
Abstract
Cells of the yeast Saccharomyces cerevisiae have an exquisite preference for high concentrations of glucose compared to other sugars or carbon sources. The likely explanation is that glucose is the best fermentable sugar, i.e., the sugar that allows the yeast to accumulate most rapidly high levels of ethanol, which are strongly inhibitory to competing microorganisms. To accomplish rapid fermentation of glucose, S. cerevisiae has evolved multiple glucose sensing and signaling pathways, which stimulate both fermentation and rapid cell proliferation. The latter is important for rapid fermentation in order to recycle the ATP generated in glycolysis to ADP. Downregulation of respiration to maximize ethanol production is accomplished by the main glucose repression pathway, in which the Snf1 protein kinase is a central regulator. It is inactivated by dephosphorylation upon glucose addition, and its reactivation upon glucose exhaustion is essential for induction of genes sustaining respiration, gluconeogenesis, and the catabolism of alternative carbon sources. Stimulation of fermentation and growth is mainly exerted by the protein kinase A pathway, which senses glucose with an extracellular and intracellular sensing mechanism that activates protein kinase A in a concerted manner through stimulation of cAMP synthesis. Sensing of other nutrients by plasma membrane transceptors integrates with this glucose-sensing mechanism to maintain high protein kinase A activity throughout fermentative growth. Induction of appropriate glucose transporters during fermentative growth is controlled by plasma membrane transporter-like proteins, which function as glucose sensors. Although detailed knowledge has been gained on the molecular mechanisms involved in glucose signaling, multiple important questions still remain.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
2.1 Introduction
Glucose is the preferred source of energy and building blocks for the yeast Saccharomyces cerevisiae. It is mainly metabolized by fermentation and also sustains the fastest growth rate in spite of producing much less ATP per mole glucose than respiration. Other sources of carbon and energy, like glycerol, ethanol, and acetate, are respired and sustain much slower growth rates. Some sugars, like galactose, are slowly fermented and partially respired. The preference of S. cerevisiae for glucose and related rapidly fermented sugars, like fructose and mannose, is manifested by the multiple regulatory pathways triggered by these sugars, which all have as main goal to stimulate both fermentation and cell proliferation (Rolland et al. 2001, 2002; Santangelo 2006). Regulation occurs at different levels: allosteric, post-translational, and transcriptional. We can distinguish different regulatory pathways, which have been elucidated in great detail during many years of focussed research. These pathways are connected to each other and to signaling pathways for other nutrients, but at this moment we understand much less about these interconnections than about the components and regulation within the pathways.
2.2 The Snf3/Rgt2 Glucose Sensors for Induction of HXT Glucose Transporter Expression
Glucose is transported into yeast cells by an extensive set of glucose transporters, which function as facilitated diffusion carriers and are encoded by the HXT genes (Ozcan and Johnston 1999; Boles and Hollenberg 1997; Bisson et al. 1993). These carriers have different affinities and catalytic activities, and their expression is adjusted according to the glucose concentration in the medium (Ozcan and Johnston 1995). The Snf3-Rgt2 regulatory pathway plays a major role in this control (Fig. 2.1). It was a breakthrough in the glucose-sensing field when two plasma membrane proteins were discovered with high sequence similarity to glucose carriers, which were unable to transport glucose and instead functioned as glucose sensors (Ozcan et al. 1996; Bisson et al. 1987). Snf3 has a high affinity while Rgt2 has a low affinity for extracellular glucose. Snf3 is required for expression of HXT2 and HXT4 in the presence of low levels of glucose, but not for induction of HXT1 by high glucose levels (Bisson et al. 1987; Ozcan and Johnston 1999). In contrast, Rgt2 seemed to exert the opposite effect by playing a vital role in the induction of HXT1 expression by high levels of glucose (Ozcan et al. 1996; Ozcan and Johnston 1999).

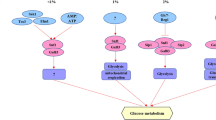
The Rgt network. When glucose binds to the glucose receptors Snf3 and Rgt2, the corepressors Std1 and Mth1 are recruited to the plasma membrane to be phosphorylated by the Yck kinases. SCFGrr1 targets phosphorylated Std1 and Mth1 to the ubiquitin conjugating complex for degradation by the proteasome. Rgt1 becomes hyperphosphorylated by PKA when glucose is present and is in this form not able to repress the HXT and HXK2 genes. When glucose becomes limited or is absent, Rgt1 becomes phosphorylated directly or indirectly by Snf1 and is recruited to the nucleus together with Mth1 and Std1. Together with the transcriptional corepressor complex Ssn6-Tup1, the Rgt1/Mth1/Std1 complex represses the transcription of the HXT and HXK2 genes
Both Snf3 and Rgt2 consist of two functional parts, a transmembrane-spanning part, which binds glucose, and a large cytosolic extension that is involved in triggering an intracellular signal to the downstream machinery (Fig. 2.1) (Marshall-Carlson et al. 1990; Moriya and Johnston 2004). The precise mechanism of how the two glucose sensors generate the glucose signal and transduce it to the intracellular machinery is not fully understood. It appears to include the phosphorylation of two signal transduction proteins, Mth1 and Std1, via Yck kinases (Robinson et al. 1992; Moriya and Johnston 2004; Babu et al. 2002; Ozcan and Johnston 1999). Yck1 and its paralog Yck2 are anchored in the plasma membrane via palmitate chains. They are activated through interaction with Snf3 and Rgt2, when these sense glucose (Babu et al. 2002; Johnston and Kim 2005; Moriya and Johnston 2004). The large C-terminal domain of the glucose sensor, Snf3 or Rgt2, facilitates interaction with the Yck kinases as well as their substrates, Mth1 and Std1 (Coons et al. 1997; Dlugai et al. 2001; Moriya and Johnston 2004). Once activated, Yck1 and Yck2 inactivate Std1 and Mth1 by phosphorylation. The latter two proteins function as inhibitors of glucose-induced HXT gene expression in the absence of glucose (Johnston and Kim 2005; Moriya and Johnston 2004). The phosphorylation of Std1 and Mth1 will cause their ubiquitination and subsequent degradation by the proteasome, which occurs through the SCF-Grr1 complex. The F-box protein Grr1 has two protein interaction domains that are essential for its function. The F-box motif interacts with Skp1, a subunit of the SCF complex. It is preceded by a leucine rich repeat domain that is necessary for substrate recruitment (Kishi et al. 1998).
Grr1 is required for glucose regulation of the transcription factor Rgt1 (Ozcan and Johnston 1999). Rgt1 exerts a repressor role on glucose-induced genes and recruits the transcriptional co-repressor complex Ssn6-Tup1. Together with Rgt1, it will condense chromatin into a repressive conformation (Edmondson et al. 1996). This process occurs at the promoters of the HXT genes and causes repression of their transcription in the absence of glucose. Transcriptional repression by Rgt1 also requires Mth1, which causes a conformational change that allows Rgt1 to bind to its recognition sites in DNA (Polish et al. 2005). In addition, the presence of Std1 is also required although it does not regulate Rgt1 binding capacity (Lakshmanan et al. 2003). Under conditions of high glucose, Rgt1 is hyperphosphorylated and this process requires Snf3 and Rgt2 as glucose sensors. It converts Rgt1 from a repressor into an activator although the latter function does not act through direct DNA binding (Kim et al. 2003; Mosley et al. 2003).
The Snf3-Rgt2 signaling pathway is connected to other glucose signaling pathways. It was discovered that Rgt1 can act as a repressor of HXK2 expression. Hxk2 is the most active hexokinase isoenzyme and is required for glucose repression through the main glucose repression pathway, also called “catabolite repression pathway” (Palomino et al. 2005). The Snf1 protein kinase, a central component of the main glucose repression pathway, seems to be involved in this process possibly through phosphorylation of Rgt1, which is essential for HXK2 repression (Palomino et al. 2006). The latter study also showed that Tpk3 relieves HXK2 repression by hyperphosphorylation of Rgt1. Tpk3 is one of the isoenzymes functioning as catalytic subunits of protein kinase A (PKA), which is the mediator of the cAMP glucose signaling pathway. Recent work has shown that Mth1 regulates the interaction between the Rgt1 repressor and the Ssn6-Tup1 co-repressor complex by modulating PKA-dependent phosphorylation of Rgt1 (Roy et al. 2013). Much more, however, has to be learned about the precise interactions of the Snf3-Rgt2 signaling pathway with other glucose signaling pathways.
2.3 Glucose Signaling Through the cAMP-PKA Pathway
2.3.1 Physiological Role of the cAMP-PKA Pathway in Nutrient Signaling
The PKA protein kinase affects a wide variety of processes in yeast, supporting its crucial role as a main cellular regulator. It is involved in control of metabolic pathways, like glycolysis and gluconeogenesis, in control of growth, proliferation, and aging of the cells, in reserve carbohydrate accumulation, stress tolerance, in developmental pathways, like pseudohyphal differentiation, invasive growth and sporulation, and multiple other pathways and processes (Santangelo 2006; Smets et al. 2010; Thevelein et al. 2000; Thevelein and de Winde 1999). The general role of PKA is stimulation of fermentative growth and inhibition of stationary-phase characteristics and other processes, like sporulation, which depend on respiration. Investigation of PKA targets in different growth conditions has revealed a striking correlation with the nutrient composition of the medium. Conditions supporting rapid fermentative growth, i.e., the presence of glucose or another rapidly fermented sugar, and a complete growth medium, are always associated with a status of the PKA targets indicating high activity of PKA in vivo. On the other hand, conditions supporting slow, respiratory growth, i.e., the presence of a nonfermentable carbon source, like glycerol, ethanol or acetate, or stationary phase conditions after carbon source depletion, are always associated with a status of the PKA targets indicating low activity of PKA in vivo. Up to this point, this correlation suggests that glucose and other fermentable sugars are activators of PKA in vivo. This has led to the concept that PKA is part of a glucose signaling pathway, which has been confirmed by the discovery of a complex glucose-sensing network controlling the level of cAMP, the second messenger that controls the activity of PKA.
Subsequently, however, a novel level of PKA regulation has been discovered following the awareness that starvation of yeast cells on a glucose-containing medium for any other single essential nutrient downregulated the PKA targets in a manner consistent with the presence of low PKA activity in vivo (Thevelein et al. 2005; Thevelein and de Winde 1999). Hence, fermentable sugar was clearly not the sole determinant for high PKA activity. Observations linking PKA targets, like trehalose and glycogen content, to conditions of starvation for specific essential nutrients on a glucose-containing medium, were already made long before the glucose-sensing role of the PKA pathway had become clear (Lillie and Pringle 1980). Subsequently, the role of other nutrients in regulating PKA activity was clearly demonstrated by experiments showing rapid activation of PKA targets after addition of nitrogen sources, phosphate, and sulfate, to appropriately starved cells on a glucose-containing medium (Hirimburegama et al. 1992; Thevelein and Beullens 1985; Thevelein 1984a). Hence, this previous work has revealed that maintenance of high PKA activity in yeast cells requires the combination of a rapidly fermented sugar and a complete growth medium, which led to the concept of a “fermentable–growth-medium” (FGM)-induced pathway for activation of PKA in vivo.
Further research on the FGM pathway led to the discovery of multiple nutrient transporters/receptors or “transceptors,” acting as sensors for activation of PKA by the other nutrients, and apparently without using cAMP as a second messenger (Kriel et al. 2011; Thevelein and Voordeckers 2009; Thevelein et al. 2005, 2008; Holsbeeks et al. 2004). Hence, in the case of the PKA pathway, integration of glucose sensing with sensing of other nutrients is very well established although the mechanistic details of the integration are not well understood.
2.3.2 Glucose Activation of cAMP Synthesis and PKA
The dramatic effects of glucose addition to yeast cells on PKA targets, like trehalose and trehalase, led already in 1974 to the discovery of the “glucose-induced cAMP signal,” a conspicuous and drastic, but very transient spike in the cAMP level that happens in the first 1–3 min after addition of glucose to respiring yeast cells (either growing or stationary-phase cells) (van der Plaat 1974). This cAMP signal activates PKA, which then phosphorylates target proteins like trehalase. This enzyme shows a conspicuous and rapid increase in activity, as measured in cell extracts, following the cAMP signal. Initially, nonspecific mechanisms were evaluated as possible triggers for the glucose-induced cAMP signal (Mazon et al. 1982; Purwin et al. 1982; Thevelein 1984a; Tortora et al. 1982). The observation that intracellular acidification caused a strong and persistent accumulation of cAMP in yeast cells (Purwin et al. 1986; Thevelein 1991; Caspani et al. 1985), while glucose addition triggered a rapid, transient drop in the intracellular pH, led to the suggestion that the cAMP signal was caused by the glucose-induced transient intracellular acidification. This explanation as well as other suggestions of transient plasma membrane depolarization or increases in ATP, the substrate of adenylate cyclase, were contradicted by a variety of experimental approaches (Thevelein et al. 1987a, b).
Like previously established for mammalian PKA, yeast PKA is also a heterotetrameric protein consisting of two catalytic and two regulatory subunits. The catalytic subunits are encoded by TPK1, TPK2, and TPK3, while the regulatory subunits are encoded by BCY1 (Toda et al. 1987a, b). Binding of the second messenger cAMP to the regulatory subunit Bcy1 causes its dissociation from the Tpk1-3 catalytic subunits, resulting in activation of PKA (Kuret et al. 1988) (Fig. 2.2). The three catalytic subunits have redundant functions for some phenotypes and specific functions for other phenotypes. For instance, any TPK gene can sustain viability of the cells while the absence of all three is lethal (Thevelein and de Winde 1999). On the other hand, pseudohyphal growth induction is stimulated by Tpk2 but counteracted by Tpk3 (Robertson and Fink 1998), while mitochondrial biogenesis is specifically stimulated by Tpk3 (Chevtzoff et al. 2010).

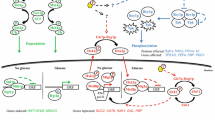
The cAMP-PKA pathway in S. cerevisiae. Glucose activates PKA via two different pathways. When glucose is transported and phosphorylated, it activates the Ras proteins by increasing their GTP/GDP loading state. The mechanism involved is not known. Active Ras will consequently activate Cyr1, the adenylate cyclase of yeast. Cyr1 catalyzes the synthesis of cAMP from ATP. This second messenger is able to bind to the regulatory subunit of PKA, Bcy1, thereby dissociating it from the catalytic subunits, Tpk1, Tpk2, and Tpk3. These are then able to phosphorylate downstream targets and regulate in this way protein activity and gene expression. Extracellular glucose can also activate PKA through the glucose-sensing G-protein-coupled receptor Gpr1. This receptor triggers activation of the G-protein Gpa2, of which the intrinsic GTPase activity is stimulated by Rgs2. Active Gpa2 in turn activates Cyr1 with the generation of cAMP as a consequence. Gpa2 can also inhibit the Krh proteins, thereby, activating PKA through the adenylate cyclase bypass pathway
Synthesis of the second messenger molecule cAMP from ATP is catalyzed by adenylate cyclase, which is encoded by CYR1/CDC35 (Kataoka et al. 1985; Matsumoto et al. 1982). The activity of adenylate cyclase is controlled in yeast by two distinct G-protein systems, the Ras1,2 proteins (Toda et al. 1985; Broek et al. 1985) and Gpa2, a homolog of the Gα subunit of the heterotrimeric G-proteins (Nakafuku et al. 1988; Lorenz and Heitman 1997; Kubler et al. 1997) (Fig. 2.2). This led to the discovery that these G-protein systems are involved in intracellular and extracellular glucose sensing, respectively (Rolland et al. 2000).
