
Abstract
Metformin, an inexpensive, well-tolerated oral agent that is commonly used in the first-line treatment for type 2 diabetes, has become the focus of intense research as a potential anticancer agent. This research reflects a convergence of epidemiologic, clinical, and preclinical evidence, suggesting that metformin may lower cancer risk in diabetics and improve outcomes of many common cancers. Notably, metformin mediates an approximately 30 % reduction in the lifetime risk of cancer in diabetic patients. There is growing recognition that metformin may act (1) directly on cancer cells, primarily by impacting mitochondrial respiration leading to the activation of the AMP-activated protein kinase (AMPK), which controls energy homeostasis in cells, but also through other mechanisms or (2) indirectly on the host metabolism, largely through AMPK-mediated reduction in hepatic gluconeogenesis, leading to reduced circulating insulin levels and decreased insulin/IGF-1 receptor-mediated activation of the PI3K pathway. Support for this comes from the observation that metformin inhibits cancer cell growth in vitro and delays the onset of tobacco carcinogen-induced lung cancer in mice and that metformin and its analog phenformin delay spontaneous tumor development cancer-prone transgenic mice. The potential for both direct antitumor effects and indirect host-mediated effects has sparked enormous interest, but has led to added challenges in translating preclinical findings to the clinical setting. Nonetheless, the accumulation of evidence has been sufficient to justify initiation of clinical trials of metformin as an anticancer agent in the clinical setting, including a large-scale adjuvant study in breast cancer, with additional studies planned.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others
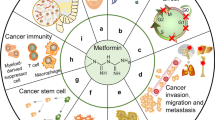
Keywords
1 Introduction
Diabetes mellitus (DM) is a metabolic disorder that is characterized by chronic hyperglycemia and aberrant carbohydrate, fat, and protein metabolism that result from defects in insulin secretion, insulin action, or both. It represents a major global health problem that has been recognized and treated for centuries. Since the Middle Ages, Europeans have treated “thirst and frequent urination”, two symptoms of DM patients, using extracts of Galega officinalis, an herbaceous plant that was later found to contain active components such as galegine, a guanidine derivate. In the 1920s, galegine was found to exhibit the ability to decrease blood sugar and insulin resistance. In the 1950s, biguanide derivates such as phenformin and metformin were introduced into the clinic and represented an important milestone in the development of oral antidiabetic pharmacotherapy. Phenformin has been withdrawn from usage in the United States and in most other countries since 1978 due to rare fatal cases of lactic acidosis, whereas metformin still represents a preferred front-line agent for antidiabetic therapy alongside diet and exercise because of its transient GI toxicity and very rare severe lactic acidosis toxicity [80].
Recently, a series of studies and meta-analyses have shown an increased risk of cancer in DM patients. In particular, meta-analyses have revealed a strong association between diabetes and cancers of the pancreas or liver, the main organs implicated during the deregulation of the metabolic equilibrium that is typical in DM [80]. Altered metabolic pathologies, such as hyperglycemia and hyperinsulinemia, as well as other DM-associated factors, such as obesity and high saturated fat diets, are also independent risk factors for cancer, illustrating a close correlation between the two diseases [39].
In contrast, several retrospective epidemiological reports have highlighted that the use of metformin in diabetic patients correlated with a reduced lifetime incidence of cancer. Moreover, metabolic reprogramming is now considered an emerging critical hallmark of cancer and represents a target for anticancer therapy [73]. In this regard, among the 24 provocative questions posed last year by NCI director Harold Varmus on a dedicated Web site,Footnote 1 which aim to identify perplexing problems to guide progress in cancer research, a critical one is how we determine the mechanism by which some drugs, commonly and chronically used for other indications (such as metformin and others), can protect against cancer incidence and mortality [9]. Thus, is metformin a diabetic drug for cancer or it is a cancer drug for diabetics? [53]. In this chapter, we will review all of these aspects in an effort to navigate through this highly debated field and to answer some of these questions. Thus, it is not surprising to observe that in the last 3 years, an increasing number of studies about metformin and cancer have been published on PubMed (Fig. 1).

The graph shows the increasing number of citations regarding the relationship between metformin and cancer, recorded in PubMed in the last 20 years (adjusted at May 2012)
2 Cancer and Metabolism
The increased glucose uptake of cancer cells is a phenomenon that was first described by the Nobel Prize winner Otto Warburg in the 1920s. The so-called Warburg effect is the observation that the metabolism of most tumor cells is characterized by increased glycolysis that is maintained even during conditions of high oxygen tension (i.e., aerobic glycolysis), followed by elevated lactate production levels. This metabolic status contrasts with the low glycolytic rates associated with the oxidation of mitochondrial pyruvate that are exhibited by most normal cells [12]. Moreover, Warburg suggested that this altered metabolism is a fundamental cause of cancer. At present, we know that cancer cells use the elevated amounts of glucose as a carbon source for anabolic reactions to drive glycolytic intermediates into various biosynthetic pathways that are essential for cancer cell growth [47]. Notably, as the alteration in glucose metabolism occurs early during carcinogenesis, the Warburg effect represents the basis of the current usage of positron emission tomography (PET) for the diagnosis of cancer using radiolabeled deoxyglucose, which exhibits increased uptake by both primary and metastatic cancer cells [25, 79].
The preferential use of aerobic glycolysis facilitates the survival of the highly proliferative cells during conditions of continuously changing oxygen tension, which is otherwise lethal for the normal cells that depend on oxidative phosphorylation for ATP production [68]. Moreover, cancer cells, by producing lactic acid, affect cellular pH, which can promote tumor invasion and suppression of anticancer immune effectors. Finally, tumors can metabolize glucose via the pentose phosphate system to generate NADPH, which can contribute to fatty acid synthesis and defend against chemotherapeutic agents [25, 73].
Although the Warburg effect has been known for many years, the precise molecular mechanisms underlying this phenomenon remain unknown. Consequently, it is also difficult to identify a selective target for anticancer approaches. Some reports have suggested that hexokinase, which is a metabolic enzyme that catalyzes the conversion of glucose to glucose-6-phosphate and which is the rate-limiting first step in the glycolytic pathway, might represent an anticancer target [87]. In addition, nicotinamide phosphoribosyltransferase (Nampt) and its product, nicotinamide adenine dinucleotide (NAD), which play crucial roles in the regulation of several metabolic reactions that are implicated in the glycolytic pathway as well as in the regulation of factors that affect both tumor progression and the inflammatory response (e.g., sirtuins), might represent therapeutic targets [24].
