
Abstract
Heat shock transcription factor 1 (HSF1) is a transcription factor that is activated upon the exposure of cells to various types of proteotoxic stress, such as heat shock stress and oxidative stress, which induces the expression of various molecular chaperones. HSF1-induced molecular chaperones, including heat shock protein 40 (Hsp40) and Hsp70, suppress protein misfolding through binding to structurally unstable proteins and thereby protect cells from proteotoxic stress. Therefore, activation of HSF1 is considered as a therapeutic approach against a group of neurodegenerative diseases that are caused by protein misfolding, including Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, and the polyglutamine diseases. In fact, many compounds that activate HSF1 have been tested for their potential as therapeutic agents against neurodegenerative diseases. In this chapter, we introduce various HSF1-activating compounds, their mechanisms of activation of HSF1, and their therapeutic effects against neurodegenerative diseases.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Heat shock transcription factor 1
- Molecular chaperones
- Neurodegenerative diseases
- Polyglutamine diseases
- Amyotrophic lateral sclerosis
- Parkinson’s disease
1 Introduction
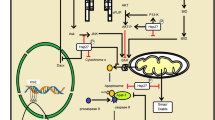
Heat shock transcription factor 1 (HSF1) is a transcription factor that protects cells from various types of proteotoxic stress, including heat shock stress and oxidative stress, by inducing the expression of various types of molecular chaperones. Under unstressed conditions, HSF1 is inactivated by its interaction with molecular chaperones, including heat shock protein 40 (Hsp40), Hsp70, and TCP-1 ring complex (TRiC)/chaperonin in the cytoplasm (Zou et al. 1998; Neef et al. 2014; Shi et al. 1998). Upon exposure to stress, HSF1 is quickly released from the chaperone complex, translocates into the nucleus, and binds to the heat shock element (HSE) in the promoter region of various molecular chaperone genes to induce their expression (Sarge et al. 1993; Baler et al. 1993). During this activation process, HSF1 forms a homotrimer and is posttranslationally modified, such as by phosphorylation and sumoylation (Chu et al. 1996; Hietakangas et al. 2003). After acute transcriptional activation, HSF1 is acetylated by the histone acetyltransferase p300/CBP, which attenuates the binding of HSF1 to HSEs, leading to a reduction in its transcriptional activity (Westerheide et al. 2009) (Fig. 14.1). HSF1-induced molecular chaperones, such as Hsp40, Hsp70, Hsp90, Hsp110, TRiC, and small Hsps, suppress protein misfolding through binding to structurally unstable proteins and assist the refolding of misfolded proteins.

Mechanisms of HSF1 activation by small compounds. Upon exposure to stress, HSF1 is quickly released from the chaperone complex, translocates into the nucleus, forms a homotrimer, and binds to the heat shock element (HSE) in the promoter region of various molecular chaperone genes. After its acute transcriptional activation, HSF1 is acetylated to attenuate its transcriptional activity. Various HSF1-activating compounds targeting various steps of HSF1 activation are shown in red boxes. Although paeoniflorin was reported to activate HSF1, the mechanisms remain unknown
Protein misfolding has been considered to be involved in the pathogenesis of various neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis (ALS), and the polyglutamine (polyQ) diseases. The aggregates of misfolded proteins that accumulate as inclusions in the central nervous system are a common pathological hallmark of various neurodegenerative diseases, whereas the main component protein of the inclusions is different in each disease (amyloid-β and tau in Alzheimer’s disease, α-synuclein in Parkinson’s disease, TAR DNA-binding protein 43 (TDP-43) or superoxide dismutase 1 (SOD1) in ALS, and polyQ-containing proteins in the polyQ diseases). In addition, many genetic mutations that are responsible for the inherited forms of neurodegenerative diseases confer a propensity for misfolding on the disease-causing proteins, resulting in aggregation of these proteins. These facts strongly indicate that protein misfolding is a common pathogenesis of various neurodegenerative diseases (Taylor et al. 2002). Therefore, the suppression of protein misfolding by molecular chaperones is considered to be a common therapeutic approach for the currently untreatable neurodegenerative diseases (Fig. 14.2).

Protein misfolding in the pathomechanisms of neurodegenerative diseases and its suppression by molecular chaperones. Protein misfolding has been considered as a common pathogenesis of various neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis, and the polyglutamine diseases. Molecular chaperones assist the refolding of misfolded proteins, and hence the suppression of protein misfolding by molecular chaperones is considered to be a common therapeutic approach for the currently untreatable neurodegenerative diseases
In fact, the overexpression of molecular chaperones has been demonstrated to lead to therapeutic effects in cell culture and animal models of various neurodegenerative diseases (Auluck et al. 2002; Bruening et al. 1999; Cummings et al. 1998, 2001; Klucken et al. 2004; Shimura et al. 2004). Importantly, the simultaneous expression of several molecular chaperones was shown to result in stronger therapeutic effects than the expression of a single type of chaperone, in various models of neurodegenerative diseases (Chan et al. 2000; Ishihara et al. 2003), indicating that additional and synergistic therapeutic effects are expected through the induction of multiple molecular chaperones. Therefore, the expression of multiple molecular chaperones by HSF1 activation is expected to result in the greatest benefits against neurodegenerative diseases. The first trial to investigate the effects of HSF1 activation against neurodegenerative diseases was reported in 2001. Zimarino and colleagues showed that the genetic overexpression of a constitutively active HSF1 mutant reduces polyQ aggregation through the induction of Hsp70 in a cell culture model (Rimoldi et al. 2001). A constitutively active HSF1 mutant was also shown to ameliorate the disease phenotypes of polyQ disease model mice through the induction of multiple molecular chaperones (Fujimoto et al. 2005). These facts suggest that the genetic overexpression of exogenous HSF1 would ameliorate the symptoms of patients with neurodegenerative diseases, through the induction of multiple molecular chaperones, although the expression of exogenous genes in the human brain is currently very limited owing to safety concerns and the lack of efficient methods for gene delivery.
