
Abstract
The imprinted brain theory proposes that autism spectrum disorder (ASD) represents a paternal bias in the expression of imprinted genes. This is reflected in a preference for mechanistic cognition and in the corresponding mentalistic deficits symptomatic of ASD. Psychotic spectrum disorder (PSD) would correspondingly result from an imbalance in favor of maternal and/or X-chromosome gene expression. If differences in imprinted gene expression were reflected locally in the human brain, as mouse models and other evidence suggests they are, ASD would represent not so much an “extreme male brain” as an extreme paternal one, with PSD correspondingly representing an extreme maternal brain. To the extent that copy number variation resembles imprinting and aneuploidy in nullifying or multiplying the expression of particular genes, it has been found to conform to the diametric model of mental illness peculiar to the imprinted brain theory. The fact that non-genetic factors like nutrition in pregnancy can mimic and/or interact with imprinted gene expression suggests that the theory might even be able to explain the notable effect of maternal starvation on risk of PSD—not to mention a part of the “autism epidemic” of modern affluent societies. Finally, the theory suggests that normality represents balanced cognition and that genius is an extraordinary extension of cognitive configuration in both mentalistic and mechanistic directions. Were it to prove correct, the imprinted brain theory would represent one of the biggest single advances in our understanding of the mind and of mental illness that has ever taken place and would revolutionize psychiatric diagnosis, prevention, and treatment—not to mention our understanding of epigenetics.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
The discovery of epigenetics has effectively given a new, third dimension to depth of the nature/nurture issue. Once, nature/nurture was two-dimensional to the extent that, for example, where identical twins raised apart differed significantly, it was assumed that the differences had to be the result of nurture and the similarities seemingly the result of their shared nature. But now we know this is not so and that monozygotic twins can differ epigenetically. A recent study of DNA methylation profiles in monozygotic and dizygotic twins pointed out that “molecular mechanisms of heritability may not be limited to DNA sequence differences.” Indeed, the authors speculate that because identical twins reared together or apart are generally quite similar on measures such as brain imaging, IQ, and other psychometrics, epigenetic differences between identical twins “are much more important than environment” (Kaminsky et al. 2009).
Another finding is that if one of two identical twins has a mental disorder such as autism or schizophrenia, there is a much-higher-than-average probability that the other will too. Autism spectrum disorder (ASD) is sometimes associated with genetic syndromes, such as Rett, Down, and Turner’s, phenylketonuria, tuberous sclerosis, and fragile X syndrome, where between a quarter and a half of all cases are diagnosed autistic (Aitken 2008). But neither autism nor schizophrenia obeys classical Mendelian laws of inheritance, suggesting that genetics cannot be the sole cause.
Is nurture also a factor? There is certainly good evidence for social, environmental causes of mental illnesses. Studies of the Dutch wartime famine and of the Chinese famine of 1959–1961 reported increased incidence of schizophrenia among children born just after the events (St Clair et al. 2005; Susser et al. 2008). Again, a study of two million Swedish children born between 1963 and 1983 revealed a significant link between schizophrenia and poverty in childhood. Those with four out of five measured indicators of hardship had an almost threefold greater risk of schizophrenia than those with none (Wicks et al. 2005).
At first sight, it would seem that no single theory could explain these seemingly contradictory facts—and certainly not an evolutionary or genetic one—but an attempt is under way to do exactly that. According to the so-called imprinted brain theory, the paradoxes can be explained in terms of the expression of genes and not simply their inheritance. Furthermore, it proposes that the pattern of expression in question is part of a more general phenomenon rooted in evolution and explained by conflicts over parental investment (Badcock 2008, 2009; Badcock and Crespi 2006, 2008; Crespi and Badcock 2008; Crespi et al. 2009a; Crespi 2008). The implication is that this new, third dimension of depth which epigenetics adds to the nature/nurture dichotomy is an evolved, natural one centered on nurture, understood principally as parental investment and secondarily as environmental factors which mimic, reinforce, or interact with genetics. The imprinted brain theory poses a provocative challenge to neo-Lamarckian interpretations of epigenetics (Jablonka and Lamb 1995) and as such is a crucial test case of the so-called selfish-gene neo-Darwinism (Badcock 1995).
