
Abstract
Enormous advances have been made in the last decade in understanding iron metabolism and iron homeostasis at both the cellular and the systemic level. This includes the identification of genes and proteins involved in iron transport, such as the ferric reductase DcytB, the proton-coupled ferrous (divalent) iron transporter DMT1, the iron exporter ferroportin and the membrane-bound ferroxidase hephaestin. The modulation of their translation by the iron regulatory protein (IRP) system has also been identified together with the impressive signalling cascades involved in regulating the chef d’orchestre of systemic iron homeostasis, hepcidin. However, exactly how the brain regulates fluxes and storage of iron between neurons, oligodendrocytes, astrocytes and microglial cells remains an enigma. In this review we discuss the possible mechanisms which may be involved in the transfer of iron across the blood–brain barrier(BBB), together with the possible role played by astrocytes. The consequences of iron deficiency and iron excess on brain function are described. Finally, various neurodegenerative diseases, where accumulation of iron may be important in the pathogenesis, are presented as well as the possible use of iron chelators to diminish disease progression.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
Iron (Fe) is a necessary cofactor in many metabolic processes in the central nervous system (CNS), including oxidative phosphorylation, myelin synthesis, neurotransmitter production, nitric oxide metabolism and oxygen transport. It plays an important role in electron transfer and is a cofactor for a large number of enzymes [1], including a number of key enzymes of neurotransmitter biosynthesis in brain, e.g. tyrosine hydroxylase (involved in the synthesis of catecholamines, including dopamine), tryptophan hydroxylase (involved in the synthesis of serotonin) and monoamine oxidase (involved in the metabolism of dopamine). It is essential that iron fluxes and storage within the brain are controlled within very exact limits in order to have adequate supplies for such metabolic processes but not ‘too much’, which could exacerbate damage via Fenton chemistry.
A brief overview of iron metabolism and homeostasis
Iron is transported throughout the circulation bound to the iron transport protein transferrin (Tf), which binds two atoms of Fe3+ as diferric transferrin \( \left( {{{\text{Fe}}^{3+}}_{2}{-}{\text{Tf}}} \right) \). Such iron is delivered to cells via the transferrin-to-cell cycle (Fig. 1) and binds to its receptor, and the complex is invaginated into clathrin-coated pits, which fuse with the target membranes of endosomes, delivering the vesicle contents into the interior of this organelle. The pH of the endosome is reduced to around 5–6 by the action of an adenosine triphosphate (ATP)-dependent proton pump. At this pH, iron is released from the transferrin–receptor complex as Fe3+Tf2, in a TfR-facilitated process (Fig. 1). It is proposed that TfR binding stimulates iron release from \( \left( {{{\text{Fe}}^{3+}}_{2}{-}{\text{Tf}}} \right) \) at acidic pH by protonation of the bound carbonate, and by stabilization of the Tf molecule in the apo-Tf (iron-free) conformation. The divalent cation transporter DMT1 then ensures the transport of iron out of the endosome into the cytoplasm, presumably after reduction of Fe3+ to Fe2+, by a member of the six transmembrane epithelial antigen of the prostate (STEAP) family of metalloreductases [2]. The cytoplasmic iron can then be transferred to the mitochondria for use in haem and iron–sulphur cluster synthesis, or stored in cytosolic ferritin. Unlike most other protein, a ligand taken up by receptor-mediated endocytosis, i.e. apotransferrin, retains high affinity for its receptor at acidic pH values, and is recycled back to the plasma membrane, where, at the slightly alkaline extracellular pH, it dissociates from its receptor and goes off into the circulation in search of further iron. This sequence of events constitutes the transferrin-to-cell cycle, which ensures iron uptake by cells that have transferrin receptors.

The transferrin cycle. Holotransferrin (HOLO-TF) binds to transferrin receptors (TFR) at the cell surface. The complexes localize to clathrin-coated pits, which invaginate to initiate endocytosis. Specialized endosomes form, and are acidified by a proton pump. At the acidic pH, iron is released from transferrin and is co-transported with protons out of the endosomes by the divalent metal ion transporter DCT1. Apotransferrin (APO-TF) bound to TFR is returned to the cell membrane, where at neutral pH it dissociates to participate in further rounds of iron delivery. In non-erythroid cells, iron is stored as ferritin and haemosiderin [47]
There is only one identified pathway for cellular iron export, from the basolateral membrane of duodenal enterocytes, from macrophages, hepatocytes and a number of other cell types. This involves ferroportin, which, together with hepcidin, plays a key role in systemic iron homeostasis. About a quarter of total body iron is stored in macrophages and hepatocytes in a readily mobilised form, mostly as ferritin for red blood cell formation (erythropoiesis). Mammalian ferritins are heteropolymers, made up of two subunit types, H and L. Whereas H-subunits have a ferroxidase activity, catalysing the oxidation of two Fe2+ atoms to Fe3+, L subunits appear to be involved in the nucleation of the mineral iron core.
The regulation of cellular iron homeostasis is to a large degree controlled at the level of the translation of the messenger RNAs (mRNAs) of proteins involved in cellular iron metabolism. The key players in this post-transcriptional regulation are two iron regulatory proteins (IRP1 and IRP2), which function as cytosolic iron sensors. In conditions of iron deficiency, IRPs bind with high affinity (K D ≈ 20–100 pM) to stem loops, known as iron regulatory elements (IREs), in mRNAs encoding the regulated proteins (Fig. 2). When the IREs are in the 5′-untranslated region (UTR) of the mRNA, as is the case for ferritin and ferroportin,Footnote 1 binding to IRPs prevents initiation of translation. In contrast, in the case of the transferrin receptor and DMT1, where the IREs are in the 3′-UTR, binding of the IRPs to the mRNAs protects them against degradation by nucleases. This results in increased iron uptake and blockage of iron storage and export. When iron is abundant, the IRPs are no longer active in binding, allowing ferritin and ferroportin mRNAs to be translated and resulting in the down-regulation of transferrin receptor and DMT1 synthesis as a result of the nuclease-catalysed degradation of their mRNAs. Under these conditions, IRP1 acquires aconitase activity, associated with the incorporation of a 4Fe–4S cluster, whereas IRP2 is condemned, after ubiquitination, to degradation in the proteasome.

Outline of translational regulation of mRNAs of a number of proteins involved in iron metabolism in low and high iron. IRPs bind to IREs located in either the 5′- or 3′-UTRs of specific mRNAs. During low-iron conditions, IRP1 and IRP2 bind with high affinity to 5′-IREs and to the five 3′-IREs in Tf R mRNA, resulting in the translational repression of 5′-IRE-containing mRNAs and the stabilization of the Tf R mRNA. During high-iron conditions, IRPs lose their affinity for IREs, increasing translation of 5′-IRE-containing mRNAs and mediating degradation of the Tf R mRNA. Increased iron levels result in the conversion of the IRP1 RNA binding form into the [4Fe–4S] cluster c-acon form, while increased iron and/or haem levels mediate IRP2 proteasomal degradation [1]
We now have an increasingly detailed understanding of how systemic iron homeostasis is regulated (Fig. 3). The first index of iron loading, increased transferrin saturation, leads to increased levels of \( \left( {{{\text{Fe}}^{3+}}_{2}{-}{\text{Tf}}} \right) \), which is detected by the liver via a complex pathway involving HFE, TFR2 and haemojuvelin (HJV). Hepatocytes respond to this signal by increased expression of the HAMP gene, resulting in increased secretion of the regulatory peptide hepcidin. Circulating hepcidin blocks dietary iron uptake by duodenal enterocytes and iron recycling from macrophages, in both cases through internalization of ferroportin, which therefore blocks iron export. The outcome of this is to decrease serum iron levels, leading logically to the feedback response of down-regulating hepcidin synthesis and secretion. This once again allows ferroportin to be displayed on the surfaces of enterocytes and macrophages, allowing them once again to export iron into the circulation.

