
Abstract
One of the important gastric physiologies is the endocrine function. Among peptide hormones produced in the stomach, gastrin and somatostatin which are produced from G and D cell, respectively, are closely related with acid secretion, while ghrelin and leptin are known to be involved in gut motility as well as the regulation of appetite and body weight. Since Helicobacter pylori (H. pylori) is the major etiologic agent of chronic active gastritis, H. pylori infection can alter gastric hormone production. While the effect of H. pylori infection and its eradication on G and D cells and subsequent acid secretion has been relatively well investigated, its effect on ghrelin and leptin levels has been inconclusive. This discrepancy originates from different measurement methodology or existence of several types in one hormone. In addition, the different stage of H. pylori infection or site of the infection may cause a different result on ghrelin levels in the stomach and blood similar to the case of acid secretion. H. pylori infection seems to raise gastric leptin production, but its effect on circulating level is not much. Understanding the effect of H. pylori infection and eradication on gastric hormonal changes might provide a critical clinical implication for the management of gastrointestinal diseases as well as obesity or eating disorder.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Although gut is not the classical endocrine tissues such as the pituitary, thyroid, and adrenal glands, it produces an array of peptide hormones which are important for both enteric and non-enteric physiology [1]. That is, enteroendocrine cells are distributed throughout the gastrointestinal tract, but a significant proportion of endocrine hormones which are involved in appetite regulation or intestinal motility are produced and secreted by the gastric mucosa.
Traditionally, gastrin and somatostatin have been well investigated related with gastric acid secretion. Both endocrine and paracrine mediators also exert control over the maintenance of gastric secretory functions. Gastrin is released from the G cells of the antral mucosa and travels through the bloodstream to the corpus where gastrin stimulates enterochromaffin-like (ECL) cells to secrete histamine which, in turn, stimulates the parietal cells to secrete acid. In contrast, somatostatin (SST) and prostaglandins inhibit acid secretion [2].
Other two hormones, ghrelin and leptin which are secreted from the stomach, play critical roles in controlling appetite and satiety. Ghrelin is a 28-amino acid peptide hormone, primarily produced in, and secreted from, the gastric mucosa [3]. Meanwhile, leptin is a hormone mainly produced by adipose tissues with modulatory effects on feeding behavior and weight control [4]. However, recently stomach has been also identified as an important source of leptin. Both ghrelin and leptin act in an autocrine/paracrine manner and can travel through blood and act in the central nerve system (CNS).
Helicobacter pylori (H. pylori) is a predominant etiologic factor of gastric inflammation leading to atrophy of gastric glands. Its infection of the human gastric mucosa alters the normal gastric physiology. Especially, cytokines deregulate secretion of gastric hormones including gastrin, SST, ghrelin, and leptin during the inflammation. Finally, altered gut–brain interactions by this deregulation of these hormones underlie gastrointestinal (GI) symptom generation. Furthermore, the change of these hormones can induce GI disorder. This chapter aims to review the data on hormones which are produced in stomach, focusing on ghrelin and the effect of H. pylori infection and its eradication on local and circulating levels of these hormones.
2 Ghrelin
2.1 Production of Ghrelin
2.1.1 Source of Ghrelin
The stomach is considered as the major source of circulating ghrelin, since severe reduction in blood ghrelin levels is observed in patients that undergone gastrectomy [5]. Ghrelin is produced in oxyntic cells that are prominent in the corpus of stomach. The ghrelin-producing cells are located from the base to the neck of the glands. It is also secreted from the small intestine and the colon [6]. In addition, ghrelin is also expressed in the hypothalamus [7], the pituitary [8], and several tissues in the periphery [9].
2.1.2 Process of Ghrelin Production
Although the major active product of the ghrelin gene is the 28-amino acid peptide acylated at Ser3 with C8:0, recent studies have revealed that the ghrelin gene can generate various molecules besides ghrelin, which include des-acyl ghrelin [10].
The human ghrelin gene, located on the short arm of chromosome 3 (3p25–26), is composed of five exons and four introns [11]. There are two different transcriptional initiation sites in the ghrelin gene, resulting in two distinct mRNA transcripts: transcript-A and transcript-B (Fig. 6.1). The main ghrelin mRNA transcript in human codes for a 117-amino acid long peptide: preproghrelin (1–117). The signal peptide sequence, preproghrelin (1–23) of preproghrelin (1–117), is cleaved to form proghrelin (1–94). A series of posttranslational steps, including the process of protease cleavage and acyl modification of the ghrelin precursor peptide, results in the production of mature ghrelin peptides (acyl and des-acyl ghrelin) or other ghrelin gene-associated peptides (C-ghrelin and obestatin).

Structure of the human ghrelin gene and processing steps from the ghrelin gene to acyl ghrelin, des-acyl ghrelin, or other ghrelin-associated peptides (Adapted from Nishi et al. [11]). The human ghrelin gene is composed of four exons. The major mRNA transcript (transcript-A) of the ghrelin gene is translated into a 117-amino acid ghrelin precursor: preproghrelin (1–117). The signal peptide sequence, preproghrelin (1–23) of preproghrelin (1–117), is cleaved to form proghrelin (1–94). A series of posttranslational steps, including the process of protease cleavage and acyl modification of the ghrelin precursor peptide, results in the production of mature ghrelin peptides (acyl and des-acyl ghrelin) or other ghrelin gene-associated peptides (C-ghrelin and obestatin). Besides these peptides, several other ghrelin gene-derived peptides with or without acyl modification (des-Gln14-ghrelin, ΔEx3-C-ghrelin, etc.) are predicted to be produced from the splicing variants of the ghrelin gene transcripts. GOAT ghrelin-O-acyltransferase
The ghrelin peptide is acylated by the enzyme ghrelin O-acyltransferase (GOAT) [12], which is expressed predominantly in the stomach, gut, and pancreas but also at other sites [13]. This acyl modification of ghrelin is easily cleaved during sample extraction. Thus, acyl ghrelin should be isolated from blood specimens by adding ethylenediaminetetraacetic acid (EDTA) with aprotinin or p-hydroxymercuribenzoic acid, separating the plasma by centrifugation and immediate acidification before freezing at −80 °C to ensure the stability of acyl ghrelin during storage. The nonacylated form of ghrelin, without octanoic acid modification at Ser3 residue, des-acyl ghrelin is also present at significant level in both the stomach and blood [10, 14]. Des-acyl ghrelin is the most abundant ghrelin-related molecule in the body, comprising 80–90 % of the total circulating ghrelin, and has a longer half-life.
Besides these peptides, several other ghrelin gene-derived peptides with or without acyl modification (des-Gln 14-ghrelin, ΔEx3-C-ghrelin, etc.) are predicted to be produced from the splicing variants of the ghrelin gene transcripts [11].
2.2 Regulation of Ghrelin
Many factors which affect serum ghrelin levels were listed in Table 6.1 [15].
2.2.1 Posttranslational Modification of Ghrelin Precursor Protein
As above mentioned, GOAT is a member of the family membrane-bound O-acyltransferases which shows highly specific expression in the gastric mucosa. This discovery manifests its significance in the regulation of ghrelin secretion, because the amount and activity of GOAT likely affect the level of acyl ghrelin. An in vitro study has demonstrated that GOAT activity could be inhibited potently by an octanoylated ghrelin pentapeptide and other end products [16], suggesting the existence of a negative feedback regulation on the production of acyl ghrelin. Plasma esterases have been reported to des-acylate acyl ghrelin, whereas plasma proteases account for the degradation of circulating ghrelin [17]. The circulating level of ghrelin is determined by the balance among its secretion rate, degradation rate, and clearance rate.
2.2.2 Nutrients Regulating Ghrelin Expression and Secretion
Glucose markedly inhibits ghrelin secretion (Table 6.1). That is, oral infusion of glucose can decrease the plasma concentration of total ghrelin 30 min after ingestion in humans [18] and in rats [19]. Ingestion of crude fiber has the similar effect with glucose [19]. Insulin-induced hypoglycemia upregulates ghrelin mRNA expression [20] and serum acyl ghrelin level [21] in the stomach. With regard to fatty acid, it appears that the effects of fatty acids and triglycerides on ghrelin secretion are dependent on the length of their chain. Generally, lipid ingestion leads to a smaller decline in ghrelin relative to the administration of glucose or amino acids [22]. This observation may explain the weight gain effect of high-fat diet. Oral ingestion of a physiological dose of essential amino acids leads to a continuous rise in serum ghrelin level in humans [23].
2.2.3 Hormones Regulating Ghrelin Expression and Secretion
2.2.3.1 Insulin
In rats, gastric artery perfusion of insulin inhibits ghrelin release from isolated stomach tissue significantly (Table 6.1) [24]. In humans, infusion of insulin significantly decreases plasma ghrelin level [25]. Fasting plasma acyl ghrelin level is negatively related to insulin concentration [21]. This inhibitory effect of insulin may underlie the suppression of glucose on ghrelin and the inverse relationship between body weight and ghrelin level.
2.2.3.2 Glucagon
Glucagon may contribute to the preprandial surge of ghrelin. Plasma acyl ghrelin concentration rises transiently, while des-acyl ghrelin increases persistently after administration of glucagon in rats [26].
2.2.3.3 Growth Hormone/Insulin-Like Growth Factor-1 (IGF-1)
Growth hormone exerts a negative feedback action on ghrelin production and secretion. Administration of growth hormone in cultured rat gastric tissue time dependently inhibits total ghrelin secretion [27]. Contrary to the growth hormone, administration of recombinant human IGF-1 in severely undernutritioned patients elevates plasma total ghrelin concentration [28].
2.2.3.4 Somatostatin
Somatostatin (SST) probably inhibits ghrelin synthesis directly (Table 6.1). Plasma acyl and total ghrelin levels fall after the infusion of somatostatin or octreotide, somatostatin analog [29]. Since ghrelin increases the level of somatostatin in plasma [30], the inhibitory effect of somatostatin on ghrelin may be considered as a negative feedback modulation.
2.2.3.5 Leptin
It is generally agreed that leptin inhibits ghrelin synthesis (Table 6.1). Leptin is mainly synthesized and secreted by adipose tissue. Leptin concentration in obese is significantly higher than normal, whereas ghrelin is lower [31]. Leptin correlates with ghrelin in a complex pattern, which depends on the body weight (normal or obesity) and insulin sensitivity or insulin concentration. As shown by recent studies, ghrelin mRNA increases in the stomach during fasting, whereas leptin and leptin mRNA decrease [32]. Leptin dose-dependently inhibits ghrelin transcription in vitro [32] and decreases ghrelin release from isolated rat stomach [24]. Central leptin gene therapy decreases plasma leptin level and increases ghrelin level significantly in the mouse fed with high-fat diet [33], indicating that leptin exerts its inhibition on ghrelin secretion only in peripheral tissues. Thus, peripheral, especially gastric, leptin probably represses ghrelin expression through its receptor in gastric mucosa cells.
2.2.3.6 Estrogen
Many studies report that estrogen upregulates ghrelin level. Administration of estrogen elevates plasma total ghrelin concentration in female patients with anorexia nervosa [28]. Ghrelin mRNA level rises significantly after estrogen administration in cultured stomach cells [34]. However, there also exist discrepant results. Estrogen replacement therapy in postmenopausal women induces serum total and acyl ghrelin secretion only to an insignificant extent or even decreases serum total ghrelin level [15]. Variable methods (i.e., per oral or transdermal), duration for estrogen administration, and physiological status could attribute to these contradictable results.
2.2.4 Autonomic Nervous System Regulating Ghrelin Expression and Secretion
Autonomic nervous system, especially the parasympathetic nerve, plays an important role in the regulation of ghrelin (Table 6.1). Excitation of the vagus nerve can stimulate ghrelin secretion. In rats and humans, ghrelin level rises after administration of muscarinic agonists and falls after administration of muscarinic antagonists [35].
2.2.5 Physiological Status
Ghrelin level is negatively correlated with body mass index (BMI) in humans. Fasting acyl ghrelin [36] and total ghrelin [37] are significantly lower in the aged population than in the youth (Table 6.1). However, this age-dependent decline of ghrelin is not observed in the obese population [38]. Many studies report an elevated serum ghrelin level in female subjects relative to male ones. Serum total ghrelin level is about threefold higher in women during the late follicular stage of the cycle than in men [39].
2.2.6 H. pylori Infection
H. pylori-induced chronic gastritis is characterized by chronic inflammatory changes in the gastric mucosa leading to extensive mucosal atrophy and eventual epithelial metaplasia. It is speculated that destruction of oxyntic mucosa could result in a negative correlation with local or circulating ghrelin level. This will be further discussed in Sect. 6.2.4.
2.3 Role of Ghrelin
2.3.1 The Mechanisms of Action of Acyl Ghrelin
Acyl ghrelin has been identified as the endogenous ligand of the growth hormone secretagogue receptor (GHS-R) [3]. It crosses the blood–brain barrier in both directions using a saturable transport system that requires the presence of the unique octanoyl residue of the ghrelin molecule [40]. GHS-Rs are also widely expressed in the central nervous system. They are found in the pituitary, brainstem, and hypothalamus, whereas peripheral receptor expression has been described in the myocardium, GI tract, adipose tissue, liver, kidney, placenta, and the T cells [41]. Acyl ghrelin acts in GHS-R on vagal afferent nerve fibers in the stomach [42], which transmit this signal to the nucleus of the solitary tract (NTS) (Fig. 6.2). From the NTS, the information is projected to the arcuate nucleus (ARC) of the hypothalamus, where neuropeptide Y (NPY) neurons are activated (Fig. 6.2). The NPYY2 and/or Y4 receptor in the CNS may be involved in upper GI motility because Y2 and Y4 receptor agonists can induce phase III-like contractions in the duodenum when given to animals in the fed state [43]. From the ARC, the signal is finally transmitted to the dorsal motor nucleus of the vagus nerve (DVC, dorsal vagal complex) and via vagal efferent fibers, and fasted motor activity is induced in the gut (Fig. 6.2) [44].