The increased de novo biosynthesis of fatty acids has also been considered a crucial metabolic alteration of cancer cells and is associated with the hyperactivity of lipogenic enzymes such as ATP citrate lyase (ACL), acetyl-CoA carboxylase (ACC), and fatty acid synthase (FAS) [48]. FAS is overexpressed in many cancers [81], and chemical inhibitors of FAS have been shown to decrease proliferation and increase apoptosis in cancer cells [48]. Recent studies have suggested that in cancer cells, the Warburg effect might depend on mitochondrial uncoupling—the abrogation of ATP synthesis in response to mitochondrial membrane potential in cancer cells—leading to a decreased pyruvate flux through the Krebs cycle and a shift in the oxidation of non-glucose carbon sources (e.g., fatty acids) to maintain mitochondrial integrity and function [71]. However, other reports have suggested that the Warburg effect does not necessarily involve in permanent mitochondrial dysfunction [87]. Interestingly, both uncoupled mitochondria, which render cells more resistant to cytotoxic insults, and increased fatty acid oxidation, which has been linked to chemoresistance, might represent additional therapeutic targets for cancer treatment.
Another relevant target for the metabolic reprogramming of cancer cells is the hypoxia-inducible factor 1α (HIF-1α), a transcription factor that mediates the hypoxia-induced gene expression changes that are thought to be adaptive for cells upon exposure to a reduced oxygen environment. Such genes encompass those involved in glycolysis and include the glucose transporters (GLUT1 and GLUT3). Moreover, recent reports have demonstrated that HIF-1α induces pyruvate dehydrogenase kinase 1 and 3 (PDK1-3) expression, which facilitates the phosphorylation and inhibition of pyruvate dehydrogenase and, consequently, mitochondrial respiration in cancer cells [44, 52, 65].
In summary, the switching of cellular metabolism from mitochondrial respiration to glycolysis drives cancer transformation and progression as well as chemoresistance and should therefore be considered a crucial target for anticancer therapy.
3 Diabetes and Cancer
DM comprises a group of metabolic disorders, which include two predominant subtypes—types 1 and 2—that are characterized by different metabolic activities. Type 1 diabetes (5–10 % of all diabetics) is associated with the complete absence of endogenous insulin that is attributed to the autoimmune destruction of insulin-secreting β-pancreatic cells and requires the exogenous administration of insulin. In contrast, type 2 diabetes (90 % of all diabetics) is characterized by the long-term presence of hyperglycemia and hyperinsulinemia associated with insulin resistance in the peripheral tissues. In the latter subtype of diabetes, treatment with exogenous insulin is required only when the β-pancreatic cells become non-functional [39, 80]. Currently, a large variety of drugs are available for the treatment for DM, including insulin, insulin analogs, and insulin secretagogues that function by compensating for the lack of insulin production by the patient’s β-pancreatic cells. Another class of drugs includes insulin sensitizers, such as the oral antidiabetic metformin or the thiazolidinediones, which can overcome insulin resistance. Lastly, glucosidase inhibitors, such as acarbose, function therapeutically by acting on carbohydrate digestion to prevent the development of post-prandial hyperglycemia. Generally, all of the classes of diabetic medications are coupled with recommended lifestyle changes including diet and exercise, which might decrease or prevent the obesity that is often associated with DM [62].
DM is associated with several complications, such as retinopathy, nephropathy, cardiovascular diseases, and, as reported in several studies, with an increased risk of cancer.
Several meta-analyses have evaluated the relative risk (RR) of cancer in both case-control and cohort studies of diabetics, demonstrating a mild increase in cancer (and in cancer mortality). Although this increase applies to both solid and hematological malignancies, it is more evident for certain site-specific cancers. Elevated relative risk (RR) has been shown for pancreatic (RR 2.50, 95 % CI 1.8–3.5) and liver (RR 1.94, 95 % CI 1.53–2.46) cancers, which are the main organs involved in the development of DM [22, 37].
The precise relationship between diabetes and pancreatic cancer is difficult to delineate because previous meta-analyses did not distinguish whether diabetes was a preexisting condition that promoted the development of exocrine pancreatic cancer or if it was a consequence of cancer.
Recently, the diagnosis of new-onset diabetes has been used as a potential early diagnostic indicator for pancreatic cancer in screening programs enrolling middle-aged patients. In elderly patients, it represents a high probability indicator of pancreatic cancer, with a 3 year risk that is eight times higher in diabetic than in non-diabetic patients of similar age and sex. Additional studies have demonstrated that new-onset diabetes is an early event that is attributed to cytokine production by pancreatic tumors rather than to alterations in normal pancreatic tissues [15, 64]. Interestingly, the RR remained higher for new-onset diabetic patients when the meta-analyses were adjusted for age, race, and cigarette smoking as well as for post-load glucose levels. These findings indicated that hyperglycemia and prediabetic status might represent risk factors for pancreatic cancer and that insulin plays a prominent role in promoting cancer progression. However, insulin alone is insufficient to promote cancer progression because the pancreatic cells in type 1 diabetic patients are not exposed to increased insulin levels compared to other tissues [64, 80].
The increased incidence of liver cancer and, in particular, of hepatocellular carcinoma (HCC) in diabetic patients is mainly due to the elevated insulin levels that the liver cells are exposed to via portal circulation. This condition is evident in both physiological and pathological situations, particularly in type 2 DM, which is characterized by exacerbated states of hyperinsulinemia and insulin resistance. Significantly, in type 1 diabetic patients treated with exogenous insulin, the insulin levels in the liver cells are the same as those in other organs. As has been previously described for pancreatic cancer, the elevated insulin levels in liver cancer are insufficient to explain the correlation between cancer and diabetes. Therefore, several epidemiological studies have analyzed other factors. For instance, hepatic steatosis and cirrhosis, as well as hepatitis B and C infections, have been implicated as connecting factors between the two diseases. Similarly, non-alcoholic fatty liver disease (NAFLD) might represent a main cancer risk factor in obese diabetic patients and in 80 % of type 2 diabetic patients [16, 18].
In conclusion, the relationship between diabetes and cancer remains unclear and requires re-evaluation because DM is not a singular disease. Rather, DM is a group of metabolic disorders in which each disorder is characterized by its own metabolic and hormonal abnormalities that affect patients differently. Thus, it is difficult to consider diabetic patients as a homogeneous cohort, and further studies are required to understand the complex relationship between cancer and diabetes.
Moreover, a group of confounding factors exist which are based on lifestyle, such as lack of physical activity, obesity, smoking, sex, ethnicity, comorbidity, duration of treatment, quality of metabolic control, and number of antidiabetic drugs changed during the treatments. Such factors might influence the meta-analysis reports. In this regard, a recent study has demonstrated that whereas insulin levels might represent a physiological indicator for early follow-up (first 5 years after diagnosis), the obesity-related factors, such as leptin, might represent an important marker over time for long-term follow-up of breast cancer [27].