Regulation of systemic iron homeostasis. Increased diferric transferrin, Fe2–Tf, is detected by the liver via an as-yet unknown complex regulatory pathway involving HFE, TFR2 and HJV. Hepatocytes respond to this signal by inducing HAMP expression and hepcidin secretion. Circulating hepcidin acts in turn to diminish dietary iron absorption by enterocytes and iron recycling by macrophages through internalization of ferroportin, which blocks iron export. As a consequence, serum iron decreases. As a feedback response, hepcidin synthesis is down-regulated, which allows ferroportin molecules to be displayed on the surface of the target cells [1]
The blood–brain barrier (BBB)
The brain is unique among all the organs of the body, being hidden behind a relatively poorly permeable vascular barrier, which limits its access to plasma nutrients, such as iron. It is generally accepted that iron transport into the brain mostly involves the transferrin-to-cell cycle, by the use of transferrin receptors within epithelial cells lining the blood–brain barrier(BBB), although the precise mechanism of iron transfer is still uncertain. The BBB proper (Fig. 4) is essentially composed of cerebral capillary endothelial cells, joined by tight junctions, a basal lamina, pericytes and astrocyte endfoot processes.

Cell associations at the BBB [98]
The types of cells found at the BBB and their associations are illustrated in Fig. 4. The endothelial cells form tight junctions, thereby sealing the paracellular pathway between the cells, such that substances which enter the brain must use dedicated endothelial cell transport systems, either passive or active transporters, which include the ATP binding cassette (ABC) transporters, as well as by transcytosis. Unlike other blood vessel epithelia, the BBB epithelia express different receptors at the luminal membrane (facing the circulation) compared with the abluminal membrane, which is surrounded by astrocyte endfeet, neuronal processes and interstitial fluid. Pericytes are distributed along the length of the cerebral capillaries, partially surrounding the endothelium. Both the cerebral endothelial cells and the pericytes are enclosed by the local basement membrane, forming a distinct perivascular extracellular matrix (basal lamina 1, BL1), different from the extracellular matrix of the glial endfeet bounding the brain parenchyma (BL2). Foot processes from astrocytes form a complex network surrounding the capillaries. Microglia, the resident immunocompetent cells of the brain, are also found in the vicinity of the BBB. Since no brain cell is further than about 25 μm from a capillary, once the BBB is crossed, diffusion distances for solutes to neurons, astrocytes and glial cells are short.
Mechanisms for iron transport into and within the brain
Endothelial cells make up the blood–brain barrier(BBB) and express transferrin receptors (transferrin receptor 1) on the luminal side of the capillaries. Although iron deficiency does not increase its expression [3], it is suggested that, in the latter situation, perhaps the cycling rate of endosomes containing the diferric transferrin receptor is increased.
The first step in the transport of iron from the luminal to the abluminal side of the blood capillary endothelial cells (BCECs) involves diferric transferrin binding to transferrin receptors expressed at the luminal membrane of the BCECs, followed by receptor-mediated endocytosis, with iron released from transferrin within the endosome by the slightly acidic pH. Iron could then be released from the endosomes, with recycling of apotransferrin to the luminal side of the endothelial cells and its ultimate release into the plasma [4, 5]. However, there is some controversy as to whether DMT1, which is required to transport Fe(II) out of the endosome, is present or not in BCECs (blood capillary endothelial cells) [6–8]. Therefore, it has been suggested that BCECs mediate iron transport into the brain by segregating iron from transferrin without the involvement of DMT1 [9]. On account of its ubiquitous expression pattern, transient receptor potential mucolipin 1 might also play an important role in the endolysosomal iron release in the BVEC [10]. It has been suggested that the interactions between BCECs and astrocytes might facilitate iron transport into the brain (Fig. 5) [11]. Subsequent to the binding of the iron-transferrin at the luminal surface of the BCEC, the \( {{{\text{Fe}}^{3+}}_{2}{-}{\text{Tf}}} \)–transferrin receptor complex would be internalized in an endosome, which is then possibly transported towards the abluminal side of the BCEC. However, the release of iron from such transferrin receptor complexes at the abluminal side remains very hypothetical. Iron bound to its receptor would then be exposed to the local microenvironment, e.g. hydrogen ions, ATP, other nucleotides and citrate [12, 13], which would bind the iron and lead to its release from transferrin. However, this would appear to be a very non-specific process. The apotransferrin would remain bound to its receptor, which has a high affinity for apotransferrin at acidic pH, and would be recycled back to the luminal cell surface, where it would be released from the transferrin receptor and returned to the circulation [11]. In addition, it is suggested that there may be a Tf-independent mechanism, since hypotransferrinaemic mice have normal amounts of iron in brain, as well as a mechanism for the entry of non-transferrin-bound iron. Since ferroportin is the only known iron exporter, iron may be exported into the brain interstitial fluid via ferroportin. This iron protein has been shown by immunohistochemistry to be present in the BBB, although its exact membrane localization is unclear [14]. Complexes of iron bound to citrate or ATP could then circulate in brain extracellular fluid to be taken up by other cell types or to bind to transferrin. Transferrin synthesised by the oligodendrocytes in the brain will bind the majority of iron that traverses the blood–brain barrier(BBB) after the oxidation of the iron, possibly by a glycophosphoinositide-linked caeruloplasmin found in astrocytic foot processes that surround brain endothelial cells. Since astrocytes have intimate contact with the abluminal side of the BBB, it is considered that they may play an important role as iron importers into the brain. Receptors for H ferritin receptors have been identified on the brain micro-vasculature, which might imply a role for this iron protein in iron transport. This could be an alternative pathway to that of transferrin [15].

Cross-section of a brain capillary demonstrating the close interaction between brain capillary endothelial cells (BCECs) and astrocytic endfeet (left). The area marked with a rectangle is shown on the right to demonstrate the possible interactions between BCECs and astrocytes which facilitate iron transport into the brain [11]
Neuronal iron homeostasis
A model for neuronal uptake and export of iron is presented in Fig. 6. An astrocytic endfoot forming intimate contact with the neuron is also shown and as such could play a critical role as a gate-keeper in regulating brain iron absorption and metabolism at the junction of the BBB. Subsequent to binding of iron-transferrin to the transferrin receptor at the cell surface, iron is transported into the neuron bound to transferrin. The resulting endosome contains DMT1 which facilitates iron transport across the endosomal membrane into the cytosol. Ferroportin is present in neurons and could therefore represent the mechanism by which iron is exported from such cells [8]. It has been suggested that iron can also enter the neuron (the interstitium contains ascorbic acid which can maintain iron in the ferrous iron) as a low-molecular-weight form bound to citrate or ATP possibly via non-vesicular import mechanisms or voltage-gated Ca2+ channels. Transferrin receptors are expressed on central neurons, as well as DMT1 and TRPML-1. Since both of these latter proteins transport Fe2+, a ferric reductase must exist within the endosomes and lysosomes of neurons. Under normal circumstances little iron is stored in the neurons, indicating that iron is taken up for rapid utilisation or secreted via ferroportin.