The effects of acyl ghrelin via the brain–gut axis (Adapted from Fujimiya et al. [44]). ARC arcuate nucleus, DVC dorsal vagal complex, GHS-R growth hormone secretagogue receptors, NPY neuropeptide Y
2.3.2 The Representable Roles of Acyl Ghrelin
2.3.2.1 Acyl Ghrelin Is a Signal for Hunger
Acyl ghrelin is considered as a short regulator of food intake in both animals and humans [45, 46]. In rats, acute and chronic administration of ghrelin enhances food intake and weight gain [47, 48]. Peripheral administration of ghrelin produced a 28 % increase of food intake in normal-weight healthy volunteers [45]. The subjects who received exogenous ghrelin reported an increase in appetite and showed a higher caloric intake than after placebo [49].
2.3.2.2 Acyl Ghrelin Influences Gut Motility
As previously mentioned, in humans, ghrelin stimulates gastric motility [50] and acid secretion [51]. This fasted motor activity of the GI tract has been considered to play a role of a mechanical cleansing of the stomach and the intestine in preparation for the next meal. In healthy volunteers, the peripheral administration of ghrelin induces the occurrence of phase III of the migrating motor complex after about 20 min. Moreover, it induces a premature phase III originating in the stomach about 14 min after its injection [52]. A positive correlation was reported between preprandial ghrelin concentration and gastric emptying time. The duration of gastric emptying is considered as an important factor for the duration of satiety [53, 54].
2.3.2.3 Ghrelin and GI Disease
Preprandial ghrelin levels have significantly decreased in patients with functional dyspepsia (FD) with delayed gastric emptying in Lee’s report [55]. Moreover, low preprandial ghrelin levels were observed in patients with dysmotility-like FD [55]. Shindo et al. [56] also reported that the maximum gastric emptying time, Tmax, was significantly prolonged with significant lower acyl ghrelin levels in postprandial distress syndrome (PDS) patients not in epigastric pain syndrome (EPS) patients. Lower acyl ghrelin levels were also found in nonerosive reflux disease patients [56]. This correlation between acyl ghrelin levels and Tmax in the PDS patients indicates acyl ghrelin’s role in gastric emptying of PDS patients [56]. Interestingly, El-Salhy et al. [57] demonstrated that ghrelin-producing cells were suppressed in constipation-predominant IBS patients, while diarrhea-predominant IBS patients have significantly higher ghrelin-positive cells in the oxyntic mucosa compared with normal controls. Since there was no difference either in the plasma levels or gastric contents between IBS groups and control subjects, it could be speculated that some compensatory mechanism may exist and disruption of this regulation could result in IBS symptoms.
Furthermore, ghrelin was suggested to be an important biomarker for activity in IBD patients. Serum ghrelin levels were found to be higher in patients with ulcerative colitis and also higher in patients with ileal Crohn’s disease compared with colonic disease patients [58]. Ghrelin is also significantly elevated in active IBD patients and positively correlated with serum inflammatory markers such as tumor necrosis factor-α (TNF-α), C-reactive protein (CRP), erythrocyte sedimentation rate (ESR), and sedimentation fibrinogen [59].
2.3.2.4 Acyl Ghrelin Is Associated with GI Malignancy
Significant reduction of ghrelin mRNA and peptide expression was found in esophagogastric adenocarcinomas compared to adjacent nonneoplastic gastric mucosa. This finding suggests that ghrelin production was suppressed due to damage to normal ghrelin-secreting mucosa from adenocarcinoma [60]. An et al. [61] also reported that lower ghrelin levels were present in differentiated tumor tissue than in undifferentiated tissue. These findings suggested that the development of cancer may lead to an inability to produce ghrelin, which is also influenced by the state of differentiation. In colorectal cancer studies, Waseem et al. [62] showed that colorectal cancer cells excessively secrete ghrelin in vitro to promote proliferation. Malignant colorectal tissue samples also showed enhanced stage-dependent expression of ghrelin. However, expression of ghrelin and its functional receptor (GHS-R1a) were suppressed in advanced state, poorly differentiated tumors. GHS-R1a expression was lost in malignant colorectal cells, while GHS-R1b expression was enhanced [62].
2.3.2.5 Glucose Control
The ghrelin system, using both the acylated and des-acylated molecules, is actively involved in the acute and the long-term control of glucose metabolism and insulin concentrations [63]. It has been demonstrated that glucose output by primary hepatocytes is time- and dose-dependently stimulated by acyl ghrelin and inhibited by des-acyl ghrelin. Apparently, the two forms of peptides must be considered as separate hormones able to modify each other’s actions on glucose handling [41].
2.3.3 The Function of Des-acyl Ghrelin
Des-acyl ghrelin was first identified as the inactive form of ghrelin unable to bind to GHS-R. Some authors suggested that des-acyl ghrelin might have an anorexigenic activity that is contrary to the orexigenic activity of acylated ghrelin [64]. Conversely, a recent study showed that both ghrelin and des-acyl ghrelin function as orexigenic peptides in the hypothalamus [65]. Des-acyl ghrelin was later proposed to have nonendocrine functions including cardioprotective, antiproliferative, and adipogenic activities and antagonizing octanoyl-ghrelin-induced effects on insulin secretion and blood glucose levels in humans. Furthermore, it has been reported that des-acyl ghrelin is able to antagonize acyl ghrelin-induced glucose output. These actions might be mediated by a different receptor than GHS-R1a, which is not expressed in the hepatocytes. In humans, the acute administration of acyl ghrelin induced a rapid rise of glucose and insulin levels [66]. In contrast, des-acyl ghrelin prevented the acyl ghrelin-induced rise of insulin and glucose when coadministered [66]. Further studies examining the physiological interaction of des-acyl ghrelin and its targets will help to highlight the roles of ghrelin-related peptides in the regulation of feeding and energy homeostasis.
2.4 The Effect of H. pylori Infection on Ghrelin
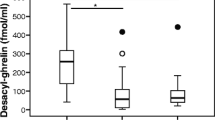
Since ghrelin is predominantly produced in the stomach, many researchers evaluated the effect of H. pylori infection on ghrelin expression in the gastric mucosa and blood. Theoretically, H. pylori-related gastritis may progress to atrophy with loss of oxyntic glands leading to a negative effect on ghrelin production in the stomach. Among the studies of this theme, Isomoto et al. [67] most well demonstrated the stepwise changes of ghrelin mRNA, peptide, and its blood level according to H. pylori infection status. The gastric ghrelin mRNA expression level of H. pylori-positive patients (1.64 ± 1.27 in arbitrary units) was significantly lower than in H. pylori-negative subjects (4.87 ± 4.1, P < 0.0001). A similar trend was noted for ghrelin peptide contents (31.2 ± 27.5 vs 81.2 ± 64.1 ng/mg protein, respectively, P < 0.0001). Although there was no significant difference in the number of ghrelin-immunoreactive cells/mm2 in terms of H. pylori status, plasma ghrelin concentrations in H. pylori-infected patients (144.6 ± 7.8.8 fmol/ml) were significantly lower than in uninfected subjects (196.1 ± 97.2, P < 0.05) and increased following the cure of the infection. In the study, plasma ghrelin levels correlated positively with the expression levels of ghrelin mRNA and peptide products. There was a significant stepwise decrease in gastric ghrelin mRNA expression, peptide contents, and density of ghrelin-immunoreactive cells with progression of histological severity of glandular atrophy in the corpus. However, there have been other different results and they will be discussed in the following Sects. (6.2.4.1, 6.2.4.2 and 6.2.5).
2.4.1 Ghrelin in Gastric Mucosa in Regard to H. pylori Infection
Comparisons of ghrelin levels in specimens from the stomach between H. pylori-infected and H. pylori-noninfected subjects have showed rather variable results (Table 6.2) [67–79]. First of all, the reports were heterogeneous in terms of diseases, specimen, and assay methods. Direct assays of ghrelin levels in gastric fluid samples [67, 69, 70, 73] and radioimmunoassay in gastric mucosa [67, 73] from infected and noninfected subjects were highly variable (Table 6.2). However, ghrelin mRNA expression looks like a more reliable method for assessment of gastric ghrelin. In most studies, mRNA expression was lower in H. pylori-positive subjects compared with H. pylori-negative subjects (Table 6.2) [67, 72, 74, 76]. Quantitative assessment of ghrelin-immunoreactive cells also showed relatively consistent results, fewer ghrelin-producing cells in H. pylori-positive subjects [67, 76, 78, 80, 81], indicating that the number of ghrelin-producing cells might decrease proportionally to the severity of H. pylori gastritis [67, 80, 81].
2.4.2 Ghrelin in Blood in Regard to H. pylori Infection
The circulating level of ghrelin is determined by the balance among its secretion rate, degradation rate, and clearance rate. While plasma esterases des-acylate acyl ghrelin, plasma proteases degrade circulating ghrelin. Therefore, the effect of H. pylori on circulating level of ghrelin seems like not simple. Consequently, studies which have compared blood ghrelin levels between H. pylori-infected and H. pylori-noninfected subjects showed inconsistent results (Table 6.3). Recently, Nweneka and Prentice [98] concluded that circulating ghrelin was significantly lower in H. pylori-positive subjects in a meta-analysis. Nevertheless, H. pylori-associated gastric ulcer was reported to be associated with high plasma ghrelin, but atrophic gastritis in H. pylori-positive subjects [99], or even in H. pylori-negative subjects [100], showed the lowest plasma ghrelin levels. These results suggest that H. pylori infection may have different effects on circulating levels of ghrelin according to different stage or disease of the infection.
Most researchers presume that total ghrelin levels were considered as a good surrogate marker not only for des-acyl but also acylated ghrelin. Majority of these studies in which only total ghrelin levels (not acylated ghrelin) were measured determined that H. pylori infection decreases plasma ghrelin levels. Few studies have evaluated acyl ghrelin for this theme, and the findings are again contradictory. In a Japanese data [96], the acylated ghrelin/total ghrelin ratio as well as plasma acyl ghrelin levels were reduced. However, Italian study [97] showed a significant increase in acyl ghrelin and the ratio of acylated ghrelin/total ghrelin for which the authors speculated a compensatory increase in the acylation process in response to a loss of total ghrelin secretion.
These controversial results might be also related with the uncertain optimum method of plasma ghrelin measurement. Several methods with different sensitivity such as radioimmunoassay or enzyme immunoassay/enzyme-linked immunosorbent assay have been used (Table 6.4) [101]. However, more basically, there are numerous factors in addition to atrophy that could affect plasma ghrelin concentration, including gastrin, IGF-1, obesity, insulin resistance, hyperinsulinemia, cholesterol, and urine excretion (Table 6.1), and H. pylori infection may not be the only major determinant that affects plasma ghrelin concentration.
Taken together, to conclude the issue of the formation of different types of ghrelin as well as the true effect of H. pylori infection on blood ghrelin levels, further well-designed research is needed.
2.5 The Effect of Eradication of H. pylori on Ghrelin Regarding FD Symptom
2.5.1 The Effect of Eradication of H. pylori on Ghrelin in the Gastric mRNA or Blood Samples
Comparisons on the gastric ghrelin parameters before and after eradication have not been much performed (Table 6.2). Three studies demonstrated that H. pylori eradication increases ghrelin mRNA (including one randomized controlled study in which a control group did not receive eradication) [68, 71, 75]. Osawa et al. [75] demonstrated median preproghrelin mRNA expression was increased nearly fourfold, 12 weeks after H. pylori eradication in the study with 134 subjects with successful eradication. Two Korean studies showed an increased ghrelin mRNA after cure of the H. pylori infection [68, 71]. The number of ghrelin-immunoreactive cells also increased in 50 H. pylori-eradicated patients although atrophy and intestinal metaplasia were not significantly changed [78]. However, Choe et al. [73] failed to demonstrate a change in ghrelin expression 4weeks after the eradication (Table 6.2).
The effect of H. pylori eradication on circulating level of ghrelin was more evaluated than on the gastric levels, since the systemic level of ghrelin is an attractive issue to other fields than gastroenterology. Unfortunately, the results were found to be still inconsistent (Table 6.5). At first, Nwokolo et al. [102] reported a rise in circulating plasma ghrelin levels following eradication of H. pylori in their study of 12 healthy subjects. Further studies reported elevated levels of blood ghrelin after the cure of H. pylori [71, 96, 103]. However, subsequent studies found no change [67, 68, 73, 82, 83, 89] or even decrease [75, 92] in circulating ghrelin levels. Nweneka and Prentice [98] performed a meta-analysis that showed that H. pylori eradication does not have an effect on circulating ghrelin levels. Nevertheless, pre-eradication elevation of ghrelin may be a predictor of a fall in plasma levels post-eradication [76, 98].
2.5.2 The Effect of Eradication of H. pylori on Ghrelin Regarding Functional Dyspepsia Symptom
Possible mechanisms by which H. pylori may elicit dyspeptic symptoms include alterations of gastric motility and endocrine and acid secretory abnormalities. Since ghrelin regulates acid secretion, appetite, and gastrointestinal motility, whether H. pylori infection or eradication could affect ghrelin levels might be closely related with the role of H. pylori in developing FD. Even though there have been some studies which evaluated the association between plasma ghrelin level and FD, there was no study which analyzed the effect of H. pylori infection status or its eradication on the change of both ghrelin levels and dyspeptic symptoms, so far (Table 6.6).
Two studies reported that fasting total ghrelin levels were significantly lower in patients with dysmotility-like FD than healthy volunteers [55, 104]. In addition, Shindo et al. [56] also showed significantly lower plasma acyl ghrelin levels in patients with PDS compared with healthy volunteers. On the contrary, Nishizawa et al. [105] reported that plasma acyl ghrelin levels were significantly higher in each dysmotility or ulcer-like dyspepsia group not in the nonspecific type compared with the control group, but H. pylori infection status was not considered in his study [105]. Two other studies with only women showed no difference between control and FD groups [106, 107]. It suggests the importance of classification of exact subgroup in the research regarding the relationship between ghrelin and FD. Acyl ghrelin may be closely associated with PDS than EPS.