2 Genomic Imprinting: The Epigenetics of Nurture
One way in which our understanding of nurture has been transformed and placed on a secure scientific and quantitative footing is via its definition as parental investment in terms of additions to an offspring’s survival and/or reproductive success at a cost to the remainder of its parent’s survival and/or reproductive success. The sexes can also be defined in terms of their contribution to offspring’s reproductive success, with an almost universal anisogamy evident in the contrast between the microscopic and highly mobile male gamete (sperm or pollen cell) and the relatively massive and immobile female one (ovum or ovule). Mammals in particular are characterized by an extreme asymmetry between the sexes in this respect, thanks to gestation and lactation being exclusive to females and male parental investment being minor or negligible in many species. As a result, the vast majority of mammalian species are polygynous, with males investing primarily in mating effort and females primarily in parental investment (Trivers 1972).
Such striking asymmetry between the sexes where reproductive success is concerned may explain the finding that some genes in mammals are only expressed from one allele depending on its parent of origin (Haig 2002). More surprisingly still, many of the genes in question are strategic, controlling ones like IGF2, and most are found to be expressed in critical organs—specifically the placenta and brain (Barlow 1995; De Chiara et al. 1991; Reik and Surani 1997). However, conflicts between maternal and paternal genetic self-interest might explain why, for example, IGF2 is often paternally active and maternally imprinted or silenced. Since growth of the offspring benefits both parents but only the mother pays the costs involved in gestation and lactation, this growth-factor gene is predominantly expressed only from the father’s copy. Indeed, in mice a maternally expressed but paternally imprinted gene, Igf2r effectively contradicts murine Igf2 by creating receptors which act as non-growth-inducing sinks for the hormone (Haig and Graham 1991). (For a review of the evidence, see Haig (2004).)
Where humans are concerned, researchers have argued that Silver-Russell syndrome (SRS) and Beckwith-Wiedemann syndrome (BWS) “may now be regarded as two diseases caused by opposite (epi)genetic disturbances of the same chromosomal region displaying opposite clinical pictures” (Eggermann et al. 2005). The region to which they refer includes IGF2, and symptoms associated with SRS feature pre- and postnatal growth retardation arguably associated with non-expression of IGF2, while BWS symptoms include generalized pre- and postnatal overgrowth, arguably accounted for by biparental expression of IGF2 (Holm et al. 1993).
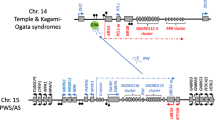
Nor is this an isolated case: similar diametrically opposite symptoms are associated with reversed imprinting of the same genes on chromosome 15 in Angelman syndrome (AS) and Prader-Willi syndrome (PWS). AS is associated with increased expression of paternal and/or reduced expression of maternal genes at 15q11–13 and PWS with the opposite pattern of expression. In some variants of PWS, the entire maternal chromosome 15 is duplicated. In other cases, duplication of the paternal chromosome 15 occurs (Nicholls et al. 1998). Significantly in view of the conflict theory of imprinting, AS children are notably demanding: hyperactive, sleepless, effusive, and prolonged sucklers with a low pleasure threshold (“paroxysms of laughter” is one of the diagnostic criteria of what is otherwise known as “happy puppet syndrome”) and an unusual readiness to smile. Indeed, Brown and Consedine (Brown and Consedine 2004) suggested that smiling produced by infants with Angelman syndrome may reflect a patri-gene adaptation to extract resources from the mother, as smiling positively correlates with night feeding and gaining positive social attention from caregivers (for an independent anticipation of the same insight, see Badcock (1999)). By contrast, PWS infants—those with the maternal bias—are the opposite: inactive, sleepy, and poor sucklers with a high pain threshold (such that they often develop severe skin lesions as a result of picking at scabs normal children would leave alone) (Angelman 1965; Nicholls et al. 1998).
