
Abstract
Activity-dependent modifications in synaptic efficacy, such as long-term depression (LTD) and long-term potentiation (LTP), represent key cellular substrates for adaptive motor control and procedural memory. The impairment of these two forms of synaptic plasticity in the nucleus striatum could account for the onset and the progression of motor and cognitive symptoms of Parkinson’s disease (PD), characterized by the massive degeneration of dopaminergic neurons. In fact, both LTD and LTP are peculiarly controlled and modulated by dopaminergic transmission coming from nigrostriatal terminals.
Changes in corticostriatal and nigrostriatal neuronal excitability may influence profoundly the threshold for the induction of synaptic plasticity, and changes in striatal synaptic transmission efficacy are supposed to play a role in the occurrence of PD symptoms. Understanding of these maladaptive forms of synaptic plasticity has mostly come from the analysis of experimental animal models of PD. A series of cellular and synaptic alterations occur in the striatum of experimental parkinsonism in response to the massive dopaminergic loss. In particular, dysfunctions in trafficking and subunit composition of glutamatergic NMDA receptors on striatal efferent neurons contribute to the clinical features of the experimental parkinsonism.
Interestingly, it has become increasingly evident that in striatal spiny neurons, the correct assembly of NMDA receptor complex at the postsynaptic site is a major player in early phases of PD, and it is sensitive to distinct degrees of DA denervation. The molecular defects at the basis of PD progression may be not confined just at the postsynaptic neuron: accumulating evidences have recently shown that the genes linked to PD play a critical role at the presynaptic site. DA release into the synaptic cleft relies on a proper presynaptic vesicular transport; impairment of SV trafficking, modification of DA flow, and altered presynaptic plasticity have been described in several PD animal models. Furthermore, an impaired DA turnover has been described in presymptomatic PD patients. Thus, given the pathological events occurring precociously at the synapses of PD patients, post- and presynaptic sites may represent an adequate target for early therapeutic intervention.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
1 Introduction
Parkinson’s disease (PD) is one of the most frequent human neurodegenerative disorders associated with the process of cerebral aging. PD physiopathology is linked to a widespread process of degeneration of dopamine (DA)-secreting neurons in the substantia nigra pars compacta (SNc), with the consequent loss of the neurons projecting to the striatum (Lang and Lozano 1998a, b). The parkinsonian symptoms appear when brain levels of DA reach the 70–80% of the normal levels. The main clinical features of PD are the direct consequences of a dysfunction occurring within both the striatum and the entire basal ganglia system (Calabresi et al. 2007). Bradykinesia, rigidity, tremor at rest, postural instability, micrographia, and shuffling gait represent the principal motor symptoms that allow the diagnosis of PD (Jankovic 2008). The clinical detection of these motor symptoms is often accompanied by autonomic, cognitive, and psychiatric problems (Calabresi et al. 2006; Kehagia et al. 2010). Rare forms of PD resulted from missense mutations of α-synuclein as well as increased expression of normal α-synuclein are characterized by early onset and autosomal-dominant inheritance (Polymeropoulos et al. 1997; Singleton et al. 2003). Intracytoplasmic inclusions called Lewy bodies and the progressive loss of DA-containing neurons in the SNc represent the main neuropathological features of the genetic forms of PD (Spillantini et al. 1998; Dickson et al. 2009; Schulz-Schaeffer 2010).
Mutations in seven genes have been implicated in various forms of familial parkinsonism. Two autosomal-dominant genes (α-synuclein and LRRK2) and three autosomal-recessive genes (Parkin, DJ-1, and PINK1) have been definitively associated with inherited PD (Nussbaum and Polymeropoulos 1997; Polymeropoulos et al. 1997; Healy et al. 2004; Paisan-Ruiz et al. 2004; Valente et al. 2004). As well as these, other mutations have been reported in UCHL-1, synphilin-1, and NR4A2 that may or may not be biologically significant (Leroy et al. 1998; Le et al. 2003; Marx et al. 2003). Synaptic loss is one of the major neurobiological dysfunction occurring in several neurological diseases (Wishart et al. 2006), for example, synaptic failure happens in a very early phase in both patients and animal models during the progression of Alzheimer’s disease (Selkoe 2002).
2 Parkinson’s Disease and Presynaptic Dysfunction
Accumulating evidence has convincingly demonstrated that the genes linked to PD play a critical role at the presynaptic site. α-synuclein is a 140–amino acid protein present in almost all subcellular compartments but particularly enriched in the presynaptic terminals where it is loosely associated with the distal reserve pool of synaptic vesicles (Lavedan 1998; Yu et al. 2007). Structural and functional studies have shown that α-synuclein is involved in the trafficking of synaptic vesicles. In fact, the presynaptic boutons of cultures lacking α-synuclein presented a marked reduction in the number of vesicles present in the distal pool although the number of vesicles docked at the synaptic plasma remained unaltered (Murphy et al. 2000). Accordingly, α-synuclein knockout (KO) mice showed a marked decrease in the pool of undocked synaptic vesicles and significantly impaired hippocampal response to long-lasting low-frequency stimulation (Cabin et al. 2002). Furthermore, α-synuclein KO mice are characterized by an increased evoked DA release: these observations might imply that α-synuclein normally acts as a negative regulator of DA neurotransmission in an activity-dependent fashion (Abeliovich et al. 2000). Strikingly, overexpression of α-synuclein inhibits neurotransmitter release affecting specifically the size of the synaptic vesicle recycling pool (Nemani et al. 2010). Thus, α-synuclein seems to be deeply implicated in the synaptic vesicle trafficking required for a proper presynaptic DA release by keeping low the amount of DA within the presynaptic bouton (Sidhu et al. 2004; Yu et al. 2005). Given that cytosolic DA might be converted into highly reactive oxidative molecules, it can be speculated that pathological mutations or aggregation of α-synuclein might prejudice normal α-synuclein functions. This impairment may bring to accumulation of DA and thus to the generation of toxic moieties. Interestingly, also DJ-1 and PINK1 KO mice exhibit presynaptic defects. DJ-1 is a redox-sensitive molecular chaperone, and it has been proposed that it inhibits protein aggregation, including α-synuclein formations (Shendelman et al. 2004; Wilson et al. 2004; Moore et al. 2006; Gasser 2009). DJ-1 is expressed widely throughout the tissues, and it is subcellularly localized to the cytosol, mitochondrial matrix, and intermembrane space (Zhang et al. 2005). Acute slice preparation from DJ-1 KO mice showed a reduce DA overflow and impaired LTD. Furthermore, the mice had a poor performance in terms of spontaneous activities and generalized hypokinesia in open field (Goldberg et al. 2003, 2005). DJ-1 has been reported to sustain also hippocampal LTD consolidation, suggesting a potential involvement for this protein in modulating hippocampal dependent cognitive dysfunctions reported in PD (Wang et al. 2008). PINK1 instead is a serine/threonine kinase localized in the mitochondria (Silvestri et al. 2005; Zhou et al. 2008). If PINK1 KO mice failed to exhibit any major abnormality, they showed clear deficits in nigrostriatal DA neurotransmission. Robust evidence supports the conclusion that loss of PINK1 function causes a selective impairment in exocytotic DA release (Kitada et al. 2007; Gispert et al. 2009). Actual knowledge about PINK1 suggests that it may reside in the mitochondria but, given that its kinase domain faces the cytosol, it may have extramitochondrial phosphotargets (Silvestri et al. 2005). Therefore, it might be argued that PINK1 can modify via phosphorylation the activity of proteins involved in DA release. Noteworthy, it has been demonstrated that Parkin, PINK1, and DJ-1 interact physically and functionally. In fact, these three proteins form a ternary complex that promotes ubiquitination and degradation of aberrantly expressed and heat shock–induced Parkin substrates, as Parkin itself and synphilin-1. Pathogenic mutants might reduce the activity of the degradative complex (Xiong et al. 2009).