In our study, plasma acyl ghrelin was lower in PDS than in control but not in EPS type [108]. However, plasma des-acyl ghrelin and gastric mRNA expression of preproghrelin did not show any significant difference [108]. Decreased expression of gastric preproghrelin mRNA in the subjects with atrophy was also shown. Importantly, plasma acyl ghrelin level and preproghrelin mRNA expression in the corpus were elevated 1 year after the eradication of H. pylori [108]. Moreover, the symptom improvement correlated with the upregulation of plasma acyl ghrelin but not with gastric ghrelin mRNA [108]. These results suggest that plasma acyl ghrelin could be one of the causal factors of dyspepsia, especially PDS, and anti-H. pylori therapy may be considered with a priority as a H. pylori-associated functional dyspepsia.
In spite of strong positive correlations among total, acyl, and des-acyl ghrelin, only a weak positive correlation between ghrelin mRNA and plasma acyl ghrelin was demonstrated in our study. As a result, some subjects with lower expression levels of ghrelin mRNA or reduced plasma total ghrelin did not show dyspeptic symptom. Moreover, at 1-year follow-up after H. pylori eradication, some subjects with increased ghrelin mRNA did not show upregulation of plasma acyl ghrelin in our study. This strongly suggests that there might not be a certain correlation between gastric mRNA and blood ghrelin (total or acyl) levels. Previously, Osawa et al. [75] also reported no correlation among the changes of plasma ghrelin or gastric preproghrelin mRNA or the number of ghrelin-positive cells after the H. pylori cure. Furthermore, after H. pylori cure plasma ghrelin concentration was more strongly influenced by body weight change than by the increase in gastric preproghrelin mRNA or the number of ghrelin-producing cells [75]. According to our study, the upregulation of plasma acyl ghrelin does not seem to be only epiphenomenon following the resolution of H. pylori infection, since there were some correlations between symptom improvement and the increase in plasma acyl ghrelin even in H. pylori-negative patients. Even though there was no significant difference in prevalence of H. pylori positivity between the control group and FD group, reduced plasma acyl ghrelin levels may attribute to dyspeptic symptoms in some subset of H. pylori-infected FD patients. Taken together, further studies are necessary regarding the dynamics of gastric ghrelin production and regulation of its circulating levels. In addition, it is very important to identify the true effect of H. pylori eradication on the gastric and circulating ghrelin levels as well as on the symptom changes in patients with FD.
3 Leptin
3.1 Production of Leptin
Leptin was discovered in 1994 as a hormone produced by adipose tissue with a modulatory effect on feeding behavior and weight control [4]. Leptin is a product of the obese (Ob) gene, which is located on chromosome 7 in humans and acts through its receptor Ob-R. Interestingly, the stomach has been identified as an important source of leptin [4], and leptin-producing cells were found to be localized in the lower half of the fundic glands, a site similar to that of the pepsinogen-secreting chief cells. The soluble isoform of its receptor (Ob-R) is secreted by chief cells in the gastric mucosa [109]. Moreover, intestines do express membrane-bound leptin receptors on their brush border [4, 109, 110] (Fig. 6.3). Collectively, gastric exocrine and endocrine secretions of leptin constitute a gastroenteric axis to coordinate its roles in the GI tract [110].

Schematic drawing that illustrates the secretion of leptin by the adipocyte and the gastric chief cell (Adapted from Cammisotto et al. [110]). Both types of cell secrete the leptin receptor. Both the leptin and the leptin receptor are synthesized in the rough endoplasmic reticulum, transferred to the Golgi apparatus, and packaged into either small vesicles (adipocytes) or secretory granules (gastric cells). At the level of the trans-cisternae of the Golgi and in the secretory granules, leptin binds its soluble receptor to form the leptin–leptin receptor complex. This complex is discharged by both cell types through an exocytotic event. The adipose tissue secretes toward the blood circulation, while the gastric cells secrete in an exocrine fashion into the gastric juice. Leptin in the gastric juice is vehiculated to the duodenal lumen. In the duodenum leptin–leptin receptor complex is internalized and separated. The leptin is channeled toward the trans-Golgi cisternae where it binds a newly synthesized soluble leptin receptor. The complex reaches the blood circulation. RER rough endoplasmic reticulum, CNS central nervous system, N nucleus
3.2 Regulation and Role of Gastric Leptin
3.2.1 Regulation of Gastric Leptin
Gastric leptin is sensitive to the nutritional status of the body. Fasting for 48 h induced a decrease in gastric leptin expression and content in rats [32]. Food intake quickly depletes gastric leptin, while sustained feeding stimulates the leptin gene expression and leptin synthesis [32, 111]. These observations are similar in rodents and humans [111].
Leptin mRNA expression in rat gastric mucosa is upregulated by sucrose-rich but not by fat-rich diets [112]. Fasted rats refed with a carbohydrate-rich diet have their gastric leptin synthesis increased [46]. Intravenous infusions of cholecystokinin (CCK), pentagastrin, or secretin trigger leptin release into the gastric juice [109]. Insulin also stimulates gastric leptin secretion, but this effect is dependent on the integrity of the vagal nerve [113]. Finally, leptin secretion seems to downregulate its own production in the stomach, as Zucker fa/fa rats with no functional leptin receptor are characterized by an upregulation of gastric leptin mRNA and gastric leptin content [114]. Interestingly, leptin infusion in rats leads to an increase in histamine gastric tissue content, which may suggest a counter-regulatory process [115].
3.2.2 Role of Gastric Leptin
Leptin derived from the stomach can be distinguished from adipocyte leptin through its rapid increased secretion following a meal and through its exocrine secretion (i.e., mainly in the gastric lumen). While the gastric mucosa secretes leptin within minutes after the beginning of food intake, it takes several hours for adipocytes to release significant amounts of leptin. Differences in time frame of secretion between adipose tissue and gastric mucosa may reflect different roles. Gastric leptin remains stable in gastric juice even at pH 2. Moss and Calam [116] also suggests that it could act locally in the stomach to affect gastric functions.
It has been considered that gastric leptin may exert paracrine effects within the gastric mucosa or stimulating vagal afferents to signal the central nervous system [109]. Gastric leptin controls food intake and satiety sensations by acting on the stomach itself. It potentiates the effect of CCK by slowing gastric emptying and promoting gastric distension [117], not directly affecting the CNS [118]. It also stimulates the production of glucagon-like peptides 1 and 2 (GLP1 and GLP2) that inhibit gastric emptying [119, 120].
Leptin receptor isoforms have been found in the rat nodose ganglion, which contains the cell bodies of vagal afferent neurons, and in the vagus nerve proper [121, 122]. Signals arising from the upper gastrointestinal tract are conveyed by the viscero-sensitive vagal afferent neurons to the nucleus of the solitary tract (NTS), then to the hypothalamus where they participate in the process of meal-induced termination of food intake [123] (Fig. 6.4). CCK secreted from duodenal endocrine I cells typically functions as one of these short-term satiety signals via activation of the CCK-1 receptor [124].

Model of the action of gastric leptin (Adapted from Guilmeau et al. [111]). Leptin (secreted by the stomach) and CCK can be considered as short-term gastrointestinal signals in the control of feeding. These signals locally activate their receptors on vagal terminals to generate signals that are processed in the NTS and the paraventricular nucleus (PVN) of the hypothalamus. The stomach-derived leptin secreted in the lumen enters the intestine in an active form. CCK-R cholecystokinin receptor, DMNX dorsomotor nucleus of the vagus nerve, Ob leptin, Ob-R leptin receptors
On the other hand, exocrine luminal leptin has been shown to act directly on intestinal cells through their specific receptors present on enterocyte microvilli. It regulates the transport of nutrients and enhances di- and tripeptide uptake by increasing the number of PepT1 transporters on microvilli [110].
In short, while adipose leptin acts on the long term mainly through its interactions with the central nervous system, gastric leptin acts locally directly on the gastric mucosa for regulating food intake. Gastric leptin influences both the intestinal track and the central nervous system (Fig. 6.4).
3.3 Leptin and H. pylori
Changes in gastric and serum leptin levels in H. pylori-infected patients have been the matter of several investigations. H. pylori infection significantly increased gastric leptin expression, and the cure of the infection significantly reduced this expression with a concomitant increase in BMI [125]. H. pylori-infected gastric mucosa was found to be capable of releasing larger amounts of leptin mRNA than that without H. pylori infection [4, 72]. Although the majority of studies have shown an increase of gastric leptin mRNA in H. pylori-infected subjects (Table 6.7), still there is a discrepancy [70, 74].
Leptin-secreting endocrine cells are present in the gastric mucosa, but they are few in number and scattered in the gastric mucosa close to blood capillaries [111]. These cells do not reach gastric lumen but secrete leptin to blood circulation. Since adipocytes are predominant source of leptin, whether H. pylori infection could affect circulating levels of leptin could be more complex (Table 6.8). It has been reported that rapid increase in the concentration of plasma leptin in response to CCK was involved in the mobilization of a gastric leptin store [126]. Similar to gastric leptin mRNA, higher serum leptin levels were reported in the subjects with H. pylori infection compared with H. pylori-negative subjects [70, 85, 127]. However, many studies reported no significant difference in plasma leptin according to H. pylori infection status [4, 72, 74, 84, 94]. Similarly, serum leptin level did not change significantly after curing H. pylori infection [102, 125].
3.4 Leptin in Comparison with Ghrelin in the GI Tract
Effects of ghrelin on appetite and observed changes in ghrelin levels in response to fasting or eating are opposite to those of leptin [128]. Ghrelin induces adiposity in adipose tissues, increases appetite, and initiates eating behavior [129]. Circadian changes in the level of circulating ghrelin and leptin are reciprocal [130]. Leptin and ghrelin also have contradictory effects on intestinal inflammation. For example, ghrelin has ameliorated inflammation in the GI tract in a mouse model of colitis [131]. Ghrelin’s effect on GI tract motility is also generally opposite to that of leptin. Ghrelin stimulates gastric acid secretion and motility and accelerates gastric emptying and small intestinal transit [132]. In contrast, leptin has decreased the expression and secretion of ghrelin from gastric mucosa [24]. Interestingly, leptin cells are adjacent to ghrelin cells in the gastric mucosa, surrounding ghrelin cells in the lower half of the stomach, possibly providing a paracrine regulation of ghrelin secretion [128]. Interestingly, ghrelin shows some effects on the gut that are similar to those of leptin. For example, ghrelin decreases the rate of apoptosis in the intestinal cells [133], ameliorates ischemic–reperfusion injury, and decreases the permeability of the intestine in case of shock [129]. Further studies are required to elucidate the nature of the interaction between ghrelin and leptin in health and disease, clearly.
4 Gastrin and Somatostatin Related with Acid Secretion
One of the important functions in gastric physiology is to regulate and sustain acid secretion to sterilize ingested nutrients in which several hormones are involved. There are three regulatory molecules that stimulate acid secretion (acetylcholine, histamine, and gastrin) and one regulatory molecule that inhibits acid secretion (somatostatin). Acetylcholine is a neurotransmitter that is released by enteric neurons, while histamine is a paracrine that is released from enterochromaffin-like (ECL) cells. Gastrin is a hormone that is released by G cells, and somatostatin (SST) is produced from D cells in the stomach (Fig. 6.5) [134]. Acetylcholine and histamine directly stimulate parietal cells to increase acid secretion. Gastrin stimulates acid secretion by stimulating histamine release from ECL cells. Gastrin also has a direct effect on parietal cells, which stimulates parietal cells’ proliferation. In oxyntic glands of the gastric body and fundus, SST-releasing cells are anatomically and functionally coupled to parietal and ECL cells. When the pH of the stomach gets too low, somatostatin secretion is stimulated. Somatostatin inhibits acid secretion by direct effects on parietal cells and also by inhibiting release of the positive regulators, histamine and gastrin. Inversely, endogenous gastrin release ceased when the pH of the perfusate dropped below 2.5. Gastrin release is suppressed primarily by direct contact of acid with the antrum [135]. Taken together, acid secretion and physiology of G and D cells are closely related to each other, and the effect of H. pylori on these cells needs to be understood in association with the acid secretory system. This part focuses on the two major acid regulatory hormones, gastrin and somatostatin, and acid secretion related with H. pylori infection.

Gastrin release is stimulated by extramural cholinergic and intramural cholinergic and non-cholinergic factors and inhibited by somatostatin (Adapted from Kaneko et al. [134]). Somatostatin release is stimulated by luminal (acid), paracrine, and hormonal factors (gastrin), and by intramural non-cholinergic factors, and is inhibited by extramural cholinergic factors. CCK cholecystokinin, PYY peptide YY, CGRP calcitonin gene-related peptide, VIP vasoactive intestinal peptide, GRP gastrin-releasing peptide, Ach acetylcholine
4.1 Gastrin and H. pylori Infection
Levi et al. [136] reported that both basal/stimulated acid secretion and basal/meal-stimulated plasma gastrin levels were significantly higher in H. pylori-positive duodenal ulcer (DU) patients (n = 25) compared with H. pylori-negative DU patients (n = 6), in which the phenomenon has been named as “the gastrin link.” Their data clearly demonstrated that H. pylori infection-induced hypergastrinemia was followed by an increase in acid secretion in DU patients. Eradication of H. pylori reduced the output of gastrin in the stomach [137].
4.2 Somatostatin and H. pylori Infection
SST is an originally discovered in sheep hypothalamus, which showed an inhibitory effect on growth hormone release. In mammals, the GI tract and pancreas contain the largest amounts of somatostatin. Most of the gastrointestinal SST immunoreactivity is confined to the mucosal layer [138], where epithelial endocrine cells, D cells, are localized.
