
Abstract
Gastrointestinal (GI) hormones are a family of peptides secreted by endocrine cells located in the GI tract. Currently, GI hormones group more than 50 hormone genes that give rise to a multitude of bioactive peptides. All GI hormones play a key role in communicating cells within the GI tract in order to regulate and coordinate numerous GI functions, including secretion, absorption, and digestion, as well as motility. In addition, around a dozen of GI hormones are also able to play a role regulating glycemia and body weight. Here, we focus on some of the key GI hormones that are believed to play a relevant role in the control of the energy homeostasis: ghrelin, cholecystokinin (CCK), glucagon-like peptide-1 (GLP-1), and peptide tyrosine-tyrosine (PYY), oxyntomodulin (OXM), pancreatic polypeptide (PP), and somatostatin (SST). We briefly review their physiological role, and we discuss their potential implications in the pathophysiology of obesity.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

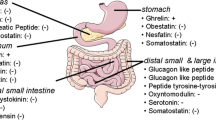

1 Introduction: General Aspects of Gastrointestinal Hormones
Gastrointestinal (GI) hormones are produced by specialized enteroendocrine cells that are located in the epithelial layer throughout the whole GI tract. All together, these cells form the biggest endocrine organ of the body and produce the largest number of hormones (Ahlman and Nilsson 2001; Valle 2014). Enteroendocrine cells are characterized as being either open- or closed-type. Open-type cells display direct contact with the GI lumen and are able to sense molecules present in the luminal content, such as nutrients, which play a major regulatory role on hormone secretion. In contrast, closed-type cells lack connection with the lumen because their apical side is enclosed by epithelial cells. Closed-type cells are mainly regulated by molecules coming from the GI capillaries or by autonomic activity. Interestingly, some enteroendocrine cells display long cytoplasmic basal processes that are filled with secretory granules and extend beneath the absorptive epithelium in order to facilitate the communication with adjacent cells.
The biosynthesis of GI hormones is a complex cellular process, which involves the initial biosynthesis of polypeptide precursors that are further cleaved, sorted, and post-translationally modified within the regulated secretory pathway in order to generate the bioactive peptides (Perello and Nillni 2007; Wren and Bloom 2007). Initially, pre-prohormones are synthesized on membrane-bound ribosomes, by which they are translocated into the lumen of the rough endoplasmic reticulum. In the endoplasmic reticulum, the signal peptide of the pre-prohormone is removed by a signal peptidase. Then, the newly synthesized prohormone is transported to the Golgi complex, which is the branch point where trafficking pathways emanate in order to generate the mature secretory granules that store the mature GI hormones. During this vectorial transport, the prohormones are subjected to post-translational modifications, which vary for the different GI hormones, in order to generate the bioactive peptides. Some frequent post-translational modifications affecting GI hormones include (1) the cleavage of the prohormones at the C-terminus of paired basic residues by proteases named prohormone convertases (PC), such as PC1/3 or PC2; (2) trimming of the C-terminal basic residues of the prohormone-derived peptides by exopeptidases, such as carboxypeptidases; and (3) carboxy-terminal amidation by the peptidylglycine α-amidating monooxygenase enzyme (PAM). In addition, the biosynthesis of some GI hormones involves specific post-translational modifications, such as sulfation or octanoylation, which are described below for each case. The post-translational modifications of the GI hormones are essential not only for peptide stability but also to ensure specific binding to their corresponding receptors.
GI hormones stored in secretory granules are released upon cell stimulation. GI hormone secretion mainly occurs in the basolateral side of enteroendocrine cells and seems to be regulated by nutrients, which activate specific nutrient receptors or transporters, as well as by different hormones and neurotransmitters (Steinert et al. 2017; Valle 2014). The secretion of all GI hormones is a reciprocally coordinated process, and their local action strongly controls the secretion of each other, as described below. In addition, the GI motility affects the secretion of some GI hormones. Secreted GI hormones can act locally on either vagal afferents and/or nearby cells or diffuse into GI capillaries to further reach the systemic circulation and act on distant tissues. Notably, the GI hormone levels acting locally are much higher than the levels in other organs, such as the brain. In addition, it is important to note that the half-life of most GI hormones is very short (ranging between 1 and 20 min), suggesting that their inactivation is another level that regulates their concentration.
The action of the GI hormones described in this chapter is mediated through G protein-coupled receptors (GPCRs), which contain an extracellular amino terminus, seven lipophilic transmembrane domains, and an intracellular carboxyl terminus (Mace et al. 2015; Valle 2014). Upon binding of the specific GI hormone, the GPCR increases its affinity for a downstream G protein that transduces the signal to an intracellular event. G proteins consist of a trimer of alpha, beta, and gamma subunits and are classified according to their alpha subunit type in Gαs, Gαi/o, Gαq/11, and Gα12/13 G proteins. Upon GPCR activation, the beta and gamma subunits dissociate from the alpha subunit, which exchanges a molecule of guanosine diphosphate for guanosine triphosphate (GTP). The GTP-bound alpha subunit regulates specific downstream signaling cascades. The Gαs or Gαi/o pathways display a stimulatory or inhibitory effect, respectively, on the activity of adenylate cyclases that catalyze the conversion of cytosolic adenosine triphosphate to cyclic adenosine monophosphate (cAMP), a second messenger that activates the protein kinase A. The target of the Gαq/11 pathway is phospholipase C, which cleavages phosphatidylinositol 4,5-bisphosphate into the second messengers inositol (1,4,5)-trisphosphate (IP3) and diacylglycerol (DAG). IP3 acts at the endoplasmic reticulum to elicit Ca2+ release, while DAG diffuses along the plasma membrane where it activates protein kinase C. Elevated intracellular Ca2+ also activates calmodulins, which in turn activate Ca2+/calmodulin-dependent kinases. The Gα12/13 pathway regulates cell processes through guanine nucleotide exchange factors that activate the cytosolic small Rho GTPase. Usually each GI hormone mainly activates one type of signaling cascade; however, some GI hormones activate multiple signaling cascades, which greatly increase the complexity of their function. In addition to the classical signaling pathways, some GI hormones have been shown to activate G protein-independent pathways, including the mitogen-activated protein kinase and extracellular signal-regulated kinase signaling pathways.
The GPCRs for GI hormones mediate their actions in the peripheral tissues as well as in the central nervous system (CNS). In addition to integrating GI functions, some GI hormones play an important role in the regulation of body energy homeostasis. The mechanisms by which some GI hormones regulate energy homeostasis are diverse and involve control of meal size, meal timing, hedonic aspects of eating, and adiposity as well as the regulation of the meal-related glycemia. Here, we focus on four GI hormones: ghrelin, cholecystokinin (CCK), glucagon-like peptide-1 (GLP-1), and peptide tyrosine-tyrosine (PYY), which likely play a relevant role in the control of the energy homeostasis. In addition, we briefly review the role of oxyntomodulin (OXM), pancreatic polypeptide (PP), and somatostatin (SST), which may also play a role on energy balance.
2 Ghrelin
Ghrelin is a 28-residue octanoylated peptide hormone discovered by Kojima in 1999 (Kojima et al. 1999). Ghrelin is the only known secreted peptide modified by an O-octanoylation; in addition, ghrelin is the only peptide hormone known to increase food intake (Kojima and Kangawa 2010). In contrast to other GI hormones that mainly impact on meal size and frequency, ghrelin seems also relevant for the long-term control of energy intake and body weight regulation as its plasma levels are inversely related to body adiposity in healthy-weight, obese, and weight-reduced humans (Cummings 2006; Perello and Zigman 2012). Ghrelin regulates both homeostatic and hedonic aspects of eating (Perello and Zigman 2012). Ghrelin may also contribute to glycemic control and, to a lesser extent, to the regulation of the GI motility (Steinert et al. 2017). Figure 7.1 depicts the GI regions producing ghrelin as well as the key targets and the main actions of this GI hormone.

Ghrelin production and effects on energy homeostasis. Ghrelin is produced in the gastric mucosa (extent indicated by intensity of light blue shading). The pathways by which ghrelin exerts its effects on energy balance are mainly central but might also involve signaling through GI vagal afferents (solid and dashed light blue arrows, respectively)
Ghrelin is predominantly synthesized by closed-type enteroendocrine ghrelin cells located within the gastric oxyntic fundic mucosa, previously referred to as P/D1-type cells in humans and A like-type cells in rodents (Kojima and Kangawa 2010). The synthesis of ghrelin is atypical as it involves the enzyme ghrelin O-acyltransferase (GOAT), which catalyzes ghrelin octanoylation. GOAT is present in the endoplasmic reticulum and acylates proghrelin before being translocated to the Golgi (Gutierrez et al. 2008; Yang et al. 2008). In the Golgi complex, the octanoylated proghrelin is cleaved by PC1/3. Since the 28 residues of ghrelin sequence immediately follow the signal peptide, the N-terminal fragment generated by cleavage of proghrelin produces bioactive ghrelin, whereas the C-terminal fragment further yields a peptide named obestatin (Takahashi et al. 2009; Zhu et al. 2006). Initial reports indicated that obestatin had anorexigenic effects; however, further studies could not confirm such observations, and its role on energy balance is still under debate (Gourcerol et al. 2007; Nogueiras et al. 2007; Zhang et al. 2005). Ghrelin is also secreted as a des-octanoylated version, named des-acyl ghrelin, which circulates even at higher levels than ghrelin because it is also produced in plasma by ghrelin deacylation (De Vriese et al. 2004). Some studies suggest that des-acyl ghrelin displays some actions in a GHSR-independent manner; however, no specific receptor for des-acyl ghrelin has yet been reported, and, as a consequence, the role of this peptide remains uncertain (Callaghan and Furness 2014).
Ghrelin levels display a surge before meals, decline after meals, and then increase gradually until the next preprandial peak (Cummings 2006). In addition, ghrelin levels are elevated in energy deficit conditions, such as fasting, malnutrition, anorexia nervosa, or cachexia (Cummings 2006). Since the half-life of plasma ghrelin is very short (Akamizu et al. 2004), the dynamics of ghrelin levels highly depends on its secretion. Ghrelin levels after meals are reduced by the consumption of all three macronutrients, although carbohydrates and proteins are more potent than isoenergetic lipid loads (Foster-Schubert et al. 2008; Steinert et al. 2017). The post-meal inhibition of ghrelin secretion is not due to signals arriving from the gastric lumen, since ghrelin cells are closed-type. Still, ghrelin cells express several nutrient-sensing receptors suggesting that ghrelin secretion is regulated by circulating nutrients (Steinert et al. 2017). In support of this notion, it has been shown that ghrelin secretion in humans is unaffected by intraduodenal glucose infusion but is inhibited when glucose reaches distal to the duodenum and proximal jejunum (Little et al. 2006). Post-meal ghrelin secretion is also inhibited by GI hormones that are recruited by nutrient ingestion. In particular, intravenous infusions of CCK or PYY reduce ghrelin levels in humans, whereas GLP-1 infusions fail to affect it (Batterham et al. 2003; Brennan et al. 2007; Degen et al. 2005). Other hormones that contribute to the post-meal inhibition of ghrelin secretion include insulin that decreases ghrelin levels and is necessary for the post-meal ghrelin reduction (Flanagan et al. 2003; Mohlig et al. 2002). In contrast, leptin fails to affect ghrelin levels (Chan et al. 2004). The autonomic nervous system does not seem to mediate the post-meal ghrelin inhibition but is critical for stimulating ghrelin secretion during fasting in both humans and animals (Broglio et al. 2004; Hosoda and Kangawa 2008; Zhao et al. 2010).