A model for neuronal uptake and export of iron. An astrocytic endfoot forming intimate contact with the neuron is also shown. Subsequent to binding of iron-transferrin to the transferrin receptor at the cell surface, iron is transported into the neuron bound to transferrin. The resulting endosome contains divalent metal transporter 1 that facilitates iron transport across the endosomal membrane into the cytosol, while pumping protons into the endosome. Astrocytes contain caeruloplasmin that exhibits ferroxidase activity, which is capable of oxidising ferrous iron to ferric iron. The ferric iron can enter the neuron in a low-molecular-weight form such as iron bound to citrate or ATP. The neuron expresses the iron exporter ferroportin that transports ferrous iron out of the cell. The interstitium contains ascorbic acid that can bind and thereby neutralize the toxicity of ferrous iron [11]
Glial cell iron homeostasis
Astrocytes
As already stated, astrocytes may play an important role in regulating iron absorption and metabolism at the BBB. It is unclear whether they express transferrin receptors or transferrin [16]. However, significant amounts of non-transferrin-bound iron may be taken up by these cells. In cultured astrocytes, Lane et al. [17] demonstrated that at least half of the accumulated iron is initially reduced by effluxed ascorbate and then imported via DMT1 in ascorbate-replete astrocytes, thereby confirming their role as an important contributor to iron homeostasis. Astrocytes contain caeruloplasmin (Cp) attached to their endfeet membranes by a glycophosphoinositide (GPI) linkage, which exhibits ferroxidase activity, capable of oxidising ferrous iron to ferric iron and thereby facilitating its binding to transferrin in the brain interstitial fluid. This GPI-Cp co-localizes on the astrocyte cell surface with the divalent metal transporter ferroportin and is physically associated with ferroportin. Ferroportin alone is unable to efflux iron from astrocytes in the absence of GPI-Cp or Cp [18]. The ferroxidase activity is required for the stability of cell surface ferroportin in cells expressing GPI-caeruloplasmin [19].
Oligodendrocytes
Since there is a high demand of iron for myelination, most of the histologically detectable iron in the brain is present in oligodendrocytes. Iron may be taken up into oligodendrocytes via the ferritin receptor Tim-2s as well as in its non-transferrin-bound form via DMT1 or other non-vesicular iron import mechanisms [16]. These cells are the predominant producers of transferrin [20].
Microglia
The control of iron homeostasis within microglia remains undefined–other phagocytic cells such as macrophages take up iron via transferrin receptors and release iron via ferroportin [21]. It is of interest that both microglia and iron deposits accumulate at the site of damage in many neurodegenerative diseases such as Alzheimer’s and Parkinson’s. Whether these accumulations are a cause or effect of the disease is currently unknown. In preliminary studies we have shown that the expression of the mRNAs of transferrin receptor 1 and ferroportin were significantly down-regulated in response to lipopolysaccharide (LPS) treatment in immortalised microglial cells (N9), while that of divalent metal ion transporter DMT1 showed no change in expression under control conditions and after LPS treatment [21]. However, the results of real-time polymerase chain reaction (RT-PCR) studies indicated that hepcidin was not produced by microglia either under control conditions or in response to LPS treatment after 24 h. This might suggest that iron homeostasis is under the control of alternative mechanisms in microglia, for instance cytokines such as interleukin (IL)-1 or IL-6. Of course, the lack of hepcidin production does not preclude the action of hepcidin from other sources, both in the brain and elsewhere. In addition, other studies that have detected hepcidin production in response to LPS treatment noted that hepcidin induction was transient and was often undetectable at 24–36 h post LPS treatment. Therefore, it cannot be ruled out that hepcidin was produced but was undetectable at 24 h post LPS exposure. Whether iron accumulates in activated microglia (which could explain the association between increased iron stores and microglia activation in specific brain regions of Parkinson’s and Alzheimer’s patients) remains unknown.
Importance of iron in the developing foetus
In pioneering studies, it was shown that iron deficiency could affect infant behaviour and development [22, 23]. Such a role for iron in neurocognitive and neurobehavioural development is highlighted by our growing understanding of the biology of developmental iron deficiency [24]. Iron requirements are expected to exceed iron intake during the first 6–18 months of postnatal life at a time of rapid neural development, during which morphological, biochemical and bioenergetic alterations may influence the way in which the brain functions in later life [25, 26]. Iron deficiency either in utero or in early postnatal life can result in abnormal cerebral development, because iron is essential for proper neurogenesis and differentiation of certain brain cells and brain regions: recent studies of iron-deficient rodents clearly identify altered morphology in the hippocampus and striatum [27–29]. This includes decreased arborization of dendrites and alteration in the location and functioning of oligodendrocytes, the cells responsible for myelin formation. Iron deficiency results in persistent alterations in both the composition and amounts of myelin in white matter, which do not return to normal levels later in life [30, 31]. Iron deficiency also affects dopamine and noradrenaline metabolism [32, 33].
In various epidemiological studies it is reported that children with iron-deficiency anaemia have poorer performance on tests of some specific cognitive functions. Animal experiments have identified some of the defects of reduced iron availability on brain function. These include post-translational changes (which result in a failure of iron incorporation into protein structures, which are subsequently degraded), vulnerability of the developing hippocampus (with loss of the neuronal metabolic marker cytochrome c oxidase) and altered dendritic structure. Iron deficiency will also have a direct effect on myelin, including a decrease in myelin lipids and proteins, as well as neurotransmitter systems, since iron is essential for a number of enzymes including tryptophan hydroxylase (serotonin) and tyrosine hydroxylase (norepinephrine and dopamine). Long-term follow-up studies of iron deficiency in the human infant brain indicate that such alterations in myelination result in slower conduction in both the auditory and visual systems. Both of these sensory systems are rapidly myelinating during the period of iron deficiency and are critical for learning and social interaction. Together with the reduced energy, impaired glial function and altered activation of monoamine circuits, this may alter experience-dependent processes, which are critical to brain structure and function during early development.
It is well known that there is specificity and selectivity for brain iron acquisition, which is emphasised by the fact that certain brain regions respond to iron deficiency and iron depletion more aggressively than other regions. For example, the densities of TfRs in different regions of human brain vary widely [34] and do not seem to correlate with the published iron content of these brain regions [35]. Of particular interest is the fact that substantia nigra (SN) iron content is not high in young rats, but that during iron repletion a dramatic increase of iron is observed in this brain region, thereby indicating that iron is targeted to this specific brain region [36]. High rates of iron accumulation occur in different brain regions at different ages of postnatal life, indicating that shortage at these important stages of development might be of significance in later life. The iron requirements by the brain are much greater than the iron uptake into the tissue, indicating that most brain iron is derived from recycling behind the blood–brain barrier(BBB) comparable to what occurs in the periphery. This would indicate that, following birth, iron recycling rather than uptake from the circulation may be the major source of iron. It is still under debate as to whether impaired iron homeostasis may be a primary cause of many neurological diseases. As yet, no major defects have been identified. One of the major points of debates concerns the amount of iron that might be transported from the systemic circulation into the brain. Earlier studies indicated that people with high circulating iron, saturated transferrin and high levels of low-molecular-weight iron, e.g. genetic haemochromatosis and thalassaemic patients, did not show increased brain iron. For example, in a study of Italian population, the most common HFE mutations, H63D and C282Y, were not associated with the individual risk to develop Parkinson’s disease (PD), although no indications of their iron loading were given [37]. However, in another smaller study of only 66 people, the results indicated that these genetic variants in iron metabolism genes can influence brain iron levels in men [38]. In one further study, the H63D polymorphism was overrepresented in individuals with sporadic amyotrophic lateral sclerosis (ALS) [odds ratio 1.85, confidence interval (CI) 1.35–2.54] [39], but there was no association with cognitive decline [40]. Furthermore, a large, well-powered meta-analysis of eight studies comprising in total 758 cases and 626 controls failed to find a significant association between Alzheimer’s disease and any haemochromatosis HFE genotype [41]. Such studies now need to be extended to include patients with high iron loading, e.g. thalassaemia, such that correlations can be made between systemic iron overload, brain iron content, cognitive impairment and neurodegeneration.