In the antrum, the D cells have apical membranes that are exposed to the lumen (“open cells”). In the corpus, the D cells are of the “closed” type; they are not exposed to the luminal surface of the mucosa [139]. SST plays an important role in the regulation of gastric acid secretion by inhibiting gastrin release. In the antral mucosa, the open D cell releases SST in response to increased acidity in the gastric lumen. Because the apical surface of D cells opens onto the gastric lumen, changes in pH may be sensed directly through chemoreceptors on the apical membranes.
The effect of H. pylori infection on gastric levels of SST has been relatively well investigated compared with ghrelin and leptin. SST release cannot be exactly assessed by measuring plasma levels, because SST is released from many organs and destroyed locally [139]. Instead, SST levels in both gastric mucosa and juice reflect the local SST regulation [140]. Antral SST concentrations were decreased in H. pylori-infected patients, but not the corpus [141] which has been demonstrated by measuring the SST concentration in the biopsy specimens from both sites [142, 143]. Moss et al. [144] demonstrated that eradication of H. pylori from patients with duodenal ulcer caused an approximately twofold increase in SST mRNA in antral, but not in corpus biopsies. They also showed an increase in D cell numbers after H. pylori eradication in subjects with active duodenal ulcer [144].
4.3 Acid Secretion and H. pylori Infection
The effect of H. pylori on acid secretion depends on the stage of infection or predominant location where the infection occurs. That is, once H. pylori infection is established, transient hypochlorhydria occurs. Chronic infection in the antrum causes upregulation of gastrin and subsequent elevated acid secretion, while chronic infection in the corpus leads to impaired acid secretion by direct suppression of H+-K+-ATPase or involvement of cytokines such as IL-1β or TNF-α. Therefore, the direct inhibition of H+-K+-ATPase, indirect inhibition through cytokines, and loss of parietal cells by ongoing inflammation were three mechanisms for the low acid secretion.
The recent progress of H. pylori-dependent molecular mechanism of acid secretion is stated in the next sections.
4.3.1 Mechanism for Hypochlorhydria by Acute H. pylori Infection
In the acute phase of H. pylori infection, transient hypochlorhydria occurs [145], while low acidic status continues from several weeks to months. Low acid secretion by acute H. pylori infection was demonstrated in the absence of loss of parietal cells [146], impaired permeability of gastric mucosa [146], and glandular atrophy [147]. Moreover, IL-1β which is produced by neutrophils and inhibits acid secretion was not involved in this stage. Instead, this low acid secretion in the acute stage is likely to result from direct contact with parietal cells by H. pylori or its product [148, 149]. Studies about ultrastructure of stomach reported that H. pylori was observed adjacent to parietal cells and even detected in secretory canaliculi of these cells [150, 151]. In human, H. pylori infection also suppressed acid secretion via histamine, acetylcholine, and cAMP [152, 153], and this inhibition was resolved soon after the eradication of H. pylori [154]. This transient inhibition of acid secretion in the acute phase facilitates the successful settlement of H. pylori in the stomach.
4.3.2 The Effect of H. pylori Infection on H+-K+-ATPase
H+-K+-ATPase has α-subunit (HKα) and β-subunit. H. pylori-infected gastric mucosa or gastric epithelial cell lines showed the inhibition of HKα promoter activity in the endogenous or transfected H+-K+-ATPase [145]. Specifically, H. pylori inserts its protein to gastric cells through the type IV secretion system (T4SS), which look like cylindrical channel, and this inserted protein upregulated NF-κB. Interestingly, the site which NF-κB combined in the promoter of H+-K+-ATPase was identified, and this fusion of NF-κB p50 homodimer resulted in repression of the transcription of HKα [145].
CagA protein encoded by cag pathogenicity island (cag PAI); CagL, CagE, and Cag M, which consist of the T4SS; and lytic transglycosylase are mechanistically involved in NF-κB activation and repression of HKα transcription (Fig. 6.6). CagL, a T4SS pilus component, binds to the integrin α5 β 1 to mediate translocation of virulence factors into the host cell and initiate signaling. During acute H. pylori infection, CagL dissociates ADAM 17 (a disintegrin and a metalloprotease 17) from the integrin α5 β 1 complex and stimulates ADAM17-dependent release of heparin-binding epidermal growth factor (HB-EGF), EGF receptor (EGFR) stimulation, ERK1/2 kinase activation, and NF-κB-mediated repression of HKα [145].

Schematic illustration of a corporal gland in the human stomach focusing on the acid-secreting region (Adapted from Smolka et al. [145]). The location of various cell types in a gland of the human corpus was indicated in different colors. A gastric parietal cell is enlarged to the right, showing Helicobacter pylori interacting with integrins through CagL, injecting CagA and possibly the bacterial peptidoglycan-derived glycosylated tripeptide GM-3, leading to the activation of diverse host signaling pathways. The consequent mobilization of nuclear factor-κB (NF-κB) p50 homodimers to the nucleus results in the repression of gastric H, K-adenosine triphosphatase (H, K-ATPase) α subunit transcription and the inhibition of acid secretion as indicated ADAM 17, a disintegrin and a metalloprotease 17, EGFR EGF receptor, HB-EGF heparin-binding epidermal growth factor, T4SS type IV secretion system
4.3.3 The Effect of Acid Secretion of IL-1β and TNF-α
As noted above, the inflammation via multiple cytokines seems to be a key mechanism for the changes in the endocrine system by H. pylori infection. It has been explained that the destroyed acid homeostasis influences the distribution of H. pylori in the stomach itself and finally the degree of gastritis. That is, H. pylori infection could diminish the number or function of D cells in the antrum, while it elevates the gastrin secretion of G cells via IL-8, IL-1β, or TNF-α [155, 156]. This disorganized endocrine system dumps excessive amount of acid into duodenum leading to duodenal ulcer. On the other hand, upregulation of IL-1β and TNF-α by H. pylori infection strongly suppresses acid secretion [157], and at the same time, IL-1β reduces secretion of histamine from ECL cells [158]. This makes H. pylori thrive in the corpus and destroy parietal cells and finally aggravating impaired acid secretion. H. pylori infection, simultaneously, enhances the gastrin release. Consequently, the gastrin level in blood was further accelerated due to reduced SST secretion, since IL-1β and TNF-α inhibit SST release during the Th1 immune response [159]. This chronic low acidic milieu accompanying atrophy and elevated gastrin has been considered to be a good condition for developing gastric cancer.
4.3.4 The Changes of Acid Secretion After H. pylori Eradication
Progression to certain disease by H .pylori infection is known to be determined by the degree of acid secretion when the organism invades the stomach. In the case of subjects with high acid secretion, H. pylori escapes the corpus and settles in the antrum leading to antrum-dominant gastritis with excessive gastrin release. By contrast, when the organism comes into the subjects with low acid secretion, it migrates into the corpus with adequate acidity. This subsequently causes atrophy of parietal cells and aggravates hypochlorhydria. These diverse situations when the H. pylori infection occurs inevitably draw different effects on acid secretion after H. pylori eradication. Basically, the changes of acid secretion after H. pylori eradication depend on the degree of inflammation in the corpus. More specifically, in the setting of antrum-dominant gastritis with intact corporal glands in spite of the infection, the eradication reduces gastrin and subsequently acid secretion, as well. On the contrary, in the setting of severe corporal inflammation, the inhibition of parietal cells by H. pylori disappears, leading to the elevated acid secretion [159].
Several studies reported immediate increase in acid secretion after administration of anti-H. pylori agents [154, 159]. These results came from the termination of direct contact of parietal cells with H. pylori or its products [148, 149] and reduction of IL-1β or TNF-α which represses the acid secretion. Osawa et al. [160] reported that mRNA of H+-K+-ATPase increased 3 months after H. pylori eradication even in severe atrophy without changes in the number of parietal cells. The changes of genes encoding H2 receptors, muscarinic M3 receptor, and anion exchanger 2, which are involved in acid secretion export in the basal and apical sides of parietal cells, were not observed [160], while the increase in H+-K+-ATPase and reduction in the concentration of IL-1β were demonstrated. This result suggests that upregulation of H+-K+-ATPase and reduction in the concentration of IL-1β are attributable to the short-term increase in acid secretion after H. pylori eradication.
This increase of acid secretion has continued until 5 years after H. pylori eradication [161]. Nine of 23 subjects showed the restoration of normal acid secretion at 7-month follow-up after the eradication, and its steady and significant increase has been observed until 2 years after the cure [161]. However, more than 2 years after the eradication, an additional increase of acid secretory function was not found, and most subjects showed lower optimal levels of acid secretion compared with healthy control subjects [161]. This suggests numeric restoration of the parietal cells might have only minor effect on increasing acid secretion.
5 Mechanisms Underlying H. pylori-Induced Hormone Change in the Stomach
H. pylori may control the production of gastric hormones directly or indirectly. In this section, possible mechanisms of H. pylori-induced hormonal change will be discussed.
5.1 Urease and Ammonia
Levi et al. [136] originally proposed that the alkaline condition generated locally by H. pylori urease increased gastrin release. Measurements of the pH in the gastric mucus layer have shown that H. pylori infection causes a more alkaline milieu, although the difference is only 0.3–0.8 of a pH point [162]. Long-term exposure of the antral mucosa to elevated levels of ammonia in the gastric juice induced G-cell hyperfunction in rats [163]. An inverse correlation between gastric juice ammonia levels and antral SST concentrations was also observed in humans [141]. In addition, 4-week-long oral treatment with 0.01 % ammonia, which was clinically estimated as the concentration in the gastric juice in patients with H. pylori infection, decreased the release of SST and the number of D cells in the rat stomach [164]. However, acute infection of H. pylori can lead to different results in D-cell secretion. Previously, increasing intragastric urea did not elevate gastrin in infected persons [165]. Inhibition of urease by acetohydroxamic acid or bismuth plus antibiotics did not decrease gastrin release in a short-term experiment [166].
5.2 Lipopolysaccharide
After a 24-h incubation with H. pylori-derived culture broth, reduced ghrelin expression in gastric biopsies from H. pylori-negative subjects has been reported [167]. Piotrowski et al. [168] demonstrated that the binding of SST to its receptor on gastric mucosal cell membranes was inhibited by LPS from H. pylori, suggesting that H. pylori, through its LPS, is capable of interfering with SST regulatory effects on gastric mucosal G-cell function. It has been demonstrated that LPS suppressed fasted plasma ghrelin through production of IL-1 and prostaglandin and that exogenous ghrelin can normalize LPS-induced-altered digestive functions [169]. The expression and activity of GOAT under H. pylori infection, chronic gastritis, and gastric atrophy is an important issue. Two hours after LPS administration, significantly greater decrease of acyl ghrelin than des-acyl ghrelin has been reported [170]. The rapid decrease in plasma GOAT levels and slightly increased gastric GOAT protein levels at 2 h after the injection suggest that the inhibition of gastric GOAT release and an important role of circulating GOAT in the formation of acyl ghrelin during H. pylori infection are still to be investigated.
5.3 Somatostatin Receptors
SST acts through a family of homologous receptors, SST receptors (SSTRs). SSTR-2, which is the most extensively investigated of the five SSTRs, has been thought to be involved in gastric secretion. SSTR-2-positive cells were co-localized in 85 % of G cells and one-third of D cells [171]. The percentage of SSTR-2-labeling cells among D cells was significantly increased in rat antrum after 2–4-week treatments with 0.01 % ammonia solution. Ammonia may cause a decrease in the inhibitory potency of SST on G-cell function not only through a decrease in D-cell number but also through the additional inhibition of SST release, via an SSTR-2 in the residual D cells, in a paracrine manner, resulting in an increase in serum gastrin levels in rats [171]. Retallack et al. [172] speculated that a decrease in SSTR-2 mRNA on G cells may be responsible for the H. pylori infection-induced increase in gastrin secretion from the G-cell-rich fraction of H. pylori-infected human antral cell preparations.
5.4 Inflammatory Mediators
After the successful establishment of H. pylori infection, inflammatory responses were followed in the host mucosa. Since most of gastric hormone-producing cells are localized from base to neck of glands, where H. pylori rarely approaches, H. pylori may not directly affect release of these hormones, but it could influence the production of the peptide via inflammatory mediators.
It has been reported that a series of the cytokines are increased in H. pylori gastritis: interleukin (IL)-1, IL-6, and IL-8, TNF-α, interferon-γ, macrophage inflammatory protein-1α, and platelet-activating factor [173, 174]. Cytokines have a key role in the disruption of acid homeostasis through affecting G or D cells (Fig. 6.7a) [159]. T helper (Th)1 cytokines such as interferon (IFN)-γ [175, 176] and the pro-inflammatory cytokines IL-1β, TNF-α, and IL-8 [177, 178] stimulate gastrin secretion from cultured G cells. IL-8 increases gastrin secretion from isolated canine G cells [176, 177]. TNF-α directly affects G cells in dogs and humans to increase gastric secretion [179]. The secretion of somatostatin from D cells, which negatively regulates gastrin, is inhibited by the pro-inflammatory cytokines TNF-α [180] and IFN-γ [175] but stimulated by the Th2 cytokine IL-4 (Fig. 6.7a) [175]. The polarization of T-cell responses in the gastric mucosa thus impacts on physiological changes induced by H. pylori infection. In addition, the pro-inflammatory cytokines IL-1β and TNF-α are potent inhibitors of acid secretion by parietal cells [157], and IL-1β also decreases histamine release from ECL cells (Fig. 6.7b) [181].

Cytokine-mediated alterations in gastrin and somatostatin secretion (a) and enterochromaffin-like (ECL) and parietal (P) cell function (b) in H. pylori infection (Adapted from Peek et al. [159]). (a) IFN-γ from Th1 helper cells, IL-8, and TNF-α stimulate gastrin secretion from G cells. In contrast, IFN-γ inhibits somatostatin release from D cells, decreasing its inhibitory effect on gastrin. IL-4 stimulates somatostatin secretion, which may be a potential mechanism by which helminth coinfection, and stimulation of Th2 responses, can protect against the development of corpus atrophy. (b) IL-1β suppresses ECL cell histamine release, and IL-1β and TNF-α inhibit acid secretion from parietal cells PMN, polymorphonuclear cell
By contrast, eradication of H. pylori has been reported to induce a decrease in antral IL-8 levels and an improvement in histological inflammatory findings [182]. Antral SST concentrations were significantly increased after eradication therapy [183]. A negative correlation has been demonstrated between antral SST concentrations and IL-8 secretion in organ cultures of mucosal biopsies [184]. These findings suggest that certain mutual interactions between IL-8 and SST might be present in H. pylori infection in humans. In addition, a close correlation between an increase in gastric SST levels and the normalization of neutrophil infiltration indicated peptide inflammation interactions in H. pylori-induced gastritis [183].
