
Abstract
The liver detoxifies and excretes both exogenous xenobiotics, such as drugs and environmental toxins, and endogenous substances, including ammonia and bilirubin. Efficient processing and elimination of these substances prevent cellular injury, including CNS toxicity in the case of ammonia and bilirubin. Hepatic synthesis, secretion and re-uptake of bile acids is an exquisitely regulated process that allows modulation of cholesterol metabolism and efficient absorption of the products of fat digestion, as well as compensatory trafficking of bile acids out of the hepatocyte in the face of bile acid retention as occurs in cholestasis. Nuclear receptor transcription factors tightly regulate these processes and offer potential therapeutic targets for new approaches to treatment of a variety of diseases. This chapter reviews these important functions of the liver, underscoring another aspect of its central role in human physiology.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
The liver detoxifies and excretes both exogenous xenobiotics, such as drugs and environmental toxins, and endogenous substances, including ammonia and bilirubin. Efficient processing and elimination of these substances prevent cellular injury, including CNS toxicity in the case of ammonia and bilirubin. Hepatic synthesis, secretion and re-uptake of bile acids is an exquisitely regulated process that allows modulation of cholesterol metabolism and efficient absorption of the products of fat digestion, as well as compensatory trafficking of bile acids out of the hepatocyte in the face of bile acid retention as occurs in cholestasis. Nuclear receptor transcription factors tightly regulate these processes and offer potential therapeutic targets for new approaches to treatment of a variety of diseases. This chapter reviews these important functions of the liver, underscoring another aspect of its central role in human physiology.
2 Urea Synthesis and Ammonia Metabolism
The urea cycle (Fig. 12.1) is responsible for clearing the majority of the body’s excess nitrogen and producing arginine for protein synthesis or conversion to urea. All five enzymes are located primarily in the zone 1 periportal hepatocytes . The first two enzymes are in the mitochondrial matrix and the other three are in the cytosol. As shown in Fig. 12.1, ammonia is the common substrate for entry into the cycle. Ammonia is derived from several sources in the body, including the degradation of amino acids, amines, and nucleic acids. Ammonia is also a byproduct of bacterial degradation of urea in the gastrointestinal tract, which diffuses into the bowel lumen from the bloodstream. Urease-producing bacteria in the lumen of the colon split the urea, as well as luminal amino acids, into CO2 and ammonia. The ammonia then diffuses back into the bloodstream into the portal circulation and is cleared by the liver , predominantly on the first pass. Glutamate is produced in large quantities from the catabolism of amino acids. Glutamate serves as a major source of urea by undergoing transamination in the liver to aspartate, which then enters the urea cycle. Although several organs produce urea, the liver is the only quantitatively important organ. Skeletal muscle is capable of ammonia detoxification by the reaction of glutamate with ammonia to produce glutamine . Ammonia clearance by skeletal muscle is the reason for the higher ammonia concentration in arterial than in venous blood. In skeletal muscle, protein catabolism generates large amounts of ammonia resulting in formation of glutamate by transamination, and the glutamate is converted to alanine. Alanine is transported to the liver, where glutamate is regenerated by transamination of alanine to pyruvate. Ammonia is then released from the glutamate and enters the urea cycle. The pyruvate is a substrate for gluconeogenesis to produce glucose, which may circulate back to the muscle for energy production.

General scheme of ammonia metabolism in the liver . Solid arrows, pathways of ammonia utilization; broken arrows, pathways of ammonia formation (Reproduced with permission from Ampola [1])
The urea cycle is subject to both short- and long-term regulation. Short-term regulation is accomplished by intracellular substrate levels. In long-term regulation, total hepatic content of the five urea cycle enzymes is proportional to protein intake and rate of urea excretion. All five urea cycle enzymes may be induced by glucagon and glucocorticoids . The rate-limiting enzyme in vivo is probably carbamylphosphate synthetase I.
Ammonia is toxic to the central nervous system , and in disorders characterized by hyperammonemia, there is increased movement of ammonia across the blood–brain barrier. The cells most susceptible to ammonia toxicity are the astrocytic or glial cells. Astrocytes are the site of ammonia detoxification in the central nervous system with glutamine production from glutamate, a major excitatory neurotransmitter in the brain. In fact, cerebrospinal fluid glutamine levels parallel the severity of neurologic dysfunction. As the level of glutamate declines, it is replenished by condensation of ammonia with α-ketoglutarate. Depletion of α-ketoglutarate results in impaired mitochondrial glucose oxidation. Ammonia may also adversely affect neurotransmitter function and directly interfere with cellular energy production. As the degree of hyperammonemia increases, neurologic function progressively deteriorates with onset of deepening coma and finally development of cerebral edema, which is often a terminal event.
Inherited defects in the urea cycle enzymes may result in hyperammonemia and neurologic dysfunction, often to a severe degree. Inherited defects in all five enzymes of the cycle, as well as N-acetylglutamate synthetase (which produces an essential cofactor for carbamyl phosphate synthetase), have been described. There may be a spectrum of severity, depending on the specific defect in the enzyme and whether there is partial residual function. In severe cases, seizures, coma, and death may occur in the neonatal period, if untreated.
3 Hepatic Encephalopathy
Hepatic encephalopathy may be classified as two general types. In patients with cirrhosis , portal hypertension, and large portosystemic shunts, ammonia from intestinal sources is not cleared effectively on first-pass through the liver . Also, the relief of portal hypertension by surgical creation of a portosystemic shunt or placement of a transjugular intrahepatic portosystemic shunt (TIPS) may precipitate encephalopathy. This ineffective clearance, combined with generally poor hepatic function, may result in chronic encephalopathy, punctuated by acute exacerbations often precipitated by gastrointestinal bleeding from esophageal varices (resulting in an increased intestinal ammonia load), infection, excessive protein intake, or overzealous diuresis to treat ascites . Chronic pathologic changes in astrocytes in the central nervous system may be present. This type of encephalopathy is referred to as portosystemic encephalopathy . The other category of encephalopathy is observed in the patient with acute, severe liver disease (usually fulminant hepatitis ) who develops progressive encephalopathy due primarily to severe liver dysfunction with inadequate ammonia detoxification . Acute liver failure with encephalopathy often rapidly progresses to cerebral edema and death. The cerebral edema is thought to be a direct result of ammonia toxicity. In fact, the edema may actually be a result of the osmotic effect of ammonia-induced elevation of brain glutamine levels.
It should be stressed that in encephalopathy associated with liver disease, other factors may contribute to the central nervous system dysfunction in addition to hyperammonemia. In fact, the degree of hyperammonemia does not always correlate with the degree of encephalopathy, suggesting that other mechanisms may be involved. In acute liver failure, other neurotoxic substances, such as mercaptans and short-chain fatty acids , may play a role. There is evidence that hepatic encephalopathy may result in part from the action of endogenous substances that bind to the GABA-benzodiazepine receptor complex in the brain. This is supported clinically by the observation that portosystemic encephalopathy and encephalopathy associated with acute liver failure may improve transiently with treatment with benzodiazepine receptor antagonists. However, there are limited data to support their clinical use. There may be increased entry of ammonia into the central nervous system due to increased permeability of the blood–brain barrier and conversion of positively charged ammonium ion to ammonia, which more readily crosses the blood–brain barrier, secondary to changes in blood pH. Accumulation of glutamine in the brain results in enhanced central nervous system uptake of tryptophan , which is converted into both neuroactive and neurotoxic metabolites, including serotonin and tryptamine.