There is a single ghrelin receptor, named the growth hormone secretagogue receptor type 1a (GHSR) (Cruz and Smith 2008; Howard et al. 1996). GHSR displays some uncommon features that greatly increase the complexity of its function. GHSR is the GPCR with the highest known constitutive activity, signaling about 50% of the maximum activity in the absence of ghrelin (Holst et al. 2003). Interestingly, ghrelin-evoked activation of GHSR mainly recruits Gαq/11 signaling pathway while the ghrelin-independent activity of GHSR not only activates Gαq/11 signaling but also involves Gαi/o signaling (Holst et al. 2003; Lopez Soto et al. 2015). Additionally, GHSR heterodimerizes with other GPCRs, and such interaction mutually impacts on the signaling pathways of each receptor (Schellekens et al. 2015). GHSR is mainly expressed in the pituitary as well as in some regions of the CNS, which mediates ghrelin actions on food intake, gastric function, and glucose homeostasis (Cruz and Smith 2008).
Ghrelin administration increases gastric emptying; however, it is uncertain if endogenous patterns of ghrelin levels are sufficient to affect gastric emptying in humans (Jones et al. 2012; Levin et al. 2006; Steinert et al. 2017). Pre-meal ghrelin levels correlate with hunger sensations and meal size, and administration of ghrelin to humans strongly increases food intake; still, the causal role of ghrelin in hunger remains unclear (Cummings 2006). Studies in rodents have shown that ghrelin plays a key role in the regulation of food intake (Perello and Zigman 2012). The main target of the orexigenic actions of ghrelin are the neuropeptide Y (NPY)-producing neurons of the arcuate nucleus (ARC) that express high levels of GHSR (Cabral et al. 2016; Perello and Raingo 2014). In contrast to rodents, intact vagal afferents seem to be required for ghrelin-induced food intake in humans (Arnold et al. 2006; le Roux et al. 2005). Additionally, ghrelin affects rewarding aspects of eating by modulating the activity of the mesolimbic pathway (Perello and Zigman 2012). By acting at the central level, ghrelin also promotes adiposity (Al Massadi et al. 2017). On the other hand, ghrelin affects the glycemic control by activating a variety of mechanisms that tend to increase glucose levels. Intravenous infusion of ghrelin reduces insulin levels in response to glucose infusion and increases growth hormone and cortisol levels, without any effect on glucagon or epinephrine levels (Tong et al. 2013). Studies in mice indicate that the insulin-inhibitory and glucagon-stimulatory effects of ghrelin involve a direct pancreatic action (Chuang et al. 2011; Kurashina et al. 2015).
3 Cholecystokinin
CCK was discovered at the beginning of the twentieth century by Bayliss and Starling (Bayliss and Starling 1902). CCK is structurally related to gastrin: they share a 5-residue sequence at the C-terminal that includes an amidated phenylalanine that is key to their bioactivity (Eysselein et al. 1986). Although gastrin is believed to have evolved from CCK by gene duplication, it does not regulate food intake in mammals but rather gastric acid secretion. CCK is secreted after meals in response to the products of carbohydrate, lipid, and protein digestion, and it is recognized as the best-established GI endocrine satiation signal in humans. In addition, CCK contributes to the control of meal-related glycemia indirectly, via its effect on gastric emptying, as well as directly via control of hepatic glucose production (Steinert et al. 2017). Figure 7.2 depicts the GI regions producing CCK as well as the key targets and the main actions on energy balance of this GI hormone.

CCK production and effects on energy homeostasis. GI-derived CCK is secreted in the proximal small intestine (production level indicated by intensity of green shading) and exerts its effects on energy balance primarily by direct action on the stomach and also by signaling through vagal afferents (solid green arrows). Potential direct central actions might also be present (dashed green arrow). The thickness of the arrows indicates the relative importance of each pathway
CCK is produced by open-type cells traditionally denominated I cells, which are found in the proximal part of the small intestine (duodenum and jejunum). For the CCK synthesis, pre-proCCK is initially cleaved and yields the signal peptide and the proCCK polypeptide. Then, three out of four tyrosine residues in proCCK are sulfated by protein tyrosine sulfotransferase in the Golgi complex, and sulfated proCCK is sorted into immature secretory granules (Beinfeld 2003). The structure of proCCK allows cleavage by PC1/3, PC2, and PC5 at six monobasic motifs, leading to the formation of CCK-58, CCK-39, CCK-33, CCK-22, and CCK-8 (Beinfeld 2003). These CCK forms share the sulfated 8 residues at their C-terminal side, which is trimmed by carboxypeptidases and amidated by PAM. The most abundant CCK form in the intestine is CCK-58, followed by CCK-39, CCK-33, and CCK-8, whereas in the brain CCK-8 is the most abundant form (Eysselein et al. 1986). The fact that CCK-8 is the major form in the brain may allow a fast degradation after release, while the larger forms might be more difficult to degrade by the kidney and liver leading to a longer biological half-life in circulation (Beinfeld 2003).
CCK secretion is directly and indirectly stimulated by intraluminal nutrients, which are sensed by a variety of nutrient receptors expressed on the apical surface of I cells (Steinert et al. 2017). In humans, CCK secretion is highly elicited by lipids and proteins and to a lesser extent by carbohydrates (Dockray 2009). Additionally, CCK secretion is stimulated by the CCK-releasing factors “pancreatic monitor peptide” and “intestinal luminal CCK-releasing factor” (Steinert et al. 2017; Wang et al. 2002).
Two CCK receptors have been identified and named CCK-1R and CCK-2R (Dufresne et al. 2006). Interestingly, the ligand binding domain of CCK receptors is different. CCK-1R is highly specific for sulfated CCK forms, whereas the CCK-2R shows only a 10- to 20-fold higher preference for the sulfated forms and binds to gastrin with similar affinity (Huang et al. 1989; Miller and Desai 2016). CCK receptors predominantly couple to Gαq/11 signaling pathway (Miller and Desai 2016; Wank 1995). In rats, CCK-1R is mainly expressed in the GI tract and participates in the digestive process, while CCK-2R is mostly present in the CNS, where it is involved in satiety (Wank 1995). Both CCK receptors are present in vagal afferent fibers projecting to the nucleus of the solitary tract (NTS) in the brainstem, which controls food intake and autonomic functions (Corp et al. 1993).
CCK is a potent regulator of the GI function. In particular, CCK reduces gastric acid secretion and gastric emptying (Whited et al. 2006), while it stimulates gallbladder contraction (Liddle et al. 1985) and exocrine pancreas secretion (Adler et al. 1991). In addition, CCK is the best-established GI endocrine satiation signal in humans playing a major role in the regulation of meal size (Lieverse et al. 1995). Studies in rodents suggest that CCK inhibits eating via both local and endocrine mechanisms; however, it seems that CCK mainly stimulates satiation in humans via activation of CCK-1R on afferent vagal sensory neurons (Lieverse et al. 1995; Steinert et al. 2017; Schwartz et al. 1993). The role of CCK in meal termination does not seem to be translated into regulation of long-term energy intake since the reduction in meal size induced by CCK before every meal is compensated for by an increase in meal frequency (Woods 2004). Notably, many actions of CCK have been studied using synthetic CCK-8, which is relatively easy to synthesize and strongly binds to CCK-1R. However, the dominant circulating form of CCK is CCK-58 (Eysselein et al. 1986), and several studies have revealed that CCK-8 and CCK-58 elicit a different pattern of biological actions on pancreatic secretion, gastric motility (Reeve et al. 1996; Yamamoto et al. 2005), meal size reduction (Owyang and Heldsinger 2011), and extension of the interval between meals (Overduin et al. 2014). Animal studies indicate that CCK affects glucose metabolism by reducing hepatic glucose production via a vagal-vagal reflex (Cheung et al. 2009). Nonetheless, CCK does not seem to play a major role in the glycemic control in humans (Steinert et al. 2017).
4 Glucagon-Like Peptide-1
GLP-1 is a GI hormone derived from the pre-proglucagon gene that increases in response to meals. GLP-1 inhibits food intake and gastric emptying; however, it is best known for its potent inhibitory effect on the meal-related increases in glycemia (Holst 2007). Remarkably, the pre-proglucagon gene is also expressed in the pancreas and the brain, where proglucagon is subject to a different post-translational processing and generates different products (Orskov et al. 1986). Specifically, proglucagon is cleaved by PC2 in pancreatic α cells, whereas it is cleaved by PC1/3 in the GI and the brain (Holst 2007). In the pancreas, the main products of proglucagon are proglucagon1–30 (or glicentin-related pancreatic peptide, GRPP, which is inactive), proglucagon33–61 (glucagon itself), and proglucagon72–158 (or major proglucagon fragment, which is also inactive). In the brain, proglucagon is produced by a subset of neurons of the NTS and gives rise to GLP-1 in the same fashion as described below for the GI tract (Trapp and Hisadome 2011). Figure 7.3 depicts the GI regions producing GLP-1 as well as the key targets, the extracellular cleavage, and the main actions of this GI hormone.

GLP-1 production and effects on energy homeostasis. GLP-1 is secreted in the distal small intestine (level of production indicated by intensity of orange shading). GLP-1 effects on energy homeostasis are exerted by direct action on the endocrine pancreas but also by signaling through vagal afferents (solid orange arrows). Potential direct central actions have also been suggested (dashed orange arrows). Note that upon secretion GLP-1 is heavily degraded to inactive forms by DPP-4 (fading orange arrow). The thickness of the arrows indicates the relative importance of each pathway
In the GI tract, GLP-1 is produced by open-type L cells located in the distal jejunum and ileum (Mojsov et al. 1986). In L cells, proglucagon is processed to generate GRPP, proglucagon33–69 (also known as OXM), proglucagon78–107 (GLP-1), and proglucagon126–158 (GLP-2) (Holst 2007). In humans, the GLP-1 peptide corresponds to the sequence of proglucagon78–107 amide, and it is designated GLP-1 (7–36amide), which is the main circulating form (Holst 2007). In rodents, however, about half of the GLP-1 appears as the Gly-extended form or GLP-1 (7–37). The Gly corresponding to proglucagon108 serves as substrate for amidation, but the biological consequences of this reaction are unclear, as GLP-1 (7–36amide) and GLP-1 (7–37) display similar bioactivities and overall metabolism. After release, both GLP-1 forms are cleaved to the inactive forms GLP-1(9–36amide) and GLP-1(9–37) by the enzyme dipeptidyl peptidase 4 (DPP-4), which is a membrane glycoprotein found locally in the GI tract, in circulation, and in the liver. Due to its inactivation, only about 10–15% of the newly secreted GLP-1 reaches the systemic circulation in its intact form, suggesting that GLP-1 mainly acts locally rather than in an endocrine manner (Holst 2007).
The L cells have microvilli protruding into the intestinal lumen that sense nutrients, which regulate GLP-1 secretion (Holst 2007). GLP-1 secretion is stimulated by dietary carbohydrates and fats (Parker et al. 2010; Hansen et al. 2011). Human studies revealed that the lowest GLP-1 levels can be seen after overnight fasting; they increase rapidly during meals and usually do not return to the morning level between meals. While oral loads of carbohydrates usually result in monophasic increases in GLP-1 levels, mixed-nutrient meals induce biphasic patterns, with secondary peaks after 60–120 min (Carr et al. 2010; Steinert et al. 2017).
