP Deficiency
Orthotopic liver transplantation usually yields a good outcome in PFIC2 patients. However, since the first case was documented by Keitel et al. [62], several cases have been reported of PFIC2 children with recurring progressive intrahepatic cholestasis in the presence of an autoantibody against BSEP after liver transplantation [63–66]. Generation of a polyclonal antibody to target the first extracellular loop of BSEP may therefore be responsible for inhibiting BSEP function [66].
3 NTCP
3.1 Characteristics
NTCP is a glycoprotein of approximately 38 kDa, consisting of 349 amino acids [67, 68]. NTCP is localized to the sinusoidal membrane of hepatocytes and functions as an electrogenic sodium-solute cotransporter [69]. Major substrates of NTCP include glycine- and taurine-conjugated bile acids, but unconjugated and sulfated bile acids can still be transported to some extent [70–72].
Other sinusoidal transporters, including organic anion transporting polypeptides (OATP) 1B1 (SLCO1B1) and OATP1B3 (SLCO1B3), are able to transport conjugated bile acids in a sodium-independent manner, as well as unconjugated species [73]. The significance of NTCP in hepatic bile acid uptake is unknown due to a lack of NTCP-null patients. Recently a case of NTCP deficiency was documented [74], a 5-year-old girl manifesting conjugated hypercholanemia without any sign of liver injury. Sequencing of the NTCP gene revealed a single homozygous nonsynonymous point mutation (c.755G>A, p. R252H). The R252H mutation resulted in a marked reduction in TCA uptake, along with a lack of plasma membrane expression when it was expressed in HEK293T cells. This indicates that NTCP is the major transporter for hepatocellular uptake of conjugated bile acids. However, serum bile acid concentrations were unexpectedly normal in the majority of Slc10a1 −/− mice [75], suggesting differences in NTCP contribution among species.
3.2 Transcriptional Regulation
NCTP regulation is important for suppressing further influx of potentially toxic bile acids into hepatocytes and is repressed in patients with inflammation-induced cholestasis [76] and advanced PBC [77], as well as in several cholestatic animal models [78–80]. Although its transcriptional regulation is mediated by bile acids, hormones such as estrogen and prolactin, and pro-inflammatory cytokines may also be involved, depending on the species (reviewed in [81]). Bile acids repress NTCP transcription through FXR activation, which in turn induces small heterodimer partner (SHP). SHP inhibits NTCP upregulation by competing with coactivators for binding to hepatocyte nuclear factor 4 alpha (HNF-4α) and RXRα [80] and by suppressing retinoic acid receptor alpha (RARα) in rats [82] and glucocorticoid receptor (GR) and peroxisome proliferator-activated receptor-gamma coactivator-1 alpha (PGC-1α) in humans [83]. However, an additional SHP-independent pathway probably exists, since Ntcp repression was not completely abolished in Shp −/− mice fed with CA [84].
NTCP expression is also regulated posttranslationally. Insertion of NTCP into the plasma membrane by cAMP is mediated by the phosphoinositide-3-kinase signaling pathway [85, 86] and protein phosphatase 2B (PP2B)-induced dephosphorylation of NTCP [87]. In contrast, taurochenodeoxycholic acid (TCDCA) decreases sinusoidal NTCP expression by inducing NTCP endocytosis in a PKC- and PP2B-dependent manner [88].
3.3 NTCP as a Receptor for Hepatitis B and D Virus (HBV and HDV)
NTCP is attracting much attention as a functional receptor for HBV and HDV [89]. The large surface protein pre-S1 domain of HBV is a key determinant for receptor binding. In vitro studies demonstrated that myrcludex B, a myristoylated lipopeptide derived from the pre-S1 domain, blocked bile acid uptake by NTCP [90], while taurine or glycine conjugates of CA and UDCA inhibited HBV infection [13]. CYP7A1, the rate-limiting enzyme that synthesizes bile acids from cholesterol, was induced in human liver chimeric mice that were infected with HBV or were given myrcludex B and in liver biopsy samples from HBV-infected patients [91]. This may be a compensatory response against reduced bile acid uptake by HBV binding to NTCP. Interestingly, the NTCP variant p. S267F (c.800C>T, s2296651), which exhibits reduced bile acid transport capacity and has only been observed among Asians [92], is protective against HBV chronic infection [93] as well as progression to cirrhosis and hepatocellular carcinoma in patients with chronic hepatitis B [94].
4 Other Basolateral Bile Acid Transporters
Retrograde bile acid elimination from the basolateral membrane of hepatocytes to the sinusoid represents a rescue mechanism for avoiding intracellular bile acid overload. The relevant transporters include OSTα/OSTβ, MRP3 (ABCC3) and MRP4 (ABCC4).