Clinically, the presentation of the first stage of hepatic encephalopathy may be subtle. There may be personality changes and inversion of sleep patterns. The patient may exhibit a short attention span and easy forgetfulness. With portosystemic encephalopathy , it is now recognized that very mild impairment may affect social skills and emotional behavior, and evaluation of driving skills may be a practical index to diagnose what is termed minimal hepatic encephalopathy (MHE). An early physical sign of hepatic encephalopathy is asterixis. This sign represents an inability to maintain posture and is usually elicited by asking the patient to dorsiflex the hand with the arm extended. After several seconds a positive sign is characterized by the hand falling forward followed by a brisk recovery, thereby resulting in the typical “flapping” tremor. This sign is dependent upon the patient being able to understand and follow directions, and is highly variable over short periods of time, even in the same individual. Asterixis is not specific for hepatic encephalopathy and may be elicited in other disorders, including uremia. In stage 2, the individual becomes lethargic and disoriented with other abnormal reflexes in addition to asterixis. In stage 3, the patient is somnolent, but arousable. However, when awake the patient is unable to communicate or perform mental tasks such as calculation. Stage 4 encephalopathy is characterized by total inability to arouse the patient accompanied by a positive Babinski sign and decerebrate posturing, and usually immediately precedes the development of cerebral edema and death. Abnormal changes on electroencephalogram (EEG) are detectable as early as stage 1 and generally worsen in parallel with clinical progression to stage 4.
Treatment of hyperammonemia should be directed toward reducing the production of ammonia and enhancing its clearance. Moderate protein restriction had been recommended in the past. However, since restriction results in enhanced catabolism of endogenous protein, and a significant positive effect on outcomes has yet to be proven, such restriction is not currently recommended. Branched-chain amino acid supplementation may have a modest beneficial effect, but is prohibitively expensive. Attention should be directed to sources of gastrointestinal bleeding, such as esophageal varices , which may result in a large intestinal ammonia load. Lactulose is a non-absorbable carbohydrate administered enterally and is fermented by colonic bacteria to form organic acids, which lower the luminal pH. Since only ammonia in the form of NH3 is readily absorbed, an increase in the NH4 +/NH3 ratio results in trapping of ammonia in the gut and elimination in the feces. Also, the osmotic property of lactulose results in a significant laxative effect and may effectively clear ammoniagenic substances from the gastrointestinal tract. The non-absorbable antibiotic, rifaximin , is used to suppress intestinal ammonia-forming bacteria. In urgent situations, high ammonia levels may also be cleared by hemodialysis or exchange transfusion. Use of external liver support systems has generally not significantly impacted outcomes. The molecular absorbent recirculating system (MARS) has shown some benefit in more severe cases. In cases of encephalopathy associated with liver failure, liver transplantation may result in prompt resolution of severe encephalopathy, providing that cerebral edema has not developed.
4 Hepatic Detoxification
The liver is responsible for the biotransformation and clearance of a variety of endogenous and exogenous toxic compounds. Exogenous organic substances entering the body that have no nutritive value for energy production, structural function, or enzyme cofactor function are called xenobiotics. The liver is capable of numerous reactions which generally serve to make the substance to be eliminated more polar and water soluble to subsequently be excreted in bile or urine (Phase I reactions: oxidoreductases and hydrolases) and to make the substance less toxic (Phase II reactions: transferases). As will also be demonstrated, these reactions do not always result in detoxification , but may actually form toxic intermediary metabolites. Apical canalicular excretion of these biotransformed molecules into bile for elimination or basolateral membrane transport into the circulation to be cleared by the kidney occurs via specific ATP-binding cassette (ABC) transporters and is termed Phase III transport.
4.1 Phase I Reactions
The enzyme systems responsible for oxidation and reduction reactions are components of Phase I biotransformation. Dehydrogenases catalyze hydrogen transfer from a substrate to a hydrogen receptor, frequently a pyridine nucleotide. Examples include alcohol dehydrogenases, cytosolic enzymes, which catalyze NAD-dependent oxidation of alcohols to aldehydes or ketones by the following reaction:
Aldehyde dehydrogenases are present in mitochondrial, microsomal, and cytosolic hepatocyte fractions. The production of aldehydes from alcohols in the above reaction is reversible. However, conversion of the aldehydes to ketones by aldehyde dehydrogenases is essentially irreversible:
Another dehydrogenase is dihydrodiol dehydrogenase, which catalyzes the NADP+-dependent oxidation of metabolites of polycyclic aromatic hydrocarbon compounds.
Hepatic reductases include aldehyde and ketone reductases, quinone reductases, nitro and nitroso reductases, azoreductases, N-oxide reductases, and sulfoxide reductases. Substrates for these enzymes are drugs and xenobiotics, including warfarin , aflatoxin, adriamycin, chloramphenicol, and dimethylsulfoxide. Oxidases catalyze transfer of electrons from substrate to oxygen, generating either hydrogen peroxide or superoxide anions, and include aldehyde oxidases and monoamine oxidases.
The hepatic mono-oxygenases are microsomal enzymes that catalyze reactions in which one atom of oxygen is incorporated into product and the other is reduced to water:
The cytochrome P450-dependent mono-oxygenases have been the most intensively studied. Although these enzymes are found in many tissues throughout the body, they are most abundant in the liver . Within the hepatocyte this system is located primarily in the smooth endoplasmic reticulum. It is also found in the nuclear membrane, where this fraction is probably more important in carcinogenesis. The system derives its name from “P” which stands for “pigment” and “450” which indicates the absorption wavelength in nm at which a maximum occurs with the binding of carbon monoxide under anaerobic conditions. This mono-oxygenase system consists of a flavoprotein and a family of hemoproteins. The scheme for the function of this complex is shown in Fig. 12.2. In the first step, the oxidized (Fe3+) form of the enzyme binds the reduced substrate. In the rate-limiting second step, the complexed cytochrome is reduced to the Fe2+ form by a flavoprotein, NADPH-cytochrome P450 reductase, with NADPH as the ultimate electron donor. In step three, the reduced cytochrome-drug complex combines with molecular oxygen to yield a ternary intermediate in which the iron is again oxidized to the Fe3+ state. In the fourth step, the ternary complex decomposes to yield the free oxidized cytochrome, the oxidized form of the substrate, and water. The entire cycle involves a two-electron transfer and consumption of one mole of NADPH and one mole of molecular oxygen per mole of drug reduced. The components of this system are integral membrane proteins. Reconstitution of the proteins in vitro requires the presence of phospholipid for a catalytically active complex. These mono-oxygenases are subject to genetic variation and can be induced or inhibited by xenobiotics, which may affect the metabolism of certain drugs.

The cytochrome P450-dependent mono-oxygenase redox cycle. In the first step, the oxidized (Fe3+) form of the enzyme binds the reduced drug substrate. In the rate-limiting second step, the complexed cytochrome is reduced to the Fe2+ form by a flavoprotein, NADPH-cytochrome P450 reductase, with NADPH as the ultimate electron donor. In step three, the reduced cytochrome-drug complex combines with molecular oxygen to yield a ternary intermediate in which the iron is again oxidized to the Fe3+ state. In the fourth step, the ternary complex decomposes to yield the free oxidized cytochrome, the oxidized form of the drug, and water
Hydrolytic reactions involve substrates that contain ester linkages. Hydrolases cleave ester linkages with addition of a molecule of water:
The products of this hydrolysis, an acid and an alcohol, are more water-soluble than the parent ester. In addition to increasing water solubility, this reaction often results in detoxification of the xenobiotic. The reaction products may also be substrates for Phase II reactions. Xenobiotics subject to hydrolysis include aspirin, meperidine, malathion, and nerve gas.
4.2 Phase II Reactions
The general scheme for conjugation reactions is as follows:
One of the most common conjugation reactions is glucuronidation . Glucuronic acid is transferred from UDP-glucuronic acid (UDPGA) to the hydroxyl or carboxyl group on the acceptor substrate molecule to form a β-glucuronide. Glucuronidation generally not only enhances water solubility, but also detoxifies the compound. Substrates for this reaction include acetaminophen, morphine, chloramphenicol, furosemide, sulfisoxazole, and valproic acid. As discussed subsequently, bilirubin, an endogenous waste product of heme catabolism, is a major substrate for conjugation with glucuronide. The glucuronidation capacity of the liver depends on the rate of synthesis of UDPGA from UDP-glucose , which requires adequate carbohydrate reserves. Another Phase II conjugation reaction is sulfation. Conjugation with sulfate generally results in compounds that are less toxic and more water-soluble. Substrates for this reaction include alcohols, bile acids, and phenolic compounds.