The GLP-1 receptor (GLP1R) belongs to the glucagon receptor family, which is a group of GPCRs that also includes the glucagon receptor (GCGR), the GLP-2 receptor, and the gastric inhibitory polypeptide (GIP) receptor (Mayo et al. 2003). GLP1R binds both GLP-1 and glucagon. GLP1R predominantly couples to the Gαs pathways (Drucker et al. 1987; Fehmann et al. 1995) but may also activate other Gα pathways (Wheeler et al. 1993). GLP1R is expressed in pancreatic islets, throughout the whole GI tract (Holst 2007) and in the brain, especially in vagal afferents and other areas implicated in food intake and energy balance regulation (Alvarez et al. 2005).
The most acknowledged effect of intestinal GLP-1 is the reduction of meal-related increases in glycemia, an effect that mainly takes place by stimulating insulin secretion, inhibiting glucagon secretion, and slowing gastric emptying (Holst 2007; Sandoval and D’Alessio 2015; Drucker et al. 1987). GLP-1, together with GIP, plays a major role in the incretin effect, which refers to the direct effect of some GI hormones on the β-cells to enhance glucose-stimulated insulin secretion and accounts for around 50–70% of the insulin release after carbohydrate intake (Mayo et al. 2003). In healthy subjects, GLP-1 may also contribute to glycemic control in the fasting state mainly via the regulation of gastric emptying and glucagon secretion (Steinert et al. 2017; Vilsboll et al. 2003; Nauck et al. 1997). Regarding food intake control, peripheral administration of GLP-1 inhibits eating in many species, including humans (Barrera et al. 2011; Williams 2009). The mechanism by which intestinal GLP-1 reduces food intake in humans remains unsettled, but likely involves its action at both vagal sensory afferents and CNS targets (Orskov et al. 1996). In rodents, GLP1R is expressed in both the ARC and the NTS, and GLP-1 seems to directly recruit NTS neurons and indirectly act on ARC neurons via ascending pathways (Wren and Bloom 2007). As described above, the CNS also produces GLP-1 that likely contributes to the regulation of energy homeostasis (Barrera et al. 2011; Trapp and Hisadome 2011).
5 PYY
PYY was first described in 1980 (Tatemoto and Mutt 1980) and belongs to the PP-fold family that also includes PP and NPY, all of which present a similar U-shaped tertiary structure and sequence homology (Wynne and Bloom 2006). PYY is released in response to meals and takes part in the local regulation of GI activity by slowing GI motility. PYY may also play a role in reducing food intake and controlling meal-related glycemia (Batterham et al. 2002; Chandarana et al. 2013). Figure 7.4 depicts the GI regions producing PYY as well as the key targets, the extracellular cleavage and the main actions of this GI hormone.

PYY production and effects on energy homeostasis. PYY is secreted from the distal intestine (extent indicated by intensity of yellow shading) and promptly undergoes a cleavage to its main active form by DPP-4 (intermediate yellow arrow). PYY effects on energy balance are primarily exerted by signaling through vagal afferents but also involve direct central signaling (solid outgoing yellow arrows). The thickness of the arrows indicates the relative importance of each pathway
PYY is produced by L cells, which can also co-secrete CCK, GLP-1, glicentin, OXM, and GIP to a varying extent (Spreckley and Murphy 2015; Steinert et al. 2017). L cells are located in the distal intestine, mainly in the rectum, followed by the colon and ileum (Wynne and Bloom 2006). Additionally, PYY is expressed in the endocrine pancreas and in some neurons of the CNS (Spreckley and Murphy 2015). As described for the other GI hormone precursors, pre-proPYY is initially cleaved by the signal peptidase and generates proPYY, which contains the PYY sequence at its N-terminal end. Thus, proPYY is further cleaved by PCs and the two basic residues trimmed by carboxypeptidases in order to generate PYY1–36-Gly. Finally, the C-terminal end of PYY is amidated, a modification required for its bioactivity, in order to generate mature PYY1–36 (Stanley et al. 2004). Two major circulating forms of PYY exist: the full-length PYY1–36, which is the secreted form, and a truncated PYY3–36 form that results from the DPP-4-mediated N-terminal cleavage, which mainly occurs in the intestinal lamina propria, liver, capillary endothelium, and blood (Spreckley and Murphy 2015; Wynne and Bloom 2006). PYY3–36 is the predominant form of PYY in circulation and is usually regarded as the active form (Manning and Batterham 2014; Wynne and Bloom 2006).
PYY exhibits a two-phase release profile following a meal. The first-phase response is mediated via neuroendocrine reflexes and likely involves CCK and GLP-1, while the second is likely driven by sensing of nutrients in the distal GI tract (Spreckley and Murphy 2015; Svendsen et al. 2015). PYY levels start rising few minutes after meals and reach the peak 60–90 min after meals, with levels proportional to caloric intake and dependent on ingested macronutrient composition, as larger and more sustained PYY elevations are induced by lipid ingestion as compared to carbohydrate ingestion (Manning and Batterham 2014; Spreckley and Murphy 2015; Steinert et al. 2017). The half-life of PYY in humans is short, but the sustained postprandial release increases its levels for several hours (Spreckley and Murphy 2015). Thus, it takes several hours after evening meals to reach typical morning fasting levels (Steinert et al. 2017). Interestingly, postprandial profiles of active PYY and GLP-1 are often dissimilar because DPP-4 activates PYY, but inactivates GLP-1 (Mortensen et al. 2003).
PYY-related peptides bind to several members of the Y receptor (YR) family, all of which are GPCRs that couple to Gαi/o pathway (Stadlbauer et al. 2015). Specifically, PYY1–36 activates Y1R, Y2R, Y4R, and Y5R, while PYY3–36 selectively targets Y2R (Dumont et al. 1995). Importantly, Y2R is expressed in a number of regions along the GI tract and in vagal afferents, as well as in several brain areas including the ARC (Steinert et al. 2017). In neurons, Y2R is located at presynaptic terminals, and its activation inhibits neurotransmitter release (Stadlbauer et al. 2015).
PYY reduces gastric emptying and acid secretion, increases ileal absorption, and delays gallbladder and pancreatic secretion (Kirchner et al. 2010; Spreckley and Murphy 2015; Wynne and Bloom 2006). Thus, PYY is part of the feedback mechanism known as the “ileal break,” which is elicited by the presence of unabsorbed dietary components in the distal GI tract and is aimed to slow proximal GI motility in order to facilitate efficient digestion and uptake of nutrients (Imamura 2002; Spreckley and Murphy 2015). PYY may also act as a satiety signal (Manning and Batterham 2014; Stanley et al. 2004; Wynne and Bloom 2006) since PYY3–36 administration reduces food intake in both rodents and humans (Batterham et al. 2002; Manning and Batterham 2014). This anorexigenic effect seems to occur via Y2R and involves the inhibition of both orexigenic ARC NPY neurons and vagal afferents fibers (Stadlbauer et al. 2015; Stanley et al. 2004; Wynne and Bloom 2006; Chaudhri et al. 2006). PYY administration affects glycemia, but the physiological relevance of endogenous PYY on glucose homeostasis in humans is still a matter of debate (Manning and Batterham 2014; Persaud and Bewick 2014; Steinert et al. 2017).
6 Other Gastrointestinal Hormones
Oxyntomodulin
First reported in 1981 (Bataille et al. 1981), OXM is a 37-residue peptide derived from the post-translational processing of the proglucagon precursor. OXM is produced by L cells of the small intestine, and its distribution along the GI tract mirrors that of GLP-1 (Holst 2007; Wynne and Bloom 2006; Chaudhri et al. 2006). Similarly to GLP-1, OXM is also produced in the pancreas and the brain (Stanley et al. 2004). OXM binds to both GLP1R and GCGR, although its affinity is almost two orders of magnitude lower than their native ligands (Spreckley and Murphy 2015). Furthermore, OXM elicits a biased GLP1R response as compared with GLP-1, as it has less preference toward the Gαs pathway as compared to the ERK pathway (Pocai 2014). Similarly to PYY and GLP-1, OXM is released after meals in an extent that depends on the caloric intake and macronutrient composition, being mainly stimulated by fat content (Huda et al. 2006). The half-life of OXM is short, being rapidly degraded in circulation by DPP-4, in a similar fashion as GLP-1 (Yi et al. 2015). OXM inhibits gastric secretion, pancreatic exocrine secretion, and gastric emptying (Field et al. 2008). OXM also inhibits food intake, in part, due to the suppression of ghrelin levels (Wren and Bloom 2007). OXM might be involved in long-term energy balance in humans since its repeated administration reduces body weight as a result of both reduction in food intake and increase in energy expenditure (Field et al. 2008). Despite OXM binds to both GLP1R and GCGR, it mainly inhibits appetite via the GLP1R since co-administration of OXM and a GLP1R antagonist blocks the anorectic actions of OXM (Dakin et al. 2001; Pocai 2014). In contrast to GLP-1, OXM is thought to inhibit food intake by acting directly at the ARC level (Wren and Bloom 2007). This possibility together with the additional effect of OXM on GCGR may explain the reason why this GI hormone is as potent as GLP-1 to reduce food intake despite its lower affinity for the GLP1R (Wynne and Bloom 2006). OXM also exhibits incretin activity albeit modest compared to that of GLP-1 (Du et al. 2012).
Pancreatic Polypeptide
PP is an amidated 36-residue peptide that belongs to the PP-fold family and was discovered in 1975 from pancreatic extracts (Kimmel et al. 1975; Chaudhri et al. 2006). PP is mainly produced in specialized pancreatic islets cells, called F cells, and to a lesser extent in the exocrine pancreas, colon, and rectum (Huda et al. 2006; Khandekar et al. 2015; Wren and Bloom 2007). As all members of the PP-fold family, PP binds to YR showing the highest affinity for Y4R but also binding to Y5R and Y1R, all of which are expressed in the CNS (Huda et al. 2006). The PP production is under vagal control (Lean and Malkova 2016), and its effects are also mainly mediated by the vagus nerve (Choudhury et al. 2016). PP is released after meals, in an amount proportional to the calories ingested, and in response to hypoglycemia, exercise, gastric distension, and elevations in gastrin, secretin, and motilin (Field et al. 2008; Huda et al. 2006). Similarly to PYY, PP has a short half-life but its levels remain elevated for several hours after meals (Chaudhri et al. 2006; Choudhury et al. 2016). In humans, peripheral administration of PP inhibits gastric emptying, exocrine pancreatic secretion, and gallbladder motility and acutely decreases food intake (Lean and Malkova 2016; Wren and Bloom 2007). In rodents, peripheral administration of PP also reduces food intake and gastric emptying, and these effects seem to occur via hypothalamic actions but also require the integrity of the vagal system (Asakawa et al. 2003; Khandekar et al. 2015). PP inhibits glucagon release through the activation of Y1R in pancreatic α cells, and delays the postprandial rise in insulin (Aragon et al. 2015; Chaudhri et al. 2006).