On the other hand, it had remained unclear as to whether endogenous HSF1 is activated in the brains of patients with protein misfolding neurodegenerative diseases. Interestingly, Bates and colleagues found that the expression levels of molecular chaperones unexpectedly decreased with disease progression in polyQ disease model mice (Hay et al. 2004), suggesting that endogenous HSF1 is gradually inactivated in the brains of these mice. Subsequently, Nukina and colleagues revealed that endogenous HSF1 is inactivated in the brains of polyQ disease model mice (Yamanaka et al. 2008), consistent with the inactivation of HSF1 under chronic heat shock stress (Kline and Morimoto 1997). These facts strongly suggest that endogenous HSF1 is inactivated in the brains of patients with protein misfolding diseases, which are under chronic proteotoxic stress, and hence the activation of endogenous HSF1 is expected to exert therapeutic effects for patients with neurodegenerative diseases.
In this chapter, we focus on small compounds that activate endogenous HSF1 and lead to the induction of molecular chaperones (Fig. 14.1), which are promising candidates as a common therapy against neurodegenerative diseases.
2 HSF1 Activation by Hsp90 Inhibitors
The molecular chaperone Hsp90 binds to many proteins, including steroid hormone receptors, protein kinases, and transcription factors, to stabilize them under unstressed conditions. Hsp90 also binds to the regulatory domain of HSF1 to keep HSF1 as an inactive monomer in the cytoplasm, and hence the pharmacological inhibition of Hsp90 is able to activate HSF1.
Geldanamycin, a benzoquinone ansamycin antitumor antibiotic, has been shown to specifically bind to the N-terminal ATP-binding pocket of Hsp90 and inhibit its activity, resulting in the activation of HSF1, leading to the induction of molecular chaperones (Dehner et al. 2003; Whitesell et al. 1994; Zou et al. 1998; Roe et al. 1999). In 2001, Wanker and colleagues reported for the first time the therapeutic effect of the pharmacological induction of molecular chaperones in a neurodegenerative disease model (Sittler et al. 2001). They showed that treatment with geldanamycin suppresses aggregation of the polyQ protein through the induction of multiple molecular chaperones in a cell culture model. A decrease in the number of polyQ aggregates by geldanamycin treatment was also subsequently reported in brain slice cultures derived from polyQ disease model mice (Hay et al. 2004). We also demonstrated that the oral administration of geldanamycin and 17-(allylamino)-17-demethoxygeldanamycin (17-AAG), a less toxic derivative of geldanamycin, suppresses not only the aggregation of polyQ proteins but also neurodegeneration in polyQ disease model flies, through HSF1-mediated induction of multiple molecular chaperones (Fujikake et al. 2008), indicating that geldanamycin and its derivative are effective in vivo. In addition, Bonini and colleagues showed the therapeutic effects of geldanamycin on the progressive loss of dopaminergic neurons in Parkinson’s disease model flies (Auluck and Bonini 2002). Furthermore, the cytotoxicity of mutant SOD1 in a cell culture model of ALS was shown to be reduced by geldanamycin treatment (Batulan et al. 2006). These facts suggest that geldanamycin and its derivatives have the potential to be developed as common therapeutic agents against various neurodegenerative diseases.
Radicicol, an antifungal macrolactone antibiotic, was also shown to activate HSF1 similarly to geldanamycin by inhibiting Hsp90 through its direct binding to the N-terminal domain of Hsp90 (Schulte et al. 1998; Roe et al. 1999; Bagatell et al. 2000). Radicicol was shown to decrease the number of polyQ aggregates in brain slice cultures derived from polyQ disease model mice (Hay et al. 2004). We also demonstrated that radicicol suppresses neurodegeneration as well as the aggregation of polyQ proteins in flies (Fujikake et al. 2008). Therefore, the potential of radicicol to be developed as a therapeutic agent is similar to that of geldanamycin.
HSP990 is a compound that was developed by Novartis as an inhibitor of the ATPase activity of Hsp90, by binding to the N-terminal ATP-binding pocket of Hsp90 (Machajewski et al. 2007). Bates and colleagues demonstrated that oral administration of HSP990 transiently improves the motor performance and reduces the aggregates of polyQ disease model mice at early stages of disease, through the induction of molecular chaperones via the activation of HSF1 (Labbadia et al. 2011). However, the induction of molecular chaperones as well as the therapeutic effects of HSP990 gradually diminished with disease progression, although HSF1 was dissociated from Hsp90, phosphorylated, and localized in the nucleus of neurons. They demonstrated that the binding of HSF1 to HSE is significantly decreased in the brains of polyQ disease model mice and that the chromatin structure and acetylation of histones are altered with disease progression, suggesting that the accessibility of HSF1 to the HSE sequence on the genome is decreased by chronic proteotoxic stress in neurodegenerative diseases. These data suggest that oral administration of HSP990 is effective against neurodegenerative diseases at least early stages of the diseases.
3 HSF1 Activation by an Hsp70 Inhibitor
The molecular chaperone Hsp70 has a major role in assisting the proper folding of newly synthesized polypeptides and in preventing the misfolding of mature proteins (Hendrick and Hartl 1995). Upon exposure to stress, Hsp70 expression is quickly induced by activated HSF1, resulting in the binding of Hsp70 to structurally unstable proteins, to prevent the misfolding of these proteins. Hsp70 also binds to the transactivation domain of HSF1 to disassemble the active HSF1 trimer into inactive monomers and to stabilize the monomers (Abravaya et al. 1992; Mosser et al. 1993; Shi et al. 1998).