Although some studies link PWS with autism (Milner et al. 2005; Veltman et al. 2004), all cases of maternal uniparental disomy chromosome 15 PWS known to the Cambridge Prader-Willi study were diagnosed with a psychotic spectrum disorder (PSD) in adulthood. As the researchers who demonstrated it point out, this is the only example of such a direct and apparently absolute relationship between a specific genetic abnormality and psychotic illness known at the present time (Whittington and Holland 2004). AS children, by contrast, tend to be diagnosed with ASD (Bonati et al. 2007; Steffenburg et al. 1996) (although there are contrary assertions (Veltman et al. 2005)), and in one sample of 87 BWS cases (double expression of paternally active IGF2), 6.8 % of the sample had been diagnosed autistic (Kent et al. 2008)—in other words approximately seven times the highest estimate of prevalence of ASD in the general population, which is about 1 % (Baird et al. 2006).
Further examples of such genomic sister syndromes associated with duplication versus deletion of the same genomic regions have recently been described by Crespi, Summers, and Dorus. Velocardiofacial syndrome deletion 22q11 carries the second highest known risk of PSD after maternal uniparental disomy PWS, with about 30 % being diagnosed schizophrenic. Duplication of the same region, however, has been linked with ASD. Williams syndrome cases with deletion at 7q11 have visuospatial deficits, but are hyper-social and highly verbal to the point of being described as possessing “cocktail party” skills and also show heightened levels of anxiety and phobias. By contrast, duplication of the same region is associated with spared visuospatial skills but severe language impairment, ASD, and seizures (which are commonly associated with ASD). Similarly, Smith-Magenis syndrome, which features deletions at 17p11, shows evidence of good verbal skills, high sociability, and a tendency to PSD, while duplication of the same region in Potocki-Lupski syndrome is associated with high risk of ASD and seizures (Crespi et al. 2009b).
X-chromosome genes resemble maternally active autosomal ones for the simple reason that female mammals have two X chromosomes to the male’s one and that all mothers are female. This means that selection for female-benefiting traits acts twice as often on X genes as it does for male-benefiting X-chromosome traits. As a result, X-chromosome aneuploidies which increase the representation of X-chromosome genes resemble maternally active autosomal ones and shift the balance of gene expression in favor of the mother and against the father (Haig 2006).
Crespi, Summers, and Dorus also point out that the short arm of the X chromosome has a high concentration of genes involved in psychosis and micro-deletions on the long-arm link to autism. There is a three times greater incidence of schizophrenia in Turner’s cases with mosaic karyotypes mixing XO, XX, and XXX, and true X trisomy is linked to schizophrenia, with X trisomics having brain imaging similar to that of schizophrenics. In Klinefelter’s syndrome (XXY) there is a four to ten times heightened risk of psychosis, with more positive female-typical symptoms (auditory hallucinations and paranoia) and a female-typical age of onset along with neuroanatomy similar to that seen in schizophrenia. As Crespi, Summers, and Dorus note, these examples “suggest that diametric copy-number alterations can, like diametric alterations to imprinted genes, generate contrasting phenotypes associated with autistic-spectrum and psychotic-spectrum conditions” (Crespi et al. 2009b).
3 The Imprinted Brain and the Epigenetics of Mental Conflict
Genes build brains to provide real-time responses to environmental challenges in motile organisms which cannot be predicted or directly encoded in DNA (Badcock 2000, pp. 69–71). However, sexual reproduction implies that the organism’s genes originate in two parents of the opposite sex with conflicting or contradictory genetic self-interests—for example, as a result of the asymmetries in parental investment and mating effort mentioned above. The consequence is that such conflicts and contradictions are likely to be built into the brain before birth and fought out throughout life—and nowhere more so than in mammals with large brains and evolved minds such as human beings (Hamilton 1996, pp. 133–5).