Somatostatin
SST is a cyclic peptide that was first identified in 1973 in hypothalamic extracts (Brazeau et al. 1973). SST displays a widespread distribution in the body, being produced in the GI tract, as well as in the brain, peripheral nerves, the pancreas, and the retina (Kumar and Grant 2010). Most of the circulating SST derives from the GI tract, where it is released from D cells as well as from intrinsic neurons located in the stomach, intestines, and pancreas (Low 2004). Interestingly, proSST can yield two bioactive products: a short 14-residue form (SST-14) and a longer 28-residue form (SST-28) that contain an extension at the N-terminus (Kumar and Grant 2010). Both SST forms present a disulfide bond between cysteine residues at positions 1 and 12. SST-28 predominates in the intestinal mucosal cells, while SST-14 predominates in the pancreas, the stomach, and neural tissues. SST binds to five GPCRs, named SSTR1–5. SSTRs 1 to 4 bind SST-14 and SST-28 with similar affinity, while SSTR5 binds SST-28 with five- to tenfold higher affinity (Rai et al. 2015). All SSTRs are coupled to the Gαi pathway and are widely distributed in the body, although SSTR2 and SSTR5 are predominantly expressed in the peripheral tissues (Low 2004). SST secretion is increased by a combination of nutritional, hormonal, and neural signals after meals (Low 2004), and SST levels can remain elevated postprandially for few hours, despite SST in plasma displays a very short half-life (Rai et al. 2015). Well known for its inhibitory physiological actions in multiple targets, SST can function via endocrine, paracrine, or neurocrine pathways. SST is involved in a variety of effects, and it is unclear whether its effects on the energy balance are direct or indirect, as SST inhibits the release of numerous hormones (Rai et al. 2015). SST inhibits the secretion of some GI hormones (CCK, ghrelin, GLP-1, GIP, secretin), gastric emptying, and GI motility while also inhibiting insulin and glucagon secretion from the pancreas (Rai et al. 2015). The effect of SST on food intake is unclear as studies in rodents have found inconsistent results (Fenske et al. 2012; Rai et al. 2015). Still, the relevance of SST in the regulation of energy balance remains uncertain (Rai et al. 2015; Steinert et al. 2017).
7 Gastrointestinal Hormones and Obesity
The understanding of the role of the GI hormones on the regulation of the mechanisms controlling energy balance has notably improved over the last decades. As summarized in Table 7.1, it is now clear that CCK, GLP-1, and PYY, which are secreted after eating, reduce food intake mainly by affecting the timing and size of individual meals, while ghrelin, which is secreted before eating, may also play a tonic role controlling the long-term body weight. Additionally, some GI hormones also regulate glucose homeostasis by affecting insulin secretion and/or sensitivity as well as by affecting gastric emptying, which contributes to the regulation of meal-related glycemia. Thus, GLP-1 strongly controls meal-related glycemia via a variety of mechanisms, while CCK and PYY decrease gastric emptying but do not seem to impact on other aspects of glycemic control. In contrast, ghrelin promotes mechanisms that increase glycemia and only slightly increases gastric emptying. Given the key role of these GI hormones in the regulation of the energy homeostasis, it is likely that they are also involved in the pathophysiology of obesity, which is the most significant growing health concern worldwide. Currently, the only treatment that produces long-term weight loss and improves obesity-related comorbid conditions in severely obese patients is bariatric surgery, particularly Roux-en-Y gastric bypass (RYGB) (Sjostrom et al. 2007). Although the mechanisms involved in the bariatric surgery-mediated remission of obesity are still uncertain, it is well-established that the levels of various GI hormones are altered and that such changes correlate with the beneficial effects of the procedure (Steinert et al. 2017). These observations have highlighted the necessity to improve our understanding of the role of the GI hormones in the pathophysiology of obesity in order to further explore their therapeutic manipulation. In this section, we briefly review possible links between the above described GI hormones and obesity as well as their potential as pharmaceutical targets.
Little association has been found between ghrelin or GHSR gene mutations and obesity in humans (Gueorguiev et al. 2009; Liu et al. 2011); however, ghrelin gene polymorphisms were associated with body mass index variation in some human populations (Li et al. 2014). Most obese patients display low ghrelin levels and a blunted increase of nocturnal ghrelin levels, as compared to normal subjects (Hillman et al. 2011; Tschop et al. 2001). In addition, some studies report that obese patients show a blunted postprandial decrease of ghrelin levels, which likely increases the time they feel hungry and contributes to the pathophysiology of obesity (le Roux et al. 2005; Morpurgo et al. 2003; Yang et al. 2009). On the other hand, ghrelin levels rise in obese patients after diet-induced weight loss, and such increase seems to be involved in the rebound weight gain usually observed in dieters (Cummings et al. 2002). Patients that undergo RYGB display reduced fasting and post-meal ghrelin levels in the first weeks after surgery, but the longer-term effects are controversial (Cummings and Shannon 2003; Steinert et al. 2017; Beckman et al. 2010). Patients that go through sleeve gastrectomy show a dramatic decrease of body weight and display almost undetectable ghrelin levels (Peterli et al. 2012). These evidences suggested that pharmacological manipulations of ghrelin signaling may be a potential anti-obesity strategy. However, no therapy targeting this system has been shown to be successful up to date. Among other reasons, the complexity of the GHSR biology, including the fact that ligands of the receptor can function as agonist, antagonist, or inverse agonists in a biased fashion (M’Kadmi et al. 2015), has become an intrinsic limitation hard to overcome.
Human CCK-1R polymorphisms have been associated with meal size, total food intake, and body weight alterations, suggesting that CCK system is involved in the pathophysiology of obesity (Steinert et al. 2017). However, it is currently controversial if CCK levels are altered in obesity, as one study found reduced fasting CCK levels in obese patients, which could contribute to overeating, while other studies could not confirm these findings (Baranowska et al. 2000; Brennan et al. 2012; Stewart et al. 2011). Interestingly, postprandial CCK levels have been reported to be normal or increased following RYGB in different studies indicating that this system may contribute to the early satiation seen in these patients (Steinert et al. 2017). The reason for this observation is unclear, as the ingested nutrients fail to contact the majority of the I cells after the surgical procedure, although this suggests that CCK system may represent a target for obesity treatment (Steinert et al. 2017). Many attempts have been performed in order to reduce appetite by pharmacological manipulations of the CCK system. In rats, continuous CCK administration rapidly becomes ineffective, while intermittent CCK administration before each meal effectively reduces meal size; nevertheless, animals compensate daily food intake by increasing meal frequency (Crawley and Beinfeld 1983; Woods 2004). Similarly, intravenous CCK reduces food intake in humans, but repeated administration of an orally available CCK-1R agonist to obese patients failed to reduce body weight (Jordan et al. 2008; Kissileff et al. 1981). Although several CCK-1R agonists have been developed, research is still ongoing and no active agents have yet reached clinical practice (Miller and Desai 2016).
A polymorphism in GLP1R has been associated with elevated body mass index in some populations, suggesting that defects in GLP-1 signaling could contribute to obesity risk (Li et al. 2014; Steinert et al. 2017). Most, but not all, studies have shown that post-meal GLP-1 levels are reduced in obese patients, suggesting that defects in GLP-1 system may contribute to overeating in obesity (Steinert et al. 2017). Interestingly, the incretin effect of GLP-1 is diminished, but still present, in obese patients, and GLP-1 is a crucial contributor for glycemic control in individuals with insulin resistance or diabetes (Aulinger et al. 2015; Bagger et al. 2011). Human and animal studies have shown that RYGB fails to affect fasting GLP-1 levels but substantially increases postprandial GLP-1 levels, which seem to play a major beneficial effect of RYGB (Rhee et al. 2012). It is still a matter of debate if GLP-1 is involved in the reduction of eating and weight loss seen after RYGB; however, compelling evidence indicates that GLP-1 contributes to the beneficial effects of RYGB on glycemic regulation in humans (Steinert et al. 2017). GLP-1 is currently the most successful GI hormone exploited as a therapeutic target, mainly in relation with glycemic control. The pharmacological strategies include the DPP-4-resistant GLP1R agonist exendin-4, which is a natural peptide component of Gila monster saliva, and its synthetic versions exenatide and liraglutide, which are GLP-1 analogs with an acylated side chain (Troke et al. 2014). Alternatively, DPP-4 inhibitors, such as sitagliptin and vildagliptin, are used to increase the endogenous GLP-1 levels. These DPP-4 inhibitors are currently licensed for the treatment of type 2 diabetes mellitus but fail to reduce body weight, likely because DPP-4 inhibition also impacts on the generation of bioactive PYY(3–36) (Field et al. 2008). Thus, the utility of the GLP-1 system as a target for weight loss therapies remains limited, and it seems unlikely that GLP-1-related monotherapy will be used for the treatment of obesity (Troke et al. 2014).
The relationship between PYY levels and obesity is uncertain. Some studies found that obese patients display lower basal PYY levels and a blunted meal-induced increase; however, this findings could not be confirmed by others (Stanley et al. 2004; Stadlbauer et al. 2015). As seen for GLP-1, obese patients that underwent RYGB show markedly increased meal-stimulated PYY levels (Manning and Batterham 2014) that, together with GLP-1 elevations, may contribute to the reduction in food intake following surgery (Karra et al. 2009; Manning and Batterham 2014). This observations as well as many rodent studies have supported the notion that the PYY system may be pharmacologically targeted as an anti-obesity therapy (Karra et al. 2009). Interestingly, PYY equally reduced food intake in lean or obese subjects in clinical trials; however, the rapid rise of PYY levels induced by either intranasal or oral preparations of PYY(3–36) was shown to induce nausea and vomiting (Field et al. 2008). Thus, it remains unclear if PYY-based therapies will be available to treat obesity.
The potential link between OXM, PP, or SST and obesity in humans is currently uncertain. Few data are available regarding the OXM levels or effects in obese patients (Huda et al. 2006). Similarly to PYY and GLP-1, OXM is exaggeratedly increased after meals in patients that underwent RYGB surgery, and such increase has been linked to the loss of appetite found after surgery (Pocai 2014; Huda et al. 2006). The beneficial actions of OXM on food intake, energy expenditure, and body weight make it a promising target for the treatment of obesity. Given the short half-life of OXM, future modified versions of this peptide, resistant to degradation, may be potential therapeutic candidates to improve glycemic control and suppress appetite in obese patients (Pocai 2014; Spreckley and Murphy 2015). Regarding PP, some studies found that obese patients display lower fasting PP levels, as compared to lean subjects, but this observation could not be confirmed by other studies (Lean and Malkova 2016). The effect of weight loss on PP levels in obese individuals has also been inconsistent (Lean and Malkova 2016). Currently, there is no anti-obesity treatment targeting the PP system. However, the ability of PP to strongly suppress appetite and promote weight loss in humans has made this GI hormone an attractive therapeutic target, and several PP analogs have been generated and successfully tested in animal models (Choudhury et al. 2016; Troke et al. 2014). Thus, some novel compounds targeting the PP system may be soon tested for human use. Notably, the potent anorectic effect of PP seen in rodents suggests that Y4R agonists may be useful to treat obesity (Troke et al. 2014). Regarding the SST system, it seems unlikely to develop an anti-obesity therapy based on this system given its pleiotropic functions (Rai et al. 2015).
In summary, no anti-obesity drug based on the manipulation of GI hormones currently exists on the market mainly because of lack of evidence of sustained body weight loss. Despite the potent effect of some GI hormones on appetite, their failure as therapeutic drugs is most likely due to the existence of many associated and redundant compensatory mechanisms that control energy homeostasis. In addition, the short-term effect of most GI hormones in physiological conditions adds some pharmacological challenges that need to be overcome in long-acting compounds. More recently, the use of combined therapies that take advantage of two or even more compounds targeting different GI hormonal systems has emerged as an exciting possibility that is currently under study (Troke et al. 2014). Hopefully, future innovative investigations will be able to develop GI hormone-based therapies to provide an effective treatment for obesity.
Didactic Elements
-
Q1.
Which are the main GI hormones that regulate energy balance?
-
Q2.
In which part of the GI are ghrelin, PYY, GLP-1, and CCK mainly produced?
-
Q3.
Which GI hormones are more important in the regulation of the appetite?
-
Q4.
Which GI hormones are more important in the regulation of the postprandial glycaemia?
-
Q5.
Which GI hormones increase their bioactivity by proteolysis once they are secreted?