Even though the relationship between these cytokines and gastric production of ghrelin or leptin has been far less evaluated, it is assumed that cytokines have been involved in the reduced production of gastric ghrelin and enhanced leptin synthesis. H. pylori-induced gastritis is thought to be associated with a strong activation of both Th1 and Th17 cells [167]. It is possible that factors released by H. pylori stimulate macrophages and dendritic cells which, in turn, produce factors driving Th1 and Th17 responses. The possible mechanism of H. pylori-related impairment of ghrelin synthesis in the stomach was illustrated in Fig. 6.8 [167].

Downregulation of ghrelin expression in the stomach during Helicobacter pylori infection (Adapted from Paoluzi et al. [167]). Epithelial damage and gastritis induced by Helicobacter pylori determine a diminished expression of ghrelin which, in turn, sustains the ongoing T helper (Th) 1 cells’ response. Downregulation of ghrelin is also followed by a reduced release of prostaglandin E2 (PGE2) and interleukin (IL)-10 which, together with pro-inflammatory factors as IL-1b, contribute to the detrimental immune response and damage in the stomach APC antigen-presenting cell
It has also been reported that multiple cytokines and inflammation raise circulating leptin levels. In experimental animals, blood leptin levels are acutely increased by inflammatory stimuli, such as endotoxin, LPS, and turpentine, and by the administration of pro-inflammatory cytokines such as TNF-α and IL-1 [185, 186]. In rats, elevated leptin levels in blood are present during infection with the nematode Nippostrongylus brasiliensis and in the course of intestinal inflammation [187, 188]. Leptin itself can induce a shift of T cells in ob/ob mice to a predominantly Th1 response by increasing interferon-γ and IL-2 and decreasing IL-4 cytokine production in vivo [189]. In colonic mucosa of patients with ulcerative colitis, elevated levels of leptin and leptin receptors were reported [190]. Against this background, it is likely that H. pylori infection could raise gastric leptin production. Since most of the present evidences reported relationships between inflammation and only systemic circulating leptin levels, whether gastric leptin has a similar pro-inflammatory effect as circulating leptin or this condition is involved in other GI disorder needs further evaluation.
5.5 Mucosal Atrophy and Bacterial Factors of H. pylori
H. pylori is a predominant etiologic factor of gastric inflammation and atrophy of gastric glands. It is assumed that atrophic gastritis may cause the loss of ghrelin-producing cells. This hypothesis has been supported by studies showing lower levels of gastric [75, 76] and circulating ghrelin [96] in patients with atrophic gastritis. It has been also reported that serum ghrelin level represents the most sensitive and specific noninvasive marker for selecting patients at high risk for atrophic body gastritis [191]. Probably, the extent of gastric atrophy and the duration of the infection may play a key role in modulation of ghrelin levels by H. pylori [80]. The atrophic changes especially in oxyntic glands were thought to be a resulting state from an array of inflammatory processes, and this leads to a negative effect of gastric hormone production. Inversely, it seems that recovery of ghrelin-producing capacity after H. pylori eradication might depend on the recovery from numeric or functional damage of the oxyntic glands.
Moreover, it is not surprising that certain strains may produce greater changes in endocrine function as these strains are more likely to cause clinical diseases. A major factor in highly virulent H. pylori isolates is the cag pathogenicity island (PAI), a 40-kb DNA segment that encodes about 32 proteins. Some of them including accessory Cag proteins are forming components of a type IV secretion system (T4SS) [145]. For example, CagL, a structural component of the T4SS pilus in pathogenic H. pylori, is essential to pathogenesis because its deletion abolishes almost completely H. pylori’s ability to induce host cell secretion of the pro-inflammatory cytokine IL-8 [192].
Among patients who were infected with cytotoxin-associated gene A (cagA)-positive strain have higher plasma gastrin concentrations than those who with cagA-negative strain [193]. Those with the s1/m1 variant of the vacuolating cytotoxin (vacA) gene, which is more likely to cause ulcers, have less antral SST peptide than those with the less virulent type s2/m2 [194]. Kim et al. [195] demonstrated that hypergastrinemia, with a decrease in the number of antral D cells in H. pylori-associated gastritis, is relevant to the presence of CagA. These findings seem to support the concept that D-cell deficiency may be relevant in toxigenic H. pylori-associated chronic active gastritis [196].
Similarly, the effect of H. pylori on ghrelin production was different according to H. pylori virulence. Patients with type I strain H. pylori, expressing CagA and VacA, have lower circulating ghrelin levels than those with the less virulent type II strain [77].
5.6 Neuron
It has been recently reported that acute administration of H. pylori is capable of inhibiting acid secretion directly as well as indirectly by activating intramural CGRP sensory neurons coupled to stimulation of SST and inhibition of histamine secretion [197]. The reciprocal changes in SST and histamine secretion were due to release of CGRP from sensory neurons.
Since H. pylori is present in the upper regions of gastric mucosa, whereas SST and ECL cells are located from base to the neck of glands, it is speculated that activation of CGRP sensory neurons may be one of the explanations to how initial patchy superficial colonization of the stomach can induce acute hypochlorhydria.
5.7 Secondary Endocrine Changes to Gastric Acid
H. pylori-infected patients with hyposecretion of acid tend to have corpus gastritis that is believed to be related to reduction in acid secretion brought about by a specific H. pylori product or by inflammatory cytokines, including IL-1β and TNF-α, which inhibit parietal cells [157]. IL-1β also inhibits ECL cells [181]. Finally, H. pylori infection accelerates the development of corpus atrophy, which further diminishes acid secretion through the loss of parietal cells [198]. A recent study clearly demonstrated that H. pylori (cagA-positive) infection induced a decrease in acid secretion and an increase in serum gastrin, with these phenomena returning to control levels after treatment with an IL-1 receptor antagonist in Mongolian gerbils [147]. Because gastrin is a physiological stimulant of acid secretion, a decrease in intragastric acidity induced by H. pylori-related corpus gastritis with atrophy might precede the gastrin release.
Interestingly, the acid-stimulating effect of ghrelin has been postulated by gastrin release [199] or vagal stimulation [200]. Indeed, Lee et al. [199] reported that gastrin was released in response to ghrelin. Whether a negative feedback to ghrelin production by elevated acid secretion might exist has not been evaluated. The ghrelin-producing cells, ECL cells, and D cells interspersed with each other in the oxyntic mucosa [201] suggests that there may be a functional link between them.
Finally, the possible mechanisms discussed in Sect. 6.4 as an underling H. pylori-induced gastric gastrin and somatostatin change are summarized in Fig. 6.9 [134].

Mechanisms speculated that are to underlie Helicobacter pylori-induced gut peptide change (Adapted from Kaneko et al. [134]). PMN polymorphonuclear leukocyte, IL interleukin, LPS lipopolysaccharide, TNF-α tumor necrosis factor-α, SSTR-2 somatostatin receptor subtype 2
6 Possible Reasons for Discrepancy in the Effect of H. pylori on the Levels of Gut Hormones
The change of physiology of G and D cells during H. pylori infection and after its eradication has been relatively well evaluated, while those of ghrelin and leptin are still under debate.
With regard to circulating ghrelin, the most often raised hypothesis for the inconsistent results is the compensatory release from other organs. However, since ghrelin is predominantly secreted from the stomach, its products derived from other tissues are unlikely to be sufficient to compensate for the altered plasma ghrelin dynamics. Another explanation may be due to differences in populations (age, race, geography, gender, BMI, diet, and overall health), extent of disease (e.g., whether atrophy is present or not), and H. pylori strain. This could partially result from the use of different immunoassay methods to measure ghrelin. The fact that ghrelin degrades to different kinds of ghrelin could be the fundamental cause on this discrepant results [101]. In the case of gastric ghrelin, different sites of gastritis or atrophy might make H. pylori infection draw different effects. As the same story, a different degree of recovery from damage or diverse duration after the completion of H. pylori eradication, probably, have different influences on ghrelin levels after cure of the infection. It has been reported that H. pylori infection alters gastric and blood level of ghrelin separately at different time points in the Mongolian gerbil model [202]. To confirm these hypotheses, serial measurements of blood ghrelin for long time after the eradication are needed.
On the other hand, gastric leptin levels according to H. pylori infection have been far less investigated compared with ghrelin. Although the exact mechanism is not clear, most studies have reported an increase in gastric leptin levels when subjects were infected by H. pylori. Nonetheless, as the primary contributor of circulating leptin is exclusively the adipose tissue, plasma leptin levels had strongly positive correlations with BMI, irrespective of H. pylori status.
7 Conclusion
Gut hormones such as ghrelin, leptin, gastrin, and SST could be regulated by neuronal, hormonal, and immune process under H. pylori infection. Understanding the changes of acid secretion related with H. pylori infection could give basic insights into identifying why the same H. pylori causes different outcomes among gastric ulcer, duodenal ulcer, gastric cancer, or asymptomatic histologic gastritis. In particular, ghrelin and leptin are involved not only in gastrointestinal physiology but in systemic energy regulation including adiposity, appetite, or circulation. Investigating these hormonal dynamics altered by H. pylori infection could provide an important clinical implication in the prevention and treatment of illnesses including obesity, functional dyspepsia, and GI cancer and give proper evidences of eradicating this microorganism.
References
Jeffery PL, McGuckin MA, Linden SK. Endocrine impact of Helicobacter pylori: focus on ghrelin and ghrelin o-acyltransferase. World J Gastroenterol. 2011;17:1249–60.
Ban S. In: Shepherd NA, Warren BF, Williams GT, Greenson JK, Lauwers GY, Novelli MR, editors. Stomach: Morson and Dawson’s gastrointestinal pathology. 5th ed. Hoboken: Wiley-Blackwell; 2013. p. 89–103.
Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402:656–60.
Breidert M, Miehlke S, Glasow A, Orban Z, Stolte M, Ehninger G, et al. Leptin and its receptor in normal human gastric mucosa and in Helicobacter pylori-associated gastritis. Scand J Gastroenterol. 1999;34:954–61.
Ariyasu H, Takaya K, Tagami T, Ogawa Y, Hosoda K, Akamizu T, et al. Stomach is a major source of circulating ghrelin, and feeding state determines plasma ghrelin-like immunoreactivity levels in humans. J Clin Endocrinol Metab. 2001;86:4753–8.
Date Y, Kojima M, Hosoda H, Sawaguchi A, Mondal MS, Suganuma T, et al. Ghrelin, a novel growth hormone-releasing acylated peptide, is synthesized in a distinct endocrine cell type in the gastrointestinal tracts of rats and humans 1. Endocrinology. 2000;141:4255–61.
Kamegai J, Tamura H, Shimizu T, Ishii S, Sugihara H, Wakabayashi I. Central effect of ghrelin, an endogenous growth hormone secretagogue, on hypothalamic peptide gene expression. Endocrinology. 2000;141:4797–800.
Korbonits M, Bustin SA, Kojima M, Jordan S, Adams EF, Lowe DG, et al. The expression of the growth hormone secretagogue receptor ligand ghrelin in normal and abnormal human pituitary and other neuroendocrine tumors 1. J Clin Endocrinol Metab. 2001;86:881–7.
Papotti M, Ghè C, Cassoni P, Catapano F, Deghenghi R, Ghigo E, et al. Growth hormone secretagogue binding sites in peripheral human tissues 1. J Clin Endocrinol Metab. 2000;85:3803–7.
Hosoda H, Kojima M, Matsuo H, Kangawa K. Ghrelin and des-acyl ghrelin: two major forms of rat ghrelin peptide in gastrointestinal tissue. Biochem Biophys Res Commun. 2000;279:909–13.
Nishi Y, Yoh J, Hiejima H, Kojima M. Structures and molecular forms of the ghrelin-family peptides. Peptides. 2011;32:2175–82.
Gutierrez JA, Solenberg PJ, Perkins DR, Willency JA, Knierman MD, Jin Z, et al. Ghrelin octanoylation mediated by an orphan lipid transferase. Proc Natl Acad Sci. 2008;105:6320–5.
Kang K, Schmahl J, Lee J-M, Garcia K, Patil K, Chen A, et al. Mouse ghrelin-O-acyltransferase (GOAT) plays a critical role in bile acid reabsorption. FASEB J. 2012;26:259–71.
Stengel A, Keire D, Goebel M, Evilevitch L, Wiggins B, Taché Y, et al. The RAPID method for blood processing yields new insight in plasma concentrations and molecular forms of circulating gut peptides. Endocrinology. 2009;150:5113–8.
Yin X, Li Y, Xu G, An W, Zhang W. Ghrelin fluctuation, what determines its production? Acta Biochim Biophys Sin. 2009;41:188–97.
Yang J, Zhao T-J, Goldstein JL, Brown MS. Inhibition of ghrelin O-acyltransferase (GOAT) by octanoylated pentapeptides. Proc Natl Acad Sci. 2008;105(31):10750–5.
Nishi Y, Hiejima H, Mifune H, Sato T, Kangawa K, Kojima M. Developmental changes in the pattern of ghrelin’s acyl modification and the levels of acyl-modified ghrelins in murine stomach. Endocrinology. 2005;146:2709–15.
Baldelli R, Bellone S, Castellino N, Petri A, Rapa A, Vivenza D, et al. Oral glucose load inhibits circulating ghrelin levels to the same extent in normal and obese children. Clin Endocrinol. 2006;64:255–9.