Glutathione (GSH) is a tripeptide (γ-glutamyl-cysteinyl-glycine ) which participates in substitution reactions that are catalyzed by glutathione S-transferases, whereby the GSH displaces a group on a xenobiotic, which generally has an electrophilic center. Electrophilic compounds are particularly toxic to cells because of their propensity to react spontaneously with -SH groups on proteins or amine groups on nucleic acids, leading to cell necrosis or carcinogenesis. Substrates for this reaction include nitroglycerin, the diuretic ethacrynic acid, certain insecticides, and sulfobromophthalein (BSP). The GSH conjugates are generally excreted into bile. However, they may be reabsorbed in the intestine and subsequently taken up by the kidney, where the enzyme γ-glutamyl-transpeptidase (GGTP) transfers the γ-glutamyl group of the conjugate to an amino acid, leaving the cysteine-glycine conjugate. A dipeptidase then removes the glycine, producing the cysteine conjugate. This conjugate may be excreted in the urine or acetylated to mercapturic acid in the liver or kidney. The mercapturic acids may be excreted in urine, and to a lesser extent, in bile. Under certain conditions, such as acetaminophen overdosage to be discussed subsequently, the GSH stores may be depleted and the potential for toxicity greatly increases.
Methylation is another Phase II reaction, which serves mainly to detoxify and actually may render some substrates less water-soluble. In this reaction the methyl group of S-adenosylmethionine is transferred to an acceptor xenobiotic that contains a polyphenol, amine, or sulfhydryl group. Substrates for this reaction include norepinephrine , epinephrine , dopamine, and tyramine and other catachols after hydroxylation by the cytochrome P450 system.
5 Bioactivation and Hepatocellular Injury
Although the biotransformation reactions just described generally function to detoxify and enhance the elimination of xenobiotics, there are instances in which the substrates are actually rendered more toxic or carcinogenic. There are five classes of such biotransformation. The xenobiotic may be transformed into a stable, but toxic metabolite. Examples include acetonitrile and methoxyflurane. A second class involves transformation into a reactive, electrophilic metabolite. Acetaminophen and furosemide are in this class. Third, the substrate may be converted to a free radical, as in the case of carbon tetrachloride. Fourth, the biotransformation reaction may lead to the formation of reduced oxygen metabolites, as occurs with paraquat and quinones. Finally, the biotransformation reaction itself may generate a metabolic derangement within the cell, as in the case of galactosamine-induced liver injury, where there is a depression of uracil nucleotide dependent biosynthesis of nucleic acids, glycolipids, glycoproteins , and glycogen, which results in cell organelle injury.
Review of the biochemical events occurring in acetaminophen toxicity serves as an excellent example of these metabolic pathways. These events are summarized in Fig. 12.3. When taken in appropriate doses, most of the acetaminophen is conjugated with sulfate and glucuronic acid and eliminated. About 5-15 % is converted to a reactive, electrophilic intermediate, N-acetyl-p-benzoquinone imine (NAPQI), which reacts with sulfhydryl groups in proteins. The NAPQI is normally immediately conjugated with glutathione before it can do any damage to the cell and is converted into a mercapturic acid for mainly renal elimination. However, in the case of an overdose the conjugation pathway is saturated and available glutathione is quickly depleted, allowing accumulation of significant amounts of NAPQI. The NAPQI causes cell injury and death resulting in the hepatic necrosis seen with acetaminophen overdose, which can result in liver failure and death. Obviously, one therapeutic strategy would be repletion of the liver’s glutathione stores. Although this cannot be done directly, another successful approach is to administer N-acetylcysteine (Mucomist R), which restores glutathione levels by enhancing synthesis. If administered early enough in the course of intoxication, this intervention can effectively prevent significant liver injury.

Hepatic metabolic pathways in acetaminophen overdose. GSH glutathione, NAPQI N-acetyl-p-benzoquinone imine (Reproduced with permission from Vessey [20])
6 Hepatic Bilirubin Metabolism
Bilirubin is a pigment derived from the catabolism of heme. It is a toxic waste product, which undergoes hepatic biotransformation and excretion in bile. Bilirubin has an open-chain tetrapyrrole structure with eight side chains (Fig. 12.4). The IX-α isomer is the most abundant naturally occurring isomer of bilirubin. From the chemical structure of bilirubin, one would predict a water-soluble compound at physiological pH. However, in reality, unconjugated bilirubin has only very limited water solubility. This phenomenon may be explained by the presence of extensive internal hydrogen bonding within the molecule as shown in Fig. 12.5. This results in the involvement of all of the polar groups of the molecule in the hydrogen bonding, so that none are available for hydration.

The four isomeric forms of bilirubin. The IX-α isomer is the most abundant naturally occurring form. (M, CH3; V, CH2 = CH2; P, CH2-CH2-COOH.) (Reproduced with permission from Odell [14])

Molecular structure of internally hydrogen-bonded bilirubin IX-α. Open, double-contoured, and filled circles represent carbon, nitrogen, and oxygen atoms, respectively. Hydrogen atoms are not shown. The six intramolecular hydrogen bonds are indicated by broken lines (Reproduced with permission from Blanckaert and Fevery [2])
Normally, serum bilirubin levels are very low, and unconjugated bilirubin predominates. In normal individuals, men have slightly higher serum bilirubin levels (0.6 mg/dL) than females (0.4 mg/dL), and any level above 1.0 mg/dL is considered abnormal. Jaundice with the appearance of the pigment in sclerae, skin, and mucus membranes generally is noted when the bilirubin level exceeds 3 mg/dL in adults and 6 mg/dL in infants.
The major site of bilirubin toxicity is the central nervous system , particularly in the newborn infant. Bilirubin inhibits central nervous system RNA and protein synthesis, uncouples oxidative phosphorylation, and inhibits ATPase activity in brain mitochondria. Clinically, bilirubin toxicity can produce severe central nervous system dysfunction resulting in permanent injury or death. Accumulation of bilirubin in the brain, primarily in the basal ganglia, hippocampus, and cochlear and cerebellar nuclei, with associated toxicity is termed kernicterus .
Bilirubin is the major degradation product of heme in mammals. Degradation of hemoglobin from senescent red blood cells by reticuloendothelial cells in the spleen accounts for 80 % of bilirubin production. The other 20 % is derived from ineffective erythropoiesis in the bone marrow, as well as the turnover of heme-containing proteins and myoglobin. The series of reactions responsible for the conversion of heme into bilirubin is shown in Fig. 12.6. The first step is the conversion of heme to biliverdin mediated by the enzyme heme oxygenase, which exists as three isoforms. The activity of this microsomal enzyme is highest in the spleen and Kupffer cells in the liver and requires NADPH and oxygen. This is the only reaction in the body that produces carbon monoxide, which is excreted in expired air and may be quantitated as an index of bilirubin production. Carbon monoxide is also an important signaling molecule and functions as a potent vasodilator. Interestingly, biliverdin is a non-toxic, water-soluble compound and is the end-product of heme catabolism in birds and other non-mammalian vertebrates. However, in mammals biliverdin is converted to bilirubin, which is toxic and requires energy-dependent biotransformation for excretion. Conversion of biliverdin to bilirubin also occurs in reticuloendothelial cells by biliverdin reductase, a cytosolic enzyme that requires NADPH, and utilizes NADH at lower pH. Both bilirubin and biliverdin may have beneficial effects as anti-oxidants under certain conditions and may affect progression of atherosclerosis.