Ageing and metal-based neurodegeneration
It is becoming apparent that the function of the BBB is affected by a number of patho-physiological factors such as age and inflammation. Immune cells are able to penetrate the BBB, either at the endothelial blood–brain barrier(BBB) or the epithelial blood CFS barrier. Inflammation as well as reactive oxygen species (ROS) and reactive nitrogen species (RNS) can acutely disrupt BBB at tight junctions. 4-Hydroxynonenal (HNE), a second messenger of free radicals, is present in the BBB under pathological conditions and could make the endothelial part of the BBB permeable. As yet, little is known about the expression pattern of transferrin receptors in the capillaries of aging brains. Modulation of P-glycoproteins at the BBB may be an important factor to improve drug delivery, as well as to augment CNS protection from challengers from the peripheral system.
Multiple structural and functional changes occur in the ageing brain, often accompanied by changes in the immune function, as well as an elevation of brain iron in specific brain regions, e.g. in the putamen, motor cortex, prefrontal cortex, sensory cortex and thalamus, which is possibly localized within H- and L-ferritin and neuromelanin. Recent studies have identified various adverse effects of such iron accumulations on cognitive decline; for example, magnetic resonance imaging (MRI) quantified iron deposits in the basal ganglia of 143 non-demented subjects at mean ages of 11, 70 and 72 years and showed that higher iron deposits were associated with lower cognitive ability [42], higher iron burdens in the putamen and caudate nucleus were associated with lower scores on dementia rating scales and longer reaction times [43], while assessment of genomic integrity in a small number of brain samples from elderly individuals identified a higher number of single-strand and double-strand DNA breaks as the concentration of iron and copper increased in the hippocampus and frontal cortex [44]. In contrast, when brain iron deposition was reduced in the globus pallidus, substantia nigra, red nucleus and temporal cortex in old rhesus monkeys receiving a calorie-restricted diet [45], there were reductions in both age-related inflammation and oxidative damage. Recent studies have suggested that indices of iron homeostasis in the blood, e.g. serum iron and transferrin saturation, as well as hepatic iron content may be correlated with iron content of various brain regions, when the latter was assessed by MRI proton traverse relaxation time, R2 [46].
Oxidaive stress in Alzheimer’s disease and Parkinson’s disease
Ill-placed excessive amounts of iron, either in specific brain cellular constituents such as mitochondria or in specific brain regions, could lead to the generation of toxic free radicals leading to neurodegenerative diseases. Over the last decade, it has become widely accepted that inflammation, associated with dysfunction of metal ion homeostasis (Fe, Cu, Zn) accompanied by concomitant oxidative stress, is a key factor in a large number of neurodegenerative diseases such as Parkinson’s disease, Alzheimer’s disease, Huntington’s disease, multiple sclerosis, Friedreich’s ataxia and others [47]. Support comes from the observation that Alzheimer’s disease (AD), Parkinson’s disease (PD) and many other neurodegenerative diseases are characterised by increased levels of iron in specific regions of the brain. Furthermore, iron increases appear to predominate in motor areas of the brain, such as the basal ganglia, which may explain the motor deterioration observed in many neurodegenerative diseases [16].
The ‘metal-based neurodegeneration’ hypothesis can be described by the following postulates:
-
1.
Redox-active metal ions (Fe, Cu), present within specific brain regions, can generate oxidative stress by production of ROS;
-
2.
ROS then cause peroxidation of polyunsaturated fatty acids in membrane phospholipids;
-
3.
This in turn leads to the formation of reactive aldehydes (Fig. 7);
Fig. 7 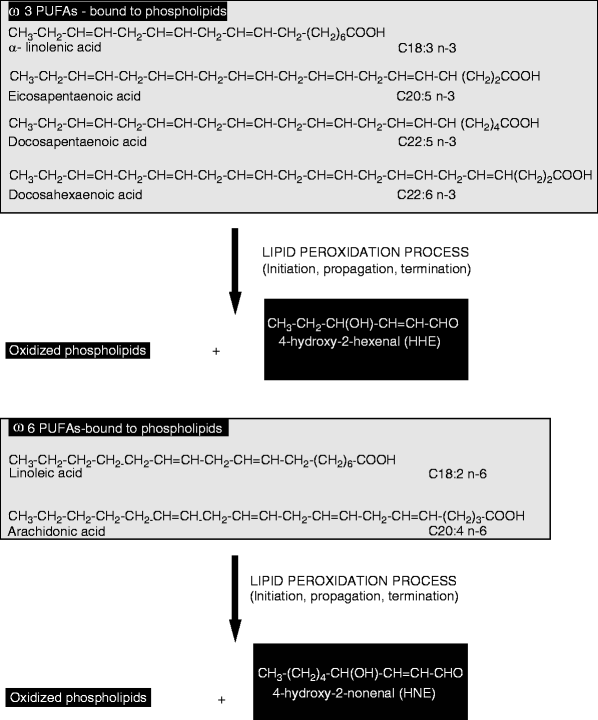
Schematic diagram of reactive hydroxy-alkenals generated during lipid peroxidation of n-3 and n-6 polyunsaturated fatty acids [50]
-
4.
The reactive aldehydes, together with other oxidative processes, react with proteins to generate carbonyl functions, which damage proteins (Fig. 8);
Fig. 8 
Production of protein carbonyls (aldehydes and ketones) [47]
-
5.
Damaged, misfolded proteins aggregate, overwhelming the cytosolic ubiquitin/proteasome protein degradation system, and accumulate within intracellular inclusion bodies;
-
6.
Such intracellular inclusion bodies are found in a great many neurodegenerative diseases (Alzheimer’s, Parkinson’s, ALS, Huntington’s etc.).


Although both ROS and RNS are involved in physiologically relevant signalling pathways, there is considerable evidence that, in situations of oxidative stress, they are associated with a number of neurodegenerative pathologies. Oxidative stress refers to a situation where elevated levels of ROS are observed, and can result from a variety of conditions that represent either increased ROS production or a decreased level of antioxidant defence. In the case of stimulation of ROS production by macrophages during the innate immune response to bacterial infection, the ROS so generated act in a protective manner. However, dysregulation of ROS levels in a variety of tissues, notably in the brain, has been linked to a growing number of inflammatory and age-associated diseases. During oxidative stress, the oxidation of cellular components results in the modification of DNA, proteins, lipids and carbohydrates, and the resulting oxidative damage is frequently associated with cell death either by necrosis or by apoptosis [48].
