
Abstract
Cochliobolus is a young genus, in the class Dothideomycetes, which includes closely related plant pathogenic and saprophytic fungal species. In this review, genome similarities and differences among sequenced Cochliobolus pathogens that cause different diseases on different hosts are detailed. Gene content and genome organization are highly similar within the group and pathogens of the same host are not more similar to each other than those with different hosts. Instead, overarching genetic patterns follow phylogenetic lines. Classical and functional genetic research using Cochliobolus species has identified genes for secondary metabolism, management of iron and oxidative stress, and signaling as being involved in virulence. The genomic inventories and phylogenetic contexts of these genes, as well as of genes encoding protein effectors and cytochrome P450s, are compared across the genus, providing new insights into the evolution of host-specific virulence. Categorization of genes for secondary metabolism, according to distribution throughout the genus, is particularly revealing with unique genes tending to encode enzymes that biosynthesize metabolites involved in virulence. Additionally, genomic analysis identified many genes that encode unique small, secreted proteins, which could act as effectors in the plant host. Finally, new avenues for research paved by genomic analysis are highlighted.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
2.1 Introduction
2.1.1 Agricultural Biology of the Genus
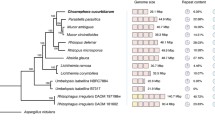
Cochliobolus spp. are young, closely related species (<20 MYA, Ohm et al. 2012), which make them ideal for comparative studies (Fig. 2.1, Table 2.1). The genus divides phylogenetically into two groups each associated with a distinct anamorphic stage. The first group, which encompasses the majority of known aggressive pathogenic species with significant impact on host crops, has a Bipolaris asexual stage while the second group has a Curvularia asexual stage (Sivanesan 1987). To comply with the International Code of Nomenclature for algae, fungi, and plants (McNeil et al. 2012), a discussion is underway in the community as to whether the name Bipolaris/Curvularia or Cochliobolus should be used to align with the “one name one fungus” recommendation. Most contemporary genetic, molecular, and genomic research on virulence determinants and reproductive development of the group has employed the Cochliobolus designation. The first group of species includes the necrotrophic corn pathogens, Cochliobolus heterostrophus and Cochliobolus carbonum, the oat pathogen, Cochliobolus victoriae, the rice pathogen, Cochliobolus miyabeanus, the sorghum pathogen, Bipolaris sorghicola, and the sugarcane pathogen, Bipolaris sacchari (Figs. 2.1 and 2.2, Table 2.1). Cochliobolus lunatus, also a pathogen of sorghum, falls in the second group (Figs. 2.1 and 2.2). The only species with a known hemibiotrophic lifestyle is the generalized cereal and grass pathogen, Cochliobolus sativus, which belongs to the first group. Some of these species, i.e., C. lunatus, can act as opportunistic human pathogens.

Phylogenetic tree showing distribution of Cochliobolus species. Cochliobolus species fall into two distinct groups, boxed in two shades of gray. “C” Cochliobolus, “B” Bipolaris, “Cu” Curvularia. Host plants are indicated. All Cochliobolus species not designated as homothallic are heterothallic, while those indicated with a “B” or “Cu” have no known sexual stage. Numbers after species name are isolate designations. Tree constructed by M. Berbee, University of British Columbia, using GPD and ITS sequences. Genera sister to Cochliobolus are indicated

Cochliobolus species and their disease phenotypes. All species, except C. victoriae, cause lesions on host leaves and in some cases on other plant tissues. C. heterostrophus race T produces T-toxin (arrow) which increases virulence on Tcms corn; C. carbonum race 1 and C. victoriae produce the HSTs HC-toxin and victorin, respectively, which are required for pathogenicity on the host. Pots contain resistant (left) and susceptible (right) oat seeds inoculated with a slurry of C. victoriae—Note none of the susceptible oat seeds germinated (extreme right). Image of C. lunatus from maizedoctor.cimmyt.org/index.php?option=com_content&t
The best-studied necrotrophic Cochliobolus spp. are notorious for their ability to evolve novel, highly virulent races producing host-selective toxins (HSTs) associated with the capacity of their producers to cause diseases on cereal crops that were bred, inadvertently, for susceptibility to the HST-producing pathogen (Yoder 1980; Turgeon and Baker 2007) (Table 2.1, Fig. 2.2). For example, in 1970, race T, a novel race of C. heterostrophus (Bipolaris maydis), caused a major epidemic of Southern Corn Leaf Blight (SCLB) that destroyed more than 15 % of the maize crop on the US eastern seaboard (Ullstrup 1970). Race T is genetically distinct from race O, first described in 1925 (Drechsler 1925), in that it uniquely carries genes for biosynthesis of T-toxin, an HST essential for high virulence (Yoder 1980) to Texas male sterile cytoplasm (Tcms) maize (Turgeon and Lu 2000).
Cochliobolus victoriae (Bipolaris victoriae), causal agent of Victoria Blight of oats , produces the chlorinated cyclic pentapeptide HST, victorin, rendering it highly virulent to oats carrying the dominant Vb allele (Fig. 2.2, Table 2.1) (Litzenberger 1949). The Vb-associated trait, susceptibility to C. victoriae, and a Pc-2-associated trait, resistance to Puccinia coronata, cannot be separated genetically (Lorang et al. 2012). Recent work with Arabidopsis revealed an NB-LRR-type resistance protein (LOV1) , guarding a thioredoxin protein target (TRX-h5) , that when activated confers susceptibility to C. victoriae and victorin (Lorang et al. 2004, 2007). Victorin thus acts by co-opting effector triggered defenses against the biotroph, P. coronata, to promote susceptibility to a necrotroph.
In contrast to the dominant plant host genes required for susceptibility to C. heterostrophus and C. victoriae, susceptibility to Northern Corn Leaf Spot caused by C. carbonum (Bipolaris zeicola ) is conferred by a homozygous recessive maize gene(s) (Johal and Briggs 1992; Multani et al. 1998). C. carbonum race 1 produces the cyclic-tetrapeptide HST, HC-toxin , which is specifically active, as is the fungus itself, against corn with the naturally occurring or mutant genotype hmhm (Fig. 2.2, Table 2.1) (Yoder 1980; Walton 1987, 1996). The site of action of HC-toxin in susceptible corn is histone deacetylase ; it is hypothesized that HC-toxin acts to promote infection of maize of genotype hm1hm1 by inhibiting this enzyme, resulting in the accumulation of hyperacetylated core histones. This then alters expression of genes encoding regulatory proteins involved in plant defense (Ransom and Walton 1997; Walton 2006). C. carbonum races 2 and 3 do not produce the toxin.
Cochliobolus sativus (Bipolaris sorokiniana), a hemibiotroph and less specialized cereal pathogen, causes diseases of roots (common root rot), leaves (spot blotch), and spikes (black point or kernel blight) of cereals (mainly barley and wheat) (Fig. 2.2, Table 2.1) (Mathre 1997; Weise 1987). Three C. sativus pathotypes (0, 1, and 2) have been described (Valjavec-Gratian and Steffenson 1997) based on differential virulence patterns on three barley genotypes (ND5883, Bowman, and NDB112). Pathotype 0 isolates show low virulence on all three barley genotypes. Pathotype 1 isolates show high virulence on ND5883 but low virulence on other barley genotypes. Pathotype 2 isolates show high virulence on Bowman but low virulence on ND5883 and NDB112. Genetic analysis and molecular mapping indicates that a single locus, VHv1, controls high virulence of the pathotype 2 isolate ND90Pr on Bowman (Valjavec Gratian and Steffenson 1997; Zhong et al. 2002). The VHv1 locus is unique to pathotype 2 and encodes two nonribosomal peptide synthetases (NRPSs) , one of which when deleted, drastically reduces virulence of pathotype 2 on cultivar Bowman (Condon et al. 2013).
Cochliobolus miyabeanus (Bipolaris oryzae) is the causal agent of brown spot of rice which contributed, along with a cyclone and tidal waves, to the Bengal rice famine of 1942/1943 that resulted in starvation of more than two million people (Dasgupta 1984) (Fig. 2.2, Table 2.1). The interaction between rice and C. miyabeanus is inadequately understood from the perspective of genetic and molecular mechanisms and no HST has been correlated with the ability of C. miyabeanus to cause disease.
Cochliobolus lunatus (Curvularia lunata) is a pathogen of sorghum (Fig. 2.2, Table 2.1) (Thakur et al. 2006) and is also known to be an opportunistic human pathogen (Thakur et al. 2006; Manamgoda et al. 2011, 2012). The sequenced strain (m118, MUCL 38696) was selected originally as a pilot organism for steroid biotransformation (Vitas et al. 1994, 1995) in the laboratories of Friedrich Schiller University, Jena, Germany. This, and another strain, C. lunata AT46, have been utilized widely for steroid transformation (Rozman et al. 1996).
2.1.2 Reproductive Biology
Sexual Cochliobolus species can be self-sterile (heterothallic , requiring genetically distinct partners) or self-fertile (homothallic , no partner required) (Fig. 2.1, Table 2.2). As in most ascomycetes, a single mating type locus (MAT) controls the ability to reproduce sexually and in Cochliobolus, all heterothallic species have either MAT1-1 or MAT1-2 (but never both) in different individuals whereas all homothallic species carry both MAT1-1 and MAT1-2 in the same nucleus of an individual (Turgeon et al. 1993). Asexual species (i.e., those with no known sexual cycle), such as B. sacchari, also are found in the group. It is well documented that asexual species also carry MAT genes (Sharon et al. 1996). Thus, Cochliobolus spp. are an excellent choice for comparisons of reproductive mechanisms in asexual, heterothallic and homothallic species within a closely related group of species in the same genus (Fig. 2.3) (Turgeon and Debuchy 2007; Debuchy and Turgeon 2006; Yun et al. 1999).