OSTα comprises 340 amino acids, forms a heterodimer with OSTβ comprising 128 amino acids, and is expressed at the basolateral membrane of ileal enterocytes, hepatocytes, and cholangiocytes. Co-expression is necessary for stable expression of OSTα and OSTβ and their delivery to the plasma membrane [95, 96]. In enterocytes, OSTα/OSTβ is responsible for excreting bile acids into the portal circulation to achieve enterohepatic circulation under physiological conditions. In hepatocytes, this transporter is upregulated to transport excess bile acids back to the sinusoidal compartment in cholestasis. Hepatic OSTα/OSTβ expression was increased in patients with advanced PBC [97]. Upregulation was also observed in mice following common bile duct ligation (CBDL) [97], ANIT treatment [98], and CA feeding [99], which was dependent on FXR [100, 101].
Mrp3 and Mrp4 were upregulated in CBDL mice independently of FXR [102]. Mrp3 −/− mice had normal bile acid transport function [103], whereas Mrp4 −/− mice exhibited an impaired cytoprotective response to CBDL-induced cholestasis [104]. MRP4, but not MRP3, was upregulated in patients with PFIC2 and PFIC3 [105], suggesting that MRP4 plays an important role in the compensatory reaction to cholestatic liver injury. Regulatory nuclear receptors include constitutive androstane receptor (CAR), pregnane X receptor (PXR) and vitamin D receptor (VDR) for MRP3, and CAR and peroxisome proliferator-activated receptors α (PPARα) for MRP4 (reviewed in [81]).
5 Bile Acid Transporters in Cholangiocytes
Cholangiocytes play an important role in bile formation by secreting bicarbonate and water and possess transport systems for the influx and efflux of bile acids. Unconjugated bile acids possibly enter cholangiocytes via passive diffusion, whereas conjugated bile acids are absorbed by ASBT [106], which is also expressed in the terminal ileum to absorb bile acids from the intestinal lumen. Bile acid secretion from the basolateral membrane into the peribiliary plexus is mediated by OSTα/OSTβ, MRP3, and a truncated ASBT (t-ASBT) [106–110]. These transport systems may play a limited role under normal physiological conditions; however, “cholehepatic shunting” [111], which bypasses enterohepatic circulation along with bile duct proliferation, may help to reduce bile acid overload in cholestasis due to bile duct obstruction.
6 Conclusions
In this review, the characteristics and regulatory systems of hepatobiliary bile acid transporters are presented. Bile acids are among the most essential molecules to organisms. The majority of bile acids are recycled through the enterohepatic circulation, and their cellular concentration is tightly regulated by refined feedback mechanisms. Impaired function of bile acid transporters causes various types of liver injury and may be responsible for other diseases for which their causality is not yet known. Nuclear receptors regulating bile acid transporters are attractive therapeutic targets, and clinical trials for obeticholic acid are ongoing. Further understanding of bile acid transporters will likely lead to new therapeutic options for intractable liver diseases.
References
Strautnieks SS, Kagalwalla AF, Tanner MS, Knisely AS, Bull L, Freimer N, et al. Identification of a locus for progressive familial intrahepatic cholestasis PFIC2 on chromosome 2q24. Am J Hum Genet. 1997;61(3):630–3. doi:10.1086/515501.
Gerloff T, Stieger B, Hagenbuch B, Madon J, Landmann L, Roth J, et al. The sister of P-glycoprotein represents the canalicular bile salt export pump of mammalian liver. J Biol Chem. 1998;273(16):10046–50.
Strautnieks SS, Bull LN, Knisely AS, Kocoshis SA, Dahl N, Arnell H, et al. A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet. 1998;20(3):233–8. doi:10.1038/3034.
Green RM, Hoda F, Ward KL. Molecular cloning and characterization of the murine bile salt export pump. Gene. 2000;241(1):117–23.
Mochizuki K, Kagawa T, Numari A, Harris MJ, Itoh J, Watanabe N, et al. Two N-linked glycans are required to maintain the transport activity of the bile salt export pump (ABCB11) in MDCK II cells. Am J Physiol Gastrointest Liver Physiol. 2007;292(3):G818–28. doi:10.1152/ajpgi.00415.2006.
Kipp H, Arias IM. Newly synthesized canalicular ABC transporters are directly targeted from the Golgi to the hepatocyte apical domain in rat liver. J Biol Chem. 2000;275(21):15917–25. doi:10.1074/jbc.M909875199.
Kipp H, Pichetshote N, Arias IM. Transporters on demand: intrahepatic pools of canalicular ATP binding cassette transporters in rat liver. J Biol Chem. 2001;276(10):7218–24. doi:10.1074/jbc.M007794200.
Wakabayashi Y, Lippincott-Schwartz J, Arias IM. Intracellular trafficking of bile salt export pump (ABCB11) in polarized hepatic cells: constitutive cycling between the canalicular membrane and rab11-positive endosomes. Mol Biol Cell. 2004;15(7):3485–96. doi:10.1091/mbc.E03-10-0737.
Ortiz DF, Moseley J, Calderon G, Swift AL, Li S, Arias IM. Identification of HAX-1 as a protein that binds bile salt export protein and regulates its abundance in the apical membrane of Madin-Darby canine kidney cells. J Biol Chem. 2004;279(31):32761–70. doi:10.1074/jbc.M404337200.