At the molecular level, several mechanisms might account for the tumor growth observed in diabetic patients. Diabetes can promote carcinogenesis through the action of insulin and its complex downstream signaling network, which induces not only a modulation of metabolic pathways but also a modulation of mitogenic signaling via the following two distinct mechanisms: (a) systemic mechanisms attributed to specific alterations including hyperglycemia and hyperinsulinemia and (b) site-specific mechanisms that affect specific organs [80]. Interestingly, hyperinsulinemia can promote tumorigenesis directly by activating the insulin receptor (IR) in epithelial tissues or indirectly by influencing the levels of other modulators such as insulin growth factors (IGFs), sex hormones, and inflammatory mediators. For instance, when insulin binds to the A isoform of IR, which is predominantly expressed in cancer cells, it can trigger mitogenic signaling pathways that act through adaptor proteins such as IR substrates (IRS1-4). This signaling results in the activation of the mitogen-activated protein kinase (MAPK) pathway—activation that is preserved in the presence of insulin resistance—and in the induction of survival signaling that is mediated by PI3K, by AKT, and by the mammalian target of rapamycin (mTOR) [39, 55]. As mentioned above, insulin can also act indirectly via IGFs and their cognate receptors (IGF-Rs). Insulin resistance and elevated levels of insulin can displace IGFs from insulin growth factor binding proteins (IGFBPs), resulting in increased levels of free IGFs, which can constitutively promote tumor growth and cancer progression. Breast cancer cell lines have been observed to exhibit an interaction between insulin with the IR-A homodimer, the isoform of the IR that is widely expressed in this tumor, and insulin with the IR-A/IGF1R heterodimer to stimulate tumorigenesis and to promote survival pathways [6, 56, 66]. Moreover, the polymorphic form of IGF1R has been demonstrated to be associated with non-small-cell lung cancer, whereas a polymorphism of IGF2R is correlated with gastric cancer risk [36, 86]. Furthermore, the activation of IGF1R/IR has been detected at elevated levels in breast cancers, independently of the specific tumor subtype (e.g., luminal, triple negative, or Her2+) and is associated with poor survival and resistance to targeted therapies, including those targeting the estrogen receptor (ER) or the Her family members. Moreover, the authors suggested that a specific IGF-IR tyrosine kinase inhibitor, BMS-536924, can be used to overcome such resistance and to promote improved survival, independently of breast cancer subtype [50].
Ultimately, elevated insulin levels are associated with insulin resistance and can promote tumorigenesis by increasing the free estrogen levels produced in the ovaries, by blocking sex-hormone-binding globulin, and by increasing the conversion of androgen to estrogen in adipose tissue [39, 80].
4 Metformin and Cancer
4.1 Epidemiological Studies
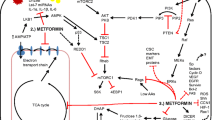
Metformin (1, 1-dimethylbiguanide hydrochloride) is a biguanide derivative and belongs to a class of oral hypoglycemic agents. Metformin acts principally on hepatocytes, myocytes, adipocytes, and β cells in the pancreas (Fig. 2). In the liver, metformin inhibits hepatic glucose production while increasing insulin sensitivity in the peripheral tissues, leading to increased glucose uptake and usage by the skeletal muscle and adipose tissues. The primary effects are a reduction in plasma insulin and glucose levels, followed by an enhancement of blood glucose control and a decreased incidence of complications that are correlated with diabetes [8, 20]. At the molecular level, the principal metabolic mediator of the glucose-lowering effects of metformin is the activation of AMPK, a serine–threonine kinase that is involved in the regulation of cellular energy metabolism [45, 67] (see in Sect. 5). Metformin is one of the most widely prescribed front-line drugs for the treatment for type 2 diabetes because of its relatively low cost and reputation as a safe drug, as well as its effects in cardiovascular disease prevention [7]. The mild-to-moderate toxicity of metformin treatment in terms of gastrointestinal disturbances, such as nausea, vomiting, and diarrhea, might be prevented by dose reduction. Although the incidence is rare, lactic acidosis is the most severe adverse event, occurring predominantly in elderly patients who present with hepatic, cardiac, or renal comorbidities [20].

Effects of metformin on various tissues in patients with diabetes mellitus type 2, leading to downregulation of ATP-consuming pathways and increase in ATP-generating pathways
In the past decade, epidemiological observations have suggested that the use of metformin in DM patients is correlated with a reduction in cancer incidence. A case-control study conducted by Evans et al. on 923 cases of cancer in 11,876 newly diagnosed type 2 diabetic patients revealed for the first time that the overall cancer incidence was lower in diabetic patients treated with metformin compared to patients treated with other drugs. Moreover, the duration of treatment and number of prescriptions further influenced the cancer incidence: The increased duration of treatment and number of prescriptions correlated with a lower incidence of cancer [23].
Since this study, an increasing number of retrospective analyses have been performed. In 2006, Bowker et al. performed a population study on administrative data derived from 5. 4 year follow-ups of a cohort of 10,309 diabetic patients who received different treatments (metformin, sulfonylureas and/or insulin). This study also demonstrated reduced cancer deaths in patients who were treated with metformin alone without insulin compared to those who were treated with sulfonylureas regardless of insulin treatment [11, 62]. Unfortunately, an untreated control group was not included in this analysis, so whether the cancer risk was reduced by metformin or increased by insulin cannot be definitively asserted.
Other observational studies reported similar trends [3, 60], and interestingly, the association of metformin with reduced incidence/mortality of cancer was also maintained in the presence of insulin or after the adjustment of insulin doses, suggesting that metformin exerts an insulin-independent indirect mechanism of action [60].
Recently, several meta-analyses have been performed to understand the role of metformin in cancer incidence/mortality. Decensi et al. examined 11 epidemiological studies (5 comprehensive studies of all cancers and 6 studies of single cancer sites) that were extrapolated from a comprehensive literature search, which was not subjected to language or time restriction. The data derived from the analyses demonstrated that metformin treatment is associated with a 31 % reduction in cancer incidence and mortality and that this reduction exhibits a dose-response trend. Interestingly, a significant correlation between cancer risk and metformin treatment was observed in hepatocellular and pancreatic cancer patients, whereas no significant correlation has been reported in patients presenting with colon, breast, or prostate cancers, suggesting that metformin elicits cancer site-specific effects [19]. Recently, another study of 1,353 type 2 diabetic patients confirmed that after a median follow-up of 9.6 years, metformin exerted anticancer protective effects in a dose-dependent manner [1, 49].
An additional meta-analysis, which was performed on large population-based data from different countries, confirmed that metformin reduced the incidence and mortality of cancer at any site by approximately 33 %, with the variability in reduction depending on the site. Metformin treatment decreased the risk of colon, lung, and liver cancers but did not affect the development of hormone-dependent prostate and breast cancers or of pancreatic or gastric cancers [61].
Retrospective analyses have also been performed to understand the role of metformin on the development of metastasis, as well as whether this drug could improve the efficacy of chemotherapy. In particular, an analysis of diabetic patients with lung cancer revealed that the use of metformin reduced the occurrence of metastatic disease [57, 58] and improved chemotherapeutic outcomes [78]. Similarly, Jiralerspong observed that metformin increased the pathological complete response in breast cancer patients receiving neoadjuvant chemotherapy. Unfortunately, metformin did not improve the estimated 3 year relapse-free survival rate [1, 42]. In contrast, recently, metformin has been shown not to induce any beneficial effect on overall survival, on disease-free survival, or on the development of distal metastasis in triple-negative breast cancer cells [4].
