
Abstract
Human tissues differ by the type and the amount of energy substrate that they use and store, as dictated by their genetically programmed “metabolic rigidity” which defines the expression of tissue-specific catabolic and anabolic enzymes. For instance, adipocytes and neurons cannot perform fat oxidation, while cardiomyocytes utilize mainly fatty acids for ATP synthesis. These programs evolve during embryogenesis, with a typical burst of oxidative metabolism at birth. Bioenergetic homeostasis is regulated at the cellular level by effectors such as insulin and at the organismal level by the metabolic coupling between “energy donor” tissues, as the adipose tissue and the liver, and “receivers” and the muscle and the brain. In contrast to normal tissues, most tumors develop a dramatic “metabolic flexibility,” which allows them to consume whatever energy substrate is available. Yet, recent findings suggest that this flexibility is restricted by specific anabolic needs, so that alternative energy-transducing pathways are not selected randomly. Cancer cells also avoid the metabolic control by insulin, growth factors, and energy substrate starvation or excess by typical mutations in the MAPK, PI3K, RAS, and related pathways. Metabolic flexibility is made possible by the molecular rewiring of several metabolic pathways, which undergo branching, truncation, and reversal, as extensively documented for the Krebs cycle in the last decade. Like normal tissues, tumors can take advantage of metabolic coupling with other tissues, although this phenomenon remains poorly described. A related bioenergetic peculiarity of cancer cells concerns the metabolic coupling which occurs inside the tumors, between hypoxic-glycolytic and oxygenated-oxidative cancer cells. Undoubtedly, the fundamental studies on metabolic rigidity in normal tissues and on metabolic flexibility in cancer cells have revealed novel means of bioenergetic control, such as the essential signaling activity of the oncometabolites succinate, fumarate, oxoglutarate, or acetyl-CoA on HIF1α and NRF2 transcription factors, but also histone acetylases, respectively. The well-known regulation of OXPHOS by citrate, NADH, F1,6BP, Ca2+, and ATP on glycolytic enzymes IDH, PDH, and CIV also operates in cancer cells. Besides energy homeostasis, the study of cancer cell metabolic remodeling has shed light on the close links between the modalities of energy transduction and the capacity to synthesize amino acids or lipids necessary for tumor growth. Metabolic remodeling is not restricted to cancer and has been investigated in genetically inherited metabolic disorders as mitochondrial diseases or in the multifactorial metabolic syndrome. The results demonstrated the existence of alternative pathways of ATP synthesis and the effect of oxidative stress on TCA truncation via aconitase cleavage. Nowadays, large-scale analyses of comprehensive proteomics, transcriptomics, and metabolomics, potentially supported by computer modeling of metabolism and genetic approaches of subpathways validation, allow the deciphering of the fine changes in cell metabolism, both in physiology and pathology. Patterns of metabolic deviations and the corresponding genetic signatures can be confirmed by flux analyses using 13C-labeled isotopes of relevant metabolites. Analysis of deviant metabolism can identify potential targets so that therapeutic strategies could be derived from this knowledge, and ongoing preclinical studies aim to alter the alternative metabolic pathways in cancer.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Energy Metabolism, Control, and Regulation
The basic principles of energy metabolism regulation were deciphered in the late 1950s with the work of Warburg, Lenhinger, Krebs, Chance, Petersen, Weinhouse, and Vaupel among several others (Scheffler 1999; Weinhouse 1956). The regulation of controlling enzymes belonging to glycolysis, PDH complex, and Krebs cycle, all involved in ATP synthesis, mostly occurs by metabolic intermediates as ATP itself, citrate, F1, 6BP, and Pi. Another level of regulation of mitochondrial energy fluxes, as ATP synthesis or respiration was identified by Chance and Williams in the 1950s with the so-called respiratory control by ADP (Chance and Williams 1956; Cogliati et al. 2013). Thereafter, a large number of additional molecular regulations of oxidative phosphorylation (OXPHOS) were identified, as the recently discovered OPA1-dependent stabilization of the respiratory supercomplexes (Cogliati et al. 2013), the ATP synthase-dependent assembly of complex III (Ostojić et al. 2013), and the energy state-dependent RHEB-induced control of mitochondrial turnover (Melser et al. 2013). Consideration of the numerous means to regulate ATP transduction in the cell led to the notion of a “multistep control” of energy metabolism (Benard et al. 2010). More recently, the regulation of energy metabolism was closely linked, in a mutual way, with the control of cell growth and division. For instance, a signaling pathway central to cell biology and governed by the HiF1α transcription factor was shown to mediate a shift in a subunit of respiratory chain complex IV (Fukuda et al. 2007). Conversely, succinate accumulation in the cytosol is capable of inhibiting HiF1α degradation and to promote its stabilization (Pollard et al. 2005). Likewise, the AMP-activated protein kinase (AMPK) pathway stimulates the expression of several OXPHOS proteins when ATP needs are increased, as testified by a higher ADP/ATP ratio in the cytosol (Hardie et al. 2003). Another central pathway is the control of energy metabolism is the PGC1α pathway, a transcription co-activator, which participates in the stimulation of oxidative phosphorylation in cooperation with ERR-α or to the induction of gluconeogenesis in cooperation with HNFAα (Lustig et al. 2011). The RAS protein, involved in the control of cell mitogenic activities, also controls oxidative phosphorylation, both in cancer and noncancer tissues (Wei et al. 2012; Palorini et al. 2013; Gough et al. 2009; Telang et al. 2007). A role in modulation of OXPHOS capacity was also discovered for MYC and for p53, both of which play central roles in the control of cell growth and division, leading to the emerging concept of oncobioenergetics (Jose and Rossignol 2013). Central to this review article, a new layer of upper- or meta-regulation of energy metabolism was identified with the discovery of rewiring of metabolic circuits, governed by genetic determinants connected or not with changes in cell microenvironment. In particular, this upper level of bioenergetic control makes the link between catabolism and anabolism, thereby providing a more integrated view of cell metabolism plasticity. Prior to discussing the molecular bases and the physiology of metabolic remodeling, we provide below a rapid overview on cellular bioenergetics.