Abbreviations
- ARC:
-
Arcuate nucleus
- cAMP:
-
Cyclic adenosine monophosphate
- CCK:
-
Cholecystokinin
- CCK-1R:
-
Cholecystokinin receptor 1
- CCK-2R:
-
Cholecystokinin receptor 2
- CNS:
-
Central nervous system
- DAG:
-
Diacylglycerol
- DPP-4:
-
Dipeptidyl peptidase 4
- GCGR:
-
Glucagon receptors
- GHSR:
-
Growth hormone secretagogue receptor type 1a
- GI:
-
Gastrointestinal
- GIP:
-
Gastric inhibitory polypeptide
- GLP-1:
-
Glucagon-like peptide-1
- GLP1R:
-
Glucagon-like peptide-1 receptor
- GLP-2:
-
Glucagon-like peptide-2
- GOAT:
-
Ghrelin O-acyltransferase
- GPCRs:
-
G protein-coupled receptors
- GRPP:
-
Glicentin-related pancreatic peptide
- GTP:
-
Guanosine triphosphate
- IP3:
-
Inositol (1,4,5)-trisphosphate
- NPY:
-
Neuropeptide Y
- NTS:
-
Nucleus of the solitary tract
- OXM:
-
Oxyntomodulin
- PAM:
-
Peptidylglycine α-amidating monooxygenase enzyme
- PC:
-
Prohormone convertase
- PP:
-
Pancreatic polypeptide
- PYY:
-
Peptide tyrosine-tyrosine
- RYGB:
-
Roux-en-Y gastric bypass
- SST:
-
Somatostatin
- SSTR:
-
Somatostatin receptor
- YR:
-
Neuropeptide Y receptor
References
Adler, G., Beglinger, C., Braun, U., Reinshagen, M., Koop, I., Schafmayer, A., Rovati, L., & Arnold, R. (1991). Interaction of the cholinergic system and cholecystokinin in the regulation of endogenous and exogenous stimulation of pancreatic secretion in humans. Gastroenterology, 100(2), 537–543.
Ahlman, H. N. (2001). The gut as the largest endocrine organ in the body. Annals of Oncology: Official Journal of the European Society for Medical Oncology, 12(Suppl 2), S63–S68.
Akamizu, T., Takaya, K., Irako, T., Hosoda, H., Teramukai, S., Matsuyama, A., Tada, H., Miura, K., Shimizu, A., Fukushima, M., Yokode, M., Tanaka, K., & Kangawa, K. (2004). Pharmacokinetics, safety, and endocrine and appetite effects of ghrelin administration in young healthy subjects. European Journal of Endocrinology, 150(4), 447–455.
Al Massadi, O., Lopez, M., Tschop, M., Dieguez, C., & Nogueiras, R. (2017). Current understanding of the hypothalamic ghrelin pathways inducing appetite and adiposity. Trends in Neurosciences. https://doi.org/10.1016/j.tins.2016.12.003.
Alvarez, E., Martinez, M. D., Roncero, I., Chowen, J. A., Garcia-Cuartero, B., Gispert, J. D., Sanz, C., Vazquez, P., Maldonado, A., de Caceres, J., Desco, M., Pozo, M. A., & Blazquez, E. (2005). The expression of GLP-1 receptor mRNA and protein allows the effect of GLP-1 on glucose metabolism in the human hypothalamus and brainstem. Journal of Neurochemistry, 92(4), 798–806. https://doi.org/10.1111/j.1471-4159.2004.02914.x.
Aragon, F., Karaca, M., Novials, A., Maldonado, R., Maechler, P., & Rubi, B. (2015). Pancreatic polypeptide regulates glucagon release through PPYR1 receptors expressed in mouse and human alpha-cells. Biochimica et Biophysica Acta, 1850(2), 343–351. https://doi.org/10.1016/j.bbagen.2014.11.005.
Arnold, M., Mura, A., Langhans, W., & Geary, N. (2006). Gut vagal afferents are not necessary for the eating-stimulatory effect of intraperitoneally injected ghrelin in the rat. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 26(43), 11052–11060. https://doi.org/10.1523/JNEUROSCI.2606-06.2006.
Asakawa, A., Inui, A., Yuzuriha, H., Ueno, N., Katsuura, G., Fujimiya, M., Fujino, M. A., Niijima, A., Meguid, M. M., & Kasuga, M. (2003). Characterization of the effects of pancreatic polypeptide in the regulation of energy balance. Gastroenterology, 124(5), 1325–1336.
Aulinger, B. A., Vahl, T. P., Wilson-Perez, H. E., Prigeon, R. L., & D’Alessio, D. A. (2015). Beta-cell sensitivity to GLP-1 in healthy humans is variable and proportional to insulin sensitivity. The Journal of Clinical Endocrinology and Metabolism, 100(6), 2489–2496. https://doi.org/10.1210/jc.2014-4009.
Bagger, J. I., Knop, F. K., Lund, A., Vestergaard, H., Holst, J. J., & Vilsboll, T. (2011). Impaired regulation of the incretin effect in patients with type 2 diabetes. The Journal of Clinical Endocrinology and Metabolism, 96(3), 737–745. https://doi.org/10.1210/jc.2010-2435.
Baranowska, B., Radzikowska, M., Wasilewska-Dziubinska, E., Roguski, K., & Borowiec, M. (2000). Disturbed release of gastrointestinal peptides in anorexia nervosa and in obesity. Diabetes, Obesity & Metabolism, 2(2), 99–103.
Barrera, J. G., Sandoval, D. A., D’Alessio, D. A., & Seeley, R. J. (2011). GLP-1 and energy balance: An integrated model of short-term and long-term control. Nature Reviews Endocrinology, 7(9), 507–516. https://doi.org/10.1038/nrendo.2011.77.
Bataille, D., Gespach, C., Tatemoto, K., Marie, J. C., Coudray, A. M., Rosselin, G., & Mutt, V. (1981). Bioactive enteroglucagon (oxyntomodulin): Present knowledge on its chemical structure and its biological activities. Peptides, 2(Suppl 2), 41–44.
Batterham, R. L., Cohen, M. A., Ellis, S. M., Le Roux, C. W., Withers, D. J., Frost, G. S., Ghatei, M. A., & Bloom, S. R. (2003). Inhibition of food intake in obese subjects by peptide YY3-36. The New England Journal of Medicine, 349(10), 941–948. https://doi.org/10.1056/NEJMoa030204.
Batterham, R. L., Cowley, M. A., Small, C. J., Herzog, H., Cohen, M. A., Dakin, C. L., Wren, A. M., Brynes, A. E., Low, M. J., Ghatei, M. A., Cone, R. D., & Bloom, S. R. (2002). Gut hormone PYY(3-36) physiologically inhibits food intake. Nature, 418(6898), 650–654. https://doi.org/10.1038/nature02666.
Bayliss, W. M., & Starling, E. H. (1902). The mechanism of pancreatic secretion. The Journal of Physiology, 28(5), 325–353.
Beckman, L. M., Beckman, T. R., & Earthman, C. P. (2010). Changes in gastrointestinal hormones and leptin after roux-en-Y gastric bypass procedure: A review. Journal of the American Dietetic Association, 110(4), 571–584. https://doi.org/10.1016/j.jada.2009.12.023.
Beinfeld, M. C. (2003). Biosynthesis and processing of pro CCK: Recent progress and future challenges. Life Sciences, 72(7), 747–757.
Brazeau, P., Vale, W., Burgus, R., Ling, N., Butcher, M., Rivier, J., & Guillemin, R. (1973). Hypothalamic polypeptide that inhibits the secretion of immunoreactive pituitary growth hormone. Science, 179(4068), 77–79.
Brennan, I. M., Luscombe-Marsh, N. D., Seimon, R. V., Otto, B., Horowitz, M., Wishart, J. M., & Feinle-Bisset, C. (2012). Effects of fat, protein, and carbohydrate and protein load on appetite, plasma cholecystokinin, peptide YY, and ghrelin, and energy intake in lean and obese men. American Journal of Physiology Gastrointestinal and Liver Physiology, 303(1), G129–G140. https://doi.org/10.1152/ajpgi.00478.2011.
Brennan, I. M., Otto, B., Feltrin, K. L., Meyer, J. H., Horowitz, M., & Feinle-Bisset, C. (2007). Intravenous CCK-8, but not GLP-1, suppresses ghrelin and stimulates PYY release in healthy men. Peptides, 28(3), 607–611. https://doi.org/10.1016/j.peptides.2006.10.014.
Broglio, F., Gottero, C., Van Koetsveld, P., Prodam, F., Destefanis, S., Benso, A., Gauna, C., Hofland, L., Arvat, E., van der Lely, A. J., & Ghigo, E. (2004). Acetylcholine regulates ghrelin secretion in humans. The Journal of Clinical Endocrinology and Metabolism, 89(5), 2429–2433. https://doi.org/10.1210/jc.2003-031517.
Cabral, A., Portiansky, E., Sanchez-Jaramillo, E., Zigman, J. M., & Perello, M. (2016). Ghrelin activates hypophysiotropic corticotropin-releasing factor neurons independently of the arcuate nucleus. Psychoneuroendocrinology, 67, 27–39. https://doi.org/10.1016/j.psyneuen.2016.01.027.
Callaghan, B., & Furness, J. B. (2014). Novel and conventional receptors for ghrelin, desacyl-ghrelin, and pharmacologically related compounds. Pharmacological Reviews, 66(4), 984–1001. https://doi.org/10.1124/pr.113.008433.
Carr, R. D., Larsen, M. O., Jelic, K., Lindgren, O., Vikman, J., Holst, J. J., Deacon, C. F., & Ahren, B. (2010). Secretion and dipeptidyl peptidase-4-mediated metabolism of incretin hormones after a mixed meal or glucose ingestion in obese compared to lean, nondiabetic men. The Journal of Clinical Endocrinology and Metabolism, 95(2), 872–878. https://doi.org/10.1210/jc.2009-2054.
Corp, E. S., McQuade, J., Moran, T. H., & Smith, G. P. (1993). Characterization of type a and type B CCK receptor binding sites in rat vagus nerve. Brain Research, 623(1), 161–166.
Crawley, J. N., & Beinfeld, M. C. (1983). Rapid development of tolerance to the behavioural actions of cholecystokinin. Nature, 302(5910), 703–706.
Cruz, C. R., & Smith, R. G. (2008). The growth hormone secretagogue receptor. Vitamins and Hormones, 77, 47–88. https://doi.org/10.1016/S0083-6729(06)77004-2.
Cummings, D. E. (2006). Ghrelin and the short- and long-term regulation of appetite and body weight. Physiology & Behavior, 89(1), 71–84. https://doi.org/10.1016/j.physbeh.2006.05.022.
Cummings, D. E., & Shannon, M. H. (2003). Ghrelin and gastric bypass: Is there a hormonal contribution to surgical weight loss? The Journal of Clinical Endocrinology and Metabolism, 88(7), 2999–3002. https://doi.org/10.1210/jc.2003-030705.
Cummings, D. E., Weigle, D. S., Frayo, R. S., Breen, P. A., Ma, M. K., Dellinger, E. P., & Purnell, J. Q. (2002). Plasma ghrelin levels after diet-induced weight loss or gastric bypass surgery. The New England Journal of Medicine, 346(21), 1623–1630. https://doi.org/10.1056/NEJMoa012908.
Chan, J. L., Bullen, J., Lee, J. H., Yiannakouris, N., & Mantzoros, C. S. (2004). Ghrelin levels are not regulated by recombinant leptin administration and/or three days of fasting in healthy subjects. The Journal of Clinical Endocrinology and Metabolism, 89(1), 335–343. https://doi.org/10.1210/jc.2003-031412.