Guo Z-F, Ren A-J, Zheng X, Qin Y-W, Cheng F, Zhang J, et al. Different responses of circulating ghrelin, obestatin levels to fasting, re-feeding and different food compositions, and their local expressions in rats. Peptides. 2008;29:1247–54.
Toshinai K, Mondal MS, Nakazato M, Date Y, Murakami N, Kojima M, et al. Upregulation of ghrelin expression in the stomach upon fasting, insulin-induced hypoglycemia, and leptin administration. Biochem Biophy Res Commun. 2001;281:1220–5.
Kim SW, Kim KW, Shin CS, Park dJ, Park KS, Cho BY. Acylated ghrelin secretion is acutely suppressed by oral glucose load or insulin-induced hypoglycemia independently of basal growth hormone secretion in humans. Horm Res. 2006;67:211–9.
Overduin J, Frayo RS, Grill HJ, Kaplan JM, Cummings DE. Role of the duodenum and macronutrient type in ghrelin regulation. Endocrinology. 2005;146:845–50.
Knerr I, Gröschl M, Rascher W, Rauh M. Endocrine effects of food intake: insulin, ghrelin, and leptin responses to a single bolus of essential amino acids in humans. Ann Nutr Metab. 2002;47:312–8.
Kamegai J, Tamura H, Shimizu T, Ishii S, Sugihara H, Oikawa S. Effects of insulin, leptin, and glucagon on ghrelin secretion from isolated perfused rat stomach. Regul Pept. 2004;119:77–81.
Flanagan DE, Evans ML, Monsod TP, Rife F, Heptulla RA, Tamborlane WV, et al. The influence of insulin on circulating ghrelin. Am J Physiol Endocrinol Metab. 2003;284:E313–E6.
Katayama T, Shimamoto S, Oda H, Nakahara K, Kangawa K, Murakami N. Glucagon receptor expression and glucagon stimulation of ghrelin secretion in rat stomach. Biochem Biophy Res Commun. 2007;357:865–70.
Seoane L, Al-Massadi O, Barreiro F, Dieguez C, Casanueva F. Growth hormone and somatostatin directly inhibit gastric ghrelin secretion. An in vitro organ culture system. J Endocrinol Investig. 2007;30:RC22–RC5.
Grinspoon S, Miller KK, Herzog DB, Grieco KA, Klibanski A. Effects of estrogen and recombinant human insulin-like growth factor-I on ghrelin secretion in severe undernutrition. J Clin Endocrinol Metab. 2004;89:3988–93.
Shimada M, Date Y, Mondal MS, Toshinai K, Shimbara T, Fukunaga K, et al. Somatostatin suppresses ghrelin secretion from the rat stomach. Biochem Biophys Res Commun. 2003;302:520–5.
Arosio M, Ronchi CL, Gebbia C, Cappiello V, Beck-Peccoz P, Peracchi M. Stimulatory effects of ghrelin on circulating somatostatin and pancreatic polypeptide levels. J Clin Endocrinol Metab. 2003;88:701–4.
Tschöp M, Weyer C, Tataranni PA, Devanarayan V, Ravussin E, Heiman ML. Circulating ghrelin levels are decreased in human obesity. Diabetes. 2001;50:707–9.
Zhao Z, Sakata I, Okubo Y, Koike K, Kangawa K, Sakai T. Gastric leptin, but not estrogen and somatostatin, contributes to the elevation of ghrelin mRNA expression level in fasted rats. J Endocrinol. 2008;196:529–38.
Dube MG, Beretta E, Dhillon H, Ueno N, Kalra PS, Kalra SP. Central leptin gene therapy blocks high-fat diet-induced weight gain, hyperleptinemia, and hyperinsulinemia increase in serum ghrelin levels. Diabetes. 2002;51:1729–36.
Sakata I, Tanaka T, Yamazaki M, Tanizaki T, Zheng Z, Sakai T. Gastric estrogen directly induces ghrelin expression and production in the rat stomach. J Endocrinol. 2006;190:749–57.
Broglio F, Gottero C, Van Koetsveld P, Prodam F, Destefanis S, Benso A, et al. Acetylcholine regulates ghrelin secretion in humans. J Clin Endocrinol Metab. 2004;89:2429–33.
Di Francesco V, Fantin F, Residori L, Bissoli L, Micciolo R, Zivelonghi A, et al. Effect of age on the dynamics of acylated ghrelin in fasting conditions and in response to a meal. J Am Geriatr Soc. 2008;56:1369–70.
Kozakowski J, Rabijewski M, Zgliczyński W. Ghrelin-growth hormone releasing and orexigenic hormone in men declines with age, insulin and with decrease in testosterone concentration. Neuro Endocrinol Lett. 2008;29:100–6.
Schutte AE, Huisman HW, Schutte R, van Rooyen JM, Malan L, Malan NT. Aging influences the level and functions of fasting plasma ghrelin levels: the POWIRS-Study. Regul Pept. 2007;139:65–71.
Barkan AL, Dimaraki EV, Jessup SK, Symons KV, Ermolenko M, Jaffe CA. Ghrelin secretion in humans is sexually dimorphic, suppressed by somatostatin, and not affected by the ambient growth hormone levels. J Clin Endocrinol Metab. 2003;88:2180–4.
Horvath TL, Diano S, Sotonyi P, Heiman M, Tschop M. Minireview: ghrelin and the regulation of energy balance – a hypothalamic perspective. Endocrinology. 2001;142:4163–9.
Perboni S, Inui A. Appetite and gastrointestinal motility: role of ghrelin-family peptides. Clin Nutr. 2010;29:227–34.
Patterson M. Ghrelin enhances gastric emptying in diabetic gastroparesis: a double blind, placebo controlled, crossover study. Gut. 2005;54:1693–8.
Fujimiya M, Asakawa A, Ataka K, Kato I, Inui A. Different effects of ghrelin, des-acyl ghrelin and obestatin on gastroduodenal motility in conscious rats. World J Gastroenterol. 2008;14:6318–26.
Fujimiya M, Asakawa A, Ataka K, Chen C-Y, Kato I, Inui A. Ghrelin, des-acyl ghrelin, and obestatin: regulatory roles on the gastrointestinal motility. Int J Pept. 2010. doi:10.1155/2010/305192.
Akamizu T, Takaya K, Irako T, Hosoda H, Teramukai S, Matsuyama A, et al. Pharmacokinetics, safety, and endocrine and appetite effects of ghrelin administration in young healthy subjects. Eur J Endocrinol. 2004;150:447–55.
Sánchez J, Oliver P, Palou A, Picó C. The inhibition of gastric ghrelin production by food intake in rats is dependent on the type of macronutrient. Endocrinology. 2004;145:5049–55.
Tschöp M, Smiley DL, Heiman ML. Ghrelin induces adiposity in rodents. Nature. 2000;407:908–13.
Nakazato M, Murakami N, Date Y, Kojima M, Matsuo H, Kangawa K, et al. A role for ghrelin in the central regulation of feeding. Nature. 2001;409:194–8.
Wren A, Seal L, Cohen M, Brynes A, Frost G, Murphy K, et al. Ghrelin enhances appetite and increases food intake in humans. J Clin Endocrinol Metab. 2001;86:5992–5.
Inui A, Asakawa A, Bowers CY, Mantovani G, Laviano A, Meguid MM, et al. Ghrelin, appetite, and gastric motility: the emerging role of the stomach as an endocrine organ. FASEB J. 2004;18:439–56.
Tack J, Depoortere I, Bisschops R, Verbeke K, Janssens J, Peeters T. Influence of ghrelin on gastric emptying and meal‐related symptoms in idiopathic gastroparesis. Aliment Pharmacol Ther. 2005;22:847–53.
Peeters T. Central and peripheral mechanisms by which ghrelin regulates gut motility. J Physol Pharmacol. 2003;54:95–103.
Hosoda H, Kojima M, Kangawa K. Ghrelin and the regulation of food intake and energy balance. Mol Interv. 2002;2:494–503.
Fujino K, Inui A, Asakawa A, Kihara N, Fujimura M, Fujimiya M. Ghrelin induces fasted motor activity of the gastrointestinal tract in conscious fed rats. J Physiol. 2003;550:227–40.
Lee KJ, Cha DY, Cheon SJ, Yeo M, Cho SW. Plasma ghrelin levels and their relationship with gastric emptying in patients with dysmotility-like functional dyspepsia. Digestion. 2009;80:58–63.
Shindo T, Futagami S, Hiratsuka T, Horie A, Hamamoto T, Ueki N, et al. Comparison of gastric emptying and plasma ghrelin levels in patients with functional dyspepsia and non-erosive reflux disease. Digestion. 2009;79:65–72.
El-Salhy M, Lillebø E, Reinemo A, Salmelid L. Ghrelin in patients with irritable bowel syndrome. Int J Mol Med. 2009;23:703–7.
Karmiris K, Koutroubakis IE, Xidakis C, Polychronaki M, Voudouri T, Kouroumalis EA. Circulating levels of leptin, adiponectin, resistin, and ghrelin in inflammatory bowel disease. Inflamm Bowel Dis. 2006;12:100–5.
Ates Y, Degertekin B, Erdil A, Yaman H, Dagalp K. Serum ghrelin levels in inflammatory bowel disease with relation to disease activity and nutritional status. Dig Dis Sci. 2008;53:2215–21.
Mottershead M, Karteris E, Barclay JY, Suortamo S, Newbold M, Randeva H, et al. Immunohistochemical and quantitative mRNA assessment of ghrelin expression in gastric and oesophageal adenocarcinoma. J Clin Pathol. 2007;60:405–9.
An JY, Choi M-G, Noh JH, Sohn TS, Jin D-K, Kim S. Clinical significance of ghrelin concentration of plasma and tumor tissue in patients with gastric cancer. J Surg Res. 2007;143:344–9.
Waseem T, Ahmad F, Azam M, Qureshi MA. Role of ghrelin axis in colorectal cancer: a novel association. Peptides. 2008;29:1369–76.
Broglio F, Gottero C, Prodam F, Gauna C, Muccioli G, Papotti M, et al. Non-acylated ghrelin counteracts the metabolic but not the neuroendocrine response to acylated ghrelin in humans. J Clin Endocrinol Metab. 2004;89:3062–5.
Chen C-Y, Chao Y, Chang F-Y, Chien EJ, Lee S-D, Doong M-L. Intracisternal des-acyl ghrelin inhibits food intake and non-nutrient gastric emptying in conscious rats. Int J Mol Med. 2005;16:695–9.
Toshinai K, Yamaguchi H, Sun Y, Smith RG, Yamanaka A, Sakurai T, et al. Des-acyl ghrelin induces food intake by a mechanism independent of the growth hormone secretagogue receptor. Endocrinology. 2006;147:2306–14.
Lucidi P, Murdolo G, Di Loreto C, Parlanti N, De Cicco A, Fatone C, et al. Metabolic and endocrine effects of physiological increments in plasma ghrelin concentrations. Nutr Metab Cardiovasc Dis. 2005;15:410–7.
Isomoto H, Ueno H, Saenko VA, Mondal MS, Nishi Y, Kawano N, et al. Impact of Helicobacter pylori infection on gastric and plasma ghrelin dynamics in humans. Am J Gastroenterol. 2005;100:1711–20.
Lee ES, Yoon YS, Park C-Y, Kim H-S, Um TH, Baik HW, et al. Eradication of Helicobacter pylori increases ghrelin mRNA expression in the gastric mucosa. J Kor Med Sci. 2010;25:265–71.
Stec-Michalska K, Malicki S, Michalski B, Peczek L, Wisniewska-Jarosinska M, Nawrot B. Gastric ghrelin in relation to gender, stomach topography and Helicobacter pylori in dyspeptic patients. World J Gastroenterol. 2009;15:5409–17.
Roper J, Francois F, Shue PL, Mourad MS, Pei Z, Olivares de Perez AZ, et al. Leptin and ghrelin in relation to Helicobacter pylori status in adult males. J Clin Endocrinol Metab. 2008;93:2350–7.
Jang EJ, Park SW, Park JS, Park SJ, Hahm KB, Paik SY. The influence of the eradication of Helicobacter pylori on gastric ghrelin, appetite, and body mass index in patients with peptic ulcer disease. J Gastroenterol Hepatol. 2008;23(Supple2):S278–S85.
Jun DW, Lee OY, Lee YY, Choi HS, Kim TH, Yoon BC. Correlation between gastrointestinal symptoms and gastric leptin and ghrelin expression in patients with gastritis. Dig Dis Sci. 2007;52:2866–72.
Choe YH, Lee JH, Lee HJ, Paik KH, Jin DK, Song SY, et al. Ghrelin levels in gastric mucosa before and after Eradication of Helicobacter pylori. Gut Liver. 2007;1:132–7.
Salles N, Ménard A, Georges A, Salzmann M, De Ledinghen V, De Mascarel A, et al. Effects of Helicobacter pylori infection on gut appetite peptide (leptin, ghrelin) expression in elderly inpatients. J Gerontol A Biol Sci Med Sci. 2006;61:1144–50.
Osawa H, Kita H, Ohnishi H, Nakazato M, Date Y, Bowlus CL, et al. Changes in plasma ghrelin levels, gastric ghrelin production, and body weight after Helicobacter pylori cure. J Gastroenterol. 2006;41:954–61.
Osawa H, Nakazato M, Date Y, Kita H, Ohnishi H, Ueno H, et al. Impaired production of gastric ghrelin in chronic gastritis associated with Helicobacter pylori. J Clin Endocrinol Metab. 2005;90:10–6.
Isomoto H, Nishi Y, Ohnita K, Mizuta Y, Kohno S, Ueno H, et al. The relationship between plasma and gastric ghrelin levels and strain diversity in Helicobacter pylori virulence. Am J Gastroenterol. 2005;100:1425–7.
Tatsuguchi A, Miyake K, Gudis K, Futagami S, Tsukui T, Wada K, et al. Effect of Helicobacter pylori infection on ghrelin expression in human gastric mucosa. Am J Gastroenterol. 2004;99:2121–7.