Reproductive stages of C. heterostrophus. a Portion of a mating plate containing a senescent corn leaf inoculated with a pigmented MAT1-1 strain and an albino MAT1-2 strain. Both black and white pseudothecia are formed indicating both strains are hermaphroditic. b Two pseudothecia that have been squeezed to release asci (arrow). c A single ascus containing ascospores (tetrad) and a single conidium. d A single ascospore and a single conidium. e Several asci containing tetrads with varying numbers of ascospores
The coexistence of heterothallic and homothallic species in the same genus is common to many classes of ascomycete and coexistence of both mating type genes in the same nucleus is common to most homothallic species, including Cochliobolus. For Cochliobolus spp., a self-sterile to self-fertile evolution is well supported since all homothallic Cochliobolus spp. are polyphyletic, their MAT genes are diverse in structure and arose independently, while all heterothallic MAT genes are conserved in structure; plus, molecular evidence exists for recombination mechanisms (Yun et al. 1999; Inderbitzin et al. 2005). Functional analyses (Yun et al. 1999; Lu et al. 2011) indicate that MAT genes can be transferred from heterothallic to homothallic species or vice versa and retain function, although there are yet to be understood nuances associated with fertility. Where causes of asexuality have been studied functionally, asexual species such as B. sacchari have been found to be asexual for reasons not associated with the MAT genes themselves; their MAT genes are fully functional in mat null strains of C. heterostrophus (Sharon et al. 1996).
C. carbonum and C. victoriae are capable of crossing to each other (Scheffer et al. 1967; Christiansen et al. 1998). We have hypothesized that C. victoriae may have evolved from a MAT1-2 C. carbonum strain (Christiansen et al. 1998). This is supported by our finding that all extant strains of C. victoriae are MAT1-2 and female sterile. As noted below, C. victoriae and C. carbonum share an intermediate number of SNPs at the whole-genome level compared to C. heterostrophus inter- and intra-species comparisons, in support of this close relationship. Table 2.2 is a summary of mating attributes for the Cochliobolus species with sequenced mating type loci.
2.1.3 Genetic Tools
Cochliobolus spp. are easily grown in culture, produce abundant asexual spores (except C. lunatus), and can be stored for long periods of time in glycerol or silica gels (Yoder 1988). They also have an efficient sexual stage readily produced in the laboratory in 3 weeks (Fig. 2.3) (Leach et al. 1982), and are easily transformed (Turgeon et al. 2010). Targeted gene deletion using PCR fragments is highly efficient (Turgeon et al. 2010; Wirsel et al. 1996; Catlett et al. 2003a). Chromosomes can be resolved using pulsed-field gel electrophoresis (Kodama et al. 1999; Tzeng et al. 1992).
In this review, we compare genome similarities and differences among sequenced Cochliobolus pathogens, with particular emphasis on strain and species-unique sequences, virulence determinants (secondary metabolites, iron and oxidative stress), mechanisms of reproduction, and signaling.
2.2 Genome Structure
2.2.1 Genome Sequence Comparisons
Five strains of C. heterostrophus and one strain each of C. victoriae, C. carbonum, C. miyabeanus, C. sativus, and C. lunatus were sequenced by the Joint Genome Institute (JGI) . Two C. heterostrophus strains and the C. sativus strain were fully sequenced, while the remaining genomes were sequenced using Illumina and assembled de novo using Velvet or AllPathsLG (http://genome.jgi.doe.gov/programs/fungi/index.jsf). The highly inbred C. heterostrophus race O lab strain C5 was used as the reference sequence for all comparisons, as it is the most complete, consisting of only 68 scaffolds. Three additional C. sativus strains have been sequenced recently, but are not discussed here (Zhong unpublished).
Overall sequence assembly and annotation statistics are presented in Table 2.3. All Cochliobolus genomes are in the 31–37 Mb range with an estimated gene content of 12,000–13,300. Gene content and genome organization are highly similar within this group of fungi, although less so for C. lunatus. In contrast, comparative analysis of C. heterostrophus and C. sativus in the context of 15 more distantly related Dothideomycetes genomes (Ohm et al. 2012) revealed significant variation.
The relative scale of conservation at the nucleotide level, compared to C. heterostrophus C5, was used as an estimation of similarity. Most of the C. heterostrophus race T strain C4 assembly could be aligned and only 1,584 SNPs were found between the two strains (Fig. 2.4). This remarkable level of conservation is molecular verification of the highly inbred nature of the two strains, achieved through generations of successive backcrossing. Note, since race O strain C5 is the reference to which all sequences are aligned, alignment of C4–C5 excludes the 1.2 Mb of Tox1 DNA that is unique to race T. In contrast to results with the inbred strains, comparison of each C. heterostrophus field strain to C5 revealed roughly 10 times more SNPs than the same comparison with C4 and comparison of each Cochliobolus species revealed roughly 10 times more SNPs than did any C. heterostrophus field strain. Thus, there is a clear diminishing gradation of similarity at the whole-genome level as comparisons move from inbred strains to field strains within a species, to across species. As expected, based on phylogenetic distance (Fig. 2.1), C. lunatus appears to be the most diverged species, as only 20 % of its genome could be aligned to reference C5, compared to ~75 % for other Cochliobolus species (Fig. 2.4).

Relative conservation of Cochliobolus species and C. heterostrophus strains to C. heterostrophus C5 reference. Each genome in this study was aligned, pairwise, to the C. heterostrophus C5 assembly using the MUMmer DNAdiff tool (Kurtz et al. 2004), and data were plotted logarithmically. The majority of each Cochliobolus species genome could be aligned (dark gray bars) to C. heterostrophus C5, except for C. lunatus. SNPs called between aligned regions (light gray bars) demonstrate that the inbred C. heterostrophus C5 and C4 strains are highly similar and C. heterostrophus field strains are more similar to C. heterostrophus strain C5 than to any other Cochliobolus species. SNPs/100 kb of aligned sequence (white bars) support this trend and show C. lunatus is the most dissimilar to C. heterostrophus of the Cochliobolus species, which fits with phylogenetic placement (Fig. 2.1). Data are displayed relative to the total query assembly size (black bar)
Most significantly, at the species level, a total of 11.76 Mb present in all C. heterostrophus genomes was missing from C. victoriae, C. carbonum, C. sativus, and C. miyabeanus (C. lunatus was excluded from this analysis). Only 1.6 Mb of this was in segments larger than 5 kb in the alignment to C5. Most of the sequence that separates C. heterostrophus from other species, therefore, is not the result of large wholesale insertions or deletions of DNA, but from a more piecewise gain and loss. We and others (Hane et al. 2011; Goodwin et al. 2011; Rouxel et al. 2011) have recently coined the term mesosynteny (Ohm et al. 2012) to describe organizational conservation between species. Genetic content is conserved across chromosomes, but not colinearly. It seems possible that our findings with Cochliobolus, showing that many small, scattered differences sum to significant quantitative differences (i.e., 25 % dissimilar), could be the product of the same mechanisms.
Pathogens of the same host (e.g., C. carbonum and C. heterostrophus on maize) were not more similar to each other than those with different hosts. Instead, overarching genetic patterns followed phylogenetic lines. A telling example of this is our finding that C. carbonum and C. victoriae have fewer SNPs between them than revealed in comparisons between other pairs of Cochliobolus species. These comparisons support our previously reported hypothesis that C. victoriae arose from a MAT1-2 strain of a non-HC-toxin-producing strain of C. carbonum and is expected therefore to be more closely related to it than to other species (Christiansen et al. 1998). Given that the Pleosporaceae arose as a group less than 23–17 MYA (see Fig. 2.1 in Ohm et al. (2012)) and the genus Cochliobolus is young in the Pleosporaceae group, genome comparisons provide us with an overall picture of a timeline of how genome diversity varies with speciation.
Less than 1 year after the comprehensive analyses of 18 genomes in Ohm et al. (2012) and Condon et al. (2013) were published, the number of sequenced Dothideomycete genomes has doubled in the JGI Mycocosm (http://genome.jgi.doe.gov/programs/fungi/index.jsf). As more genera are sequenced to the same depth as Cochliobolus, the close similarity seen among Cochliobolus species can be compared to relationships among suites of species taxa within other genera. Attempting to align separate Aspergillus species, using our methodologies, for example, would yield poor alignments, as they are much more distantly related to each other. Indeed—studies that identify syntenic genomic regions between Aspergillus species do so with a much lower threshold for similarity and conservation (Fedorova et al. 2008).
2.2.2 Insights from Genome Comparisons
2.2.2.1 Secondary Metabolism
Armed with the knowledge that most of the best known aggressive Cochliobolus pathogens are necrotrophs and that high virulence/pathogenicity of the most devastating of these is associated with secondary metabolite production in the form of HSTs biosynthesized by NRPS and PKSs, we extracted all NRPS and PKS encoding genes from all 6 species (10 strains). Number of NPSs per genome, ranged from 9–25, while number of PKSs ranged from 15–27 (Table 2.3). Comparative analyses revealed that the suites of these genes are astoundingly diverse among species but remarkably conserved among isolates of the same species, whether inbred or field strains, except for defining examples that generally map to unique genomic regions. Functional analysis of several of these strain-unique PKSs and NPSs reveals a strong correlation with a role in virulence as hinted at decades earlier with, e.g., the PKS genes for T-toxin production in C. heterostrophus race T and the genes for HC-toxin production by race 1 of C. carbonum, which are not found in any other Cochliobolus species.