Mutations in LRRK2 gene account for up to 13% of familial PD cases compatible with dominant inheritance (Paisan-Ruiz et al. 2004, 2008; Zimprich et al. 2004) and 1–2% of sporadic PD patients, thus suggesting this protein as the most significant player in PD pathogenesis identified to date (Aasly et al. 2005; Berg et al. 2005; Taylor et al. 2006). Clinically and pathologically, the features of LRRK2-associated parkinsonism are often indistinguishable from idiopathic PD, although pathologic variability exists even within PARK8-linked kindred, ranging from nigral neuronal loss only to general neuronal loss with α-synuclein, ubiquitin, or tau inclusions [reviewed in (Whaley et al. 2006)]. Furthermore, the neuropathology demonstrated in postmortem brain examinations of patients with LRRK2 mutations most often involves synucleinopathy, but occasionally tauopathy, suggesting a role for LRRK2 that is upstream of protein inclusion pathology (Zimprich et al. 2004; Taymans and Cookson 2010; Wider et al. 2010). The LRRK2 protein has a molecular weight of approximately 280 kDa and contains several domains including a Ras/GTPase like (Roc), a C-terminal of Roc (COR), a kinase (similar to mitogen-activated protein kinase), and a WD40 domain (Bosgraaf and Van Haastert 2003; Guo et al. 2006). Phylogenetically, the LRRK2 kinase domain belongs to the TKL (tyrosine like kinases) and shows high similarity to mixed lineage kinases (MLKs) (Manning et al. 2002; Marin 2006). Few LRRK2 substrates, including moesin, 4E-BP, MKKs, tubulin beta, and α-synuclein, have been found so far in in vitro assays (Jaleel et al. 2007; Imai et al. 2008; Gillardon 2009; Gloeckner et al. 2009; Qing et al. 2009). Several single nucleotide alterations have been identified in LRRK2 (Lesage et al. 2005; Mata et al. 2005), covering all functional domains, but only five missense mutations clearly segregate with PD in large family studies (Goldwurm et al. 2005; Bonifati 2006a, b). Disease-segregating mutations in LRRK2 have been reported in the kinase domain (G2019S, I2020T), in the Roc domain (R1441C/G), and in the COR domain (Y1699C) [reviewed in Mata et al. (2005)].
The most common mutation found in western countries kindred, G2019S, falls in the kinase domain and increases LRRK2 kinase activity while mutations in the Roc domain appear to decrease the GTPase activity of LRRK2 to affect protein dimerization and to slightly increase kinase activity [reviewed in more detail in (Moore 2008)]. The G2019S mutation has been identified also in parkinsonian patients with no family history of disease (Gilks et al. 2005; Healy et al. 2008); other LRRK2 variants affecting kinase activity appear to be important risk factors in two genome-wide association studies of sporadic PD (Simon-Sanchez et al. 2009). Although studies show little concordance regarding the level of LRRK2 mRNA/protein expression in the SN, LRRK2 protein expression has been demonstrated in tyrosine-hydroxylase positive neurons of the SNc and in medium-sized spiny neurons of the striatum (Galter et al. 2006; Melrose et al. 2006; Higashi et al. 2007a, b). Cortical regions that are affected in dementia associated with PD, including pyramidal neurons of the cerebral cortex and of Ammon’s horn, also demonstrate relatively high levels of LRRK2 (Biskup et al. 2006; Higashi et al. 2007b). At the subcellular level, precedent studies showed LRRK2 is mainly associated with mitochondria but also with multiple vesicles structure, including synaptic vesicles (Biskup et al. 2006). Despite its predominance in PD, the physiological function of LRRK2 is not known, and therefore, its precise role in the etiology of PD is far from being understood.
Neurotransmission defects have been repeatedly observed in different LRRK2 models (Li et al. 2009; Tong et al. 2009; Xiong et al. 2009; Li et al. 2010). Functional impairments in nigrostriatal dopaminergic innervation and degeneration of the nigrostriatal projections have been demonstrated in R1441C-LRRK2 homozygous knock-in mice (Tong et al. 2009) and in R1441C-LRRK2 BAC transgenic mice (Li et al. 2009), respectively. G2019S BAC transgenic mice show deficiencies in striatal dopamine release and enhanced striatal tau immunoreactivity without dopaminergic neuron loss in the substantia nigra (Li et al. 2010). Recent studies have enlightened that LRRK2 acts directly at the secretory and endocytic molecular machinery (Shin et al. 2008; Xiong et al. 2010). Finally, it has been shown that electrophysiological properties as well as proper vesicular trafficking and spatial distribution in the presynaptic pool depend on the presence of LRRK2 as an integral part of presynaptic protein complex (Piccoli et al. 2011). Presynaptic proteins – NSF, AP-2 complex subunits, SV2A, synapsin, syntaxin 1 (Piccoli et al. 2011), and Rab5b (Shin et al. 2008) – as well as actin (Meixner et al. 2010) have been found to interact, at least in vitro, with LRRK2 (Fig 24.1). These proteins have been previously described as key elements of synaptic vesicle trafficking. NSF catalyzes the release of the SNARE complex (SNAP 25, syntaxin 1, and VAMP) and allows the first step of the endocytic cycle where also Rab5 proteins are called in action. The clathrin complex [clathrin, AP-2 adaptor complex, and accessory proteins as dynamin and AP180] constitutes one of the major pathways for SV recycling from the membrane to the resting pool (RP). The control of storage and mobilization of SV in the RRP depends instead on the synaptic vesicle glycoproteins SV2A and B while synapsins are thought to immobilize SV in the RP by cross-linking vesicles to the actin cytoskeleton. Strikingly, an increased DA turnover has been noticed in presymptomatic LRRK2 mutation carriers (Sossi et al. 2010). Increased turnover might arise as a compensatory mechanism to counteract DA-neurons loss (Adams et al. 2005), but it has also been suggested that increased DA turnover might by itself contribute to disease progression secondary to DA-associated toxicity (Smith et al. 2002; Zigmond et al. 2002). Therefore, accumulating evidences suggest that synaptic dysfunction is a primary effect of LRRK2 gene mutations and that synaptic failure is intimately involved in LRRK2 due PD pathogenesis.
3 Postsynaptic Dysfunction in Parkinson’s Disease
The natural history of PD is complex and involves differential mechanisms during its various clinical phases. Most of the evidence on pathogenic pathways in PD has been obtained using experimental models of complete striatal DA depletion mimicking advanced PD such as rats lesioned with 6-hydroxydopamine (6-OHDA) (Schwarting and Huston 1996) and macaques lesioned with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (Jenner and Marsden 1986).
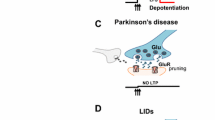
The massive denervation of the dopaminergic nigrostriatal terminals, as observed in advanced PD, is associated to maladaptive plasticity (Calabresi et al. 2007), alteration of striatal dendritic spines (Anglade et al. 1996; Day et al. 2006), and changes of glutamatergic signaling (Betarbet et al. 2000; Picconi et al. 2004). In advanced PD, spontaneous excitatory glutamatergic synaptic activity can be dramatically altered. These pathological events may also alter the amplitude and the direction of long-term changes of excitatory transmission induced by repetitive synaptic activation. Moreover, changes in neuronal phasic and/or tonic firing discharge may occur. Even slight changes in corticostriatal and nigrostriatal neuronal excitability may influence profoundly the threshold for the induction of synaptic plasticity. Changes in striatal synaptic transmission efficacy are supposed to be the cellular basis for such complex integrative functions (Calabresi et al. 2006, 2007), and experimental findings show that short- and long-term changes in corticostriatal synaptic plasticity may play a role in PD (Gubellini et al. 2002; Picconi et al. 2003).
Two cardinal features of PD pathophysiology are represented by the alteration of glutamatergic synapses paradoxically accompanied by the described increase of glutamatergic transmission within the striatum. The real mechanisms underlying this increased excitatory drive remains unknown. Recently, the synaptic changes in both corticostriatal and thalamostriatal afferents have been studied in MPTP-treated monkeys taking as main markers the vesicular glutamate transporters (vGluTs) 1 and 2 (Raju et al. 2008). This study demonstrates the increased presence of vGluT1 in the striatum of MPTP monkeys without any significant change in the pattern of synaptic connectivity. However, a clear degree of synaptic reorganization of the thalamostriatal system has been found. These findings suggest a differential degree of plasticity between the two systems in parkinsonian primates.
In the last decades have been described and extensively studied two forms of striatal synaptic plasticity (long-term depression (LTD) and long-term potentiation (LTP)) thought to underlie cognitive performance both in vitro (Calabresi et al. 1992b, c; Lovinger et al. 1993; Walsh 1993; Walsh and Dunia 1993; Partridge et al. 2000) and in vivo (Charpier and Deniau 1997; Reynolds and Wickens 2000; Mahon et al. 2004).
A high-frequency stimulation (HFS) protocol of the corticostriatal fibers (Calabresi et al. 1992b, c; Lovinger et al. 1993) allows to induce both forms of synaptic plasticity, the type of the long-lasting changes being critically dependent upon the level of membrane depolarization and on the ionotropic glutamate receptor subtype activated during the HFS. A third form of synaptic plasticity (depotentiation) results from the reversal of an established LTP by the application of a low-frequency stimulation of corticostriatal fibers (O’Dell and Kandel 1994; Picconi et al. 2003).