Chan W, Calderon G, Swift AL, Moseley J, Li S, Hosoya H, et al. Myosin II regulatory light chain is required for trafficking of bile salt export protein to the apical membrane in Madin-Darby canine kidney cells. J Biol Chem. 2005;280(25):23741–7. doi:10.1074/jbc.M502767200.
Hayashi H, Inamura K, Aida K, Naoi S, Horikawa R, Nagasaka H, et al. AP2 adaptor complex mediates bile salt export pump internalization and modulates its hepatocanalicular expression and transport function. Hepatology. 2012;55(6):1889–900. doi:10.1002/hep.25591.
Lam P, Xu S, Soroka CJ, Boyer JL. A C-terminal tyrosine-based motif in the bile salt export pump directs clathrin-dependent endocytosis. Hepatology. 2012;55(6):1901–11. doi:10.1002/hep.25523.
Kubitz R, Sutfels G, Kuhlkamp T, Kolling R, Haussinger D. Trafficking of the bile salt export pump from the Golgi to the canalicular membrane is regulated by the p38 MAP kinase. Gastroenterology. 2004;126(2):541–53.
Hayashi H, Sugiyama Y. Short-chain ubiquitination is associated with the degradation rate of a cell-surface-resident bile salt export pump (BSEP/ABCB11). Mol Pharmacol. 2009;75(1):143–50. doi:10.1124/mol.108.049288.
Byrne JA, Strautnieks SS, Mieli-Vergani G, Higgins CF, Linton KJ, Thompson RJ. The human bile salt export pump: characterization of substrate specificity and identification of inhibitors. Gastroenterology. 2002;123(5):1649–58.
Noe J, Stieger B, Meier PJ. Functional expression of the canalicular bile salt export pump of human liver. Gastroenterology. 2002;123(5):1659–66.
Hayashi H, Takada T, Suzuki H, Onuki R, Hofmann AF, Sugiyama Y. Transport by vesicles of glycine- and taurine-conjugated bile salts and taurolithocholate 3-sulfate: a comparison of human BSEP with rat Bsep. Biochim Biophys Acta. 2005;1738(1–3):54–62. doi:10.1016/j.bbalip.2005.10.006.
Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, et al. Identification of a nuclear receptor for bile acids. Science. 1999;284(5418):1362–5.
Ananthanarayanan M, Balasubramanian N, Makishima M, Mangelsdorf DJ, Suchy FJ. Human bile salt export pump promoter is transactivated by the farnesoid X receptor/bile acid receptor. J Biol Chem. 2001;276(31):28857–65. doi:10.1074/jbc.M011610200.
Plass JR, Mol O, Heegsma J, Geuken M, Faber KN, Jansen PL, et al. Farnesoid X receptor and bile salts are involved in transcriptional regulation of the gene encoding the human bile salt export pump. Hepatology. 2002;35(3):589–96. doi:10.1053/jhep.2002.31724.
Pellicciari R, Fiorucci S, Camaioni E, Clerici C, Costantino G, Maloney PR, et al. 6alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J Med Chem. 2002;45(17):3569–72.
Rizzo G, Passeri D, De Franco F, Ciaccioli G, Donadio L, Rizzo G, et al. Functional characterization of the semisynthetic bile acid derivative INT-767, a dual farnesoid X receptor and TGR5 agonist. Mol Pharmacol. 2010;78(4):617–30. doi:10.1124/mol.110.064501.
Yu J, Lo JL, Huang L, Zhao A, Metzger E, Adams A, et al. Lithocholic acid decreases expression of bile salt export pump through farnesoid X receptor antagonist activity. J Biol Chem. 2002;277(35):31441–7. doi:10.1074/jbc.M200474200.
Sayin SI, Wahlstrom A, Felin J, Jantti S, Marschall HU, Bamberg K, et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013;17(2):225–35. doi:10.1016/j.cmet.2013.01.003.
Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102(6):731–44.
Gomez-Ospina N, Potter CJ, Xiao R, Manickam K, Kim MS, Kim KH, et al. Mutations in the nuclear bile acid receptor FXR cause progressive familial intrahepatic cholestasis. Nat Commun. 2016;7:10713. doi:10.1038/ncomms10713.
Song X, Kaimal R, Yan B, Deng R. Liver receptor homolog 1 transcriptionally regulates human bile salt export pump expression. J Lipid Res. 2008;49(5):973–84. doi:10.1194/jlr.M700417-JLR200.
Mataki C, Magnier BC, Houten SM, Annicotte JS, Argmann C, Thomas C, et al. Compromised intestinal lipid absorption in mice with a liver-specific deficiency of liver receptor homolog 1. Mol Cell Biol. 2007;27(23):8330–9. doi:10.1128/MCB.00852-07.
Weerachayaphorn J, Cai SY, Soroka CJ, Boyer JL. Nuclear factor erythroid 2-related factor 2 is a positive regulator of human bile salt export pump expression. Hepatology. 2009;50(5):1588–96. doi:10.1002/hep.23151.