A precedent for this expectation can be found in mice. Chimeric mice can be engineered to express a preponderance of one parent’s genes as opposed to the other, androgenetic (AG) chimeras expressing the father’s or parthenogenetic (PG) chimeras expressing the mother’s, and staining can be incorporated to show where these genes are expressed in the developing embryo (Allen et al. 1995). As the conflict theory of imprinting would lead you to expect, the resulting AG embryos are larger than normal (excepting the brain) and have massive placentas. By contrast, PG embryos are small overall (except for the brain, where, interestingly, Igf2 is maternally active and paternally imprinted (Gregg et al. 2010)) and have little or no placenta (Keverne et al. 1996). Naturally occurring triploid human fetuses with a double set of the mother’s genes and one of the father’s are small except for the head, show a retardation of growth, and have small placentas. Those with a double set of their father’s genes and a single set of the mother’s are well grown except for the head and have a large placenta (Hannah et al. 2002; Newton 2001).
Such experiments also show that while genes from both parents are equally expressed in the brain stem of mice as Mendelian norms might suggest, PG cells are found in large numbers in the cerebral cortex (and the underlying striatum) and in the forebrain, but very few are found in the so-called limbic system (MacLean 1996)—especially in the hypothalamus. This is true of mature, fully grown mice but even more so of fetuses where there is a complete absence of PG cells in the hypothalamus. In both cases, PG cells are found to be particularly clustered in the frontal lobes of the cortex. AG cells, by contrast, are the exact opposite: these are found in the hypothalamus and limbic system, but not in the cerebral cortex. The few that are found in the forebrain tissue of embryos do not proliferate and are subsequently eliminated (Allen et al. 1995).
More recent research on normal, fully developed mouse brains found imprinting at 1,300 different places in the murine genome. Three hundred and forty-seven non-sex chromosome genes were found to have sex-specific imprints: in other words, these genes not only were limited to being expressed from one parent but were further limited by the sex of the offspring in which they found themselves. For example, Il18, a gene linked to multiple sclerosis (a disease which predominates in women and runs down the maternal line of descent) was found to be preferentially expressed from the mother in the female brain but not in the male. In the hypothalamus, sex-specific imprinted genes were found in females, suggesting, as the authors point out, “parental influence over the hypothalamic function of daughters” (Gregg et al. 2010).
In the same study it was found that the mother’s genes made a greater contribution during development, but that the father’s contributed more in adulthood. Furthermore, 40–50 % more neurons expressed the mother’s X chromosome as compared to the father’s in the prefrontal and other parts of the cortex. By contrast, there was no difference in X-chromosome expression in the hypothalamus. As the authors point out, there are many genes involved with brain function on the X in both mouse and man, and as we have just seen, theory suggests that maternally biased inheritance of X-chromosome genes serves maternal genetic self-interest. As the authors of the latest research on the mouse brain conclude, “parental expression bias emerges as a major mode of epigenetic regulation in the brain” (Gregg et al. 2010).
Hints of a similar pattern of maternal expression in the frontal cortex in humans are found in a study by Goos and Silverman. Intra-familial correlations on cognitive tests involving occipital, temporal, parietal, and frontal lobe functions in 65 families found that abilities mediated by frontal, parietal, and temporal lobes—but not occipital lobes—were more closely correlated between children and mothers than between children and their fathers (Goos and Silverman 2006). The prefrontal cortex tends to be larger in women, while elements of the limbic system such as the amygdala and hippocampus tend to be larger in men and larger still in autism but smaller in schizophrenia (Gur et al. 2004; Mendrek 2007).