Compared to other brain areas, in which synaptic plasticity has been extensively studied, the striatum has the peculiar feature of receiving a massive dopaminergic input arising from SNc. Accordingly, a unique characteristic of striatal LTD is the requirement of DA receptor activation by endogenous DA (Calabresi et al. 2007). In fact, this form of synaptic plasticity is lost after massive DA denervation both in 6-OHDA rats (Calabresi et al. 1992c) and MPTP-treated monkeys (Quik et al. 2006).
The absence of LTD in the striatum of parkinsonian animals can be attributed to the failed activation of DA receptors during the induction phase of this form of synaptic plasticity. LTD, in fact, can be restored after DA denervation by ensuring DA receptor activation through the application of exogenous DA or by the coactivation of both D1 and D2 receptors (Calabresi et al. 1992a, 2007). Similarly, massive nigrostriatal denervation blocks corticostriatal LTP (Picconi et al. 2003; Calabresi et al. 2007). Interestingly, a “balanced” DA/DARPP-32 pathway is required for the corticostriatal system to be able to express both LTD and LTP (Calabresi et al. 2000).
It is of interest to note that distinct degrees of DA denervation may differentially affect the induction and maintenance of these two distinct and opposite forms of corticostriatal synaptic changes (Paille et al. 2010). An incomplete DA denervation does not affect corticostriatal LTD which is, however, abolished by a complete lesion suggesting that a low, although critical, level of DA is required for this form of synaptic plasticity. Conversely, an incomplete DA denervation dramatically alters the maintenance of LTP confirming a critical role of this form of synaptic plasticity in the early motor parkinsonian symptoms (Paille et al. 2010).
Recently, to understand the early synaptic mechanisms occurring in PD, the striatal dysfunctions have been studied in mice overexpressing human A53T-alfa-synuclein (Kurz et al. 2010). A53T-alfa-synuclein overexpressing mice, in their advanced stage, present dysfunctional DA neurotransmission and consequently an impaired striatal LTD, confirming, once more, the relevant role of an intact and correct balance in the dopaminergic nigrostriatal transmission for a physiological synaptic activity.
The pathophysiological picture emerging from the last years of experimental approach shows that the strength of glutamatergic signals from the cortex to the striatum might be dynamically regulated during the progression of the disease. In fact, bidirectional changes in corticostriatal synaptic plasticity are critically controlled by the different degree of nigral denervation which influences the endogenous DA levels and the assembly of striatal N-methyl-D-aspartate (NMDA)–type glutamate receptor subunits.
NMDA receptors are glutamate ion channels and represent the key elements in the regulation of synaptic function in the central nervous system. They resulted from the coassembly of three different receptor subunit families: NMDA receptor 1 (NR1), NR2A-NR2D, and NR3A-NR3B (Dingledine et al. 1999; Nishi et al. 2001). NMDA receptors are highly permeable to Ca2+, and its influx through the receptor channel is essential for the synaptogenesis, the synaptic remodeling, and the long-lasting changes in synaptic efficacy such as synaptic plasticity (Collingridge et al. 2004).
In the neuronal synapses, NMDA receptors are clustered in the postsynaptic density (PSD) that consists of numerous scaffolding cytoskeletal and signaling proteins, some of which are in close contact with the cytoplasmic domain of glutamate ionotropic receptors in the postsynaptic membrane (Kennedy 2000; Gardoni et al. 2001). This accumulation of NMDA receptors at the postsynaptic compartment ensures a rapid response to neurotransmitter release and provides a molecular mechanism for linking the transmembrane ion flux to the signaling machinery responsible for specific second messenger pathways. Among the protein complex governing the response of the signaling cascade, the α-calcium-calmodulin-dependent protein kinase II (α-CaMKII) is directly linked to the NR2A/NR2B subunits (Gardoni et al. 1998; Strack et al. 2000) and competes in NR2A binding with PSD-95 (Gardoni et al. 2001). Interestingly, CaMKII- and tyrosine-dependent phosphorylation of NMDA receptors is altered in experimental model of PD (Oh et al. 1999).
In the striatum as well as in other brain areas, LTP requires activation of NMDA receptors (Calabresi et al. 1992b, 2007; Collingridge and Bliss 1995; Malenka and Bear 2004). Interestingly, it has become increasingly evident that in striatal spiny neurons, NMDA receptor complex is also profoundly altered in experimental PD (Ulas and Cotman 1996; Dunah and Standaert 2001).
Early studies evaluated NMDA receptor abundance, composition, and phosphorylation in advanced model of PD. In the DA, denervated striatum has been found a decreased level of NR1 and NR2B subunits in striatal membranes, while the abundance of NR2A was unchanged (Ulas and Cotman 1996; Dunah and Standaert 2001). Further studies in the 6-OHDA model showed similar results and associated to alterations in synaptic plasticity (Picconi et al. 2003, 2004; Gardoni et al. 2006). In particular, NR2B subunit was specifically reduced in the synaptic density from advanced parkinsonian rats when compared with sham-lesioned rats in the absence of parallel alterations of NR1 and NR2A (Picconi et al. 2003, 2004; Gardoni et al. 2006). Interestingly, these molecular alterations have been further confirmed in parkinsonian macaques (Hallett et al. 2005). Hallett’s group shows that in the striatum of MPTP-lesioned macaques the DA depletion induces massive changes in the levels of striatal NMDA receptor proteins, such as a reduction in the abundance of NR1 and NR2B but not NR2A subunit. Moreover, in the denervated striatum of parkinsonian animals, the alteration of NMDA receptor subunit localization at synaptic sites is accompanied by a decreased recruitment of PSD-95 to NR2A–NR2B subunits; these events are paralleled by an increased activation of the pool of α-CaMKII associated to the NMDA receptor complex (Picconi et al. 2004). Further, other studies reported that experimental Parkinsonism in rats appears to be associated with decreased synaptic membrane localization and increased vesicular localization of PSD-95 and SAP97 members of the PSD-MAGUK family (Nash et al. 2005) that could account for dysregulation of NMDA receptors at synapses.
While in advanced parkinsonism LTP is completely lost and this synaptic alteration is coupled to specifically reduced levels of NR2B subunits in the PSD compartment (Gardoni et al. 2006), the picture found in the early parkinsonian rats is quite different.
As mentioned above, the incomplete DA denervation dramatically alters the maintenance of LTP. This synaptic alteration recorded in striatal spiny neurons is also accompanied by a dramatic increase in the NR2A NMDA receptor subunits in the striatal synapses, suggesting the presence of a profound rearrangement of the receptor complex composition (Paille et al. 2010). These profound differences in NMDA receptors in the postsynaptic compartment of partially versus fully lesioned rats suggest that NR2-type regulatory subunits are sensitive to plastic changes induced by the differential degree of DA denervation.
Moreover, NMDA receptor subunits NR2A and NR2B interact with membrane-associated guanylate kinases (MAGUK); this interaction governs their trafficking and clustering at synaptic sites (Kim and Sheng 2004). The analysis of PSD-95, SAP97, and SAP102 in the postsynaptic compartment reveals a significant reduction of the three proteins in advanced parkinsonian rats compared with sham-operated rats (Gardoni et al. 2006). In contrast, in early parkinsonian animals, the level of these proteins is the same as in the sham-operated animals, suggesting that in this model of “early” PD, no alteration of MAGUK protein distribution at the synapse is present. These data suggest that the NR2A subunit level at the synaptic site is a major player in early phases of PD, and it is sensitive to distinct degrees of DA denervation; thus, it may represent an adequate target for early therapeutic intervention.
In the PSD, other important receptors included in the glutamatergic ionotropic receptors class, and mediating the functions of glutamate, are represented by alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, tetrameric proteins composed of subunits GluR1-4. Upon binding with glutamate, synaptic AMPA receptors induce membrane depolarization and after removing magnesium (Mg2+) block from NMDA allow to reduce the threshold to induce long-term increases of the synaptic responses. AMPA receptor–dependent depolarization also opens L-type calcium (Ca2+) channels and leads to activation of CRE elements that are responsible for gene transcription.