When ROS are generated by redox trace metals in the proximity of membrane phospholipids, they initiate the peroxidation of polyunsaturated acyl chains of phospholipids or n-6 polyunsaturated fatty acids (PUFA) (Fig. 7). The lipid hydroperoxides are highly susceptible to breakdown through non-enzymatic Hock cleavage, forming a variety of lipid-derived α,β-unsaturated 4-hydroxyaldehydes of which the most prominent is 4-hydroxynonenal (HNE) [49]. 4-hydroxynonenal (HNE) is the main aldehyde formed during lipid peroxidation of n-6 polyunsaturated fatty acids, such as linoleic acid C18:2 n-6 and arachidonic acid C20:4 n-6, whereas peroxidation of n-3 polyunsaturated fatty acids such as α-linolenic acid C18:3 n-3 and docosahexaenoic acid C22:6 n-3 generates a closely related compound, 4-hydroxy-2-hexenal (HHE) [50]. The mechanisms by which these 4-hydroxyalkenals might be formed from membrane phospholipid PUFAs have been recently reviewed [51].
HNE was initially recognised as the product of lipid peroxidation with the greatest toxicological potential; it is subsequently considered to be one of the most reliable markers of oxidative stress: It can also trigger signalling events in a physiological context as well as acting as a growth modulating factor [52]. Immunohistochemical studies show the presence of HNE in neurofibrillary tangles and senile plaques in Alzheimer’s disease (AD), in the cytoplasm of the residual motor neurons in sporadic amyotrophic lateral sclerosis (ALS), in Lewy bodies in neocortical and brain stem neurons in Parkinson’s disease (PD) and in diffuse Lewy bodies disease (DLBD) [53]. 4-HNE is relatively stable in vivo and has been proposed to be one of the key mediators of the damage resulting from exposure to reactive oxygen and nitrogen species.
During oxidative stress, numerous post-translational modifications of proteins have been characterised resulting either from direct oxidation of amino acid residues by highly reactive oxygen species (that are formed during normal metabolism), or through the conversion of lipid and carbohydrate derivatives to compounds that react with functional groups on proteins [54]. A significant portion of these ROS-induced post-translational modifications result in the formation of reactive protein carbonyl derivatives, generically termed ‘protein carbonylation’. The level of carbonyl groups in proteins is widely used as a marker of oxidative protein damage [55, 56].
Direct oxidation of certain amino acid side-chains in proteins (proline, arginine, lysine and threonine) or oxidative cleavage of the protein backbone [55] can lead to the formation of protein carbonyl derivatives (Fig. 8). Methionine and cysteine can be directly oxidised. Unlike other types of modification (except cysteine oxidation), oxidation of methionine residues to methionine sulphoxide is reversible; thus, cyclic oxidation and reduction of methionine residues leads to consumption of ROS and thereby increases the resistance of proteins to oxidation [57]. Carbonyl groups can also be introduced into proteins by addition of reactive carbonyl compounds (ketoamines, ketoaldehydes and deoxyosones) produced by a complex series of reactions between reducing sugars or their oxidation products with the amino groups of lysine residues in proteins, by mechanisms known as glycation and glyoxidation [57]. Because of their electron-withdrawing functional groups, the double bond of 4-HNE and other α,β-unsaturated aldehydes serves as a site for Michael addition with the sulphur atom of cysteine, the imidizole nitrogen of histidine and, to a lesser extent, the amine nitrogen of lysine [58]. After forming Michael adducts, the aldehyde moiety may in some cases undergo Schiff base formation with amines of adjacent lysines, producing intra- and/or intermolecular cross-linking [59, 60]. Recent studies have suggested that such protein carbonylation from lipid-derived aldehydes is more prevalent than that formed via direct amino acid side-chain oxidation [58].
Peroxynitrite is able to oxidise methionine residues and to nitrate tyrosine residues in proteins; however, this depends on the availability of CO2, which stimulates the nitration of tyrosine residues but inhibits the oxidation of methionine residues [55]. Nitration of tyrosine residues may contribute significantly to peroxynitrite toxicity, since nitration will prevent the phosphorylation or nucleotidylation of key tyrosine residues in enzymes which are regulated by phosphorylation/adenylation, thereby seriously compromising one of the most important mechanisms of cellular regulation and signal transduction.
ROS can also readily attack DNA, generating a variety of DNA lesions, such as oxidised bases, abasic sites and single- and double-strand breaks. If not properly removed, DNA damage can be potentially dangerous, leading to mutagenesis and/or cell death, especially in the case of lesions that block the progression of DNA/RNA polymerases.
Iron involvement in Alzheimer’s disease and Parkinson’s disease
There is an increasing body of evidence that elevated iron levels are found in particular brain regions in specificneurological diseases including Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD) and Friedreich’s ataxia (FA). We summarize some of the findings below.
As life expectancy in the developed world increases, there is a significant increase in the development of mild cognitive impairment (dementia) which can progress to Alzheimer’s disease (AD). Over 24 million people worldwide suffer from some form of dementia, and estimates are that, by 2040, 80 million people will be demented, with Alzheimer’s disease accounting for some 60% of all dementia. AD is the most common cause of age-related neurodegeneration, affecting memory and behaviour centres of the brain. This progressive loss of cognitive and behavioural functions is associated with the temporal and frontal lobes of the brain [61]. The classical patho-physiological hallmarks are the presence of toxic insoluble aggregates of amyloid-β peptide (Aβ) in extracellular senile plaques and of neurofibrillary tangles (NFT) created by the hyperphosphorylation and subsequent aggregation of the microtubule-associated protein, tau, associated with the loss of cortical neurones [62]. Changes in the levels of iron, ferritin and transferrin have been reported in areas of the brain associated with centres of memory and thought processing which are lost in the clinical development of AD, notably the hippocampus and the cerebral cortex [63, 64], with iron accumulating more rapidly than ferritin in areas of particular neurodegeneration [65]. Another important pathological finding in AD is that the iron accumulation occurs in the same brain regions characterised by Aβ deposition [36]. It has been suggested therefore that abnormal deposition of iron in AD plaques associated with β-amyloid can mediate free-radical-related neurotoxicity [66].
Aβ is derived by the proteolytic cleavage of the amyloid precursor protein (APP), a type 1 transmembrane glycoprotein. APP is cleaved by three types of proteases, the α-, β- and γ-secretases. The majority of APP is processed in the non-amyloidogenic pathway (Fig. 9); APP is first cleaved by α-secretase within the amyloid-β protein (Aβ) domain, leading to release of the neuroprotective extracellular soluble sAβPPsα fragment, and precluding Aβ generation. The membrane-anchored α-carboxy terminal fragment (αCTF) is then cleaved by γ-secretase within the membrane, releasing the p3 peptide and the APP intracellular domain (AICD). Alternatively, amyloidogenesis takes place when APP is first cleaved by β-secretase, producing AβPP. Aβ and AICD are generated upon cleavage by γ-secretase of the βCTF fragment retained in the membrane. Therefore, stimulation of the α-secretase pathway attenuates Aβ accumulation in the brain and amyloid formation [67]. The processing of both α- and β-secretases is modulated by furin, a member of the subtilisin-like proprotein convertase family which catalyses the cleavage of precursor proteins into their biologically active forms [68]. Furin is also involved in modulation of systemic iron homeostasis through the production of soluble haemojuvelin (HJV) [69], an antagonist of bone morphogenic protein (BMP)-mediated activation of hepcidin [70], an important regulator of iron homeostasis. Furin transcription is modulated by cellular iron levels and by hypoxia [69, 71]. Excess iron decreases furin protein levels and therefore impairs the production of soluble HJV. In contrast, iron deficiency or hypoxia up-regulates furin activity, thereby increasing the production of soluble HJV, and blocking hepcidin activation [69]. This has led to the hypothesis, illustrated in Fig. 9, that iron regulation of furin may play a role in AD [72]. Increased levels of iron in the brain could down-regulate furin protein levels, thereby impairing the ability of α-secretase to generate the neuroprotective sAβPPsα fragment, thereby activating the amyloidogenic pathway, leading to Aβ production and ultimately neurodegeneration. In addition, the iron-dependent production of ROS could shift the IRP1 to its IRE-binding form [73], thereby increasing cellular iron uptake via the transferrin receptor, creating a vicious circle which would progressively increase the intracellular iron content, further down-regulating furin and shifting the secretase equilibrium in favour of Aβ production. Furin mRNA levels in the brains of AD patients and the AD animal model, Tg2576 mice, were significantly lower than those in controls. Moreover, injection of furin-adenovirus into Tg2576 mouse brains markedly increased α-secretase activity and reduced β-amyloid protein (Aβ) production in infected brain regions [74]. Further support for the connection between iron metabolism and AD comes from the identification of a functional IRE in the 5′-UTR of the amyloid precursor protein mRNA. As is the case of ferritin, APP levels increase in the presence of iron and decrease upon addition of an iron chelator in neuroblastoma cells [75]. Increased APP formation in parallel with inhibition of α-secretase activity would favour Aβ deposition. Recently it has been shown that APP possesses ferroxidase activity, oxidising Fe2+ incorporating Fe3+ into transferrin and shows an interaction with ferroportin in specific cells which lack caeruloplasmin, HEK293T cells [76]. Further studies are clearly necessary to ascertain this important function of APP in health and disease in man.