Pathways of biliverdin production from heme by chain-opening and bilirubin production by reduction of biliverdin (Reproduced with permission from Odell [15])
After its formation in reticuloendothelial cells, bilirubin is released into the bloodstream, where it is transported bound to albumin via one primary and two secondary binding sites. This binding prevents the diffusion of free bilirubin into tissues, particularly the central nervous system , thereby preventing toxicity. However, other ligands such as drugs (salicylates, sulfonamides, furosemide, etc.) and fatty acids can displace bilirubin from albumin binding sites and precipitate toxicity.
At the hepatocyte plasma membrane, bilirubin enters the hepatocyte via a specific uptake mechanism, as shown in Fig. 12.7. Once inside the cell, bilirubin binds to a group of cytosolic carrier proteins, glutathione-S-transferases (GSTs), also termed ligandin or Y protein, keeping the bilirubin in solution and within the cell. The bilirubin is then conjugated with glucuronide by the microsomal enzyme, bilirubin UDP-glucuronosyltransferase (UGT1A1). The resulting bilirubin monoglucuronide and diglucuronide are excreted at the canalicular membrane as a component of bile by the ATP-dependent transporter MRP2 (ABCC2). The transcription factors, constitutive androstane receptor (CAR) and pregnane X receptor (PXR), regulate the various proteins involved in bilirubin uptake, glucuronidation and secretion. Conjugation renders bilirubin non-toxic, water-soluble, and non-absorbable from the gastrointestinal tract. In the distal small intestinal and colonic lumen, the conjugated bilirubin is converted to urobilinogens by bacteria and eliminated in stool. However, a small fraction of these tetrapyrroles may be reabsorbed and re-excreted in bile, and the kidney excretes a small fraction.

Summary of hepatic transport and metabolism of bilirubin. During its carriage in plasma, bilirubin is strongly, but reversibly, bound to albumin. In hepatic sinusoids, this complex comes in direct contact with the basolateral domain of the hepatocyte plasma membrane through fenestrae of the specialized hepatic endothelial cells. At the hepatocyte surface, dissociation of the albumin–bilirubin complex occurs, and bilirubin enters hepatocytes by a specific uptake mechanism(s). A fraction of the bilirubin is also derived from catabolism of hepatocellular heme proteins. Storage within the hepatocyte is accomplished by binding of bilirubin to a group of cytosolic proteins, glutathione-S-transferases (GSTs) (also termed ligandin or Y-protein). Binding to these proteins keeps bilirubin in solution and inhibits its efflux from the cell, thereby increasing the net uptake. Conjugation of bilirubin in the endoplasmic reticulum is catalyzed by bilirubin-uridinediphosphoglucuronate glucuronosyltransferase (UGT1A1), forming bilirubin monoglucuronide and diglucuronide. Both conjugates may bind to GSTs in the cytosol. Conjugation is obligatory for efficient transport across the bile canaliculus. Bilirubin glucuronides are secreted across the bile canaliculus by an energy-consuming process mediated by MRP2 (previously termed cMOAT). This process is normally rate-limiting in bilirubin throughput, and is shared by other organic anions, but not bile salts (Reproduced with permission from Elsevier [17])
In the fetus, the placenta is the only available mechanism for clearance of waste products. Therefore, conjugated bilirubin excreted by the liver into the intestinal lumen is deconjugated by an intestinal β-glucuronidase and reabsorbed to be ultimately cleared by the placenta. Normal infants may experience a transient unconjugated hyperbilirubinemia in the first week of life caused by immaturity of bilirubin conjugation, as well as enhanced enterohepatic circulation of bilirubin, potentially due to postpartum persistence of intestinal β-glucuronidase activity. Newborn infants may develop unconjugated hyperbilirubinemia as a consequence of breastfeeding. There appears to be a factor in certain breast milks, which enhances the enterohepatic circulation of bilirubin leading to the unconjugated hyperbilirubinemia of “breast milk jaundice”.
Disorders of bilirubin metabolism may occur at several of the metabolic steps just described. Unconjugated hyperbilirubinemia may occur when there is an overproduction of bilirubin from red blood cell hemolysis, as may occur in the newborn with a maternal-fetal blood group incompatibility or an extravascular accumulation of blood, such as a hematoma. There may be defective bilirubin conjugation. In the Crigler-Najjar syndrome , there is a genetic defect in UGT1A1. In autosomal recessively inherited type I of this syndrome, there is complete absence of enzyme activity, resulting in severe hyperbilirubinemia and often death from kernicterus . In type II there is reduced, but not absent, transferase activity. This residual activity is inducible by phenobarbital treatment, and this form of the disorder usually has a more favorable prognosis, although kernicterus can occasionally occur.
The Gilbert syndrome is a benign condition, which is relatively common (5–10 % of the Caucasian population) and results in mild elevation of the serum unconjugated bilirubin level. This elevation and associated visible icterus is often precipitated or exacerbated by fasting, stress, or illness. This autosomal dominant condition results from a genetic defect in the promoter of the UGT1A1 gene resulting in reduced transcription and moderate impairment of conjugation activity. Other benign disorders of bilirubin metabolism include the Dubin-Johnson and Rotor syndromes. The Dubin-Johnson syndrome , caused by genetic defects in the canalicular MRP2 transporter, is characterized by the presence of intermittent mild jaundice with a predominantly conjugated hyperbilirubinemia. There is also accumulation of a dark pigment in the liver primarily in lysosomes. Rotor syndrome resembles Dubin-Johnson syndrome except the liver pigment is lacking, and recent evidence suggests presence of simultaneous defects in both basolateral membrane organic anion transporting polypeptides OATP1B1 and OATP1B3 . Both conditions exhibit an autosomal recessive mode of inheritance.
In conditions such as intra- and extrahepatic cholestasis and hepatitis, there is failure to excrete conjugated bilirubin, which is regurgitated into the bloodstream resulting in conjugated hyperbilirubinemia. Unlike unconjugated bilirubin, conjugated bilirubin is filtered by the kidney and excreted in the urine resulting in the dark urine, which is characteristic of these disorders. Another serum bilirubin fraction found in cholestatic liver disease is delta-bilirubin , which consists of conjugated bilirubin covalently bound to albumin.
There are several therapeutic modalities used to treat unconjugated hyperbilirubinemia, particularly in the neonate. Phototherapy, which involves exposure of the individual to light (the blue portion of the spectrum appears to be most effective), results in production of water-soluble photooxidation products, as well as photoisomerization of unconjugated bilirubin with disruption of the internal hydrogen bonding. This results in increased water solubility and elimination of these isomers in bile and urine. This treatment is less effective in older children and adults (such as those with Crigler-Najjar syndrome , type 1) due to the higher body mass to surface area ratio in these individuals. In extreme cases, exchange transfusion or plasmapheresis may be employed to rapidly reduce the unconjugated bilirubin level. Ultimately, patients with Crigler-Najjar syndrome, type 1, require liver transplantation to correct the hepatic enzymatic defect. The enterohepatic circulation of bilirubin may be interrupted by frequent feeding. Also, oral administration of substances that bind bilirubin, such as agar, will interrupt the enterohepatic circulation, but has not achieved wide use. Pharmacologic induction of UGT1A1 may reduce serum bilirubin levels. Substances such as Sn-protoporphyrin, which block the first enzyme involved in bilirubin production, heme oxygenase, have shown some effectiveness, but are not widely used. Experimental treatments currently under development include hepatocyte transplantation and gene therapy.