Recently, a critical role of AMPA receptors in PD has been shown (Lee et al. 2008). Lee and colleagues found that paraquat, a putative causative agent for PD, inhibits postsynaptic AMPA receptors on dopaminergic neurons in the SNc. However, there is still no general consensus on the mechanism underlying dysregulation of AMPA receptor distribution or composition changes in PD. GluR1 subunit of AMPA receptor has not found changed in the striatum of parkinsonian rats (Bernard et al. 1996; Betarbet et al. 2000), while GluR1 immunoreactivity is increased in the caudate and putamen of MPTP monkeys (Betarbet et al. 2000). Evidence has been provided that GluR1 immunoreactivity is decreased in striatal spiny neurons (Lai et al. 2003) and in striatal membrane fractions of parkinsonian rats (Gardoni et al. 2006); on the contrary, no alteration of GluR1 levels in the postsynaptic density has been found in 6-OHDA-lesioned rats (Picconi et al. 2004).
4 Conclusions
Given the correlation recently described between LRRK2 and α-synuclein (Lin et al. 2009; Carballo-Carbajal et al. 2010), the impact of α-synuclein on synaptic vesicle recycling (Fortin et al. 2010; Nemani et al. 2010), and the functional links among DJ-1, Parkin, PINK1, and α-synuclein (Shendelman et al. 2004; Xiong et al. 2009), the regulation of DA release might arise as one the main biological pathway compromised during PD onset. The molecular mechanisms underlying these synaptic transmission defects, however, remain largely elusive. Although little is known about the precise mechanisms of exocytotic DA release, it likely uses a similar mechanism as glutamatergic synapses, in which release is energy-dependent, is mediated by the SNARE-dependent fusion of synaptic vesicles and is triggered by Ca2+ binding to synaptotagmins. Synaptic vesicles undergo in the nerve terminal to high-frequency trafficking cycles thanks to the presence of extremely specialized machinery, allowing very rapid triggering and switching off of synaptic vesicle exocytosis in response to depolarization-evoked Ca2+ influx. A major goal in neurobiology in recent years has been to gain insight into the molecular machinery that mediates neurotransmitter release. More than 1,000 proteins function in the presynaptic nerve terminal, and hundreds are thought to participate in exo-endocytosis. The processes are finely tuned and depend on the interaction between protein expressed on SV membranes and protein expressed on the presynaptic membranes (Rizo and Rosenmund 2008; Sudhof and Rothman 2009).
This complex network of interaction is plastically shaped by posttranslational modifications: the presynaptic modulation of neurotransmitter release is in fact altered by protein kinases and protein phosphatases (Turner et al. 1999; Fdez and Hilfiker 2006) and by protein degradation (Ehlers 2003; Yao et al. 2007). One possibility worth to be explored is that PD-related proteins alter SV trafficking via modification of presynaptic proteins.
Cellular and postsynaptic alterations occurring in the striatum of experimental parkinsonism in response to the massive dopaminergic loss may lead to synaptic dysfunction and corticostriatal transmission instabilities. In particular, maladaptive forms of synaptic plasticity consequently to the alteration in the subunit composition of glutamatergic ionotropic receptors, that is, NMDA receptors, contribute to the clinical features of PD. Interestingly, it has become increasingly evident that the correct assembly of NMDA receptor complex at the synaptic site is a major player in early phases of PD and it is sensitive to distinct degrees of DA denervation; thus, it may represent an adequate target for early therapeutic intervention.

Model of LRRK2 function at the presynaptic site. Given the interaction between LRRK2, cytoskeletal elements, and presynaptic proteins (Shin et al. 2008; Meixner et al. 2010; Piccoli et al. 2011), it has been proposed LRRK2 is part of the molecular complex that controls SV fusion rate. It might modulate SV storage in the RP and SV trafficking between the RP and the membrane. (a) SV actively cycles between the RRP and the RP, even if the major part of SV belongs to an apparently inactive resting pool. (1) SV is maintained in the RP by synapsin-actin cytoskeleton interaction. (2) SV2A and calcium-dependent secretion activator 1 (CASP1) convert the vesicles into fusion-responsive state. (3) SNAREs dock SV to the presynaptic membrane in preparation for fusion. (4) After fusion-pore opening, vesicle-fusing ATPase (NSF) disrupts the SNARE complex releasing SV. (5) SV recycles to the RP mainly through clathrin-coated pits endocytosis (5). (b) Impairment of LRRK2 levels/function might impair the functionality of the exo-endo machinery. In absence of LRRK2, SV might not properly cycle between the (1) RRP and (2) the RP. (3) The reduction of the molecular constrain represented by LRRK2 and LRRK2-associated protein might increase SV probability to reach the membrane and fuse (In the cartoon are depicted only presynaptic proteins putatively interacting with LRRK2)
References
Aasly, J. O., Toft, M., Fernandez-Mata, I., Kachergus, J., Hulihan, M., White, L. R., & Farrer, M. (2005). Clinical features of LRRK2-associated Parkinson’s disease in central Norway. Annals of Neurology, 57, 762–765.
Abeliovich, A., Schmitz, Y., Farinas, I., Choi-Lundberg, D., Ho, W. H., Castillo, P. E., Shinsky, N., Verdugo, J. M., Armanini, M., Ryan, A., Hynes, M., Phillips, H., Sulzer, D., & Rosenthal, A. (2000). Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron, 25, 239–252.
Adams, J. R., van Netten, H., Schulzer, M., Mak, E., McKenzie, J., Strongosky, A., Sossi, V., Ruth, T. J., Lee, C. S., Farrer, M., Gasser, T., Uitti, R. J., Calne, D. B., Wszolek, Z. K., & Stoessl, A. J. (2005). PET in LRRK2 mutations: Comparison to sporadic Parkinson’s disease and evidence for presymptomatic compensation. Brain, 128, 2777–2785.
Anglade, P., Mouatt-Prigent, A., Agid, Y., & Hirsch, E. (1996). Synaptic plasticity in the caudate nucleus of patients with Parkinson’s disease. Neurodegeneration, 5, 121–128.
Berg, D., Schweitzer, K., Leitner, P., Zimprich, A., Lichtner, P., Belcredi, P., Brussel, T., Schulte, C., Maass, S., & Nagele, T. (2005). Type and frequency of mutations in the LRRK2 gene in familial and sporadic Parkinson’s disease*. Brain, 128, 3000–3011.
Bernard, V., Gardiol, A., Faucheux, B., Bloch, B., Agid, Y., & Hirsch, E. C. (1996). Expression of glutamate receptors in the human and rat basal ganglia: Effect of the dopaminergic denervation on AMPA receptor gene expression in the striatopallidal complex in Parkinson’s disease and rat with 6-OHDA lesion. The Journal of Comparative Neurology, 368, 553–568.
Betarbet, R., Porter, R. H., & Greenamyre, J. T. (2000). GluR1 glutamate receptor subunit is regulated differentially in the primate basal ganglia following nigrostriatal dopamine denervation. Journal of Neurochemistry, 74, 1166–1174.
Biskup, S., Moore, D. J., Celsi, F., Higashi, S., West, A. B., Andrabi, S. A., Kurkinen, K., Yu, S. W., Savitt, J. M., Waldvogel, H. J., Faull, R. L., Emson, P. C., Torp, R., Ottersen, O. P., Dawson, T. M., & Dawson, V. L. (2006). Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Annals of Neurology, 60, 557–569.
Bonifati, V. (2006a). Parkinson’s disease: The LRRK2-G2019S mutation: Opening a novel era in Parkinson’s disease genetics. European Journal of Human Genetics, 14, 1061–1062.
Bonifati, V. (2006b). The pleomorphic pathology of inherited Parkinson’s disease: Lessons from LRRK2. Current Neurology and Neuroscience Reports, 6, 355–357.
Bosgraaf, L., & Van Haastert, P. J. (2003). Roc, a Ras/GTPase domain in complex proteins. Biochimica et Biophysica Acta, 1643, 5–10.
Cabin, D. E., Shimazu, K., Murphy, D., Cole, N. B., Gottschalk, W., McIlwain, K. L., Orrison, B., Chen, A., Ellis, C. E., Paylor, R., Lu, B., & Nussbaum, R. L. (2002). Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. The Journal of Neuroscience, 22, 8797–8807.
Calabresi, P., Maj, R., Mercuri, N. B., & Bernardi, G. (1992a). Coactivation of D1 and D2 dopamine receptors is required for long-term synaptic depression in the striatum. Neuroscience Letters, 142, 95–99.
Calabresi, P., Pisani, A., Mercuri, N. B., & Bernardi, G. (1992b). Long-term potentiation in the striatum is unmasked by removing the voltage-dependent magnesium block of NMDA receptor channels. European Journal of Neuroscience, 4, 929–935.
Calabresi, P., Picconi, B., Parnetti, L., & Di Filippo, M. (2006). A convergent model for cognitive dysfunctions in Parkinson’s disease: The critical dopamine-acetylcholine synaptic balance. Lancet Neurology, 5, 974–983.