Furin activity and the fate of AψPP cleavage by ψ- and ψ?-secretases. Low cellular iron levels are thought to increase furin activity, stimulating the non-amyloidogenic pathway. In contrast, high cellular iron levels decrease furin activity and may activate the amyloidogenic pathway [93]
PD is the second most common neurodegenerative disease after AD, affecting about 1 % of the population older than 60 years. Unlike AD, which affects memory and behaviour centres in the brain, PD is characterised by progressive loss of control over voluntary movement. The characteristic symptoms (bradykinesia, rigidity, tremor and loss of balance) arise from progressive loss of dopaminergic neurons (neurons which synthesise and release dopamine) in the substantia nigra pars compacta (SNPC), located in the mid-brain [77].
In the brains of Parkinson’s disease (PD) patients there is a specific elevation of iron in the substantia nigra and the lateral globus pallidus, by approximately two-fold in comparison with age-matched controls (reviewed in [35, 36]). This is in marked contrast to other iron storage diseases, such as untreated genetic haemochromatosis and thalassaemia patients, where 10–20-fold iron increases in iron stores must be attained before clinical abnormalities occur [78]. Increased iron levels are also found by phase microscopy in individual dopaminergic neurones of PD patients [79].
A second characteristic hallmark of PD is the presence, within dopaminergic neurons, axons and synapses of the substantia nigra, of intracellular, eosinophilic proteinaceous aggregates called Lewy bodies, which are composed mostly of α-synuclein [80], but also contain ubiquitin, tyrosine hydroxylase and IRP2 [35]. Many studies have shown that iron promotes the aggregation of α-synuclein [81–84] while chelation of free iron with desferrioxamine blocks α-synuclein aggregation [82–85]. Iron is also found to accumulate within Lewy bodies in the brains of PD patients [86–88].
The role of iron in PD is outlined in Fig. 10 [36]. In the brain interstitial fluid, iron is transported bound to transferrin (Tf), which is absorbed by neuronal cells via transferrin receptor (TfR1)-mediated endocytosis. Iron levels are elevated in the neurons of the substantia nigra in PD. Iron is normally stored in ferritin, but in PD, ferritin levels are found to be inappropriately low. If the capacity of the neurons to store iron is exceeded, potentially toxic free iron will accumulate. Similarly, neuromelanin, a dark pigment produced by dopaminergic neurons which binds free iron, is also found to be decreased in PD. Iron also promotes conformational changes within parkin and α-synuclein, which cause their aggregation. Iron is an important cofactor of tyrosine hydroxylase (TH), which is involved in dopamine biosynthesis, and of monoamine oxidase (MAO), an enzyme which is involved in dopamine metabolism. Hydrogen peroxide, generated in this reaction, can be converted to reaction oxygen species (ROS) by ‘free’ iron.

Iron induces oxidative stress in PD [36]
Therapeutic considerations
From the description of the pathologies of aged brains and neurodegenerative disease it can be observed that there are certain common factors between each of these pathologies which include inflammation, oxidative stress and increased iron accumulation. Therefore, therapeutic agents that could reduce (a) the inflammatory signalling process, (b) oxidative stress and (c) excessive iron accumulation could show efficacy in reducing the progression of the disease, for which there is currently no known cure.
Neuroinflammation is considered to be an important contributor to pathogenesis of neurological disorders, with microglial activation as a hallmark of neuroinflammation. Microglia, a subset of the glial cells, are the resident macrophage population in the CNS and are regarded as the resident immunocompetent effector cells of innate immunity in the brain. In the adult brain, under normal conditions, the blood–brain barrier(BBB) prevents molecules from gaining access to the vascular lumen. However, molecules of the systemic innate immune system are able to stimulate immune cells of the brain as well as the neuronal populations. Microglia are considered to be primary mediators of neuroinflammation. In the healthy adult brain they exist in a non-activated state displaying a ramified morphology and minimal expression of surface antigens. The activation of microglia is an early event in neurodegenerative process when they are rapidly transformed to a reactive phenotype and release cytotoxic pro-inflammatory molecules, including oxygen radicals, NO, glutamate, cytokines and prostaglandins which can have a detrimental effect on other neural cells. Their activation involves NFκB, which may involve neuronal–microglial interactions [89]. Therefore, inhibition of microglia-mediated neuroinflammation may present a promising therapeutic target for neurological disorders. Various compounds such as resveratrol, a non-flavonoid polyphenol rich in red wine and grapes [90], and taurine [91] have been shown to have antioxidant and anti-inflammatory properties, respectively, in animal models of neurodegeneration.
Disease-modifying therapies aimed at removing excess iron, without affecting iron-containing enzymes involved in neurotransmitter function, could be part of the therapeutic approaches utilised to prevent the progression of PD. There has been considerable speculation as to whether such chelators could be utilised in the treatment of neurodegenerative disease, where there are marginal increases in toxic iron in specific brain regions. However, in our earlier studies it was demonstrated that both DFO and deferiprone, a hydroxypyridone, were able to traverse the BBB and chelate iron in various brain regions in the ferrocene model of brain iron overload [92]. Subsequently, in more recent experiments where DFO was administered to experimental animals with intracranial haemorrhage, mortality was decreased [93, 94]. In addition, DFO administration to control animals had no significant effect on either iron homeostasis or haematological parameters. Several commercially available iron chelators, e.g. deferrioxamine (DFO–hexadentate), deferiprone (bidentate) and deferasirox (tridentate), which are clinically effective for the treatment of peripheral iron overload disorders, could potentially be utilised to treat PD. Although early studies indicated that the BBB is relatively impermeable to DFO [95], in our recent studies of these three chelators it was shown that each of them could penetrate the BBB and prevent the loss of tyrosine hydroxylase granules as well as improving dopamine levels in the 6-hydroxy dopamine model of Parkinson’s disease [96].
Other therapies which incorporate iron chelators into drug delivery systems, e.g. liposomes and nanoparticles, may improve the targeting of these compounds to regions of the brain with excessive iron deposition. However, the toxicity associated with these approaches needs to be clarified. Chelators bound to transferrin receptors may also be a viable drug delivery system.
