
Abstract
A penetration enhancer is by definition anything that is used to improve the delivery of a chemical substance across some biological/biochemical barrier. For the present purposes, we are interested in chemical-, as opposed to, physical-, electrical-, or mechanical-based approaches to improving the delivery of a drug into or through the skin barrier by improving dermal or transdermal delivery, respectively, or collectively by improving the topical delivery of a drug. Enhanced topical delivery using chemical approaches can be achieved by making transient derivatives of drugs, prodrugs, that exhibit optimized (a balance of) lipid and aqueous solubilities that increase their solubility in the first few layers of the skin. Increased lipid and aqueous solubilities of drugs containing polar functional groups can usually be accomplished by making a prodrug to mask the polar functional group. Increased water solubility of more lipid soluble drugs can be accomplished by using promoieties that contain water solubilizing functional groups. These same chemical-based approaches could also be used to improve the delivery of cosmeceuticals, but there the task would be specifically only for improving their dermal delivery.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Roberts-Sloan equation
- Potts-Guy equation
- Water solubility
- Lipid solubility
- Biphasic solubility
- Diol solubility
- Amine solubility
- Oxyethylene solubility
- Soft alkyl
- Acyl
1 Introduction
1.1 Push Versus Pull Mechanisms for Penetration Enhancers
From a mechanistic point of view, there are two general ways to accomplish the task of improving topical delivery using a chemical-based approach. The first approach is to increase the “push” of the vehicle components on the drug to drive it into the skin (Kadir et al. 1987). One way to increase the “push” of the vehicle is to use vehicle components in which the drug is more soluble but which are more volatile than the other components. Evaporation of the volatile components after application of the drug-vehicle combination leaves a supersaturated solution of the drug in a state of heightened thermodynamic activity in the vehicle (α VEH) (Coldman et al. 1969), that is, α VEH greater than one. The second approach is to increase the “pull” on the drug into the skin by components of the vehicle that have permeated the skin and have decreased the resistance of the skin to permeation by the drug (Kadir et al. 1987) or increased the solubility of the drug in the skin, S M1: these components interact with the skin. Such components of the vehicle do not have to permeate the skin faster than the drug. However, another way to increase the “pull” on the drug by components of the vehicle is to use components that do permeate the skin faster than the drug and pull the drug along with them – a “drag” effect (Friend and Smedley 1993).
The basis for the two chemical-based approaches to enhancing topical delivery (decreasing the solubility of the drug in the vehicle and increasing its solubility in the skin; “push” and “pull,” respectively) lies in the form of the equation that describes flux. The flux, J, of the drug through skin is directly related to the concentration of the drug in the first layer of the skin, C M1, from Fick’s Law: J = (C M1 − C Mn) D/L where C Mn is the concentration of the drug in the last layer of skin (and is assumed to approach zero at steady-state), D is the diffusion coefficient of the drug in the skin, and L is the thickness of the membrane. The concentration of the drug in the skin, C M1, is generated from its equilibrium with the concentration of the drug in the vehicle, C VEH, through the product of its partition coefficient between the two phases K M1:VEH and C VEH. The concentration of the drug in the skin approaches its saturated solubility in the skin, S M1, and a thermodynamic activity (α M1) of one when C VEH approaches the saturated solubility of the drug in a noninteractive vehicle, S VEH; that is, α VEH also is one. The flux J is now the maximum possible flux from noninteractive vehicles, J M, and Fick’s law can be written as Eq. 10.1. Regardless of the value for S VEH, the highest concentration of drug in the skin that is possible from a drug applied in a noninteractive vehicle is S M1. As S VEH increases K M1: VEH tends to decrease and as S VEH decreases K M1: VEH tends to increase. S M1 can only be increased by using an interactive component in the vehicle that changes the solubilizing capacity of the skin, the “pull,” or by increasing the thermodynamic activity of the drug in the vehicle, α VEH, so that it is greater than one, the “push,” and hence the activity of the drug in the skin, α M1, is also greater than one.
2 Basis for Prodrugs as Penetration Enhancers
Although increasing the “push” can be easily accomplished by manipulating the components of the vehicle in which the drug is applied (its formulation), increasing the “pull” can be more easily accomplished using a prodrug approach that changes the solubility properties of the drug. A prodrug is a chemically or enzymatically reversible derivative of a parent drug that improves the physicochemical or biological properties of the parent drug molecule to overcome some intrinsic problem associated with its therapeutic use: in this case, poor solubility in the skin and hence low topical delivery (Sloan 1992). The particular combination of functional groups that is added to the parent drug is called the promoiety, and the reversible connection between the promoiety and the parent drug is called the enabling functional group. A prodrug approach, then, can be envisaged as a 1:1 molecular combination of the drug and a promoiety that contains functional groups that will increase its solubility in the skin (Sloan and Wasdo 2003). This prodrug approach stands in sharp contrast to most formulation approaches where large molar excesses of penetration enhancers as vehicle components are routinely needed to increase S M1 for the drug.
What are the properties of the functional groups in the promoiety which, when added to the parent drug, could be reasonably expected to cause an increase in S M1 of the resulting prodrug compared to the parent drug and hence to cause an increase in its maximum flux, J M? Since it is difficult to measure S M1 of the prodrug, it is more convenient to measure its J M in diffusion cell experiments and assume, based on Fick’s law Eq. 10.1, that there is a direct relationship between increased J M and increased S M1. Using increases in J M as the criterion for increased S M1, it has been observed for quite some time that for homologous series of more lipophilic prodrugs that the more water soluble members of the series gave the greatest increase in J M and not the more lipid soluble members (Sloan 1989, 1992; Sloan et al. 1984). In order to account for these qualitative observations, S M1 in Fick’s law Eq. 10.1 was expanded mathematically to include dependence on solubility in a lipid, S LIPID, and in water, S AQ. This form of Fick’s law is the Roberts-Sloan (RS) Eq. 10.2 (Roberts and Sloan 1999): a transformation of the popular, but very specific, Potts-Guy (PG) Eq. 10.3 (Potts and Guy 1992) into more general, useful terms.