Tanaka Y, Aleksunes LM, Cui YJ, Klaassen CD. ANIT-induced intrahepatic cholestasis alters hepatobiliary transporter expression via Nrf2-dependent and independent signaling. Toxicol Sci. 2009;108(2):247–57. doi:10.1093/toxsci/kfp020.
Song X, Vasilenko A, Chen Y, Valanejad L, Verma R, Yan B, et al. Transcriptional dynamics of bile salt export pump during pregnancy: mechanisms and implications in intrahepatic cholestasis of pregnancy. Hepatology. 2014;60(6):1993–2007. doi:10.1002/hep.27171.
Morotti RA, Suchy FJ, Magid MS. Progressive familial intrahepatic cholestasis (PFIC) type 1, 2, and 3: a review of the liver pathology findings. Semin Liver Dis. 2011;31(1):3–10. doi:10.1055/s-0031-1272831.
van Mil SW, van der Woerd WL, van der Brugge G, Sturm E, Jansen PL, Bull LN, et al. Benign recurrent intrahepatic cholestasis type 2 is caused by mutations in ABCB11. Gastroenterology. 2004;127(2):379–84.
Strautnieks SS, Byrne JA, Pawlikowska L, Cebecauerova D, Rayner A, Dutton L, et al. Severe bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology. 2008;134(4):1203–14. doi:10.1053/j.gastro.2008.01.038.
Byrne JA, Strautnieks SS, Ihrke G, Pagani F, Knisely AS, Linton KJ, et al. Missense mutations and single nucleotide polymorphisms in ABCB11 impair bile salt export pump processing and function or disrupt pre-messenger RNA splicing. Hepatology. 2009;49(2):553–67. doi:10.1002/hep.22683.
Davit-Spraul A, Fabre M, Branchereau S, Baussan C, Gonzales E, Stieger B, et al. ATP8B1 and ABCB11 analysis in 62 children with normal gamma-glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): phenotypic differences between PFIC1 and PFIC2 and natural history. Hepatology. 2010;51(5):1645–55. doi:10.1002/hep.23539.
Jansen PL, Strautnieks SS, Jacquemin E, Hadchouel M, Sokal EM, Hooiveld GJ, et al. Hepatocanalicular bile salt export pump deficiency in patients with progressive familial intrahepatic cholestasis. Gastroenterology. 1999;117(6):1370–9.
Wang L, Soroka CJ, Boyer JL. The role of bile salt export pump mutations in progressive familial intrahepatic cholestasis type II. J Clin Invest. 2002;110(7):965–72. doi:10.1172/JCI15968.
Plass JR, Mol O, Heegsma J, Geuken M, de Bruin J, Elling G, et al. A progressive familial intrahepatic cholestasis type 2 mutation causes an unstable, temperature-sensitive bile salt export pump. J Hepatol. 2004;40(1):24–30.
Hayashi H, Takada T, Suzuki H, Akita H, Sugiyama Y. Two common PFIC2 mutations are associated with the impaired membrane trafficking of BSEP/ABCB11. Hepatology. 2005;41(4):916–24. doi:10.1002/hep.20627.
Lam P, Pearson CL, Soroka CJ, Xu S, Mennone A, Boyer JL. Levels of plasma membrane expression in progressive and benign mutations of the bile salt export pump (Bsep/Abcb11) correlate with severity of cholestatic diseases. Am J Phys Cell Physiol. 2007;293(5):C1709–16. doi:10.1152/ajpcell.00327.2007.
Kagawa T, Watanabe N, Mochizuki K, Numari A, Ikeno Y, Itoh J, et al. Phenotypic differences in PFIC2 and BRIC2 correlate with protein stability of mutant Bsep and impaired taurocholate secretion in MDCK II cells. Am J Physiol Gastrointest Liver Physiol. 2008;294(1):G58–67. doi:10.1152/ajpgi.00367.2007.
van Ooteghem NA, Klomp LW, van Berge-Henegouwen GP, Houwen RH. Benign recurrent intrahepatic cholestasis progressing to progressive familial intrahepatic cholestasis: low GGT cholestasis is a clinical continuum. J Hepatol. 2002;36(3):439–43.
Cai SY, Gautam S, Nguyen T, Soroka CJ, Rahner C, Boyer JL. ATP8B1 deficiency disrupts the bile canalicular membrane bilayer structure in hepatocytes, but FXR expression and activity are maintained. Gastroenterology. 2009;136(3):1060–9. doi:10.1053/j.gastro.2008.10.025.
Paulusma CC, de Waart DR, Kunne C, Mok KS, Elferink RP. Activity of the bile salt export pump (ABCB11) is critically dependent on canalicular membrane cholesterol content. J Biol Chem. 2009;284(15):9947–54. doi:10.1074/jbc.M808667200.
Girard M, Lacaille F, Verkarre V, Mategot R, Feldmann G, Grodet A, et al. MYO5B and bile salt export pump contribute to cholestatic liver disorder in microvillous inclusion disease. Hepatology. 2014;60(1):301–10. doi:10.1002/hep.26974.