In most human tissues, mitochondria provide the energy necessary for cell growth and biological activities. It has been estimated that about 90 % of mammalian oxygen consumption is mitochondrial, which primarily serves to synthesize ATP, although in variable levels according to the tissue considered and the organism’s activity status. Mitochondria intervene in the ultimate phase of cellular catabolism, following the enzymatic reactions of intermediate metabolism that degrade carbohydrates, fats, and proteins into smaller molecules such as pyruvate, fatty acids, and amino acids, respectively (Fig. 1.1). Mitochondria further transform these energetic elements into NADH and/or FADH2, through β-oxidation and the Krebs cycle. Those reduced equivalents are then degraded by the mitochondrial respiratory chain in a global energy converting process called oxidative phosphorylation (OXPHOS) where the electrons liberated by the oxidation of NADH and FADH2 are passed along a series of carriers regrouped under the name of “respiratory chain” or “electron transport chain” (ETC) and ultimately transferred to molecular oxygen (Fig. 1.2). ETC is located in the mitochondrial inner membrane, with an enrichment in the cristae. ETC consists of four enzyme complexes (complexes I–IV) and two mobile electron carriers (coenzyme Q and cytochrome c). These complexes are composed of numerous subunits encoded by both nuclear genes and mitochondrial DNA, with the exception of complex II (nuclear only). It was demonstrated that these complexes assemble into supramolecular assemblies called “supercomplexes” or respirasome (Schägger and Pfeiffer 2000; Schagger 2001). It is still debated whether some complexes, as complex I, can be found alone or if all are embedded in supercomplexes. In addition to the classic ETC components, other proteins are involved in the oxidation of nutrient-derived reduced equivalents and the subsequent reduction of coenzyme Q, used for ultimate ATP synthesis. This is the case for the electron-flavoprotein system, composed of the ETF and the ETF-QO, which connect fatty acid oxidation and coenzyme Q reduction. The glycerol-3-phosphate dehydrogenase, which oxidizes cytosolic NADH to reduce mitochondrial FAD, also supports oxidative phosphorylation and participates to REDOX homeostasis. Lastly, the NADH-shuttling system, as the malate-aspartate shuttle, also supports OXPHOS and REDOX homeostasis via the delivery of cytosolic NADH to the mitochondrial matrix. The oxidation of NADH or FADH2 by complex I or complex II, respectively, triggers the transfer of electrons from complex I (or II) to complex IV and mediates the extrusion of protons from the matrix to the intermembrane space, thus generating an electrochemical gradient of protons (ΔûH +) which is finally used by the F1F0 ATP synthase (i.e., complex V) to produce adenosine triphosphate (ATP), the main energetic currency of the cell. This gradient has two components: an electric potential (ΔΨ) and a chemical potential (ΔμH +) that can also be expressed as a pH gradient (ΔpH). According to the chemiosmotic theory proposed by Peter Mitchell (1961), ΔûH + = ΔΨ − ZΔpH, with Z = −2.303 RT/F.

Overview of energy metabolism pathways. In this chapter we refer to the production of biological energy in the form of adenosine triphosphate or ATP. This process occurs primarily through glycolysis, the end product of which is pyruvate and through subsequent oxidative phosphorylation. In most tissues, the pyruvate enters the mitochondrion and generates acetyl-CoA which is further oxidized at the level of the Krebs cycle to produce ATP, NADH, and FADH2. The latter reduced equivalents are further oxidized by the respiratory chain to generate ATP via chemiosmosis, at the level of the F1F0 ATP synthase. This second mechanism of ATP production is referred to as oxidative phosphorylation. The Krebs cycle can also process alpha-ketoglutarate formed from glutamine, via glutaminolysis, or acetyl-CoA generated from fatty acids beta-oxidation. Those anaplerotic pathways are of particular importance in cancer cells. The citrate produced in the Krebs cycle can also escape this cycle (truncated Krebs cycle) and serve for lipid synthesis. As discussed in this chapter, this canonic description of energy metabolism does not apply to several cancers where the pathways are truncated (glycolysis and Krebs), rewired (anaplerotic entries from canonical or noncanonical glutaminolysis), and branched (lipid or serine synthesis from glycolysis). Therefore, while the pentose phosphate pathway and the Krebs cycle generate both reducing equivalents (NADH, NADPH, FADH2), ATP and GTP used for energy needs, these pathways also produce intermediates such as 3PG used for biosynthesis. Some metabolites such as fumarate can also modulate transcription factors as NRF2 while oxoglutarate can serve as substrate for HIF1α degradation and acetyl-CoA for histone acetylation. This figure illustrates the close link between catabolism, anabolism, and genetic/epigenetic regulations. 3-PG 3-phosphoglyceric acid; PPP pentose phosphate pathway; TCA tricarboxylic acid cycle, i.e., Krebs cycle; ETC electron transport chain

The respiratory chain. For mammals, the respiratory chain consists of four enzyme complexes (complexes I–IV) and two intermediary substrates (coenzyme Q and cytochrome c). The NADH, H+, and FADH2 produced by the intermediate metabolism are oxidized further by the mitochondrial respiratory chain to establish an electrochemical gradient of protons, which is finally used by the F1F0-ATP synthase (complex V) to produce ATP, the only form of energy used by the cell. In this simple representation of the respiratory chain, the supramolecular organization (supercomplexes, dimers) is not shown. Of importance for this chapter, electrons can also be delivered to the respiration chain at the level of coenzyme Q by the ETF system or by the glycerol 3 phosphate dehydrogenase system. The respiratory chain can generate reactive oxygen species, and current research in the field of cancer metabolism indicates that such feature plays a role in metabolic remodeling, notably in metastasis. Uncoupling proteins can be expressed in the inner mitochondrial membrane to modulate ROS production. Also, different isoforms of complex IV subunits were found in cancer cells (COX4-1 and COX-2), depending on HIF1α stabilization. Lastly, mutations in mtDNA, impacting respiratory chain complexes activity, were found in a large number of tumors. ETF electron-transferring-flavoprotein dehydrogenase, mtDNA mitochondrial DNA, nDNA nuclear DNA
Under physiological conditions, mitochondrial energy production can alternate between two energy steady states: basically, at state 4 (also denominated the “leak respiration state”), respiration is slow and ATP is not produced (ΔΨ is high), while during state 3, respiration is faster and ATP is largely produced (ΔΨ is lower). In particular conditions, such as mitochondrial inner membrane permeabilization or the use of a chemical uncoupler, ΔΨ can be totally dispersed. As a consequence, respiration is accelerated and ATP production annihilated. The inhibition of respiratory chain complexes also generally decreases ΔΨ. Under physiological conditions, it is considered that mitochondria produce ATP in an intermediate state lying between state 3 and state 4. As shown by E. Gnaiger, respiration strongly depends on the availability of energy substrates which are multiple and can cooperate at the level of the Q-junction, thereby determining the value of the apparent maximal (uncoupled) respiration (Gnaiger 2009). ATP is the only form of energy used by the cell, and when produced in the mitochondrion, it is exported to the cytosol by the adenine nucleotide translocators (ANT1-4) in exchange for cytosolic ADP. Generally, the transport of energy metabolites, nucleotides, and cofactors in and out of the mitochondrial matrix is performed by specific transporters located in the inner membrane (Palmieri and Pierri 2010). These carriers can consume the membrane electrochemical gradient or not, depending on their mechanism of transport (electroneutral or electrogenic). A large part of OXPHOS regulation occurs at the level of these carriers, as shown by the control of the glutamate-aspartate shuttle (SLC25A12 also named Aralar or AGC1) by calcium (Fig. 1.3) and the “Gas pedal” model proposed by Frank Gellerich (Gellerich et al. 2013). Studies of metabolic control also showed that a large part of the control of mitochondrial respiration is located at the level of substrate carriers (Rossignol et al. 2000).