Comparing the inventories of secondary metabolism genes across several closely related species yields key insights (Figs. 2.5 and 2.6). The first insight is that broadly conserved NRPS s or PKSs are most likely to produce metabolites of biological function central to the fungal cell itself. NPS2, NPS6, NPS4, NPS10, and PKS18 are C. heterostrophus NPS and PKS genes conserved across all Cochliobolus species (Figs. 2.5, 2.6 and 2.7). Functional studies of C. heterostrophus mutants deleted for these genes demonstrate that the metabolites produced by the conserved biosynthetic enzymes affect developmental processes such as sexual and asexual development, morphology, hydrophobicity of colony surfaces, as well as stress (oxidative, iron, etc.) management (Figs. 2.6 and 2.7). Properly defining the scope of inclusion for this inference is essential—across the 18 Dothideomycetes examined in Ohm et al. (2012), only NPS10 is conserved in all, despite the importance of these metabolites in Cochliobolus species. This finding is in agreement with the earlier hypotheses (Bushley and Turgeon 2010) that NPS10 is among the more ancestral NRPSs. The product of NPS10 is not known, however, C. heterostrophus mutants are sensitive to oxidative stress. C. heterostrophus NPS2 is responsible for siderophore biosynthesis and intracellular iron storage and is conserved in 17 out of the 18 Dothideomycetes examined in Ohm et al. (2012, Table S19) (Fig. 2.6). NPS6 is present in 11 of the 18 genomes and is responsible for extracellular siderophore biosynthesis and thus competition for iron in the plant–fungal interaction (Fig. 2.6). NPS6 has been shown to be involved in virulence of C. heterostrophus to corn, of C. miyabeanus to rice, of A. brassicicola to Arabidopsis thaliana, and of Fusarium graminearum to wheat. It is also required for in vitro oxidative stress management (Oide et al. 2006). NPS4 makes an unknown product, but is present in 10 of the 18 genomes. C. heterostrophus, A. brassicicola, and F. graminearum nps4 mutant colony surfaces are hydrophilic, rather than hydrophobic like wild type (Fig. 2.7) (Turgeon et al. 2008). PKS18, responsible for melanin biosynthesis , is conserved in all Cochliobolus species and was reported as conserved in 17 of 18 genomes in the study of Ohm et al. (2012). We have subsequently observed that A. brassicicola, the species missing PKS18, does in fact posses the gene (Fig. 2.6).

Cartoon of cross-species phylogenomic analyses of individual AMP binding domains from NRPS proteins. NRPS AMP domains were extracted from all five C. heterostrophus and from the C. victoriae, C. carbonum, C. miyabeanus, C. sativus,, and Setosphaeria turcica genomes. Members of the reference set of previously annotated C. heterostrophus NRPS AMP domains (Lee et al. 2005; Bushley and Turgeon 2010) were used as benchmarks for branches. Branches of the full phylogenetic tree are collapsed according to clustering with the reference set of C. heterostrophus AMP domains. Presence in each of the five C. heterostrophus strains, Cochliobolus species, and S. turcica is noted by “*”, AMP domains not grouping with the previously annotated C. heterostrophus set are labeled as “newly revealed” or “new, groups with known metabolite.” NPS1, NPS3 AMPs are labeled as diversity generating (Fig. 2.9). C. carbonum HTS1 AMPs are indicated, as is the C. sativus pathotype 2 NRPS discussed in text (Fig. 2.8)

Cartoon of cross-species phylogenomic analyses of individual ketosynthase (KS) domains from PKS proteins. The KS domains were extracted from all five C. heterostrophus and from the C. victoriae, C. carbonum, C. miyabeanus, C. sativus and S. turcica genomes. PKS designations match the C. heterostrophus set. KS domains not grouping with the previously annotated C. heterostrophus set are labeled as “New_1 through _10.” Highly conserved PKS18, encoding the PKS for melanin biosynthesis and the unique PKSs (PKS1, PKS2) for C. heterostrophus race T T-toxin production are indicated

The NRPS AMP domain tree (Fig. 2.5) and highly conserved AMPs. See text NPS2 consists of four AMP domains that group together and produce the hexapeptide intracellular siderophore, ferricrocin, responsible for iron storage within cells. When deleted, sexual reproduction (ascus formation, right) is absent. NPS4 consists of four AMP domains, only two of which group together. Product is unknown but lack of NPS4 converts colony surfaces from hydrophobic to hydrophilic (middle). NPS6 consists of one complete and one incomplete AMP domain for production of the tripeptide extracellular siderophore, coprogen, which when absent impacts ability to acquire iron, resist oxidative stress (left), and reduces wild-type virulence (bottom). There are two copies of NPS12 which has no known phenotype. AAR is alpha-aminoadipate reductase responsible for lysine biosynthesis in fungi. For each NPS, the number after the period refers to a particular AMP domain in the protein, starting from the N terminal end
The second key insight is that genes encoded by genes “unique” to a particular species or strain of a species, encode enzymes that are likely biosynthesizing secondary metabolites involved in virulence (Fig. 2.8). A canonical example is the identification of a group of C. sativus pathotype 2-specific AMP domains (Fig. 2.5), one of which (ID 115356) when deleted, drastically reduces virulence on cultivar Bowman (Fig. 2.8). Another example is the C. heterostrophus race T- specific PKS1 and PKS2 genes (Fig. 2.6). These two polyketide synthases are responsible for production of T-toxin in race T and high virulence to Tcms maize and have long been described as unique to race T based on DNA–DNA hybridization blots. Phylogenetic analyses of PKS KS domains confirmed that they are not found in any other Cochliobolus species (Fig. 2.6).

The NRPS AMP domain tree (Fig. 2.5) and unique AMPs. See Figs. 2.5 and 2.7 for labeling. An example of a unique NRPS in C. sativus, associated with virulence of the strain on a particular cultivar of the host is shown (left). Barley cv. Bowman was inoculated with wild type (ND90Pr) and a mutant lacking the gene corresponding to protein ID 115356 shows reduced virulence. Right Susceptible (S) and resistant (R) maize inoculated with C. carbonum race 1, which produces the HST HC-toxin
A third example is the genes encoding C. carbonum HC-toxin (Fig. 2.5). C. carbonum race 1 is the only Cochliobolus species to possess HTS1, the NRPS (4 AMP domains) responsible for producing HC-toxin (Fig. 2.8). Wider genome resources, however, uncover candidate orthologs for all 4 AMP domains plus other genes associated with biosynthesis of HC-toxin, in Setosphaeria turcica, Alternaria jesenkae, and Pyrenophora tritici-repentis and Fusarium semitectum (Manning et al. 2013; Condon et al. 2013). The metabolites produced by the first three of these HTS1 orthologous clusters have not been identified and they may not be HC-toxin (C22H34N4O6). Fusarium semitectum, for example, has the HTS1 ortholog, APS1, however this is the core NRPS for biosynthesis of apicidin (C34H49N5O6), a structurally different metabolite with the same biological activity as HC-toxin (both are histone deacetylase inhibitors) (Jin et al. 2010). Whether or not the other species produce HC-toxin, the discovery of these HTS1 orthologs furthers our understanding of evolution of genes associated with HSTs in the fungal–plant interaction. Like apicidin and HC-toxin, orthologs may have profound medicinal application (Jin et al. 2010; Han et al. 2000). Thus, HST genes that were originally thought to be unique to the producer, like those for HC-toxin in race 1 of C. carbonum may prove not to be. As more and more genome sequences become available, it is even likelier that genes, such as HTS1, are not unique but are spottily distributed with candidate orthologs in distant and/or isolated branches of the fungal phylogenetic tree.
As orthologs are discovered in more and more species, horizontal gene transfer may be a less enticing hypothesis—or, it may be the best explanation, depending on the distribution. The alternative hypothesis for rare distribution among species is rapid selective duplication and loss (Kroken et al. 2003; Bushley and Turgeon 2010). By diversifying inventories of HSTs or effectors, pathogens prevent hosts from developing a single resistant genotype. It is possible that uncharacterized members of the pool of uniquely distributed secondary metabolism genes act as HSTs in undiscovered contexts. Their anonymity may relate more to the fact that the corresponding host target, or host itself, is not widely deployed in agriculture, and therefore, the pathogenic potential of these metabolites is not known to us.
The third insight comes from our species inventory of NRPS genes and may weigh on the last point above. The AMP domains comprising C. heterostrophus NPS1, NPS3, NPS13 NRPS proteins indicate a complex evolutionary history (Condon et al. 2013) (Fig. 2.5). On the whole protein level, the complete C. heterostrophus NPS1 (trimodular) and NPS3 (tetramodular) domain sets are either present or absent in other species (Fig. 2.9). NPS1 is intact in C. victoriae, C. carbonum, and C. lunatus, while NPS3 is intact in C. miyabeanus and C. sativus, but absent from the other genomes. Mono-modular C. heterostrophus NPS13 is found only in C. heterostrophus. NPS1, NPS3, and NPS13 protein AMP domains are expanded discontinuously resulting in a suite of novel proteins which may be mono- or multi-modular (Fig. 2.9). All of the AMP domains that comprise these proteins form two separate clades in the phylogenetic tree of Cochliobolus AMP domains (Figs. 2.5 and 2.9).

NPS1, NPS3, and NPS13 are examples of NRPS proteins encoded by highly recombinogenic and expanded NPS genes. Full AMP domain phylogenetic tree (Condon et al. 2013) is cartooned at left. The reference NPS1, NPS3, and NPS13 proteins are cartooned bottom left. AMP domains corresponding to these proteins are completely conserved in the five strains of C. heterostrophus, but show discontinuous presence in all other Cochliobolus species (Fig. 2.5) and Setosphaeria. Note some AMP domains from NPS1 to NPS3 group at the top of the tree (AMPs 2 and 4, white box), while the rest group at the bottom of the tree (AMPs 1 and 3, hatched box); NPS13 AMP1 (black) also groups at the bottom of the tree. Branches correspond to individual AMP domains which group together and the particular corresponding AMP domain is depicted on the right of the diagram. Note collection of novel NRPSs composed of NPS1, NPS3, and NPS13 AMPs, at bottom
We speculate that this group of AMP domains is a hotbed of evolutionary activity. Domains are rapidly duplicated, swapped, recombined, and genes are gained and lost. Future studies on the evolutionary signatures of different clades could help support this hypothesis. If the idea holds, it could explain how some NRPS are found in such a patchwork distribution throughout a phylogeny.