Dixon PH, van Mil SW, Chambers J, Strautnieks S, Thompson RJ, Lammert F, et al. Contribution of variant alleles of ABCB11 to susceptibility to intrahepatic cholestasis of pregnancy. Gut. 2009;58(4):537–44. doi:10.1136/gut.2008.159541.
Dixon PH, Wadsworth CA, Chambers J, Donnelly J, Cooley S, Buckley R, et al. A comprehensive analysis of common genetic variation around six candidate loci for intrahepatic cholestasis of pregnancy. Am J Gastroenterol. 2014;109(1):76–84. doi:10.1038/ajg.2013.406.
Meier Y, Pauli-Magnus C, Zanger UM, Klein K, Schaeffeler E, Nussler AK, et al. Interindividual variability of canalicular ATP-binding-cassette (ABC)-transporter expression in human liver. Hepatology. 2006;44(1):62–74. doi:10.1002/hep.21214.
Ho RH, Leake BF, Kilkenny DM, Meyer Zu Schwabedissen HE, Glaeser H, Kroetz DL, et al. Polymorphic variants in the human bile salt export pump (BSEP; ABCB11): functional characterization and interindividual variability. Pharmacogenet Genomics. 2010;20(1):45–57. doi:10.1097/FPC.0b013e3283349eb0.
Lang C, Meier Y, Stieger B, Beuers U, Lang T, Kerb R, et al. Mutations and polymorphisms in the bile salt export pump and the multidrug resistance protein 3 associated with drug-induced liver injury. Pharmacogenet Genomics. 2007;17(1):47–60. doi:10.1097/01.fpc.0000230418.28091.76.
Kagawa T, Hirose S, Arase Y, Oka A, Anzai K, Tsuruya K, et al. No contribution of the ABCB11 p.444A polymorphism in Japanese patients with drug-induced cholestasis. Drug Metab Dispos. 2015; doi:10.1124/dmd.114.061325.
Aleo MD, Luo Y, Swiss R, Bonin PD, Potter DM, Will Y. Human drug-induced liver injury severity is highly associated with dual inhibition of liver mitochondrial function and bile salt export pump. Hepatology. 2014;60(3):1015–22. doi:10.1002/hep.27206.
Beuers U, Nathanson MH, Isales CM, Boyer JL. Tauroursodeoxycholic acid stimulates hepatocellular exocytosis and mobilizes extracellular Ca++ mechanisms defective in cholestasis. J Clin Invest. 1993;92(6):2984–93. doi:10.1172/JCI116921.
Kurz AK, Graf D, Schmitt M, Vom Dahl S, Haussinger D. Tauroursodesoxycholate-induced choleresis involves p38(MAPK) activation and translocation of the bile salt export pump in rats. Gastroenterology. 2001;121(2):407–19.
Kagawa T, Orii R, Hirose S, Arase Y, Shiraishi K, Mizutani A, et al. Ursodeoxycholic acid stabilizes the bile salt export pump in the apical membrane in MDCK II cells. J Gastroenterol. 2013; doi:10.1007/s00535-013-0833-y.
Varma S, Revencu N, Stephenne X, Scheers I, Smets F, Beleza-Meireles A, et al. Retargeting of bile salt export pump and favorable outcome in children with progressive familial intrahepatic cholestasis type 2. Hepatology. 2015;62(1):198–206. doi:10.1002/hep.27834.
Hayashi H, Sugiyama Y. 4-phenylbutyrate enhances the cell surface expression and the transport capacity of wild-type and mutated bile salt export pumps. Hepatology. 2007;45(6):1506–16. doi:10.1002/hep.21630.
Naoi S, Hayashi H, Inoue T, Tanikawa K, Igarashi K, Nagasaka H, et al. Improved liver function and relieved pruritus after 4-phenylbutyrate therapy in a patient with progressive familial intrahepatic cholestasis type 2. J Pediatr. 2014;164(5):1219–27. e3 doi:10.1016/j.jpeds.2013.12.032.
Gonzales E, Grosse B, Schuller B, Davit-Spraul A, Conti F, Guettier C, et al. Targeted pharmacotherapy in progressive familial intrahepatic cholestasis type 2: evidence for improvement of cholestasis with 4-phenylbutyrate. Hepatology. 2015;62(2):558–66. doi:10.1002/hep.27767.
Hayashi H, Naoi S, Hirose Y, Matsuzaka Y, Tanikawa K, Igarashi K, et al. Successful treatment with 4-phenylbutyrate in a patient with benign recurrent intrahepatic cholestasis type 2 refractory to biliary drainage and bilirubin absorption. Hepatol Res. 2016;46(2):192–200. doi:10.1111/hepr.12561.
Keitel V, Burdelski M, Vojnisek Z, Schmitt L, Haussinger D, Kubitz R. De novo bile salt transporter antibodies as a possible cause of recurrent graft failure after liver transplantation: a novel mechanism of cholestasis. Hepatology. 2009;50(2):510–7. doi:10.1002/hep.23083.