When a database of those homologous series of more lipid soluble prodrugs (n = 42) comprised of their molecular weights, MW, their solubilities in isopropyl myristate (IPM), S IPM (S IPM = S LIPID in Eq. 10.2), and in water, S AQ, and their maximum fluxes from IPM through hairless mouse skin, J MMIPM, were collected and fitted to Eq. 10.2, the values for the coefficients were x = −0.211, y = 0.534, z = 0.00364, and r 2 = 0.937 (Roberts and Sloan 1999). The size of the J MMIPM database has since been increased to n = 94, and the values for the coefficients are now x = −0.377, y = 0.527, z = 0.00346, and r 2 = 0.900 (Majumdar et al. 2012). The maximum fluxes of prodrugs and non-prodrug through human skin in vitro and in vivo, respectively, from mineral oil (MO), J MHMO, their solubilities in mineral oil, S MO (S MO = S LIPID in Eq. 10.2), and in water, S AQ, and their MW also gave good fit to Eq. 10.2: x = −1.83, y = 0.462, z = 0.00153, and r 2 = 0.80 for n = 30 prodrugs (Sloan et al. 2011); x = −1.459, y = 0.72, z = 0.00013, and r 2 = 0.934 for n = 10 nonsteriodal anti-inflammatory drugs (Wenkers and Lippold 1999; Roberts and Sloan 2001). Thus, good fits to Eq. 10.2 are obtained if the vehicle is a lipid (IPM or MO) and the lipid solubility of the permeant, S LIPID, and S AQ are independent valuables.
A similar strong dependence of maximum flux through hairless mouse from water, J MMAQ, on S IPM (S IPM = S LIPID in Eq. 10.2) and S AQ for some of the members of the n = 94 J MMIPM database was observed where x = −2.30, y = 0.575, z = 0.0016, and r 2 = 0.903 for n = 32 (Sloan et al. 2003; Wasdo et al. 2009). Also a strong dependence of maximum flux through human skin in vitro from water, J MHAQ, on the solubilities of the permeants in octanol, S OCT (S OCT = S LIPID in Eq. 10.2) and S AQ, was observed where x = −2.506, y = 0.538, z = 0.00402, and r 2 = 0.839 for n = 185 (Juntunen et al. 2008). Even maximum flux through silicone membranes from water, J MPAQ, for some of the members of the n = 94 J MMIPM database was found to be dependent on S IPM (S IPM = S LIPID in Eq. 10.2) and S AQ where x = −1.837, y = 0.742, z = 0.00435, and r 2 = 0.86 for n = 38 (Synovec et al. 2013). Thus, good fits to Eq. 10.2 are obtained regardless of whether the membrane is mouse, human, or silicone and regardless of whether the vehicle is a lipid or aqueous. Since the solubilities of the permeant in a lipid and in water are both necessary to define maximum flux, functional groups should be incorporated into the promoieties of prodrugs that can ideally increase both lipid and aqueous solubilities to increase maximum flux and by inference S M1.
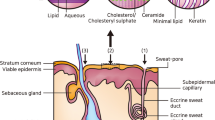
The reason that increasing both lipid and aqueous solubilities of the drug is important to increasing its solubility in skin, and hence its topical delivery, can be found in the structure of the barrier to topical delivery – the intercellular compartment of the stratum corneum (SC). The intercellular compartment consists of lamellar double bilayers comprised of lipid components such as ceramides, cholesterol, and fatty acids which have polar groups attached to them. These polar head groups have water associated with them so that for a permeant to cross these bilayers perpendicular to the axis of the bilayers, it must alternately cross lipid and aqueous phases (Sloan and Wasdo 2003; Sloan et al. 1984, 2011a, b). Thus, a balance of solubility in both lipid and aqueous phases by the drug (or increased lipid and aqueous solubility by its prodrug) is necessary for its most efficient permeation of the intercellular compartment of the SC. The agreement between the experimentally measurable physicochemical parameters in the theoretically derived Roberts-Sloan equation and in the biochemically based biphasic solubility model (Sloan et al. 2011a, b) for the barrier to permeation is encouraging.
3 Acyl Versus Soft Alkyl Promoieties
The promoieties that have been used to increase lipid and aqueous solubilities can be divided into two types based on whether they are attached directly to the functional group in the parent drug that is to be modified or indirectly through a methylene or vinylogous methylene (aryl methylene) spacer (Sloan 1989, 1992; Sloan and Wasdo 2003). In each type, the enabling functional group is usually a carbonyl-type functional group because of its sensitivity to cleavage by chemical or enzymatic hydrolysis. Generally these types have been referred as acyl and soft alkyl-type promoieties, respectively. Cleavage of the acyl-type promoiety regenerates the parent drug directly while cleavage of the acyl group in the soft alkyl promoiety generates an intermediate drug–X–CHR–X′H from drug–X–CHR–X′–(C = X″)–X′′′R′: X, X′, X″, and X′′′ can be O, N, or S and R and R′ can be alkyl or aryl. The intermediate is designed to be intrinsically unstable and undergo rapid and complete chemical hydrolysis to the parent drug–X–H. The advantage of the soft alkyl prodrug approach is that the stability of the prodrug (as well as its attendant physicochemical properties) is not limited by the functional group in the parent drug to which it is attached. Generally, changing X will change the biochemical and/or pharmacological activity of the drug, but changing X′ to obtain a more or less stable or more or less soluble prodrug will not. Of course X″ and X′′′ can be changed in the same ways that they could have been if an acyl prodrug approach had been used.