7 Hepatic Bile Acid Metabolism
Bile formation and excretion are major functions of the liver . Bile is secreted by the hepatocyte canaliculus, undergoes modification in the bile ducts, is concentrated and stored in the gallbladder, and is ultimately excreted into the duodenum. Biotransformed xenobiotics and endogenous waste products, such as bilirubin, as well as bile acids and biliary lipids, are secreted in bile. Of the total solutes in bile, bile acids constitute 67 %; phospholipids, 22 %; cholesterol, 4 %; proteins, 4.5 %; and bilirubin, 0.3 %. Bile acids are amphiphilic molecules, that is, one side of the molecule is hydrophilic and the other is hydrophobic. This property of bile acids allows them to act as detergents in the solubilization of the products of lipid digestion to facilitate intestinal absorption. In an aqueous environment, bile acids are in a monomeric form until they reach their critical micellar concentration, approximately 1-5 mM, when they aggregate as micelles and incorporate cholesterol and phospholipid (Fig. 12.8). Once in the gut lumen, products of dietary lipid digestion, including monoacylglycerols, long-chain free fatty acids, lysophospholipids, and fat-soluble vitamins, are incorporated to form mixed micelles to allow these products to efficiently traverse the diffusion barrier of the unstirred water layer and gain access to the enterocyte microvillus membrane for absorption.

Molecular models of the major biliary lipid molecules with depictions of the average sizes and structures of simple and mixed micelles found in human bile. BS, bile salt; L, lecithin; Ch, cholesterol; Rh, mean hydrodynamic radius; Å, Ångstrom unit. Monomeric solubilities are given in SI units. With typical gallbladder lipid compositions, simple bile salt and mixed bile salt-lecithin micelles coexist in a ratio of about 1:5. The site of attachment for cholesterol molecules on simple micelles is on the exterior (hydrophilic) surface. In mixed micelles cholesterol is solubilized within the micelles. The unilamellar vesicles/micelles ratio depends on the bile salt and cholesterol content of the bile, being greatest in bile with low bile salt and high and high cholesterol content (Reproduced with permission from Carey and Duane [4])
In addition to their role in lipid absorption, bile acids have other functions as well. They participate in cholesterol balance, since bile acids are synthesized from cholesterol in a tightly regulated fashion. Although the focus of this chapter will be the role of bile acids as regulatory molecules for their synthesis in liver and their enterohepatic circulation, bile acids are also important signaling molecules in the regulation of gut motility and carbohydrate metabolism. Bile acid mixed micelles have an impact on the gut microbiome through bacteriostatic action in the proximal intestine and induction of genes in the distal intestine that inhibit microbial growth. Proper regulation of the composition of gallbladder bile, including the concentration of bile acids, prevents the formation of gallstones.
The primary bile acids synthesized in the liver are cholic acid and chenodeoxycholic acid . They are synthesized from cholesterol as shown in Fig. 12.9. The rate-limiting step in bile acid synthesis is catalyzed by the microsomal enzyme, cholesterol 7α-hydroxylase (CYP7A1). A minor (10 %) pathway in humans involves sterol 12α-hydroxylase (CYP8B1, not shown) that regulates the amount of cholic acid synthesized and thereby determines the ratio of cholic acid to chenodeoxycholic acid, which modulates the composition and hydrophobicity of the bile acid pool. Prior to canalicular secretion, bile acids are conjugated with either glycine or taurine , which lowers their pKa resulting in more effective lipid solubilization in the duodenum. Taurine conjugates predominate in infants, and glycine conjugates are the most abundant later in life. In addition to the primary bile acids, cholic acid and chenodeoxycholic acid, which are synthesized by the hepatocyte, there are secondary and tertiary bile acids in the bile acid pool (Fig. 12.9). Secondary bile acids are produced by biotransformation of primary bile acids by intestinal bacteria. Cholic acid is converted to deoxycholic acid, chenodeoxycholic acid is converted to both lithocholic acid and 7-oxo-lithocholic acid . Tertiary bile acids are formed by both the liver and intestinal bacteria from secondary bile acids. Lithocholic acid is sulfated to form sulfolithocholic acid , and 7-oxo-lithocholic acid is converted to ursodeoxycholic acid . Sulfolithocholic acid is poorly absorbed and rapidly lost from the enterohepatic circulation.

Major primary, secondary, and tertiary (modified secondary) bile salts of humans with sites of synthesis and metabolism (Reproduced with permission from Carey and Duane [5])
After canalicular secretion, bile undergoes modification as it passes through the ductular system. Biliary tract epithelium is capable of absorption and secretion of electrolytes and water by active and passive processes. The hormone secretin is capable of stimulating secretion by bile ducts and ductules. The gallbladder concentrates bile five to tenfold by active absorption of cations and chloride/bicarbonate ions coupled with passive water movement. During fasting the gallbladder stores and concentrates bile. Post-prandially, the gallbladder contracts in response to the release of cholecystokinin and empties its contents into the duodenum. The relative percentages of bile acids, cholesterol, and phospholipid are normally maintained in a relatively narrow range in bile (Fig. 12.10). If this balance is disturbed such that the percentage of cholesterol exceeds this range, cholesterol gallstones may form in the gallbladder, where bile is normally concentrated and there is relative stasis. Also, in conditions characterized by increased bilirubin production, such as hemolytic anemias, there is a propensity to form bilirubin gallstones.

Relative lipid compositions as functions of micellar, one-phase metastable (supersaturated micelles), and two-phase metastable (supersaturated micelles plus vesicles) zones of human hepatic and gallbladder biles (Reproduced with permission from Carey and Duane [6])
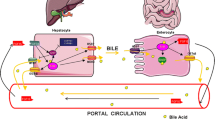
Bile acids undergo an enterohepatic circulation as summarized in Fig. 12.11. After participation in lipid absorption, bile acids are actively absorbed in the distal small intestine via an ileal high-affinity transporter. A small fraction of bile acids may be passively absorbed in the proximal small intestine, particularly under conditions of abnormally low intraluminal pH. The absorbed bile acids are secreted into portal blood where they are bound to albumin and lipoproteins. There is greater than 90 % extraction of the bile acids from the portal blood on the first pass through the liver via a high-affinity hepatocyte membrane transporter (Fig. 12.11). Therefore, under normal conditions, the concentration of bile acids in peripheral blood is extremely low. These bile acids may be re-secreted into bile and make repeated passes through the enterohepatic circulation. In adults, it is estimated that the bile acid pool circulates 8–10 times per day and approximately two times with each meal. Conservation of bile acids during this cycling is highly efficient, and only small amounts of the bile acid pool (400–600 mg/day) are lost each day into stool. This small amount is important, however, since this represents the only route by which cholesterol (the precursor of bile acid synthesis) is eliminated from the body. This loss is compensated for by an increase in hepatic synthesis.

Enterohepatic circulation of bile salts showing typical kinetic values for healthy human adults. (Reproduced with permission from Carey and Duane [5])
The various transporters involved in hepatocyte basolateral bile acid uptake and canalicular secretion, modification of bile by the bile ducts, and ileal uptake of bile acids have been identified (Fig. 12.12). Their nomenclature, function and associated diseases caused by genetic mutations are shown in Table 12.1. At the canalicular membrane, conjugated bile acids are secreted by BSEP. The other major components of bile, phosphatidylcholine and cholesterol, are secreted via the phospholipid flippase , MDR3, and the heterodimer, ABCG5/ABCG8, respectively. Anionic conjugates, including conjugated bilirubin, are secreted by MRP2. The gene ATP8B1 encodes a protein termed FIC1, an aminophospholipid transporter, that is defective in genetic cholestasis syndromes. The electrolyte and water content in bile is modified in the bile ducts after secretion through multiple transport processes including water movement via aquaporin (AQ), chloride secretion by CFTR and chloride/bicarbonate exchange by AE2. After participation in solubilization of the products of lipid digestion, bile acids are actively taken up by the luminal membrane of ileal enterocytes by ASBT and secreted from the basolateral aspect of the cell by OST-αβ. The bile acids then enter the portal venous blood and are transported back to the liver , where the majority are extracted on first pass by the hepatocyte basolateral membrane transporter, NCTP, and may again participate in the enterohepatic circulation. As indicated in Fig. 12.12, biliary cholesterol also undergoes enterohepatic recycling.