2.3.3 The Ras Proteins and Their Role in Intracellular Glucose Sensing
Discovery of the yeast Ras proteins The yeast Ras proteins were discovered based on sequence similarity with the mammalian RAS oncogenes (Kataoka et al. 1984; Powers et al. 1984; Tatchell et al. 1984). The purpose was to use yeast as a model system to identify the elusive physiological function of the mammalian RAS gene products. Deletion of both RAS genes in yeast was lethal because it caused cell cycle arrest in G1 and entrance into stationary phase, similar to cells starved for nutrients. A specific category of temperature-sensitive cell cycle mutants (cdc mutants), including the cdc35 mutant, also arrested at the restrictive temperature at the same point in the cell cycle (Hayles and Nurse 1986). This suggested that the function of these gene products was related to that of Ras. Cloning of CYR1/CDC35 revealed that it encodes adenylate cyclase (Kataoka et al. 1985; Matsumoto et al. 1982), and subsequent work showed that the yeast Ras proteins act as essential G-proteins for yeast adenylate cyclase (Toda et al. 1985). This work formed the basis for the further elucidation of the cAMP-PKA pathway in yeast, but it failed to deliver originally expected insight on two points. First, in mammalian cells, the Ras proteins do not act on adenylate cyclase (Beckner et al. 1985) and the yeast work therefore failed to help identify the mammalian Ras target. Second, in spite of many efforts no upstream activators of Cdc25 could be found, which would have pointed to the physiological signal being transmitted by the Ras proteins in yeast. Hence, the original goal of using yeast as a model system to understand the physiological function of the mammalian Ras proteins as signal transmission proteins and thus to shed light on their oncogenic mechanism was not fulfilled.
Ras and its regulatory proteins In spite of this, detailed analysis of the Ras proteins and their direct, physical regulators in yeast revealed strong conservation with the system in mammalian cells. The yeast Ras1 and Ras2 proteins share more than 70 % amino acid similarity and approximately 90 % similarity in their 180 N-terminal residues (Powers et al. 1984; Kataoka et al. 1984), and these 180 amino acids are also highly conserved in the human Ras proteins. The Ras proteins are monomeric GTPases whose activity depends on GDP/GTP exchange and GTP hydrolysis (Broach and Deschenes 1990). The activity of monomeric GTPases is displayed as a binary switch. When GTP is bound, the Ras proteins are activated and stimulate cAMP synthesis by activating Cyr1/adenylate cyclase (Matsumoto et al. 1982). Conversely, when Ras-bound GTP is hydrolysed to GDP by the intrinsic Ras GTPase activity, it switches to the inactivated state. Mammalian Ras oncogene products usually contain mutations that render the protein constitutively active, for instance by reducing the intrinsic GTPase activity. A major example is Rasval12, in which glycine12 is converted into a valine residue. The corresponding mutation was engineered into the yeast Ras2 protein, which resulted in the Ras2 val19 protein, which is also constitutively active in yeast (Broek et al. 1985). It causes higher cAMP levels and PKA activity, which is detrimental to the cells when they grow on non- or poorly fermentable carbon sources or enter into stationary phase. Originally, the failure of this mutant to arrest properly at the start site in the G1 phase of the cell cycle upon nitrogen starvation was ascribed to its oncogenic character, causing defective cell cycle control, but was later attributed to its inability to complete the cell cycle because of deficient internal amino acid stores (Markwardt et al. 1995).
Ras activity is modulated by stimulation of guanine nucleotide exchange and stimulation of the intrinsic GTPase activity (Fig. 2.2). Cloning of the CDC25 gene by complementation of another temperature-sensitive mutant that arrested at the restrictive temperature like nutrient-starved cells, showed that it encodes an essential guanine nucleotide exchange factor (GEF) of Ras (Broek et al. 1987; Camonis et al. 1986; Jones et al. 1991). Later work also identified a homolog of CDC25, SDC25, but this gene contains an inactivating nonsense mutation in the S288c background causing CDC25 to be essential (Boy-Marcotte et al. 1996; Damak et al. 1991). In the W303 lab strain, deletion of CDC25 is not lethal under growth conditions in which SDC25 is expressed (Folch-Mallol et al. 2004; Boy-Marcotte et al. 1996). These GEF proteins only bind and thereby stabilize the open nucleotide-free state of Ras (Lai et al. 1993; Haney and Broach 1994). Because the cytosolic concentration of GTP is higher than that of GDP in well-energized cells, nucleotide-free Ras will be loaded preferentially with GTP when it binds a new nucleotide, leading to activation of Ras. GTP enters Ras together with one molecule of Mg2+, which creates a GTP-Mg2+ complex that will close the Ras protein and stabilize its active conformation (Pai et al. 1990; Farnsworth and Feig 1991). The C-terminus of Cdc25 includes the catalytic domain and a membrane localization signal, while the N-terminus contains an SH3 domain that regulates Ras interaction with adenylate cyclase (Daniel 1986; Garreau et al. 1996; Mintzer and Field 1999). The C-terminus of Cdc25 shows very high sequence similarity with the human Ras GEF factor hSos1. The catalytic part of hSos1 is referred to as the Cdc25 domain (Boguski and McCormick 1993).
Inactivation of the Ras proteins occurs through their intrinsic GTPase activity. However, without aid this reaction is very slow, and therefore it is stimulated by two GTPase activating proteins (GAPs), Ira1 and Ira2 (Tanaka et al. 1990). These proteins stick an arginine finger into the catalytic site of Ras, which decreases the activation energy for hydrolysis of the γ-phosphate from GTP (Kotting et al. 2008). Ira1 and Ira2 are among the largest proteins present in yeast (3,093 and 3,080 amino acids, respectively) and, in addition to their GTPase activating function, they show further regulatory functions (Tanaka et al. 1990). Ira1, for instance, was found to interact with Cyr1 and seems to be necessary for its membrane localization (Mitts et al. 1991). Tfs1 was found to inhibit Ira2, but not Ira1 (Chautard et al. 2004). Deletion of Ira1 or Ira2 can suppress lethality caused by deletion of CDC25, just like the presence of a constitutively active allele of Ras. This is consistent with higher activity of Ras in ira1 and ira2 deletion strains (Tanaka et al. 1990).
The essential character of Cyr1/Cdc35/adenylate cyclase as well as its regulators Ras1 and Ras2, or Cdc25 and Sdc25, for cell viability in all tested genetic backgrounds, indicates that a critical concentration of cAMP is essential for cell growth in yeast and more specifically for progression over the START site in the G1 phase of the cell cycle and prevention of precocious entrance into the stationary phase G0 (Boy-Marcotte et al. 1998; Broach and Deschenes 1990; Ptacek et al. 2005; Smith et al. 1998; Thevelein 1994). Since nutrient starvation also prevents progression through G1 and forces cells into G0, this suggested that the Cdc25-Ras-adenylate cyclase system responds to nutrient availability (Thevelein 1994; Thevelein et al. 2000). The precise connection between glucose and cAMP, however, was not revealed in cell cycle studies but rather by research on glucose regulation of storage carbohydrate metabolism (Thevelein 1991; Thevelein and de Winde 1999). Whether there is a mechanistic connection between the availability of all the other nutrients, besides glucose and related rapidly fermented sugars, and cAMP synthesis remains unclear up to today. In this respect, it is important to realize that a critical level of PKA activity may be required for growth rather than a critical concentration of cAMP per se. In the presence of a basal level of cAMP, other regulators, such as the kelch repeat proteins Krh1 and Krh2, may modulate PKA activity (Peeters et al. 2007).
Another protein involved in activation of Cyr1/adenylate cyclase by Ras is Srv2 (Fedor-Chaiken et al. 1990; Field et al. 1990). It is bound to Cyr1 (and therefore also called CAP or cyclase-associated protein) and also binds to actin, which facilitates the interaction between Cyr1 and Ras. Its main task, however, appears to be in the regulation of the actin skeleton in yeast, although there is also evidence that modulation of the actin cytoskeleton can cause hyperactivation of Ras (Gourlay and Ayscough 2006).
Glucose activation of Ras and its role in glucose activation of cAMP synthesis Investigation of the glucose-induced cAMP signal in different mutants in yeast glycolysis revealed that glucose phosphorylation was essential for the glucose-induced cAMP signal (Beullens et al. 1988). This suggested that the trigger for this process was an intracellular event originating in intracellular glucose catabolism. Subsequently, evidence was provided that the Ras proteins were involved in mediating the glucose-induced cAMP signal, which indicated for the first time a connection between glucose sensing and Ras (Mbonyi et al. 1988). Combined with the previous finding, it suggested that Ras is activated by one or more factors generated in glucose catabolism. Other evidence for involvement of Ras in glucose-induced cAMP signaling has later been provided. Ras is anchored in the plasma membrane via palmitoylation and farnesylation of the two cysteine residues at positions 318 and 319, respectively. Membrane targeting of Ras is not required for maintenance of a basal level of cAMP and thus for sustaining viability, but is required for rapid glucose-induced cAMP signaling (Bhattacharya et al. 1995). Evidence for involvement of Cdc25 and especially its C-terminus in glucose-induced cAMP signaling was also reported, strengthening the evidence for a role of the Ras proteins as signal transducers in glucose-induced cAMP signaling (van Aelst et al. 1990, 1991). In the absence of glucose, Cdc25 is also located at the plasma membrane, and adenylate cyclase, although not an intrinsic membrane protein in yeast, also associates with the plasma membrane. This configuration of Cdc25, Ras, and adenylate cyclase at the plasma membrane appears to be important for rapid glucose-induced cAMP signaling and its loss may play a role in the rapid decrease of the cAMP level after the initial surge. The increase in cAMP activates PKA, which hyperphosphorylates Cdc25 resulting in its translocation to the cytosol and reduction of its ability to activate Ras (Gross et al. 1992; Dong and Bai 2011; Jian et al. 2010).
Direct measurement of the GTP/GDP loading state on Ras after addition of glucose, however, failed to reveal any increase in GTP, as opposed to intracellular acidification, which triggered a rapid and huge increase in Ras-GTP (Colombo et al. 1998). For technical reasons, these experiments required overexpression of Ras, and subsequent work, using a more sensitive assay for Ras-GTP based on the interaction of mammalian Ras with the Ras-binding domain of Raf, revealed that the overexpression of Ras, possibly through a feedback inhibition mechanism, prevented detection of the glucose-induced increase in the Ras-GTP level (Colombo et al. 2004). In the same work, it was shown that glucose activation of Ras requires glucose phosphorylation, again linking glucose catabolism with activation of Ras. How glucose catabolism causes activation of Ras is still not clear today.
The establishment of Ras activation by glucose catabolism in yeast brings us back to the original aim of the studies of Ras in yeast. The purpose was to understand the physiological role of the oncogenic Ras protein in mammalian cells with a goal of finding an explanation for its role in induction of cancer. The absence of the Ras—adenylate cyclase connection in mammalian cells (Beckner et al. 1985) suggested that yeast Ras had a different function compared to mammalian Ras and that yeast, therefore, was not a good model system to learn about Ras functionality, which made the interest in the yeast Ras system by mammalian researchers fade away. However, cancer cells and yeast cells present a striking similarity in the related so-called Warburg and Crabtree effects (Diaz-Ruiz et al. 2011). As opposed to other eukaryotic cells, cancer cells and (in the presence of a high concentration of fermentable sugar) yeast cells favor fermentation over respiration in the presence of oxygen and also show the most rapid proliferation when fermenting in spite of the fact that fermentation delivers much less ATP compared to respiration. Whether the high glycolytic flux in cancer cells is a consequence or a cause of the cancerous state has been a matter of much debate and is still not clear (Upadhyay et al. 2013). In this respect, the connection between glucose catabolism and activation of the oncogenic Ras protein in yeast might still serve as a valuable model system to understand the Warburg effect in cancer cells and to make a distinction between high fermentation activity as a consequence or a cause of cancer.
2.3.4 The Gpr1-Gpa2 GPCR System and Its Role in Extracellular Glucose Sensing
The observation that the Ras proteins were not activated after glucose addition in cells overexpressing Ras stimulated the search for an alternative G-protein involved in glucose-induced cAMP signaling. This led to the discovery of a G-protein-coupled receptor (GPCR) system that senses extracellular glucose and is dependent on the intracellular glucose-sensing system that activates Ras for stimulation of adenylate cyclase and cAMP signaling (Thevelein and de Winde 1999).
The GPCR system is composed of the receptor, Gpr1, and its Gα protein Gpa2 (Fig. 2.2). Gpr1 has the typical structure of a GPCR with seven transmembrane domains but little sequence similarity to other GPCR families (Kraakman et al. 1999; Xue et al. 1998; Yun et al. 1997). Together with its homologues in other fungi, it represents a separate subfamily in the large GPCR superfamily (Graul and Sadee 2001). Glucose and sucrose, but not fructose, mannose, galactose, or other sugars, act as ligands of the Gpr1 receptor, with sucrose having much higher affinity (±1 mM) compared to glucose (±20 mM) (Lemaire et al. 2004). The sugar specificity of Gpr1 indicates that fructose- and mannose-induced cAMP signaling are exclusively mediated by the intracellular sugar catabolism-dependent activation of Ras. The glucose sensitivity fits with the concentrations of glucose that cause full stimulation of fermentative growth in yeast, while the high sensitivity for sucrose suggests that detection of low sucrose concentrations may be important for survival in the natural habitat of yeast (Van de Velde and Thevelein 2008). Deletion of Gpr1 is not lethal and causes delayed activation of the cAMP-PKA signaling pathway upon addition of glucose (Kraakman et al. 1999). Whereas extracellular glucose signaling through the Gpr1–Gpa2 system is entirely dependent on intracellular activation of Ras by glucose catabolism, the opposite is not true, and therefore glucose still causes stimulation of the cAMP-PKA pathway in the absence of Gpr1 or Gpa2 (Rolland et al. 2000). A constitutively active allele of Ras2 also causes a stronger effect on gene expression controlled by the cAMP-PKA pathway compared to a constitutively active allele of Gpa2 (Wang et al. 2004) consistent with the Ras system having a more dominant effect on adenylate cyclase than the Gpr1–Gpa2 GPCR system. Gpr1 was discovered in two independent ways. The C-terminus of Gpr1 was isolated in two hybrid screens with Gpa2, and a mutant with delayed glucose-induced stimulation of PKA targets turned out to have a nonsense mutation in Gpr1 (Kraakman et al. 1999; Xue et al. 1998).
Gpa2 is a member of the Gα family of heterotrimeric G-proteins (Nakafuku et al. 1988; Kubler et al. 1997). It was the first member of this family that does not function in association with a classical Gβ and Gγ subunit (Peeters et al. 2007; Hoffman 2007). Deletion of Gpa2 is not lethal; it delays glucose-induced stimulation of the cAMP-PKA pathway and affects other PKA-dependent phenotypes like pseudohyphal growth (Nakafuku et al. 1988; Kubler et al. 1997; Colombo et al. 1998). In general, deletion of Gpa2 seems to cause stronger phenotypic effects than deletion of Gpr1, which may hint to additional regulation at the level of Gpa2. The intrinsic GTPase activity of Gpa2 is stimulated by the RGS2 gene product, which thus acts as an inhibitor of signaling (Versele et al. 1999). Gpa2 is anchored in the plasma membrane via myristoylation and palmitoylation of its N-terminus (Harashima and Heitman 2005).