In 1996, geranylgeranylacetone (GGA), an antiulcer drug, was demonstrated to induce molecular chaperones via the activation of HSF1 (Hirakawa et al. 1996). About 10 years later, GGA was shown to bind to and inhibit Hsp70, leading to the dissociation of HSF1 from Hsp70, resulting in the activation of HSF1 (Otaka et al. 2007). Sobue and colleagues showed that the oral administration of GGA induces the expression of multiple molecular chaperones in the spinal cord of polyQ disease model mice, which suppresses the accumulation of polyQ proteins, resulting in the improvement of motor performance (Katsuno et al. 2005). GGA is considered as a strong candidate agent for patients with neurodegenerative diseases, since it has already been used as an antiulcer drug for humans.
4 HSF1 Activation by a TRiC Inhibitor
TRiC is a eukaryotic chaperonin composed of eight paralogous subunits (Tcp1 and Cct2-8) (Lopez et al. 2015). Although TRiC was considered to specifically assist the folding of cytoskeletal proteins, such as α-tubulin and actin, it was later estimated to interact with 6–7 % of cytosolic proteins, probably to assist their folding (Yam et al. 2008). TRiC directly binds to HSF1 and suppresses its activation, similarly to Hsp90 and Hsp70.
HSF1A, a benzyl pyrazole derivative, was recently identified as an HSF1-activating compound by a humanized yeast screen and was confirmed to also activate HSF1 in human cells (Neef et al. 2010, 2011). HSF1A has been shown to interact with at least four TRiC subunits (Tcp1, Cct2, Cct5, and Cct8) and to mildly inhibit its chaperone activity, leading to the activation of HSF1 (Neef et al. 2014). Treatment with HSF1A was shown to reduce the aggregates and cytotoxicity of polyQ proteins in a cell culture model and to attenuate a disease phenotype in polyQ diseases model flies (Neef et al. 2010). These facts indicate that TRiC is a new target to develop HSF1-mediated therapies against neurodegenerative diseases.
5 Prolongation of HSF1 Activity
Silent information regulator 2 (Sir2), an NAD-dependent protein deacetylase in budding yeast, was found to be a determinant of their lifespan (Kaeberlein et al. 1999). The overexpression of Sir2 slowed the aging process in yeast, whereas Sir2 mutants had a shortened lifespan. Subsequently, an increase in lifespan by the overexpression of Sir2 orthologs has been demonstrated in worms and flies (Rogina and Helfand 2004; Tissenbaum and Guarente 2001). Furthermore, the overexpression of sirtuin 1 (Sirt1), a mammalian ortholog of Sir2, results in healthy aging in mice, such as a reduction in DNA damage and fewer spontaneous carcinomas and sarcomas (Herranz et al. 2010). Morimoto and colleagues demonstrated that the DNA-binding domain of HSF1 is acetylated after its acute transcriptional activation, to reduce its transcriptional activity (Westerheide et al. 2009). Importantly, they further showed that Sirt1 deacetylates the acetylated HSF1, leading to its prolonged transcriptional activity, which may be involved in the mechanisms of longevity and healthy aging by Sirt1.
By screening small compound libraries for modulators of Sirt1 activity, resveratrol, a polyphenol found in red wine, was identified as an activator of Sirt1 (Howitz et al. 2003). Resveratrol treatment extended the lifespan of budding yeast in a Sirt1-dependent manner and conferred resistance to ionizing radiation in human cultured cells, accompanied with the deacetylation of p53, which is a known target of Sirt1. Treatment of ALS model mice with resveratrol by chronic intraperitoneal injection was reported to successfully extend their lifespan, decrease motor neuron death, and delay the onset of muscle weakness (Han et al. 2012). Although the effect of resveratrol on protein aggregation was not investigated, the authors demonstrated a decrease in acetylated HSF1 accompanied by the induction of Hsp27 and Hsp70 in the spinal cord of the resveratrol-treated ALS model mice, suggesting that the therapeutic effects of resveratrol are mediated by prolonged HSF1 activity.
6 Enhancement of HSF1 Activity
Bimoclomol, a derivative of hydroxylamine, has been reported to enhance the induction of molecular chaperones in cultured cells upon heat shock stress, whereas it does not induce molecular chaperones under unstressed conditions (Vigh et al. 1997). Although the specific mechanisms by which bimoclomol enhances the induction of molecular chaperones are not fully understood, Csermely and colleagues reported that bimoclomol directly binds to HSF1, resulting in significant prolongation of the binding of HSF1 to HSE (Hargitai et al. 2003). On the other hand, Vigh and colleagues reported that bimoclomol interacts with acidic lipids and modifies the fluidity of the plasma membrane, similarly to the effect of heat shock stress on the membrane, leading to an enhancement of HSF1 activity (Torok et al. 2003).
Arimoclomol, an orally active derivative of bimoclomol, has been shown to induce molecular chaperones in the pancreas of rats with pancreatitis, upon its intragastric administration (Rakonczay et al. 2002). Arimoclomol has also been shown to induce multiple molecular chaperones via the activation of HSF1 in the spinal cord upon intraperitoneal injection, resulting in the alleviation of disease symptoms of ALS model mice (Kieran et al. 2004). Furthermore, the lifespan of ALS model mice was extended by arimoclomol treatment, when treatment was started at the time of symptom onset (Kieran et al. 2004), indicating that this compound is expected to exert therapeutic effects in the early stages of ALS. In a phase II clinical trial, arimoclomol was shown to be safe and well tolerated in patients with ALS and confirmed to be delivered to the cerebrospinal fluid upon oral administration (Cudkowicz et al. 2008). Currently, a phase II/III clinical trial of arimoclomol to ALS patients is ongoing (ClinicalTrials.gov Identifier: NCT00706147).