Chandarana, K., Gelegen, C., Irvine, E. E., Choudhury, A. I., Amouyal, C., Andreelli, F., Withers, D. J., & Batterham, R. L. (2013). Peripheral activation of the Y2-receptor promotes secretion of GLP-1 and improves glucose tolerance. Molecular Metabolism, 2(3), 142–152. https://doi.org/10.1016/j.molmet.2013.03.001.
Chaudhri, O., Small, C., & Bloom, S. (2006). Gastrointestinal hormones regulating appetite. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 361(1471), 1187–1209. https://doi.org/10.1098/rstb.2006.1856.
Cheung, G. W., Kokorovic, A., Lam, C. K., Chari, M., & Lam, T. K. (2009). Intestinal cholecystokinin controls glucose production through a neuronal network. Cell Metabolism, 10(2), 99–109. https://doi.org/10.1016/j.cmet.2009.07.005.
Choudhury, S. M., Tan, T. M., & Bloom, S. R. (2016). Gastrointestinal hormones and their role in obesity. Current Opinion in Endocrinology, Diabetes, and Obesity, 23(1), 18–22. https://doi.org/10.1097/MED.0000000000000216.
Chuang, J. C., Sakata, I., Kohno, D., Perello, M., Osborne-Lawrence, S., Repa, J. J., & Zigman, J. M. (2011). Ghrelin directly stimulates glucagon secretion from pancreatic alpha-cells. Molecular Endocrinology, 25(9), 1600–1611. https://doi.org/10.1210/me.2011-1001.
Dakin, C. L., Gunn, I., Small, C. J., Edwards, C. M., Hay, D. L., Smith, D. M., Ghatei, M. A., & Bloom, S. R. (2001). Oxyntomodulin inhibits food intake in the rat. Endocrinology, 142(10), 4244–4250. https://doi.org/10.1210/endo.142.10.8430.
De Vriese, C., Gregoire, F., Lema-Kisoka, R., Waelbroeck, M., Robberecht, P., & Delporte, C. (2004). Ghrelin degradation by serum and tissue homogenates: Identification of the cleavage sites. Endocrinology, 145(11), 4997–5005. https://doi.org/10.1210/en.2004-0569.
Degen, L., Oesch, S., Casanova, M., Graf, S., Ketterer, S., Drewe, J., & Beglinger, C. (2005). Effect of peptide YY3-36 on food intake in humans. Gastroenterology, 129(5), 1430–1436. https://doi.org/10.1053/j.gastro.2005.09.001.
Dockray, G. J. (2009). Cholecystokinin and gut-brain signalling. Regulatory Peptides, 155(1–3), 6–10. https://doi.org/10.1016/j.regpep.2009.03.015.
Drucker, D. J., Philippe, J., Mojsov, S., Chick, W. L., & Habener, J. F. (1987). Glucagon-like peptide I stimulates insulin gene expression and increases cyclic AMP levels in a rat islet cell line. Proceedings of the National Academy of Sciences of the United States of America, 84(10), 3434–3438.
Du, X., Kosinski, J. R., Lao, J., Shen, X., Petrov, A., Chicchi, G. G., Eiermann, G. J., & Pocai, A. (2012). Differential effects of oxyntomodulin and GLP-1 on glucose metabolism. American Journal of Physiology. Endocrinology and Metabolism, 303(2), E265–E271. https://doi.org/10.1152/ajpendo.00142.2012.
Dufresne, M., Seva, C., & Fourmy, D. (2006). Cholecystokinin and gastrin receptors. Physiological Reviews, 86(3), 805–847. https://doi.org/10.1152/physrev.00014.2005.
Dumont, Y., Fournier, A., St-Pierre, S., & Quirion, R. (1995). Characterization of neuropeptide Y binding sites in rat brain membrane preparations using [125I][Leu31,Pro34]peptide YY and [125I]peptide YY3-36 as selective Y1 and Y2 radioligands. The Journal of Pharmacology and Experimental Therapeutics, 272(2), 673–680.
Eysselein, V. E., Reeve, J. R., Jr., & Eberlein, G. (1986). Cholecystokinin--gene structure, and molecular forms in tissue and blood. Zeitschrift fur Gastroenterologie, 24(10), 645–659.
Fehmann, H. C., Goke, R., & Goke, B. (1995). Cell and molecular biology of the incretin hormones glucagon-like peptide-I and glucose-dependent insulin releasing polypeptide. Endocrine Reviews, 16(3), 390–410. https://doi.org/10.1210/edrv-16-3-390.
Fenske, W. K., Bueter, M., Miras, A. D., Ghatei, M. A., Bloom, S. R., & le Roux, C. W. (2012). Exogenous peptide YY3-36 and Exendin-4 further decrease food intake, whereas octreotide increases food intake in rats after roux-en-Y gastric bypass. International Journal of Obesity, 36(3), 379–384. https://doi.org/10.1038/ijo.2011.126.
Field, B. C. T., Wren, A. M., Cooke, D., & Bloom, S. R. (2008). Gut hormones as potential new targets for appetite regulation and the treatment of obesity. Drugs, 68(2), 147–163.
Flanagan, D. E., Evans, M. L., Monsod, T. P., Rife, F., Heptulla, R. A., Tamborlane, W. V., & Sherwin, R. S. (2003). The influence of insulin on circulating ghrelin. American Journal of Physiology. Endocrinology and Metabolism, 284(2), E313–E316. https://doi.org/10.1152/ajpendo.00569.2001.
Foster-Schubert, K. E., Overduin, J., Prudom, C. E., Liu, J., Callahan, H. S., Gaylinn, B. D., Thorner, M. O., & Cummings, D. E. (2008). Acyl and total ghrelin are suppressed strongly by ingested proteins, weakly by lipids, and biphasically by carbohydrates. The Journal of Clinical Endocrinology and Metabolism, 93(5), 1971–1979. https://doi.org/10.1210/jc.2007-2289.
Gourcerol, G., Coskun, T., Craft, L. S., Mayer, J. P., Heiman, M. L., Wang, L., Million, M., St-Pierre, D. H., & Tache, Y. (2007). Preproghrelin-derived peptide, obestatin, fails to influence food intake in lean or obese rodents. Obesity (Silver Spring), 15(11), 2643–2652. https://doi.org/10.1038/oby.2007.316.
Gueorguiev, M., Lecoeur, C., Meyre, D., Benzinou, M., Mein, C. A., Hinney, A., Vatin, V., Weill, J., Heude, B., Hebebrand, J., Grossman, A. B., Korbonits, M., & Froguel, P. (2009). Association studies on ghrelin and ghrelin receptor gene polymorphisms with obesity. Obesity (Silver Spring), 17(4), 745–754. https://doi.org/10.1038/oby.2008.589.
Gutierrez, J. A., Solenberg, P. J., Perkins, D. R., Willency, J. A., Knierman, M. D., Jin, Z., Witcher, D. R., Luo, S., Onyia, J. E., & Hale, J. E. (2008). Ghrelin octanoylation mediated by an orphan lipid transferase. Proceedings of the National Academy of Sciences of the United States of America, 105(17), 6320–6325. https://doi.org/10.1073/pnas.0800708105.
Hansen, K. B., Rosenkilde, M. M., Knop, F. K., Wellner, N., Diep, T. A., Rehfeld, J. F., Andersen, U. B., Holst, J. J., & Hansen, H. S. (2011). 2-Oleoyl glycerol is a GPR119 agonist and signals GLP-1 release in humans. The Journal of Clinical Endocrinology and Metabolism, 96(9), E1409–E1417. https://doi.org/10.1210/jc.2011-0647.
Hillman, J. B., Tong, J., & Tschop, M. (2011). Ghrelin biology and its role in weight-related disorders. Discovery Medicine, 11(61), 521–528.
Holst, B., Cygankiewicz, A., Jensen, T. H., Ankersen, M., & Schwartz, T. W. (2003). High constitutive signaling of the ghrelin receptor--identification of a potent inverse agonist. Molecular Endocrinology, 17(11), 2201–2210. https://doi.org/10.1210/me.2003-0069.
Holst, J. J. (2007). The physiology of glucagon-like peptide 1. Physiological Reviews, 87(4), 1409–1439. https://doi.org/10.1152/physrev.00034.2006.
Hosoda, H., & Kangawa, K. (2008). The autonomic nervous system regulates gastric ghrelin secretion in rats. Regulatory Peptides, 146(1–3), 12–18. https://doi.org/10.1016/j.regpep.2007.07.005.
Howard, A. D., Feighner, S. D., Cully, D. F., Arena, J. P., Liberator, P. A., Rosenblum, C. I., Hamelin, M., Hreniuk, D. L., Palyha, O. C., Anderson, J., Paress, P. S., Diaz, C., Chou, M., Liu, K. K., McKee, K. K., Pong, S. S., Chaung, L. Y., Elbrecht, A., Dashkevicz, M., Heavens, R., Rigby, M., Sirinathsinghji, D. J., Dean, D. C., Melillo, D. G., Patchett, A. A., Nargund, R., Griffin, P. R., DeMartino, J. A., Gupta, S. K., Schaeffer, J. M., Smith, R. G., & Van der Ploeg, L. H. (1996). A receptor in pituitary and hypothalamus that functions in growth hormone release. Science, 273(5277), 974–977.
Huang, S. C., Yu, D. H., Wank, S. A., Mantey, S., Gardner, J. D., & Jensen, R. T. (1989). Importance of sulfation of gastrin or cholecystokinin (CCK) on affinity for gastrin and CCK receptors. Peptides, 10(4), 785–789.
Huda, M. S. B., Wilding, J. P. H., & Pinkney, J. H. (2006). Gut peptides and the regulation of appetite. Obesity Reviews, 7(2), 163–182. https://doi.org/10.1111/j.1467-789X.2006.00245.x.
Imamura, M. (2002). Effects of surgical manipulation of the intestine on peptide YY and its physiology. Peptides, 23(2), 403–407.
Jones, R. B., McKie, S., Astbury, N., Little, T. J., Tivey, S., Lassman, D. J., McLaughlin, J., Luckman, S., Williams, S. R., Dockray, G. J., & Thompson, D. G. (2012). Functional neuroimaging demonstrates that ghrelin inhibits the central nervous system response to ingested lipid. Gut, 61(11), 1543–1551. https://doi.org/10.1136/gutjnl-2011-301323.
Jordan, J., Greenway, F. L., Leiter, L. A., Li, Z., Jacobson, P., Murphy, K., Hill, J., Kler, L., & Aftring, R. P. (2008). Stimulation of cholecystokinin-a receptors with GI181771X does not cause weight loss in overweight or obese patients. Clinical Pharmacology and Therapeutics, 83(2), 281–287. https://doi.org/10.1038/sj.clpt.6100272.
Karra, E., Chandarana, K., & Batterham, R. L. (2009). The role of peptide YY in appetite regulation and obesity. The Journal of Physiology, 587(1), 19–25. https://doi.org/10.1113/jphysiol.2008.164269.
Khandekar, N., Berning, B. A., Sainsbury, A., & Lin, S. (2015). The role of pancreatic polypeptide in the regulation of energy homeostasis. Molecular and Cellular Endocrinology, 418(Pt 1), 33–41. https://doi.org/10.1016/j.mce.2015.06.028.
Kimmel, J. R., Hayden, L. J., & Pollock, H. G. (1975). Isolation and characterization of a new pancreatic polypeptide hormone. The Journal of Biological Chemistry, 250(24), 9369–9376.
Kirchner, H., Tong, J., Tschöp, M. H., & Pfluger, P. T. (2010). Ghrelin and PYY in the regulation of energy balance and metabolism: Lessons from mouse mutants. American Journal of Physiology. Endocrinology and Metabolism, 298(5), E909–E919. https://doi.org/10.1152/ajpendo.00191.2009.