The observation that Gpa2 functions without classical β and γ subunits has led to intensive research and also much debate concerning possible alternative Gβ and Gγ proteins. Initially, the kelch repeat proteins, Krh1 and Krh2, were proposed as alternative Gβ subunits (and called Gpb2 and Gpb1) and Gpg1 was proposed to be the γ subunit of Gpa2. Krh1 and Krh2 have a seven-kelch repeat structure, which results in a conformation very similar to the seven-WD-40 repeat structure of Gβ proteins, and physically binds to Gpa2 (Harashima and Heitman 2002, 2005). This initial suggestion was contradicted by later, more extensive work (Hoffman 2007; Niranjan et al. 2007). Krh1 and Krh2 do not interact with Gpa2 in a way that would be expected from a genuine Gβ replacement subunit. Deletion of Krh1 and Krh2 causes a high PKA phenotype, but this is apparently not due to relief of inhibition on Gpa2, as would be expected for a genuine Gβ protein. Krh1 and Krh2 directly interact with the catalytic subunits of PKA, Tpk1-3, and stimulate their interaction with the regulatory subunit, Bcy1, causing a higher cAMP level to be required for their dissociation. Krh1 and Krh2 promote the phosphorylation of the Bcy1 regulatory subunit of PKA and this produces a form of Bcy1 that is more stable and more effective as an inhibitor catalytic subunits (Budhwar et al. 2010). Hence, inactivation of Krh1 and Krh2 causes higher PKA activity in the presence of the same cAMP concentration. Gpa2 appears to inhibit Krh1 and Krh2, creating a bypass pathway for activation of adenylate cyclase, directly from the Gα protein Gpa2 to PKA (Batlle et al. 2003; Lu and Hirsch 2005; Peeters et al. 2006; Niranjan et al. 2007). Krh1 and Krh2 were also shown to function as regulators of the Ras GAPs, Ira1, and Ira2, either by stabilizing the proteins (Harashima et al. 2006) or target them for degradation (Phan et al. 2010). Asc1, another protein with seven-WD-40 repeats that binds most tightly to the GDP-loaded Gpa2 protein, has also been proposed as an alternative Gβ subunit (Zeller et al. 2007). There remain many questions concerning the precise role of Krh1 and Krh2 and the two G-protein signaling modules, Ras and Gpa2, in the control of cAMP synthesis and PKA activity in yeast.
2.3.5 Downstream Targets of PKA
Negative feedback regulation of PKA on cAMP synthesis Yeast strains with reduced PKA activity display huge increases in the basal cAMP level (Nikawa et al. 1987). This suggested that PKA downregulates cAMP synthesis by negative feedback regulation. This phenomenon also explains why the glucose-induced cAMP increase is very short-lived and actually occurs as a sharp cAMP signal. The extent of the glucose-induced cAMP signal is inversely correlated with the activity of PKA, and strains with attenuated PKA activity display large glucose-induced cAMP increases (Mbonyi et al. 1990). In a strain with elevated PKA activity, the cAMP signal is completely suppressed. This explains the seemingly contradictory finding that in a yeast strain devoid of the two cAMP phosphodiesterases the glucose-induced cAMP signal is virtually absent, rather than strongly enhanced (Ma et al. 1999). In spite of many efforts, the main target of the negative feedback regulation still remains elusive. Several targets have been proposed, including Ras and Cdc25. Mutagenesis of Ser214 to alanine (Ras2S214A) caused phenotypes consistent with higher activity of the cAMP-PKA pathway and also resulted in a higher basal level of cAMP and stronger glucose-induced cAMP signaling (Xiaojia and Jian 2010). However, the increase in the basal cAMP level was very limited compared to the huge cAMP increases in the Tpk-attenuated strains, indicating that phosphorylation of Ras cannot be the main target of the negative feedback regulation. As previously mentioned, glucose-induced hyperphosphorylation of Cdc25 resulting in its translocation from the plasma membrane to the cytosol and hence, reduced ability to activate Ras, may also form part of the negative feedback regulation mechanism (Gross et al. 1992; Dong and Bai 2011; Jian et al. 2010). Moreover, it has been shown that the Ras2 guanine nucleotide exchange activity of Cdc25 in vitro is inhibited by phosphorylation, due to downregulation of the association between Cdc25 and GTP-bound Ras2 (Dong and Bai 2011; Jian et al. 2010). Based on these data, it was suggested that PKA causes negative feedback regulation on cAMP synthesis through phosphorylation of Cdc25 (Jian et al. 2010). Putative phosphorylation sites in Cdc25 have been eliminated, and multiple truncations of the protein were made with various effects on the basal cAMP level or on glucose-induced cAMP signaling, but in all cases these changes were limited and never even approached the huge increase in cAMP as observed in Tpk-attenuated strains (Schomerus et al. 1990; van Aelst et al. 1990, 1991).
The low-affinity cAMP phosphodiesterase, Pde1, was shown to have a specific function in downregulating glucose-induced cAMP signaling, whereas the high-affinity cAMP phosphodiesterase, Pde2, controls the basal cAMP level in the cell. Pde1 is a target of PKA, and inactivation of its PKA phosphorylation site, Ser252, caused a higher glucose-induced cAMP signal (Ma et al. 1999). Pde2 is also regulated by PKA (Hu et al. 2010). The half-life of Pde2 seems to be increased in strains growing on glucose or strains with a high PKA phenotype. Pde2 localization in these strains is mainly in the nucleus. In contrast, in derepressed cells or strains with an attenuated PKA phenotype, Pde2 protein levels are lower and it is distributed over the nucleus and cytoplasm. Neither mutagenesis of the PKA phosphorylation site in Pde1 nor mutagenesis of any other putative target of PKA negative feedback regulation has resulted in a strain with equally high cAMP hyperaccumulation as in a tpk-attenuated strain. This seems to indicate that the main target of PKA negative feedback regulation has not been identified yet or that there are multiple parallel targets.
Post-translational targets of PKA in storage carbohydrate metabolism and glycolysis The first cellular target of the cAMP-PKA pathway identified was storage carbohydrate metabolism. Yeast has two storage carbohydrates, glycogen and trehalose, of which the second also serves as a stress protectant sugar. Trehalose appears most important for long-term survival in stationary phase cells and likely also in ascospores since these are devoid of glycogen (Thevelein 1984b). Addition of glucose to derepressed yeast cells, i.e., cells growing on a nonfermentable carbon source, glucose-starved stationary phase cells or ascospores, causes rapid mobilization of trehalose and glycogen, which is mediated by activation of the PKA pathway. Neutral trehalase was probably the first PKA target identified in yeast. It is within a few minutes activated after glucose addition to glucose-deprived cells (van der Plaat 1974), which is due to phosphorylation by PKA on several sites of the enzyme and binding of 14-3-3 proteins to the phosphorylated sites (Schepers et al. 2012; App and Holzer 1989). Mutants with reduced or constitutively enhanced activation of PKA show similarly reduced or constitutively elevated trehalase activity (Hirimburegama et al. 1992; Durnez et al. 1994; Giots et al. 2003; Mbonyi et al. 1990; Thevelein and Beullens 1985; Van Nuland et al. 2006). Glycogen synthase is downregulated by phosphorylation, while glycogen phosphorylase is activated by phosphorylation. Although it is well established that PKA activity in vivo is inversely correlated with the glycogen level and that both enzymes are phosphorylated by PKA in vitro, the precise contribution of direct phosphorylation by PKA of these enzymes is not very clear (Francois and Hers 1988; Hardy and Roach 1993; Francois and Parrou 2001; Wilson et al. 2010).
A second well-characterized target activated by PKA is 6-phosphofructo-2-kinase, which synthesizes fructose-2,6-bisphosphate, an allosteric activator of phosphofructokinase 1 and allosteric inhibitor of fructose-1,6-bisphosphatase (Dihazi et al. 2003; Noda et al. 1984). Fructose-1,6-bisphosphatase is also directly inactivated through phosphorylation by PKA (Pohlig and Holzer 1985). Through these mechanisms, activation of PKA stimulates glycolysis and fermentation, while it inhibits gluconeogenesis. Additional stimulation of glycolysis occurs through phosphorylation of pyruvate kinase (Cytrynska et al. 2001; Portela et al. 2006). This fits with the conclusion that fermentatively growing cells have high PKA activity while respiratively growing cells have low PKA activity.
Transcription factors as direct and indirect targets of PKA PKA has dramatic effects on the expression of a wide variety of genes involved in energy metabolism, cell cycle progression, stress response, ribosomal biogenesis and accumulation of the storage carbohydrate glycogen, and the storage and stress protectant sugar trehalose (Boy-Marcotte et al. 1998; Broach and Deschenes 1990; Ptacek et al. 2005; Smith et al. 1998; Thevelein 1994). Ninety percentage of the transcriptional remodeling of the cell in response to glucose is mediated via the G-proteins Ras1, Ras2, and Gpa2, which act in a redundant manner through activation of the cAMP-PKA pathway (Wang et al. 2004).
Since PKA activity is high in cells growing on glucose or other rapidly fermented sugars, i.e., glucose-repressed cells, and it is low in cells growing on nonfermentable carbon sources or glucose-starved, i.e., glucose-derepressed cells, there has initially been confusion between the function of the main glucose repression pathway and the PKA pathway in repression of transcription. Initially, the genes regulated by both pathways appeared to be very similar. The distinction between the two sets of transcription targets, however, can be made based on the fact that the main glucose repression pathway is only regulated by glucose or related rapidly fermented sugars, whereas the PKA pathway is also regulated by all other essential nutrients. Hence, when yeast cells are starved on a glucose-containing medium for another essential nutrient, e.g., nitrogen or phosphate, the main glucose repression pathway will remain active and the cells glucose repressed as long as there is a sufficient level of glucose in the medium. The PKA pathway, on the other hand, will be downregulated when the cells enter stationary phase and its target genes therefore will either no longer be repressed or induced. This does not preclude that the expression of some genes, like GSY2, encoding glycogen synthase, is regulated both by the main glucose repression and the PKA pathway (Wilson et al. 2010).
PKA controls the transcription factors Msn2, Msn4, and Gis1 by direct phosphorylation but also through control of protein kinases Rim15 and Yak1. Msn2 and Msn4 mediate the induction of a set of stress responsive genes, which contain so-called STRE elements (STress Response Element) in their promoters (Boy-Marcotte et al. 1998; Estruch and Carlson 1993; Martinez-Pastor et al. 1996; Schmitt and McEntee 1996; Smith et al. 1998). The STRE element consists of a pentameric core of CCCCT (Wieser et al. 1991). Glucose-induced activation of PKA triggers phosphorylation of Msn2 and Msn4, which blocks their translocation toward the nucleus and in this way inhibits targeted gene expression. As a result, high PKA activity counteracts the stress response and thus prevents establishment of high stress tolerance in yeast cells (Gorner et al. 1998, 2002). Deletion of both Msn2 and Msn4 suppresses the lethality caused by inactivation of the cAMP-PKA pathway, e.g., it can rescue a tpk-null strain or a ras1∆ ras2∆ strain (Smith et al. 1998), which reflects the importance of Msn2/Msn4-dependent targets for control of cell proliferation.
The Gis1 transcription factor supports expression of another set of genes through the PDS (Post-Diauxic Shift) element T(T/A)AGGGAT in their promoter (Pedruzzi et al. 2000; Zhang et al. 2009). These genes are expressed during the diauxic shift, and their regulation is not dependent on Msn2 or on Msn4 (Boy-Marcotte et al. 1998). However, most genes containing the PDS consensus sequence also contain one or more STRE consensus sequences.
The Rim15 and Yak1 protein kinases are positive effectors of gene expression and regulate the activity of the transcription factors Msn2, Msn4, and Gis1 (Garrett and Broach 1989; Garrett et al. 1991; Reinders et al. 1998). Rim15 is a glucose-repressible protein kinase (Vidan and Mitchell 1997) that is inhibited by PKA via direct phosphorylation. The deletion of RIM15 can also suppress the lethality caused by the loss of PKA activity (Reinders et al. 1998). This protein acts as an activator of STRE-controlled gene expression during entry into stationary phase (G0). The induction of genes during the diauxic shift via Rim15 is almost entirely mediated via the Msn2, Msn4, and Gis1 transcription factors (Cameroni et al. 2004; Pedruzzi et al. 2000; Reinders et al. 1998). Yak1 and PKA have an antagonistic effect on cell cycle progression through G1 (Garrett and Broach 1989; Garrett et al. 1991). Expression of the protein kinase Yak1 is controlled in a Msn2/Msn4-dependent manner (Smith et al. 1998). The deletion of YAK1 rescues lethal PKA deletion, i.e., it renders a tpk-null strain viable (Garrett and Broach 1989) and the activation of Yak1 is directly counteracted by PKA phosphorylation (Lee et al. 2008). Yak1 in turn can activate Msn2 by direct phosphorylation and in this way provides a positive feedback loop upon glucose limitation (Lee et al. 2008). Nuclear localization of Yak1 is promoted by glucose availability, while glucose limitation causes phosphorylation of Pop2, a substrate of the Yak1 protein kinase, and a regulator of transcription of many genes (Moriya et al. 2001). In addition, upon glucose starvation, Bcy1 is phosphorylated by Yak1 and restricted to the cytoplasm (Griffioen et al. 2001; Werner-Washburne et al. 1991). PKA thus counteracts stationary phase and stress response-related gene expression in at least two ways, by phosphorylation of the transcription factors and by phosphorylation of protein kinases required for proper activity of the same transcription factors.
PKA also plays a role in the transcriptional induction of genes upon addition of glucose. This has been investigated most intensively for the glucose-induced upshift in expression of the ribosomal protein genes (Herruer et al. 1987; Griffioen et al. 1994; Kraakman et al. 1993). In general, expression of ribosomal protein genes is strongly correlated with the growth rate of the cells. The glucose-induced upshift was claimed not to involve cAMP signaling. PKA was shown to promote expression of the ribosomal protein genes through the transcription factor Sfp1. Under optimal growth conditions, Sfp1 is localized in the nucleus, bound to the promoters of ribosomal protein genes, and helps promote ribosomal protein gene expression. When glucose gets depleted, Sfp1 is released from ribosomal protein gene promoters and leaves the nucleus, resulting in downregulation of ribosomal protein gene expression (Marion et al. 2004).
Although it has been known for a long time that inactivation of the Ras-cAMP-PKA pathway causes arrest in the G1 phase of the cell cycle and permanent entry into G0, the underlying mechanism is not well understood. Recent work has shown that Whi3, a negative regulator of the G1 cyclins, is inhibited through phosphorylation by PKA on Ser568. Phosphorylation of Whi3 by PKA leads to decreased interaction with CLN3 G1 cyclin mRNA and is required for the promotion of G1/S progression, implicating Whi3 in PKA regulation of cell cycle control (Mizunuma et al. 2013).