7 Inhibition of HSF1 Degradation
Riluzole, a compound that acts against glutamate toxicity, is the clinically approved agent for the treatment of ALS. Although the therapeutic effects of riluzole were believed to be from its protective effects against glutamate toxicity (Bensimon et al. 1994), the effect of riluzole on HSF1 has recently been clarified. Treatment of cultured cells with riluzole was reported to accelerate the induction of Hsp70 upon heat shock stress by increasing the amount of HSF1, whereas riluzole treatment under unstressed conditions only increased the amount of steady-state HSF1 and did not induce the activation of HSF1 (Yang et al. 2008; Liu et al. 2011). Since the expression of HSF1 mRNA was not altered, riluzole is thought to inhibit degradation of the HSF1 protein. These results suggest that the increase in the amount of HSF1 may be involved in the therapeutic effects of riluzole against ALS.
8 Medicinal Plants
Root extracts from the Celastraceae family are used for the treatment of fever, chills, joint pain, inflammation, edema, rheumatoid arthritis, and bacterial infection. Celastrol, which is isolated from the root extract of the Celastraceae family, was identified as an inducer of molecular chaperones through HSF1 activation (Westerheide et al. 2004). Although the mechanisms by which celastrol activates HSF1 are not fully understood, celastrol is thought to inhibit the binding of Hsp90 to ATP without affecting the N-terminal ATP-binding pocket of Hsp90 (Zhang et al. 2009; Hieronymus et al. 2006). Treatment of a cell culture model of the polyQ diseases with celastrol was reported to induce Hsp70 in an HSF1-dependent manner and to suppress polyQ aggregation, leading to a reduction in cell death (Zhang and Sarge 2007). However, since celastrol was reported to have multiple targets, such as Cdc37, IKKβ, and the proteasome, and, furthermore, since it directly suppresses the aggregation of purified polyQ proteins in vitro, other mechanisms may also contribute to its therapeutic effects (Yang et al. 2006; Lee et al. 2006; Sreeramulu et al. 2009; Zhang and Sarge 2007; Wang et al. 2005).
Paeonia lactiflora is a medicinal plant used for nourishing blood, alleviating pain, reducing irritability, and treating liver disease and cancer. Paeoniflorin, one of the main compounds extracted from Paeonia lactiflora, was reported to induce multiple molecular chaperones via the activation of HSF1, although the mechanisms remain unknown (Yan et al. 2004). Furthermore, treatment with paeoniflorin was reported to induce molecular chaperones in a polyQ disease cell culture model, resulting in the suppression of polyQ protein aggregation (Chang et al. 2013). In addition, intraperitoneal injection of paeoniflorin to polyQ disease model mice was shown to induce the expression of not only molecular chaperones but also nuclear factor-YA (NF-YA), a transcriptional factor (Tohnai et al. 2014). NF-YA increases the expression levels of transcription factor EB (TFEB) and carboxyl-terminus of Hsc70-interacting proteins (CHIP), which are proteins involved in the lysosome and proteasome protein degradation system, respectively (Sardiello et al. 2009; Stankiewicz et al. 2010), leading to the degradation of polyQ proteins, resulting in the amelioration of motor performance and extension of lifespan of polyQ disease model mice. These results suggest that paeoniflorin targets not only HSF1 but also other molecules, demonstrating its additional therapeutic effects against neurodegenerative diseases.
9 Dexamethasone
Dexamethasone, a synthetic corticosteroid used as an antiinflammatory agent, was shown to induce Hsp70 but not Hsp27 and Hsp60 via the activation of HSF1 in rat cardiac myocytes (Sun et al. 2000). Jana and colleagues found that the expression levels of HSF1 mRNA and protein are decreased in the eyes and brains of polyQ disease model flies and mice, respectively (Maheshwari et al. 2014). They tested the effects of dexamethasone on these models and found that dexamethasone treatment increases the expression level of HSF1 to a similar level to that in wild-type mice. Furthermore, dexamethasone not only increased the expression levels of HSF1 but also induced HSF1 activation in the brains of a mouse model of the polyQ diseases, leading to a decrease in the number of aggregates and an improvement in motor performance through the induction of Hsp70. Although the mechanisms by which dexamethasone increases the expression level of HSF1 and induces its activation are unclear, they showed that dexamethasone reduces the expression level of Hsp90, which probably contributes to the activation of HSF1.
10 Inhibition of Histone Deacetylase
Heat shock stress induces not only the activation of HSF1 but also the acetylation of histones, which alters the chromatin structure in the promoter region of molecular chaperone genes. The acetylation of core histones weakens the interaction of histones with DNA, leading to the open chromatin structure that confers high accessibility for transcription factors to the target sequence on the genome. In fact, hyperacetylation of histones by treatment with histone deacetylase (HDAC) inhibitors has been reported to increase the accessibility of HSF1 to the HSE sequence and to induce the expression of molecular chaperones (Chen et al. 2002; Zhao et al. 2005; Marinova et al. 2011; Ovakim and Heikkila 2003). Therefore, HDAC inhibitors can be regarded as activators of HSF1-mediated induction of molecular chaperones in trans. Treatment with HDAC inhibitors, such as trichostatin A and sodium phenylbutyrate, has indeed been shown to exert therapeutic effects against various models of neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s diseases, ALS, and the polyQ diseases (Ricobaraza et al. 2009; Kontopoulos et al. 2006; Ryu et al. 2005; Steffan et al. 2001; Minamiyama et al. 2004), although the contribution of HSF1 to these effects was not investigated. These facts suggest that induction of molecular chaperones via HSF1 may contribute to the therapeutic effects of HDAC inhibitors against neurodegenerative disease models.
11 Future Perspectives
Here we introduced various studies investigating the therapeutic effects of various small compounds that induce the activation of HSF1, for the treatment of various neurodegenerative diseases involving protein misfolding. To date, several HSF1-activating compounds were demonstrated as potential therapeutic agents using mouse models of various neurodegenerative diseases. However, we note here that in almost all studies showing the therapeutic effects of HSF1 activators in mouse models, treatments were started before disease onset. Bates and colleagues demonstrated an important issue, namely, that the induction of molecular chaperones is gradually diminished with disease progression in the brains of neurodegenerative disease model mice, through alterations in chromatin structure, which decreases the accessibility of activated HSF1 to the HSE sequence (Labbadia et al. 2011). Considering that the majority of patients with neurodegenerative diseases are diagnosed after disease onset, the chromatin structure of the HSE sequence in patients’ brains may already be altered, which would interfere with activated HSF1 accessing the HSE, and hence this issue should be solved toward developing a therapy for human patients.