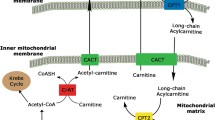
Glutamine import in the mitochondrion. Mitochondria from mammals differ from those of yeast in numerous ways, one being the capacity to import NADH to fuel the respiratory chain. In yeast, a NADH transporter exists. In mammals, the NADH produced by glycolysis and other cytosolic reactions must enter the mitochondrion to be reoxidized by the respiratory chain, and this occurs by NADH-shuttle systems. The malate-aspartate shuttle is shown here. The cytosolic malate dehydrogenase consumes NADH to produce malate from oxaloacetate. This malate enters the mitochondrion (in exchange with alpha-ketoglutarate) where it is converted back into oxaloacetate and NADH. Oxaloacetate is transformed to aspartate by consuming glutamate, using the enzyme glutamate-aspartate aminotransferase. This glutamate is imported by the glutamate-aspartate antiporter (SLC25A12, Aralar, AGC1), so that aspartate is exported to the cytosol where it is converted to oxaloacetate. The net effect of this system in NADH entry in the mitochondrion. GOT1 and -2, cytoplasmic and mitochondrial aspartate aminotransferase, respectively; MDH1 and -2, cytosolic and mitochondrial malate dehydrogenase, respectively; SLC solute carrier family, DHAP dihydroxyacetone phosphate, G3P glyceraldehyde 3-phosphate
Therefore, the regulation of mitochondrial energy production at the level of ETC is concerted and multisite (Fig. 1.4) since modulations have been described at the level of the individual complexes, membrane leak, respirasome cohesion, or carrier activity. One should add to this molecular description the numerous signaling pathways that modulate OXPHOS properties (AMPK, HIF1α, PGC1α, RAS, MYC, …); this has been reviewed elsewhere (Jose et al. 2013). To conclude, the different levels of OXPHOS regulation include (1) the direct modulation of respiratory chain kinetic parameters, (2) modulation of OXPHOS intrinsic efficiency by changes in the basal proton conductance or the induced proton conductance, (3) possible changes in the morphological state of the mitochondrial compartment, (4) modulation of mitochondrial biogenesis and degradation, and (5) in situ regulation of mitochondrial heterogeneity by the cellular and the mitochondrial microenvironment. Most of these regulatory mechanisms of mitochondrial energy transduction were discovered at the level of the respiratory chain and its surrounding lipidic environment. Below, we discuss a wider level of bioenergetic regulation (6) which considers a large-scale modification of the metabolic pathways involved in catabolism and anabolism.

Multisite regulation of OXPHOS. The regulation of ATP production occurs at several sites depicted along the arrow. (1) Regulation of mitobiogenesis (mitochondrial content and mtDNA levels) occurs according to energy needs by, among others, PGC1α, Sirt1, AMPK, and ERRα. Agents shown to affect these processes include AICAR, resveratrol, and bezafibrate. Downregulation occurs via RHEB-dependent bulk degradation or Parkin-dependent targeted degradation. (2) At the level of the electron transport chain (ETC), isoforms and expression levels can differ between tissues (the heart and liver isoforms of COX, for instance) and/or according to current conditions; moreover, supercomplex cohesion can vary, and complexes can harbor different posttranslational modifications, e.g., acetylation which is regulated by sirtuins. Sirtuin inhibitors, some vitamins, and cannabinoids may impact such modifications. (3) OXPHOS efficiency and the coupling of ATP synthesis to NADH and FADH2 oxidation further depend on the intrinsic properties of the proton pumps (slipping), on the ATP synthase, and on the membrane permeability to protons (uncoupling and decoupling can occur). Lipid peroxidation can hamper efficiency; this may be countered by antioxidants. (4) ATP synthesis is modulated also by mitochondrial network dynamics, involving fusion and fission of mitochondria and the overall shape and motility of the mitochondrial network, either fused or fragmented. Moreover, the mitochondrial membrane composition which impacts its fluidity and leakiness to protons can also modulate ATP synthesis; diet is suggested to affect these properties. The roles of fusion and fission proteins as bioenergetic modulators are not yet clear. Mdivi-1 is a small-molecule inhibitor of mitochondrial division. (5) OXPHOS networks or interactions: the principles of mitochondrial bioenergetics and pharmacology must be considered when trying to analyze or extrapolate genotype-phenotype relationships. For instance, the biochemical threshold effect defines a value of inhibition of individual ETC complexes above which the overall flux of respiration will collapse. This value is high (around 70 %) and varies between tissues. The control coefficient of ETC complexes is also different in different tissues, providing a biochemical basis for the tissue specificity of mitochondrial disorders. Another level of bioenergetics regulation concerns energy substrate delivery to the chain, with the phenomena of channeling, metabolic remodeling, and hormonal control of glucose and lipids catabolism. (6) A recent layer of bioenergetics control was found in the form of metabolic remodeling as extensively discussed in this chapter. Genetic or environmentally mediated toxic alterations of each of these levels have been found in human diseases, e.g., mitochondrial diseases, rare motoneuron disorders, metabolic syndrome, and neurodegenerative diseases. Drugs are also being developed to stimulate energy transduction at each of these levels. (7) In particular in cancer research, the role of the microenvironment and pathological tissue in regulation of bioenergetics is attracting increasing attention, for instance, regarding the plasticity of the cancer cell in adapting to various conditions of substrate availability and hypoxia
2 Metabolic Remodeling in Physiology and Metabolic Disorders
The different tissues of the human organism demonstrate a preference for particular energy substrates, dictated by their enzymatic equipment, linked with their physiological function. The adipocytes involved in fat storage do not contain the machinery for β-oxidation, so that fatty acids are not consumed. Likewise, the brain relies mostly on glucose and lactate, with a limited capacity to oxidize fat. Conversely, the heart favors fatty acid oxidation for ATP synthesis, while the skeletal muscle (depending on the type of fibers) consumes glucose, glycerol, fatty acids, ketones, and then amino acids as valine. The liver serves for energy storage in the form of glycerol and also fat. Therefore, tissue-specific differences in the molecular organization of the metabolic pathways determine the metabolic abilities of these tissues, as well as their storage capacity, both qualitatively and quantitatively (Fig. 1.5). Physiology can also adapt to variations in nutrient availability, as metabolic coupling exists between organs, as observed in conditions of fasting. In such situation, the liver can liberate ketone bodies which will be consumed by the brain. Intermediates of amino acids degradation, as 3-hydroxybutyrate, can also be used for gluconeogenesis in different tissues under such conditions. Studies performed in the 1970s revealed genetic mechanisms of metabolic control, as well as described the regulatory role of different hormones, as insulin, leptin, and ghrelin. Therefore, metabolic flexibility can be modulated at the level of the cell, the tissue, or the whole organism by interrelated mechanisms. As discussed below, such regulatory circuits involved in the control of metabolic plasticity could serve for therapeutic intervention in situations of excess food intake or genetic disorders, as suggested for the metabolic syndrome but also for cancer.