Jara P, Hierro L, Martinez-Fernandez P, Alvarez-Doforno R, Yanez F, Diaz MC, et al. Recurrence of bile salt export pump deficiency after liver transplantation. N Engl J Med. 2009;361(14):1359–67. doi:10.1056/NEJMoa0901075.
Maggiore G, Gonzales E, Sciveres M, Redon MJ, Grosse B, Stieger B, et al. Relapsing features of bile salt export pump deficiency after liver transplantation in two patients with progressive familial intrahepatic cholestasis type 2. J Hepatol. 2010;53(5):981–6. doi:10.1016/j.jhep.2010.05.025.
Lin HC, Alvarez L, Laroche G, Melin-Aldana H, Pfeifer K, Schwarz K, et al. Rituximab as therapy for the recurrence of bile salt export pump deficiency after liver transplantation. Liver Transpl. 2013;19(12):1403–10. doi:10.1002/lt.23754.
Stindt J, Kluge S, Droge C, Keitel V, Stross C, Baumann U, et al. Bile salt export pump-reactive antibodies form a polyclonal, multi-inhibitory response in antibody-induced bile salt export pump deficiency. Hepatology. 2016;63(2):524–37. doi:10.1002/hep.28311.
Boyer JL, Ng OC, Ananthanarayanan M, Hofmann AF, Schteingart CD, Hagenbuch B, et al. Expression and characterization of a functional rat liver Na+ bile acid cotransport system in COS-7 cells. Am J Phys. 1994;266(3 Pt 1):G382–7.
Hagenbuch B, Meier PJ. Molecular cloning, chromosomal localization, and functional characterization of a human liver Na+/bile acid cotransporter. J Clin Invest. 1994;93(3):1326–31. doi:10.1172/JCI117091.
Hagenbuch B, Meier PJ. Sinusoidal (basolateral) bile salt uptake systems of hepatocytes. Semin Liver Dis. 1996;16(2):129–36. doi:10.1055/s-2007-1007226.
Hata S, Wang P, Eftychiou N, Ananthanarayanan M, Batta A, Salen G, et al. Substrate specificities of rat oatp1 and ntcp: implications for hepatic organic anion uptake. Am J Physiol Gastrointest Liver Physiol. 2003;285(5):G829–39. doi:10.1152/ajpgi.00352.2002.
Mita S, Suzuki H, Akita H, Stieger B, Meier PJ, Hofmann AF, et al. Vectorial transport of bile salts across MDCK cells expressing both rat Na+−taurocholate cotransporting polypeptide and rat bile salt export pump. Am J Physiol Gastrointest Liver Physiol. 2005;288(1):G159–67. doi:10.1152/ajpgi.00360.2003.
Mita S, Suzuki H, Akita H, Hayashi H, Onuki R, Hofmann AF, et al. Vectorial transport of unconjugated and conjugated bile salts by monolayers of LLC-PK1 cells doubly transfected with human NTCP and BSEP or with rat Ntcp and Bsep. Am J Physiol Gastrointest Liver Physiol. 2006;290(3):G550–6. doi:10.1152/ajpgi.00364.2005.
Hagenbuch B, Meier PJ. The superfamily of organic anion transporting polypeptides. Biochim Biophys Acta. 2003;1609(1):1–18.
Vaz FM, Paulusma CC, Huidekoper H, de Ru M, Lim C, Koster J, et al. Sodium taurocholate cotransporting polypeptide (SLC10A1) deficiency: conjugated hypercholanemia without a clear clinical phenotype. Hepatology. 2015;61(1):260–7. doi:10.1002/hep.27240.
Slijepcevic D, Kaufman C, Wichers CG, Gilglioni EH, Lempp FA, Duijst S, et al. Impaired uptake of conjugated bile acids and hepatitis b virus pres1-binding in na(+) – taurocholate cotransporting polypeptide knockout mice. Hepatology. 2015;62(1):207–19. doi:10.1002/hep.27694.
Zollner G, Fickert P, Zenz R, Fuchsbichler A, Stumptner C, Kenner L, et al. Hepatobiliary transporter expression in percutaneous liver biopsies of patients with cholestatic liver diseases. Hepatology. 2001;33(3):633–46. doi:10.1053/jhep.2001.22646.
Zollner G, Fickert P, Silbert D, Fuchsbichler A, Marschall HU, Zatloukal K, et al. Adaptive changes in hepatobiliary transporter expression in primary biliary cirrhosis. J Hepatol. 2003;38(6):717–27.
Gartung C, Ananthanarayanan M, Rahman MA, Schuele S, Nundy S, Soroka CJ, et al. Down-regulation of expression and function of the rat liver Na+/bile acid cotransporter in extrahepatic cholestasis. Gastroenterology. 1996;110(1):199–209.
Trauner M, Arrese M, Lee H, Boyer JL, Karpen SJ. Endotoxin downregulates rat hepatic ntcp gene expression via decreased activity of critical transcription factors. J Clin Invest. 1998;101(10):2092–100. doi:10.1172/JCI1680.