Human liver transporter proteins involved in bile formation. Transporter proteins located in the basolateral membrane are responsible for the uptake of bile acids (NTCP), bulky organic anions, uncharged compounds (OATPs), and cations (OATPs, OCT1). Transporter proteins located in the canalicular membrane are responsible for the biliary secretion of compounds such as bile acids, phosphatidylcholine, cholesterol, bilirubin conjugates, and oxidized and reduced glutathione. These transporter proteins comprise the bile salt transporter, BSEP, the phosphatidylcholine translocator, MDR3, the anionic conjugate transporter, MRP2, and the multidrug transporter, MDR1 (not shown). The organic anion transporters, MRP3, MRP4, and OST-α/β, are present at very low levels in normal hepatocytes but are up-regulated during cholestasis. ABCG5/G8 are two half-transporters (half the molecular mass of regular ABC transporters) and as a heterodimer function as cholesterol and plant sterol transporters. Fibroblast growth factor 19 (FGF19) is synthesized in the terminal ileum upon binding of bile acids to FXR. Niemann-Pick C1-Like 1 (NPC1L1 ) protein is the intestinal cholesterol transporter. Both bile acids and cholesterol participate in an enterohepatic cycle (Reproduced with permission from Elsevier [12])
Integration and regulation of all aspects of the enterohepatic circulation are accomplished via nuclear receptors that modulate target gene transcription (Fig. 12.13). Ileal bile acid uptake by ASBT activates farnesoid X receptor (FXR) that induces expression of fibroblast growth factor 19 (FGF19). Then FGF19 is transported to the liver via the portal blood and binds to a basolateral hepatocyte membrane receptor (FGFR4/βKlotho complex) and down-regulates bile acid synthesis through MAPK/ERK1/2 signaling and stabilization of small heterodimer protein (SHP) to suppress activation of transcription of the CYP7A1 gene by liver receptor homolog-1 (LRH-1) and hepatocyte nuclear factor-α (HNF-4 α). Also, intracellular bile acids, including those extracted from portal blood, may also inhibit synthesis via FXR activation, which induces SHP expression to down-regulate CYP7A1. CYP8B1 is also regulated via SHP.

Regulation of bile acid synthesis via the enterohepatic circulation. In the liver , bile acids activate FXR, which induces SHP expression. SHP then inhibits LRH-1 and HNF-4α activation of CYP7A1 and CYP8B1. In the major physiological pathway, intestinal bile acids are taken up by the ASBT and activate FXR to induce FGF19 expression. FGF19 is then carried to the liver where it signals via a receptor tyrosine kinase, the FGFR4:βKlotho complex, and the MAPK/ERK1/2 pathway to inhibit CYP7A1 transcription. At the liver, FGF19 may also signal via FGFR4:βKlotho to stabilize SHP protein and further reduce CYP7A1 expression (Reproduced with permission from Dawson [9])
The newborn infant, especially those born prematurely, have developmental immaturity at several steps in the enterohepatic cycling of bile acids. There is reduced hepatocyte bile acid synthesis, conjugation, uptake, and secretion. Also, ileal uptake of bile acids is reduced and not demonstrable until 3 weeks of age. These factors lead to a reduced bile acid pool size in the newborn. Defective hepatic bile acid uptake and secretion lead to slightly elevated serum bile acids and “physiologic cholestasis of infancy.” These deficiencies also lead to decreased intraluminal bile acid concentrations and transient fat malabsorption in the newborn, which is even more striking in the premature infant.
7.1 Disease States Characterized by Disturbances in Bile Acid Metabolism and Enterohepatic Circulation
7.1.1 Cholestasis
Cholestasis is defined as bile excretory failure and may be the result of a variety of disorders. Cholestasis may be broadly classified as intrahepatic (or hepatocellular) or extrahepatic (or obstructive). Examples of intrahepatic cholestasis would include certain types of drug-induced liver injury, inherited cholestatic syndromes such as Alagille syndrome , and idiopathic neonatal hepatitis. Gram negative endotoxin inhibits bile acid transporters and may cause cholestasis during sepsis or even in infants with gram negative bacterial urinary tract infections. Examples of extrahepatic cholestasis include bile duct obstruction by gallstones, choledochal cyst, primary sclerosing cholangitis , and biliary atresia in infants. In chronic cholestasis there is regurgitation of bile acids and biliary lipids into the bloodstream, which leads to several abnormalities. Accumulation of bile acids in the skin causes severe itching or pruritis , which can be quite debilitating. As previously described, lipoprotein metabolism is perturbed, and hyperlipidemia may be quite striking, particularly cholesterol levels. An abnormal lipoprotein particle enriched in free cholesterol and phospholipid called lipoprotein-X accumulates in the circulation. Cutaneous xanthomata often result from the hyperlipidemia. Lack of adequate intraluminal bile acid concentrations results in dietary fat malabsorption , including fat-soluble vitamins. Jaundice and dark urine may be present due to conjugated hyperbilirubinemia. It is interesting that in several chronic cholestatic conditions, the serum bilirubin level may be normal or slightly elevated in the face of a markedly elevated serum bile acid level.
There is evidence that bile acids, which accumulate during cholestasis, particularly hydrophobic bile acids such as lithocholic acid, may perpetuate or exacerbate the underlying liver disease. During cholestasis associated with retention of bile acids and conjugated bilirubin within the hepatocyte, basolateral OST-αβ is up-regulated via FXR to transport potentially toxic bile acids out of the cell. Also, basolateral MRP3 and MRP4 are up-regulated by pregnane X receptor (PXR) to transport bilirubin conjugates and bile acids, respectively, out of the hepatocyte.
As would be expected, genetic defects in bile acid transporters lead to clinical cholestatic syndromes (Table 12.1). Progressive familial intrahepatic cholestasis (PFIC) comprises a group of these disorders that has led to identification of some of these transporters and better understanding of their function. Patients with PFIC are characterized by the presence of cholestasis early in life that is progressive usually leading to need for transplantation. Pruritis, often severe, and growth retardation are features.
7.1.2 Type 1 PFIC
Type 1 PFIC results from mutations in the gene encoding FIC1 (ATP8B1), a protein that translocates aminophospholipid from the outer to inner leaflets of the canalicular membrane. Type 1 PFIC is also associated with diarrhea thought to be due to the expression of defective FIC1 in the small intestine . This type of PFIC was formerly known as Byler disease after the Amish kindred in whom it was first described. Mutations in ATP8B1 also may result in a much milder phenotype, benign recurrent intrahepatic cholestasis (BRIC) characterized by recurrent episodes of mild to severe cholestasis with intervening asymptomatic periods.
7.1.3 Type 2 PFIC
Type 2 PFIC is due to mutations in the bile salt export pump, BSEP (ABCB11), and is similar to type 1, but with more rapid progression to need for transplantation.
7.1.4 Type 3 PFIC
Type 3 PFIC is caused by mutations in the gene for MDR3 (ABCB4), a canalicular phospholipid flippase responsible for secretion of phospholipid into bile. The resultant abnormal bile devoid of phospholipid permits bile acids to cause injury to hepatocytes and cholangiocytes resulting in cholestasis. Progression may be variable depending on the specific mutation, although chronic cholestasis, marked fibrosis and liver failure usually result. Mutations in ABCB4 may also be related to intrahepatic cholestasis of pregnancy and gallstone formation. A subset of patients with CFTR mutations and cystic fibrosis also develop a form of liver disease called focal biliary cirrhosis , and some mutations in this gene may be responsible for the development of primary sclerosing cholangitis in the absence of the classic phenotype of cystic fibrosis.
7.2 Treatment of Cholestasis due to Bile Transporter Mutations
Patients with cholestasis due to bile transporter mutations represent challenges to management of their disease. Oral administration of ursodeoxycholic acid , a non-toxic, water-soluble bile acid, may result in improvement through its ability to increase bile flow and displace toxic bile acids in the bile acid pool. This bile acid also has anti-inflammatory and anti-apoptotic properties. It is a minor tertiary bile acid in humans, but is the major bile acid in bears; thus the name. Another approach is to orally administer a bile acid binding resin , such as cholestyramine, or perform a surgical external biliary diversion. Both of these measures chronically eliminate bile acids from the body and reduce the size of the bile acid pool. However, response to both of these treatments is variable, and compliance with bile acid sequestrants is often poor and biliary diversion may be associated with complications.