One plausible strategy is the rearrangement of the altered chromatin structure, allowing it to be accessed by HSF1. Huang and colleagues clearly showed that HDAC inhibitor treatment rearranges the chromatin structure, increasing the accessibility of HSF1 to the HSE sequence (Chen et al. 2002). Hence, a combination of HSF1-activating compounds and HDAC inhibitors is expected to show synergistic effects against the neurodegenerative diseases. In fact, Marsh and colleagues showed that combinatorial treatment with an Hsp90 inhibitor (geldanamycin) and an HDAC inhibitor (suberoylanilide hydroxamic acid) results in much greater suppression of neurodegeneration than treatment with each compound alone in polyQ disease model flies, although the induction of molecular chaperones was not examined (Agrawal et al. 2005). This robust suppression may be caused by the efficient induction of molecular chaperones via the activation of HSF1 both in cis and in trans.
Another strategy is based on the novel concept of exosome-mediated intercellular chaperone transmission, which contributes to the maintenance of protein homeostasis at the organismal level. We recently reported that molecular chaperones, including Hsp40 and Hsp70, are secreted from cells via exosomes, transmitted to other cells, and suppress polyQ aggregation in other cultured cells (Takeuchi et al. 2015). Furthermore, the overexpression of molecular chaperones in nonneuronal tissues, such as the muscle and fat body non-cell, autonomously suppresses neurodegeneration in the eyes of polyQ disease model flies, probably through intercellular chaperone transmission. Therefore, our study indicates that even if molecular chaperones cannot be induced in the brain through the activation of HSF1, their induction in peripheral tissues or supplying chaperone-containing exosomes from the periphery is expected to suppress protein misfolding in patients’ brains in a non-cell autonomous manner.
In summary, the pharmacological activation of HSF1 is a promising therapeutic approach against various protein misfolding neurodegenerative diseases. Toward developing HSF1-mediated therapies against neurodegenerative diseases, additional studies toward understanding the mechanisms of activation of HSF1 under chronic proteotoxic stress should be performed in the future.
References
Abravaya K, Myers MP, Murphy SP, Morimoto RI (1992) The human heat shock protein hsp70 interacts with HSF, the transcription factor that regulates heat shock gene expression. Genes Dev 6(7):1153–1164
Agrawal N, Pallos J, Slepko N, Apostol BL, Bodai L, Chang LW, Chiang AS, Thompson LM, Marsh JL (2005) Identification of combinatorial drug regimens for treatment of Huntington’s disease using Drosophila. Proc Natl Acad Sci U S A 102(10):3777–3781
Auluck PK, Bonini NM (2002) Pharmacological prevention of Parkinson disease in Drosophila. Nat Med 8(11):1185–1186
Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM (2002) Chaperone suppression of α-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science 295(5556):865–868
Bagatell R, Paine-Murrieta GD, Taylor CW, Pulcini EJ, Akinaga S, Benjamin IJ, Whitesell L (2000) Induction of a heat shock factor 1-dependent stress response alters the cytotoxic activity of hsp90-binding agents. Clin Cancer Res 6(8):3312–3318
Baler R, Dahl G, Voellmy R (1993) Activation of human heat shock genes is accompanied by oligomerization, modification, and rapid translocation of heat shock transcription factor HSF1. Mol Cell Biol 13(4):2486–2496
Batulan Z, Taylor DM, Aarons RJ, Minotti S, Doroudchi MM, Nalbantoglu J, Durham HD (2006) Induction of multiple heat shock proteins and neuroprotection in a primary culture model of familial amyotrophic lateral sclerosis. Neurobiol Dis 24(2):213–225
Bensimon G, Lacomblez L, Meininger V (1994) A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med 330(9):585–591
Bruening W, Roy J, Giasson B, Figlewicz DA, Mushynski WE, Durham HD (1999) Up-regulation of protein chaperones preserves viability of cells expressing toxic Cu/Zn-superoxide dismutase mutants associated with amyotrophic lateral sclerosis. J Neurochem 72(2):693–699
Chan HY, Warrick JM, Gray-Board GL, Paulson HL, Bonini NM (2000) Mechanisms of chaperone suppression of polyglutamine disease: selectivity, synergy and modulation of protein solubility in Drosophila. Hum Mol Genet 9(19):2811–2820
Chang KH, Chen WL, Lee LC, Lin CH, Kung PJ, Lin TH, Wu YC, Wu YR, Chen YC, Lee-Chen GJ, Chen CM (2013) Aqueous extract of Paeonia lactiflora and paeoniflorin as aggregation reducers targeting chaperones in cell models of spinocerebellar ataxia 3. Evid Based Complement Alternat Med 2013:471659
Chen T, Sun H, Lu J, Zhao Y, Tao D, Li X, Huang B (2002) Histone acetylation is involved in hsp70 gene transcription regulation in Drosophila melanogaster. Arch Biochem Biophys 408(2):171–176
Chu B, Soncin F, Price BD, Stevenson MA, Calderwood SK (1996) Sequential phosphorylation by mitogen-activated protein kinase and glycogen synthase kinase 3 represses transcriptional activation by heat shock factor-1. J Biol Chem 271(48):30847–30857