2.2.2.2 Iron and Oxidative Stress
Among the NRPSs involved in running the fungal cell itself are those biosynthesizing intracellular and extracellular siderophores for iron chelation. Iron is indispensable for virtually all organisms (Winkelmann 1991) and is involved in many fundamental biochemical reactions (respiration, the TCA cycle). It is also required for success as a pathogen. Iron can occur either in reduced ferrous (Fe2+) or oxidized ferric (Fe3+) form; this capacity to gain or lose electrons makes iron a major redox mediator. Iron has the potential to catalyze the Fenton/Haber Weiss reactions (Fenton 1894) generating highly cytotoxic ROS. Hence, mechanisms that sequester iron in cells are critical for survival. Paradoxically, although iron is essential, bioavailable forms are very limited in aerobic environments (Neilands and Leong 1986; Lesuisse and Labbe 1994; Haas 2003). Therefore, efficient and competitive iron-uptake mechanisms are also critical to survival of all organisms, including fungi during infection of plants. For this, fungi employ a variety of strategies, including two high-affinity uptake mechanisms, siderophore-assisted mobilization, and non-siderophore reductive iron assimilation (RIA) (Schrettl et al. 2004; Oide et al. 2006; Wolpert et al. 2011). As noted, with their strong iron-binding activity, siderophores function both in acquisition and in storage/sequestration of iron (Neubauer et al. 2000; Oide et al. 2006). Fungal (and bacterial) siderophores are biosynthesized by multi-modular NRPSs (encoded by NPS2 and NPS6, previous section) (Fig. 2.7) (Oide et al. 2006). The alternative high-affinity iron-chelating mechanism in fungi, RIA , is a three step process in which ferric iron is reduced by a metalloreductase (Fre1p) extracellularly, and then the ferrous iron is oxidized by an iron multicopper oxidase (Fet3p) that is coupled to a high-affinity iron permease (Ftr1p) for transport across the plasma membrane to the cytosol (Haas 2003). To a first approximation, necrotrophs , such as most Cochliobolus species, rely on extracellular siderophores for in planta iron acquisition, while (hemi)biotrophs tend to use the RIA mechanism of iron gathering.
We have generated many C. heterostrophus mutants lacking iron or oxidative stress related genes (Fig. 2.7). Associated phenotypes are shown in Table 2.4. NPS6 is a virulence factor for several pathogens (Oide et al. 2006). nps6 mutants still have the RIA route available and also still produce the intracellular siderophore, ferricrocin, made by the product of the NRPS encoding gene, NPS2. Ferricrocin is not required for virulence of C. heterostrophus, but is required for sexual reproduction (Fig. 2.7). nps2nps6 double mutants exhibit a greater reduction in virulence and impairment in sexual development than single nps6 or nps2 mutants (Fig. 2.7). Triple iron acquisition and storage mutants (nps2nps6ftr1) are almost avirulent, but do attach to and penetrate the host (Condon, Turgeon unpublished). nps6 mutants are also hypersensitive to oxidative stress (Fig. 2.7) and there is a gradation of sensitivity of the single, double, and triple mutants, with the latter being the most sensitive.
Double mutants lacking ChAP1 (Lev et al. 2005), a gene encoding a redox-regulated transcription factor and NPS6 (Chap1nps6), or lacking ChAP1 and the iron-sensitive transcription factor Sre1 (Chap1sre1) have been constructed (Table 2.4) and tested for oxidative stress and virulence. Chap1nps6 mutants are more sensitive to oxidative stress than either parent, while Chap1sre1 mutants partially rescue the Chap1 oxidant-sensitive phenotype. Double mutant phenotypes are consistent with a model in which sequestering of iron by the NPS6 siderophore defends the fungal pathogen against oxidative stress .
2.2.2.3 The CYPome of Cochliobolus spp.
The published Cochliobolus genome manuscript (Condon et al. 2013) did not include C. lunatus, a species, as indicated in the Introduction, that has been used as a workhorse for steroid biosynthesis centered on the activity of cytochromes P450 (CYPs) . CYPs, a superfamily of heme-containing monooxygenases, are ubiquitously present in all kingdoms of life with fungi having the second largest number after plants. Some are involved in primary metabolism and are indispensable for normal development and homeostasis or in allowing fungi to live on particular carbon sources. Others are involved in xenobiotic metabolism and provide defense against natural products, while still others are associated with genes for secondary metabolite production and the biosynthesis of pigments, antioxidants, defense compounds, and toxins.
Despite the fact that CYPs play roles in hydroxylation and oxidation processes leading to degradation, detoxification, and syntheses of compounds crucial for life or for niche survival, the substrates on which they act are largely unknown. To identify P450s and annotate those associated with secondary metabolite gene clusters across Cochliobolus species, we searched gene models for annotations with the PF00067 (P450 superfamily) domain. Almost one thousand predicted P450s (943) were identified across six Cochliobolus species, averaging ~135 P450s per species and represents ~1 % of the total gene catalog (Table 2.3). This tally is comparable to the number in Aspergillus nidulans (version AN.3, CADRE (Kelly et al. 2009)) and other Aspergillus species (~125 P450s per species). The CYPome of the Dothideomycete Mycosphaerella graminicola has fewer (82 P450s plus one pseudogene) (Newsome et al. 2013). P450s in close proximity to secondary metabolism backbone genes (such as NPS or PKS genes) may be involved in secondary metabolite biosynthesis. NPS or PKS genes were located near 13–17 % of C. heterostrophus P450s, slightly lower than when this analysis was done for A. nidulans, 29 % (32 of 111 functional P450s) (Kelly et al. 2009). No preference was observed in the association of P450s with mono- or multi-modular NPS genes.
It is difficult and in most instances impossible, to predict the specific functions of the CYPs from their sequence similarities or even their association with PKSs or NPSs in gene clusters, as it is known that a single amino acid change can significantly alter metabolic capabilities. These difficulties, in combination with the abundance of P450s, make phylogenetic analyses an essential first step for studying these crucial genes.
2.2.2.4 Small Secreted Proteins (SSP)
A search for candidate effector proteins that are cysteine rich (>2 % cysteine), small (<200 amino acids), predicted to be secreted (using Phobius (Kall et al. 2007)), and without transmembrane domains revealed between 143 and 289 SSPs per Cochliobolus (Table 2.3) (Condon et al. 2013). An all-versus-all BLAST analysis to determine if SSPs were strain or species-unique revealed that few candidate C. heterostrophus SSPs were unique to any particular strain within the species. Among species, C. sativus had the most isolate-unique SSPs, containing 167 candidates (Condon et al. 2013). As this is the only Cochliobolus strain thought to act as a hemibiotroph , it is interesting that it contains more SSPs, and more unique SSPs, than the necrotrophic isolates, although this is only a correlation at this point. As is typical with candidate effectors, functional domain predictions were lacking, with only 37 candidates having some predicted function, generally involved in cell wall or extracellular matrix function. An additional 23 candidates were conserved in other fungi outside of the Dothideomycetes. The remaining 120 predicted candidates were featureless and seemingly unique to the Dothideomycetes (Condon et al. 2013). Cochliobolus heterostrophus strain C5 SSP predicted candidates were rich in SNP calls to other Cochliobolus genomes: 101 candidate SSPs had SNPs with at least one other Cochliobolus genome (Condon et al. 2013).
In our all-versus-all BLAST analysis, only 6 of the 180 C. heterostrophus C5 SSPs were found in all 10 strains examined and 14 were unique to strain C5 (Condon et al. 2013). The presence or absence of most SSPs did not fall into easily categorized bins such as C. heterostrophus-specific, or maize-pathogens only. Instead, SSPs were present and absent in no particular pattern across the genomes. 115 SSPs were present in at least one other species (C. victoriae, C. miyabeanus, C. carbonum), with seven found in all species, and 27 in all Cochliobolus species. Unlike those in some phytopathogens, such as Leptosphaeria maculans (Rouxel et al. 2011), SSP-encoding genes did not occur in clusters; candidates seldom were located within 10 kb of each other. It has become clear in recent years that necrotrophs, like (hemi)biotrophs, also use effectors to manipulate specific targets in the host cell for the benefit of the pathogen. Unlike (hemi)biotrophs, however, the aim seems to be to trigger host defenses or cell death, rather than circumvent these processes. Two clear examples are victorin produced by the necrotroph C. victoriae and ToxA produced by the necrotrophs Pyrenophora tritici-repentis (Ciuffetti et al. 1998) and Stagonospora nodorum (Friesen et al. 2008). The extent to which necrotrophs employ effectors is an exciting and unknown frontier. Do necrotrophic effector molecules always aim to trip host defenses, or do some act more according to (hemi)biotrophic principles, quelling host defense response and intercepting signaling? As for their metabolic origins, are (presumably) secondarily encoded molecules like victorin the norm, or do necrotrophs utilize small cysteine rich ribosomally encoded effector proteins typical of hemi(biotrophic) interactions? The bioinformatics analysis described above is an earnest attempt to break ground answering these questions. The limitations of this approach, however, cannot be overstated. Bioinformatically predicted SSPs require in planta expression or protein secretion data, or functional knockout data, before they can be considered bona fide effectors. SSPs are small, and typically lack predicted functional domains—a trait they share with miscalled ORFs. Secretion prediction is also an imprecise technique with many false-positive and false-negative predictions. That our SSP inventories are larger for known hemibiotrophs than necrotrophs seems to suggest that we are indeed including at least some effectors in our prediction.