The difference in the average volume of the orbitofrontal cortex between men and women accounts for about half of the variance in antisocial behavior between the sexes (Jones 2008), and reduced frontal volume is associated with antisocial behavior and psychopathy (Gur et al. 2002). If maternal genes are predominantly expressed in the prefrontal cortex, we might be justified in thinking of it as a critical part of the maternal brain. If so, then we could see the hypothalamus, amygdala, and other parts of the lower or limbic brain as paternal for parallel reasons: paternal genes are mainly expressed there, and these regions are also proportionately larger in men (Goldstein et al. 2001). The limbic system is sometimes called “the emotional brain” (LeDoux 1996) and certainly contains centers concerned with gratification of basic drives, appetite, and gut reactions such as fear, pleasure, and disgust. Bearing this in mind, we can now begin to see that the relation between the paternal and maternal brains is reminiscent of that between paternally active and maternally active genes, such as Igf2 and Igf2r. (Indeed, in man IGF2R is linked to high intelligence (Chorney et al. 1998), although it is not imprinted (Killian et al. 2001).) The paternal brain could be seen as serving the father’s genetic interest in the offspring’s growth and consumption of resources, but as we can also now see, the maternal brain—and the prefrontal cortex in particular—could equally be seen as serving maternal genetic self-interest to the extent that it is able to inhibit, control, and contain the paternal brain.
4 The Battle of the Sexes in the Brain and the Epigenetics of Mental Illness
According to the so-called extreme male brain theory of autism (Baron-Cohen 2002), ASD represents a pathological hypertrophy of typical male cognitive tendencies, variously described as systemizing (Baron-Cohen et al. 2009) or mechanistic (Badcock 2004) in contrast to typical female ones described correspondingly as empathizing (Baron-Cohen 2005) or mentalistic (Badcock 2004). High-functioning male autistics certainly outnumber female ones by at least four to one, but paradoxically for the extreme male brain theory, the sex ratio is much closer to unity where severe ASD is concerned (Baron-Cohen et al. 2005). If autism were indeed an extreme male brain condition, you would expect the exact opposite: more males with severe symptoms and more females with mild ones. This anomaly—not to mention the paradox of how females could be said to have “extreme male” brains—is readily resolved by the imprinted brain theory. According to this way of looking at things, ASD is caused not by an extreme male brain but by a bias in gene expression in the brain in favor of paternally expressed imprinted genes.
Because the mother’s genes are equally present in all her offspring, her genetic self-interest is best served by cooperation and family unity. Any net benefit from social behavior among her offspring is also a benefit to the ultimate reproductive success of her genes invested equally in all of them (Trivers 1974). Thanks to gestation and lactation, the mother is biologically the prime nurturer, and so it serves her interests to be able to nurture, educate, and instruct her children—for example, to teach them their “mother tongue” and then use it to program their thinking in ways she approves. By these means the mother can indoctrinate, condition, and socialize her offspring in behavior that is likely to benefit her equitable genetic investment in all of them.
The father, on the other hand, need make no obligatory biological contribution to his offspring beyond a single sperm, and other children of the same mother need not share his genes: Mother’s baby—father’s? Maybe! As a result, we have seen that the father’s genes build parts of the brain that tend to motivate self-interested, instinctual, and nonsocial behavior: the limbic system. The father’s genetic self-interest is not necessarily served by his child seeing things its mother’s way—for example, in making sacrifices for siblings to which its paternal genes may not be related in any way whatsoever. The verbal deficits of autism would be explained by the fact that the paternal brain—alias the limbic system—“eludes the grasp of the intellect because its animalist, primitive structure makes it impossible to communicate in verbal terms” (MacLean 1996, p. 455).
As Crespi has pointed out (Crespi 2007), “The origin of speech and language is arguably the most important transition in the evolution of modern humans.” He adds that there is now good evidence that one gene in particular—FOXP2—is critical both to the mirror neuron system in humans and to articulate speech. Mirror neurons fire when a person sees someone else performing an act which larger-scale firing of motor neurons in the same region would produce in the observer. As such, they have been seen as part of the neural basis of empathy, and there are averagely more of them in women than in men and fewest of all in autistics, whose deficits in empathy have been claimed to epitomize the disorder (Baron-Cohen 2005).