Cudkowicz ME, Shefner JM, Simpson E, Grasso D, Yu H, Zhang H, Shui A, Schoenfeld D, Brown RH, Wieland S, Barber JR, Northeast ALSC (2008) Arimoclomol at dosages up to 300 mg/day is well tolerated and safe in amyotrophic lateral sclerosis. Muscle Nerve 38(1):837–844
Cummings CJ, Mancini MA, Antalffy B, DeFranco DB, Orr HT, Zoghbi HY (1998) Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet 19(2):148–154
Cummings CJ, Sun Y, Opal P, Antalffy B, Mestril R, Orr HT, Dillmann WH, Zoghbi HY (2001) Over-expression of inducible HSP70 chaperone suppresses neuropathology and improves motor function in SCA1 mice. Hum Mol Genet 10(14):1511–1518
Dehner A, Furrer J, Richter K, Schuster I, Buchner J, Kessler H (2003) NMR chemical shift perturbation study of the N-terminal domain of Hsp90 upon binding of ADP, AMP-PNP, geldanamycin, and radicicol. Chembiochem 4(9):870–877
Fujikake N, Nagai Y, Popiel HA, Okamoto Y, Yamaguchi M, Toda T (2008) Heat shock transcription factor 1-activating compounds suppress polyglutamine-induced neurodegeneration through induction of multiple molecular chaperones. J Biol Chem 283(38):26188–26197
Fujimoto M, Takaki E, Hayashi T, Kitaura Y, Tanaka Y, Inouye S, Nakai A (2005) Active HSF1 significantly suppresses polyglutamine aggregate formation in cellular and mouse models. J Biol Chem 280(41):34908–34916
Han S, Choi JR, Soon Shin K, Kang SJ (2012) Resveratrol upregulated heat shock proteins and extended the survival of G93A-SOD1 mice. Brain Res 1483:112–117
Hargitai J, Lewis H, Boros I, Racz T, Fiser A, Kurucz I, Benjamin I, Vigh L, Penzes Z, Csermely P, Latchman DS (2003) Bimoclomol, a heat shock protein co-inducer, acts by the prolonged activation of heat shock factor-1. Biochem Biophys Res Commun 307(3):689–695
Hay DG, Sathasivam K, Tobaben S, Stahl B, Marber M, Mestril R, Mahal A, Smith DL, Woodman B, Bates GP (2004) Progressive decrease in chaperone protein levels in a mouse model of Huntington’s disease and induction of stress proteins as a therapeutic approach. Hum Mol Genet 13(13):1389–1405
Hendrick JP, Hartl FU (1995) The role of molecular chaperones in protein folding. FASEB J 9(15):1559–1569
Herranz D, Munoz-Martin M, Canamero M, Mulero F, Martinez-Pastor B, Fernandez-Capetillo O, Serrano M (2010) Sirt1 improves healthy aging and protects from metabolic syndrome-associated cancer. Nat Commun 1:3
Hieronymus H, Lamb J, Ross KN, Peng XP, Clement C, Rodina A, Nieto M, Du J, Stegmaier K, Raj SM, Maloney KN, Clardy J, Hahn WC, Chiosis G, Golub TR (2006) Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell 10(4):321–330
Hietakangas V, Ahlskog JK, Jakobsson AM, Hellesuo M, Sahlberg NM, Holmberg CI, Mikhailov A, Palvimo JJ, Pirkkala L, Sistonen L (2003) Phosphorylation of serine 303 is a prerequisite for the stress-inducible SUMO modification of heat shock factor 1. Mol Cell Biol 23(8):2953–2968
Hirakawa T, Rokutan K, Nikawa T, Kishi K (1996) Geranylgeranylacetone induces heat shock proteins in cultured guinea pig gastric mucosal cells and rat gastric mucosa. Gastroenterology 111(2):345–357
Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, Scherer B, Sinclair DA (2003) Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 425(6954):191–196
Ishihara K, Yamagishi N, Saito Y, Adachi H, Kobayashi Y, Sobue G, Ohtsuka K, Hatayama T (2003) Hsp105α suppresses the aggregation of truncated androgen receptor with expanded CAG repeats and cell toxicity. J Biol Chem 278(27):25143–25150
Kaeberlein M, McVey M, Guarente L (1999) The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev 13(19):2570–2580
Katsuno M, Sang C, Adachi H, Minamiyama M, Waza M, Tanaka F, Doyu M, Sobue G (2005) Pharmacological induction of heat-shock proteins alleviates polyglutamine-mediated motor neuron disease. Proc Natl Acad Sci U S A 102(46):16801–16806
Kieran D, Kalmar B, Dick JR, Riddoch-Contreras J, Burnstock G, Greensmith L (2004) Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat Med 10(4):402–405
Kline MP, Morimoto RI (1997) Repression of the heat shock factor 1 transcriptional activation domain is modulated by constitutive phosphorylation. Mol Cell Biol 17(4):2107–2115
Klucken J, Shin Y, Masliah E, Hyman BT, McLean PJ (2004) Hsp70 reduces α-synuclein aggregation and toxicity. J Biol Chem 279(24):25497–25502
Kontopoulos E, Parvin JD, Feany MB (2006) α-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum Mol Genet 15(20):3012–3023
Labbadia J, Cunliffe H, Weiss A, Katsyuba E, Sathasivam K, Seredenina T, Woodman B, Moussaoui S, Frentzel S, Luthi-Carter R, Paganetti P, Bates GP (2011) Altered chromatin architecture underlies progressive impairment of the heat shock response in mouse models of Huntington disease. J Clin Invest 121(8):3306–3319
Lee JH, Koo TH, Yoon H, Jung HS, Jin HZ, Lee K, Hong YS, Lee JJ (2006) Inhibition of NF-kappa B activation through targeting I kappa B kinase by celastrol, a quinone methide triterpenoid. Biochem Pharmacol 72(10):1311–1321
Liu AY, Mathur R, Mei N, Langhammer CG, Babiarz B, Firestein BL (2011) Neuroprotective drug riluzole amplifies the heat shock factor 1 (HSF1)- and glutamate transporter 1 (GLT1)-dependent cytoprotective mechanisms for neuronal survival. J Biol Chem 286(4):2785–2794
Lopez T, Dalton K, Frydman J (2015) The mechanism and function of group II chaperonins. J Mol Biol 427(18):2919–2930