7.2.1 Ileal Dysfunction or Resection
Individuals with inflammatory disease of the ileum , such as Crohn’s disease, or an ileal resection, as in infants requiring resection secondary to necrotizing enterocolitis, have defective ileal bile acid uptake and bile acid malabsorption . Bile acids may enter the colon and cause a secretory diarrhea mediated by a cAMP-dependent mechanism. With increased fecal loss of bile acids, there is a compensatory increase in bile acid synthesis, and the bile acid pool size is maintained. These patients may be treated with oral administration of bile acid binding resins, which bind the malabsorbed bile acids to allow elimination in stool and prevent the associated diarrhea. If the inflammatory process is very severe or the amount of resected ileum is extensive, the bile acid loss may exceed the capacity of the liver to increase synthesis to compensate for the loss. In this situation there is a reduction is the bile acid pool size and resultant reduction in the intraluminal bile acid concentration, often to below the critical micellar concentration. This results in malabsorption of dietary fat and fat-soluble vitamins.
7.2.2 Intestinal Blind Loop Syndrome
In conditions that result in stasis of proximal intestinal contents such as blind loop syndrome, bacterial overgrowth may occur resulting in deconjugation of bile acids by primarily anaerobic bacteria. Unconjugated bile acids are readily passively absorbed leading to a deficient intraluminal bile acid concentration and fat malabsorption . These unconjugated bile acids may also exert a toxic effect on the intestinal mucosa , leading to exacerbation of malabsorption and diarrhea . Oral administration of broad-spectrum non-absorbable antibiotics may result in clinical improvement.
Clinical Correlations
Case Study 1
A 22-year-old male comes to your office with the complaint of noticing “yellow eyes” intermittently throughout his life, generally associated with an acute febrile illness with decreased appetite and food intake. Apparently, blood tests had been performed in childhood, and his parents had been told that he had “mild hepatitis”, since the only abnormality reported was an elevated bilirubin. He otherwise has been healthy with normal growth and development and no other complaints. There is no history of blood exposure or exposure to anyone with hepatitis. Family history is significant for similar symptoms in his father who was killed in a traffic accident at age 32 years. The present problem prompting a visit to you is the patient’s inability to obtain health insurance coverage for a pre-existing “recurrent hepatitis”. The physical examination is normal. No jaundice is noted. There is no hepatosplenomegaly or ascites . There are no findings to suggest chronic liver disease or cirrhosis .
Laboratory studies: Hematocrit 46 %; hemoglobin 15.1 g/dL; reticulocyte count 1.1 % (0.8–2.5); white blood cell count 7,900 cells/mm3 (4,500–11,000); 220, 000 platelets/mm3 (150,000–350,000); prothrombin time 12.2 s (control 12.4 s); albumin 4.9 g/dL (3.7–5.6); total bilirubin 1.8 mg/dL (0.1–1.0); direct bilirubin 0.2 mg/dL (0–0.2); ALT 22 IU/L (10–30), AST 24 IU/L (15–30); GGTP 21 IU/L (0–25); alkaline phosphatase 61 IU/L (30–100); urinalysis is negative for bilirubin and shows normal urobilinogen.
Questions:
-
1.
What is this patient’s most likely diagnosis?
Answer: The presence of a non-hemolytic (normal Hgb, reticulocyte count, and urine urobilinogen) unconjugated hyperbilirubinemia in an adult, which is exaggerated with the occurrence of an acute illness, would strongly suggest the diagnosis of the Gilbert syndrome . This is especially true with the positive family history, since there appears to be autosomal dominant inheritance.
-
2.
Why is the patient not icteric now?
Answer: Even though individuals with the Gilbert syndrome have an exaggeration of their indirect hyperbilirubinemia with acute illness or fasting, the baseline level is generally above normal, as in this individual. However, adults do not generally demonstrate visible jaundice until the serum bilirubin level reaches 2.0 mg/dL. Fasting or illness is thought to elevate the bilirubin level further by reducing the availability of glucose for formation of UDP-glucuronic acid by UDP-glucose dehydrogenase, thereby reducing bilirubin conjugation. However, the precise mechanism is not known.
-
3.
How would you confirm the diagnosis?
Answer: The Gilbert syndrome is generally considered a diagnosis of exclusion with mild unconjugated hyperbilirubinemia in an otherwise healthy individual without evidence of hemolysis or structural liver disease. A past approach has involved measuring bilirubin levels in the parents after a 48-h fast to uncover the affected parent. However, this approach has not been shown to be reliable. In this case, the presumably affected parent is deceased. In most situations, the present evaluation would be adequate. However, the measurement of the ratio of monoglucuronide to diglucuronide bilirubin conjugates in duodenal bile may be confirmatory if an increased ratio of monoconjugates to diconjugates is found. This same finding is also present in the Crigler-Najjar syndrome , type II. Genetic testing is also available for the Gilbert syndrome mutation.
-
4.
How would you counsel this patient?
Answer: He should be reassured that the Gilbert syndrome is a completely benign disorder and should not affect his health or activity in any way.
Case Study 2
A 28-year-old female has a history of Crohn’s disease involving the terminal ileum for the past 8 years. Six months ago she underwent resection of an unknown length of her terminal ileum secondary to intractable inflammation and enterocolic fistula formation. Her recovery from surgery was uneventful. She initially had an ileostomy, which was later reconnected. She has continued to have watery diarrhea with 6–7 large watery stools per day. No mucus or blood has been noted in the stool. Otherwise, she has been afebrile and has had no other symptoms. A contrast radiographic study of the area of the anastomosis shows no evidence of recurrent disease. A colonoscopy with ileoscopy also shows normal mucosa without evidence of recurrent Crohn’s disease which is confirmed by biopsies. Physical examination is unremarkable except for well-healed abdominal surgical scars.
Laboratory studies: Hematocrit 30 %; hemoglobin 10.2 g/dL (RBC indices are macrocytic); reticulocyte count 0.4 % (0.8–2.5); white blood cell count 5,400 cells/mm3 (4,500–11,000); 233, 000 platelets/mm3 (150,000–350,000); erythrocyte sedimentation rate 4 mm/h (0–20); albumin 4.2 g/dL (3.7–5.6); serum iron 150 μg/dL (50–170); serum folate 2.2.ng/L (1.8–9.0); serum vitamin B12 19 pg/mL (30–785); stool negative for white blood cells, occult blood, Giardia-specific antigen, Clostridium difficile toxin; stool culture negative for enteric pathogens.
Questions:
-
1.
What is the most likely cause of the diarrhea and how would you confirm it?
Answer: A recurrence of Crohn’s disease proximal to the surgical anastomosis is ruled out by radiographic studies and endoscopy with biopsy. Additionally, the ESR and serum albumin are normal. Stool studies and cultures are negative and make an infectious etiology unlikely. The fact that the patient is vitamin B12-deficient with associated megaloblastic anemia suggests that a significant amount of ileum , the site of absorption of this vitamin, was resected. Therefore, deficient ileal absorption of bile acids with a resulting colonic secretory diarrhea is a strong possibility. A therapeutic trial of oral cholestyramine (a bile acid binding resin ) is instituted and results in resolution of the diarrhea.
-
2.
What is another complication of deficient ileal absorption of bile acids?
Answer: If the liver is unable to compensate for the chronic loss of bile acids from the bile acid pool by increased synthesis, bile acid deficiency will result. This deficiency can result in a luminal bile acid concentration below the critical micellar concentration with resultant fat and fat-soluble vitamin malabsorption .