In conclusion, several studies have suggested the use of metformin as an anticancer drug, but some limitations need to be considered. The majority of studies were retrospective, and the data were not obtained from population-based registries but rather from clinical and hospital data. Moreover, diabetic patients received a variety of treatments and were not randomized either for the administration of metformin or for objective criteria (for example, some patients with a history of cancer were included in cancer risk studies). Furthermore, an imbalance in important cancer risks and prognostic factors was evident. Nonetheless, such observational studies have suggested the plausible antitumor effects of metformin and have provided the basis for the initiation of prospective clinical trials.
4.2 Mechanisms of Action and Preclinical Studies
Several mechanisms of action have been proposed to explain the antitumor effects of metformin, although at the molecular level, the main effect is the activation of AMPK [21, 63]. AMPK acts as a cellular sensor of metabolism and stresses, such as hypoxia, oxidative stress, ischemia, and others, which lead to an increased ratio of AMP:ATP and the consequential increase in AMPK activation [20]. AMPK could be activated by metformin via the following three independent mechanisms: (1) by LKB1 (liver kinase B1), which induces phosphorylation of Thr 172 in the α catalytic subunit of AMPK [51]; (2) indirectly through the inhibition of complex I of the respiratory chain; and (3) by the activation of other inhibitors of mitochondrial ATP synthesis, such as oligomycin. The blockade of complex I of the respiratory chain results in low oxygen consumption, followed by modulation of NAD+/NADH ratios and an increase in the AMP:ATP ratio, resulting in increased AMPK activation [1].. In mammals, another important kinase is Ca2+/calmodulin-activated kinase kinase (CaMKK2), which activates an AMPK alternative pathway in response to increases in intracellular Ca2+ without altering the AMP:ATP ratio [31]. During physiological conditions, activated AMPK exerts its hypoglycemic action on the liver, β-pancreatic cells, and muscle and adipose tissue, where it enhances glucose uptake by inducing elevated levels of Glut1. AMPK also induces ATP-generating pathways, such as glycolysis, while inhibiting the ATP-consuming pathways such as gluconeogenesis, glycogen synthesis, and cholesterol synthesis [39, 45, 67]. The activation of AMPK is also crucial for the induction of the oxidative catabolism of glucose and fatty acids as well as for the regulation of mitochondrial biogenesis. Furthermore, the activation of AMPK also results in the inhibition of protein synthesis by blocking the mTOR pathway [31]. Overall, the targeting of AMPK elicits both a hypoglycemic effect and a direct effect on several pathways involved in tumor development.
Based on these findings, AMPK might be considered as a tumor suppressor. Notably, the upstream kinase LKB1 has been demonstrated to be a tumor suppressor gene that is frequently mutated in solid tumors. The LKB1 gene exhibits loss of function mutations in approximately 30 % of non-small-cell lung cancers and in 20 % of cervical cancers [40, 83]. Mutations in LKB1 have been demonstrated to influence cell growth and cell cycle progression, as well as cell polarity [30, 82]. Moreover, LKB1 is often comutated with kRAS in non-small-cell lung cancers, inducing an increase in tumor incidence and metastasis [30]. The inhibition of AMPK results in B-Raf V600E mutation-harboring melanomas, in which LKB1 is phosphorylated at two C-terminal sites [88] and is unable to activate AMPK. Moreover, the hyperactivation of AKT is frequently found in many tumors and induces the phosphorylation of AMPK at Ser485, which inhibits the phosphorylation and activation of AMPK by LKB1 [34]. The relevance of AMPK as a favorable prognostic marker has been shown by another study, in which the concomitant increases in pAMPK and pMAPK3/1 were implicated as prognostic markers of favorable outcome for the treatment of colorectal cancer patients, suggesting a possible interaction of these two pathways and new therapeutic strategies [2].
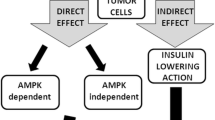
The anticancer effects of metformin have been proposed to be exerted via the following two distinct mechanisms: an indirect insulin-dependent effect and a tumor-direct insulin-independent effect (Fig. 3). The indirect effect of metformin is exerted on the liver, where metformin inhibits hepatic gluconeogenesis by inducing the activation of AMPK, followed by a reduction in circulating levels of glucose and insulin. In vitro and in vivo studies have reported that metformin, similar to other biguanides, induces the hyperactivation of 4E-binding protein in glucose-deprived cells, leading to inhibition of the unfolded protein response and strong inhibition of the mTOR pathway [56]. Moreover, Buzzai and colleagues demonstrated that in colorectal cell lines, glucose deprivation induced p53-dependent autophagy by the activation of AMPK in response to metformin [14]. In contrast, p53 status does not appear to be relevant for the metformin sensitivity of breast cancer cells. In response to the deprivation of glucose, metformin can induce apoptosis by both caspase-dependent and caspase-independent mechanisms concomitantly with changes in mitochondrial morphology and membrane permeability that depend on the cellular energy collapse that is related to the depletion of NAD+ levels [90]. Furthermore, pancreatic cancer cells have recently been shown to exhibit crosstalk between insulin/IGF1 receptors and G-protein-coupled receptors (GPCRs), which leads to increased cell growth and survival signaling induced by mTORC1, ERK, and PI3 K pathways. Because ERK and PI3 K are well-defined effectors of KRAS, which is mutated in 90 % of pancreatic cancers, these crosstalk results are reinforced by that mutation. Interestingly, metformin has been observed to disrupt the crosstalk between IR/IGF1R and GPCRs, reducing cellular proliferation in both cell lines [46] and xenograft models [70].

Metformin exerts its antitumor effects by insulin-dependent and insulin-independent mechanisms. Metformin can suppress cell growth by inactivation of AKT–mTOR pathway and/or may act on both AMPK and mitochondrial complex I, leading, respectively, to increase in apoptosis and downregulation of protein translation and inhibition of cell growth
The indirect effect of metformin has been confirmed in a study by Memmott et al., who observed in vivo that metformin prevented tobacco carcinogen-induced lung tumorigenesis. In particular, metformin has been shown to reduce tumor burden and simultaneously to markedly downregulate mTOR in tumors. Interestingly, metformin inhibits mTOR by activating AMPK in the liver, but not in the lung, where it indirectly downregulates IR/IGF1R and decreases AKT [59].