Empathy is important in language—at the very least you need to understand what other people are trying to say. But Crespi goes on to note that evidence that FOXP2 is imprinted and predominantly expressed from the paternal chromosome reminds us that language is also much concerned with self-assertion—and in the case of young children, with demands on the parents. “By this hypothesis, articulate human speech evolved as it develops, predominantly in the context of mother-offspring interactions, which are permeated by a complex mix of cooperation and conflict” (Crespi 2007).
Recently, Brown (2011) has added a new twist in his contribution to what he calls “The parental antagonism theory of language evolution.” Like Crespi, he notes that imprinted patri-genes will select for language skills related to extracting resources from the mother early in development, but adds that matri-genes will select for language skills related to cooperation with the mother and kin later in development. And of course, the mother is much more likely to be the primary caregiver than the father throughout childhood. Brown suggests that this may explain why although only about 2 % of human genes overall are imprinted, about 35 % involved with language are subject to such differential expression by parent of origin. Brown notes that loss of FOXP2 is linked to expressive but not receptive verbal dyspraxia, just as the parental antagonism theory would predict. Moreover, he also points out that Turner’s syndrome cases with a paternal X chromosome are more likely to have hearing impairments which may filter out maternal speech. Again, and as predicted by its maternal bias, he notes that the X chromosome has more language-related loci (29 %) than expected by chance and that maternal genes of humans and chimps are more distant than paternal, suggesting recent maternally mediated selection for social learning.
So-called mands (verbal demands and requests) result from aversion and deprivation and should be paternal in origin if they followed the precedent set by resource-demanding genes like IGF2. Interestingly in this respect, children diagnosed with AS do indeed show high levels of mands, and the condition is indeed caused by a paternal bias on chromosome 15. Brown adds that in Williams syndrome you see hyper-social behavior with high levels of receptivity and cooperativeness—just as its associated maternal bias on chromosome 7 would lead you to expect (Brown in press).
According to such a view of the matter (Badcock and Crespi 2006), autism could be the consequence of the failure of the maternal brain in this respect and the notable impulsiveness, compulsiveness, and contrariness of autistics the inevitable result of the paternal brain’s corresponding success. The striking social deficits seen in autism would seem to fit the idea that paternal genetic self-interest underlies the disorder because autistic children seem perversely committed to doing things their own way, in their own time, and for their own selves. If they can learn at all, they usually refuse to do so in the way adults think they should and inevitably pose a severe challenge to any caregiver (who in our evolutionary past would predominantly have been the mother and her relatives). Certainly, the reduced empathy, uncooperativeness, and insistence of routine seen in autism hardly contribute to easy parenting. Indeed, there is evidence that in experimental animals, failure to cope with change is a central characteristic of paternal brain lesions, and a persuasive case can be made for the limbic system being centrally involved in the problems associated with autism (Lathe 2006).
The same reasoning would certainly explain why the brain systems that malfunction in autism seem to be critical to a child’s social interaction with its mother (Maestro et al. 2002; Zwaigenbaum et al. 2005). Indeed, when paternal brain centers such as the hypothalamus and amygdala are active in dreams, aggressive impulses on the part of the dreamer emerge. However, when what I am calling maternal brain centers are activated in dreaming (the forebrain and neocortex), aggressive impulses are inhibited and cooperative and pro-social ones expressed (McNamara et al. 2005). As you would predict if autism was indeed caused by enhanced expression of the father’s genes, proliferative overgrowth of the placenta is found at three times the normal rate in autism (Anderson et al. 2007), and studies suggest that autistics—and males in particular—are heavier than normal at birth and have elevated levels of growth hormones such as IGF2 (Mills et al. 2007), confirming that they are indeed predisposed to consume more than usual of the mother’s resources (Sugie et al. 2005). Again, there is evidence that autistics show early brain growth during gestation at the expense of the mother and have a 100-fold greater risk of neurofibromatosis (in other words, pathological overgrowth in the form of benign nerve-tissue tumors) along with genetic alterations relating to the PI3K (phosphatidylinositol 3-kinase) pathway resulting in greater vulnerability to cancer—despite the fact that autistics smoke less (Crespi and Badcock 2008).