Metabolic rigidity and metabolic flexibility. The different tissues present with specificities in the type of substrate primarily used for energy synthesis. A metabolic coupling between tissues exists to cope with this metabolic rigidity. The different tissues also differ in the form of energy they store. When a normal tissue undergoes cancer transformation, this metabolic rigidity is lost and the phenomenon of metabolic flexibility is observed. This is not a general rule as this feature also depends on the bioenergetics environment of the cancer cells
Metabolic syndrome is a multisystemic disease with a complex pathophysiology. One determinant is excess food intake which triggers increased (visceral) fat storage, insulin resistance, and metabolic alteration at the cellular level. Mitochondrial deficiency was observed in models of metabolic syndrome where a reduction of the respiratory rate and an increased production of reactive oxygen species were reported (Curtis et al. 2012). Accordingly, ROS-induced alterations of mitochondrial proteins were also described in the metabolic syndrome, as S-glutathionylations and carbonylations (Curtis et al. 2012). Interestingly, proteomic studies revealed that adaptative processes occurred in different diseased tissues to counter those ROS-induced damages (Peinado et al. 2014). The observed upregulation of enzymes involved in ROS and aldehydes detoxification strengthened the hypothesis of oxidative stress in the pathophysiology of the metabolic syndrome. Murphy and colleagues recently proposed that an excess of ROS could participate in the metabolic remodeling described in tissues from patients with the metabolic syndrome (James et al. 2012). Such remodeling includes the cleavage of aconitase, a TCA enzyme which normally transforms citrate in isocitrate. When aconitase is cleaved (Bulteau et al. 2003), citrate accumulates in the matrix and leaks out from the mitochondrion by a dedicated carrier. In the cytosol, the ATP citrate lyase (ACLY) cleaves citrate in acetyl-CoA and oxaloacetate, so that lipid synthesis can proceed from acetyl-CoA (Hatzivassiliou et al. 2005), while oxaloacetate can enter neoglucogenesis. This circuit of truncated Krebs cycle can accommodate constant overfueling of the mitochondrion by pyruvate generated from carbohydrates. The alteration of the Krebs cycle toward anabolism was also observed in some cancer cells, where truncation of the Krebs cycle allows lipid synthesis from citrate, produced either from pyruvate (as in metabolic syndrome) or from glutamine (as in cancer) (Fig. 1.6).

Examples of metabolic remodeling. In addition to the classic Warburg effect (high glycolysis and poor OXPHOS), other types of metabolic remodeling were recently described in cancer cells. They include lipid synthesis from glutamine or oxidative tumors deriving ATP from fatty acid oxidation and from amino acid degradation. This is discussed in the text
In the case of cancer, there are no excess food intake, but a strong demand for both ATP production and various biosyntheses as lipids. The role of ACLY goes beyond lipid synthesis, as the acetyl-CoA generated by this enzyme can serve for histone acetylation, and the subsequent regulation of bioenergetic genes expression, as LDHA (Wellen et al. 2009). This loop of bioenergetic control evidenced in cancer was not studied in metabolic syndrome. Another example of metabolic remodeling in metabolic disorders was described in genetic mitochondrial diseases (Saada 2014). The group of M.J. Falk recently investigated the common downstream effects of primary respiratory chain dysfunction on global gene expression and pathway regulation. To this end, a bioinformatic analysis of transcriptome datasets from all publicly available studies of respiratory dysfunction resulting from genetic disorders, acute pathophysiologic processes, or environmental toxins was developed.
This analysis revealed the cellular and tissue adaptative response to mitochondrial dysfunction, which identified several commonly dysregulated genes across diverse mitochondrial diseases etiologies, models, and tissue types (Zhang and Falk 2014; Zhang et al. 2013). In particular, the so-called integrated nutrient-sensing signaling network (NSSN) centered on the AKT/mTORC pathways appeared to be one central mediator of the cellular response to respiratory chain dysfunction. NSSN includes the AMPK (low energy sensor), mTORC1 (cell growth regulator by balancing cytosolic protein synthesis and autophagy), SREBP (lipid homeostasis), FOXO1 (glucose homeostasis), and PPAR family transcription factors (lipid metabolism), as well as YY1/PGC1α (mitochondrial ribosome biogenesis) and HIF1α (hypoxia response) transcription factors. The metabolic remodeling suggested by such transcriptomic analyses and the associated GSEA and KEGG functional analyses revealed a modulation of the genes involved in fatty acid and amino acid metabolism, as a central feature of metabolic remodeling in mitochondrial diseases. This type of study indicates that cells or tissues carrying an ETC defect do not simply rely of the Pasteur effect to activate glycolysis to generate ATP, but that a more profound metabolic remodeling occurs to fulfill other needs that remain to be identified, in order to propose innovative therapeutic approaches. A recent study on resveratrol showed that fibroblasts from patients carrying a complex I or a complex IV defect can be rescued at the level of respiration and ATP synthesis by treatment with this drug (Lopes Costa et al. 2014). In this study, two types of patients were identified as responders or nonresponders. The differences between these patients are not well understood at the molecular level, and a thorough proteomic analysis of metabolic remodeling could provide such information.
3 Molecular Basis of the Metabolic Flexibility of Tumors
At the molecular level, metabolic flexibility relies on the rewiring of existing metabolic pathways and the synthesis/degradation of metabolic “pathway switching proteins” or “alternative pathway enhancing proteins” which allow an efficient rerouting of metabolites selected by cellular needs. A thorough investigation of the molecular and signaling mechanisms underlying cancer cells (and other metabolically diseased cells) metabolic remodeling could allow to identify “pathway switching proteins” and “alternative pathway enhancing proteins,” which could be considered as innovative targets for the metabolic therapy of cancer. One strategy to alter the metabolic flexibility of cancer cells resides in the ability to block the catalytic function of PSPs or APEPs. As shown in Fig. 1.6, different types of metabolic remodeling have been reported. The first observation by Otto Warburg revealed that some tumors consumed large amount of glucose without a parallel consumption of oxygen but increased production of lactate (Warburg 1930). Following these seminal findings, many studies addressed the molecular basis of the Warburg effect (Jose and Rossignol 2013). Several mechanisms were identified, as the stimulation of glycolysis by oncogenes (the so-called high glycolysis), notably via the expression of rapid fetal-like isoforms. The Warburg effect also raised the hypothesis of dysfunctional mitochondria, to explain why pyruvate was not degraded by the Krebs cycle. During his Nobel Prize Lecture, at Lindau, Germany, in 1966, Otto Warburg stated that “The prime cause of cancer is the replacement of the respiration of oxygen in normal body cells by a fermentation of sugar.” From the early 1990s until now, molecular mechanisms were discovered at the level of the mitochondrion to explain the Warburg effect, with the inhibition of PDH by PDK1 overexpression (controlled by HIF1α), the reduction of mitochondrial biogenesis (notably triggered by p53 inactivation), or the inhibition of respiratory chain activity [also triggered by mutant p53 via SCO2 and by HIF1 α through a COX4-1/2 subunit isoform shift (Fukuda et al. 2007)].