2.3.6 The PKA-Related Protein Kinase Sch9
The Sch9 protein kinase was originally discovered as a multicopy suppressor of lethality caused by inactivation of the cAMP-PKA pathway, i.e., as a suppressor of a cdc25 ts strain (Toda et al. 1988). Although much new information on Sch9 has been obtained since then, including evidence for requirement of Sch9 in different nutrient signaling processes (Zaman et al. 2008), its precise role in nutrient signaling remains enigmatic. Sch9 is a serine/threonine kinase and is part of the AGC kinase family (including protein kinase A, G and C). The sequence of Sch9 shows high similarity with other AGC protein kinases like Tpk1, 2, and 3 (Toda et al. 1988). Overexpression of SCH9 also suppresses other lethal PKA mutations like the tpk triple deletion strain, cyr1∆ or ras1∆ ras2∆. This is probably due to the fact that Sch9 regulates a similar set of genes as the Ras-cAMP-PKA pathway (Jorgensen et al. 2002). For example, overexpression of SCH9 induces expression of ribosomal protein genes and represses genes involved in carboxylic acid metabolism (Zaman et al. 2008). Sch9 affects the PKA pathway since its deletion causes increased PKA activity in derepressed cells (Crauwels et al. 1997), which is probably mediated by controlling the localization and phosphorylation of Bcy1, the regulatory subunit of PKA. In repressed cells, Bcy1 is almost entirely localized in the nucleus. However, when yeast is grown on nonfermentable carbon sources, Bcy1 is observed both in the nucleus and in the cytoplasm (Griffioen et al. 2000, 2001). Deletion of SCH9 causes constitutive nuclear localization of Bcy1, even in cells growing on glycerol (Zhang et al. 2011; Zhang and Gao 2012). Also the feedback regulation of Cdc25 by PKA phosphorylation seems to be controlled by Sch9 (Zhang et al. 2011). Although these studies provided evidence for direct involvement of Sch9 in control of PKA, other studies indicated that PKA and Sch9 also work in parallel, with either the same or different effects on specific phenotypes (Roosen et al. 2005). Sch9 also directly phosphorylates Rim15, which causes its inhibition by preventing its nuclear accumulation. Proper entrance into G0 requires release of both PKA-mediated inhibition of its protein kinase activity and Sch9-mediated inhibition of its nuclear accumulation (Pedruzzi et al. 2003; Wanke et al. 2008).
Sch9 itself is a phosphoprotein, and its phosphorylation state is dramatically decreased upon carbon, nitrogen, and phosphate starvation. It has been shown that the rapamycin-sensitive, nutrient-responsive TORC1 (target of rapamycin complex 1) protein kinase causes activation of Sch9 by direct phosphorylation of its C-terminal part when nutrients are available (Urban et al. 2007). This activation leads to enhanced expression of ribosomal protein genes, stimulates ribosome biogenesis and translation initiation, and prevents entry into the G0 phase (Urban et al. 2007; Huber et al. 2009, 2011). Sch9 is also phosphorylated and activated by the Snf1 protein kinase complex (Lu et al. 2011). Also the Pkh1 and Pkh2 protein kinases, which are involved in nutrient and stress signaling, are able to phosphorylate Sch9 (Roelants et al. 2004). Most likely, there are also other yet unknown kinases involved in the phosphorylation and regulation of Sch9.
2.4 The Main Glucose Repression Pathway
Another major regulator of cellular homeostasis in yeast carbon metabolism is the main glucose repression pathway. This pathway is responsible for the downregulation of respiration and the utilization of alternative sugars in the presence of glucose or related fermentable sugars, like fructose and mannose. In a typical aerobic yeast culture on glucose, the yeast will first grow rapidly by fermentation on the glucose, a phase in which respiration is repressed and ethanol accumulated. In this phase, the main glucose repression pathway is active and the cells are said to be glucose repressed. When the glucose concentration drops to a low level, the cells show a transient growth arrest, called diauxic shift, during which the enzymes for respiration and utilization of ethanol are being derepressed. Subsequently, the derepressed cells start to consume the ethanol utilizing respiration. In this phase, they grow much more slowly than during the first fermentation phase. When the ethanol is depleted, the cells enter stationary phase and remain derepressed. In this phase, they utilize storage carbohydrates (trehalose and glycogen) with respiration.
The Snf1 protein kinase is a major player in the main glucose repression pathway. It is an ortholog of the AMPK kinase family in mammalian cells. Snf1 acts in the sensing of glucose limitation (less than ±20 mM) and allows the cells to grow on less-preferred sugars, like sucrose and galactose, and on nonfermentable carbon sources, like ethanol and glycerol (Hedbacker and Carlson 2008; Zaman et al. 2008). Snf1 stands for “Sucrose Non Fermenting,” a name allocated to the snf1 mutant strain since it was unable to ferment sucrose but still able to ferment glucose (Carlson et al. 1981). The snf1 mutant showed a defect in the expression of SUC2, which encodes invertase, an enzyme that catalyzes the conversion of sucrose into glucose and fructose (Neigeborn and Carlson 1984). The snf4 mutant had the same phenotype and was also discovered in a screen for genes affecting the regulation of SUC2 gene expression (Neigeborn and Carlson 1984). Subsequent work showed that Snf1 is part of a serine/threonine protein kinase complex with a heterotrimeric structure: it contains one catalytic α subunit (encoded by SNF1), one of three β subunits (encoded by SIP1, SIP2, and GAL83), and one regulatory γ subunit (encoded by SNF4) (Celenza and Carlson 1984, 1986).
The Snf1 protein kinase complex is regulated in different ways (Fig. 2.3). Activation of Snf1 occurs upon glucose limitation through phosphorylation by upstream protein kinases, release of autoinhibition by Snf4, and through control of its subcellular localization, which is regulated by the β subunits (Celenza et al. 1989; Celenza and Carlson 1989; Jiang and Carlson 1996; Leech et al. 2003). Three protein kinases with related kinase domains, Sak1, Elm1, and Tos3, activate Snf1 by phosphorylation of Thr210. These kinases display high similarity and exert overlapping functions, so that abolishment of Snf1 activity in vivo is only observed in the triple mutant (Hong et al. 2003; Sutherland et al. 2003). The three upstream protein kinases are not affected by a drop in the external glucose level (Rubenstein et al. 2008), and glucose sensing for downregulation of Snf1 must therefore be mediated by another mechanism. The activity of Snf1 is downregulated by dephosphorylation, mediated by Protein Phosphatase 1 (PP1). The catalytic subunit of this enzyme is encoded by GLC7. It has multiple regulatory subunits that target the catalytic domain to specific substrates, of which the Reg1 regulatory subunit plays a role in the downregulation of Snf1 and thus in control of the main glucose repression pathway (Feng et al. 1991; Tu and Carlson 1995; Tu et al. 1996). In a reg1∆ mutant, Snf1 is constitutively phosphorylated and active (McCartney and Schmidt 2001).

The main glucose repression pathway in S. cerevisiae. In the inactive state, the regulatory domain (RD) of Snf1 covers the kinase domain of the catalytic domain (KD) thereby autoinhibiting it. In the absence of glucose, Snf4 can counteract the inhibition thereby opening up the complex. This open complex is phosphorylated by the redundant kinases Sak1, Elm1, and Tos3. The open phosphorylated Snf1/Snf4 complex is the active state and phosphorylates downstream targets. Upon glucose addition, the Snf1 complex is dephosphorylated by the Protein Phosphatase 1 (PP1) catalytic subunit Glc7, as controlled by its regulatory subunit Reg1. Glucose phosphorylation, possibly through activation of PKA, is probably responsible for PP1 activation. Active Snf1 complex is localized by its β subunits (Sip1, Sip2, and Gal83). Sip1 localizes the Snf1 complex toward the vacuole, Sip2 keeps the Snf1 complex in the cytoplasm, and Gal83 (the most abundant β) translocates the Snf1 complex toward the nucleus. In the nucleus, Snf1 phosphorylates Mig1, thereby inhibiting its repression of many target genes. Snf1 also phosphorylates the transcription factors Sip4 and Cat8 causing their activation
The control of Snf1 activity via phosphorylation/dephosphorylation is tightly connected with a second way of regulation, which is mediated by Snf4, the γ subunit of the Snf1 complex. Interaction between Snf1 and Snf4 is regulated by glucose availability. When glucose levels are low, Snf1 is phosphorylated on Thr210 and is then able to interact with Snf4. This leads to an open and active conformation of the complex, and thereby releases the autoinhibition caused by the regulatory domain of Snf1 (Celenza and Carlson 1989; Jiang and Carlson 1996; Estruch et al. 1992; Ludin et al. 1998). The active Snf1 kinase complex phosphorylates Reg1, thereby stabilizing the interaction between Snf1 and Reg1-Glc7 (Sanz et al. 2000). Upon glucose addition, Glc7 dephosphorylates Reg1 and subsequently dephosphorylates Snf1, causing its inactivation. The dephosphorylation of Reg1 by Glc7 seems to require Hxk2 activity (Sanz et al. 2000). Deletion of HXK2 leads to an Snf1 kinase complex that is trapped in the active conformation. The hxk2 mutant lacks glucose repression, and overexpression of REG1 suppresses this defect (Sanz et al. 2000). The dephosphorylation of the Snf1 complex seems to stimulate its conversion from an open, active conformation to a closed, inactive autoinhibitory conformation (Ludin et al. 1998). The autoinhibitory state of the complex is thus restored by the dephosphorylation of Snf1 by Glc7. New evidence has shown that Reg1 can also bind to Snf1 independently of Glc7 (Elbing et al. 2006), and binding of Reg1 to Snf1 seems to use the same site in Reg1 as binding of Glc7 to Reg1 (Tabba et al. 2010), suggesting competition between the binding of Glc7 and Snf1 with Reg1.
Recent studies have identified Sit4 as a second phosphatase involved in the deactivation of Snf1 by dephosphorylation (Ruiz et al. 2011). The intracellular ADP concentration is also involved in the regulation of Snf1. Increased concentrations of ADP protect Snf1 from dephosphorylation by binding to Snf4 (Chandrashekarappa et al. 2011; Mayer et al. 2011). This contrasts with regulation of its mammalian homolog, which is protected from dephosphorylation by both high AMP and ADP levels (Davies et al. 1995; Xiao et al. 2011).
How glucose is sensed for regulation of the main glucose repression pathway has remained enigmatic in spite of the many detailed studies of this pathway. Also the discovery of the three upstream kinases of Snf1 did not bring an answer to this question, since they do not appear to be regulated by glucose availability (Rubenstein et al. 2008). All evidence, on the other hand, points to regulation of Snf1 dephosphorylation by glucose availability. Recent work may finally have brought an answer to this question. It revealed that addition of glucose to derepressed yeast cells triggers a rapid increase in the intrinsic activity of the PP1 protein phosphatase and that this activation depends on the regulatory subunits Reg1 and Shp1. Deletion of Shp1 also caused strong derepression of the invertase gene SUC2. Rapid glucose-induced activation of PP1 was dependent on activation of the PKA pathway (Castermans et al. 2012). There has been other evidence for interaction between the PKA pathway and the main glucose repression pathway. The deletion of IRA1, IRA2, or BCY1, which causes constitutive activation of the PKA pathway, causes reduced activation of the Snf1 kinase complex and suppresses the slow-growth phenotype of a reg1 mutant. Conversely, downregulation of the PKA pathway by deletion of GPR1 caused elevated Snf1 kinase activation (Barrett et al. 2012).
Finally, the activity of the Snf1 complex is also regulated by control of its intracellular localization as a function of glucose availability. When glucose concentrations are high, Snf1 and the three β subunits reside in the cytosol. Upon glucose limitation, the different β subunits direct the Snf1 kinase complex to different locations within the cell. Gal83 is the most abundant β subunit and is involved in the translocation of active Snf1 toward the nucleus (Vincent et al. 2001; Hedbacker et al. 2004a). Sip1 is involved in localization toward the vacuolar membrane, but in glucose-grown cells the maintenance of the cytosolic Sip1 localization is dependent on PKA activity (Hedbacker et al. 2004b). Sip2 is required to keep the Snf1 kinase complex in the cytoplasm (Vincent et al. 2001).
The activation of the Snf1 kinase complex has multiple functions. The complex can be translocated in a Gal83-mediated way toward the nucleus to affect the expression of a set of genes involved in the metabolism of alternative carbon sources, gluconeogenesis, respiration, transport, and meiosis (Hedbacker and Carlson 2008; Schuller 2003; Zaman et al. 2009). This set of genes is only small compared with the much more extensive changes in gene expression triggered by the Ras-cAMP-PKA pathway. In addition, a large part of the genes repressed after inactivation of Snf1 is also repressed by activation of the Ras-cAMP-PKA pathway (Zaman et al. 2009). This reflects the possible cooperation of PKA with the Snf1 kinase complex at least under certain conditions in affecting a similar set of cellular functions (Thompson-Jaeger et al. 1991; Hubbard et al. 1992).
Mig1 is the main transcription factor downstream in the glucose repression pathway (Nehlin et al. 1991; Nehlin and Ronne 1990). It is involved in glucose repression of at least 90 different genes, mostly required for the metabolism of alternative carbon sources (Klein et al. 1998; Lutfiyya et al. 1998). Snf1 phosphorylates the Mig1 transcriptional repressor and thereby promotes its nuclear export, causing derepression of Mig1-controlled genes. Mig1 also recruits the transcriptional co-repressor complex Ssn6-Tup1 (Treitel and Carlson 1995). Hxk2 is translocated toward the nucleus in a Mig1-dependent way and is part of the Mig1 repressor complex (Ahuatzi et al. 2007). For interaction between Mig1 and Hxk2, the serine at position 311 of Mig1 seems to be important. This site is the major Snf1 phosphorylation site and promotes nuclear export of Mig1 after phosphorylation. Hxk2 binds to this site thereby inhibiting Snf1-dependent phosphorylation of Mig1 (Ahuatzi et al. 2007).
Snf1 also positively regulates the transcriptional activators Cat8 and Sip4 (Lesage et al. 1996; Rahner et al. 1999; Hiesinger et al. 2001). These two transcriptional activators bind specifically to carbon source responsive elements (CSRE) under glucose-limiting conditions (Vincent and Carlson 1998). When activated, they induce the expression of genes involved in gluconeogenesis, respiration, and the glyoxylate cycle (Santangelo 2006). SIP4 has a CSRE element in its promoter and is expressed upon activation of Cat8 by Snf1 phosphorylation (Vincent and Carlson 1998). The expression of CAT8 in turn, is repressed by Mig1 (Hedges et al. 1995; Randez-Gil et al. 1997). Besides regulating gene transcription, Snf1 also regulates through phosphorylation proteins involved in fatty acid metabolism, carbohydrate storage, and transport (Hedbacker and Carlson 2008). For instance, Snf1 phosphorylates and inactivates acetyl-CoA carboxylase (Acc1). This results in blocked fatty acid biosynthesis under glucose-limiting conditions (Woods et al. 1994).
2.5 Conclusions
The exquisite preference of the yeast S. cerevisiae for glucose as carbon source is reflected in the multiple, sophisticated mechanisms that it has developed to detect the presence of glucose and to adjust various cellular functions accordingly. Two types of plasma membrane glucose sensors have been discovered first in S. cerevisiae: transporter homologues, which have developed into nontransporting glucose sensors, and a glucose-sensing GPCR. The concerted action of extracellular and intracellular glucose sensing has also been demonstrated and elucidated for the first time in S. cerevisiae. The Snf1 protein kinase has been discovered in S. cerevisiae as a central element of a glucose signaling pathway and has served as a model for investigation of the related AMP-activated kinase in other organisms. Elucidation of the enigmatic role of Ras in yeast glucose signaling may have important consequences for understanding aberrant glucose metabolism in tumor cells. We predict that glucose regulation of major protein phosphatases will reveal many novel and important aspects about glucose signaling and its interplay with other signal transduction pathways and mechanisms of cellular regulation.