Conclusions
The cause of the enhanced brain iron content in PD remains unknown but may be attributable to a variety of factors which include changes in iron release mechanisms across the BBB, or perhaps more likely, a dysregulation of iron homeostatic control in the substantia nigra. In our recent studies [97], mRNA was isolated from two regions within the substantia nigra and from the cortex of Parkinson’s brain, and the expression of a number of iron genes was compared with those from control post mortem material. Our global conclusions, in line with previous studies, are that, in human PD brain, cells of the SN behave phenotypically as if they were Fe deficient. Iron uptake systems are up-regulated, whereas iron storage in ferritin is down-regulated.
We conclude that excess iron accumulation in specific brain regions, albeit relatively slight when compared with other iron-accumulating diseases such as haemochromatosis and thalassaemia, is implicated in neurodegenerative disorders, notably in AD and PD as we have illustrated above. The mechanisms involved in these processes remain to be established, but the involvement of iron in PD and AD presents us with what could be an extremely precious therapeutic possibility of using iron chelators for its removal [47]. Clearly identifying mechanisms which are involved in brain iron accumulation will facilitate new therapeutic interventions to reduce the progression of age-related neurodegenerative diseases.
References
Crichton RR (2008) Biological chemistry, an introduction. Elsevier, Amsterdam, p 369
Ohgami RS, Campagne DR, McDonald A, Fleming MD (2006) Blood 108:1388
Moos T, Oates PS, Morgan EH (1998) J Comp Neurol 31:420
Moos T, Morgan EH (2002) Dev Neurosci 24:99
Rouault TA, Cooperman S (2006) Semin Pediatr Neurol 13:142
Burdo JR, Menzies SL, Simpson IA, Garrick LM, Garrick MD, Dolan KG, Haile DJ, Beard JL, Connor JR (2001) J Neurosci Res 66:1198
Moos T, Morgan EH (2004) J Neurochem 88:233
Moos T, Rosengren Nielsen T (2006) Semin Pediatr Neurol 13:149
Moos T, Skjoerringe T, Gosk S, Morgan EH (2006) J Neurochem 98:1946
Dong XP, Cheng X, Mills E, Delling M, Wang F, Kurz T, Xu H (2008) Nature 455:992
Moos T, Rosengren Nielsen T, Skj½rringe T, Morgan EH (2007) J Neurochem 103:1730
Morgan EH (1977) Biochim Biophys Acta 499:169
Morgan EH (1979) Biochim Biophys Acta 580:312
Wu LJ, Leenders AG, Cooperman S, Meyron-Holtz E, Smith S, Land W, Tsai RY, Berger UV, Sheng ZH, Rouault TA (2004) Brain Res 1001:108
Fischer J, Devraj K, Ingram J, Slagle-Webb B, Madhankumar AB, Liu X, Klinger M, Simpson IA, Connor JR (2007) Am J Physiol Cell Physiol 293:C641
Mills E, Dong X-P, Wang F, Xu H (2010) Future Med Chem 2:51
Lane DJR, Robinson SR, Czerwinska H, Bishop GM, Lawen A (2011) Biochem J 432:123–132
Jeong SY, David S (2003) J Biol Chem 278:27144
De Domenico I, Ward DM, di Patti MC, Jeong SY, David S, Musci G, Kaplan J (2007) EMBO J 26:2823
Todorich B, Pasquini JM, Garcia CI, Paez PM, Connor JR (2009) Glia 57:467
Ward RJ, Crichton RR, Taylor DL, Corte LD, Srai SK, Dexter DT (2010) J Neural Transm. doi:10.1007/s00702-010-0479-3
Dallman PR, Beutler E, Finch CA (1978) Br J Haematol 40:179
Dallman PR, Siimes MA, Stekel A (1980) Am J Clin Nutr 33:86
Beard JL (2008) J Nutr 138:2534
Lozoff B, Beard J, Connor J, Barbara F, Georgieff M, Schallert T (2006) Nutr Rev 64:S34
Rao R, Georgieff MK (2007) Semin Fetal Neonatal Med 12:54
Rao R, Tkac I, Townsend EL, Gruetter R, Georgieff MK (2003) J Nutr 133:3215
Rao R, Tkac I, Townsend EL, Ennis K, Gruetter R, Georgieff MK (2007) J Cerebr Blood Flow Metab 27:872
Ward KL, Tkac I, Jing Y, Felt B, Beard J, Connor J, Schallert T, Georgieff MK, Rao R (2007) J Nutr 137:1043
Beard JL, Wiesinger JA, Connor JR (2003) Dev Neurosci 25:308
Ortiz E, Pasquini JM, Thompson K, Felt B, Butkus G, Beard J, Connor JR (2004) J Neurosci Res 77:681
Beard J, Erikson KM, Jones BC (2003) J Nutr 133:1174
Burhans MS, Dailey C, Beard Z, Wiesinger J, Murray-Kolb L, Jones BC, Beard JL (2005) Nutr Neurosci 8:31
Bradbury MWB (1997) J Neurochem 67:443
G–tz ME, Double K, Gerlach M, Youdim MB, Riederer P (2004) Ann N Y Acad Sci 1012:193
Zecca L, Youdim MB, Riederer P, Connor JR, Crichton RR (2004) Nat Rev Neurosci 5:863
Biasiotto G, Goldwurm S, Finazzi D, Tunesi S, Zecchinelli A, Sironi F, Pezzoli G, Arosio P (2008) Parkinsonism Relat Disord 14:426
Bartzokis G, Lu PH, Tishler TA, Peters DG, Kosenko A, Barrall KA, Finn JP, Villablanca P, Laub G, Altshuler LL, Geschwind DH, Mintz J, Neely E, Connor JR (2010) J Alzheimers Dis 20:333
Goodhall EF, Greenway MJ, van Marion I, Carrol CB, Hardiman O, Morrison KE (2005) Neurology 65:934
Johnstone D, Milward EA (2010) J Neurochem 113:1387
Ellervik C, Birgens H, Tybjaerg-Hansen A, Nordestgaard BG (2007) Hepatology 46:1071
Penke L, Valdés Hernandéz MC, Maniega SM, Gow AJ, Murray C, Starr JM, Bastin ME, Deary IJ, Wardlaw JM (2010) Neurobiol Aging. doi:10.1016/j.neurobiolaging.2010.04.032
Sullivan EV, Adalsteinsson E, Rohlfing T, Pfefferbaum A (2009) Brain Imaging Behav 3:167
Vasudevaraju P, Bharathi TJ, Shamasundar NM, Subba Rao K, Balaraj BM, Ksj R (2010) Indian J Psychiatry 52:140
Kastman EK, Willette AA, Coe CL, Bendlin BB, Kosmatka KJ, McLaren DG, Xu G, Canu E, Field AS, Alexander AL, Voytko ML, Beasley TM, Colman RJ, Weindruch RH, Johnson SC (2010) J Neurosci 30:7940
House MJ, St Pierre TG, Milward EA, Bruce DG, Olynyk JK (2010) Magn Reson Med 63:275
Crichton RR, Ward RJ (2006) Metal-based neurodegeneration. From molecular mechanisms to therapeutic strategies. Wiley, Chichester, p 227