4 Mechanisms for Penetration Enhancement
4.1 Decrease Crystal Lattice Energy by Masking Hydrogen Bond Donor Functional Groups
Regardless of whether the prodrug is derived from an acyl or soft alkyl-type promoiety, there are two general mechanisms by which both types of promoieties can increase both lipid and aqueous solubilities. The first mechanism has its basis in decreasing the crystal lattice energy of the parent drug by modifying polar groups capable of forming intermolecular hydrogen bonds. In many if not most drug molecules, the X in drug–X–H is a heteroatom which causes X–H to be polarized because of the difference in electronegativities between X and H. This polarized drug–X–H bond is capable of forming intermolecular hydrogen bonds within the crystal lattice which leads to low solubilities especially in lipids but also frequently in water. The polarization is further attenuated if an electron withdrawing carbonyl-type functional group is attached to X–H to give drug–(O = C)–X–H. Examples of this type of drug molecule, which can be measurably but not highly ionized at physiological pH, include heterocycles such as 5-flurouracil (5-FU) (drug–(O = C)–NH) and 6-mercaptopurine (6-MP) (drug–(S = C)–NH) which are very high melting and exhibit low solubilities in both water and lipids. In other examples such as parent drugs containing a carboxylic acid functional group (drug–(O = C) –OH), the functional group is so highly polarized that it becomes highly ionized at physiological pH which does not allow it to readily cross the lipid phase of the alternating lipid-aqueous phases of the biological barrier. An important class of drugs that belong to this category is the nonsteroidal anti-inflammatory drugs. Another example of this class are the nucleotide-based drugs where the highly ionized functional group is a phosphate group. Simply masking the hydrogen bond donating abilities of the functional group by replacing the H in the drug–X–H with either an acyl or soft alkyl group decreases the melting point (mp) and increases the lipid solubility (S LIPID) as well as frequently increasing the aqueous solubility (S AQ) of the prodrug compared to the parent drug, especially for the shorter alkyl chain members of a homologous series (Sloan 1989).
Examples of the results that can be obtained by masking the polar functional groups in drugs to increase S LIPID (S IPM) and S AQ and to increase topical delivery of the parent drug are several prodrugs of 5-FU.
The mp, S AQ, S IPM, log partition coefficients between IPM and pH 4.0 buffer (log K IPM:AQ), and rates of delivery of total 5-FU containing species through hairless mouse skin from an IPM vehicle in vitro (J MMIPM) for four different series of prodrug of 5-FU are given in Table 10.1: three acyl types and a one soft alkyl type. The first acyl type of prodrug of 5-FU that was evaluated for its ability to increase the delivery of 5-FU was the alkylaminocarbonyl-5-FU (1-AAC-5-FU) prodrugs (Table 10.1 and Fig. 10.1). Initially only the longer alkyl chain members of the series were evaluated (4–6) (Sasaki et al. 1990), but subsequently the shorter alkyl chain members (1–3) were evaluated, and one of them, 3, was found to give the greatest increase in the delivery of the total 5-FU containing species, J MMIPM (Sloan et al. 1993). All of the 1-AAC-5-FU prodrugs exhibited lower mp than 5-FU and all of them were more soluble in IPM than 5-FU: from 6 times for 1 to almost 1,000 times for 6. However, the most lipid soluble member evaluated, 6, gave only 0.25 times the flux of 5-FU. None of the 1-AAC-5-FU prodrugs was even as soluble in water as 5-FU, and the C3 member (3), not the shortest alkyl chain member of the series (1), gave the highest S AQ value: only 0.11 times S AQ for 5-FU. The C3 member also gave the greatest increase in J MMIPM values for the series, albeit only three times. Thus, as predicted (Sloan 1992, 1989; Sloan and Wasdo 2003; Sloan et al. 1984), for a more lipid soluble homologous series of prodrugs, the more water soluble member gave the highest J MMIPM value. The low increase in J MMIPM can be attributed to the low S AQ values exhibited by the 1-ACC-5-FU prodrugs compared to subsequent series, and the low S AQ values can be attributed to the fact that one of the hydrogen bond donor functional groups, (O = C)–NH, in 5-FU was merely replaced with another hydrogen bond donor group, N–(O = C) –NH, in the promoiety. The potential for forming intermolecular hydrogen bonds was not decreased significantly and the added alkyl group in the promoiety further depressed S AQ

Chemical structures of prodrugs for compounds 1-54
.