Case Study 3
A 16-year-old white female is brought to the emergency room after admitting to her parents that she attempted suicide by ingesting an unknown number of acetaminophen tablets approximately 6 h ago. The patient is otherwise healthy, but does have a past history of depression and has been seeing a psychologist. Her physical examination is unremarkable. She is afebrile with normal vital signs. She is anicteric and in no distress. She is alert and oriented. Her abdomen is soft and non-distended. Her liver span is 10 cm by percussion without splenomegaly . Her neurological examination is normal without asterixis.
Laboratory studies: Hematocrit 38 %; hemoglobin 18.1 g/dL; white blood cell count 9,900 cells/mm3 (4,500–11,000); 305, 000 platelets/mm3 (150,000–350,000); prothrombin time 11.0 s (control 12.4 s); albumin 4.6 g/dL (3.7–5.6); total bilirubin 0.8 mg/dL (0.1–1.0); direct bilirubin 0.2 mg/dL (0–0.2); ALT 96 IU/L (10–30), AST 110 IU/L (15–30); GGTP 21 IU/L (6–29); alkaline phosphatase 230 IU/L (61–264); urinalysis is negative for bilirubin and shows normal urobilinogen; toxicology screen positive for fluoxetine, which the patient takes for depression, and acetaminophen.
Questions:
-
1.
What is the next step?
Answer: An acetaminophen level is measured and is 200 μg/mL 8 h post estimated ingestion. The Rumack-Matthew nomogram is consulted to determine the risk for significant hepatotoxicity and need for treatment based on serum level and time post-ingestion. The patient’s level is in the range for toxicity, and treatment is indicated. The time frame for effective gastric lavage and administration of activated charcoal is past. N-acetylcysteine is administered by nasogastric tube per protocol that includes a loading dose and every 4 h dosing based on body mass for a total of 72 h. The patient is admitted with an intravenous line for hydration, and is closely monitored with vital sign and neurological checks, as well as serial laboratory testing.
-
2.
What is the expected clinical course?
Answer: The clinical course of acetaminophen intoxication may be divided into four phases:
-
Phase 1, 30 min to 24 h after ingestion, during which there may be minimal or no symptoms and subclinical rise in transaminases.
-
Phase 2, 18-72 h post-ingestion, during which patients may have nausea and vomiting, right upper quadrant pain, and decreased urine output.
-
Phase 3, hepatic phase, 72-96 h post-ingestion, is associated with continued nausea and vomiting, abdominal pain and tender liver . Hepatic necrosis may become obvious with markedly elevated transaminases, jaundice, coagulopathy, hypoglycemia and hepatic encephalopathy . Acute renal failure may occur, followed by death from multiorgan failure. Transplantation should be considered.
-
Phase 4, recovery phase, 4 days to 3 weeks post-ingestion, and patients who survive usually have complete resolution of symptoms and hepatic recovery.
In the case of this patient, her liver tests peaked at between 72 and 96 h post ingestion with AST 12,000 IU/L and ALT 10,700 IU/L, bilirubin total 3.7 mg/dL, direct 2.9 mg/dL, prothrombin time 15.5 s, and normal blood ammonia level. The patient experienced nausea, vomiting and abdominal pain, but remained alert with no signs of hepatic encephalopathy . She subsequently made an uneventful recovery.
-
-
3.
To what do you attribute the patient’s recovery?
Answer: The features of this case that contributed to full recovery included a young, otherwise healthy patient, a single acute ingestion, and rapid institution of N-acetylcysteine therapy to restore hepatic glutathione levels to deplete toxic NAPQI levels. The acetaminophen level and Rumack-Matthew nomogram are useful in guiding therapy, but are not helpful if there is multiple chronic dosing or the time of ingestion is unknown. Studies of the use other markers for potential hepatotoxicity, such as serum levels of the degradation products of acetaminophen protein adducts, are ongoing and may prove useful for clinical management.
Further Reading
Ampola MG (1994) In: Arias IM, Boyer JL, Fausto N, Jakoby WB, Schachter DA, Shafritz DA (eds) The liver: biology and pathobiology, 3rd edn. Raven Press, New York, p 366
Blanckaert N, Fevery J (1990) In: Zakim D, Boyer TD (eds) Hepatology: a textbook of liver disease, 2nd edn. W. B. Saunders Co., Ltd, Philadelphia, p 263
Brites D (2012) The evolving landscape of neurotoxicity by unconjugated bilirubin: role of glial cells and inflammation. Front Pharmacol 3:88
Carey MC, Duane WC (1994) In: Arias IM, Boyer JL, Fausto N, Jakoby WB, Schachter DA, Shafritz DA (eds) The liver: biology and pathobiology, 3rd edn. Raven Press, New York, p 728
Carey MC, Duane WC (1994) In: Arias IM, Boyer JL, Fausto N, Jakoby WB, Schachter DA, Shafritz DA (eds) The liver: biology and pathobiology, 3rd edn. Raven Press, New York, p 722
Carey MC, Duane WC (1994) In: Arias IM, Boyer JL, Fausto N, Jakoby WB, Schachter DA, Shafritz DA (eds) The liver: biology and pathobiology, 3rd edn. Raven Press, New York, p 749
Carey EJ, Lindor KD (2012) Current pharmacotherapy for cholestatic liver disease. Expert Opin Pharmacother 13(17):2473–2484
Dawson PA (2011) Role of the intestinal bile acid transporters in bile acid and drug disposition. Handb Exp Pharmacol 201:169–203
Dawson PA (2012) In: Johnson LR (ed) Physiology of the gastrointestinal tract, 5th edn. Academic Press, New York, p 1467
Imam MH, Gossard AA, Sinakos E, Lindor KD (2012) Pathogenesis and management of pruritus in cholestatic liver disease. J Gastroenterol Hepatol 27(7):1150–1158
Jacquemin E (2012) Progressive familial intrahepatic cholestasis. Clin Res Hepatol Gastroenterol 36(Suppl 1):S26–S35
Jansen LMJ, Beuers U, Oude Elferink RPJ (2012) In: Boyer TD, Manns MP, Sanyal AJ (eds) Hepatology: a textbook of liver disease, 6th edn. Elsevier Saunders, Philadelphia, p 49
Montagnese S, De Pitta C, De Rui M et al (2013) Sleep-wake abnormalities in patients with cirrhosis. Hepatology 59:705–712
Odell GB (1980) Neonatal hyperbilirubinemia. Grune and Stratton, New York, p 5
Odell GB (1980) Neonatal hyperbilirubinemia. Grune and Stratton, New York, p 4
Poh Z, Chang PE (2012) A current review of the diagnostic and treatment strategies of hepatic encephalopathy. Int J Hepatol 2012:480309
Roy-Chowdhury J, Roy-Chowdhury N (2012) In: Boyer TD, Manns MP, Sanyal AJ (eds) Hepatology: a textbook of liver disease, 6th edn. Elsevier Saunders, Philadelphia, p 1087
Sticova E, Jirsa M (2013) New insights in bilirubin metabolism and their clinical implications. World J Gastroenterol: WJG 19(38):6398–6407
Trauner M, Baghdasaryan A, Claudel T et al (2011) Targeting nuclear bile acid receptors for liver disease. Dig Dis 29(1):98–102
Vessey DA (1990) In: Zakim D, Boyer TD (eds) Hepatology: a textbook of liver disease, 2nd edn. W. B. Saunders Co., Ltd, Philadelphia, p 228
Wagner M, Zollner G, Trauner M (2011) Nuclear receptors in liver disease. Hepatology 53(3):1023–1034
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Black, D.D. (2014). Biotransformation, Elimination and Bile Acid Metabolism. In: Leung, P. (eds) The Gastrointestinal System. Springer, Dordrecht. https://doi.org/10.1007/978-94-017-8771-0_12
Download citation
DOI: https://doi.org/10.1007/978-94-017-8771-0_12
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-017-8770-3
Online ISBN: 978-94-017-8771-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)