Machajewski T, Shafer C, McBride C, inventors; Novartis Pharma AG, assignee (2007) 2–amino–7,8–dihydro–6H–pyrido[4,3–D]pyrimidine–5–one compounds. US Patent 20070123546. 31 May 2007
Maheshwari M, Bhutani S, Das A, Mukherjee R, Sharma A, Kino Y, Nukina N, Jana NR (2014) Dexamethasone induces heat shock response and slows down disease progression in mouse and fly models of Huntington’s disease. Hum Mol Genet 23(10):2737–2751
Marinova Z, Leng Y, Leeds P, Chuang DM (2011) Histone deacetylase inhibition alters histone methylation associated with heat shock protein 70 promoter modifications in astrocytes and neurons. Neuropharmacology 60(7–8):1109–1115
Minamiyama M, Katsuno M, Adachi H, Waza M, Sang C, Kobayashi Y, Tanaka F, Doyu M, Inukai A, Sobue G (2004) Sodium butyrate ameliorates phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Hum Mol Genet 13(11):1183–1192
Mosser DD, Duchaine J, Massie B (1993) The DNA-binding activity of the human heat shock transcription factor is regulated in vivo by hsp70. Mol Cell Biol 13(9):5427–5438
Neef DW, Turski ML, Thiele DJ (2010) Modulation of heat shock transcription factor 1 as a therapeutic target for small molecule intervention in neurodegenerative disease. PLoS Biol 8(1), e1000291
Neef DW, Jaeger AM, Thiele DJ (2011) Heat shock transcription factor 1 as a therapeutic target in neurodegenerative diseases. Nat Rev Drug Discov 10(12):930–944
Neef DW, Jaeger AM, Gomez-Pastor R, Willmund F, Frydman J, Thiele DJ (2014) A direct regulatory interaction between chaperonin TRiC and stress-responsive transcription factor HSF1. Cell Rep 9(3):955–966
Otaka M, Yamamoto S, Ogasawara K, Takaoka Y, Noguchi S, Miyazaki T, Nakai A, Odashima M, Matsuhashi T, Watanabe S, Itoh H (2007) The induction mechanism of the molecular chaperone HSP70 in the gastric mucosa by Geranylgeranylacetone (HSP-inducer). Biochem Biophys Res Commun 353(2):399–404
Ovakim DH, Heikkila JJ (2003) Effect of histone deacetylase inhibitors on heat shock protein gene expression during Xenopus development. Genesis 36(2):88–96
Rakonczay Z Jr, Ivanyi B, Varga I, Boros I, Jednakovits A, Nemeth I, Lonovics J, Takacs T (2002) Nontoxic heat shock protein coinducer BRX-220 protects against acute pancreatitis in rats. Free Radic Biol Med 32(12):1283–1292
Ricobaraza A, Cuadrado-Tejedor M, Perez-Mediavilla A, Frechilla D, Del Rio J, Garcia-Osta A (2009) Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an Alzheimer’s disease mouse model. Neuropsychopharmacology 34(7):1721–1732
Rimoldi M, Servadio A, Zimarino V (2001) Analysis of heat shock transcription factor for suppression of polyglutamine toxicity. Brain Res Bull 56(3–4):353–362
Roe SM, Prodromou C, O’Brien R, Ladbury JE, Piper PW, Pearl LH (1999) Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J Med Chem 42(2):260–266
Rogina B, Helfand SL (2004) Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci U S A 101(45):15998–16003
Ryu H, Smith K, Camelo SI, Carreras I, Lee J, Iglesias AH, Dangond F, Cormier KA, Cudkowicz ME, Brown RH Jr, Ferrante RJ (2005) Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J Neurochem 93(5):1087–1098
Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, Banfi S, Parenti G, Cattaneo E, Ballabio A (2009) A gene network regulating lysosomal biogenesis and function. Science 325(5939):473–477
Sarge KD, Murphy SP, Morimoto RI (1993) Activation of heat shock gene transcription by heat shock factor 1 involves oligomerization, acquisition of DNA-binding activity, and nuclear localization and can occur in the absence of stress. Mol Cell Biol 13(3):1392–1407
Schulte TW, Akinaga S, Soga S, Sullivan W, Stensgard B, Toft D, Neckers LM (1998) Antibiotic radicicol binds to the N-terminal domain of Hsp90 and shares important biologic activities with geldanamycin. Cell Stress Chaperones 3(2):100–108
Shi Y, Mosser DD, Morimoto RI (1998) Molecular chaperones as HSF1-specific transcriptional repressors. Genes Dev 12(5):654–666
Shimura H, Miura-Shimura Y, Kosik KS (2004) Binding of tau to heat shock protein 27 leads to decreased concentration of hyperphosphorylated tau and enhanced cell survival. J Biol Chem 279(17):17957–17962
Sittler A, Lurz R, Lueder G, Priller J, Lehrach H, Hayer-Hartl MK, Hartl FU, Wanker EE (2001) Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington’s disease. Hum Mol Genet 10(12):1307–1315
Sreeramulu S, Gande SL, Gobel M, Schwalbe H (2009) Molecular mechanism of inhibition of the human protein complex Hsp90-Cdc37, a kinome chaperone-cochaperone, by triterpene celastrol. Angew Chem Int Ed Engl 48(32):5853–5855
Stankiewicz M, Nikolay R, Rybin V, Mayer MP (2010) CHIP participates in protein triage decisions by preferentially ubiquitinating Hsp70-bound substrates. FEBS J 277(16):3353–3367
Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM (2001) Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 413(6857):739–743
Sun L, Chang J, Kirchhoff SR, Knowlton AA (2000) Activation of HSF and selective increase in heat-shock proteins by acute dexamethasone treatment. Am J Physiol Heart Circ Physiol 278(4):H1091–H1097
Takeuchi T, Suzuki M, Fujikake N, Popiel HA, Kikuchi H, Futaki S, Wada K, Nagai Y (2015) Intercellular chaperone transmission via exosomes contributes to maintenance of protein homeostasis at the organismal level. Proc Natl Acad Sci U S A 112(19):E2497–E2506