The second acyl type of prodrug of 5-FU that was evaluated was the alkyloxycarbonyl-5-FU (1-AOC-5-FU) prodrugs (Table 10.1, Fig. 10.1) (Beall et al. 1994). In this series the hydrogen bond donating group in the parent drug has not been replaced with another hydrogen bond donating group in the promoiety so the mp are somewhat lower than the corresponding members in the 1-AAC-5-FU series except for the C8 member of the series. Consequently, the members of the 1-AOC-5-FU series were also somewhat more soluble in IPM than the members of the 1-AAC-5-FU series except for the C8 member, 12; and the worst member of the series in terms of increased S IPM was 43 times instead of 6 times more soluble in IPM than 5-FU. However, the big difference between the two series was in the S AQ values. Not only were two members of the series more water soluble than 5-FU, 7 and 8 (1.3 and 2 times, respectively), but they were all more water soluble than the corresponding members of the 1-AAC5-FU series (from 30 to 4.3 times). Thus, since the 1-AOC-5-FU series was more soluble in lipids and in water, as predicted (Sloan 1989, 1992; Sloan et al. 1984), they delivered more total 5-FU species through hairless mouse skin than the 1-AAC-5-FU series (from 3 to 12.5 times). Also, as predicted (Sloan 1989, 1992; Sloan et al. 1984) the C2 member, 8, which was the most water soluble member of the series gave the greatest increase in J MMIPM compared to 5-FU (24.7 times), and not the most lipid soluble member of the series, 11. The next most water soluble member, 7, gave the next greatest increase in J MMIPM compared to 5-FU (11 times).
Based on previous literature, the 1-AOC series was expected to be more stable than the 1-AAC series of prodrugs of 5-FU. Whereas the amount of intact prodrug delivered by the 1-AAC series was in the 6–10 % range, the amount delivered by the 1-AOC series was in the 40–70 % range and was up to 90 % for the best performing member of the series, 8. If delivery through the skin and subsequent slower release of 5-FU systemically was the target of topical delivery, then the members of the 1-AOC-5-FU series performed well. On the other hand, if delivery into the skin was the target, then a more rapidly hydrolyzing type of prodrug of 5-FU would be required.
The third acyl type of prodrug 5-FU that was evaluated was the alkylcarbonyl-5-FU (1-AC-5-FU) prodrugs (Table 10.1, Fig. 10.1) (Beall et al. 1996). The members of this series were known to hydrolyze quite rapidly (t 1/2 = 3–5 min), so it was expected that only 5-FU would be delivered through the skin. This expectation was realized, and only 5-FU and no intact prodrug was observed in the receptor phase after application of 1-AC-5-FU prodrugs in IPM in diffusion cell experiments. All of the members of the 1-AC series were much more soluble in IPM than 5-FU (355–2,300 times), and one member, C1 (13), was more soluble in water than 5-FU (1.4 times). However, direct comparisons between the 1-AC series and either the 1-AOC or the 1-AAC series based only on the alkyl chain length in the promoiety would be misleading without taking into account the added heteroatom in the latter two series. For example, we will compare the OC1 member (7) of the 1-AOC series with the C2 member of the 1-AC series (14), the OC2 with the C3, the OC3 with the C4, the OC4 with the C5, and the OC6 with the C7. Using these interseries comparisons, the members of the 1-AC series were more soluble in IPM (1.3–17 times) than those of the 1-AOC series, except for 18 compared to 11. On the other hand, the members of the 1-AOC series were more soluble in water (2.4–33 times) than those of the 1-AC series, and as predicted (Sloan 1989, 1992; Sloan et al. 1984) they all gave higher J MMIPM values than the corresponding members of the 1-AC series, except for OC1, 7, versus C2, 14. Prodrug 7 was only 2.4 times more soluble in water than 14, while 14 was 17 times more soluble in IPM than 7. Prodrug 14 exhibited a somewhat better balance of S AQ and S IPM than 7 and gave a higher J MMIPM value (1.6 times). However, within the 1-AC series the C1 member, 13, which was the more water soluble member of the series and not one of the more lipid soluble members, gave the greatest enhancement in J MMIPM (39 times that of 5-FU).
In the 1-AC series the effect of the mp on solubilities and ultimately on flux can be readily illustrated. The C3 member of the series, 15, exhibited a higher mp than either the shorter, 14, or longer alkyl chain member, 16, and hence exhibited a lower S IPM value than those members. The S AQ value for 15 also dropped off more rapidly than expected as did its J MMIPM value. On the other hand, the log K values appeared normally spaced and the methylene π values derived from the log K values only varied by 10 %: π = 0.59 ± 0.05. Thus, log K values are no substitute for experimental solubilities for purposes of predicting trends in J M.
The example of the use of a soft alkyl prodrug in the designs of prodrugs to increase S IPM and S AQ and to increase the topical delivery of the parent drug is also a 5-FU prodrug: the 1-alkylcarbonyloxymethyl-5-FU (1-ACOM-5-FU) prodrugs (Table 10.1, Fig. 10.1) (Taylor and Sloan 1998). As expected each of the 1-ACOM-5-FU prodrugs exhibited a lower mp than 5-FU since a hydrogen bond donor group had been masked in the prodrug. Also as expected each was much more soluble in IPM than 5-FU (67–302 times), and there were members, 19 and 20, that were more soluble in water than 5-FU (2.1 and 1.9 times, respectively). As predicted (Sloan 1989, 1992; Sloan et al. 1984) 19 and 20 were the members that gave the greatest enhancement in J MMIPM (12 and 16 times, respectively) and not the more lipid soluble, longer alkyl chain members of the series. However, to compare members of the 1-ACOM series with members of any one of the 1-acyl series, the added heteroatom and methylene spacer in the 1-ACOM series needs to be taken into account. Thus, comparison should be made between the C1 member of the 1-ACOM series, 19, and the C3 member of the 1-AC, 15; or the C2 member of the 1-AOC series, 8, the C2 member of the 1-ACOM series, 20, and the C4 member of the 1-AC series, 16; or the C3 member of the 1-AOC series, 9, etc. Using these interseries comparisons, the members of the 1-ACOM series were less soluble in IPM but much more soluble in water (15.0–48.0 times) than the members of the 1-AC series, and their J MMIPM values were greater except for the comparison between 23 and 18 where the J MMIPM values were equivalent. On the other hand, although the members of the 1-ACOM series were less soluble in IPM than the members of the 1-AOC series, in this comparison only two members of the 1-ACOM series, 20 and 21, were substantially more soluble in water (4.0 and 1.8 times, respectively) and hence gave greater J MMIPM values than the corresponding members of the 1-AOC series. In the comparison of 19 and 8, the S AQ values were very close and 8 was four times more lipid soluble, so 8 gave a two times greater increase in J MMIPM. Similarly, 11 was 2.2 times more water soluble and ten times more IPM soluble than 23, so 11 gave a three times greater increase in J MMIPM.