Calabresi, P., Picconi, B., Tozzi, A., & Di Filippo, M. (2007). Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends in Neurosciences, 30, 211–219.
Calabresi, P., Maj, R., Pisani, A., Mercuri, N. B., & Bernardi, G. (1992c). Long-term synaptic depression in the striatum: Physiological and pharmacological characterization. The Journal of Neuroscience, 12, 4224–4233.
Calabresi, P., Gubellini, P., Centonze, D., Picconi, B., Bernardi, G., Chergui, K., Svenningsson, P., Fienberg, A. A., & Greengard, P. (2000). Dopamine and cAMP-regulated phosphoprotein 32 kDa controls both striatal long-term depression and long-term potentiation, opposing forms of synaptic plasticity. The Journal of Neuroscience, 20, 8443–8451.
Carballo-Carbajal, I., Weber-Endress, S., Rovelli, G., Chan, D., Wolozin, B., Klein, C. L., Patenge, N., Gasser, T., & Kahle, P. J. (2010). Leucine-rich repeat kinase 2 induces alpha-synuclein expression via the extracellular signal-regulated kinase pathway. Cellular Signalling, 22, 821–827.
Charpier, S., & Deniau, J. M. (1997). In vivo activity-dependent plasticity at cortico-striatal connections: Evidence for physiological long-term potentiation. Proceedings of the National Academy of Sciences of the United States of America, 94, 7036–7040.
Collingridge, G. L., & Bliss, T. V. (1995). Memories of NMDA receptors and LTP. Trends in Neurosciences, 18, 54–56.
Collingridge, G. L., Isaac, J. T., & Wang, Y. T. (2004). Receptor trafficking and synaptic plasticity. Nature Reviews: Neuroscience, 5, 952–962.
Day, M., Wang, Z., Ding, J., An, X., Ingham, C. A., Shering, A. F., Wokosin, D., Ilijic, E., Sun, Z., Sampson, A. R., Mugnaini, E., Deutch, A. Y., Sesack, S. R., Arbuthnott, G. W., & Surmeier, D. J. (2006). Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nature Neuroscience, 9, 251–259.
Dickson, D. W., Braak, H., Duda, J. E., Duyckaerts, C., Gasser, T., Halliday, G. M., Hardy, J., Leverenz, J. B., Del Tredici, K., Wszolek, Z. K., & Litvan, I. (2009). Neuropathological assessment of Parkinson’s disease: Refining the diagnostic criteria. Lancet Neurology, 8, 1150–1157.
Dingledine, R., Borges, K., Bowie, D., & Traynelis, S. F. (1999). The glutamate receptor ion channels. Pharmacological Reviews, 51, 7–61.
Dunah, A. W., & Standaert, D. G. (2001). Dopamine D1 receptor-dependent trafficking of striatal NMDA glutamate receptors to the postsynaptic membrane. The Journal of Neuroscience, 21, 5546–5558.
Ehlers, M. D. (2003). Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nature Neuroscience, 6, 231–242.
Fdez, E., & Hilfiker, S. (2006). Vesicle pools and synapsins: New insights into old enigmas. Brain Cell Biology, 35, 107–115.
Fortin, D. L., Nemani, V. M., Nakamura, K., & Edwards, R. H. (2010). The behavior of alpha-synuclein in neurons. Movement Disorders, 25(Suppl 1), S21–26.
Galter, D., Westerlund, M., Carmine, A., Lindqvist, E., Sydow, O., & Olson, L. (2006). LRRK2 expression linked to dopamine-innervated areas. Annals of Neurology, 59, 714–719.
Gardoni, F., Caputi, A., Cimino, M., Pastorino, L., Cattabeni, F., & Di Luca, M. (1998). Calcium/calmodulin-dependent protein kinase II is associated with NR2A/B subunits of NMDA receptor in postsynaptic densities. Journal of Neurochemistry, 71, 1733–1741.
Gardoni, F., Schrama, L. H., Kamal, A., Gispen, W. H., Cattabeni, F., & Di Luca, M. (2001). Hippocampal synaptic plasticity involves competition between Ca2+/calmodulin-dependent protein kinase II and postsynaptic density 95 for binding to the NR2A subunit of the NMDA receptor. The Journal of Neuroscience, 21, 1501–1509.
Gardoni, F., Picconi, B., Ghiglieri, V., Polli, F., Bagetta, V., Bernardi, G., Cattabeni, F., Di Luca, M., & Calabresi, P. (2006). A critical interaction between NR2B and MAGUK in L-DOPA induced dyskinesia. The Journal of Neuroscience, 26, 2914–2922.
Gasser, T. (2009). Molecular pathogenesis of Parkinson disease: Insights from genetic studies. Expert Reviews in Molecular Medicine, 11, e22.
Gilks, W. P., Abou-Sleiman, P. M., Gandhi, S., Jain, S., Singleton, A., Lees, A. J., Shaw, K., Bhatia, K. P., Bonifati, V., Quinn, N. P., Lynch, J., Healy, D. G., Holton, J. L., Revesz, T., & Wood, N. W. (2005). A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet, 365, 415–416.
Gillardon, F. (2009). Leucine-rich repeat kinase 2 phosphorylates brain tubulin-beta isoforms and modulates microtubule stability – a point of convergence in parkinsonian neurodegeneration? Journal of Neurochemistry, 110, 1514–1522.
Gispert, S., Ricciardi, F., Kurz, A., Azizov, M., Hoepken, H. H., Becker, D., Voos, W., Leuner, K., Muller, W. E., Kudin, A. P., Kunz, W. S., Zimmermann, A., Roeper, J., Wenzel, D., Jendrach, M., Garcia-Arencibia, M., Fernandez-Ruiz, J., Huber, L., Rohrer, H., Barrera, M., Reichert, A. S., Rub, U., Chen, A., Nussbaum, R. L., & Auburger, G. (2009). Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PLoS One, 4, e5777.
Gloeckner, C. J., Schumacher, A., Boldt, K., & Ueffing, M. (2009). The Parkinson disease-associated protein kinase LRRK2 exhibits MAPKKK activity and phosphorylates MKK3/6 and MKK4/7, in vitro. Journal of Neurochemistry, 109, 959–968.
Goldberg, M. S., Pisani, A., Haburcak, M., Vortherms, T. A., Kitada, T., Costa, C., Tong, Y., Martella, G., Tscherter, A., Martins, A., Bernardi, G., Roth, B. L., Pothos, E. N., Calabresi, P., & Shen, J. (2005). Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron, 45, 489–496.
Goldberg, M. S., Fleming, S. M., Palacino, J. J., Cepeda, C., Lam, H. A., Bhatnagar, A., Meloni, E. G., Wu, N., Ackerson, L. C., Klapstein, G. J., Gajendiran, M., Roth, B. L., Chesselet, M. F., Maidment, N. T., Levine, M. S., & Shen, J. (2003). Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. The Journal of Biological Chemistry, 278, 43628–43635.
Goldwurm, S., Di Fonzo, A., Simons, E. J., Rohe, C. F., Zini, M., Canesi, M., Tesei, S., Zecchinelli, A., Antonini, A., Mariani, C., Meucci, N., Sacilotto, G., Sironi, F., Salani, G., Ferreira, J., Chien, H. F., Fabrizio, E., Vanacore, N., Dalla Libera, A., Stocchi, F., Diroma, C., Lamberti, P., Sampaio, C., Meco, G., Barbosa, E., Bertoli-Avella, A. M., Breedveld, G. J., Oostra, B. A., Pezzoli, G., & Bonifati, V. (2005). The G6055A (G2019S) mutation in LRRK2 is frequent in both early and late onset Parkinson’s disease and originates from a common ancestor. Journal of Medical Genetics, 42, e65.
Gubellini, P., Picconi, B., Bari, M., Battista, N., Calabresi, P., Centonze, D., Bernardi, G., Finazzi-Agro, A., & Maccarrone, M. (2002). Experimental Parkinsonism alters endocannabinoid degradation: Implications for striatal glutamatergic transmission. The Journal of Neuroscience, 22, 6900–6907.
Guo, L., Wang, W., & Chen, S. G. (2006). Leucine-rich repeat kinase 2: Relevance to Parkinson’s disease. The International Journal of Biochemistry & Cell Biology, 38, 1469–1475.
Hallett, P. J., Dunah, A. W., Ravenscroft, P., Zhou, S., Bezard, E., Crossman, A. R., Brotchie, J. M., & Standaert, D. G. (2005). Alterations of striatal NMDA receptor subunits associated with the development of dyskinesia in the MPTP-lesioned primate model of Parkinson’s disease. Neuropharmacology, 48, 503–516.