Yet, it is very important to mention here that not all cancer cells conform to the Warburg effect and that some cancer cells represent an opposite phenotype, i.e., with enhancement of the OXPHOS system (Fig. 1.6). As we discussed in a previous article, a large body of evidence indicates the existence of oxidative cancer cells and tumors both in vitro and in vivo (Jose et al. 2011a). Already in 1976, Reitzer LJ reported that “in HeLa cells glutamine provides more than half of the cellular energy by aerobic oxidation from citric acid cycle metabolism when glucose is present.” Likewise, the idea that glucose, glutamine, hydroxybutyrate, or palmitate can serve both for energy production and anabolism (lipid and cholesterol synthesis) was experimentally tested (Morton et al. 1976). This work demonstrated that freshly excised Morris hepatomas can oxidize palmitate and hydroxybutyrate to produce ATP. The molecular determinants of this oxidative phenotype include the activation of mitochondrial biogenesis, the stimulation of fatty acid oxidation, the stimulation of canonical or noncanonical glutaminolysis, and the activation of amino acid degradation pathways. The “oxidative phenotype” of cancer cells illustrated in Fig. 1.6 (bottom panel where ATP is produced by OXPHOS from fatty acid oxidation or glutamine oxidation) was found in lymphomas, melanomas, glioblastomas, and breast cancer. Between these two extreme phenotypes, i.e., the Warburg (glycolytic) and the “oxidative,” other types of metabolic remodeling were recently described. Indeed, looking at the anabolic side, studies revealed the existence of Krebs cycle truncation aiming at the conversion of glutamine to lipids, via citrate extrusion from the mitochondrion and production of acetyl-CoA using the enzyme ATP citrate lyase.
Two modes of glutamine utilization have been described, with the Krebs cycle running in the textbook direction “clockwise” or in the deviant mode “anticlockwise” (Mullen et al. 2012; Metallo et al. 2012; Fendt et al. 2013; DeBerardinis et al. 2007). The first mode requires anaplerotic entry of glutamine carbons in the TCA at the level of oxoglutarate and the normal route toward citrate, which require the use of acetyl-CoA derived from pyruvate. The second mode of glutaminolysis (anticlockwise) is made possible by the accumulation of oxoglutarate and the presence of high levels of NADH, as found in situations of defective oxoglutarate dehydrogenase and succinate dehydrogenase. Reversal of the truncated TCA is facilitated by the isoform shift of IDH from IDH2 to IDH3 which consumes NADH. Little is known on the metabolic remodeling which utilizes fatty acids as carbon substrates or amino acids as valine, since the acetyl-CoA produced by β-oxidation could serve for ketogenesis but also other means, as reentry in the TCA. It may be pointed out here that most of the metabolic deviations were discovered during attempts to understand the link between mutations in TCA cycle enzymes such as SDH, IDH1/2, and FH and a predisposition to tumors as diverse as hereditary paragangliomas (Niemann and Muller 2000), leiomyomas (Pollard et al. 2005), and glioblastomas multiforme (Parsons et al. 2008). The fate of branched-chain amino acids or ketone bodies is also poorly described, despite reports that indicate the use of such energy sources by different types of tumors (Martinez-Outschoorn et al. 2012). Strong advances in the field of metabolic remodeling were presented by the group of R. DeBerardinis who investigated the fate of glucose and glutamine in different types of cancers, both in vitro and in vivo, even in human subjects (Marin-Valencia et al. 2012a). In 2012, this group reported that glucose oxidation by the mitochondrion is active in glioblastomas, as measured in the mouse brain in vivo (Mullen et al. 2012). Interestingly, glucose was converted to CO2 and glutamine. Analysis of human gliomas xenografts also showed that glutamine can regenerate glucose through neoglucogenesis, evidencing the complexity of tumor metabolic remodeling. As mentioned in the introduction, neoglucogenesis typically occurs in the liver, while here malignant brain cells are capable of doing so from glutamine. The study of brain tumors metabolism by NMR showed that 13C glucose is converted to lactate, glycine, glutamate, and glutamine, indicating again the importance of glutamine synthesis in brain tumors (Marin-Valencia et al. 2012b).
4 The Signaling Pathways Involved in Metabolic Remodeling
The understanding of metabolic remodeling in cancer cells and other diseases also requires the investigation of the signaling mechanisms involved in pathways switching. So far, many determinants have been found to explain the Warburg type of metabolic remodeling, with well-described roles for HIF1α, MYC, p53, PTEN, PI3K, Akt, LKB1, and AMPK on most glycolytic enzymes and LDH. At the level of the mitochondrion, PDH inhibition depends on HIF1α, and glutaminolysis is activated by MYC, as well as lipid synthesis from citrate (at the level of ACLY). A general negative regulator of the Warburg effect is AMPK, in agreement with the frequent alteration of the LKB1-AMPK axis in cancer cells (Faubert et al. 2013). So far, no cancer-related dysregulation has been discovered at the level of the newly identified pyruvate carrier (Herzig et al. 2012), which could participate in the onset of the Warburg effect.
Recently, some signals and genes involved in anabolic stimulation, and in connection with glycolysis, were discovered. It was found that when cancer cells are confronted with serine deprivation, they activate the mTOR pathway to stimulate PKM2 protein synthesis, which in turn shifts glycolysis to its anabolic mode, thereby providing serine through the phosphorylated pathway (Ye et al. 2012). It was further discovered that in addition to mTOR, the protein TP53, a target of p53 tumor suppressor, spares the available serine for glutathione synthesis and limits other utilizations (Maddocks et al. 2013). In addition to mTOR and P53, the HIF1α pathway was shown to control PKM2 and PDK1 expression under hypoxia. The former protein drives anabolism from glycolysis, while the latter blocks fueling of the Krebs cycle with acetyl-CoA derived from glucose. The oncogene N-MYC was recently shown to induce a large-scale remodeling of energy metabolism in human cancer cells, with an activation of fatty acids oxidation concomitant with a stimulation of glycolysis (Zirath et al. 2013). Analysis of C-MYC bioenergetics properties also showed that activation of this oncogene stimulates OXPHOS, redirects glutamine toward lipids synthesis, and gives the preference to glutamine instead of glucose to fuel the energetic machinery (Wise et al. 2008).