Thus, the general mechanism for increasing lipid and aqueous solubilities of a drug by decreasing its ability to form intermolecular hydrogen bonds in the crystal lattice can be very effective (11–40 times enhancement of flux). But it is essential to evaluate the shorter alkyl chain members of any series to be considered because those are the members that are most likely to be more water soluble as well as more lipid soluble. In the examples based on 5-FU, the increases in flux realized with these acyl and soft alkyl prodrug approaches are more than sufficient to enlarge the indicated use of topical 5-FU from treating only actinic keratoses of the scalp (Dillaha et al. 1965) to treating recalcitrant psoriasis on less permeable areas of the body (Tsuji and Sugai 1972).
4.2 Incorporation of Water Solubility Enhancing Functional Groups into Promoiety
The second general mechanism by which acyl and soft alkyl promoieties can be used to increase the lipid and aqueous solubilities of prodrugs compared to their parent drugs is to incorporate polar, water solubilizing groups into their promoieties. In the examples illustrating the previous mechanism, the primary effect of the prodrug modification was to increase lipid solubility because the promoiety contained only an enabling functional group and a simple alkyl group. Although large increases in S IPM were realized for all members of homologous series, increases in S AQ were usually modest (less than two times) and only for the shorter alkyl chain members. In the examples illustrating the second general mechanism, the promoiety contains an additional amine, amide, ether, or diol functional group which in retrospect could have been designed specifically to increase S AQ. However, in most examples S AQ values were not available from the original references.
The first example is the use of a diol functional group in the promoiety to increase the S AQ of the prodrug and hence J M for the delivery of the parent drug.
Although the stated rationale was that more hydrophilic prodrugs could overcome the perceived rate limiting contribution of the aqueous viable epidermis part of the barrier to permeation of the skin by highly lipophlic drugs (Friend et al. 1988), the success of such prodrugs would also support a model for permeation where alternating lipid-aqueous barriers must be crossed in the intercellular compartment of the SC (Sloan et al. 1984, Sloan et al. 2011a, b. In Table 10.2 the mp (°C), solubilities in mixtures of ethanol and water (S VEH), log K between octanol, and pH 7.4 buffer (log K OCT:AQ) and fluxes of total species delivered from suspensions in ethanol and water (VEH) through rat skin in vitro (J MHVEH) are given for the evaluation of four acyl prodrugs of levonorgestrel.
Two of the prodrugs in Table 10.2 (Fig. 10.1) were simple alkylcarbonyl prodrugs: 26 and 27. Neither was representative of the shorter alkyl chain members of the series which would have had the greatest potential for increased aqueous as well as lipid solubility. Since 26 and 27 were both more soluble in 95 % ethanol than levonorgestrel, 25, was soluble in 100 % ethanol, it is reasonable to assume they would also be more soluble in octanol and hence be defined as more lipophilic than 25. Since partition coefficients for 26 and 27 could not be obtained because no 26 or 27 could be measured in the aqueous phase (while 25 could), it is reasonable to assume that 26 and 27 were less hydrophilic than 25. Finally, since the flux of 25 from various ethanol and water (40–100 %) mixtures did not vary significantly (applications of ethanol and water mixtures did not change S M1), it can be assumed that delivery of total species containing 25 by the prodrugs from widely different ethanol and water mixtures can be compared to the average flux generated by the application of 25 (0.00020 μmol cm−2 h−1) in ethanol and water mixtures. Thus, 26 and 27, which were more soluble in lipids but estimated to be less soluble in water, gave 3 and 1.3 times greater J MHVEH values, respectively, than 25. Only 25 was observed in the receptor phases.
By comparison, since the two prodrugs containing a diol functional group in the promoiety, 28 and 29, were both more soluble in an ethanol and water mixture that was primarily aqueous in composition (40 % ethanol) than 25 was in 100 % ethanol, it can be reasonably assumed that 28 and 29 were more soluble in water than 25. In addition, since 28 and 29 exhibit log K OCT:AQ that were comparable to that of 25 and were more soluble in water than 25, it can be reasonably assumed that 28 and 29 were more soluble in octanol than 25, that is, more lipophilic. Thus, since 28 and 29 were more soluble in a lipid and in water than their parent drug, as predicted (Sloan 1989, 1992; Sloan et al. 1984), they gave much larger increases in JMHVEH than the simple alkylcarbonyl prodrugs that were only more soluble in a lipid (31 and 15 times, respectively). However, because of their greater stabilities as carbonate esters, they delivered mostly intact prodrug through the skin (80 and 96 %, respectively).