Taylor JP, Hardy J, Fischbeck KH (2002) Toxic proteins in neurodegenerative disease. Science 296(5575):1991–1995
Tissenbaum HA, Guarente L (2001) Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature 410(6825):227–230
Tohnai G, Adachi H, Katsuno M, Doi H, Matsumoto S, Kondo N, Miyazaki Y, Iida M, Nakatsuji H, Qiang Q, Ding Y, Watanabe H, Yamamoto M, Ohtsuka K, Sobue G (2014) Paeoniflorin eliminates a mutant AR via NF-YA-dependent proteolysis in spinal and bulbar muscular atrophy. Hum Mol Genet 23(13):3552–3565
Torok Z, Tsvetkova NM, Balogh G, Horvath I, Nagy E, Penzes Z, Hargitai J, Bensaude O, Csermely P, Crowe JH, Maresca B, Vigh L (2003) Heat shock protein coinducers with no effect on protein denaturation specifically modulate the membrane lipid phase. Proc Natl Acad Sci U S A 100(6):3131–3136
Vigh L, Literati PN, Horvath I, Torok Z, Balogh G, Glatz A, Kovacs E, Boros I, Ferdinandy P, Farkas B, Jaszlits L, Jednakovits A, Koranyi L, Maresca B (1997) Bimoclomol: a nontoxic, hydroxylamine derivative with stress protein-inducing activity and cytoprotective effects. Nat Med 3(10):1150–1154
Wang J, Gines S, MacDonald ME, Gusella JF (2005) Reversal of a full-length mutant huntingtin neuronal cell phenotype by chemical inhibitors of polyglutamine-mediated aggregation. BMC Neurosci 6:1
Westerheide SD, Bosman JD, Mbadugha BN, Kawahara TL, Matsumoto G, Kim S, Gu W, Devlin JP, Silverman RB, Morimoto RI (2004) Celastrols as inducers of the heat shock response and cytoprotection. J Biol Chem 279(53):56053–56060
Westerheide SD, Anckar J, Stevens SM Jr, Sistonen L, Morimoto RI (2009) Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science 323(5917):1063–1066
Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM (1994) Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci U S A 91(18):8324–8328
Yam AY, Xia Y, Lin HT, Burlingame A, Gerstein M, Frydman J (2008) Defining the TRiC/CCT interactome links chaperonin function to stabilization of newly made proteins with complex topologies. Nat Struct Mol Biol 15(12):1255–1262
Yamanaka T, Miyazaki H, Oyama F, Kurosawa M, Washizu C, Doi H, Nukina N (2008) Mutant Huntingtin reduces HSP70 expression through the sequestration of NF-Y transcription factor. EMBO J 27(6):827–839
Yan D, Saito K, Ohmi Y, Fujie N, Ohtsuka K (2004) Paeoniflorin, a novel heat shock protein-inducing compound. Cell Stress Chaperones 9(4):378–389
Yang H, Chen D, Cui QC, Yuan X, Dou QP (2006) Celastrol, a triterpene extracted from the Chinese “Thunder of God Vine,” is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res 66(9):4758–4765
Yang J, Bridges K, Chen KY, Liu AY (2008) Riluzole increases the amount of latent HSF1 for an amplified heat shock response and cytoprotection. PLoS One 3(8):e2864
Zhang YQ, Sarge KD (2007) Celastrol inhibits polyglutamine aggregation and toxicity though induction of the heat shock response. J Mol Med (Berl) 85(12):1421–1428
Zhang T, Li Y, Yu Y, Zou P, Jiang Y, Sun D (2009) Characterization of celastrol to inhibit hsp90 and cdc37 interaction. J Biol Chem 284(51):35381–35389
Zhao Y, Sun H, Lu J, Li X, Chen X, Tao D, Huang W, Huang B (2005) Lifespan extension and elevated hsp gene expression in Drosophila caused by histone deacetylase inhibitors. J Exp Biol 208(Pt 4):697–705
Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R (1998) Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell 94(4):471–480
Acknowledgments
We thank Dr. H. Akiko Popiel (Tokyo Medical University) for editing the manuscript. We also thank lab members and Dr. Shigeomi Shimizu (Tokyo Medical and Dental University) for their helpful discussions. This work was supported, in part, by Grants-in-Aid for Scientific Research on Priority Areas (Research on Pathomechanisms of Brain Disorders, Protein Community, and Proteolysis) to Y.N. and on Innovative Areas (Synapse and Neurocircuit Pathology) to Y.N. and Strategic Research Program for Brain Sciences (Integrated research on neuropsychiatric disorders) to Y.N. from the Ministry of Education, Culture, Sports, Science, and Technology, Japan; by Grants-in-Aid for Scientific Research (B) to Y.N. and (C) to N.F., for Challenging Exploratory Research to Y.N. and for Young Scientists (A) to T.T. from the Japan Society for the Promotion of Science, Japan; by Health and Labour Sciences Research Grants for Research on Development of New Drugs and Research on Measures for Intractable Diseases to Y.N. from the Ministry of Health, Labour, and Welfare, Japan; and by a grant from Core Research for Evolutional Science and Technology (CREST) of the Japan Science and Technology Agency to Y.N.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Japan
About this chapter
Cite this chapter
Fujikake, N., Takeuchi, T., Nagai, Y. (2016). HSF1 Activation by Small Chemical Compounds for the Treatment of Neurodegenerative Diseases. In: Nakai, A. (eds) Heat Shock Factor. Springer, Tokyo. https://doi.org/10.1007/978-4-431-55852-1_14
Download citation
DOI: https://doi.org/10.1007/978-4-431-55852-1_14
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-55850-7
Online ISBN: 978-4-431-55852-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)