2.2.2.5 Signaling
As for P450s, the published Cochliobolus genome manuscript (Condon et al. 2013) did not include a comprehensive analyses of genes associated with signaling. Because signaling mechanisms are the centerpiece of interaction biology, we have included a brief summary of annotation of relevant genes in Cochliobolus species.
Conserved signaling pathways. If two closely related pathogens infect different hosts, one might conclude that they respond to different signals and hypothesize that comparison of the genomes of Cochliobolus pathogens of different hosts would identify critical response differences. There is, however, no simple correlation. Genes encoding heterotrimeric G protein subunits, MAP kinases, and histidine kinase response regulators have been studied in C. heterostrophus since the 1990s and these studies are now facilitated by the genome projects (Horwitz et al. 1999; Lev and Horwitz 2003; Lev et al. 2009; Oide et al.; Degani et al. 2004). Two signaling pathways (MAP kinase and heterotrimeric G protein ) are shown schematically in Fig. 2.10. As in other pathogens, the C. heterostrophus core signaling proteins could be considered virulence factors because mutants are unsuccessful pathogens, but additional developmental alterations, obfuscate how exactly, signaling impacts virulence.

Illustration of two conserved signaling pathways, with corresponding gene models from C. heterostrophus. The following genes have been studied by constructing deletion mutants: MAP kinases ChHK1, MPS1, and HOG1; G protein Gα subunit CGA1, and G protein Gβ subunit CGB1. The gene models identified by reciprocal BLASTP search and/or other methods (see text) are indicated as C. heterostrophus strain C5 v2.0 protein ID numbers. Two examples of signaling pathways are shown: above, MAP kinase cascade; the vertical dashed lines indicate a tentative association into MAPK modules, by homology (there is no functional information to support this, as yet) below, a model of heterotrimeric G protein signaling in which activation of adenylyl cyclase produces cAMP, which activates protein kinase A (PKA). Heterotrimeric G protein signaling could lower or raise cAMP levels
One way that host specificity could be attained is for cell surface receptors in each species to recognize host-specific ligands, which then transmit the signal via a conserved intracellular cascade. C. heterostrophus, for example, has 21 genes predicted to encode histidine kinase sensors but only four downstream response regulators (Catlett et al. 2003b; Oide et al. 2010). The two-component pathways initiated by histidine kinase sensors in C. heterostrophus have central functions in morphogenesis, stress response, and virulence (Oide et al. 2010). Comparing histidine kinase sensors suites across Cochliobolus pathogens may reveal nuances not readily apparent when core signaling components are compared.
For heterotrimeric G protein pathways, the capacity for signals from multiple receptors to converge on a few downstream transducers may be even greater than for the two-component pathways. G protein-coupled receptors (GPCRs) are more difficult to identify bioinformatically, than the highly conserved signal transducers, but methods are improving (Xue et al. 2008; Lafon et al. 2006; Omann et al. 2012; Kim et al. 2012). To estimate the number of GPCRs in C. heterostrophus, an initial analysis was done as part of the annotation effort (Horwitz lab unpublished; Ohm et al. 2012): filtered protein models were searched with an HMM tool designed to identify GPCRs (Wistrand et al. 2006), then those with seven transmembrane segments as predicted by PHOBIUS (Kall et al. 2007) selected. An initial phylogenetic tree was constructed. The candidate sequences were used to query the NCBI database to identify those with convincing homology to transporters, or having a conserved domain indicating that they may be transporters. These sequences, as well as sequences falling on branches of the initial phylogeny together with annotated transporters or ATPases, were removed and the phylogeny was then recalculated. The analysis indicates orthologs of pheromone receptors Ste3 (1203184) and Ste2 (1215526), whose function in mating could be tested by gene deletion experiments. 20 candidates group with sequences annotated as related to the CFEM/Pth11 family in other fungi. Of these 20 candidates, three contain CFEM domains detected by Pfam, and in addition, have similarity with annotated CFEM-containing sequences. Three sequences show similarity to annotated Pth11-like sequences. These classes are proposed to be involved in pathogenicity (DeZwaan et al. 1999; Kulkarni et al. 2003). It would be of interest to compare these among Cochliobolus species for species-specific associations and, where possible, to test their function by gene deletion. This analysis provided no obvious orthologs of fungal opsins, even though two candidates were recognized previously by homology (C5 protein IDs 1195154 and 1139038, Oide and Turgeon unpublished). No members of the GPCR classes represented by Neurospora Gpr1-1, Gpr-5, and Gpr-4 (see Xue et al. 2008) were identified either, supporting our statement above that GPCRs are difficult to extract bioinformatically.
Light regulation. Not all signals are transduced from the cell surface to the nucleus by G protein and protein kinase pathways. In particular, dedicated fungal transcription factors relay information about light, pH, oxidants, and hypoxia. Once activated by the primary stimulus, these transcription factors may rely on additional regulators in order to produce the physiological output. The Neurospora circadian clock is a good example: the stress-activated MAPK (Hog1, Fig. 2.10) is activated rhythmically by the circadian oscillator (Vitalini et al. 2007). Circadian rhythmicity has not been studied in detail in any Cochliobolus species, but C. heterostrophus shows a clear banding pattern when grown under light/dark (L/D) cycles (Wu et al. 2012). A C. heterostrophus mutant lacking the ortholog of N. crassa WC1 shows defective banding with a weaker, residual banding pattern suggesting that additional photoreceptors are active. Initial evidence that the circadian clock controls Hog1 phosphorylation via the response regulator Ssk1 (N. crassa RRG-1) in C. heterostrophus comes from our finding that the L/D banding pattern is defective in hog1 and ssk1 mutants (Oide et al. 2010), similar to that of wc1 mutants (Turgeon and Horwitz labs unpublished).
Light regulation is of particular interest because it couples environmental sensing and secondary metabolism. C. heterostrophus mutants lacking key components of the velvet complex (VEL1 or LAE1), which controls reproduction and secondary metabolism produce much less T-toxin than WT in the dark (Wu et al. 2012). Light conditions could be particularly relevant because light, as the source of energy, is a critical environmental factor for the host plant. Light could synchronize gene expression of the pathogen to match the host; thus, photocontrol of plant–pathogen interactions has received recent attention (Kim et al. 2011a, b; Lee et al. 2006a, b).
Redox signals. As noted above, ChAp1, an ortholog of yeast Yap1, senses oxidants in C. heterostrophus. Yap1 homolog-mediated oxidative stress tolerance is crucial for pathogenicity of the necrotrophic fungus Alternaria alternata on citrus (Lin et al. 2009; Kim et al. 2009) and M. oryzae on rice (Guo et al. 2011). C. heterostrophus Chap1 mutants, although hypersensitive to oxidants, retain wild-type (Lev et al. 2005) or moderately reduced (Zhang et al. submitted) virulence on maize. The Botrytis cinerea Yap1 ortholog is required to resist peroxide stress in vitro, yet, Yap1 is not a virulence factor on bean, Arabidopsis, apple or tomato fruits, and its target genes are not induced on bean although H2O2 was detected (Temme and Tudzynski 2009).
There is strong genetic evidence for the involvement of multiple pathways in sensing oxidants. Loss of C. heterostrophus Hog1 (Fig. 2.10), its upstream response regulator Ssk1, or the response regulator Skn7, all result in hypersensitivity to oxidants (Oide et al. 2010). Although the oxidative burst is considered key to plant defense, it is worth noting that the ability to cope with hypoxic stress is important for pathogens of animals. Neutrophils in the mammalian immune system produce ROS, yet Aspergillus fumigatus needs the hypoxic stress response for virulence (Blatzer et al. 2011; Grahl and Cramer 2010; Willger et al. 2012). Hypoxic stress has received less attention in plant pathogens.
Signaling pathways cannot be studied in isolation and the study of oxidants provides a good example. Loss of HOG1, ChAP1, SKN7 as well as the NRPS responsible for extracellular siderophore production (Fig. 2.7), all diminish the ability of the pathogen to resist oxidative stress. Light signals are directly detected by the white collar transcription factors, but once again, the pathway is not a linear one because the global regulators Vel1 and Lae1 also participate. The light-sensing complex could be similar to that of other fungi but the details, and the genes regulated, likely hold surprises specific to Cochliobolus.
2.3 Applications from the Genome and Future Perspectives
The genomic resources available for comparative studies across the Cochliobolus genus are legion, thanks to the generous contributions of the JGI, with sequences and resources available for many different Cochliobolus species (and, for many species, multiple strains). The long history of using Cochliobolus species as model organisms allows an exciting marriage of functional work and in silico comparative genomics. As discussed in the Introduction, Cochliobolus taxa vary in their biology, host specificity, and developmental pathways. Whole-genome comparisons were startling vis-a-vis the incredible homology between most Cochliobolus species. Attempts to characterize “species-unique” sequence, found in all five C. heterostrophus strains, but no other Cochliobolus species, did not result in identification of large C. heterostrophus unique regions, but smaller differences. Uncovering the core identity of each species as it relates to their biology, therefore, is not as easy as identifying and characterizing a large obvious patch of genome. To address questions of differential biology, more refined approaches are necessary, facilitated by the history of molecular-genetic work for each species.
Molecular investigations into virulence factors have run the gamut from discovery of highly specific HSTs, to more general mechanisms involving iron and oxidative stress. Initially, secondary metabolism as a source of HSTs was considered the most compelling type of functional investigation for Cochliobolus pathogens (Lee et al. 2005; Turgeon et al. 2008). Extensive bioinformatic analyses of secondary metabolism genes occupy a large share of this chapter and these studies coupled with experimental research support our reasoning in this regard. The observation that phenotypes associated with secondary metabolism gene mutants follow phylogenetic distribution signatures provides a strong hypothesis and platform for further work. Conserved secondary metabolite clusters are likely to biosynthesize metabolites that broker basic cellular metabolism (iron gathering, oxidative stress management, etc.), while discontinuous and severely restricted gene distribution suggests niche-specific/virulence-specific function.