References
Ahuatzi D, Riera A, Pelaez R, Herrero P, Moreno F (2007) Hxk2 regulates the phosphorylation state of Mig1 and therefore its nucleocytoplasmic distribution. J Biol Chem 282(7):4485–4493. doi:10.1074/jbc.M606854200 M606854200 [pii]
App H, Holzer H (1989) Purification and characterization of neutral trehalase from the yeast ABYS1 mutant. J Biol Chem 264(29):17583–17588
Babu P, Bryan JD, Panek HR, Jordan SL, Forbrich BM, Kelley SC, Colvin RT, Robinson LC (2002) Plasma membrane localization of the Yck2p yeast casein kinase 1 isoform requires the C-terminal extension and secretory pathway function. J Cell Sci 115(Pt 24):4957–4968
Barrett L, Orlova M, Maziarz M, Kuchin S (2012) Protein kinase A contributes to the negative control of Snf1 protein kinase in Saccharomyces cerevisiae. Eukaryot Cell 11(2):119–128. doi:10.1128/EC.05061-11 EC.05061-11 [pii]
Batlle M, Lu A, Green DA, Xue Y, Hirsch JP (2003) Krh1p and Krh2p act downstream of the Gpa2p G(alpha) subunit to negatively regulate haploid invasive growth. J Cell Sci 116(Pt 4):701–710
Beckner SK, Hattori S, Shih TY (1985) The ras oncogene product p21 is not a regulatory component of adenylate cyclase. Nature 317(6032):71–72
Beullens M, Mbonyi K, Geerts L, Gladines D, Detremerie K, Jans AW, Thevelein JM (1988) Studies on the mechanism of the glucose-induced cAMP signal in glycolysis and glucose repression mutants of the yeast Saccharomyces cerevisiae. Eur J Biochem 172(1):227–231
Bhattacharya S, Chen L, Broach JR, Powers S (1995) Ras membrane targeting is essential for glucose signaling but not for viability in yeast. Proc Natl Acad Sci USA 92(7):2984–2988
Bisson LF, Coons DM, Kruckeberg AL, Lewis DA (1993) Yeast sugar transporters. Crit Rev Biochem Mol Biol 28(4):259–308. doi:10.3109/10409239309078437
Bisson LF, Neigeborn L, Carlson M, Fraenkel DG (1987) The SNF3 gene is required for high-affinity glucose transport in Saccharomyces cerevisiae. J Bacteriol 169(4):1656–1662
Boguski MS, McCormick F (1993) Proteins regulating Ras and its relatives. Nature 366(6456):643–654. doi:10.1038/366643a0
Boles E, Hollenberg CP (1997) The molecular genetics of hexose transport in yeasts. FEMS Microbiol Rev 21(1):85–111 S0168-6445(97)00052-1 [pii]
Boy-Marcotte E, Ikonomi P, Jacquet M (1996) SDC25, a dispensable Ras guanine nucleotide exchange factor of Saccharomyces cerevisiae differs from CDC25 by its regulation. Mol Biol Cell 7(4):529–539
Boy-Marcotte E, Perrot M, Bussereau F, Boucherie H, Jacquet M (1998) Msn2p and Msn4p control a large number of genes induced at the diauxic transition which are repressed by cyclic AMP in Saccharomyces cerevisiae. J Bacteriol 180(5):1044–1052
Broach JR, Deschenes RJ (1990) The function of ras genes in Saccharomyces cerevisiae. Adv Cancer Res 54:79–139
Broek D, Samiy N, Fasano O, Fujiyama A, Tamanoi F, Northup J, Wigler M (1985) Differential activation of yeast adenylate cyclase by wild-type and mutant RAS proteins. Cell 41(3):763–769 S0092-8674(85)80057-X [pii]
Broek D, Toda T, Michaeli T, Levin L, Birchmeier C, Zoller M, Powers S, Wigler M (1987) The S. cerevisiae CDC25 gene product regulates the RAS/adenylate cyclase pathway. Cell 48(5):789–799 0092-8674(87)90076-6 [pii]
Budhwar R, Lu A, Hirsch JP (2010) Nutrient control of yeast PKA activity involves opposing effects on phosphorylation of the Bcy1 regulatory subunit. Mol Biol Cell 21(21):3749–3758. doi:10.1091/mbc.E10-05-0388
Cameroni E, Hulo N, Roosen J, Winderickx J, De Virgilio C (2004) The novel yeast PAS kinase Rim 15 orchestrates G0-associated antioxidant defense mechanisms. Cell Cycle 3(4):462–468 786 [pii]
Camonis JH, Kalekine M, Gondre B, Garreau H, Boy-Marcotte E, Jacquet M (1986) Characterization, cloning and sequence analysis of the CDC25 gene which controls the cyclic AMP level of Saccharomyces cerevisiae. EMBO J 5(2):375–380
Carlson M, Osmond BC, Botstein D (1981) Mutants of yeast defective in sucrose utilization. Genetics 98(1):25–40
Caspani G, Tortora P, Hanozet GM, Guerritore A (1985) Glucose-stimulated cAMP increase may be mediated by intracellular acidification in Saccharomyces cerevisiae. FEBS Lett 186(1):75–79
Castermans D, Somers I, Kriel J, Louwet W, Wera S, Versele M, Janssens V, Thevelein JM (2012) Glucose-induced posttranslational activation of protein phosphatases PP2A and PP1 in yeast. Cell Res 22(6):1058–1077. doi:10.1038/cr.2012.20 cr201220 [pii]
Celenza JL, Carlson M (1984) Cloning and genetic mapping of SNF1, a gene required for expression of glucose-repressible genes in Saccharomyces cerevisiae. Mol Cell Biol 4(1):49–53
Celenza JL, Carlson M (1986) A yeast gene that is essential for release from glucose repression encodes a protein kinase. Science 233(4769):1175–1180
Celenza JL, Carlson M (1989) Mutational analysis of the Saccharomyces cerevisiae SNF1 protein kinase and evidence for functional interaction with the SNF4 protein. Mol Cell Biol 9(11):5034–5044
Celenza JL, Eng FJ, Carlson M (1989) Molecular analysis of the SNF4 gene of Saccharomyces cerevisiae: evidence for physical association of the SNF4 protein with the SNF1 protein kinase. Mol Cell Biol 9(11):5045–5054
Chandrashekarappa DG, McCartney RR, Schmidt MC (2011) Subunit and domain requirements for adenylate-mediated protection of Snf1 kinase activation loop from dephosphorylation. J Biol Chem 286(52):44532–44541. doi:10.1074/jbc.M111.315895 M111.315895 [pii]
Chautard H, Jacquet M, Schoentgen F, Bureaud N, Benedetti H (2004) Tfs1p, a member of the PEBP family, inhibits the Ira2p but not the Ira1p Ras GTPase-activating protein in Saccharomyces cerevisiae. Eukaryot Cell 3(2):459–470
Chevtzoff C, Yoboue ED, Galinier A, Casteilla L, Daignan-Fornier B, Rigoulet M, Devin A (2010) Reactive oxygen species-mediated regulation of mitochondrial biogenesis in the yeast Saccharomyces cerevisiae. J Biol Chem 285(3):1733–1742. doi:10.1074/jbc.M109.019570
Colombo S, Ma P, Cauwenberg L, Winderickx J, Crauwels M, Teunissen A, Nauwelaers D, de Winde JH, Gorwa MF, Colavizza D, Thevelein JM (1998) Involvement of distinct G-proteins, Gpa2 and Ras, in glucose- and intracellular acidification-induced cAMP signalling in the yeast Saccharomyces cerevisiae. EMBO J 17(12):3326–3341. doi:10.1093/emboj/17.12.3326
Colombo S, Ronchetti D, Thevelein JM, Winderickx J, Martegani E (2004) Activation state of the Ras2 protein and glucose-induced signaling in Saccharomyces cerevisiae. J Biol Chem 279(45):46715–46722. doi:10.1074/jbc.M405136200 [pii]
Coons DM, Vagnoli P, Bisson LF (1997) The C-terminal domain of Snf3p is sufficient to complement the growth defect of snf3 null mutations in Saccharomyces cerevisiae: SNF3 functions in glucose recognition. Yeast 13(1):9–20. doi:10.1002/(SICI)1097-0061(199701)13:1<9:AID-YEA51>3.0.CO;2-U [pii] 10.1002/(SICI)1097-0061(199701)13:1 < 9:AID-YEA51 > 3.0.CO;2-U
Crauwels M, Donaton MC, Pernambuco MB, Winderickx J, de Winde JH, Thevelein JM (1997) The Sch9 protein kinase in the yeast Saccharomyces cerevisiae controls cAPK activity and is required for nitrogen activation of the fermentable-growth-medium-induced (FGM) pathway. Microbiology 143(Pt 8):2627–2637
Cytrynska M, Frajnt M, Jakubowicz T (2001) Saccharomyces cerevisiae pyruvate kinase Pyk1 is PKA phosphorylation substrate in vitro. FEMS Microbiol Lett 203(2):223–227
Damak F, Boy-Marcotte E, Le-Roscouet D, Guilbaud R, Jacquet M (1991) SDC25, a CDC25-like gene which contains a RAS-activating domain and is a dispensable gene of Saccharomyces cerevisiae. Mol Cell Biol 11(1):202–212
Daniel JH (1986) The CDC25 “Start” gene of Saccharomyces cerevisiae: sequencing of the active C-terminal fragment and regional homologies with rhodopsin and cytochrome P450. Curr Genet 10(12):879–885
Davies SP, Helps NR, Cohen PT, Hardie DG (1995) 5′-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C alpha and native bovine protein phosphatase-2AC. FEBS Lett 377(3):421–425. doi:10.1016/0014-5793(95)01368-7 0014-5793(95)01368-7 [pii]
Diaz-Ruiz R, Rigoulet M, Devin A (2011) The Warburg and Crabtree effects: on the origin of cancer cell energy metabolism and of yeast glucose repression. Biochim Biophys Acta 1807(6):568–576. doi:10.1016/j.bbabio.2010.08.010
Dihazi H, Kessler R, Eschrich K (2003) Glucose-induced stimulation of the Ras-cAMP pathway in yeast leads to multiple phosphorylations and activation of 6-phosphofructo-2-kinase. Biochemistry 42(20):6275–6282. doi:10.1021/bi034167r
Dlugai S, Hippler S, Wieczorke R, Boles E (2001) Glucose-dependent and -independent signalling functions of the yeast glucose sensor Snf3. FEBS Lett 505(3):389–392 S0014-5793(01)02854-X [pii]
Dong J, Bai X (2011) The membrane localization of Ras2p and the association between Cdc25p and Ras2-GTP are regulated by protein kinase A (PKA) in the yeast Saccharomyces cerevisiae. FEBS Lett 585(8):1127–1134. doi:10.1016/j.febslet.2011.03.057 S0014-5793(11)00223-7 [pii]
Durnez P, Pernambuco MB, Oris E, Arguelles JC, Mergelsberg H, Thevelein JM (1994) Activation of trehalase during growth induction by nitrogen sources in the yeast Saccharomyces cerevisiae depends on the free catalytic subunits of cAMP-dependent protein kinase, but not on functional Ras proteins. Yeast 10(8):1049–1064. doi:10.1002/yea.320100807
Edmondson DG, Smith MM, Roth SY (1996) Repression domain of the yeast global repressor Tup1 interacts directly with histones H3 and H4. Genes Dev 10(10):1247–1259
Elbing K, McCartney RR, Schmidt MC (2006) Purification and characterization of the three Snf1-activating kinases of Saccharomyces cerevisiae. Biochem J 393(Pt 3):797–805. doi:10.1042/BJ20051213 BJ20051213 [pii]
Estruch F, Carlson M (1993) Two homologous zinc finger genes identified by multicopy suppression in a SNF1 protein kinase mutant of Saccharomyces cerevisiae. Mol Cell Biol 13(7):3872–3881
Estruch F, Treitel MA, Yang X, Carlson M (1992) N-terminal mutations modulate yeast SNF1 protein kinase function. Genetics 132(3):639–650
Farnsworth CL, Feig LA (1991) Dominant inhibitory mutations in the Mg(2 +)-binding site of RasH prevent its activation by GTP. Mol Cell Biol 11(10):4822–4829
Fedor-Chaiken M, Deschenes RJ, Broach JR (1990) SRV2, a gene required for RAS activation of adenylate cyclase in yeast. Cell 61(2):329–340
Feng ZH, Wilson SE, Peng ZY, Schlender KK, Reimann EM, Trumbly RJ (1991) The yeast GLC7 gene required for glycogen accumulation encodes a type 1 protein phosphatase. J Biol Chem 266(35):23796–23801
Field J, Vojtek A, Ballester R, Bolger G, Colicelli J, Ferguson K, Gerst J, Kataoka T, Michaeli T, Powers S et al (1990) Cloning and characterization of CAP, the S. cerevisiae gene encoding the 70 kd adenylyl cyclase-associated protein. Cell 61(2):319–327. doi:10.1099/mic.0.27144-0150/9/2865 [pii]
Folch-Mallol JL, Martinez LM, Casas SJ, Yang R, Martinez-Anaya C, Lopez L, Hernandez A, Nieto-Sotelo J (2004) New roles for CDC25 in growth control, galactose regulation and cellular differentiation in Saccharomyces cerevisiae. Microbiology 150(Pt 9):2865–2879. doi:10.1099/mic.0.27144-0150/9/2865 [pii]
Francois J, Hers HG (1988) The control of glycogen metabolism in yeast. 2. A kinetic study of the two forms of glycogen synthase and of glycogen phosphorylase and an investigation of their interconversion in a cell-free extract. Eur J Biochem 174(3):561–567
Francois J, Parrou JL (2001) Reserve carbohydrates metabolism in the yeast Saccharomyces cerevisiae. FEMS Microbiol Rev 25(1):125–145
Garreau H, Geymonat M, Renault G, Jacquet M (1996) Membrane-anchoring domains of Cdc25p, a Saccharomyces cerevisiae ras exchange factor. Biol Cell 86(2–3):93–102
Garrett S, Broach J (1989) Loss of Ras activity in Saccharomyces cerevisiae is suppressed by disruptions of a new kinase gene, YAKI, whose product may act downstream of the cAMP-dependent protein kinase. Genes Dev 3(9):1336–1348
Garrett S, Menold MM, Broach JR (1991) The Saccharomyces cerevisiae YAK1 gene encodes a protein kinase that is induced by arrest early in the cell cycle. Mol Cell Biol 11(8):4045–4052
Giots F, Donaton MC, Thevelein JM (2003) Inorganic phosphate is sensed by specific phosphate carriers and acts in concert with glucose as a nutrient signal for activation of the protein kinase A pathway in the yeast Saccharomyces cerevisiae. Mol Microbiol 47(4):1163–1181
Gorner W, Durchschlag E, Martinez-Pastor MT, Estruch F, Ammerer G, Hamilton B, Ruis H, Schuller C (1998) Nuclear localization of the C2H2 zinc finger protein Msn2p is regulated by stress and protein kinase A activity. Genes Dev 12(4):586–597
Gorner W, Durchschlag E, Wolf J, Brown EL, Ammerer G, Ruis H, Schuller C (2002) Acute glucose starvation activates the nuclear localization signal of a stress-specific yeast transcription factor. EMBO J 21(1–2):135–144. doi:10.1093/emboj/21.1.135