Healy, D. G., Wood, N. W., & Schapira, A. H. (2008). Test for LRRK2 mutations in patients with Parkinson’s disease. Practical Neurology, 8, 381–385.
Healy, D. G., Abou-Sleiman, P. M., Valente, E. M., Gilks, W. P., Bhatia, K., Quinn, N., Lees, A. J., & Wood, N. W. (2004). DJ-1 mutations in Parkinson’s disease. Journal of Neurology, Neurosurgery, and Psychiatry, 75, 144–145.
Higashi, S., Moore, D. J., Colebrooke, R. E., Biskup, S., Dawson, V. L., Arai, H., Dawson, T. M., & Emson, P. C. (2007a). Expression and localization of Parkinson’s disease-associated leucine-rich repeat kinase 2 in the mouse brain. Journal of Neurochemistry, 100, 368–381.
Higashi, S., Biskup, S., West, A. B., Trinkaus, D., Dawson, V. L., Faull, R. L., Waldvogel, H. J., Arai, H., Dawson, T. M., Moore, D. J., & Emson, P. C. (2007b). Localization of Parkinson’s disease-associated LRRK2 in normal and pathological human brain. Brain Research, 1155, 208–219.
Imai, Y., Gehrke, S., Wang, H. Q., Takahashi, R., Hasegawa, K., Oota, E., & Lu, B. (2008). Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. The EMBO Journal, 27, 2432–2443.
Jaleel, M., Nichols, R. J., Deak, M., Campbell, D. G., Gillardon, F., Knebel, A., & Alessi, D. R. (2007). LRRK2 phosphorylates moesin at threonine-558: Characterization of how Parkinson’s disease mutants affect kinase activity. The Biochemical Journal, 405, 307–317.
Jankovic, J. (2008). Parkinson’s disease: Clinical features and diagnosis. Journal of Neurology, Neurosurgery, and Psychiatry, 79, 368–376.
Jenner, P., & Marsden, C. D. (1986). The actions of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in animals as a model of Parkinson’s disease. Journal of Neural Transmission: Supplementum, 20, 11–39.
Kehagia, A. A., Barker, R. A., & Robbins, T. W. (2010). Neuropsychological and clinical heterogeneity of cognitive impairment and dementia in patients with Parkinson’s disease. Lancet Neurology, 9, 1200–1213.
Kennedy, M. B. (2000). Signal-processing machines at the postsynaptic density. Science, 290, 750–754.
Kim, E., & Sheng, M. (2004). PDZ domain proteins of synapses. Nature Reviews: Neuroscience, 5, 771–781.
Kitada, T., Pisani, A., Porter, D. R., Yamaguchi, H., Tscherter, A., Martella, G., Bonsi, P., Zhang, C., Pothos, E. N., & Shen, J. (2007). Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proceedings of the National Academy of Sciences of the United States of America, 104, 11441–11446.
Kurz, A., Double, K. L., Lastres-Becker, I., Tozzi, A., Tantucci, M., Bockhart, V., Bonin, M., Garcia-Arencibia, M., Nuber, S., Schlaudraff, F., Liss, B., Fernandez-Ruiz, J., Gerlach, M., Wullner, U., Luddens, H., Calabresi, P., Auburger, G., & Gispert, S. (2010). A53T-alpha-synuclein overexpression impairs dopamine signaling and striatal synaptic plasticity in old mice. PLoS One, 5, e11464.
Lai, S. K., Tse, Y. C., Yang, M. S., Wong, C. K., Chan, Y. S., & Yung, K. K. (2003). Gene expression of glutamate receptors GluR1 and NR1 is differentially modulated in striatal neurons in rats after 6-hydroxydopamine lesion. Neurochemistry International, 43, 639–653.
Lang, A. E., & Lozano, A. M. (1998a). Parkinson’s disease. Second of two parts. The New England Journal of Medicine, 339, 1130–1143.
Lang, A. E., & Lozano, A. M. (1998b). Parkinson’s disease. First of two parts. The New England Journal of Medicine, 339, 1044–1053.
Lavedan, C. (1998). The synuclein family. Genome Research, 8, 871–880.
Le, W. D., Xu, P., Jankovic, J., Jiang, H., Appel, S. H., Smith, R. G., & Vassilatis, D. K. (2003). Mutations in NR4A2 associated with familial Parkinson disease. Nature Genetics, 33, 85–89.
Lee, C. Y., Lee, C. H., Shih, C. C., & Liou, H. H. (2008). Paraquat inhibits postsynaptic AMPA receptors on dopaminergic neurons in the substantia nigra pars compacta. Biochemical Pharmacology, 76, 1155–1164.
Leroy, E., Boyer, R., Auburger, G., Leube, B., Ulm, G., Mezey, E., Harta, G., Brownstein, M. J., Jonnalagada, S., Chernova, T., Dehejia, A., Lavedan, C., Gasser, T., Steinbach, P. J., Wilkinson, K. D., & Polymeropoulos, M. H. (1998). The ubiquitin pathway in Parkinson’s disease. Nature, 395, 451–452.
Lesage, S., Leutenegger, A. L., Ibanez, P., Janin, S., Lohmann, E., Durr, A., & Brice, A. (2005). LRRK2 haplotype analyses in European and North African families with Parkinson disease: A common founder for the G2019S mutation dating from the 13th century. The American Society of Human Genetics, 77, 330–332.
Li, X., Patel, J. C., Wang, J., Avshalumov, M. V., Nicholson, C., Buxbaum, J. D., Elder, G. A., Rice, M. E., & Yue, Z. (2010). Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson’s disease mutation G2019S. The Journal of Neuroscience, 30, 1788–1797.
Li, Y., Liu, W., Oo, T. F., Wang, L., Tang, Y., Jackson-Lewis, V., Zhou, C., Geghman, K., Bogdanov, M., Przedborski, S., Beal, M. F., Burke, R. E., & Li, C. (2009). Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson’s disease. Nature Neuroscience, 12, 826–828.
Lin, X., Parisiadou, L., Gu, X. L., Wang, L., Shim, H., Sun, L., Xie, C., Long, C. X., Yang, W. J., Ding, J., Chen, Z. Z., Gallant, P. E., Tao-Cheng, J. H., Rudow, G., Troncoso, J. C., Liu, Z., Li, Z., & Cai, H. (2009). Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant alpha-synuclein. Neuron, 64, 807–827.
Lovinger, D. M., Tyler, E. C., & Merritt, A. (1993). Short- and long-term synaptic depression in rat neostriatum. Journal of Neurophysiology, 70, 1937–1949.
Mahon, S., Deniau, J. M., & Charpier, S. (2004). Corticostriatal plasticity: Life after the depression. Trends in Neurosciences, 27, 460–467.
Malenka, R. C., & Bear, M. F. (2004). LTP and LTD: An embarrassment of riches. Neuron, 44, 5–21.
Manning, G., Whyte, D. B., Martinez, R., Hunter, T., & Sudarsanam, S. (2002). The protein kinase complement of the human genome. Science, 298, 1912–1934.
Marin, I. (2006). The Parkinson disease gene LRRK2: Evolutionary and structural insights. Molecular Biology and Evolution, 23, 2423–2433.
Marx, F. P., Holzmann, C., Strauss, K. M., Li, L., Eberhardt, O., Gerhardt, E., Cookson, M. R., Hernandez, D., Farrer, M. J., Kachergus, J., Engelender, S., Ross, C. A., Berger, K., Schols, L., Schulz, J. B., Riess, O., & Kruger, R. (2003). Identification and functional characterization of a novel R621C mutation in the synphilin-1 gene in Parkinson’s disease. Human Molecular Genetics, 12, 1223–1231.
Mata, I. F., Kachergus, J. M., Taylor, J. P., Lincoln, S., Aasly, J., Lynch, T., Hulihan, M. M., Cobb, S. A., Wu, R. M., Lu, C. S., Lahoz, C., Wszolek, Z. K., & Farrer, M. J. (2005). Lrrk2 pathogenic substitutions in Parkinson’s disease. Neurogenetics, 6, 171–177.
Meixner, A., Boldt, K., Van Troys, M., Askenazi, M., Gloeckner, C. J., Bauer, M., Marto, J. A., Ampe, C., Kinkl, N., & Ueffing, M. (2010). A QUICK screen for Lrrk2 interaction partners–leucine-rich repeat kinase 2 is involved in actin cytoskeleton dynamics. Molecular and Cellular Proteomics, 10, M110 001172.