The second example is the use of an amide functional group in the promoiety to increase S AQ of the prodrug and hence J M for the delivery of the parent drug. The first report of the synthesis of a promoiety containing an amide functional group as part of an effort to increase topical delivery was for theophylline: 7-(N, N-diethysuccinamoyloxymethyl) theophylline (Sloan and Bodor 1982). However, the prodrug was never completely evaluated. More recently 1-alkylazacycloalkan-2-one esters of indomethacin, 30 (Bonina et al. 1991), and naproxen, 35 (Bonina et al. 1993), have been synthesized and evaluated.
In Table 10.3 (Fig. 10.1) the values of S IPM, S AQ, and rates of delivery of total species containing 30 or 35 from water through human skin in vitro (J Mt) are given. For the indomethacin series, the second member of the series, 32, was the only member of the series that exhibited a greater S AQ than indomethacin, and although it was barely as soluble in IPM as indomethacin, it caused the greatest enhancement of J MHEV (4 times). The more lipid soluble but less water soluble members gave lower enhancement of J MHAQ. For the naproxen series, the first member of the series, 36, was more soluble in water (8 times) than naproxen and was more soluble in water than the other members of the series. Prodrug 36 was also more soluble in IPM than the other members of the series but none were as soluble as naproxen. Thus, 36, which was more soluble in lipids and water than the other members of the series, gave the greatest enhancement in J MHAQ (2.7 times) as would be predicted (Sloan 1989, 1992; Sloan et al. 1984).
There are two additional observations that can be made about these two series of prodrugs which have an amide functional incorporated into the promoiety. First, although the S IPM values for the two series are comparable, the S AQ values for the naproxen series (36–39) are almost uniformly ten times greater than those for the indomethacin series (31–34), and consequently the J MHAQ values for the naproxen series are almost uniformly ten times greater. Second, although more labile soft alkyl-type prodrugs (n = 1) had been synthesized, they were never evaluated because they were considered to be too labile. On the other hand, the n = 2 prodrugs were too stable, and only 10–12 % of either parent drug was observed in the receptor phases of the diffusion cell experiments in which they were evaluated. It would have been interesting to have evaluated the n = 1 series of prodrugs using an IPM vehicle, in which they would have been stable, to determine how effective they might have been at delivering the parent drug.
The third example is the use of an amine functional group in the promoiety to increase the S AQ of the prodrug and hence J M. Again the first report of the synthesis of a promoiety containing an amine functional group as part of an effort to increase the topical delivery of a parent drug was for theophylline: 7-(N, N-dimethylaminoacetyloxymethyl) theophylline (Sloan and Bodor 1982). However, again the prodrug was never completely evaluated. More recently the 17-(4′-dimethylaminobutyrate) ester prodrug of testosterone was evaluated using a 10 % solution of the prodrug in pH 7.4 buffer (Milosovich et al. 1993). Compared to the delivery from a suspension of testosterone in pH 7.4 buffer, the prodrug was 60 times more effective at delivering testosterone. Although no solubility data were reported, a 10 % solution of the prodrug was evaluated which suggests that it is substantially more soluble in water than testosterone which was soluble only to the extent of 0.004 %. The 2-diethylaminoethyl ester prodrug of indomethacin was also evaluated by the same group (Jona et al. 1995). It was reported that the prodrug drug was 3.7 times more soluble in pH 7.4 buffer and its partition coefficient between octanol and pH 7.4 buffer was 6.2 times greater than that of indomethacin so the prodrug was also much more soluble in octanol (23 times). Thus, it was entirely predictable (Sloan 1989, 1992; Sloan et al. 1984) that the prodrug gave a 4.3 times enhancement in the delivery of total indomethacin containing species through human skin in vitro.
The fourth example is the use of an ether functional group in the promoiety to increase the S AQ of the prodrug and hence J M. There are numerous reports in the literature where polyoxyethylene (POE) esters have been used as prodrugs to enhance oral delivery (Greenwald 2001) but only a few where POE esters have been used to enhance topical delivery. One of the limiting factors associated with using data from previous reports on the use of prodrugs containing oxyethylene groups in their promoieties to enhance the topical delivery of their parent drugs to design new prodrugs is the lack of experimental values for S LIPID (S OCT, S MO, S IPM), S AQ, and K LIPID:AQ in the literature (Bonina et al. 2001). This lack of experimental solubility and K data makes it impossible to predict changes in the solubility of the prodrug, attributable to the properties of the promoiety, compared to its parent in the membrane, S M1, and hence J M in Eq. 10.2.
However, there are several examples where those experimental S LIPID and S AQ values for prodrugs containing oxyethylene groups in their promoieties have been reported together with their corresponding maximum flux values, J M. In the first example, the effect of incorporating one oxyethylene group into carbonate derivatives of acetaminophen, APAP (Fig. 10.1), on their S LIPID and S AQ was compared with the effect of incorporating an alkyl group into carbonate derivatives of APAP on their S LIPID and S AQ (Table 10.4) (Wasdo and Sloan 2004).
The resulting effect on experimental J MMIPM was predictable based on the fit of the data to Eq. 10.2 (Roberts and Sloan 1999). The best alkyl carbonate in terms of enhancing J MMIPM was the C1 derivative, and the best oxyethylene carbonate was CH2CH2OCH3. Although the CH2CH2OCH3 carbonate was equally soluble in IPM and somewhat more soluble in water than the C1 carbonate, the C1 carbonate produced the greater J M. The slightly better S AQ of the CH2CH2OCH3 carbonate was offset by its higher molecular weight which was predicted by Eq. 10.2 (Roberts and Sloan 1999) to reduce the value of J M. Note that the solubility ratio (SR) for the CH(CH3)CH2OCH3 carbonate was greater than that for the CH2CH2OCH3 carbonate derivative (log SR = 0.013 and −0.52, respectively), but it was less soluble in both IPM and water than the CH2CH2OCH3 carbonate derivative so it only produced about one tenth the maximum flux. This illustrates how misleading SR or K can be in predicting flux and indesigning optimized topical products.