These facts are all the more telling when contrasted with the corresponding situation in PSD: there, in accordance with the growth-limiting effect of maternal genes, you find intrauterine growth restriction, placental undergrowth, and higher incidence of fetal hypoxia; low birth weight, low levels of brain growth factors and smaller brain size, thinner cortex, and smaller hippocampus and amygdala; decreased risk of cancer among schizophrenics (despite increased smoking) and their first-order relatives; and reduced expression of growth factors and decreased stem cell proliferation, reduced thresholds for apoptosis (programmed cell death, e.g., if a cell becomes precancerous), and evidence of increased expression of tumor-suppressor genes in schizophrenics (Crespi and Badcock 2008) (Fig. 5.1).

The imprinted brain theory (Redrawn and modified from Crespi and Badcock (2008))
The fact that all fathers are male explains why you could mistake autism for an extreme male brain disorder. But because males and females have both paternal and maternal brains, as we are calling them, you can easily account for the fact that females as well as males can suffer from autism. More high-functioning autistics might be expected to be male if only their paternal brain were affected—perhaps driven to an extreme early in development by male sex hormones (Ingudomnukul et al. 2007; Knickmeyer et al. 2004). The intact intelligence and verbal abilities of high-functioning autistics seen in Asperger’s syndrome would therefore be the result of predominantly normal maternal brain development, while the occasional appearance of savant skills could be explained by an enhancement of characteristically male cognitive skills associated with an extreme paternal brain.
Finally, the obvious implication of all this is that if ASD is an extreme paternal brain disorder, PSD must be an extreme maternal one. Simon Baron-Cohen, who developed the extreme male brain theory of autism in recent times, explicitly rejected the proposition that psychosis might be the extreme female/paternal brain counterpart of autism. However, recent research revealed findings consistent with the imprinted brain theory, particularly those linking measures of hyper-mentalism to paranoia and positive symptoms of PSD in 70 healthy female students (Brosnan et al. 2010).
5 The Diametric Model of the Mind and Mental Illness
Mental disorders can be located along a dimension of mentalism (otherwise known as “theory of mind,” “folk psychology,” or “people skills”) defined as our evolved ability to comprehend others’ actions and behavior in purely mental terms (such as intention, belief, desire, emotion). Autistics, notoriously, are poor where mentalistic skills like inferring intention or understanding false belief are concerned. ASDs therefore belong on the hypo-mentalistic side of the continuum. However, PSD can be typified as hyper-mentalistic: paranoid schizophrenics, for example, symptomatically over-interpret intention either positively in erotomania (delusions that others are in love with you) or negatively in delusions of persecution. They also entertain bizarre false beliefs about themselves and others, and generally exhibit excessive mentalism, often enshrined in quasi-religious or mystical delusions (Badcock 2004). Indeed, the symptoms and signs of autism and psychoses like paranoid schizophrenia exhibit a remarkable pattern of antithesis similar to that seen in the sister syndromes mentioned above, such as BWS/SRS and AS/PWS (Table 5.1).
The concepts of hypo- and hyper-mentalism readily explain the last item in Table 5.1: age of onset. Typically, this is early childhood for autism but late adolescence or adulthood for schizophrenia: a difference which up until now has lacked an obvious explanation. But the fact that you need years to develop normal mentalistic skills before you can overdevelop them to the point of psychosis readily explains why the mentalistic deficits of autism are apparent in childhood and why the hyper-mentalism of psychosis can normally only become fully apparent much later.