Gokcel A, Gumurdulu Y, Kayaselcuk F, Serin E, Ozer B, Ozsahin AK, et al. Helicobacter pylori has no effect on plasma ghrelin levels. Eur J Endocrinol. 2003;148:423–6.
Liew P-L, Lee W-J, Lee Y-C, Chen W-Y. Gastric ghrelin expression associated with Helicobacter pylori infection and chronic gastritis in obese patients. Obes Surg. 2006;16:612–9.
Méndez-Sánchez N, Pichardo-Bahena R, Vásquez-Fernández F, Lezama-Mora JI, León-Canales AL, Barredo-Prieto B, et al. Effect of Helicobacter pylori infection on gastric ghrelin expression and body weight. Rev Gastroenterol Mex. 2007;72:359–64.
Isomoto H, Nakazato M, Ueno H, Date Y, Nishi Y, Mukae H, et al. Low plasma ghrelin levels in patients with Helicobacter pylori–associated gastritis. Am J Med. 2004;117:429–32.
Isomoto H, Nishi Y, Kohno S, Wen C-Y, Ueno H, Nakazato M. Impact of Helicobacter pylori infection on ghrelin and various neuroendocrine hormones in plasma. World J Gastroenterol. 2005;52:53.4–14.3.
Shiotani A, Miyanishi T, Uedo N, Iishi H. Helicobacter pylori infection is associated with reduced circulating ghrelin levels independent of body mass index. Helicobacter. 2005;10:373–8.
Konturek P, Czesnikiewicz-Guzik M, Bielanski W, Konturek S. Involvement of Helicobacter pylori infection in neuro-hormonal control of food intake. J Physiol Pharmacol. 2006;57:67–81.
Plonka M, Konturek P, Bielanski W, Pawlik T, Brzozowski T, Konturek S. Relationship between ghrelin and Helicobacter pylori infection in Polish adult shepherds and their children. Aliment Pharmacol Ther Symp Ser. 2006;2:160–8.
Plonka M, Bielanski W, Konturek S, Targosz A, Sliwowski Z, Dobrzanska M, et al. Helicobacter pylori infection and serum gastrin, ghrelin and leptin in children of Polish shepherds. Dig Liver Dis. 2006;38:91–7.
Alonso N, Granada ML, Salinas I, Reverter JL, Flores L, Ojanguren I, et al. Plasma ghrelin concentrations in type 1 diabetic patients with autoimmune atrophic gastritis. Eur J Endocrinol. 2007;157:763–9.
Cindoruk M, Yetkin I, Deger SM, Karakan T, Kan E, Unal S. Influence of H. pylori on plasma ghrelin in patients without atrophic gastritis. World J Gastroenterol. 2007;13:1595–8.
D’Onghia V, Leoncini R, Carli R, Santoro A, Giglioni S, Sorbellini F, et al. Circulating gastrin and ghrelin levels in patients with colorectal cancer: correlation with tumour stage, Helicobacter pylori infection and BMI. Biomed Pharmacother. 2007;61:137–41.
de Martel C, Haggerty TD, Corley DA, Vogelman JH, Orentreich N, Parsonnet J. Serum ghrelin levels and risk of subsequent adenocarcinoma of the esophagus. Am J Gastroenterol. 2007;102:1166–72.
Pacifico L, Anania C, Osborn JF, Ferrara E, Schiavo E, Bonamico M, et al. Long-term effects of Helicobacter pylori eradication on circulating ghrelin and leptin concentrations and body composition in prepubertal children. Eur J Endocrinol. 2008;158:323–32.
Shak JR, Roper J, Perez-Perez GI, Tseng C-h, Francois F, Gamagaris Z, et al. The effect of laparoscopic gastric banding surgery on plasma levels of appetite-control, insulinotropic, and digestive hormones. Obes Surg. 2008;18:1089–96.
Chuang CH, Sheu BS, Yang HB, Lee SC, Kao AW, Cheng HC, et al. Gender difference of circulating ghrelin and leptin concentrations in chronic Helicobacter pylori infection. Helicobacter. 2009;14:54–60.
Gao X-Y, Kuang H-Y, Liu X-M, Duan P, Yang Y, Ma Z-B. Circulating ghrelin/obestatin ratio in subjects with Helicobacter pylori infection. Nutrition. 2009;25:506–11.
Kawashima J, Ohno S, Sakurada T, Takabayashi H, Kudo M, Ro S, et al. Circulating acylated ghrelin level decreases in accordance with the extent of atrophic gastritis. J Gastroenterol. 2009;44:1046–54.
Campana D, Nori F, Pagotto U, De Iasio R, Morselli‐Labate AM, Pasquali R, et al. Plasma acylated ghrelin levels are higher in patients with chronic atrophic gastritis. Clin Endocrinol. 2007;67:761–6.
Nweneka C, Prentice A. Helicobacter pylori infection and circulating ghrelin levels – a systematic review. BMC Gastroenterol. 2011;11:7.
Suzuki H, Masaoka T, Nomoto Y, Hosoda H, Mori M, Nishizawa T, et al. Increased levels of plasma ghrelin in peptic ulcer disease. Aliment Pharmacol Ther. 2006;24(s4):120–6.
Zub-Pokrowiecka A, Rembiasz K, Konturek SJ, Budzynski A, Konturek PC, Budzynski P. Ghrelin in diseases of the gastric mucosa associated with Helicobacter pylori infection. Med Sci Monit Basic Res. 2010;16:CR493–500.
Satou M, Nakamura Y, Ando H, Sugimoto H. Understanding the functional significance of ghrelin processing and degradation. Peptides. 2011;32:2183–90.
Nwokolo C, Freshwater D, O’Hare P, Randeva H. Plasma ghrelin following cure of Helicobacter pylori. Gut. 2003;52:637–40.
Czesnikiewicz-Guzik M, Loster B, Bielanski W, Guzik TJ, Konturek PC, Zapala J, et al. Implications of oral Helicobacter pylori for the outcome of its gastric eradication therapy. J Clin Gastroenterol. 2007;41:145–51.
Takamori K, Mizuta Y, Takeshima F, Akazawa Y, Isomoto H, Ohnita K, et al. Relation among plasma ghrelin level, gastric emptying, and psychologic condition in patients with functional dyspepsia. J Clin Gastroenterol. 2007;41:477–83.
Nishizawa T, Suzuki H, Nomoto Y, Masaoka T, Hosoda H, Mori M, et al. Enhanced plasma ghrelin levels in patients with functional dyspepsia. Aliment Pharmacol Ther. 2006;24:104–10.
Shinomiya T, Fukunaga M, Akamizu T, Irako T, Yokode M, Kangawa K, et al. Plasma acylated ghrelin levels correlate with subjective symptoms of functional dyspepsia in female patient. Scand J Gastroenterol. 2005;40:648–53.
Kim YS, Lee JS, Lee TH, Cho JY, Kim JO, Kim WJ, et al. Plasma levels of acylated ghrelin in patients with functional dyspepsia. World J Gastroenterol. 2012;18:2231–7.
Choi YJ, Kim N, Kim JY, Lee DH, Jung HC. Plasma ghrelin, leptin and serotoin levels in patients with functional dyspepsia. Gastroenterology. 2013;144(Sulppl 1):S928–9.
Sobhani I, Bado A, Vissuzaine C, Buyse M, Kermorgant S, Laigneau J, et al. Leptin secretion and leptin receptor in the human stomach. Gut. 2000;47:178–83.
Cammisotto P, Bendayan M. A review on gastric leptin: the exocrine secretion of a gastric hormone. Anat cell biolol. 2012;45:1–16.
Guilmeau S, Buyse M, Bado A. Gastric leptin: a new manager of gastrointestinal function. Curr Opin Pharmacol. 2004;4:561–6.
Cinti S, De Matteis R, Pico C, Ceresi E, Obrador A, Maffeis C, et al. Secretory granules of endocrine and chief cells of human stomach mucosa contain leptin. Int J Obes. 2000;24:789–93.
Lindqvist A, de la Cour CD, Stegmark A, Håkanson R, Erlanson-Albertsson C. Overeating of palatable food is associated with blunted leptin and ghrelin responses. Regul Pept. 2005;130:123–32.
Picó C, Sánchez J, Oliver P, Palou A. Leptin production by the stomach is up‐regulated in obese (fa/fa) Zucker rats. Obes Res. 2002;10:932–8.
Erkasap N, Uzuner K, Serteser M, Köken T, Aydın Y. Gastroprotective effect of leptin on gastric mucosal injury induced by ischemia–reperfusion is related to gastric histamine content in rats. Peptides. 2003;24:1181–7.
Moss SF, Calam J. Acid secretion and sensitivity to gastrin in patients with duodenal ulcer: effect of eradication of Helicobacter pylori. Gut. 1993;34:888–92.
Moran TH, McHugh PR. Cholecystokinin suppresses food intake by inhibiting gastric emptying. Am J Physiol. 1982;242:R491–7.
Wang L, Barachina MD, Martınez V, Wei JY, Taché Y. Synergistic interaction between CCK and leptin to regulate food intake. Regul Pept. 2000;92:79–85.
Nauck MA, Niedereichholz U, Ettler R, Holst JJ, Ørskov C, Ritzel R, et al. Glucagon-like peptide 1 inhibition of gastric emptying outweighs its insulinotropic effects in healthy humans. Am J Physiol Endocrinol Metab. 1997;273:E981–E8.
Naveilhan P, Hassani H, Canals JM, Ekstrand AJ, Larefalk Å, Chhajlani V, et al. Normal feeding behavior, body weight and leptin response require the neuropeptide Y Y2 receptor. Nat Med. 1999;5:1188–93.
Andrews PL, Sanger GJ. Abdominal vagal afferent neurones: an important target for the treatment of gastrointestinal dysfunction. Curr Opin Pharmacol. 2002;2:650–6.
Burdyga G, Spiller D, Morris R, Lal S, Thompson D, Saeed S, et al. Expression of the leptin receptor in rat and human nodose ganglion neurones. Neuroscience. 2002;109:339–47.
Cammisotto P, Bendayan M. Leptin secretion by with adipose tissue and gastric mucosa. Histol Histopathol. 2007;22:199–210.
Weller A, Smith GP, Gibbs J. Endogenous cholecystokinin reduces feeding in young rats. Science. 1990;247:1589–91.
Azuma T, Suto H, Ito Y, Ohtani M, Dojo M, Kuriyama M, et al. Gastric leptin and Helicobacter pylori infection. Gut. 2001;49:324–9.
Bado A, Levasseur S, Attoub S, Kermorgant S, Laigneau J-P, Bortoluzzi M-N, et al. The stomach is a source of leptin. Nature. 1998;394:790–3.
Lankarani KB, Moghadami M, Masoumpoor M, Geramizadeh B, Omrani GR. Serum leptin level in patients with functional dyspepsia. Dig Liver Dis. 2004;36:717–21.
Zhao Z, Sakai T. Characteristic features of ghrelin cells in the gastrointestinal tract and the regulation of stomach ghrelin expression and production. World J Gastroenterol. 2008;14:6306.
Yarandi SS, Hebbar G, Sauer CG, Cole CR, Ziegler TR. Diverse roles of leptin in the gastrointestinal tract: modulation of motility, absorption, growth, and inflammation. Nutrition. 2011;27:269–75.
Cummings DE, Purnell JQ, Frayo RS, Schmidova K, Wisse BE, Weigle DS. A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes. 2001;50:1714–9.
Gonzalez–Rey E, Chorny A, Delgado M. Therapeutic action of ghrelin in a mouse model of colitis. Gastroenterology. 2006;130:1707–20.
Date Y, Nakazato M, Murakami N, Kojima M, Kangawa K, Matsukura S. Ghrelin acts in the central nervous system to stimulate gastric acid secretion. Biochem Biophys Res Commun. 2001;280:904–7.
Park JM, Kakimoto T, Kuroki T, Shiraishi R, Fujise T, Iwakiri R, et al. Suppression of intestinal mucosal apoptosis by ghrelin in fasting rats. Exp Biol Med. 2008;233:48–56.
Kaneko H, Konagaya T, Kusugami K. Helicobacter pylori and gut hormones. J Gastroenterol. 2002;37:77–86.
Itoh Z, Takeuchi S, Aizawa I, Honda R. The negative feedback mechanism of gastric acid secretion: significance of acid in the gastric juice in man and dog. Surgery. 1975;77:648–60.
Levi S, Beardshall K, Haddad G, Playford R, Ghosh P, Calam J. Campylobacter pylori and duodenal ulcers: the gastrin link. Lancet. 1989;1:1167–8.
Beardshall K, Moss S, Gill J, Levi S, Ghosh P, Playford R, et al. Suppression of Helicobacter pylori reduces gastrin releasing peptide stimulated gastrin release in duodenal ulcer patients. Gut. 1992;33:601–3.
Penman E, Wass J, Butler M, Penny E, Price J, Wu P, et al. Distribution and characterisation of immunoreactive somatostatin in human gastrointestinal tract. Regul Pept. 1983;7:53–65.
Walsh JH, Dockray GJ, Mitty RD. Gut peptides: biochemistry and physiology. Endocrinologist. 1994;4:487.
Arnold R, Lankisch P. Somatostatin and the gastrointestinal tract. Clin Gastroenterol. 1980;9:733–53.
Kaneko H, Nakada K, Mitsuma T, Uchida K, Furusawa A, Maeda Y, et al. Helicobacter pylori infection induces a decrease in immunoreactive-somatostatin concentrations of human stomach. Dig Dis Sci. 1992;37:409–16.
Zavros Y, Paterson A, Lambert J, Shulkes A. Expression of progastrin-derived peptides and somatostatin in fundus and antrum of nonulcer dyspepsia subjects with and without Helicobacter pylori infection. Dig Dis Sci. 2000;45:2058–64.