Similarly, in the second example the effect of incorporating one or two oxyethylene groups into carbamate derivatives of theophylline, Th-H (Fig. 10.1), on their experimental S IPM and S AQ values was compared with the effect of incorporating alkyl groups into carbamate derivatives of Th-H on their S IPM and S AQ values (Table 10.5) (Majumdar et al. 2012).
Again the resulting effect on experimental J MMIPM was predicted based on the fit of the data to Eq. 10.2 (Roberts and Sloan 1999). The best alkyl carbamate in terms of increasing J MMIPM was the C3 derivative and the best oxyethylene carbamate derivative was the (CH2CH2O)2CH3 derivative. The C3 alkyl carbamate was essentially equal in solubility in water to the C2 alkyl carbamate, but it was about 20 times more soluble in IPM. Therefore, the J MMIPM for the C3 alkyl carbamate was about four times that of the C1 regardless of the negative effect of its increased molecular weight predicted by Eq. 10.2 (Roberts and Sloan 1999). Although the (CH2CH2O)2CH3 carbamate derivative was only about 0.25 times as soluble in IPM as the C3 alkyl carbamate derivative, it was 11 times more soluble in water. Therefore, the J MMIPM for the (CH2CH2O)2CH3 carbamate derivative was about three times that of the C3 alkyl derivative regardless of the negative effect of its increased molecular weight. Among the oxyethylene carbamate derivatives, the (CH2CH2O)2CH3 carbamate derivative was three times more soluble in water and 30 % more soluble in IPM than the CH2CH2OCH3 carbamate derivative so, as predicted by Eq. 10.2 (Roberts and Sloan 1999), its J M value was about two times that of the CH2CH2OCH3 carbamate derivative. Although the CH(CH3)CH2OCH3 carbamate derivative was almost two times more soluble in IPM, it was only 0.40 times as soluble in water as the CH2CH2OCH3 carbamate derivative so, together with its increased molecular weight, the effect of its solubilities on J MMIPM led to its lower J MMIPM value. Again, the log SR value for the CH(CH3)CH2OCH3 carbamate derivative was much more positive than that of the other oxyethylene carbamate derivatives, but its JMMIPM value was lower, illustrating the misleading effect of SR and K in predicting flux.
In both examples, the incorporation of oxyethylene groups into the promoieties of prodrugs led to enhanced solubility properties of the prodrugs compared to their parent compounds that led to higher J M values.
5 Conclusion
Recognizing that one of the mechanisms for topical penetration enhancement involves increasing the solubility of the drug in the skin and that prodrugs increase the delivery of drugs into and through the skin by achieving the same, then it is quite clear that prodrugs constitute one type of penetration enhancer separate from formulation approaches. An even more powerful approach to enhancing topical delivery would be to use combinations of prodrugs with formulation approaches to enhancing topical delivery. So far there have been no reports of the use of such combinations except for simple one-component vehicles which have obviously not been optimized (Waranis and Sloan 1987). However, the possibilities with the use of such a combination approach would seem to be limitless.
References
Beall H, Prankerd R, Sloan KB (1994) Transdermal delivery of 5-fluorouracil (5-FU) through hairless mouse skin by 1-alkyloxycarbonyl-5-FU prodrugs: physicochemical characterization of prodrugs and correlation with transdermal delivery. Int J Pharm 111:223–233
Beall H, Prankerd R, Sloan KB (1996) Transdermal delivery of 5-fluorouracil (5-FU) by 1-alkylcarbonyl-5-FU prodrugs. Int J Pharm 129:203–210
Bonina FP, Montenegro L, DeCapraris P, Bousquet E, Tirendi S (1991) 1-Alkylazacycloalkan-2-one esters as prodrugs of indomethacin for improved delivery through human skin. Int J Pharm 77:21–29
Bonina FP, Montenegro L, Guerrera F (1993) Naproxen 1-alkylazacycloalkan-2-one esters as dermal prodrugs: in vitro evaluation. Int J Pharm 100:99–105
Bonina FP, Puglia C, Barbuzzi T, DeCapraris P, Palagiano F, Rimoli MG et al (2001) In vitro and in vivo evaluation of polyoxyethylene esters as dermal prodrugs of ketoprofen, naproxen and diclofenac. Eur J Pharm Sci 14:123–134
Coldman MF, Poulson BJ, Higuchi T (1969) Enhancement of percutaneous absorption by use of volatile: nonvolatile systems as vehicles. J Pharm Sci 58:1098–1102
Dillaha CJ, Jansen GT, Honeycutt WM, Holt GA (1965) Further studies with topical 5-fluorouracil. Arch Dermatol 92:410–417
Friend DR, Smedley SI (1993) Solvent drag in ethanol/ethyl acetate enhanced skin permeation of d-norgestrel. Int J Pharm 97:39–46
Friend D, Catz P, Heller J, Reid J, Baker R (1988) Transdermal delivery of levonorgestrel II: effect of prodrug structure on skin permeability in vitro. J Control Release 7:251–261