Melrose, H., Lincoln, S., Tyndall, G., Dickson, D., & Farrer, M. (2006). Anatomical localization of leucine-rich repeat kinase 2 in mouse brain. Neuroscience, 139, 791–794.
Moore, D. J. (2008). The biology and pathobiology of LRRK2: Implications for Parkinson’s disease. Parkinsonism & Related Disorders, 14(Suppl 2), S92–98.
Moore, D. J., Dawson, V. L., & Dawson, T. M. (2006). Lessons from Drosophila models of DJ-1 deficiency. Science of Aging Knowledge Environment, 2006, pe2.
Murphy, D. D., Rueter, S. M., Trojanowski, J. Q., & Lee, V. M. (2000). Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. The Journal of Neuroscience, 20, 3214–3220.
Nash, J. E., Johnston, T. H., Collingridge, G. L., Garner, C. C., & Brotchie, J. M. (2005). Subcellular redistribution of the synapse-associated proteins PSD-95 and SAP97 in animal models of Parkinson’s disease and L-DOPA-induced dyskinesia. The FASEB Journal, 19, 583–585.
Nemani, V. M., Lu, W., Berge, V., Nakamura, K., Onoa, B., Lee, M. K., Chaudhry, F. A., Nicoll, R. A., & Edwards, R. H. (2010). Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron, 65, 66–79.
Nishi, M., Hinds, H., Lu, H. P., Kawata, M., & Hayashi, Y. (2001). Motoneuron-specific expression of NR3B, a novel NMDA-type glutamate receptor subunit that works in a dominant-negative manner. The Journal of Neuroscience, 21, RC185.
Nussbaum, R. L., & Polymeropoulos, M. H. (1997). Genetics of Parkinson’s disease. Human Molecular Genetics, 6, 1687–1691.
O’Dell, T. J., & Kandel, E. R. (1994). Low-frequency stimulation erases LTP through an NMDA receptor-mediated activation of protein phosphatases. Learning & Memory, 1, 129–139.
Oh, J. D., Vaughan, C. L., & Chase, T. N. (1999). Effect of dopamine denervation and dopamine agonist administration on serine phosphorylation of striatal NMDA receptor subunits. Brain Research, 821, 433–442.
Paille, V., Picconi, B., Bagetta, V., Ghiglieri, V., Sgobio, C., Di Filippo, M., Viscomi, M. T., Giampa, C., Fusco, F. R., Gardoni, F., Bernardi, G., Greengard, P., Di Luca, M., & Calabresi, P. (2010). Distinct levels of dopamine denervation differentially alter striatal synaptic plasticity and NMDA receptor subunit composition. The Journal of Neuroscience, 30, 14182–14193.
Paisan-Ruiz, C., Nath, P., Washecka, N., Gibbs, J. R., & Singleton, A. B. (2008). Comprehensive analysis of LRRK2 in publicly available Parkinson’s disease cases and neurologically normal controls. Human Mutation, 29, 485–490.
Paisan-Ruiz, C., Jain, S., Evans, E. W., Gilks, W. P., Simon, J., van der Brug, M., Lopez de Munain, A., Aparicio, S., Gil, A. M., Khan, N., Johnson, J., Martinez, J. R., Nicholl, D., Carrera, I. M., Pena, A. S., de Silva, R., Lees, A., Marti-Masso, J. F., Perez-Tur, J., Wood, N. W., & Singleton, A. B. (2004). Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron, 44, 595–600.
Partridge, J. G., Tang, K. C., & Lovinger, D. M. (2000). Regional and postnatal heterogeneity of activity-dependent long-term changes in synaptic efficacy in the dorsal striatum. Journal of Neurophysiology, 84, 1422–1429.
Piccoli, G., Condliffe, S. B., Bauer, M., Giesert, F., Boldt, K., De Astis, S., Meixner, A., Sarioglu, H., Vogt-Weisenhorn, D. M., Wurst, W., Gloeckner, C. J., Matteoli, M., Sala, C., & Ueffing, M. (2011). LRRK2 controls synaptic vesicle storage and mobilization within the recycling pool. The Journal of Neuroscience, 31, 2225–2237.
Picconi, B., Centonze, D., Hakansson, K., Bernardi, G., Greengard, P., Fisone, G., Cenci, M. A., & Calabresi, P. (2003). Loss of bidirectional striatal synaptic plasticity in L-DOPA-induced dyskinesia. Nature Neuroscience, 6, 501–506.
Picconi, B., Gardoni, F., Centonze, D., Mauceri, D., Cenci, M. A., Bernardi, G., Calabresi, P., & Di Luca, M. (2004). Abnormal Ca2 + −calmodulin-dependent protein kinase II function mediates synaptic and motor deficits in experimental parkinsonism. The Journal of Neuroscience, 24, 5283–5291.
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., Pike, B., Root, H., Rubenstein, J., Boyer, R., Stenroos, E. S., Chandrasekharappa, S., Athanassiadou, A., Papapetropoulos, T., Johnson, W. G., Lazzarini, A. M., Duvoisin, R. C., Di Iorio, G., Golbe, L. I., & Nussbaum, R. L. (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science, 276, 2045–2047.
Qing, H., Wong, W., McGeer, E. G., & McGeer, P. L. (2009). Lrrk2 phosphorylates alpha synuclein at serine 129: Parkinson disease implications. Biochemical and Biophysical Research Communications, 387, 149–152.
Quik, M., Chen, L., Parameswaran, N., Xie, X., Langston, J. W., & McCallum, S. E. (2006). Chronic oral nicotine normalizes dopaminergic function and synaptic plasticity in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned primates. The Journal of Neuroscience, 26, 4681–4689.
Raju, D. V., Ahern, T. H., Shah, D. J., Wright, T. M., Standaert, D. G., Hall, R. A., & Smith, Y. (2008). Differential synaptic plasticity of the corticostriatal and thalamostriatal systems in an MPTP-treated monkey model of parkinsonism. European Journal of Neuroscience, 27, 1647–1658.
Reynolds, J. N., & Wickens, J. R. (2000). Substantia nigra dopamine regulates synaptic plasticity and membrane potential fluctuations in the rat neostriatum, in vivo. Neuroscience, 99, 199–203.
Rizo, J., & Rosenmund, C. (2008). Synaptic vesicle fusion. Nature Structural and Molecular Biology, 15, 665–674.
Schulz-Schaeffer, W. J. (2010). The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathologica, 120, 131–143.
Schwarting, R. K., & Huston, J. P. (1996). The unilateral 6-hydroxydopamine lesion model in behavioral brain research. Analysis of functional deficits, recovery and treatments. Progress in Neurobiology, 50, 275–331.
Selkoe, D. J. (2002). Alzheimer’s disease is a synaptic failure. Science, 298, 789–791.
Shendelman, S., Jonason, A., Martinat, C., Leete, T., & Abeliovich, A. (2004). DJ-1 is a redox-dependent molecular chaperone that inhibits alpha-synuclein aggregate formation. PLoS Biology, 2, e362.
Shin, N., Jeong, H., Kwon, J., Heo, H. Y., Kwon, J. J., Yun, H. J., Kim, C. H., Han, B. S., Tong, Y., Shen, J., Hatano, T., Hattori, N., Kim, K. S., Chang, S., & Seol, W. (2008). LRRK2 regulates synaptic vesicle endocytosis. Experimental Cell Research, 314, 2055–2065.
Sidhu, A., Wersinger, C., & Vernier, P. (2004). Does alpha-synuclein modulate dopaminergic synaptic content and tone at the synapse? The FASEB Journal, 18, 637–647.
Silvestri, L., Caputo, V., Bellacchio, E., Atorino, L., Dallapiccola, B., Valente, E. M., & Casari, G. (2005). Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Human Molecular Genetics, 14, 3477–3492.
Simon-Sanchez, J., Schulte, C., Bras, J. M., Sharma, M., Gibbs, J. R., Berg, D., Paisan-Ruiz, C., Lichtner, P., Scholz, S. W., Hernandez, D. G., Kruger, R., Federoff, M., Klein, C., Goate, A., Perlmutter, J., Bonin, M., Nalls, M. A., Illig, T., Gieger, C., Houlden, H., Steffens, M., Okun, M. S., Racette, B. A., Cookson, M. R., Foote, K. D., Fernandez, H. H., Traynor, B. J., Schreiber, S., Arepalli, S., Zonozi, R., Gwinn, K., van der Brug, M., Lopez, G., Chanock, S. J., Schatzkin, A., Park, Y., Hollenbeck, A., Gao, J., Huang, X., Wood, N. W., Lorenz, D., Deuschl, G., Chen, H., Riess, O., Hardy, J. A., Singleton, A. B., & Gasser, T. (2009). Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nature Genetics, 41, 1308–1312.