The inhibition of P53 tumor suppressor also triggers the stimulation of glycolysis (notably via TIGAR inhibition), along with the inhibition of oxidative phosphorylation via respiratory chain complex IV destabilization [through SCO2 (Matoba et al. 2006)]. Lastly, the discovery of oxidative tumors in lymphoma by the group of Nika Danial revealed a switch toward fatty acid utilization controlled by PPARγ alpha and a successful cancer-killing strategy (in vitro) using a PPARγ antagonist (T0070907) (Caro et al. 2012a). Of central importance for the regulation of oxidative phosphorylation, the RAS oncogene was shown to stimulate respiration, by molecular mechanisms which remain unclear (Wei et al. 2012; Gough et al. 2009; De Groof et al. 2009; Baracca et al. 2010; Weinberg et al. 2010). Cancer bioenergetics studies revealed that oxidative phosphorylation is required for K-RAS to promote tumor progression, notably through the activation of mitochondrial respiration and the subsequent production of reactive oxygen species. K-RAS also stimulates the cytosolic part of the glutamate-aspartate shuttle (GOT1, MDH1, ME1) (Son et al. 2013) used to reoxidize the cytosolic NADH.
Besides the metabolic remodeling triggered by oncogenes, variations in the microenvironment can also induce pathway rewiring or branching, as discussed above in conditions of serine deprivation or as found in situations of glutamine deprivation. Upon glutamine removal, cancer cells rely of the pyruvate carboxylase to fuel the Krebs cycle and to generate citrate by a truncated TCA (Cheng et al. 2011). The metabolic remodeling that occurs in conditions of glucose deprivation is less documented despite the fact that aglycemia is encountered in tumors [we discussed this point in a recent review (Jose et al. 2013)]. In addition to these biochemical studies, recent findings suggested a link between the metabolic remodeling of tumors and chemoresistance: a subclass of resistant melanomas undergo a shift toward OXPHOS which opened a therapeutic window by using OXPHOS inhibitors as oligomycin or BZ-423 (Roesch et al. 2013). Likewise, metabolic remodeling has implication for epigenetics, cell anchorage, metastasis, and immune response, which demonstrate the importance and complexity of its investigation.
We described above a series of metabolic remodelings found in various types of cancer cells. So far, it cannot be said that only a limited number of profiles exists in tumors, albeit with variations in the pathways utilized. Yet, one can distinguish types of remodeling patterns, the objectives of which are (1) lipid synthesis from glutamine, (2) serine synthesis from glutamine or pyruvate, (3) ATP synthesis from fatty acids or amino acids, and (4) ROS generation by the ETC. The need for the tumor to consume building blocks such as arginine and asparagine inspired the therapeutic strategy to utilize arginase and asparaginase, which proved to be efficient. Likewise, we must understand the biological significance of the observed metabolic remodeling to efficiently target pathways of vital importance for tumor growth and progression. The inhibition of fatty acid oxidation by PPARγ blockers was efficient on selected lymphomas as shown in the study of Nika Danial’s group (Caro et al. 2012b), and the inhibition of lipolysis and subsequent fatty acid oxidation by orlistat and etomoxir was efficient on a mice model of leukemia (Samudio et al. 2010). Again, these findings indicate the need for a better stratification of human cancer based on their bioenergetic and associated biosynthetic profile. Clearly, an exhaustive evaluation of tumor proteomics cannot be done for each patient, but relevant biomarkers could help to delineate the metabolic profile of a given tumor, taken as a whole, and to propose adapted metabolic therapies. Ideally, circulating biomarkers could be discovered, and the potential of metabolomics must be considered to reach that goal. We also need to better connect, if relevant, the oncogenetic signature with the metabolic remodeling pattern, to evidence potential links between a particular subgroup of tumors (e.g., RAS mutated, or EGFR mutated, or resistant to a certain therapy) and specific metabolic features. Are MYC-driven tumors more prone to glutaminase therapy? Are RAS-driven tumors more sensitive to OXPHOS-targeted approaches?
A recent study by the group of JE Sarry on the anticancer effect of the AMPK agonist metformin (Scotland et al. 2013) showed that not all leukemic cells are sensitive and that the cell capacity to perform the Pasteur effect was a good indicator of their sensitivity. Likewise, we tested the anticancer effect of AICAR on different cancer cell lines (Jose et al. 2011b, 2012) and showed that different sensitivities as well as different modes of action could be found in each cell line. This argues again in favor of requisite a metabolic profiling of tumors prior to consider a metabolic therapy.
5 Future Directions in the Field of Energy Metabolism
As argued in this review, cancer research on tumor energy metabolism partly aims at the identification of specific mechanisms used by cancer cells to transduce energy from different sources of carbons, as glucose, fatty acids, amino acids, or ketone bodies. In 2013, around 400 publications were reported in the PubMed database with the association of the terms “cancer” and “energy metabolism.” Likewise, 90 clinical trials reported in the clinicaltrial.gov database also associated these two terms. How cancer cells transduce energy is determined by several factors which include the oncogenetic profile and the microenvironment, notably the type and concentration of available energy substrates, as well as the metabolic cross-talk with surrounding cells. The modalities of energy transduction are also closely determined by anabolic needs as amino acids, lipids, and nucleic acid syntheses, which consume carbon intermediates generated by the bioenergetic pathways, thereby impacting the modalities of energy transduction and the selection of appropriate “branched pathways” according to specific needs. Metabolic remodeling is also controlled, directly or indirectly, by numerous oncogenes and tumor suppressors. Hence, to determine the metabolic profile of a tumor requires the combination of series of investigations including bioenergetics, metabolomics, proteomics, and transcriptomics. Studies in vivo or on freshly excised tumors should be preferred to in vitro analyses on cancer cells which adapt to the artificial cell culture conditions and may not retain the particularities of human tumors that we need to decipher in order to propose innovative therapeutic strategies. A global analysis of catabolism and anabolism must be undertaken on those tumor samples to determine the biological objective of tumor metabolic remodeling. If serine is one endpoint, a strategy aiming at serine deprivation in tumor could be developed, as done for arginine and asparagine in leukemias. Undoubtedly, in silico reconstruction of metabolic pathways and their deviations will help to resolve such objectives and to test the validity of different bioenergetics targets.
Moreover, the transcriptomic study of large panels of human tumors, well clinically and genetically annotated, could allow stratification of tumors in bioenergetic groups, based on the expression level of different markers involved in metabolic switches, such as ACLY, PC, and IDH2/3. The direct assessment of tumor metabolic profile in vivo will give more accurate information on which pathway to target. However, this raises the problem of tumor internal heterogeneity. While genetic studies of tumors show the coexistence of cancer cell clones of different adaptative and resistance abilities, the biochemical studies performed on tumors do not yet take into account this heterogeneity.