Greenwald RB (2001) PEG drugs: an overview. J Control Release 74:159–171
Jona JA, Dittert LW, Crooks PA, Milosovich SM, Hussain AA (1995) Design of novel prodrugs for the transdermal penetration of indomethacin. Int J Pharm 123:127–136
Juntunen J, Majumdar S, Sloan KB (2008) The effect of water solubility of solutes on their flux through human skin in vitro: a prodrug database integrated into the extended Flynn database. Int J Pharm 351:92–103
Kadir R, Stempler D, Liron Z, Cohen S (1987) Delivery of theophylline into excised human skin from alkanoic acid solutions: a “push-pull” mechanism. J Pharm Sci 76:774–779
Majumdar S, Mueller-Spaeth M, Sloan KB (2012) Prodrugs of theophylline incorporating ethyleneoxygroups in the promoiety: synthesis, characterization and transdermal delivery. AAPS PharmSciTech 13:853–862
Milosovich S, Hussain A, Dittert L, Aungst B, Hussain M (1993) Testosteronyl-4-dimethylaminobutyrate HCl: a prodrug with improved skin permeation rate. J Pharm Sci 82:227–228
Potts RO, Guy RH (1992) Predicting skin permeability. Pharm Res 9:663–669
Roberts WJ, Sloan KB (1999) Correlation of aqueous and lipid solubilities with flux of prodrugs of 5-fluorouracil, theophylline and 6-mercaptopurine: a Potts-Guy approach. J Pharm Sci 88:515–522
Roberts WJ, Sloan KB (2001) Application of the transformed Potts-Guy equation to in vivo human skin data. J Pharm Sci 90:1318–1323
Sasaki H, Takahashi T, Mori Y, Nakamura J, Shibasaki J (1990) Transdermal delivery of 5-fluorouracil and alkylcarbamoyl derivatives. Int J Pharm 60:1–9
Sloan KB (1989) Prodrugs for dermal delivery. Adv Drug Deliv Rev 3:67–101
Sloan KB (1992) Functional group considerations in the development of prodrug approaches to solving topical delivery problems. In: Sloan KB (ed) Prodrugs: topical and ocular drug delivery. Marcel Dekker, New York, pp 17–116
Sloan KB, Bodor N (1982) Hydroxymethyl and acyloxymethyl prodrugs of theophylline: enhanced delivery of polar drugs through skin. Int J Pharm 12:299–213
Sloan KB, Wasdo S (2003) Designing for topical delivery: prodrugs can make the difference. Med Res Rev 23:763–793
Sloan KB, Koch SAM, Siver KG (1984) Mannich base derivatives of theophylline and 5-fluorouracil: syntheses, properties and topical delivery characteristics. Int J Pharm 21:251–264
Sloan KB, Getz JJ, Beal HD, Prankerd R (1993) Transdermal delivery of 5-fluorouracil (5-FU) through hairless mouse skin by 1-alkylaminocarbonyl-5-FU prodrugs: physicochemical characterization of prodrugs and correlation with transdermal delivery. Int J Pharm 93:27–36
Sloan KB, Wasdo S, Ezike-Mkparu U, Murray TJ, Nichels D, Singh S et al (2003) Topical delivery of 5-fluorouracil (5-FU) and 6-mercaptopurine 6-MP) by their alkylcarbonyloxymethyl (ACOM) prodrugs from water: vehicle effects on design of prodrugs. Pharm Res 20:639–645
Sloan KB, Devarajan-Ketha H, Wasdo SC (2011a) Dermal and transdermal delivery: prodrugs. Ther Deliv 2:83–105
Sloan KB, Wasdo SC, Majundar S (2011b) Topical and transdermal delivery using prodrugs. In: Rautio J (ed) Prodrugs and targeted delivery. Wiley-VCH Verlag, Weinheim, pp 153–179
Synovec J, Wasdo SC, Sloan KB (2013) The effect of lipid and aqueous solubilities on flux of nicotinic acid esters from water through silicone membrane. Drug Dev Ind Pharm 39(9):1494–1497. doi:10.3109/03639045. 2012. 694590
Taylor HE, Sloan KB (1998) 1-Alkycarbonyloxymethyl prodrugs of 5-fluorouracil (5-FU): syntheses, physicochemical properties and topical delivery of 5-FU. J Pharm Sci 87:15–20
Tsuji T, Sugai T (1972) Topical administered fluorouracil in psoriasis. Arch Dermatol 105:208–212
Waranis RP, Sloan KB (1987) The effects of vehicles and prodrug properties and their interactions on the delivery of 6-mercaptopurine through skin: bisacyloxymethyl-6-mercaptopurine prodrugs. J Pharm Sci 76:587–595
Wasdo SC, Sloan KB (2004) Topical delivery of a model phenolic drug: alkyloxycarbonyl prodrugs of acetaminophen. Pharm Res 21:940–946
Wasdo SC, Juntunen J, Devarajan H, Sloan KB (2009) A comparison of the fit of flux through hairless mouse skin from water data to three model equations. Int J Pharm 366:65–73
Wenkers BP, Lippold BC (1999) Skin penetration of nonsteroidal antiinflammatory drugs out of lipophilic vehicle: influence of the viable epidermis. J Pharm Sci 88:1326–1331
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Sloan, K.B., Synovec, J., Wasdo, S.C. (2015). Selection of a Proper Prodrug for Penetration Enhancement. In: Dragicevic, N., Maibach, H. (eds) Percutaneous Penetration Enhancers Chemical Methods in Penetration Enhancement. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-45013-0_10
Download citation
DOI: https://doi.org/10.1007/978-3-662-45013-0_10
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-45012-3
Online ISBN: 978-3-662-45013-0
eBook Packages: MedicineMedicine (R0)