Singleton, A. B., Farrer, M., Johnson, J., Singleton, A., Hague, S., Kachergus, J., Hulihan, M., Peuralinna, T., Dutra, A., Nussbaum, R., Lincoln, S., Crawley, A., Hanson, M., Maraganore, D., Adler, C., Cookson, M. R., Muenter, M., Baptista, M., Miller, D., Blancato, J., Hardy, J., & Gwinn-Hardy, K. (2003). Alpha-Synuclein locus triplication causes Parkinson’s disease. Science, 302, 841.
Smith, A. D., Castro, S. L., & Zigmond, M. J. (2002). Stress-induced Parkinson’s disease: A working hypothesis. Physiology and Behavior, 77, 527–531.
Sossi, V., de la Fuente-Fernandez, R., Nandhagopal, R., Schulzer, M., McKenzie, J., Ruth, T. J., Aasly, J. O., Farrer, M. J., Wszolek, Z. K., & Stoessl, J. A. (2010). Dopamine turnover increases in asymptomatic LRRK2 mutations carriers. Movement Disorders, 25, 2717–2723.
Spillantini, M. G., Crowther, R. A., Jakes, R., Hasegawa, M., & Goedert, M. (1998). Alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proceedings of the National Academy of Sciences of the United States of America, 95, 6469–6473.
Strack, S., McNeill, R. B., & Colbran, R. J. (2000). Mechanism and regulation of calcium/calmodulin-dependent protein kinase II targeting to the NR2B subunit of the N-methyl-D-aspartate receptor. The Journal of Biological Chemistry, 275, 23798–23806.
Sudhof, T. C., & Rothman, J. E. (2009). Membrane fusion: Grappling with SNARE and SM proteins. Science, 323, 474–477.
Taylor, J. P., Mata, I. F., & Farrer, M. J. (2006). LRRK2: A common pathway for parkinsonism, pathogenesis and prevention? Trends in Molecular Medicine, 12, 76–82.
Taymans, J. M., & Cookson, M. R. (2010). Mechanisms in dominant parkinsonism: The toxic triangle of LRRK2, alpha-synuclein, and tau. Bioessays, 32, 227–235.
Tong, Y., Pisani, A., Martella, G., Karouani, M., Yamaguchi, H., Pothos, E. N., & Shen, J. (2009). R1441C mutation in LRRK2 impairs dopaminergic neurotransmission in mice. Proceedings of the National Academy of Sciences of the United States of America, 106, 14622–14627.
Turner, K. M., Burgoyne, R. D., & Morgan, A. (1999). Protein phosphorylation and the regulation of synaptic membrane traffic. Trends in Neurosciences, 22, 459–464.
Ulas, J., & Cotman, C. W. (1996). Dopaminergic denervation of striatum results in elevated expression of NR2A subunit. Neuroreport, 7, 1789–1793.
Valente, E. M., Abou-Sleiman, P. M., Caputo, V., Muqit, M. M., Harvey, K., Gispert, S., Ali, Z., Del Turco, D., Bentivoglio, A. R., Healy, D. G., Albanese, A., Nussbaum, R., Gonzalez-Maldonado, R., Deller, T., Salvi, S., Cortelli, P., Gilks, W. P., Latchman, D. S., Harvey, R. J., Dallapiccola, B., Auburger, G., & Wood, N. W. (2004). Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science, 304, 1158–1160.
Walsh, J. P. (1993). Depression of excitatory synaptic input in rat striatal neurons. Brain Research, 608, 123–128.
Walsh, J. P., & Dunia, R. (1993). Synaptic activation of N-methyl-D-aspartate receptors induces short-term potentiation at excitatory synapses in the striatum of the rat. Neuroscience, 57, 241–248.
Wang, Y., Chandran, J. S., Cai, H., & Mattson, M. P. (2008). DJ-1 is essential for long-term depression at hippocampal CA1 synapses. Neuromolecular Medicine, 10, 40–45.
Whaley, N. R., Uitti, R. J., Dickson, D. W., Farrer, M. J., & Wszolek, Z. K. (2006). Clinical and pathologic features of families with LRRK2-associated Parkinson’s disease. Journal of Neural Transmission. Supplementum, 70, 221–229.
Wider, C., Dickson, D. W., & Wszolek, Z. K. (2010). Leucine-rich repeat kinase 2 gene-associated disease: redefining genotype-phenotype correlation. Neurodegenerative Diseases, 7, 175–179.
Wilson, M. A., St Amour, C. V., Collins, J. L., Ringe, D., & Petsko, G. A. (2004). The 1.8-A resolution crystal structure of YDR533Cp from Saccharomyces cerevisiae: a member of the DJ-1/ThiJ/PfpI superfamily. Proceedings of the National Academy of Sciences of the United States of America, 101, 1531–1536.
Wishart TM, Parson SH, Gillingwater TH. (2006). Synaptic vulnerability in neurodegenerative disease. Journal of Neuropathology and Experimental Neurology, 65, 733–739.
Xiong, H., Wang, D., Chen, L., Choo, Y. S., Ma, H., Tang, C., Xia, K., Jiang, W., Ronai, Z., Zhuang, X., & Zhang, Z. (2009). Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. The Journal of Clinical Investigation, 119, 650–660.
Xiong, Y., Coombes, C. E., Kilaru, A., Li, X., Gitler, A. D., Bowers, W. J., Dawson, V. L., Dawson, T. M., & Moore, D. J. (2010). GTPase activity plays a key role in the pathobiology of LRRK2. PLoS Genetics, 6, e1000902.
Yao, I., Takagi, H., Ageta, H., Kahyo, T., Sato, S., Hatanaka, K., Fukuda, Y., Chiba, T., Morone, N., Yuasa, S., Inokuchi, K., Ohtsuka, T., Macgregor, G. R., Tanaka, K., & Setou, M. (2007). SCRAPPER-dependent ubiquitination of active zone protein RIM1 regulates synaptic vesicle release. Cell, 130, 943–957.
Yu, S., Ueda, K., & Chan, P. (2005). Alpha-synuclein and dopamine metabolism. Molecular Neurobiology, 31, 243–254.
Yu, S., Li, X., Liu, G., Han, J., Zhang, C., Li, Y., Xu, S., Liu, C., Gao, Y., Yang, H., Ueda, K., & Chan, P. (2007). Extensive nuclear localization of alpha-synuclein in normal rat brain neurons revealed by a novel monoclonal antibody. Neuroscience, 145, 539–555.
Zhang, L., Shimoji, M., Thomas, B., Moore, D. J., Yu, S. W., Marupudi, N. I., Torp, R., Torgner, I. A., Ottersen, O. P., Dawson, T. M., & Dawson, V. L. (2005). Mitochondrial localization of the Parkinson’s disease related protein DJ-1: Implications for pathogenesis. Human Molecular Genetics, 14, 2063–2073.
Zhou, C., Huang, Y., Shao, Y., May, J., Prou, D., Perier, C., Dauer, W., Schon, E. A., & Przedborski, S. (2008). The kinase domain of mitochondrial PINK1 faces the cytoplasm. Proceedings of the National Academy of Sciences of the United States of America, 105, 12022–12027.
Zigmond, M. J., Hastings, T. G., & Perez, R. G. (2002). Increased dopamine turnover after partial loss of dopaminergic neurons: Compensation or toxicity? Parkinsonism & Related Disorders, 8, 389–393.
Zimprich, A., Biskup, S., Leitner, P., Lichtner, P., Farrer, M., Lincoln, S., Kachergus, J., Hulihan, M., Uitti, R. J., Calne, D. B., Stoessl, A. J., Pfeiffer, R. F., Patenge, N., Carbajal, I. C., Vieregge, P., Asmus, F., Muller-Myhsok, B., Dickson, D. W., Meitinger, T., Strom, T. M., Wszolek, Z. K., & Gasser, T. (2004). Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron, 44, 601–607.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer-Verlag/WIen
About this chapter
Cite this chapter
Picconi, B., Piccoli, G., Calabresi, P. (2012). Synaptic Dysfunction in Parkinson’s Disease. In: Kreutz, M., Sala, C. (eds) Synaptic Plasticity. Advances in Experimental Medicine and Biology, vol 970. Springer, Vienna. https://doi.org/10.1007/978-3-7091-0932-8_24
Download citation
DOI: https://doi.org/10.1007/978-3-7091-0932-8_24
Published:
Publisher Name: Springer, Vienna
Print ISBN: 978-3-7091-0931-1
Online ISBN: 978-3-7091-0932-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)