The second major aim of our comparative genomics study was to consider the role small secreted proteins may play in Cochliobolus species. Unlike secondary metabolites, this was not done against the backdrop of years of genetic characterization, but rather in the broader context of plant–microbe interactions. It has long been understood that biotrophic pathogens secrete effectors, which are often small and cysteine-rich proteins that elegantly subvert host defenses and prevent cell death. The traditional necrotroph, on the other hand, was thought to use a combination of toxins (including HSTs) and “brute force” methods (cell wall-degrading cellulases, pectinases) to overpower hosts. Recent work suggests that many necrotrophic virulence factors should truly be classified as effectors . An example of this is C. victoriae’s HST, victorin, which in the presence of an NB-LRR-type protein results in host susceptibility, instead of resistance. In light of these and similar observations, the obvious question is to what extent do necrotrophs utilize effectors and do they employ small secreted proteins, as biotrophs do? Cochliobolus is a wonderful system to ask these questions, as it contains both hemibiotrophic and necrotrophic pathogens. Bioinformatic searches found SSPs in all species examined, although the number of predicted SSPs, and species-unique SSPs, was higher in hemibiotrophic species. The set of SSPs identified serves as a toehold for identifying candidate SSP effectors in necrotrophs and concomitant functional analysis has the capacity to greatly alter our perception of such pathogens.
We have also sought to discuss the functional work conducted in different Cochliobolus species on other topics pertinent to pathogenic and reproductive development, including iron metabolism, oxidative stress management, P450s, signaling components, and mating determinants. Extrapolation of rich molecular work within a single species, such as C. heterostrophus, to other closely related species, results in new hypotheses. Genomic differences among species can unearth biological phenomena that might go unnoticed examining one system alone. Each analysis, of course, must be taken with a digital grain of salt, until functional work can support a given hypothesis. Comparative bioinformatics offers us a tentative and highly valuable glimpse into the inner workings of an entire genus.
References
Arie T, Christiansen SK, Yoder OC, Turgeon BG (1997) Efficient cloning of ascomycete mating type genes by PCR amplification of the conserved MAT HMG box. Fungal Genet Biol 21(1):118–130
Blatzer M, Barker BM, Willger SD, Beckmann N, Blosser SJ, Cornish EJ, Mazurie A, Grahl N, Haas H, Cramer RA (2011) SREBP coordinates iron and ergosterol homeostasis to mediate triazole drug and hypoxia responses in the human fungal pathogen Aspergillus fumigatus. PLoS Genet 7(12):e1002374
Bushley KE, Turgeon BG (2010) Phylogenomics reveals subfamilies of fungal nonribosomal peptide synthetases and their evolutionary relationships. BMC Evol Biol 10:26
Catlett N, Lee B-N, Yoder O, Turgeon B (2003a) Split-marker recombination for efficient targeted deletion of fungal genes. Fungal Genet Newsl 50:9–11
Catlett NL, Yoder OC, Turgeon BG (2003b) Whole-genome analysis of two-component signal transduction genes in fungal pathogens. Eukaryot Cell 2(6):1151–1161
Christiansen SK, Wirsel S, Yoder OC, Turgeon BG (1998) The two Cochliobolus mating type genes are conserved among species but one of them is missing in C. victoriae. Mycol Res 102:919–929
Ciuffetti LM, Tuori RP, Gaventa JM (1998) Cloning and expression of the ToxA gene in Pyrenophora triticirepentis. In: Kohmoto K, Yoder OC (eds) Molecular genetics of host-specific toxins in plant disease, vol 13. Kluwer, Dordrecht, pp 167–175
Condon BJ, Leng Y, Wu D, Bushley KE, Ohm RA, Otillar R, Martin J, Schackwitz W, Grimwood J, Mohdzainudin N, Xue C, Wang R, Manning VA, Dhillon B, Tu ZJ, Steffenson BJ, Salamov A, Sun H, Lowry S, Labutti K, Han J, Copeland A, Lindquist E, Barry K, Schmutz J, Baker SE, Ciuffetti LM, Grigoriev IV, Zhong S, Turgeon BG (2013) Comparative genome structure, secondary metabolite, and effector coding capacity across Cochliobolus pathogens. PLoS Genet 9(1):e1003233
Dasgupta MK (1984) The Bengal famine, 1943 and the brown spot of rice–an inquiry into their relations. Hist Agric 2(3):1–18
Debuchy R, Turgeon BG (2006) Mating-type structure, evolution and function in Euascomycetes. In: Kües U, Fischer R (eds) The Mycota, vol 1., Growth, Differentiation and SexualitySpringer, Berlin, pp 293–324
Degani O, Maor R, Hadar R, Sharon A, Horwitz BA (2004) Host physiology and pathogenic variation of Cochliobolus heterostrophus strains with mutations in the G protein alpha subunit, CGA1. Appl Environ Microbiol 70(8):5005–5009
DeZwaan TM, Carroll AM, Valent B, Sweigard JA (1999) Magnaporthe grisea Pth11p is a novel plasma membrane protein that mediates appressorium differentiation in response to inductive substrate cues. Plant Cell 11(10):2013–2030
Drechsler C (1925) Leafspot of maize caused by Ophiobolus heterostrophus n. sp., the ascigerous stage of a Helminthosporium exhibiting bipolar germination. J Agr Res 31:701–726
Fedorova ND, Khaldi N, Joardar VS, Maiti R, Amedeo P, Anderson MJ, Crabtree J, Silva JC, Badger JH, Albarraq A, Angiuoli S, Bussey H, Bowyer P, Cotty PJ, Dyer PS, Egan A, Galens K, Fraser-Liggett CM, Haas BJ, Inman JM, Kent R, Lemieux S, Malavazi I, Orvis J, Roemer T, Ronning CM, Sundaram JP, Sutton G, Turner G, Venter JC, White OR, Whitty BR, Youngman P, Wolfe KH, Goldman GH, Wortman JR, Jiang B, Denning DW, Nierman WC (2008) Genomic islands in the pathogenic filamentous fungus Aspergillus fumigatus. PLoS Genet 4(4):e1000046
Fenton HJH (1894) The oxidation of tartaric acid in presence of iron. J Chem Soc Proc 10:157–158
Friesen TL, Faris JD, Solomon PS, Oliver RP (2008) Host-specific toxins: effectors of necrotrophic pathogenicity. Cell Microbiol 10(7):1421–1428
Goodwin SB, Ben M’Barek S, Dhillon B, Wittenberg AHJ, Crane CF, Hane JK, Foster AJ, Van der Lee TAJ, Grimwood J, Aerts A, Antoniw J, Bailey A, Bluhm B, Bowler J, Bristow J, van der Burgt A, Canto-Canche B, Churchill ACL, Conde-Ferraez L, Cools HJ, Coutinho PM, Csukai M, Dehal P, De Wit P, Donzelli B, van de Geest HC, Van Ham RCHJ, Hammond-Kosack KE, Henrissat B, Kilian A, Kobayashi AK, Koopmann E, Kourmpetis Y, Kuzniar A, Lindquist E, Lombard V, Maliepaard C, Martins N, Mehrabi R, Nap JPH, Ponomarenko A, Rudd JJ, Salamov A, Schmutz J, Schouten HJ, Shapiro H, Stergiopoulos I, Torriani SFF, Tu H, de Vries RP, Waalwijk C, Ware SB, Wiebenga A, Zwiers LH, Oliver RP, Grigoriev IV, Kema GHJ (2011) Finished genome of the fungal wheat pathogen Mycosphaerella graminicola reveals dispensome structure, chromosome plasticity, and stealth pathogenesis. PLoS Genetics 7(6):e1002070
Grahl N, Cramer RA Jr (2010) Regulation of hypoxia adaptation: an overlooked virulence attribute of pathogenic fungi? Med Mycol 48(1):1–15
Guo M, Chen Y, Du Y, Dong YH, Guo W, Zhai S, Zhang HF, Dong SM, Zhang ZG, Wang YC, Wang P, Zheng XB (2011) The bZIP transcription factor MoAP1 mediates the oxidative stress response and is critical for pathogenicity of the Rice Blast Fungus Magnaporthe oryzae. PLoS Pathogens 7(2):e1001302
Haas H (2003) Molecular genetics of fungal siderophore biosynthesis and uptake: the role of siderophores in iron uptake and storage. Appl Microbiol Biotechnol 62(4):316–330
Han JW, Ahn SH, Park SH, Wang SY, Bae GU, Seo DW, Kwon HK, Hong S, Lee HY, Lee YW, Lee HW (2000) Apicidin, a histone deacetylase inhibitor, inhibits proliferation of tumor cells via induction of p21(WAF1/Cip1) and gelsolin. Cancer Res 60(21):6068–6074
Hane JK, Rouxel T, Howlett BJ, Kema GH, Goodwin SB, Oliver RP (2011) A novel mode of chromosomal evolution peculiar to filamentous Ascomycete fungi. Genome Biol 12(5):R45
Horwitz BA, Sharon A, Lu SW, Ritter V, Sandrock TM, Yoder OC, Turgeon BG (1999) A G protein alpha subunit from Cochliobolus heterostrophus involved in mating and appressorium formation. Fungal Genet Biol 26(1):19–32
Inderbitzin P, Harkness J, Turgeon BG, Berbee ML (2005) Lateral transfer of mating system in Stemphylium. Proc Natl Acad Sci U S A 102(32):11390–11395