Kissileff, H. R., Pi-Sunyer, F. X., Thornton, J., & Smith, G. P. (1981). C-terminal octapeptide of cholecystokinin decreases food intake in man. The American Journal of Clinical Nutrition, 34(2), 154–160.
Kojima, M., Hosoda, H., Date, Y., Nakazato, M., Matsuo, H., & Kangawa, K. (1999). Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature, 402(6762), 656–660. https://doi.org/10.1038/45230.
Kojima, M., & Kangawa, K. (2010). Ghrelin: More than endogenous growth hormone secretagogue. Annals of the New York Academy of Sciences, 1200, 140–148. https://doi.org/10.1111/j.1749-6632.2010.05516.x.
Kumar, U., & Grant, M. (2010). Somatostatin and somatostatin receptors. Results and Problems in Cell Differentiation, 50, 137–184. https://doi.org/10.1007/400_2009_29.
Kurashina, T., Dezaki, K., Yoshida, M., Sukma Rita, R., Ito, K., Taguchi, M., Miura, R., Tominaga, M., Ishibashi, S., Kakei, M., & Yada, T. (2015). The beta-cell GHSR and downstream cAMP/TRPM2 signaling account for insulinostatic and glycemic effects of ghrelin. Scientific Reports, 5, 14041. https://doi.org/10.1038/srep14041.
le Roux, C. W., Patterson, M., Vincent, R. P., Hunt, C., Ghatei, M. A., & Bloom, S. R. (2005). Postprandial plasma ghrelin is suppressed proportional to meal calorie content in normal-weight but not obese subjects. The Journal of Clinical Endocrinology and Metabolism, 90(2), 1068–1071. https://doi.org/10.1210/jc.2004-1216.
Lean, M. E., & Malkova, D. (2016). Altered gut and adipose tissue hormones in overweight and obese individuals: Cause or consequence? International Journal of Obesity, 40(4), 622–632. https://doi.org/10.1038/ijo.2015.220.
Levin, F., Edholm, T., Schmidt, P. T., Gryback, P., Jacobsson, H., Degerblad, M., Hoybye, C., Holst, J. J., Rehfeld, J. F., Hellstrom, P. M., & Naslund, E. (2006). Ghrelin stimulates gastric emptying and hunger in normal-weight humans. The Journal of Clinical Endocrinology and Metabolism, 91(9), 3296–3302. https://doi.org/10.1210/jc.2005-2638.
Li, P., Tiwari, H. K., Lin, W. Y., Allison, D. B., Chung, W. K., Leibel, R. L., Yi, N., & Liu, N. (2014). Genetic association analysis of 30 genes related to obesity in a European American population. International Journal of Obesity, 38(5), 724–729. https://doi.org/10.1038/ijo.2013.140.
Liddle, R. A., Goldfine, I. D., Rosen, M. S., Taplitz, R. A., & Williams, J. A. (1985). Cholecystokinin bioactivity in human plasma. Molecular forms, responses to feeding, and relationship to gallbladder contraction. The Journal of Clinical Investigation, 75(4), 1144–1152. https://doi.org/10.1172/JCI111809.
Lieverse, R. J., Jansen, J. B., Masclee, A. A., & Lamers, C. B. (1995). Satiety effects of a physiological dose of cholecystokinin in humans. Gut, 36(2), 176–179.
Little, T. J., Doran, S., Meyer, J. H., Smout, A. J., O’Donovan, D. G., Wu, K. L., Jones, K. L., Wishart, J., Rayner, C. K., Horowitz, M., & Feinle-Bisset, C. (2006). The release of GLP-1 and ghrelin, but not GIP and CCK, by glucose is dependent upon the length of small intestine exposed. American Journal of Physiology. Endocrinology and Metabolism, 291(3), E647–E655. https://doi.org/10.1152/ajpendo.00099.2006.
Liu, B., Garcia, E. A., & Korbonits, M. (2011). Genetic studies on the ghrelin, growth hormone secretagogue receptor (GHSR) and ghrelin O-acyl transferase (GOAT) genes. Peptides, 32(11), 2191–2207. https://doi.org/10.1016/j.peptides.2011.09.006.
Lopez Soto, E. J., Agosti, F., Cabral, A., Mustafa, E. R., Damonte, V. M., Gandini, M. A., Rodriguez, S., Castrogiovanni, D., Felix, R., Perello, M., & Raingo, J. (2015). Constitutive and ghrelin-dependent GHSR1a activation impairs CaV2.1 and CaV2.2 currents in hypothalamic neurons. The Journal of General Physiology, 146(3), 205–219. https://doi.org/10.1085/jgp.201511383.
Low, M. J. (2004). Clinical endocrinology and metabolism. The somatostatin neuroendocrine system: Physiology and clinical relevance in gastrointestinal and pancreatic disorders. Best Practice & Research Clinical Endocrinology & Metabolism, 18(4), 607–622. https://doi.org/10.1016/j.beem.2004.08.005.
M’Kadmi, C., Leyris, J. P., Onfroy, L., Gales, C., Sauliere, A., Gagne, D., Damian, M., Mary, S., Maingot, M., Denoyelle, S., Verdie, P., Fehrentz, J. A., Martinez, J., Baneres, J. L., & Marie, J. (2015). Agonism, antagonism, and inverse Agonism Bias at the ghrelin receptor signaling. The Journal of Biological Chemistry, 290(45), 27021–27039. https://doi.org/10.1074/jbc.M115.659250.
Mace, O. J., Tehan, B., & Marshall, F. (2015). Pharmacology and physiology of gastrointestinal enteroendocrine cells. Pharmacology Research & Perspectives, 3(4), e00155. https://doi.org/10.1002/prp2.155.
Manning, S., & Batterham, R. L. (2014). The role of gut hormone peptide YY in energy and glucose homeostasis: Twelve years on. Annual Review of Physiology, 76, 585–608. https://doi.org/10.1146/annurev-physiol-021113-170404.
Mayo, K. E., Miller, L. J., Bataille, D., Dalle, S., Goke, B., Thorens, B., & Drucker, D. J. (2003). International Union of Pharmacology. XXXV. The glucagon receptor family. Pharmacological Reviews, 55(1), 167–194. https://doi.org/10.1124/pr.55.1.6.
Miller, L. J., & Desai, A. J. (2016). Metabolic actions of the type 1 cholecystokinin receptor: Its potential as a therapeutic target. Trends in Endocrinology and Metabolism: TEM, 27(9), 609–619. https://doi.org/10.1016/j.tem.2016.04.002.
Mohlig, M., Spranger, J., Otto, B., Ristow, M., Tschop, M., & Pfeiffer, A. F. (2002). Euglycemic hyperinsulinemia, but not lipid infusion, decreases circulating ghrelin levels in humans. Journal of Endocrinological Investigation, 25(11), RC36–RC38. https://doi.org/10.1007/BF03344062.
Mojsov, S., Heinrich, G., Wilson, I. B., Ravazzola, M., Orci, L., & Habener, J. F. (1986). Preproglucagon gene expression in pancreas and intestine diversifies at the level of post-translational processing. The Journal of Biological Chemistry, 261(25), 11880–11889.
Morpurgo, P. S., Resnik, M., Agosti, F., Cappiello, V., Sartorio, A., & Spada, A. (2003). Ghrelin secretion in severely obese subjects before and after a 3-week integrated body mass reduction program. Journal of Endocrinological Investigation, 26(8), 723–727. https://doi.org/10.1007/BF03347353.
Mortensen, K., Christensen, L. L., Holst, J. J., & Orskov, C. (2003). GLP-1 and GIP are colocalized in a subset of endocrine cells in the small intestine. Regulatory Peptides, 114(2–3), 189–196.
Nauck, M. A., Niedereichholz, U., Ettler, R., Holst, J. J., Orskov, C., Ritzel, R., & Schmiegel, W. H. (1997). Glucagon-like peptide 1 inhibition of gastric emptying outweighs its insulinotropic effects in healthy humans. The American Journal of Physiology, 273(5 Pt 1), E981–E988.
Nogueiras, R., Pfluger, P., Tovar, S., Arnold, M., Mitchell, S., Morris, A., Perez-Tilve, D., Vazquez, M. J., Wiedmer, P., Castaneda, T. R., DiMarchi, R., Tschop, M., Schurmann, A., Joost, H. G., Williams, L. M., Langhans, W., & Dieguez, C. (2007). Effects of obestatin on energy balance and growth hormone secretion in rodents. Endocrinology, 148(1), 21–26. https://doi.org/10.1210/en.2006-0915.
Orskov, C., Holst, J. J., Knuhtsen, S., Baldissera, F. G., Poulsen, S. S., & Nielsen, O. V. (1986). Glucagon-like peptides GLP-1 and GLP-2, predicted products of the glucagon gene, are secreted separately from pig small intestine but not pancreas. Endocrinology, 119(4), 1467–1475. https://doi.org/10.1210/endo-119-4-1467.
Orskov, C., Poulsen, S. S., Moller, M., & Holst, J. J. (1996). Glucagon-like peptide I receptors in the subfornical organ and the area postrema are accessible to circulating glucagon-like peptide I. Diabetes, 45(6), 832–835.
Overduin, J., Gibbs, J., Cummings, D. E., & Reeve, J. R., Jr. (2014). CCK-58 elicits both satiety and satiation in rats while CCK-8 elicits only satiation. Peptides, 54, 71–80. https://doi.org/10.1016/j.peptides.2014.01.008.
Owyang, C., & Heldsinger, A. (2011). Vagal control of satiety and hormonal regulation of appetite. Journal of Neurogastroenterology and Motility, 17(4), 338–348. https://doi.org/10.5056/jnm.2011.17.4.338.
Parker, H. E., Reimann, F., & Gribble, F. M. (2010). Molecular mechanisms underlying nutrient-stimulated incretin secretion. Expert Reviews in Molecular Medicine, 12, e1. https://doi.org/10.1017/S146239940900132X.
Perello, M., & Nillni, E. A. (2007). The biosynthesis and processing of neuropeptides: Lessons from prothyrotropin releasing hormone (proTRH). Frontiers in Bioscience: A Journal and Virtual Library, 12, 3554–3565.
Perello, M., & Raingo, J. (2014). Central ghrelin receptors and food intake. In J. Portelli & I. Smolders (Eds.), Central functions of the ghrelin receptor (pp. 65–88). New York: Springer. https://doi.org/10.1007/978-1-4939-0823-3_5.
Perello, M., & Zigman, J. M. (2012). The role of ghrelin in reward-based eating. Biological Psychiatry, 72(5), 347–353. https://doi.org/10.1016/j.biopsych.2012.02.016.
Persaud, S. J., & Bewick, G. A. (2014). Peptide YY: More than just an appetite regulator. Diabetologia, 57(9), 1762–1769. https://doi.org/10.1007/s00125-014-3292-y.
Peterli, R., Steinert, R. E., Woelnerhanssen, B., Peters, T., Christoffel-Courtin, C., Gass, M., Kern, B., von Fluee, M., & Beglinger, C. (2012). Metabolic and hormonal changes after laparoscopic roux-en-Y gastric bypass and sleeve gastrectomy: A randomized, prospective trial. Obesity Surgery, 22(5), 740–748. https://doi.org/10.1007/s11695-012-0622-3.
Pocai, A. (2014). Action and therapeutic potential of oxyntomodulin. Molecular Metabolism, 3(3), 241–251. https://doi.org/10.1016/j.molmet.2013.12.001.