Tzaneva M, Julianov A. Chromogranin A-, somatostatin-and serotonin-containing endocrine cells in the corporal gastric mucosa of patients with Helicobacter pylori associated chronic gastritis. Endocr Regul. 1999;33:79–82.
Moss SF, Calam J, Legon S, Bishop A, Polak J. Effect of Helicobacter pylori on gastric somatostatin in duodenal ulcer disease. Lancet. 1992;340:930–2.
Smolka AJ, Backert S. How Helicobacter pylori infection controls gastric acid secretion. J Gastroenterol. 2012;47:609–18.
Murphy F, Read N, Taylor K, Trier J. Epidemic gastritis with hypochlorhydria. Gastroenterology. 1979;76:1449–57.
Takashima M, Furuta T, Hanai H, Sugimura H, Kaneko E. Effects of Helicobacter pylori infection on gastric acid secretion and serum gastrin levels in Mongolian gerbils. Gut. 2001;48:765–73.
Hoffman JS, King WW, Fox JG, Janik D, Cave DR. Rabbit and ferret parietal cell inhibition by Helicobacter species. Dig Dis Sci. 1995;40:147–52.
Cave D, Vargas M. Effect of a Campylobacter pylori protein on acid secretion by parietal cells. Lancet. 1989;334:187–9.
Tagkalidis PP, Royce SG, Macrae FA, Bhathal PS. Selective colonization by Helicobacter pylori of the deep gastric glands and intracellular canaliculi of parietal cells in the setting of chronic proton pump inhibitor use. Eur J Gastroenterol Hepatol. 2002;14:453–6.
Chen XG, Correa P, Offerhaus J, Rodriguez E, Janney F, Hoffmann E, et al. Ultrastructure of the gastric mucosa harboring Campylobacter-like organisms. Am J Clin Pathol. 1986;86:575–82.
Jablonowski HHK, Kramer N, Geis G, Opferkuch W, Strohmeyer G. Effect of Helicobacter pylori on dbc-AMP stimulated acid secretion by human parietal cells. Hepatogastroenterology. 1994;41:546–8.
Jablonowski H, Hengels K, Kraemer N, Geis G, Opferkuch W, Strohmeyer G. Effects of Helicobacter pylori on histamine and carbachol stimulated acid secretion by human parietal cells. Gut. 1994;35:755–7.
Furuta T, Baba S, Takashima M, Shirai N, Xiao F, Futami H, et al. H+/K+ −adenosine triphosphatase mRNA in gastric fundic gland mucosa in patients infected with Helicobacter pylori. Scand J Gastroenterol. 1999;34:384–90.
Wolfe M, Nompleggi D. Cytokine inhibition of gastric acid secretion – a little goes a long way. Gastroenterology. 1992;102:2177–8.
Wallace JL, Cucala M, Mugridge K, Parente L. Secretagogue-specific effects of interleukin-1 on gastric acid secretion. Am J Physiol Gastrointest Liver Physiol. 1991;261:G559–G64.
Beales I, Calam J. Interleukin 1β and tumour necrosis factor α inhibit acid secretion in cultured rabbit parietal cells by multiple pathways. Gut. 1998;42:227–34.
Amedei A, Munarp F, Bella CD, Niccolai E, Benagiano M, Bencini L, et al. Helicobacter pylori HP0175 promotes the production of IL-23, IL-6, IL-1β AND TGF-β. Eur J Inflamm. 2013;11:261–8.
Peek RM, Crabtree JE. Helicobacter infection and gastric neoplasia. J Pathol. 2006;208:233–48.
Osawa H, Kita H, Ohnishi H, Hoshino H, Mutoh H, Ishino Y, et al. Helicobacter pylori eradication induces marked increase in H+/K + −adenosine triphosphatase expression without altering parietal cell number in human gastric mucosa. Gut. 2006;55:152–7.
Iijima K, Sekine H, Koike T, Imatani A, Ohara S, Shimosegawa T. Long-term effect of Helicobacter pylori eradication on the reversibility of acid secretion in profound hypochlorhydria. Aliment Pharmacol Ther. 2004;19:1181–8.
Moss S, Calam J. Helicobacter pylori and peptic ulcers: the present position. Gut. 1992;33:289–92.
Lichtenberger LM, Dial EJ, Romero JJ, Lechago J, Jarboe LA, Wolfe MM. Role of luminal ammonia in the development of gastropathy and hypergastrinemia in the rat. Gastroenterology. 1995;108:320–9.
Kaneko H, Uchida K, Mitsuma T, Kotera H, Nagai H, Furusawa A, et al. Effect of a long-term oral ammonia administration on immunoreactive-somatostatin concentrations of rat stomach. Nihon Shokakibyo Gakkai zasshi. 1992;89:1477–83.
Graham DY, Opekun A, Lew GM, Klein PD, Walsh JH. Helicobacter pylori-associated exaggerated gastrin release in duodenal ulcer patients. The effect of bombesin infusion and urea ingestion. Gastroenterology. 1991;100:1571–5.
El Nujumi A, Dorrian C, Chittajallu R, Neithercut W, McColl K. Effect of inhibition of Helicobacter pylori urease activity by acetohydroxamic acid on serum gastrin in duodenal ulcer subjects. Gut. 1991;32:866–70.
Paoluzi OA. Del Vecchio Giovanna Blanco RC, Monteleone I, Monteleone G, Pallone F. Impairment of ghrelin synthesis in Helicobacter pylori-colonized stomach: New clues for the pathogenesis of H. pylori-related gastric inflammation. World J Gastroenterol. 2014;20:639–46.
Piotrowski J, Majka J, Slomiany A, Slomiany B. Helicobacter pylori lipopolysaccharide inhibition of gastric mucosal somatostatin receptor. Biochem Mol Biol Int. 1995;36:491–8.
Wang L, Basa NR, Shaikh A, Luckey A, Heber D, St-Pierre DH, et al. LPS inhibits fasted plasma ghrelin levels in rats: role of IL-1 and PGs and functional implications. Am J Physiol Gastrointest Liver Physiol. 2006;291:G611–G20.
Stengel A, Goebel M, Wang L, Reeve JR, Taché Y, Lambrecht NW. Lipopolysaccharide differentially decreases plasma acyl and desacyl ghrelin levels in rats: potential role of the circulating ghrelin-acylating enzyme GOAT. Peptides. 2010;31:1689–96.
Iyo T, Kaneko H, Konagaya T, Mori S, Kotera H, Uruma M, et al. Effect of intragastric ammonia on gastrin-, somatostatin-and somatostatin receptor subtype 2 positive-cells in rat antral mucosa. Life Sci. 1999;64:2497–504.
Retallack JE CS, Ring M, Moloche RM, Hezeko U, Finlay BB, et al. Differential regulation of SSTR mRNA expression in human antral cells infected with Helicobacter pylori (abstract). Gastroenterology. 1999;116:291.
Kusugami K, Ando T, Imada A, Ina K, Ohsuga M, Shimizu T, et al. Mucosal macrophage inflammatory protein-1alpha activity in Helicobacter. J Gastroenterol Hepatol. 1999;20(6).
Blaser MJ. Hypotheses on the pathogenesis and natural history of Helicobacter pylori-induced inflammation. Gastroenterology. 1992;102:720–7.
Zavros Y, Rathinavelu S, Kao JY, Todisco A, Del Valle J, Weinstock JV, et al. Treatment of Helicobacter gastritis with IL-4 requires somatostatin. Proc Natl Acad Sci. 2003;100:12944–9.
Lehmann FS, Golodner EH, Wang J, Chen M, Avedian D, Calam J, et al. Mononuclear cells and cytokines stimulate gastrin release from canine antral cells in primary culture. Am J Physiol Gastrointest Liver Physiol. 1996;270:G783–G8.
Beales I, Blaser MJ, Srinivasan S, Calam J, Perez-Perez GI, Yamada T, et al. Effect of Helicobacter pylori products and recombinant cytokines on gastrin release from cultured canine G cells. Gastroenterology. 1997;113:465–71.
Weigert N, Schaffer K, Schusdziarra V, Classen M, Schepp W. Gastrin secretion from primary cultures of rabbit antral G cells: stimulation by inflammatory cytokines. Gastroenterology. 1996;110:147–54.
Beales I, Post L, Calam J, Yamada T, DelValle J. Tumour necrosis factor alpha stimulates gastrin release from canine and human antral G cells: possible mechanism of the Helicobacter pylori – gastrin link. Eur J Clin Invest. 1996;26:609–11.
Beales I, Calam J, Post L, Srinivasan S, Yamada T, DelValle J. Effect of transforming growth factor alpha and interleukin 8 on somatostatin release from canine fundic D cells. Gastroenterology. 1997;112:136–43.
Prinz C, Neumayer N, Mahr S, Classen M, Schepp W. Functional impairment of rat enterochromaffin-like cells by interleukin 1 beta. Gastroenterology. 1997;112:364–75.
Witteman E, Mravunac M, Becx M, Hopman W, Verschoor J, Tytgat G, et al. Improvement of gastric inflammation and resolution of epithelial damage one year after eradication of Helicobacter pylori. J Clin Pathol. 1995;48:250–6.
Yamamoto S, Kaneko H, Konagaya T, Mori S, Kotera H, Hayakawa T, et al. Interactions among gastric somatostatin, interleukin‐8 and mucosal inflammation in Helicobacter pylori‐positive peptic ulcer patients. Helicobacter. 2001;6:136–45.
Konagaya T, Kusugami K, Kaneko H, Nishio Y, Osuga M, Shimizu T, et al. Negative correlation between somatostatin levels and interleukin-8 activity in gastric antral mucosa (abstract. Gut. 1997;41Supp:1–24.
Fantuzzi G, Faggioni R. Leptin in the regulation of immunity, inflammation, and hematopoiesis. J Leukoc Biol. 2000;68:437–46.
Grunfeld C, Zhao C, Fuller J, Pollack A, Moser A, Friedman J, et al. Endotoxin and cytokines induce expression of leptin, the ob gene product, in hamsters. J Clin Invest. 1996;97:2152–7.
Barbier M, Cherbut C, Aube A, Blottiere H, Galmiche J. Elevated plasma leptin concentrations in early stages of experimental intestinal inflammation in rats. Gut. 1998;43:783–90.
Roberts H, Hardie L, Chappell L, Mercer J. Parasite-induced anorexia: leptin, insulin and corticosterone responses to infection with the nematode, Nippostrongylus brasiliensis. Parasitology. 1999;118:117–23.
Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature. 1998;394:897–901.
Sitaraman S, Liu X, Charrier L, Gu LH, Ziegler TR, Gewirtz A, et al. Colonic leptin: source of a novel proinflammatory cytokine involved in IBD. FASEB J. 2004;18:696–8.
Checchi S, Montanaro A, Pasqui L, Ciuoli C, Cevenini G, Sestini F, et al. Serum ghrelin as a marker of atrophic body gastritis in patients with parietal cell antibodies. J Clin Endocrinol Metab. 2007;92:4346–51.
Covacci A, Rappuoli R. Tyrosine-phosphorylated bacterial proteins trojan horses for the host cell. J Exp Med. 2000;191:587–92.
McColl KEL eOE, Gillen D, Ardill JES, Murray L, Crabtree JE. H. pylori induced hypergastrinemia is related to bacterial CagA status (abstract). Gastroenterology. 1997;112:215.
Queiroz D, Mendes E, Rocha G, Moura S, Oliveira A, Oliveira C. Somatostatin concentration (SC) and Helicobacter pylori (HP) vac A genotypes and cag A. Gut. 1996;39 Suppl 2:60.
Kim JH, Park HJ, Cho JS, Lee KS, Lee SI, Park IS, et al. Relationship of CagA to serum gastrin concentrations and antral G, D cell densities in Helicobacter pylori infection. Yonsei Med J. 1999;40:301–6.
Yamaoka Y, Kodama T, Kita M, Imanishi J, Kashima K, Graham D. Relation between clinical presentation, Helicobacter pylori density, interleukin 1β and 8 production, and cagA status. Gut. 1999;45:804–11.
Zaki M, Coudron PE, McCuen RW, Harrington L, Chu S, Schubert ML. H. pylori acutely inhibits gastric secretion by activating CGRP sensory neurons coupled to stimulation of somatostatin and inhibition of histamine secretion. Am J Phys Gastrointest Liver Physiol. 2013;304:G715–G22.
Kuipers E, Pena A, Festen H, Meuwissen S, Uyterlinde A, Roosendaal R, et al. Long-term sequelae of Helicobacter pylori gastritis. Lancet. 1995;345:1525–8.
Lee H-M, Wang G, Englander EW, Kojima M, Greeley Jr GH. Ghrelin, a new gastrointestinal endocrine peptide that stimulates insulin secretion: enteric distribution, ontogeny, influence of endocrine, and dietary manipulations. Endocrinology. 2002;143:185–90.
Masuda Y, Tanaka T, Inomata N, Ohnuma N, Tanaka S, Itoh Z, et al. Ghrelin stimulates gastric acid secretion and motility in rats. Biochem Biophy Res Commun. 2000;276:905–8.
de la Cour CD, Lindström E, Norlén P, Håkanson R. Ghrelin stimulates gastric emptying but is without effect on acid secretion and gastric endocrine cells. Regul Pept. 2004;120:23–32.
Suzuki H, Masaoka T, Hosoda H, Ota T, Minegishi Y, Nomura S, et al. Helicobacter pylori infection modifies gastric and plasma ghrelin dynamics in Mongolian gerbils. Gut. 2004;53:187–94.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Japan
About this chapter
Cite this chapter
Choi, Y.J., Kim, N. (2016). Ghrelin and Gut Hormone. In: Suzuki, H., Warren, R., Marshall, B. (eds) Helicobacter pylori. Springer, Tokyo. https://doi.org/10.1007/978-4-431-55705-0_6
Download citation
DOI: https://doi.org/10.1007/978-4-431-55705-0_6
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-55704-3
Online ISBN: 978-4-431-55705-0
eBook Packages: MedicineMedicine (R0)