Jin JM, Lee S, Lee J, Baek SR, Kim JC, Yun SH, Park SY, Kang SC, Lee YW (2010) Functional characterization and manipulation of the apicidin biosynthetic pathway in Fusarium semitectum. Molec Microbiol 76(2):456–466
Johal GS, Briggs SP (1992) Reductase activity encoded by the HM1 disease resistance gene in maize. Science 258(5084):985–987
Kall L, Krogh A, Sonnhammer EL (2007) Advantages of combined transmembrane topology and signal peptide prediction–the Phobius web server. Nucl Acids Res 35(Web Server issue):W429–W432
Kelly DE, Kraševec N, Mullins J, Nelson DR (2009) The CYPome (cytochrome P450 complement) of Aspergillus nidulans. Fungal Genet Biol 46:S53–S61
Kim H, Ridenour JB, Dunkle LD, Bluhm BH (2011a) Regulation of pathogenesis by light in Cercospora zeae-maydis: An updated perspective. Plant Pathol J 27(2):103–109
Kim H, Wright SJ, Park G, Ouyang SQ, Krystofova S, Borkovich KA (2012) Roles for Receptors, pheromones, G proteins, and mating type genes during sexual reproduction in Neurospora crassa. Genetics 190(4):1389–1404
Kim KH, Willger SD, Park SW, Puttikamonkul S, Grahl N, Cho Y, Mukhopadhyay B, Cramer RA, Lawrence CB (2009) TmpL, a transmembrane protein required for intracellular redox homeostasis and virulence in a plant and an animal fungal pathogen. Plos Pathogens 5(11):e1000653
Kim S, Singh P, Park J, Park S, Friedman A, Zheng T, Lee YH, Lee K (2011b) Genetic and molecular characterization of a blue light photoreceptor MGWC-1 in Magnaportha oryzae. Fungal Genet Biol 48(4):400–407
Kodama M, Rose MS, Yang G, Yun SH, Yoder OC, Turgeon BG (1999) The translocation-associated Tox1 locus of Cochliobolus heterostrophus is two genetic elements on two different chromosomes. Genetics 151(2):585–596
Kroken S, Glass NL, Taylor JW, Yoder OC, Turgeon BG (2003) Phylogenomic analysis of type I polyketide synthase genes in pathogenic and saprobic ascomycetes. Proc Natl Acad Sci U S A 100(26):15670–15675
Kulkarni RD, Kelkar HS, Dean RA (2003) An eight-cysteine-containing CFEM domain unique to a group of fungal membrane proteins. Trends in Biochem Sci 28(3):118–121
Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL (2004) Versatile and open software for comparing large genomes. Genome Biol 5(2):R12
Lafon A, Han KH, Seo JA, Yu JH, d’Enfert C (2006) G-protein and cAMP-mediated signaling in aspergilli: a genomic perspective. Fungal Genet Biol 43(7):490–502
Leach J, Lang BR, Yoder OC (1982) Methods for selection of mutants and in vitro culture of Cochliobolus heterostrophus. J Gen Microbiol 128:1719–1729
Lee BN, Kroken S, Chou DYT, Robbertse B, Yoder OC, Turgeon BG (2005) Functional analysis of all nonribosomal peptide synthetases in Cochliobolus heterostrophus reveals a factor, NPS6, involved in virulence and resistance to oxidative stress. Eukaryot Cell 4(3):545–555
Lee K, Singh P, Chung W, Ash J, Kim T, Hang L, Park S (2006a) Disease-suppressing roles of light in pathogenic interactions between Magnaporthe oryzae-Oryza sativa. Phytopathology 96(6):S66–S66
Lee K, Singh P, Chung WC, Ash J, Kim TS, Hang L, Park S (2006b) Light regulation of asexual development in the rice blast fungus, Magnaporthe oryzae. Fungal Genet Biol 43(10):694–706
Lesuisse E, Labbe P (1994) Reductive iron assimilation in Saccharomyces cerevisiae. In: Winge DR, Winkelmann G (eds) In metal ions in fungi. Marcell Dekker, New York, pp 149–178
Lev S, Hadar R, Amedeo P, Baker SE, Yoder OC, Horwitz BA (2005) Activation of an AP1-like transcription factor of the maize pathogen Cochliobolus heterostrophus in response to oxidative stress and plant signals. Eukaryot Cell 4(2):443–454
Lev S, Horwitz BA (2003) A mitogen-activated protein kinase pathway modulates the expression of two cellulase genes in Cochliobolus heterostrophus during plant infection. Plant Cell 15(4):835–844
Lev S, Tal H, Rose MS, Horwitz BA (2009) Signaling by the pathogenicity-related MAP kinase of Cochliobolus heterostrophus correlates with its local accumulation rather than phosphorylation. Mol Plant Microbe Interact 22(9):1093–1103
Lin CH, Yang SL, Chung KR (2009) The YAP1 homolog-mediated oxidative stress tolerance is crucial for pathogenicity of the necrotrophic fungus Alternaria alternata in citrus. Mol Plant Microbe Interact 22(8):942–952
Litzenberger SC (1949) Nature of susceptibility to Helminthosporium victoriae and resistance to Puccinia coronata in Victoria oats. Phytopathology 39:300–318
Lorang J, Kidarsa T, Bradford CS, Gilbert B, Curtis M, Tzeng SC, Maier CS, Wolpert TJ (2012) Tricking the guard: exploiting plant defense for disease susceptibility. Science 338(6107):659–662
Lorang JM, Carkaci-Salli N, Wolpert TJ (2004) Identification and characterization of victorin sensitivity in Arabidopsis thaliana. Mol Plant Microbe Interact 17(6):577–582
Lorang JM, Sweat TA, Wolpert TJ (2007) Plant disease susceptibility conferred by a “resistance” gene. Proc Natl Acad Sci U S A 104(37):14861–14866
Lu SW, Yun SH, Lee T, Turgeon BG (2011) Altering sexual reproductive mode by interspecific exchange of MAT loci. Fungal Genet Biol 48(7):714–724
Manamgoda DS, Cai L, Bahkali AH, Chukeatirote E, Hyde KD (2011) Cochliobolus: an overview and current status of species. Fungal Divers 51(1):3–42
Manamgoda DS, Cai L, McKenzie EHC, Crous PW, Madrid H, Chukeatirote E, Shivas RG, Tan YP, Hyde KD (2012) A phylogenetic and taxonomic re-evaluation of the Bipolaris—Cochliobolus—Curvularia Complex. Fungal Divers 56(1):131–144
Manning VA, Pandelova I, Dhillon B, Wilhelm LJ, Goodwin SB, Berlin AM, Figueroa M, Freitag M, Hane JK, Henrissat B, Holman WH, Kodira CD, Martin J, Oliver RP, Robbertse B, Schackwitz W, Schwartz DC, Spatafora JW, Turgeon BG, Yandava C, Young S, Zhou S, Zeng Q, Grigoriev IV, Ma LJ, Ciuffetti LM (2013) Comparative genomics of a plant-pathogenic fungus, Pyrenophora tritici-repentis, reveals transduplication and the impact of repeat elements on pathogenicity and population divergence. G3 (Bethesda) 3(1):41–63
Mathre DE (1997) Compendium of barley diseases, 2nd edn. APS Press, St. Paul
McNeil J, Barrie FR, Buck WR, Demoulin V, Greuter W, Hawkworth DL, Herendeen PS, Knapp S, Marhold K, Prado J, Prud’homme Van Reine WF, Smith GF, Wiersema JH, Turland NJ (2012) International code of nomenclature for algae, fungi, and plants (Melbourne Code). Regnum Vegetabile 154:232
Multani DS, Meeley RB, Paterson AH, Gray J, Briggs SP, Johal GS (1998) Plant-pathogen microevolution: molecular basis for the origin of a fungal disease in maize. Proc Natl Acad Sci U S A 95(4):1686–1691
Neilands JB, Leong SA (1986) Siderophores in relation to plant growth and disease. Annual Rev Plant Physiol Plant Mol Biol 37:187–208
Neubauer U, Nowack B, Furrer G, Schulin R (2000) Heavy metal sorption on clay minerals affected by the siderophore Desferrioxamine B. Environ Sci Technol 34(13):2749–2755
Newsome AW, Nelson D, Corran A, Kelly SL, Kelly DE (2013) The cytochrome P450 complement (CYPome) of Mycosphaerella graminicola. Biotechnol Appl Biochem 60(1):52–64
Ohm RA, Feau N, Henrissat B, Schoch CL, Horwitz BA, Barry KW, Condon BJ, Copeland AC, Dhillon B, Glaser F, Hesse CN, Kosti I, Labutti K, Lindquist EA, Lucas S, Salamov AA, Bradshaw RE, Ciuffetti L, Hamelin RC, Kema GH, Lawrence C, Scott JA, Spatafora JW, Turgeon BG, de Wit PJ, Zhong S, Goodwin SB, Grigoriev IV (2012) Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen dothideomycetes fungi. PLoS Pathog 8(12):e1003037
Oide S, Krasnoff SB, Gibson DM, Turgeon BG (2007) Intracellular siderophores are essential for ascomycete sexual development in heterothallic Cochliobolus heterostrophus and homothallic Gibberella zeae. Eukaryot Cell 6:1337–1353
Oide S, Liu J, Yun SH, Wu D, Michev A, Choi MY, Horwitz BA, Turgeon BG (2010) Histidine kinase two-component response regulator proteins regulate reproductive development, virulence, and stress responses of the fungal cereal pathogens Cochliobolus heterostrophus and Gibberella zeae. Eukaryot Cell 9(12):1867–1880
Oide S, Moeder W, Krasnoff S, Gibson D, Haas H, Yoshioka K, Turgeon BG (2006) NPS6, encoding a nonribosomal peptide synthetase involved in siderophore-mediated iron metabolism, is a conserved virulence determinant of plant pathogenic ascomycetes. Plant Cell 18(10):2836–2853