Gourlay CW, Ayscough KR (2006) Actin-induced hyperactivation of the Ras signaling pathway leads to apoptosis in Saccharomyces cerevisiae. Mol Cell Biol 26(17):6487–6501. doi:10.1128/MCB.00117-06
Graul RC, Sadee W (2001) Evolutionary relationships among G protein-coupled receptors using a clustered database approach. AAPS PharmSci 3(2):E12
Griffioen G, Anghileri P, Imre E, Baroni MD, Ruis H (2000) Nutritional control of nucleocytoplasmic localization of cAMP-dependent protein kinase catalytic and regulatory subunits in Saccharomyces cerevisiae. J Biol Chem 275(2):1449–1456
Griffioen G, Branduardi P, Ballarini A, Anghileri P, Norbeck J, Baroni MD, Ruis H (2001) Nucleocytoplasmic distribution of budding yeast protein kinase A regulatory subunit Bcy1 requires Zds1 and is regulated by Yak1-dependent phosphorylation of its targeting domain. Mol Cell Biol 21(2):511–523. doi:10.1128/MCB.21.2.511-523.2001
Griffioen G, Mager WH, Planta RJ (1994) Nutritional upshift response of ribosomal protein gene transcription in Saccharomyces cerevisiae. FEMS Microbiol Lett 123(1–2):137–144
Gross E, Goldberg D, Levitzki A (1992) Phosphorylation of the S. cerevisiae Cdc25 in response to glucose results in its dissociation from Ras. Nature 360(6406):762–765. doi:10.1038/360762a0
Haney SA, Broach JR (1994) Cdc25p, the guanine nucleotide exchange factor for the Ras proteins of Saccharomyces cerevisiae, promotes exchange by stabilizing Ras in a nucleotide-free state. J Biol Chem 269(24):16541–16548
Harashima T, Anderson S, Yates JR 3rd, Heitman J (2006) The kelch proteins Gpb1 and Gpb2 inhibit Ras activity via association with the yeast RasGAP neurofibromin homologs Ira1 and Ira2. Mol Cell 22(6):819–830. doi:10.1016/j.molcel.2006.05.011 S1097-2765(06)00324-8 [pii]
Harashima T, Heitman J (2002) The Galpha protein Gpa2 controls yeast differentiation by interacting with kelch repeat proteins that mimic Gbeta subunits. Mol Cell 10(1):163–173 S1097276502005695 [pii]
Harashima T, Heitman J (2005) Galpha subunit Gpa2 recruits kelch repeat subunits that inhibit receptor-G protein coupling during cAMP-induced dimorphic transitions in Saccharomyces cerevisiae. Mol Biol Cell 16(10):4557–4571. doi:10.1091/mbc.E05-05-0403 E05-05-0403 [pii]
Hardy TA, Roach PJ (1993) Control of yeast glycogen synthase-2 by COOH-terminal phosphorylation. J Biol Chem 268(32):23799–23805
Hayles J, Nurse P (1986) Cell cycle regulation in yeast. J Cell Sci Suppl 4:155–170
Hedbacker K, Carlson M (2008) SNF1/AMPK pathways in yeast. Front Biosci 13:2408–2420 2854 [pii]
Hedbacker K, Hong SP, Carlson M (2004a) Pak1 protein kinase regulates activation and nuclear localization of Snf1-Gal83 protein kinase. Mol Cell Biol 24(18):8255–8263. doi:10.1128/MCB.24.18.8255-8263.2004 24/18/8255 [pii]
Hedbacker K, Townley R, Carlson M (2004b) Cyclic AMP-dependent protein kinase regulates the subcellular localization of Snf1-Sip1 protein kinase. Mol Cell Biol 24(5):1836–1843
Hedges D, Proft M, Entian KD (1995) CAT8, a new zinc cluster-encoding gene necessary for derepression of gluconeogenic enzymes in the yeast Saccharomyces cerevisiae. Mol Cell Biol 15(4):1915–1922
Herruer MH, Mager WH, Woudt LP, Nieuwint RT, Wassenaar GM, Groeneveld P, Planta RJ (1987) Transcriptional control of yeast ribosomal protein synthesis during carbon-source upshift. Nucleic Acids Res 15(24):10133–10144
Hiesinger M, Roth S, Meissner E, Schuller HJ (2001) Contribution of Cat8 and Sip4 to the transcriptional activation of yeast gluconeogenic genes by carbon source-responsive elements. Curr Genet 39(2):68–76
Hirimburegama K, Durnez P, Keleman J, Oris E, Vergauwen R, Mergelsberg H, Thevelein JM (1992) Nutrient-induced activation of trehalase in nutrient-starved cells of the yeast Saccharomyces cerevisiae: cAMP is not involved as second messenger. J Gen Microbiol 138(10):2035–2043
Hoffman CS (2007) Propping up our knowledge of G protein signaling pathways: diverse functions of putative noncanonical G beta subunits in fungi. Sci STKE 370:pe3
Holsbeeks I, Lagatie O, Van Nuland A, Van de Velde S, Thevelein JM (2004) The eukaryotic plasma membrane as a nutrient-sensing device. Trends Biochem Sci 29(10):556–564. doi:10.1016/j.tibs.2004.08.010S0968-0004(04)00211-7 [pii]
Hong SP, Leiper FC, Woods A, Carling D, Carlson M (2003) Activation of yeast Snf1 and mammalian AMP-activated protein kinase by upstream kinases. Proc Natl Acad Sci USA 100(15):8839–8843. doi:10.1073/pnas.15331361001533136100 [pii]
Hu Y, Liu E, Bai X, Zhang A (2010) The localization and concentration of the PDE2-encoded high-affinity cAMP phosphodiesterase is regulated by cAMP-dependent protein kinase A in the yeast Saccharomyces cerevisiae. FEMS Yeast Res 10(2):177–187. doi:10.1111/j.1567-1364.2009.00598.x FYR598 [pii]
Hubbard EJ, Yang XL, Carlson M (1992) Relationship of the cAMP-dependent protein kinase pathway to the SNF1 protein kinase and invertase expression in Saccharomyces cerevisiae. Genetics 130(1):71–80
Huber A, Bodenmiller B, Uotila A, Stahl M, Wanka S, Gerrits B, Aebersold R, Loewith R (2009) Characterization of the rapamycin-sensitive phosphoproteome reveals that Sch9 is a central coordinator of protein synthesis. Genes Dev 23(16):1929–1943. doi:10.1101/gad.532109 23/16/1929 [pii]
Huber A, French SL, Tekotte H, Yerlikaya S, Stahl M, Perepelkina MP, Tyers M, Rougemont J, Beyer AL, Loewith R (2011) Sch9 regulates ribosome biogenesis via Stb3, Dot6 and Tod6 and the histone deacetylase complex RPD3L. EMBO J 30(15):3052–3064. doi:10.1038/emboj.2011.221 emboj2011221 [pii]
Jian D, Aili Z, Xiaojia B, Huansheng Z, Yun H (2010) Feedback regulation of Ras2 guanine nucleotide exchange factor (Ras2-GEF) activity of Cdc25p by Cdc25p phosphorylation in the yeast Saccharomyces cerevisiae. FEBS Lett 584(23):4745–4750. doi:10.1016/j.febslet.2010.11.006 S0014-5793(10)00900-2 [pii]
Jiang R, Carlson M (1996) Glucose regulates protein interactions within the yeast SNF1 protein kinase complex. Genes Dev 10(24):3105–3115
Johnston M, Kim JH (2005) Glucose as a hormone: receptor-mediated glucose sensing in the yeast Saccharomyces cerevisiae. Biochem Soc Trans 33(Pt 1):247–252. doi:10.1042/BST0330247 BST0330247 [pii]
Jones S, Vignais ML, Broach JR (1991) The CDC25 protein of Saccharomyces cerevisiae promotes exchange of guanine nucleotides bound to ras. Mol Cell Biol 11(5):2641–2646
Jorgensen P, Nishikawa JL, Breitkreutz BJ, Tyers M (2002) Systematic identification of pathways that couple cell growth and division in yeast. Science 297(5580):395–400. doi:10.1126/science.10708501070850 [pii]
Kataoka T, Broek D, Wigler M (1985) DNA sequence and characterization of the S. cerevisiae gene encoding adenylate cyclase. Cell 43(2 Pt 1):493–505 0092-8674(85)90179-5 [pii]
Kataoka T, Powers S, McGill C, Fasano O, Strathern J, Broach J, Wigler M (1984) Genetic analysis of yeast RAS1 and RAS2 genes. Cell 37(2):437–445 0092-8674(84)90374-X [pii]
Kim JH, Polish J, Johnston M (2003) Specificity and regulation of DNA binding by the yeast glucose transporter gene repressor Rgt1. Mol Cell Biol 23(15):5208–5216
Kishi T, Seno T, Yamao F (1998) Grr1 functions in the ubiquitin pathway in Saccharomyces cerevisiae through association with Skp1. Mol Gen Genet 257(2):143–148
Klein CJ, Olsson L, Nielsen J (1998) Glucose control in Saccharomyces cerevisiae: the role of Mig1 in metabolic functions. Microbiology 144(Pt 1):13–24
Kotting C, Kallenbach A, Suveyzdis Y, Wittinghofer A, Gerwert K (2008) The GAP arginine finger movement into the catalytic site of Ras increases the activation entropy. Proc Natl Acad Sci USA 105(17):6260–6265. doi:10.1073/pnas.0712095105 0712095105 [pii]
Kraakman L, Lemaire K, Ma P, Teunissen AW, Donaton MC, Van Dijck P, Winderickx J, de Winde JH, Thevelein JM (1999) A Saccharomyces cerevisiae G-protein coupled receptor, Gpr1, is specifically required for glucose activation of the cAMP pathway during the transition to growth on glucose. Mol Microbiol 32(5):1002–1012
Kraakman LS, Griffioen G, Zerp S, Groeneveld P, Thevelein JM, Mager WH, Planta RJ (1993) Growth-related expression of ribosomal protein genes in Saccharomyces cerevisiae. Mol Gen Genet 239(1–2):196–204
Kriel J, Haesendonckx S, Rubio-Texeira M, Van Zeebroeck G, Thevelein JM (2011) From transporter to transceptor: signaling from transporters provokes re-evaluation of complex trafficking and regulatory controls: endocytic internalization and intracellular trafficking of nutrient transceptors may, at least in part, be governed by their signaling function. BioEssays 33(11):870–879. doi:10.1002/bies.201100100
Kubler E, Mosch HU, Rupp S, Lisanti MP (1997) Gpa2p, a G-protein alpha-subunit, regulates growth and pseudohyphal development in Saccharomyces cerevisiae via a cAMP-dependent mechanism. J Biol Chem 272(33):20321–20323
Kuret J, Johnson KE, Nicolette C, Zoller MJ (1988) Mutagenesis of the regulatory subunit of yeast cAMP-dependent protein kinase. Isolation of site-directed mutants with altered binding affinity for catalytic subunit. J Biol Chem 263(19):9149–9154
Lai CC, Boguski M, Broek D, Powers S (1993) Influence of guanine nucleotides on complex formation between Ras and CDC25 proteins. Mol Cell Biol 13(3):1345–1352
Lakshmanan J, Mosley AL, Ozcan S (2003) Repression of transcription by Rgt1 in the absence of glucose requires Std1 and Mth1. Curr Genet 44(1):19–25. doi:10.1007/s00294-003-0423-2
Lee P, Cho BR, Joo HS, Hahn JS (2008) Yeast Yak1 kinase, a bridge between PKA and stress-responsive transcription factors, Hsf1 and Msn2/Msn4. Mol Microbiol 70(4):882–895. doi:10.1111/j.1365-2958.2008.06450.x MMI6450 [pii]
Leech A, Nath N, McCartney RR, Schmidt MC (2003) Isolation of mutations in the catalytic domain of the snf1 kinase that render its activity independent of the snf4 subunit. Eukaryot Cell 2(2):265–273
Lemaire K, Van de Velde S, Van Dijck P, Thevelein JM (2004) Glucose and sucrose act as agonist and mannose as antagonist ligands of the G protein-coupled receptor Gpr1 in the yeast Saccharomyces cerevisiae. Mol Cell 16(2):293–299. doi:10.1016/j.molcel.2004.10.004 S1097276504006100 [pii]
Lesage P, Yang X, Carlson M (1996) Yeast SNF1 protein kinase interacts with SIP4, a C6 zinc cluster transcriptional activator: a new role for SNF1 in the glucose response. Mol Cell Biol 16(5):1921–1928
Lillie SH, Pringle JR (1980) Reserve carbohydrate metabolism in Saccharomyces cerevisiae: responses to nutrient limitation. J Bacteriol 143(3):1384–1394
Lorenz MC, Heitman J (1997) Yeast pseudohyphal growth is regulated by GPA2, a G protein alpha homolog. EMBO J 16(23):7008–7018. doi:10.1093/emboj/16.23.7008
Lu A, Hirsch JP (2005) Cyclic AMP-independent regulation of protein kinase A substrate phosphorylation by Kelch repeat proteins. Eukaryot Cell 4(11):1794–1800. doi:10.1128/EC.4.11.1794-1800.2005 4/11/1794 [pii]
Lu JY, Lin YY, Sheu JC, Wu JT, Lee FJ, Chen Y, Lin MI, Chiang FT, Tai TY, Berger SL, Zhao Y, Tsai KS, Zhu H, Chuang LM, Boeke JD (2011) Acetylation of yeast AMPK controls intrinsic aging independently of caloric restriction. Cell 146(6):969–979. doi:10.1016/j.cell.2011.07.044 S0092-8674(11)00888-9 [pii]
Ludin K, Jiang R, Carlson M (1998) Glucose-regulated interaction of a regulatory subunit of protein phosphatase 1 with the Snf1 protein kinase in Saccharomyces cerevisiae. Proc Natl Acad Sci USA 95(11):6245–6250
Lutfiyya LL, Iyer VR, DeRisi J, DeVit MJ, Brown PO, Johnston M (1998) Characterization of three related glucose repressors and genes they regulate in Saccharomyces cerevisiae. Genetics 150(4):1377–1391
Ma P, Wera S, Van Dijck P, Thevelein JM (1999) The PDE1-encoded low-affinity phosphodiesterase in the yeast Saccharomyces cerevisiae has a specific function in controlling agonist-induced cAMP signaling. Mol Biol Cell 10(1):91–104
Marion RM, Regev A, Segal E, Barash Y, Koller D, Friedman N, O’Shea EK (2004) Sfp1 is a stress- and nutrient-sensitive regulator of ribosomal protein gene expression. Proc Natl Acad Sci USA 101(40):14315–14322. doi:10.1073/pnas.0405353101
Markwardt DD, Garrett JM, Eberhardy S, Heideman W (1995) Activation of the Ras/cyclic AMP pathway in the yeast Saccharomyces cerevisiae does not prevent G1 arrest in response to nitrogen starvation. J Bacteriol 177(23):6761–6765
Marshall-Carlson L, Celenza JL, Laurent BC, Carlson M (1990) Mutational analysis of the SNF3 glucose transporter of Saccharomyces cerevisiae. Mol Cell Biol 10(3):1105–1115
Martinez-Pastor MT, Marchler G, Schuller C, Marchler-Bauer A, Ruis H, Estruch F (1996) The Saccharomyces cerevisiae zinc finger proteins Msn2p and Msn4p are required for transcriptional induction through the stress response element (STRE). EMBO J 15(9):2227–2235