Lee YK, Dell H, Dowhan DH, Hadzopoulou-Cladaras M, Moore DD. The orphan nuclear receptor SHP inhibits hepatocyte nuclear factor 4 and retinoid X receptor transactivation: two mechanisms for repression. Mol Cell Biol. 2000;20(1):187–95.
Halilbasic E, Claudel T, Trauner M. Bile acid transporters and regulatory nuclear receptors in the liver and beyond. J Hepatol. 2013;58(1):155–68. doi:10.1016/j.jhep.2012.08.002.
Denson LA, Sturm E, Echevarria W, Zimmerman TL, Makishima M, Mangelsdorf DJ, et al. The orphan nuclear receptor, shp, mediates bile acid-induced inhibition of the rat bile acid transporter, ntcp. Gastroenterology. 2001;121(1):140–7.
Eloranta JJ, Jung D, Kullak-Ublick GA. The human Na+−taurocholate cotransporting polypeptide gene is activated by glucocorticoid receptor and peroxisome proliferator-activated receptor-gamma coactivator-1alpha, and suppressed by bile acids via a small heterodimer partner-dependent mechanism. Mol Endocrinol. 2006;20(1):65–79. doi:10.1210/me.2005-0159.
Wang L, Han Y, Kim CS, Lee YK, Moore DD. Resistance of SHP-null mice to bile acid-induced liver damage. J Biol Chem. 2003;278(45):44475–81. doi:10.1074/jbc.M305258200.
Webster CR, Anwer MS. Role of the PI3K/PKB signaling pathway in cAMP-mediated translocation of rat liver Ntcp. Am J Phys. 1999;277(6 Pt 1):G1165–72.
Webster CR, Srinivasulu U, Ananthanarayanan M, Suchy FJ, Anwer MS. Protein kinase B/Akt mediates cAMP – and cell swelling-stimulated Na+/taurocholate cotransport and Ntcp translocation. J Biol Chem. 2002;277(32):28578–83. doi:10.1074/jbc.M201937200.
Webster CR, Blanch C, Anwer MS. Role of PP2B in cAMP-induced dephosphorylation and translocation of NTCP. Am J Physiol Gastrointest Liver Physiol. 2002;283(1):G44–50. doi:10.1152/ajpgi.00530.2001.
Muhlfeld S, Domanova O, Berlage T, Stross C, Helmer A, Keitel V, et al. Short-term feedback regulation of bile salt uptake by bile salts in rodent liver. Hepatology. 2012;56(6):2387–97. doi:10.1002/hep.25955.
Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife. 2012;1:e00049. doi:10.7554/eLife.00049.
Ni Y, Lempp FA, Mehrle S, Nkongolo S, Kaufman C, Falth M, et al. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology. 2014;146(4):1070–83. doi:10.1053/j.gastro.2013.12.024.
Oehler N, Volz T, Bhadra OD, Kah J, Allweiss L, Giersch K, et al. Binding of hepatitis B virus to its cellular receptor alters the expression profile of genes of bile acid metabolism. Hepatology. 2014;60(5):1483–93. doi:10.1002/hep.27159.
Ho RH, Leake BF, Roberts RL, Lee W, Kim RB. Ethnicity-dependent polymorphism in Na+-taurocholate cotransporting polypeptide (SLC10A1) reveals a domain critical for bile acid substrate recognition. J Biol Chem. 2004;279(8):7213–22. doi:10.1074/jbc.M305782200.
Peng L, Zhao Q, Li Q, Li M, Li C, Xu T, et al. The p.Ser267Phe variant in SLC10A1 is associated with resistance to chronic hepatitis B. Hepatology. 2015;61(4):1251–60. doi:10.1002/hep.27608.
Hu HH, Liu J, Lin YL, Luo WS, Chu YJ, Chang CL, et al. The rs2296651 (S267F) variant on NTCP (SLC10A1) is inversely associated with chronic hepatitis B and progression to cirrhosis and hepatocellular carcinoma in patients with chronic hepatitis B. Gut. 2015; doi:10.1136/gutjnl-2015-310686.
Dawson PA, Hubbert M, Haywood J, Craddock AL, Zerangue N, Christian WV, et al. The heteromeric organic solute transporter alpha-beta, Ostalpha-Ostbeta, is an ileal basolateral bile acid transporter. J Biol Chem. 2005;280(8):6960–8. doi:10.1074/jbc.M412752200.
Li N, Cui Z, Fang F, Lee JY, Ballatori N. Heterodimerization, trafficking and membrane topology of the two proteins, Ost alpha and Ost beta, that constitute the organic solute and steroid transporter. Biochem J. 2007;407(3):363–72. doi:10.1042/BJ20070716.
Boyer JL, Trauner M, Mennone A, Soroka CJ, Cai SY, Moustafa T, et al. Upregulation of a basolateral FXR-dependent bile acid efflux transporter OSTalpha-OSTbeta in cholestasis in humans and rodents. Am J Physiol Gastrointest Liver Physiol. 2006;290(6):G1124–30. doi:10.1152/ajpgi.00539.2005.