Omann MR, Lehner S, Escobar Rodriguez C, Brunner K, Zeilinger S (2012) The seven-transmembrane receptor Gpr1 governs processes relevant for the antagonistic interaction of Trichoderma atroviride with its host. Microbiology 158(Pt 1):107–118
Ransom RF, Walton JD (1997) Histone hyperacetylation in maize in response to treatment with HC-toxin or infection by the filamentous fungus Cochliobolus carbonum. Plant Physiol 115(3):1021–1027
Rouxel T, Grandaubert J, Hane JK, Hoede C, van de Wouw AP, Couloux A, Dominguez V, Anthouard V, Bally P, Bourras S, Cozijnsen AJ, Ciuffetti LM, Degrave A, Dilmaghani A, Duret L, Fudal I, Goodwin SB, Gout L, Glaser N, Linglin J, Kema GH, Lapalu N, Lawrence CB, May K, Meyer M, Ollivier B, Poulain J, Schoch CL, Simon A, Spatafora JW, Stachowiak A, Turgeon BG, Tyler BM, Vincent D, Weissenbach J, Amselem J, Quesneville H, Oliver RP, Wincker P, Balesdent MH, Howlett BJ (2011) Effector diversification within compartments of the Leptosphaeria maculans genome affected by repeat-induced point mutations. Nat Commun 2:202
Rozman D, Hennebert GL, Kunej T, Decock C, Komel R (1996) Steroid biotransforming strains designated Cochliobolus lunatus m118 and Curvularia lunata AT46 are both Curvularia lunata var. lunata. Mycotaxon 59:489–498
Scheffer RP, Nelson RR, Ullstrup AJ (1967) Inheritance of toxin production and pathogenicity in Cochliobolus carbonum and Cochliobolus victoriae. Phytopathology 57:1288–1291
Schrettl M, Bignell E, Kragl C, Joechl C, Rogers T, Arst HN Jr, Haynes K, Haas H (2004) Siderophore biosynthesis but not reductive iron assimilation is essential for Aspergillus fumigatus virulence. J Exp Med 200(9):1213–1219
Sharon A, Yamaguchi K, Christiansen S, Horwitz BA, Yoder OC, Turgeon BG (1996) An asexual fungus has the potential for sexual development. Mol Gen Genet 251(1):60–68
Sivanesan A (1987) Graminicolous species of Bipolaris, Curvularia, Drechslera, exserohilum and their teleomorphs. C. A. B. International, Wallingford
Temme N, Tudzynski P (2009) Does Botrytis cinerea ignore H2O2-induced oxidative stress during infection? Characterization of Botrytis Activator Protein 1. Mol Plant Microbe Interact 22(8):987–998
Thakur RP, Reddy BVS, Indira S, Rao VP, Navi SS, Yang XB, Ramesh S (2006) Sorghum grain mold. Inf Bull 72:1–28
Turgeon B, Debuchy R (eds) (2007) Cochliobolus and Podospora: mechanisms of sex determination and the evolution of reproductive lifestyle. Sex in fungi: molecular determination and evolutionary implications. ASM, Washington, DC
Turgeon BG, Baker SE (2007) Genetic and genomic dissection of the Cochliobolus heterostrophus Tox1 locus controlling biosynthesis of the polyketide virulence factor T-toxin. Adv Genet 57:219–261
Turgeon BG, Bohlmann H, Ciuffetti LM, Christiansen SK, Yang G, Schafer W, Yoder OC (1993) Cloning and analysis of the mating type genes from Cochliobolus heterostrophus. Mol Gen Genet 238(1–2):270–284
Turgeon BG, Condon B, Liu J, Zhang N (2010) Protoplast transformation of filamentous fungi. Methods Mol Biol 638:3–19
Turgeon BG, Lu S-W (2000) Evolution of host specific virulence in Cochliobolus heterostrophus. In: Kronstad JW (ed) Fungal pathology. Kluwer, Dordrecht, pp 93–126
Turgeon BG, Oide S, Bushley K (2008) Creating and screening Cochliobolus heterostrophus non-ribosomal peptide synthetase mutants. MycologRes 112:200–206
Tzeng TH, Lyngholm LK, Ford CF, Bronson CR (1992) A restriction fragment length polymorphism map and electrophoretic karyotype of the fungal maize pathogen Cochliobolus heterostrophus. Genetics 130(1):81–96
Ullstrup AJ (1970) History of southern corn leaf blight. Plant Dis Reptr 54:1100–1102
Valjavec-Gratian M, Steffenson B (1997) Pathotypes of Cochliobolus sativus on barley. Plant Dis 81:1275–1278
Valjavec Gratian M, Steffenson BJ (1997) Genetics of virulence in Cochliobolus sativus and resistance in barley. Phytopathology 87(11):1140–1143
Vitalini MW, de Paula RM, Goldsmith CS, Jones CA, Borkovich KA, Bell-Pedersen D (2007) Circadian rhythmicity mediated by temporal regulation of the activity of p38 MAPK. Proc Natl Acad Sci U S A 104(46):18223–18228
Vitas M, Rozman D, Komel R, Kelly SL (1995) P450-mediated progesterone hydroxylation in Cochliobolus lunatus. J Biotechnol 42(2):145–150
Vitas M, Smith K, Rozman D, Komel R (1994) Progesterone metabolism by the filamentous fungus Cochliobolus lunatus. J Steroid Biochem Molec Biol 49(1):87–92
Walton JD (1987) Two enzymes involved in biosynthesis of the host-selective phytotoxin HC-toxin. Proc Natl Acad Sci 84:8444–8447
Walton JD (1996) Host-selective toxins: agents of compatibility. Plant Cell 8(10):1723–1733
Walton JD (2006) HC-toxin. Phytochemistry 67(14):1406–1413
Weise MV (1987) Compendium of wheat diseases, 2nd edn. APS Press, St. Paul
Willger SD, Cornish EJ, Chung D, Fleming BA, Lehmann MM, Puttikamonkul S, Cramer RA (2012) Dsc orthologs are required for hypoxia adaptation, triazole drug responses, and fungal virulence in Aspergillus fumigatus. Eukaryot Cell 11(12):1557–1567
Winkelmann G (1991) Importance of siderophores in fungal growth, sporulation and spore germination. In: Hawksworth DL (ed) Frontiers in mycology. C. A. B. International, Wallingford, pp 49–65
Wirsel S, Turgeon BG, Yoder OC (1996) Deletion of the Cochliobolus heterostrophus mating type (MAT) locus promotes function of MAT transgenes. Curr Genet 29(3):241–249
Wistrand M, Kall L, Sonnhammer ELL (2006) A general model of G protein-coupled receptor sequences and its application to detect remote homologs. Protein Sci 15(3):509–521
Wolpert T, Shiraishi T, Collmer A, Akimitsu K, Glazebrook J (eds) (2011) Cochliobolus heterostrophus and maize: a model for genome-wide integration of iron homeostasis, oxidative stress management, and virulence. Genome-enabled analysis of plant-pathogen interactions. The American Phytopathological Society, St. Paul
Wu DL, Oide S, Zhang N, Choi MY, Turgeon BG (2012) ChLae1 and ChVel1 regulate T-toxin production, virulence, oxidative stress response, and development of the maize pathogen Cochliobolus heterostrophus. PLoS Pathogens 8(2):e1002542
Xue C, Hsueh YP, Heitman J (2008) Magnificent seven: roles of G protein-coupled receptors in extracellular sensing in fungi. FEMS Microbiol Rev 32(6):1010–1032
Yoder OC (1980) Toxins in pathogenesis. Ann Rev Phytopathol 18:103–129
Yoder OC (1988) Cochliobolus heterostrophus, cause of southern corn leaf blight. In: Sidhu GS (ed) Genetics of plant pathogenic fungi, vol 6. Academic Press, San Diego, pp 93–112
Yun SH, Berbee ML, Yoder OC, Turgeon BG (1999) Evolution of the fungal self-fertile reproductive life style from self-sterile ancestors. Proc Natl Acad Sci U S A 96(10):5592–5597
Zhong S, Steffenson BJ, Martinez JP, Ciuffetti LM (2002) A molecular genetic map and electrophoretic karyotype of the plant pathogenic fungus Cochliobolus sativus. Mol Plant Microbe Interact 15(5):481–492
Robbertse B, Yoder OC, Nguyen A, Schoch C, Turgeon BG (2003) Deletion of all monofunctional catalase-encoding genes of Cochliobolus heterostrophus enhances oxidative stress sensitivity but does not affect virulence. Molec Plant Micr Inter 16:1013–1021
Acknowledgments
We gratefully acknowledge the contribution of Igor Grigoriev and colleagues at the JGI for sequencing the genomes and for their consistent interest in Cochliobolus and the Dothideomycete class this fascinating genus belongs to. Work in our labs including some unpublished data shown here was supported by the Agriculture and Food Research Initiative of USDA’s National Institute of Food and Agriculture (BGT), the US-Israel Binational Agricultural Research and Development Fund (BARD) (BH, BGT), the National Science Foundation (BGT), and the Slovenian Research Agency (NK).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Condon, B.J., Wu, D., Kraševec, N., Horwitz, B.A., Turgeon, B.G. (2014). Comparative Genomics of Cochliobolus Phytopathogens. In: Dean, R., Lichens-Park, A., Kole, C. (eds) Genomics of Plant-Associated Fungi: Monocot Pathogens. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-44053-7_2
Download citation
DOI: https://doi.org/10.1007/978-3-662-44053-7_2
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-44052-0
Online ISBN: 978-3-662-44053-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)