Dalle-Donne I, Giustarini D, Colombo R, Rossi R, Milzani A (2003) Trends Mol Med 9:169
Benedetti A, Pmpella A, Fulceri R, Romani A, Comporti M (1986) Toxicol Pathol 14:457
Catala A (2009) Chem Phys Lipids 157:1
Schneider C, Porter NA, Brash AR (2008) J Biol Chem 283:15539
Zarkovic N (2003) Mol Aspects Med 24:281
Zarkovic K (2003) Mol Aspects Med 24:293
Grimsrud PA, Xie H, Griffin TJ, Bernlohr DA (2008) J Biol Chem 283:21837
Berlett BS, Stadtman ER (1997) J Biol Chem 272:20313
Beal MF (2002) Free Radic Biol Med 32:797
Stadtman ER (2006) Free Radic Res 40:1250
Sayre LM, Lin D, Yuan Q, Zhu X, Tang X (2006) Drug Metab Rev 38:651
Oe T, Arora JS, Lee SH, Blair IA (2003) J Biol Chem 27:42098
Zhang WH, Liu J, Xu G, Yuan Q, Sayre LM (2003) Chem Res Toxicol 16:512
Bush AL, Tanzi RE (2002) Proc Natl Acad Sci USA 99:97317
Honda K, Casadesus G, Peterson RB, Perry G, Smith MA (2004) Ann NY Acad Sci 1012:179
Honda K, Smith MA, Zhu X, Baus D, Merrick WC, Tartakoff AM, Hattier T, Harris PL, Siedlak SL, Fujioka H, Liu Q, Moreira PI, Miller FP, Nunomura A, Shimohama S, Perry G (2005) J Biol Chem 280:20978
Sipe JC, Lee P, Beutler E (2002) Dev Neurosci 24:188
Connor JR, Snyder BS, Beard JL, Fine RE, Mufson EJ (1992) J Neurosci Res 31:327
Smith MA, Harris PL, Sayre LM, Perry G (1997) Proc Natl Acad Sci USA 94:9866
Tachida Y, Nakagawa K, Saito T, Saido TC, Honda T, Saito Y, Murayama S, Endo T, Sakaguchi G, Kato A, Kitazume S, Hashimoto Y (2008) J Neurochem 104:1387
Seidah NG, Chrétien M, Day R (1994) Biochimie 76:197
Silvestri L, Pagani A, Camaschella C (2008) Blood 111:924
Babitt JL, Huang FW, Wrighting DM, Xia Y, Sidis Y, Samad TA, Campagna JA, Chung RT, Schneyer AL, Woolf CJ, Andrews NC, Lin HY (2006) Nat Genet 38:531
McMahon S, Grondin F, McDonald PP, Richard DE, Dubois CM (2005) J Biol Chem 280:6561
Silvestri L, Camaschella C (2008) J Cell Mol Med 12:1548
Mueller S (2005) Biofactors 24:171
Hwang EM, Kim SK, Sohn JH, Lee JY, Kim Y, Kim YS, Mook-Jung I (2006) Biochem Biophys Res Commun 349:654
Rogers JT, Randall JD, Cahill CM, Eder PS, Huang X, Gunshin H, Leiter L, McPhee J, Sarang SS, Utsuki T, Greig NH, Lahiri DK, Tanzi RE, Bush Al, Giordano T, Gullans SR (2002) J Biol Chem 277:45518
Duce JA, Tsatsanis A, Cater MA, Janes SA, Robb E, Wikhe K, Leong SL, Perez K, Johanssen T, Greenough MA, Cho H-H, Galatis D, Moir RD, Masters CL, McLean C, Tanzi RE, Cappai R, Barnham KJ, Ciccotosto GD, Rogers JT, Bush AI (2010) Cell 142:857
Schapira AH, Olanow CW (2004) JAMA 291:358
Crichton RR (2009) Inorganic biochemistry of iron metabolism. from molecular mechanisms to clinical consequences, 3rd edn. Wiley, Chichester, p 461
Oakley AE, Collingwood JF, Dobson L, Love G, Perrott HR, Edwardson JA, Elstner M, Morris CM (2007) Neurology 68:1820
Goedert M (2001) Nat Rev Neurosci 2:492
Golts N, Snyder H, Frasier M, Theisler C, Choi P, Wolozin B (2002) J Biol Chem 277:16116
Hashimoto M, Hsu LJ, Xia Y, Takeda A, Sisk A, Sundsmo M, Masliah E (1999) Neuroreport 10:717
Münch G, Lüth HJ, Wong A, Arendt T, Hirsch E, Ravid R, Riederer P (2000) J Chem Neuroanat 20:253
Ostrerova-Golts N, Petrucelli L, Hardy J, Lee JM, Farer M, Wolozin B (2000) J Neurosci 20:6048
Sangchot P, Sharma S, Chetsawang B, Porter J, Govitrapong P, Ebadi M (2002) Dev Neurosci 24:143
Takanashi M, Mochizuki H, Yokomizo K, Hattori N, Mori H, Yamamura Y, Mizuno Y (2001) Parkinsonism Relat Disord 7:311
Castellani RJ, Siedlak SL, Perry G, Smith MA (2000) Acta Neuropathol 100:111
Arawaka S, Saito Y, Murayama S, Mori H (1998) Neurology 51:887
Kaltschmidt C, Kaltschmidt B, Lannes-Vieira J, Kreutzberg GW, Wekerle H, Baeuerle PA, Gehrmann J (1994) J Neuroimmunol 55:99
Zhang W, Murao K, Zhange X, Matsumoto K, Diah S, Okada M, Miyake K, Kawai N, Tamiya T (2010) BMC Cancer 10:593
Ward RJ, Lallemand F, de Witte P, Crichton RR, Piette J, Tipton K, Hemmings K, Pitard A, Page M, Della Corte L, Taylor D, Dexter DT (2011) Biochem Pharmacol 81:743–751
Ward RJ, Dexter D, Florence A, Aouad F, Hider R, Jenner P, Crichton RR (1995) Biochem Pharmacol 49:1821
Hua Y, Keep RF, Hoff JT, Xi G (2008) Acta Neurochir Suppl 105:3
Selim M (2009) Stroke 40:S90
Shachar DB, Kahana N, Kampel V, Warshawsky A, Youdim MB (2004) Neuropharmacology 46:254
Dexter DT, Statton S, Whitmore C, Freinbichler W, Weinberger P, Tipton KT, Della Corte L, Ward RJ, Crichton RR (2011) J Neural Trans 118:223–231
Crichton RR, Dexter DT, Ward RJ (2008) Coord Chem Rev 252:1189
Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ (2009) Neurobiol Dis 37:13
Zimmer M, Ebert BL, Neil C, Brenner K, Papaioannou I, Melas A, Tolliday N, Lamb J, Pantopoulos K, Golub T, Iliopoulos O (2008) Mol Cell 32:838
Hentze MW, Kühn LC (1996) Proc Natl Acad Sci USA 93:8175
Cho HH, Cahill CM, Vanderburg CR, Scherzer CR, Wang B, Huang X, Rogers JT (2010) J Biol Chem 285:31217
Acknowledgments
Grateful thanks are due to COST D34 for facilitating the interaction between the authors.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer-Verlag Wien
About this chapter
Cite this chapter
Crichton, R.R., Dexter, D.T., Ward, R.J. (2012). Brain iron metabolism and its perturbation in neurological diseases. In: Linert, W., Kozlowski, H. (eds) Metal Ions in Neurological Systems. Springer, Vienna. https://doi.org/10.1007/978-3-7091-1001-0_1
Download citation
DOI: https://doi.org/10.1007/978-3-7091-1001-0_1
Published:
Publisher Name: Springer, Vienna
Print ISBN: 978-3-7091-1000-3
Online ISBN: 978-3-7091-1001-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)