Matsumoto K, Uno I, Oshima Y, Ishikawa T (1982) Isolation and characterization of yeast mutants deficient in adenylate cyclase and cAMP-dependent protein kinase. Proc Natl Acad Sci USA 79(7):2355–2359
Mayer FV, Heath R, Underwood E, Sanders MJ, Carmena D, McCartney RR, Leiper FC, Xiao B, Jing C, Walker PA, Haire LF, Ogrodowicz R, Martin SR, Schmidt MC, Gamblin SJ, Carling D (2011) ADP regulates SNF1, the Saccharomyces cerevisiae homolog of AMP-activated protein kinase. Cell Metab 14(5):707–714. doi:10.1016/j.cmet.2011.09.009 S1550-4131(11)00355-X [pii]
Mazon MJ, Gancedo JM, Gancedo C (1982) Phosphorylation and inactivation of yeast fructose-bisphosphatase in vivo by glucose and by proton ionophores. A possible role for cAMP. Eur J Biochem 127(3):605–608
Mbonyi K, Beullens M, Detremerie K, Geerts L, Thevelein JM (1988) Requirement of one functional RAS gene and inability of an oncogenic ras variant to mediate the glucose-induced cyclic AMP signal in the yeast Saccharomyces cerevisiae. Mol Cell Biol 8(8):3051–3057
Mbonyi K, van Aelst L, Arguelles JC, Jans AW, Thevelein JM (1990) Glucose-induced hyperaccumulation of cyclic AMP and defective glucose repression in yeast strains with reduced activity of cyclic AMP-dependent protein kinase. Mol Cell Biol 10(9):4518–4523
McCartney RR, Schmidt MC (2001) Regulation of Snf1 kinase. Activation requires phosphorylation of threonine 210 by an upstream kinase as well as a distinct step mediated by the Snf4 subunit. J Biol Chem 276(39):36460–36466. doi:10.1074/jbc.M104418200M104418200 [pii]
Mintzer KA, Field J (1999) The SH3 domain of the S. cerevisiae Cdc25p binds adenylyl cyclase and facilitates Ras regulation of cAMP signalling. Cell Signal 11(2):127–135 S0898-6568(98)00044-8 [pii]
Mitts MR, Bradshaw-Rouse J, Heideman W (1991) Interactions between adenylate cyclase and the yeast GTPase-activating protein IRA1. Mol Cell Biol 11(9):4591–4598
Mizunuma M, Tsubakiyama R, Ogawa T, Shitamukai A, Kobayashi Y, Inai T, Kume K, Hirata D (2013) Ras/cAMP-dependent protein kinase (PKA) regulates multiple aspects of cellular events by phosphorylating the Whi3 cell cycle regulator in budding yeast. J Biol Chem 288(15):10558–10566. doi:10.1074/jbc.M112.402214
Moriya H, Johnston M (2004) Glucose sensing and signaling in Saccharomyces cerevisiae through the Rgt2 glucose sensor and casein kinase I. Proc Natl Acad Sci USA 101(6):1572–1577. doi:10.1073/pnas.03059011010305901101 [pii]
Moriya H, Shimizu-Yoshida Y, Omori A, Iwashita S, Katoh M, Sakai A (2001) Yak1p, a DYRK family kinase, translocates to the nucleus and phosphorylates yeast Pop2p in response to a glucose signal. Genes Dev 15(10):1217–1228. doi:10.1101/gad.884001
Mosley AL, Lakshmanan J, Aryal BK, Ozcan S (2003) Glucose-mediated phosphorylation converts the transcription factor Rgt1 from a repressor to an activator. J Biol Chem 278(12):10322–10327. doi:10.1074/jbc.M212802200M212802200 [pii]
Nakafuku M, Obara T, Kaibuchi K, Miyajima I, Miyajima A, Itoh H, Nakamura S, Arai K, Matsumoto K, Kaziro Y (1988) Isolation of a second yeast Saccharomyces cerevisiae gene (GPA2) coding for guanine nucleotide-binding regulatory protein: studies on its structure and possible functions. Proc Natl Acad Sci USA 85(5):1374–1378
Nehlin JO, Carlberg M, Ronne H (1991) Control of yeast GAL genes by MIG1 repressor: a transcriptional cascade in the glucose response. EMBO J 10(11):3373–3377
Nehlin JO, Ronne H (1990) Yeast MIG1 repressor is related to the mammalian early growth response and Wilms’ tumour finger proteins. EMBO J 9(9):2891–2898
Neigeborn L, Carlson M (1984) Genes affecting the regulation of SUC2 gene expression by glucose repression in Saccharomyces cerevisiae. Genetics 108(4):845–858
Nikawa J, Cameron S, Toda T, Ferguson KM, Wigler M (1987) Rigorous feedback control of cAMP levels in Saccharomyces cerevisiae. Genes Dev 1(9):931–937
Niranjan T, Guo X, Victor J, Lu A, Hirsch JP (2007) Kelch repeat protein interacts with the yeast Galpha subunit Gpa2p at a site that couples receptor binding to guanine nucleotide exchange. J Biol Chem 282(33):24231–24238. doi:10.1074/jbc.M702595200
Noda T, Hoffschulte H, Holzer H (1984) Characterization of fructose 1,6-bisphosphatase from bakers’ yeast. J Biol Chem 259(11):7191–7197
Ozcan S, Dover J, Rosenwald AG, Wolfl S, Johnston M (1996) Two glucose transporters in Saccharomyces cerevisiae are glucose sensors that generate a signal for induction of gene expression. Proc Natl Acad Sci USA 93(22):12428–12432
Ozcan S, Johnston M (1995) Three different regulatory mechanisms enable yeast hexose transporter (HXT) genes to be induced by different levels of glucose. Mol Cell Biol 15(3):1564–1572
Ozcan S, Johnston M (1999) Function and regulation of yeast hexose transporters. Microbiol Mol Biol Rev 63(3):554–569
Pai EF, Krengel U, Petsko GA, Goody RS, Kabsch W, Wittinghofer A (1990) Refined crystal structure of the triphosphate conformation of H-ras p21 at 1.35 A resolution: implications for the mechanism of GTP hydrolysis. EMBO J 9(8):2351–2359
Palomino A, Herrero P, Moreno F (2005) Rgt1, a glucose sensing transcription factor, is required for transcriptional repression of the HXK2 gene in Saccharomyces cerevisiae. Biochem J 388(Pt 2):697–703. doi:10.1042/BJ20050160 BJ20050160 [pii]
Palomino A, Herrero P, Moreno F (2006) Tpk3 and Snf1 protein kinases regulate Rgt1 association with Saccharomyces cerevisiae HXK2 promoter. Nucleic Acids Res 34(5):1427–1438. doi:10.1093/nar/gkl028 34/5/1427 [pii]
Pedruzzi I, Burckert N, Egger P, De Virgilio C (2000) Saccharomyces cerevisiae Ras/cAMP pathway controls post-diauxic shift element-dependent transcription through the zinc finger protein Gis1. EMBO J 19(11):2569–2579. doi:10.1093/emboj/19.11.2569
Pedruzzi I, Dubouloz F, Cameroni E, Wanke V, Roosen J, Winderickx J, De Virgilio C (2003) TOR and PKA signaling pathways converge on the protein kinase Rim15 to control entry into G0. Mol Cell 12(6):1607–1613 S1097276503004854 [pii]
Peeters T, Louwet W, Gelade R, Nauwelaers D, Thevelein JM, Versele M (2006) Kelch-repeat proteins interacting with the Galpha protein Gpa2 bypass adenylate cyclase for direct regulation of protein kinase A in yeast. Proc Natl Acad Sci USA 103(35):13034–13039. doi:10.1073/pnas.0509644103 0509644103 [pii]
Peeters T, Versele M, Thevelein JM (2007) Directly from Galpha to protein kinase A: the kelch repeat protein bypass of adenylate cyclase. Trends Biochem Sci 32(12):547–554. doi:10.1016/j.tibs.2007.09.011 S0968-0004(07)00262-9 [pii]
Phan VT, Ding VW, Li F, Chalkley RJ, Burlingame A, McCormick F (2010) The RasGAP proteins Ira2 and neurofibromin are negatively regulated by Gpb1 in yeast and ETEA in humans. Mol Cell Biol 30(9):2264–2279. doi:10.1128/MCB.01450-08
Pohlig G, Holzer H (1985) Phosphorylation and inactivation of yeast fructose-1,6-bisphosphatase by cyclic AMP-dependent protein kinase from yeast. J Biol Chem 260(25):13818–13823
Polish JA, Kim JH, Johnston M (2005) How the Rgt1 transcription factor of Saccharomyces cerevisiae is regulated by glucose. Genetics 169(2):583–594. doi:10.1534/genetics.104.034512 genetics.104.034512 [pii]
Portela P, Moreno S, Rossi S (2006) Characterization of yeast pyruvate kinase 1 as a protein kinase A substrate, and specificity of the phosphorylation site sequence in the whole protein. Biochem J 396(1):117–126. doi:10.1042/BJ20051642
Powers S, Kataoka T, Fasano O, Goldfarb M, Strathern J, Broach J, Wigler M (1984) Genes in S. cerevisiae encoding proteins with domains homologous to the mammalian ras proteins. Cell 36(3):607–612 0092-8674(84)90340-4 [pii]
Ptacek J, Devgan G, Michaud G, Zhu H, Zhu X, Fasolo J, Guo H, Jona G, Breitkreutz A, Sopko R, McCartney RR, Schmidt MC, Rachidi N, Lee SJ, Mah AS, Meng L, Stark MJ, Stern DF, De Virgilio C, Tyers M, Andrews B, Gerstein M, Schweitzer B, Predki PF, Snyder M (2005) Global analysis of protein phosphorylation in yeast. Nature 438(7068):679–684. doi:10.1038/nature04187 nature04187 [pii]
Purwin C, Leidig F, Holzer H (1982) Cyclic AMP-dependent phosphorylation of fructose-1,6-bisphosphatase in yeast. Biochem Biophys Res Commun 107(4):1482–1489 S0006-291X(82)80166-6 [pii]
Purwin C, Nicolay K, Scheffers WA, Holzer H (1986) Mechanism of control of adenylate cyclase activity in yeast by fermentable sugars and carbonyl cyanide m-chlorophenylhydrazone. J Biol Chem 261(19):8744–8749
Rahner A, Hiesinger M, Schuller HJ (1999) Deregulation of gluconeogenic structural genes by variants of the transcriptional activator Cat8p of the yeast Saccharomyces cerevisiae. Mol Microbiol 34(1):146–156 mmi1588 [pii]
Randez-Gil F, Bojunga N, Proft M, Entian KD (1997) Glucose derepression of gluconeogenic enzymes in Saccharomyces cerevisiae correlates with phosphorylation of the gene activator Cat8p. Mol Cell Biol 17(5):2502–2510
Reinders A, Burckert N, Boller T, Wiemken A, De Virgilio C (1998) Saccharomyces cerevisiae cAMP-dependent protein kinase controls entry into stationary phase through the Rim15p protein kinase. Genes Dev 12(18):2943–2955
Robertson LS, Fink GR (1998) The three yeast A kinases have specific signaling functions in pseudohyphal growth. Proc Natl Acad Sci USA 95(23):13783–13787
Robinson LC, Hubbard EJ, Graves PR, DePaoli-Roach AA, Roach PJ, Kung C, Haas DW, Hagedorn CH, Goebl M, Culbertson MR et al (1992) Yeast casein kinase I homologues: an essential gene pair. Proc Natl Acad Sci USA 89(1):28–32
Roelants FM, Torrance PD, Thorner J (2004) Differential roles of PDK1- and PDK2-phosphorylation sites in the yeast AGC kinases Ypk1, Pkc1 and Sch9. Microbiology 150(Pt 10):3289–3304. doi:10.1099/mic.0.27286-0 150/10/3289 [pii]
Rolland F, De Winde JH, Lemaire K, Boles E, Thevelein JM, Winderickx J (2000) Glucose-induced cAMP signalling in yeast requires both a G-protein coupled receptor system for extracellular glucose detection and a separable hexose kinase-dependent sensing process. Mol Microbiol 38(2):348–358 mmi2125 [pii]
Rolland F, Winderickx J, Thevelein JM (2001) Glucose-sensing mechanisms in eukaryotic cells. Trends Biochem Sci 26(5):310–317
Rolland F, Winderickx J, Thevelein JM (2002) Glucose-sensing and -signalling mechanisms in yeast. FEMS Yeast Res 2(2):183–201
Roosen J, Engelen K, Marchal K, Mathys J, Griffioen G, Cameroni E, Thevelein JM, De Virgilio C, De Moor B, Winderickx J (2005) PKA and Sch9 control a molecular switch important for the proper adaptation to nutrient availability. Mol Microbiol 55(3):862–880. doi:10.1111/j.1365-2958.2004.04429.x MMI4429 [pii]
Roy A, Shin YJ, Cho KH, Kim JH (2013) Mth1 regulates the interaction between the Rgt1 repressor and the Ssn6-Tup1 corepressor complex by modulating PKA-dependent phosphorylation of Rgt1. Mol Biol Cell 24(9):1493–1503. doi:10.1091/mbc.E13-01-0047
Rubenstein EM, McCartney RR, Zhang C, Shokat KM, Shirra MK, Arndt KM, Schmidt MC (2008) Access denied: Snf1 activation loop phosphorylation is controlled by availability of the phosphorylated threonine 210 to the PP1 phosphatase. J Biol Chem 283(1):222–230. doi:10.1074/jbc.M707957200 M707957200 [pii]
Ruiz A, Xu X, Carlson M (2011) Roles of two protein phosphatases, Reg1-Glc7 and Sit4, and glycogen synthesis in regulation of SNF1 protein kinase. Proc Natl Acad Sci USA 108(16):6349–6354. doi:10.1073/pnas.1102758108 1102758108 [pii]
Santangelo GM (2006) Glucose signaling in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 70(1):253–282. doi:10.1128/MMBR.70.1.253-282.2006 70/1/253 [pii]
Sanz P, Alms GR, Haystead TA, Carlson M (2000) Regulatory interactions between the Reg1-Glc7 protein phosphatase and the Snf1 protein kinase. Mol Cell Biol 20(4):1321–1328
Schepers W, Van Zeebroeck G, Pinkse M, Verhaert P, Thevelein JM (2012) In vivo phosphorylation of Ser21 and Ser83 during nutrient-induced activation of the yeast protein kinase A (PKA) target trehalase. J Biol Chem 287(53):44130–44142. doi:10.1074/jbc.M112.421503
Schmitt AP, McEntee K (1996) Msn2p, a zinc finger DNA-binding protein, is the transcriptional activator of the multistress response in Saccharomyces cerevisiae. Proc Natl Acad Sci USA 93(12):5777–5782
Schomerus C, Munder T, Kuntzel H (1990) Site-directed mutagenesis of the Saccharomyces cerevisiae CDC25 gene: effects on mitotic growth and cAMP signalling. Mol Gen Genet 223(3):426–432
Schuller HJ (2003) Transcriptional control of nonfermentative metabolism in the yeast Saccharomyces cerevisiae. Curr Genet 43(3):139–160. doi:10.1007/s00294-003-0381-8
Smets B, Ghillebert R, De Snijder P, Binda M, Swinnen E, De Virgilio C, Winderickx J (2010) Life in the midst of scarcity: adaptations to nutrient availability in Saccharomyces cerevisiae. Curr Genet 56(1):1–32. doi:10.1007/s00294-009-0287-1
Smith A, Ward MP, Garrett S (1998) Yeast PKA represses Msn2p/Msn4p-dependent gene expression to regulate growth, stress response and glycogen accumulation. EMBO J 17(13):3556–3564. doi:10.1093/emboj/17.13.3556