Rai, U., Thrimawithana, T. R., Valery, C., & Young, S. A. (2015). Therapeutic uses of somatostatin and its analogues: Current view and potential applications. Pharmacology & Therapeutics, 152, 98–110. https://doi.org/10.1016/j.pharmthera.2015.05.007.
Reeve, J. R., Jr., Eysselein, V. E., Rosenquist, G., Zeeh, J., Regner, U., Ho, F. J., Chew, P., Davis, M. T., Lee, T. D., Shively, J. E., Brazer, S. R., & Liddle, R. A. (1996). Evidence that CCK-58 has structure that influences its biological activity. The American Journal of Physiology, 270(5 Pt 1), G860–G868.
Rhee, N. A., Vilsboll, T., & Knop, F. K. (2012). Current evidence for a role of GLP-1 in roux-en-Y gastric bypass-induced remission of type 2 diabetes. Diabetes, Obesity & Metabolism, 14(4), 291–298. https://doi.org/10.1111/j.1463-1326.2011.01505.x.
Sandoval, D. A., & D’Alessio, D. A. (2015). Physiology of proglucagon peptides: Role of glucagon and GLP-1 in health and disease. Physiological Reviews, 95(2), 513–548. https://doi.org/10.1152/physrev.00013.2014.
Schellekens, H., De Francesco, P. N., Kandil, D., Theeuwes, W. F., McCarthy, T., van Oeffelen, W. E., Perello, M., Giblin, L., Dinan, T. G., & Cryan, J. F. (2015). Ghrelin’s Orexigenic effect is modulated via a serotonin 2C receptor interaction. ACS Chemical Neuroscience, 6(7), 1186–1197. https://doi.org/10.1021/cn500318q.
Schwartz, G. J., McHugh, P. R., & Moran, T. H. (1993). Gastric loads and cholecystokinin synergistically stimulate rat gastric vagal afferents. The American Journal of Physiology, 265(4 Pt 2), R872–R876.
Sjostrom, L., Narbro, K., Sjostrom, C. D., Karason, K., Larsson, B., Wedel, H., Lystig, T., Sullivan, M., Bouchard, C., Carlsson, B., Bengtsson, C., Dahlgren, S., Gummesson, A., Jacobson, P., Karlsson, J., Lindroos, A. K., Lonroth, H., Naslund, I., Olbers, T., Stenlof, K., Torgerson, J., Agren, G., & Carlsson, L. M. (2007). Effects of bariatric surgery on mortality in Swedish obese subjects. The New England Journal of Medicine, 357(8), 741–752. https://doi.org/10.1056/NEJMoa066254.
Spreckley, E., & Murphy, K. G. (2015). The L-cell in nutritional sensing and the regulation of appetite. Frontiers in Nutrition, 2, 23. https://doi.org/10.3389/fnut.2015.00023.
Stadlbauer, U., Woods, S. C., Langhans, W., & Meyer, U. (2015). PYY3-36: Beyond food intake. Frontiers in Neuroendocrinology, 38, 1–11. https://doi.org/10.1016/j.yfrne.2014.12.003.
Stanley, S., Wynne, K., & Bloom, S. (2004). Gastrointestinal satiety signals III. Glucagon-like peptide 1, oxyntomodulin, peptide YY, and pancreatic polypeptide. American Journal of Physiology Gastrointestinal and Liver Physiology, 286(5), G693–G697. https://doi.org/10.1152/ajpgi.00536.2003.
Steinert, R. E., Feinle-Bisset, C., Asarian, L., Horowitz, M., Beglinger, C., & Geary, N. (2017). Ghrelin, CCK, GLP-1, and PYY(3-36): Secretory controls and physiological roles in eating and Glycemia in health, obesity, and after RYGB. Physiological Reviews, 97(1), 411–463. https://doi.org/10.1152/physrev.00031.2014.
Stewart, J. E., Seimon, R. V., Otto, B., Keast, R. S., Clifton, P. M., & Feinle-Bisset, C. (2011). Marked differences in gustatory and gastrointestinal sensitivity to oleic acid between lean and obese men. The American Journal of Clinical Nutrition, 93(4), 703–711. https://doi.org/10.3945/ajcn.110.007583.
Svendsen, B., Pedersen, J., Albrechtsen, N. J., Hartmann, B., Torang, S., Rehfeld, J. F., Poulsen, S. S., & Holst, J. J. (2015). An analysis of cosecretion and coexpression of gut hormones from male rat proximal and distal small intestine. Endocrinology, 156(3), 847–857. https://doi.org/10.1210/en.2014-1710.
Takahashi, T., Ida, T., Sato, T., Nakashima, Y., Nakamura, Y., Tsuji, A., & Kojima, M. (2009). Production of n-octanoyl-modified ghrelin in cultured cells requires prohormone processing protease and ghrelin O-acyltransferase, as well as n-octanoic acid. Journal of Biochemistry, 146(5), 675–682. https://doi.org/10.1093/jb/mvp112.
Tatemoto, K., & Mutt, V. (1980). Isolation of two novel candidate hormones using a chemical method for finding naturally occurring polypeptides. Nature, 285(5764), 417–418.
Tong, J., Prigeon, R. L., Davis, H. W., Bidlingmaier, M., Tschop, M. H., & D’Alessio, D. (2013). Physiologic concentrations of exogenously infused ghrelin reduces insulin secretion without affecting insulin sensitivity in healthy humans. The Journal of Clinical Endocrinology and Metabolism, 98(6), 2536–2543. https://doi.org/10.1210/jc.2012-4162.
Trapp, S., & Hisadome, K. (2011). Glucagon-like peptide 1 and the brain: Central actions-central sources? Autonomic Neuroscience: Basic & Clinical, 161(1–2), 14–19. https://doi.org/10.1016/j.autneu.2010.09.008.
Troke, R. C., Tan, T. M., & Bloom, S. R. (2014). The future role of gut hormones in the treatment of obesity. Therapeutic Advances in Chronic Disease, 5(1), 4–14. https://doi.org/10.1177/2040622313506730.
Tschop, M., Weyer, C., Tataranni, P. A., Devanarayan, V., Ravussin, E., & Heiman, M. L. (2001). Circulating ghrelin levels are decreased in human obesity. Diabetes, 50(4), 707–709.
Valle, J. D. (2014). Gastrointestinal hormones in the regulation of gut function in health and disease. In Gastrointestinal anatomy and physiology (pp. 15–32). John Wiley & Sons, Ltd. https://doi.org/10.1002/9781118833001.ch2.
Vilsboll, T., Krarup, T., Madsbad, S., & Holst, J. J. (2003). Both GLP-1 and GIP are insulinotropic at basal and postprandial glucose levels and contribute nearly equally to the incretin effect of a meal in healthy subjects. Regulatory Peptides, 114(2–3), 115–121.
Wang, Y., Prpic, V., Green, G. M., Reeve, J. R., Jr., & Liddle, R. A. (2002). Luminal CCK-releasing factor stimulates CCK release from human intestinal endocrine and STC-1 cells. American Journal of Physiology Gastrointestinal and Liver Physiology, 282(1), G16–G22.
Wank, S. A. (1995). Cholecystokinin receptors. The American Journal of Physiology, 269(5 Pt 1), G628–G646.
Wheeler, M. B., Lu, M., Dillon, J. S., Leng, X. H., Chen, C., & Boyd, A. E., 3rd. (1993). Functional expression of the rat glucagon-like peptide-I receptor, evidence for coupling to both adenylyl cyclase and phospholipase-C. Endocrinology, 133(1), 57–62. https://doi.org/10.1210/endo.133.1.8391428.
Whited, K. L., Thao, D., Lloyd, K. C., Kopin, A. S., & Raybould, H. E. (2006). Targeted disruption of the murine CCK1 receptor gene reduces intestinal lipid-induced feedback inhibition of gastric function. American Journal of Physiology Gastrointestinal and Liver Physiology, 291(1), G156–G162. https://doi.org/10.1152/ajpgi.00569.2005.
Williams, D. L. (2009). Minireview: Finding the sweet spot: Peripheral versus central glucagon-like peptide 1 action in feeding and glucose homeostasis. Endocrinology, 150(7), 2997–3001. https://doi.org/10.1210/en.2009-0220.
Woods, S. C. (2004). Gastrointestinal satiety signals I. An overview of gastrointestinal signals that influence food intake. American Journal of Physiology Gastrointestinal and Liver Physiology, 286(1), G7–G13. https://doi.org/10.1152/ajpgi.00448.2003.
Wren, A. M., & Bloom, S. R. (2007). Gut hormones and appetite control. Gastroenterology, 132(6), 2116–2130. https://doi.org/10.1053/j.gastro.2007.03.048.
Wynne, K., & Bloom, S. R. (2006). The role of oxyntomodulin and peptide tyrosine-tyrosine (PYY) in appetite control. Nature Clinical Practice. Endocrinology & Metabolism, 2(11), 612–620. https://doi.org/10.1038/ncpendmet0318.
Yamamoto, M., Reeve, J. R., Jr., Keire, D. A., & Green, G. M. (2005). Water and enzyme secretion are tightly coupled in pancreatic secretion stimulated by food or CCK-58 but not by CCK-8. American Journal of Physiology Gastrointestinal and Liver Physiology, 288(5), G866–G879. https://doi.org/10.1152/ajpgi.00389.2003.
Yang, J., Brown, M. S., Liang, G., Grishin, N. V., & Goldstein, J. L. (2008). Identification of the acyltransferase that octanoylates ghrelin, an appetite-stimulating peptide hormone. Cell, 132(3), 387–396. https://doi.org/10.1016/j.cell.2008.01.017.
Yang, N., Liu, X., Ding, E. L., Xu, M., Wu, S., Liu, L., Sun, X., & Hu, F. B. (2009). Impaired ghrelin response after high-fat meals is associated with decreased satiety in obese and lean Chinese young adults. The Journal of Nutrition, 139(7), 1286–1291. https://doi.org/10.3945/jn.109.104406.
Yi, J., Warunek, D., & Craft, D. (2015). Degradation and stabilization of peptide hormones in human blood specimens. PLoS One, 10(7), e0134427. https://doi.org/10.1371/journal.pone.0134427.
Zhang, J. V., Ren, P. G., Avsian-Kretchmer, O., Luo, C. W., Rauch, R., Klein, C., & Hsueh, A. J. (2005). Obestatin, a peptide encoded by the ghrelin gene, opposes ghrelin’s effects on food intake. Science, 310(5750), 996–999. https://doi.org/10.1126/science.1117255.
Zhao, T. J., Sakata, I., Li, R. L., Liang, G., Richardson, J. A., Brown, M. S., Goldstein, J. L., & Zigman, J. M. (2010). Ghrelin secretion stimulated by {beta}1-adrenergic receptors in cultured ghrelinoma cells and in fasted mice. Proceedings of the National Academy of Sciences of the United States of America, 107(36), 15868–15873. https://doi.org/10.1073/pnas.1011116107.
Zhu, X., Cao, Y., Voogd, K., & Steiner, D. F. (2006). On the processing of proghrelin to ghrelin. The Journal of Biological Chemistry, 281(50), 38867–38870. https://doi.org/10.1074/jbc.M607955200.
Disclosure Statement
The authors have nothing to disclose.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Andreoli, M.F., De Francesco, P.N., Perello, M. (2018). Gastrointestinal Hormones Controlling Energy Homeostasis and Their Potential Role in Obesity. In: Nillni, E. (eds) Textbook of Energy Balance, Neuropeptide Hormones, and Neuroendocrine Function. Springer, Cham. https://doi.org/10.1007/978-3-319-89506-2_7
Download citation
DOI: https://doi.org/10.1007/978-3-319-89506-2_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-89505-5
Online ISBN: 978-3-319-89506-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)