Importantly, the problem of metabolic resistance will have to be considered, as suggested by the great metabolic flexibility of cancer cells. Therefore, a better definition of the link between the large-scale metabolic remodeling of tumor and their extended oncogenetic profile is required to adapt metabolic strategies that could be the more effective. In particular, to target “nodal enzymes” at the interface between catabolism and anabolism requires (1) their identification and (2) assessment of their regulating power (flux control coefficients). However, the complexity of cell metabolism in terms of numbers of reactions and biochemical intermediates strongly impedes our capacity to perform in vitro metabolic and bioenergetic analyses and thereby also the discovery of novel targets for cancer metabolic therapy. The metabolic charts indicate the existence of several subpathways potentially involved both in energy transduction and anabolism, but only some of them have been explored so far in the context of cancer adaptation to metabolic stress. The power of in silico analyses of cancer metabolism is needed to (1) identify the possible routes of energy transduction linked with anabolism; (2) select the optimal ones, with the better performance and significance for cancer metabolism; and (3) designate the enzyme with the highest control of the identified pathways.
References
Baracca A, Chiaradonna F, Sgarbi G et al (2010) Mitochondrial Complex I decrease is responsible for bioenergetic dysfunction in K-ras transformed cells. Biochim Biophys Acta 1797:314–323. doi:10.1016/j.bbabio.2009.11.006
Benard G, Bellance N, Jose C et al (2010) Multi-site control and regulation of mitochondrial energy production. Biochim Biophys Acta 1797:698–709. doi:10.1016/j.bbabio.2010.02.030, S0005-2728(10)00084-8 [pii]
Bulteau A-L, Ikeda-Saito M, Szweda LI (2003) Redox-dependent modulation of aconitase activity in intact mitochondria. Biochemistry 42:14846–14855. doi:10.1021/bi0353979
Caro P, Kishan AU, Norberg E et al (2012) Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell 22:547–560. doi:10.1016/j.ccr.2012.08.014
Chance B, Williams GR (1955) Respiratory enzymes in oxidative phosphorylation. III. The steady state. J Biol Chem 217:409–427
Chance B, Williams GR (1956) The respiratory chain and oxidative phosphorylation. Adv Enzymol Relat Areas Mol Biol 17:65–134
Cheng T, Sudderth J, Yang C et al (2011) Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc Natl Acad Sci USA 108:8674–8679. doi:10.1073/pnas.1016627108
Cogliati S, Frezza C, Soriano ME et al (2013) Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155:160–171. doi:10.1016/j.cell.2013.08.032
Curtis JM, Hahn WS, Long EK et al (2012) Protein carbonylation and metabolic control systems. Trends Endocrinol Metab 23:399–406. doi:10.1016/j.tem.2012.05.008
De Groof AJ, te Lindert MM, van Dommelen MM et al (2009) Increased OXPHOS activity precedes rise in glycolytic rate in H-RasV12/E1A transformed fibroblasts that develop a Warburg phenotype. Mol Cancer 8:54. doi:10.1186/1476-4598-8-54, 1476-4598-8-54 [pii]
DeBerardinis RJ, Mancuso A, Daikhin E et al (2007) Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA 104:19345–19350. doi:10.1073/pnas.0709747104, 0709747104 [pii]
Faubert B, Boily G, Izreig S et al (2013) AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab 17:113–124. doi:10.1016/j.cmet.2012.12.001
Fendt SM, Bell EL, Keibler MA et al (2013) Reductive glutamine metabolism is a function of the alpha-ketoglutarate to citrate ratio in cells. Nat Commun 4:2236. doi:10.1038/ncomms3236
Fukuda R, Zhang H, Kim JW et al (2007) HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 129:111–122. doi:10.1016/j.cell.2007.01.047, S0092-8674(07)00307-8 [pii]
Gellerich FN, Gizatullina Z, Gainutdinov T et al (2013) The control of brain mitochondrial energization by cytosolic calcium: the mitochondrial gas pedal. IUBMB Life 65:180–190. doi:10.1002/iub.1131
Gnaiger E (2009) Capacity of oxidative phosphorylation in human skeletal muscle: new perspectives of mitochondrial physiology. Int J Biochem Cell Biol 41:1837–1845. doi:10.1016/j.biocel.2009.03.013, S1357-2725(09)00117-4 [pii]
Gough DJ, Corlett A, Schlessinger K et al (2009) Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 324:1713–1716. doi:10.1126/science.1171721, 324/5935/1713 [pii]
Hardie DG, Scott JW, Pan DA, Hudson ER (2003) Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett 546:113–120
Hatzivassiliou G, Zhao F, Bauer DE et al (2005) ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 8:311–321
Herzig S, Raemy E, Montessuit S et al (2012) Identification and functional expression of the mitochondrial pyruvate carrier. Science 337:93–96. doi:10.1126/science.1218530
James AM, Collins Y, Logan A, Murphy MP (2012) Mitochondrial oxidative stress and the metabolic syndrome. Trends Endocrinol Metab 23:429–434. doi:10.1016/j.tem.2012.06.008
Jose C, Rossignol R (2013) Rationale for mitochondria-targeting strategies in cancer bioenergetic therapies. Int J Biochem Cell Biol 45:123–129. doi:10.1016/j.biocel.2012.07.005
Jose C, Bellance N, Rossignol R (2011a) Choosing between glycolysis and oxidative phosphorylation: a tumor’s dilemma? Biochim Biophys Acta 1807:552–561. doi:10.1016/j.bbabio.2010.10.012
Jose C, Hebert-Chatelain E, Bellance N et al (2011b) AICAR inhibits cancer cell growth and triggers cell-type distinct effects on OXPHOS biogenesis, oxidative stress and Akt activation. Biochim Biophys Acta 1807:707–718
Jose C, Bellance N, Chatelain EH et al (2012) Antiproliferative activity of levobupivacaine and aminoimidazole carboxamide ribonucleotide on human cancer cells of variable bioenergetic profile. Mitochondrion 12:100–109. doi:10.1016/j.mito.2011.03.010
Jose C, Melser S, Benard G, Rossignol R (2013) Mitoplasticity: adaptation biology of the mitochondrion to the cellular redox state in physiology and carcinogenesis. Antioxid Redox Signal 18:808–849. doi:10.1089/ars.2011.4357
Lopes Costa A, Le Bachelier C, Mathieu L et al (2014) Beneficial effects of resveratrol on respiratory chain defects in patients’ fibroblasts involve estrogen receptor and estrogen-related receptor alpha signaling. Hum Mol Genet 23:2106–2119. doi:10.1093/hmg/ddt603
Lustig Y, Ruas JL, Estall JL et al (2011) Separation of the gluconeogenic and mitochondrial functions of PGC-1{alpha} through S6 kinase. Genes Dev 25:1232–1244. doi:10.1101/gad.2054711