The direct effects of metformin on cancer growth are independent of insulin levels and might involve in the activation of AMPK, which inhibits several pathways, such as lipogenic pathways, via the suppression of SREBP-1 (sterol regulator element binding protein 1), via the regulation of the cell cycle by the phosphorylation of p53 and FOXO3a [84], or via the modulation of the estrogen-dependent pathway by the phosphorylation of CRTCS, a cAMP-responsive element binding protein (CREB)-regulated transcription coactivator [13]. The direct, AMPK-dependent effect also involves the direct action of AMPK on TSC2 and, as a consequence of its downstream signaling, reduces both protein synthesis and cell cycle progression. Interestingly, during the loss of TSC2, AMPK has been proposed to act directly on mTOR by phosphorylating the mTOR-associated protein Raptor [29]. Furthermore, a correlation between AMPK activation and adiponectin, a protein involved in metabolic signaling, has recently been demonstrated. Physiological concentrations of adiponectin were found to inhibit prostate and colon cancer growth by activating AMPK and inhibiting S6 K in patients presenting with normal weight. This finding has not been evident in obese patients who present with low levels of adiponectin, leading to reduced activation of AMPK and increased tumor growth. Consequently, the authors have suggested that metformin, by acting as an endogenous AMPK activator, can overcome resistance to adiponectin, thereby inhibiting cell growth [85].
Metformin has recently been observed to exhibit a direct action on mTOR, independently of TSC2 and AMPK [43]. That study found that in MEFs, metformin exerted an antitumor effect by inhibiting Rag, a GTPase protein involved in a direct translocation of mTORC1 to a cellular compartment that contains Rheb, a protein that induces mTOR activity [43]. To confirm this finding, a recent study demonstrated, using pancreatic cells, that metformin directly influenced the mTOR pathway in a p53-dependent manner via an AMPK-independent mechanism to increase REDD1, a negative regulator of mTOR. Moreover, the induction of REDD1 decreased cyclin D1 independently of AMPK1, which resulted in cell cycle arrest [8]. Lastly, an additional AMPK-independent tumor-direct effect of metformin could be related to the modulation of complex I of the respiratory chain and to the consequent regulation of the ATP levels that can subsequently affect apoptosis.
Based on the elevated heterogeneity of tumors, several recent studies have suggested that metformin acts selectively on a subset of cancer cells and, in particular, on cancer stem cells (CSCs) [5, 33]. Hirsch et al. [33] have demonstrated this hypothesis for the first time, observing that metformin selectively targeted the CSCs in triple-negative breast cancer cells and elicited synergistic antitumor activity in combination with doxorubicin. This finding has been confirmed recently in mouse xenografts, in which the simultaneous administration of metformin in combination with several chemotherapeutic drugs reduced tumor growth and prevented relapse in several cancer cell models, presumably by inhibiting the highly tumorigenic CSC-like cells. Moreover, the synergistic interaction of metformin with chemotherapeutic drugs has also been confirmed [38]. Interestingly, combinatorial treatment with metformin and chemotherapeutic drugs has been performed in another study, in which the combinatorial administration of metformin and paclitaxel converged on AMPK activation to reduce cell growth [69]. Recently, metformin has been demonstrated to increase the sensitivity of cancer cells to radiotherapy and to exert cytotoxicity on CSCs, overcoming their radioresistance via the activation of AMPK and the suppression of mTOR [75].
Metformin, similar to its analog biguanide phenformin, is a cation, and both the direct and indirect selective antitumor effects of metformin depend on the expression of organic cation transporters (OCT1, 2 and 3). In diabetic patients, metformin enters hepatic cells via OCT1 transporters during high levels of exposure to the drug via the hepatic portal vein. This cationic transporter is highly expressed in hepatocytes; in mice exhibiting a global knockout of OCT1, as well as polymorphisms of OCT1, the hepatic uptake of metformin is severely impaired, resulting in reduced hypoglycemic effects [41, 59, 74, 89]. Little is known about the expression of OCT1 in neoplastic cells. However, recently, OCT1 expression has been demonstrated to be highly variable in both epithelial ovarian cancer cell lines and primary human tumors. Interestingly, the knockdown of OCT1 in epithelial ovarian cancer cell lines, as well as the application of the OCT1 inhibitor quinidine, reduced the antitumor effect of metformin but did not affect the antineoplastic activity of phenformin [31, “personal communication”, 72].
4.3 Clinical Studies
Prospective and ongoing clinical trials are aimed at investigating the safety and the efficacy of metformin in cancer patients, independent of diabetic status, to analyze its role as a chemopreventive agent and to identify its biological effects (Fig. 3). More than fifty studies on the effects of metformin in cancer patients are currently registered at NCI’s cancer.gov. The majority of these trials are phase II breast cancer studies, which include biomarker analysis and the administration of metformin either as a single agent or in combination with other treatment modalities.
Limited clinical trials have been published so far regarding metformin. Hadad et al. conducted a preoperative, “window of opportunity” randomized trial, in which they showed the possible biological effects of metformin on tumor tissues. Metformin was administered to non-diabetic breast cancer patients before surgery, and the antitumor effects were compared with those of the untreated control group. Interestingly, the patients did not exhibit any quantifiable change in tumor size after 2–3 weeks of metformin treatment. However, an analysis of the tumor-derived biopsies revealed decreased insulin levels and a decrease in Ki67 staining, a marker of proliferation, indicating possible biological effects on tumor tissues [30]. In contrast, a recent study demonstrated that, overall, presurgery treatment with metformin did not affect Ki67 levels compared to treatment with the placebo arm in non-diabetic breast cancer patients. Interestingly, in a planned subgroup analysis, the effects of metformin on Ki67 could be stratified on the basis of insulin resistance according to the HOMA index (homeostasis model assessment). No correlation was observed between the insulin levels and metformin treatment in patients with HOMA indices greater than 2.8, whereas changes in Ki67 levels were noted in patients with HOMA indices less than 2.8, suggesting that metformin exerts its effects indirectly, depending on the grade of insulin resistance. Intriguingly, a similar stratification of Ki67 by HOMA index was noted in women who were overweight or obese, had abdominal obesity, or partook in moderate alcohol consumption [10].
According to observational studies in which elevated levels of peptide-C have been associated with poor outcome in non-diabetic breast cancer patients [17], Goodwin et al. [27, 28] demonstrated that metformin, when administered at a standard dose (1,500 mg/day), reduced insulin levels in non-diabetic breast cancer survivors by 22 % without relapse. Recently, the same group proposed a neoadjuvant “window of opportunity” study, in which metformin would be administered three times daily for 2–4 weeks prior to surgery. They identified the following potential predictors of metformin benefit: elevated BMI, physical inactivity, high fasting insulin as markers of host influence and tumor immunopositivity for Ki67 and TUNEL staining, and the presence of OCT1 and LKB1 as markers of tumor influence. Interestingly, a clear increase in TUNEL staining and a decrease in Ki67 have been observed to correlate with metabolic changes, following the administration of metformin [27, “personal communication”, 76].