Sutherland CM, Hawley SA, McCartney RR, Leech A, Stark MJ, Schmidt MC, Hardie DG (2003) Elm1p is one of three upstream kinases for the Saccharomyces cerevisiae SNF1 complex. Curr Biol 13(15):1299–1305 S0960982203004597 [pii]
Tabba S, Mangat S, McCartney R, Schmidt MC (2010) PP1 phosphatase-binding motif in Reg1 protein of Saccharomyces cerevisiae is required for interaction with both the PP1 phosphatase Glc7 and the Snf1 protein kinase. Cell Signal 22(7):1013–1021. doi:10.1016/j.cellsig.2010.02.003 S0898-6568(10)00056-2 [pii]
Tanaka K, Nakafuku M, Satoh T, Marshall MS, Gibbs JB, Matsumoto K, Kaziro Y, Toh-e A (1990) S. cerevisiae genes IRA1 and IRA2 encode proteins that may be functionally equivalent to mammalian ras GTPase activating protein. Cell 60(5):803–807 0092-8674(90)90094-U [pii]
Tatchell K, Chaleff DT, DeFeo-Jones D, Scolnick EM (1984) Requirement of either of a pair of ras-related genes of Saccharomyces cerevisiae for spore viability. Nature 309(5968):523–527
Thevelein JM (1984a) Cyclic-AMP content and trehalase activation in vegetative cells and ascospores of yeast. Arch Microbiol 138(1):64–67
Thevelein JM (1984b) Regulation of trehalose mobilization in fungi. Microbiol Rev 48(1):42–59
Thevelein JM (1991) Fermentable sugars and intracellular acidification as specific activators of the RAS-adenylate cyclase signalling pathway in yeast: the relationship to nutrient-induced cell cycle control. Mol Microbiol 5(6):1301–1307
Thevelein JM (1994) Signal transduction in yeast. Yeast 10(13):1753–1790
Thevelein JM, Beullens M (1985) Cyclic AMP and the stimulation of trehalase activity in the yeast Saccharomyces cerevisiae by carbon sources, nitrogen sources and inhibitors of protein synthesis. J Gen Microbiol 131(12):3199–3209
Thevelein JM, Beullens M, Honshoven F, Hoebeeck G, Detremerie K, den Hollander JA, Jans AWH (1987a) Regulation of the cAMP level in the yeast Saccharomyces cerevisiae: intracellular pH and the effect of membrane depolarizing compounds. J Gen Microbiol 133:2191–2196
Thevelein JM, Beullens M, Honshoven F, Hoebeeck G, Detremerie K, Griewel B, den Hollander JA, Jans AWH (1987b) Regulation of the cAMP level in the yeast Saccharomyces cerevisiae: the glucose-induced cAMP signal is not mediated by a transient drop in the intracellular pH. J Gen Microbiol 133:2197–2205
Thevelein JM, Bonini BM, Castermans D, Haesendonckx S, Kriel J, Louwet W, Thayumanavan P, Popova Y, Rubio-Texeira M, Schepers W, Vandormael P, Van Zeebroeck G, Verhaert P, Versele M, Voordeckers K (2008) Novel mechanisms in nutrient activation of the yeast protein kinase A pathway. Acta Microbiol Immunol Hung 55(2):75–89. doi:10.1556/AMicr.55.2008.2.1
Thevelein JM, Cauwenberg L, Colombo S, De Winde JH, Donation M, Dumortier F, Kraakman L, Lemaire K, Ma P, Nauwelaers D, Rolland F, Teunissen A, Van Dijck P, Versele M, Wera S, Winderickx J (2000) Nutrient-induced signal transduction through the protein kinase A pathway and its role in the control of metabolism, stress resistance, and growth in yeast. Enzyme Microb Technol 26(9–10):819–825 S0141022900001770 [pii]
Thevelein JM, de Winde JH (1999) Novel sensing mechanisms and targets for the cAMP-protein kinase A pathway in the yeast Saccharomyces cerevisiae. Mol Microbiol 33(5):904–918 mmi1538 [pii]
Thevelein JM, Gelade R, Holsbeeks I, Lagatie O, Popova Y, Rolland F, Stolz F, Van de Velde S, Van Dijck P, Vandormael P, Van Nuland A, Van Roey K, Van Zeebroeck G, Yan B (2005) Nutrient sensing systems for rapid activation of the protein kinase A pathway in yeast. Biochem Soc Trans 33(Pt 1):253–256. doi:10.1042/BST0330253 BST0330253 [pii]
Thevelein JM, Voordeckers K (2009) Functioning and evolutionary significance of nutrient transceptors. Mol Biol Evol 26(11):2407–2414. doi:10.1093/molbev/msp168 msp168 [pii]
Thompson-Jaeger S, Francois J, Gaughran JP, Tatchell K (1991) Deletion of SNF1 affects the nutrient response of yeast and resembles mutations which activate the adenylate cyclase pathway. Genetics 129(3):697–706
Toda T, Cameron S, Sass P, Wigler M (1988) SCH9, a gene of Saccharomyces cerevisiae that encodes a protein distinct from, but functionally and structurally related to, cAMP-dependent protein kinase catalytic subunits. Genes Dev 2(5):517–527
Toda T, Cameron S, Sass P, Zoller M, Scott JD, McMullen B, Hurwitz M, Krebs EG, Wigler M (1987a) Cloning and characterization of BCY1, a locus encoding a regulatory subunit of the cyclic AMP-dependent protein kinase in Saccharomyces cerevisiae. Mol Cell Biol 7(4):1371–1377
Toda T, Cameron S, Sass P, Zoller M, Wigler M (1987b) Three different genes in S. cerevisiae encode the catalytic subunits of the cAMP-dependent protein kinase. Cell 50(2):277–287 0092-8674(87)90223-6 [pii]
Toda T, Uno I, Ishikawa T, Powers S, Kataoka T, Broek D, Cameron S, Broach J, Matsumoto K, Wigler M (1985) In yeast, RAS proteins are controlling elements of adenylate cyclase. Cell 40(1):27–36 0092-8674(85)90305-8 [pii]
Tortora P, Burlini N, Hanozet GM, Guerritore A (1982) Effect of caffeine on glucose-induced inactivation of gluconeogenetic enzymes in Saccharomyces cerevisiae. A possible role of cyclic AMP. Eur J Biochem 126(3):617–622
Treitel MA, Carlson M (1995) Repression by SSN6-TUP1 is directed by MIG1, a repressor/activator protein. Proc Natl Acad Sci USA 92(8):3132–3136
Tu J, Carlson M (1995) REG1 binds to protein phosphatase type 1 and regulates glucose repression in Saccharomyces cerevisiae. EMBO J 14(23):5939–5946
Tu J, Song W, Carlson M (1996) Protein phosphatase type 1 interacts with proteins required for meiosis and other cellular processes in Saccharomyces cerevisiae. Mol Cell Biol 16(8):4199–4206
Upadhyay M, Samal J, Kandpal M, Singh OV, Vivekanandan P (2013) The Warburg effect: insights from the past decade. Pharmacol Ther 137(3):318–330. doi:10.1016/j.pharmthera.2012.11.003
Urban J, Soulard A, Huber A, Lippman S, Mukhopadhyay D, Deloche O, Wanke V, Anrather D, Ammerer G, Riezman H, Broach JR, De Virgilio C, Hall MN, Loewith R (2007) Sch9 is a major target of TORC1 in Saccharomyces cerevisiae. Mol Cell 26(5):663–674. doi:10.1016/j.molcel.2007.04.020 S1097-2765(07)00256-0 [pii]
van Aelst L, Boy-Marcotte E, Camonis JH, Thevelein JM, Jacquet M (1990) The C-terminal part of the CDC25 gene product plays a key role in signal transduction in the glucose-induced modulation of cAMP level in Saccharomyces cerevisiae. Eur J Biochem 193(3):675–680
van Aelst L, Jans AW, Thevelein JM (1991) Involvement of the CDC25 gene product in the signal transmission pathway of the glucose-induced RAS-mediated cAMP signal in the yeast Saccharomyces cerevisiae. J Gen Microbiol 137(2):341–349
Van de Velde S, Thevelein JM (2008) Cyclic AMP-protein kinase A and Snf1 signaling mechanisms underlie the superior potency of sucrose for induction of filamentation in Saccharomyces cerevisiae. Eukaryot Cell 7(2):286–293. doi:10.1128/EC.00276-07
van der Plaat JB (1974) Cyclic 3′,5′—adenosine monophosphate stimulates trehalose degradation in bakers’ yeast. Biochem Biophys Res Commun 56:580–587
Van Nuland A, Vandormael P, Donaton M, Alenquer M, Lourenco A, Quintino E, Versele M, Thevelein JM (2006) Ammonium permease-based sensing mechanism for rapid ammonium activation of the protein kinase A pathway in yeast. Mol Microbiol 59(5):1485–1505. doi:10.1111/j.1365-2958.2005.05043.x
Versele M, de Winde JH, Thevelein JM (1999) A novel regulator of G protein signalling in yeast, Rgs2, downregulates glucose-activation of the cAMP pathway through direct inhibition of Gpa2. EMBO J 18(20):5577–5591. doi:10.1093/emboj/18.20.5577
Vidan S, Mitchell AP (1997) Stimulation of yeast meiotic gene expression by the glucose-repressible protein kinase Rim15p. Mol Cell Biol 17(5):2688–2697
Vincent O, Carlson M (1998) Sip4, a Snf1 kinase-dependent transcriptional activator, binds to the carbon source-responsive element of gluconeogenic genes. EMBO J 17(23):7002–7008. doi:10.1093/emboj/17.23.7002
Vincent O, Kuchin S, Hong SP, Townley R, Vyas VK, Carlson M (2001) Interaction of the Srb10 kinase with Sip4, a transcriptional activator of gluconeogenic genes in Saccharomyces cerevisiae. Mol Cell Biol 21(17):5790–5796
Wang Y, Pierce M, Schneper L, Guldal CG, Zhang X, Tavazoie S, Broach JR (2004) Ras and Gpa2 mediate one branch of a redundant glucose signaling pathway in yeast. PLoS Biol 2(5):E128. doi:10.1371/journal.pbio.0020128
Wanke V, Cameroni E, Uotila A, Piccolis M, Urban J, Loewith R, De Virgilio C (2008) Caffeine extends yeast lifespan by targeting TORC1. Mol Microbiol 69(1):277–285. doi:10.1111/j.1365-2958.2008.06292.x MMI6292 [pii]
Werner-Washburne M, Brown D, Braun E (1991) Bcy1, the regulatory subunit of cAMP-dependent protein kinase in yeast, is differentially modified in response to the physiological status of the cell. J Biol Chem 266(29):19704–19709
Wieser R, Adam G, Wagner A, Schuller C, Marchler G, Ruis H, Krawiec Z, Bilinski T (1991) Heat shock factor-independent heat control of transcription of the CTT1 gene encoding the cytosolic catalase T of Saccharomyces cerevisiae. J Biol Chem 266(19):12406–12411
Wilson WA, Roach PJ, Montero M, Baroja-Fernandez E, Munoz FJ, Eydallin G, Viale AM, Pozueta-Romero J (2010) Regulation of glycogen metabolism in yeast and bacteria. FEMS Microbiol Rev 34(6):952–985. doi:10.1111/j.1574-6976.2010.00220.x
Woods A, Munday MR, Scott J, Yang X, Carlson M, Carling D (1994) Yeast SNF1 is functionally related to mammalian AMP-activated protein kinase and regulates acetyl-CoA carboxylase in vivo. J Biol Chem 269(30):19509–19515
Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, Jing C, Walker PA, Eccleston JF, Haire LF, Saiu P, Howell SA, Aasland R, Martin SR, Carling D, Gamblin SJ (2011) Structure of mammalian AMPK and its regulation by ADP. Nature 472(7342):230–233. doi:10.1038/nature09932 nature09932 [pii]
Xiaojia B, Jian D (2010) Serine214 of Ras2p plays a role in the feedback regulation of the Ras-cAMP pathway in the yeast Saccharomyces cerevisiae. FEBS Lett 584(11):2333–2338. doi:10.1016/j.febslet.2010.04.011
Xue Y, Batlle M, Hirsch JP (1998) GPR1 encodes a putative G protein-coupled receptor that associates with the Gpa2p Galpha subunit and functions in a Ras-independent pathway. EMBO J 17(7):1996–2007. doi:10.1093/emboj/17.7.1996
Yun CW, Tamaki H, Nakayama R, Yamamoto K, Kumagai H (1997) G-protein coupled receptor from yeast Saccharomyces cerevisiae. Biochem Biophys Res Commun 240(2):287–292. doi:10.1006/bbrc.1997.7649 S0006-291X(97)97649-X [pii]
Zaman S, Lippman SI, Schneper L, Slonim N, Broach JR (2009) Glucose regulates transcription in yeast through a network of signaling pathways. Mol Syst Biol 5:245. doi:10.1038/msb.2009.2 msb20092 [pii]
Zaman S, Lippman SI, Zhao X, Broach JR (2008) How Saccharomyces responds to nutrients. Annu Rev Genet 42:27–81. doi:10.1146/annurev.genet.41.110306.130206
Zeller CE, Parnell SC, Dohlman HG (2007) The RACK1 ortholog Asc1 functions as a G-protein beta subunit coupled to glucose responsiveness in yeast. J Biol Chem 282(34):25168–25176. doi:10.1074/jbc.M702569200 M702569200 [pii]
Zhang A, Gao W (2012) Mechanisms of protein kinase Sch9 regulating Bcy1 in Saccharomyces cerevisiae. FEMS Microbiol Lett 331(1):10–16. doi:10.1111/j.1574-6968.2012.02552.x
Zhang A, Shen Y, Gao W, Dong J (2011) Role of Sch9 in regulating Ras-cAMP signal pathway in Saccharomyces cerevisiae. FEBS Lett 585(19):3026–3032. doi:10.1016/j.febslet.2011.08.023 S0014-5793(11)00625-9 [pii]
Zhang N, Wu J, Oliver SG (2009) Gis1 is required for transcriptional reprogramming of carbon metabolism and the stress response during transition into stationary phase in yeast. Microbiology 155(Pt 5):1690–1698. doi:10.1099/mic.0.026377-0
Acknowledgments
Original research performed in our laboratory was supported by a predoctoral fellowship to KP from the Agency for Innovation by Science and Technology (IWT-Flanders) and by grants from the Fund for Scientific Research—Flanders, Interuniversity Attraction Poles Network P6/14 and P7/40, the Research Fund of the KU Leuven (Concerted Research Actions) and the Hercules Foundation (Flanders) to JMT.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Peeters, K., Thevelein, J.M. (2014). Glucose Sensing and Signal Transduction in Saccharomyces cerevisiae . In: Piškur, J., Compagno, C. (eds) Molecular Mechanisms in Yeast Carbon Metabolism. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-55013-3_2
Download citation
DOI: https://doi.org/10.1007/978-3-642-55013-3_2
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-55012-6
Online ISBN: 978-3-642-55013-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)