Maddocks OD, Berkers CR, Mason SM et al (2013) Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 493:542–546. doi:10.1038/nature11743
Marin-Valencia I, Yang C, Mashimo T et al (2012a) Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab 15:827–837. doi:10.1016/j.cmet.2012.05.001
Marin-Valencia I, Cho SK, Rakheja D et al (2012b) Glucose metabolism via the pentose phosphate pathway, glycolysis and Krebs cycle in an orthotopic mouse model of human brain tumors. NMR Biomed 25:1177–1186. doi:10.1002/nbm.2787
Martinez-Outschoorn UE, Lin Z, Whitaker-Menezes D et al (2012) Ketone body utilization drives tumor growth and metastasis. Cell Cycle 11:3964–3971. doi:10.4161/cc.22137
Matoba S, Kang JG, Patino WD et al (2006) p53 regulates mitochondrial respiration. Science 312:1650–1653. doi:10.1126/science.1126863, 1126863 [pii]
Melser S, Chatelain EH, Lavie J et al (2013) Rheb regulates mitophagy induced by mitochondrial energetic status. Cell Metab 17:719–730. doi:10.1016/j.cmet.2013.03.014
Metallo CM, Gameiro PA, Bell EL et al (2012) Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481:380–384. doi:10.1038/nature10602
Mitchell P (1961) Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 191:144–148
Morton R, Cunningham C, Jester R et al (1976) Alteration of mitochondrial function and lipid composition in Morris 7777 hepatoma. Cancer Res 36:3246–3254
Mullen AR, Wheaton WW, Jin ES et al (2012) Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 481:385–388. doi:10.1038/nature10642
Niemann S, Muller U (2000) Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet 26:268–270
Ostojić J, Panozzo C, Lasserre J-P et al (2013) The energetic state of mitochondria modulates complex III biogenesis through the ATP-dependent activity of Bcs1. Cell Metab 18:567–577. doi:10.1016/j.cmet.2013.08.017
Palmieri F, Pierri CL (2010) Mitochondrial metabolite transport. Essays Biochem 47:37–52. doi:10.1042/bse0470037
Palorini R, Cammarata F, Balestrieri C et al (2013) Glucose starvation induces cell death in K-ras-transformed cells by interfering with the hexosamine biosynthesis pathway and activating the unfolded protein response. Cell Death Dis 4:e732. doi:10.1038/cddis.2013.257
Parsons DW, Jones S, Zhang X et al (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321:1807–1812
Peinado JR, Diaz-Ruiz A, Frühbeck G, Malagon MM (2014) Mitochondria in metabolic disease: getting clues from proteomic studies. Proteomics 14:452–466. doi:10.1002/pmic.201300376
Pollard PJ, Briere JJ, Alam NA et al (2005) Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet 14:2231–2239
Roesch A, Vultur A, Bogeski I et al (2013) Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell 23:811–825. doi:10.1016/j.ccr.2013.05.003
Rossignol R, Letellier T, Malgat M et al (2000) Tissular variation in the control of oxidative phosphorylations, implication for mitochondrial diseases. Biochem J 347:45–53
Saada A (2014) Mitochondria: mitochondrial OXPHOS (dys) function ex vivo—the use of primary fibroblasts. Int J Biochem Cell Biol 48:60–65. doi:10.1016/j.biocel.2013.12.010
Samudio I, Harmancey R, Fiegl M et al (2010) Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest 120:142–156. doi:10.1172/JCI38942
Schagger H (2001) Blue-native gels to isolate protein complexes from mitochondria. Methods Cell Biol 65:231–244
Schägger H, Pfeiffer K (2000) Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J 19:1777–1783
Scheffler IE (1999) Mitochondria. Wiley-Liss, New York, 364 p
Scotland S, Saland E, Skuli N et al (2013) Mitochondrial energetic and AKT status mediate metabolic effects and apoptosis of metformin in human leukemic cells. Leukemia 27:2129–2138. doi:10.1038/leu.2013.107
Son J, Lyssiotis CA, Ying H et al (2013) Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496:101–105. doi:10.1038/nature12040
Telang S, Lane AN, Nelson KK et al (2007) The oncoprotein H-RasV12 increases mitochondrial metabolism. Mol Cancer 6:77. doi:10.1186/1476-4598-6-77, 1476-4598-6-77 [pii]
Warburg O (1930) Metabolism of tumors. Arnold Constable, London
Wei B-R, Simpson RM, Johann DJ et al (2012) Proteomic profiling of H-Ras-G12V induced hypertrophic cardiomyopathy in transgenic mice using comparative LC-MS analysis of thin fresh-frozen tissue sections. J Proteome Res 11:1561–1570. doi:10.1021/pr200612y
Weinberg F, Hamanaka R, Wheaton WW et al (2010) Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA 107:8788–8793. doi:10.1073/pnas.1003428107
Weinhouse S (1956) On respiratory impairment in cancer cells. Science 124:267–268
Wellen KE, Hatzivassiliou G, Sachdeva UM et al (2009) ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324:1076–1080. doi:10.1126/science.1164097
Wise DR, DeBerardinis RJ, Mancuso A et al (2008) Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA 105:18782–18787. doi:10.1073/pnas.0810199105, 0810199105 [pii]
Ye J, Mancuso A, Tong X et al (2012) Pyruvate kinase M2 promotes de novo serine synthesis to sustain mTORC1 activity and cell proliferation. Proc Natl Acad Sci USA 109:6904–6909. doi:10.1073/pnas.1204176109
Zhang Z, Falk MJ (2014) Integrated transcriptome analysis across mitochondrial disease etiologies and tissues improves understanding of common cellular adaptations to respiratory chain dysfunction. Int J Biochem Cell Biol 50:106–111. doi:10.1016/j.biocel.2014.02.012
Zhang Z, Tsukikawa M, Peng M et al (2013) Primary respiratory chain disease causes tissue-specific dysregulation of the global transcriptome and nutrient-sensing signaling network. PLoS One 8:e69282. doi:10.1371/journal.pone.0069282
Zirath H, Frenzel A, Oliynyk G et al (2013) MYC inhibition induces metabolic changes leading to accumulation of lipid droplets in tumor cells. Proc Natl Acad Sci USA 110:10258–10263. doi:10.1073/pnas.1222404110
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer-Verlag Wien
About this chapter
Cite this chapter
Obre, E., Rossignol, R. (2015). Metabolic Remodeling in Bioenergetic Disorders and Cancer. In: Mazurek, S., Shoshan, M. (eds) Tumor Cell Metabolism. Springer, Vienna. https://doi.org/10.1007/978-3-7091-1824-5_1
Download citation
DOI: https://doi.org/10.1007/978-3-7091-1824-5_1
Publisher Name: Springer, Vienna
Print ISBN: 978-3-7091-1823-8
Online ISBN: 978-3-7091-1824-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)