A chemopreventive role for metformin has been demonstrated by a short-term clinical trial performed on rectal aberrant crypt foci (ACF), which is considered as an endoscopic surrogate marker for colorectal cancer. In non-diabetic patients with ACF, treatment with metformin significantly decreased the number of ACF after 1 month of therapy compared to the control group. Moreover, the authors observed a downregulation of colonic epithelial proliferative activity in the same group, as evaluated using the proliferating cell nuclear antigen labeling index. In contrast, no significant apoptotic modulation was detected [35]. As the biological significance of ACF as a surrogate marker for colorectal cancer remains controversial, the same group has recently registered a prospective randomized controlled trial in the University Hospital Medical Information Network (UMIN) Clinical Trials Registry as UMIN000006254, in which the chemopreventive effects of metformin will be evaluated in metachronous colorectal polyps and in non-diabetic post-polypectomy patients [32].
5 Conclusions
Here, we have reported on several lines of evidence supporting a role for the biguanide metformin as an antitumor agent. Nonetheless, some issues remain unresolved, such as the principal mechanism of action of metformin (direct or indirect/host effect), the characteristics of the patient and/or tumor that can influence responses to metformin, what types of cancer respond to treatment with biguanides, and which therapeutic setting could enhance the benefits of metformin (chemoprevention, neoadjuvant, adjuvant, combined, or single-agent administration).
Several reports have suggested that the antitumor activity of metformin appears to be elicited by both direct and indirect mechanisms, through lowering insulin levels and by directly affecting the tumor tissues via both AMPK-dependent and AMPK-independent mechanisms. Interestingly, a principal mediator of metformin activity, AMPK, is defective in many tumor cells. Several mechanisms can cooperate to induce the loss of AMPK activation and consequently might affect the antitumor effects of metformin. Paradoxically, metformin might be more effective for the treatment for tumors in which AMPK activation has been lost because it can cause greater decreases in ATP levels and more apoptosis.
The mechanism of action of metformin has remained obscure due to the limitations of the preclinical models, in which higher levels of metformin, glucose, and insulin were used, compared to more physiological/clinical conditions. For example, the dose range of metformin in clinical/epidemiological studies is 250–2,250 mg/day, whereas the preclinical doses range from 45-fold increased dosages for in vivo studies to 10,000-fold increased dosages for in vitro studies, in which mM concentrations of metformin have been used. Moreover, tissue culture media contain up to 3- to 5-fold excess of glucose and up to a 40-fold excess of insulin. However, low doses of metformin (approximately 10 μM) have recently been demonstrated to be sufficient to induce moderate activation of AMPK and the consequential activation of downstream pathways [77].
Recently, a phase III randomized trial was registered to address these issues, in which the effects of metformin that was administered in combination with chemotherapy were compared to those of placebo in early breast cancer patients. In this study, the patients were stratified for hormone receptor status, for HER2 expression, and for the chemotherapeutic drugs used, such as paclitaxel, docetaxel, doxorubicin, and cyclophosphamide [26]. The primary outcome was the rate of cancer-free survival, whereas the secondary outcomes were overall survival, disease-free survival and adverse events, and factors that were associated with correlative analysis, such as weight, fasting insulin levels, and tumor tissue. The primary hypothesis of this study was that elevated fasting insulin at baseline that exhibited a significant reduction at 6 months would predict metformin benefit ([27], “personal communication”, [28]).
Which type of biguanide is best for clinical application remains unclear. For example, if direct action on the tumor is required, then the OCT1 receptor expression levels become crucial, and it might therefore be better to reconsider the use of phenformin, which does not require the presence of OCT1 receptors to penetrate the cells.
Lastly, a recent, controversial study reported that the mutant V600E Braf gene, which is present in 50 % of melanomas, conferred in vitro resistance to metformin treatment by activating RSK to prevent AMPK activation. Moreover, metformin treatment accelerated tumor growth and induced VEGF-A expression in vivo. Interestingly, combined anti-VEGF treatment synergistically reduced the tumor growth of the Braf mutant but not of the wild-type cells [54].
In conclusion, numerous studies have clearly demonstrated a new application of the “old” antidiabetic drug metformin as an anticancer drug. However, further in-depth knowledge of its mechanism of action is necessary to identify the optimal therapeutic and clinical context in which to use it as an antitumor drug.
Abbreviations
- ACC:
-
Acetyl-CoA carboxylase
- ACF:
-
Aberrant crypt foci
- ACL:
-
ATP citrate lyase
- AKT/PKB:
-
Protein kinase B
- CaMKK2:
-
Ca2+/calmodulin-activated kinase kinase
- cAMP:
-
Cyclic adenosine monophosphate
- CI:
-
Confidence interval
- CRTCS:
-
cAMP-responsive element binding protein (CREB)-regulated transcription coactivator
- CSC:
-
Cancer stem cell
- DM:
-
Diabetes mellitus
- ER:
-
Estrogen receptor
- ERK:
-
Extracellular signal-regulated kinase
- FAS:
-
Fatty acid synthase
- FOXO:
-
Forkhead box O
- GLUT:
-
Glucose transporter
- GPCR:
-
G-protein-coupled receptor
- HCC:
-
Hepatocellular carcinoma
- HIF-1α:
-
Hypoxia-inducible factor 1α
- HOMA:
-
Homeostasis model assessment
- IGF:
-
Insulin growth factor
- IGFBP:
-
Insulin growth factor binding protein
- IGF-R:
-
Insulin growth factor receptor
- IR:
-
Insulin receptor
- IRS:
-
Insulin receptor substrates
- LKB1:
-
Liver kinase B1
- MAPK:
-
Mitogen-activated protein kinase
- MEF:
-
Mouse embryonic fibroblast
- mTOR:
-
Mammalian target of rapamycin
- mTORC1:
-
mTOR complex 1
- NAFLD:
-
Non-alcoholic fatty liver disease
- Nampt:
-
Nicotinamide phosphoribosyltransferase
- OCT:
-
Organic cation transporter
- PDK1-3:
-
Pyruvate dehydrogenase kinase 1 and 3
- PET:
-
Positron emission tomography
- PI3K:
-
Phosphatidylinositol 3 kinase
- rag:
-
Recombination activating gene
- RR:
-
Relative risk
- SREBP-1:
-
Sterol regulator element binding protein 1
- TSC:
-
Tuberous sclerosis
- UMIN:
-
University Hospital Medical Information Network
- VEGF:
-
Vascular endothelial growth factor
References