Cui YJ, Aleksunes LM, Tanaka Y, Goedken MJ, Klaassen CD. Compensatory induction of liver efflux transporters in response to ANIT-induced liver injury is impaired in FXR-null mice. Toxicol Sci. 2009;110(1):47–60. doi:10.1093/toxsci/kfp094.
Zollner G, Wagner M, Moustafa T, Fickert P, Silbert D, Gumhold J, et al. Coordinated induction of bile acid detoxification and alternative elimination in mice: role of FXR-regulated organic solute transporter-alpha/beta in the adaptive response to bile acids. Am J Physiol Gastrointest Liver Physiol. 2006;290(5):G923–32. doi:10.1152/ajpgi.00490.2005.
Landrier JF, Eloranta JJ, Vavricka SR, Kullak-Ublick GA. The nuclear receptor for bile acids, FXR, transactivates human organic solute transporter-alpha and – beta genes. Am J Physiol Gastrointest Liver Physiol. 2006;290(3):G476–85. doi:10.1152/ajpgi.00430.2005.
Lee H, Zhang Y, Lee FY, Nelson SF, Gonzalez FJ, Edwards PA. FXR regulates organic solute transporters alpha and beta in the adrenal gland, kidney, and intestine. J Lipid Res. 2006;47(1):201–14. doi:10.1194/jlr.M500417-JLR200.
Wagner M, Fickert P, Zollner G, Fuchsbichler A, Silbert D, Tsybrovskyy O, et al. Role of farnesoid X receptor in determining hepatic ABC transporter expression and liver injury in bile duct-ligated mice. Gastroenterology. 2003;125(3):825–38.
Zelcer N, van de Wetering K, de Waart R, Scheffer GL, Marschall HU, Wielinga PR, et al. Mice lacking Mrp3 (Abcc3) have normal bile salt transport, but altered hepatic transport of endogenous glucuronides. J Hepatol. 2006;44(4):768–75. doi:10.1016/j.jhep.2005.07.022.
Mennone A, Soroka CJ, Cai SY, Harry K, Adachi M, Hagey L, et al. Mrp4−/− mice have an impaired cytoprotective response in obstructive cholestasis. Hepatology. 2006;43(5):1013–21. doi:10.1002/hep.21158.
Keitel V, Burdelski M, Warskulat U, Kuhlkamp T, Keppler D, Haussinger D, et al. Expression and localization of hepatobiliary transport proteins in progressive familial intrahepatic cholestasis. Hepatology. 2005;41(5):1160–72. doi:10.1002/hep.20682.
Lazaridis KN, Pham L, Tietz P, Marinelli RA, deGroen PC, Levine S, et al. Rat cholangiocytes absorb bile acids at their apical domain via the ileal sodium-dependent bile acid transporter. J Clin Invest. 1997;100(11):2714–21. doi:10.1172/JCI119816.
Kool M, van der Linden M, de Haas M, Scheffer GL, de Vree JM, Smith AJ, et al. MRP3, an organic anion transporter able to transport anti-cancer drugs. Proc Natl Acad Sci U S A. 1999;96(12):6914–9.
Lazaridis KN, Tietz P, Wu T, Kip S, Dawson PA, LaRusso NF. Alternative splicing of the rat sodium/bile acid transporter changes its cellular localization and transport properties. Proc Natl Acad Sci U S A. 2000;97(20):11092–7. doi:10.1073/pnas.200325297.
Soroka CJ, Lee JM, Azzaroli F, Boyer JL. Cellular localization and up-regulation of multidrug resistance-associated protein 3 in hepatocytes and cholangiocytes during obstructive cholestasis in rat liver. Hepatology. 2001;33(4):783–91. doi:10.1053/jhep.2001.23501.
Ballatori N, Christian WV, Lee JY, Dawson PA, Soroka CJ, Boyer JL, et al. OSTalpha-OSTbeta: a major basolateral bile acid and steroid transporter in human intestinal, renal, and biliary epithelia. Hepatology. 2005;42(6):1270–9. doi:10.1002/hep.20961.
Hofmann AF. Bile acids: trying to understand their chemistry and biology with the hope of helping patients. Hepatology. 2009;49(5):1403–18. doi:10.1002/hep.22789.
Acknowledgments
This work was supported in part by the Japan Agency for Medical Research and Development (16mk0101045s0202). I thank Ms. Satsuki Ieda and Reiko Orii for their technical assistance.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Japan KK
About this chapter
Cite this chapter
Kagawa, T. (2017). Hepatobiliary Transport of Bile Acids. In: Tazuma, S., Takikawa, H. (eds) Bile Acids in Gastroenterology. Springer, Tokyo. https://doi.org/10.1007/978-4-431-56062-3_2
Download citation
DOI: https://doi.org/10.1007/978-4-431-56062-3_2
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-56060-9
Online ISBN: 978-4-431-56062-3
eBook Packages: MedicineMedicine (R0)