Aljada A, Mousa SA (2012) Metformin and neoplasia: implications and indications. Pharmacol Ther 133:108–115
Baba Y, Nosho K, Shima K et al (2010) Prognostic significance of AMP-activated protein kinase expression and modifying effect of MAPK3/1 in colorectal cancer. Br J Cancer 103:1025–1033
Baur DM, Klotsche J, Hamnvik OP et al (2011) Type 2 diabetes mellitus and medications for type 2 diabetes mellitus are associated with risk for and mortality from cancer in a German primary care cohort. Metabolism 60:1363–1371
Bayraktar S, Hernadez-Aya LF, Lei X et al (2012) Effect of metformin on survival outcomes in diabetic patients with triple receptor-negative breast cancer. Cancer 118:1202–1211
Bednar F, Simeone DM (2012) Metformin and cancer stem cells: old drug, new targets. Cancer Prev Res (Phila) 5:351–354
Belfiore A, Frasca F (2008) IGF and insulin receptor signaling in breast cancer. J Mammary Gland Biol Neoplasia 13:381–406
Ben Sahra I, Le Marchand-Brustel Y, Tanti JF et al (2010) Metformin in cancer therapy: a new perspective for an old antidiabetic drug? Mol Cancer Ther 9:1092–1099
Ben Sahra I, Regazzetti C, Robert G et al (2011) Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res 71:4366–4372
Blagosklonny MV (2011) NCI’s provocative questions on cancer: some answers to ignite discussion. Oncotarget 2:1352–1367
Bonanni B, Puntoni M, Cazzaniga M et al (2012) Dual effect of metformin on breast cancer proliferation in a randomized presurgical trial. J Clin Oncol 30(21):2593–2600
Bowker SL, Majumdar SR, Veugelers P et al (2006) Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care 29:254–258
Brahimi-Horn MC, Chiche J, Pouyssegur J (2007) Hypoxia signalling controls metabolic demand. Curr Opin Cell Biol 19:223–229
Brown KA, Simpson ER (2010) Obesity and breast cancer: progress to understanding the relationship. Cancer Res 70:4–7
Buzzai M, Jones RG, Amaravadi RK et al (2007) Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res 67:6745–6752
Chari ST, Leibson CL, Rabe KG et al (2008) Pancreatic cancer-associated diabetes mellitus: prevalence and temporal association with diagnosis of cancer. Gastroenterology 134:95–101
Chen HF, Li CY, Chen P et al (2006) Seroprevalence of hepatitis B and C in type 2 diabetic patients. J Chin Med Assoc 69:146–152
Chlebowski RT, Aiello E, McTiernan A (2002) Weight loss in breast cancer patient management. J Clin Oncol 20:1128–1143
Davila JA, Morgan RO, Shaib Y et al (2005) Diabetes increases the risk of hepatocellular carcinoma in the United States: a population based case control study. Gut 54:533–539
Decensi A, Puntoni M, Goodwin P et al (2010) Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prev Res (Phila) 3:1451–1461
Dowling RJ, Niraula S, Stambolic V et al (2012) Metformin in cancer: translational challenges. J Mol Endocrinol 48:R31–R43
El-Mir MY, Nogueira V, Fontaine E et al (2000) Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem 275:223–228
El-Serag HB, Hampel H, Javadi F (2006) The association between diabetes and hepatocellular carcinoma: a systematic review of epidemiologic evidence. Clin Gastroenterol Hepatol 4:369–380
Evans JM, Donnelly LA, Emslie-Smith AM et al (2005) Metformin and reduced risk of cancer in diabetic patients. BMJ 330:1304–1305
Galli M, Van Gool F, Rongvaux A et al (2010) The nicotinamide phosphoribosyltransferase: a molecular link between metabolism, inflammation, and cancer. Cancer Res 70:8–11
Gatenby RA, Gillies RJ (2004) Why do cancers have high aerobic glycolysis? Nat Rev Cancer 4:891–899
Gonzalez-Angulo AM, Meric-Bernstam F (2010) Metformin: a therapeutic opportunity in breast cancer. Clin Cancer Res 16:1695–1700
Goodwin PJ, Ennis M, Pritchard KI et al (2012) Insulin- and obesity-related variables in early-stage breast cancer: correlations and time course of prognostic associations. J Clin Oncol 30:164–171
Goodwin PJ, Stambolic V, Lemieux J et al (2011) Evaluation of metformin in early breast cancer: a modification of the traditional paradigm for clinical testing of anti-cancer agents. Breast Cancer Res Treat 126:215–220
Gwinn DM, Shackelford DB, Egan DF et al (2008) AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 30:214–226
Hadad S, Iwamoto T, Jordan L et al (2011) Evidence for biological effects of metformin in operable breast cancer: a pre-operative, window-of-opportunity, randomized trial. Breast Cancer Res Treat 128:783–794
Hardie DG, Ross FA, Hawley SA (2012) AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13:251–262
Higurashi T, Takahashi H, Endo H et al (2012) Metformin efficacy and safety for colorectal polyps: a double-blind randomized controlled trial. BMC Cancer 12:118
Hirsch HA, Iliopoulos D, Tsichlis PN et al (2009) Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res 69:7507–7511
Horman S, Vertommen D, Heath R et al (2006) Insulin antagonizes ischemia-induced Thr172 phosphorylation of AMP-activated protein kinase alpha-subunits in heart via hierarchical phosphorylation of Ser485/491. J Biol Chem 281:5335–5340
Hosono K, Endo H, Takahashi H et al (2010) Metformin suppresses colorectal aberrant crypt foci in a short-term clinical trial. Cancer Prev Res (Phila) 3:1077–1083
Hoyo C, Schildkraut JM, Murphy SK et al (2009) IGF2R polymorphisms and risk of esophageal and gastric adenocarcinomas. Int J Cancer 125:2673–2678
Huxley R, Ansary-Moghaddam A, Berrington de Gonzalez A et al (2005) Type-II diabetes and pancreatic cancer: a meta-analysis of 36 studies. Br J Cancer 92:2076–2083
Iliopoulos D, Hirsch HA, Struhl K (2011) Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer Res 71:3196–3201
Jalving M, Gietema JA, Lefrandt JD et al (2010) Metformin: taking away the candy for cancer? Eur J Cancer 46:2369–2380
Ji H, Ramsey MR, Hayes DN et al (2007) LKB1 modulates lung cancer differentiation and metastasis. Nature 448:807–810
Jin HE, Hong SS, Choi MK et al (2009) Reduced antidiabetic effect of metformin and down-regulation of hepatic Oct1 in rats with ethynylestradiol-induced cholestasis. Pharm Res 26:549–559
Jiralerspong S, Palla SL, Giordano SH et al (2009) Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J Clin Oncol 27:3297–3302
Kalender A, Selvaraj A, Kim SY et al (2010) Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab 11:390–401
Kim JW, Tchernyshyov I, Semenza GL et al (2006) HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3:177–185
Kim YH, Liang H, Liu X et al (2012) AMPKalpha modulation in cancer progression: multilayer integrative analysis of the whole transcriptome in Asian gastric cancer. Cancer Res 72:2512–2521
Kisfalvi K, Eibl G, Sinnett-Smith J et al (2009) Metformin disrupts crosstalk between G protein-coupled receptor and insulin receptor signaling systems and inhibits pancreatic cancer growth. Cancer Res 69:6539–6545
Kroemer G, Pouyssegur J (2008) Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell 13:472–482
Kuhajda FP (2000) Fatty-acid synthase and human cancer: new perspectives on its role in tumor biology. Nutrition 16:202–208
Landman GW, Kleefstra N, van Hateren KJ et al (2010) Metformin associated with lower cancer mortality in type 2 diabetes: ZODIAC-16. Diabetes Care 33:322–326
Law JH, Habibi G, Hu K et al (2008) Phosphorylated insulin-like growth factor-i/insulin receptor is present in all breast cancer subtypes and is related to poor survival. Cancer Res 68:10238–10246
Long YC, Zierath JR (2006) AMP-activated protein kinase signaling in metabolic regulation. J Clin Invest 116:1776–1783
Lu CW, Lin SC, Chen KF et al (2008) Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J Biol Chem 283:28106–28114
Martin M, Marais R (2012) Metformin: a diabetes drug for cancer, or a cancer drug for diabetics? J Clin Oncol 30(21):2698–2700
Martin MJ, Hayward R, Viros A et al (2012) Metformin accelerates the growth of BRAFV600E-driven melanoma by upregulating VEGF-A. Cancer Discov 2:344–355
Massoner P, Ladurner-Rennau M, Eder IE et al (2010) Insulin-like growth factors and insulin control a multifunctional signalling network of significant importance in cancer. Br J Cancer 103:1479–1484
Matsuo J, Tsukumo Y, Saito S et al (2012) Hyperactivation of 4E-binding protein 1 as a mediator of biguanide-induced cytotoxicity during glucose deprivation. Mol Cancer Ther 11:1082–1091
Mazzone PJ (2010) Lung cancer screening: an update, discussion, and look ahead. Curr Oncol Rep 12:226–234
Mazzone PJ (2010) Preoperative evaluation of the lung cancer resection candidate. Expert Rev Respir Med 4:97–113
Memmott RM, Mercado JR, Maier CR et al (2010) Metformin prevents tobacco carcinogen–induced lung tumorigenesis. Cancer Prev Res (Phila) 3:1066–1076
Monami M, Colombi C, Balzi D et al (2011) Metformin and cancer occurrence in insulin-treated type 2 diabetic patients. Diabetes Care 34:129–131
Noto H, Goto A, Tsujimoto T et al (2012) Cancer risk in diabetic patients treated with metformin: a systematic review and meta-analysis. PLoS ONE 7:e33411
Onitilo AA, Engel JM, Glurich I et al (2012) Diabetes and cancer II: role of diabetes medications and influence of shared risk factors. Cancer Causes Control 23:991–1008
Owen MR, Doran E, Halestrap AP (2000) Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J 348(Pt 3):607–614
Pannala R, Basu A, Petersen GM et al (2009) New-onset diabetes: a potential clue to the early diagnosis of pancreatic cancer. Lancet Oncol 10:88–95
Papandreou I, Cairns RA, Fontana L et al (2006) HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 3:187–197
Pollak M (2008) Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer 8:915–928
Pollak M (2012) Metformin and pancreatic cancer: a clue requiring investigation. Clin Cancer Res 18:2723–2725
Pouyssegur J, Dayan F, Mazure NM (2006) Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 441:437–443
Rocha GZ, Dias MM, Ropelle ER et al (2011) Metformin amplifies chemotherapy-induced AMPK activation and antitumoral growth. Clin Cancer Res 17:3993–4005
Rozengurt E, Sinnett-Smith J, Kisfalvi K (2010) Crosstalk between insulin/insulin-like growth factor-1 receptors and G protein-coupled receptor signaling systems: a novel target for the antidiabetic drug metformin in pancreatic cancer. Clin Cancer Res 16:2505–2511
Samudio I, Fiegl M, Andreeff M (2009) Mitochondrial uncoupling and the Warburg effect: molecular basis for the reprogramming of cancer cell metabolism. Cancer Res 69:2163–2166
Segal ED, Yasmeen A, Beauchamp MC et al (2011) Relevance of the OCT1 transporter to the antineoplastic effect of biguanides. Biochem Biophys Res Commun 414:694–699
Seyfried TN, Shelton LM (2010) Cancer as a metabolic disease. Nutr Metab (Lond) 7:7
Shu Y, Sheardown SA, Brown C et al (2007) Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J Clin Invest 117:1422–1431
Song CW, Lee H, Dings RP et al (2012) Metformin kills and radiosensitizes cancer cells and preferentially kills cancer stem cells. Sci Rep 2:362
Stambolic V (2012). A. 2012. Chicago
Stambolic V, Woodgett JR, Fantus IG et al (2009) Utility of metformin in breast cancer treatment, is neoangiogenesis a risk factor? Breast Cancer Res Treat 114:387–389
Tan BX, Yao WX, Ge J et al (2011) Prognostic influence of metformin as first-line chemotherapy for advanced nonsmall cell lung cancer in patients with type 2 diabetes. Cancer 117:5103–5111
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033
Vigneri P, Frasca F, Sciacca L et al (2009) Diabetes and cancer. Endocr Relat Cancer 16:1103–1123
Wang HQ, Altomare DA, Skele KL et al (2005) Positive feedback regulation between AKT activation and fatty acid synthase expression in ovarian carcinoma cells. Oncogene 24:3574–3582
Williams T, Brenman JE (2008) LKB1 and AMPK in cell polarity and division. Trends Cell Biol 18:193–198
Wingo SN, Gallardo TD, Akbay EA et al (2009) Somatic LKB1 mutations promote cervical cancer progression. PLoS ONE 4:e5137
Zadra G, Priolo C, Patnaik A et al (2010) New strategies in prostate cancer: targeting lipogenic pathways and the energy sensor AMPK. Clin Cancer Res 16:3322–3328
Zakikhani M, Dowling RJ, Sonenberg N et al (2008) The effects of adiponectin and metformin on prostate and colon neoplasia involve activation of AMP-activated protein kinase. Cancer Prev Res (Phila) 1:369–375
Zhang M, Hu Z, Huang J et al (2010) A 3’-untranslated region polymorphism in IGF1 predicts survival of non-small cell lung cancer in a Chinese population. Clin Cancer Res 16:1236–1244
Zhao Y, Liu H, Riker AI et al (2011) Emerging metabolic targets in cancer therapy. Front Biosci 16:1844–1860
Zheng B, Jeong JH, Asara JM et al (2009) Oncogenic B-RAF negatively regulates the tumor suppressor LKB1 to promote melanoma cell proliferation. Mol Cell 33:237–247
Zhou G, Myers R, Li Y et al (2001) Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 108:1167–1174
Zhuang Y, Miskimins WK (2011) Metformin induces both caspase-dependent and poly(ADP-ribose) polymerase-dependent cell death in breast cancer cells. Mol Cancer Res 9:603–615
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg
About this paper
Cite this paper
Leone, A., Di Gennaro, E., Bruzzese, F., Avallone, A., Budillon, A. (2014). New Perspective for an Old Antidiabetic Drug: Metformin as Anticancer Agent. In: Zappia, V., Panico, S., Russo, G., Budillon, A., Della Ragione, F. (eds) Advances in Nutrition and Cancer. Cancer Treatment and Research, vol 159. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-38007-5_21
Download citation
DOI: https://doi.org/10.1007/978-3-642-38007-5_21
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-38006-8
Online ISBN: 978-3-642-38007-5
eBook Packages: MedicineMedicine (R0)