Recent brain imaging studies have found evidence of hyper-mentalism in the brain. The studies presented normal and paranoid schizophrenic subjects with cartoon-based tests depicting various forms of intentions while scanning their brain activity. It confirmed earlier findings (Walter et al. 2009) suggesting that the medial prefrontal cortex is peculiarly involved in the social dimension of mentalizing but not active when it is a question of purely private intentions. Private prior intentions activated only the right temporoparietal junction (TPJ) and the precuneus (a deeply buried part of the parietal cortex involved with episodic memory, visuospatial processing, and self-awareness). The left TPJ became active when there was a social dimension to communicating intention, but only if it related to the present. In contrast to normal subjects, the schizophrenics’ intentional thinking was found to be permanently active, even when unwarranted and inappropriate: for example, in relation to inanimate objects. The authors cite the diametric model (Crespi and Badcock 2008) and then add that “Adopting a similar approach, we claim that the impairments in understanding others’ intentions exhibited by paranoid patients and autistic patients, respectively, can be considered as the two extremes of a continuum” (Bara et al. 2011)—just as the imprinted brain theory proposes.
One of the most counterintuitive implications of the concept of hyper-mentalism and its relation to psychosis is that if ASD is symptomatically deficient in mentalistic skills, PSD should reveal better than normal mentalistic abilities in certain respects. Psychiatrists since Bleuler have been commenting on the uncanny ability of some PSD patients where tuning into other people’s minds and emotions is concerned. Recent experiments with borderline personality disorder (BPD) and schizotypical patients in particular have revealed better than normal skills in reading expression and detecting other’s state of mind. BPD patients get more correct responses than healthy volunteers on the Reading the Mind in the Eyes Test. But as the concept of hyper-mentalism might suggest, they do particularly well on neutral emotional expressions but tend to over-personalize their responses (Fertuck et al. 2009). When playing trust games with and without relevant facial cues, BPD players equaled normal controls in recognizing emotions and assessing fairness but were superior to normal controls when emotional cues were present and were more objective in assessing fairness (Franzen et al. 2010). Indeed, in the first laboratory study to explicitly test the diametric model, 318 students were assessed for autistic versus schizotypical (i.e., mildly psychotic) tendencies and tested for their ability on an Embedded Figures Test. The authors report that their results offer support for the claim made by the diametric model “that autistic and positive schizophrenia traits are diametrically opposed with regard to their effect on local versus global processing” (Russell-Smith et al. 2010).
Copy number variation (CNV) is the recently discovered and very surprising finding that individuals vary in the number of copies of particular genes they carry by up to 12 % of the total. CNV can result from duplication or deletion of genes and to this extent resembles imprinted gene expression, which can produce nil or double expression of normally singly expressed genes. Crespi, Stead, and Elliot used CNV, single-gene associations, growth-signaling pathways, and brain-growth outcomes to evaluate the diametric model. They found that CNV findings support the diametric model, which holds that autism and schizophrenia stand in opposition to one another: at four places in the genome, deletions predispose to one, while duplications predispose to the other. They also found that single-gene associations are inconsistent with the model which sees ASD and PSD as separate entities because schizophrenia and autism frequently share associated genes. Where brain growth was concerned, they found that autism goes with enhanced brain growth, whereas schizophrenia is characterized by reduced brain size—just as the diametric model predicts (Crespi et al. 2009a). Indeed, Shinawi et al. report independently that autism and macrocephaly observed with deletion and microcephaly seen in duplication of a site on chromosome 16 support the diametric model (Shinawi et al. 2010).
6 The Genes that Made Us Human and the Epigenesis of Culture
Schizophrenia, like autism, poses a striking paradox because, along with this high degree of heritability, it manifestly damages individuals’ survival and reproductive success. So how could the genes responsible evolve? Nevertheless—and again like autism—it persists at a prevalence of about 1 % across all human cultures (Tamminga and Holcomb 2005). A proposed solution is that genetic liability to schizophrenia has evolved as a consequence of selection for human traits involving social cognition, creativity, and language (Crow 1997; Horrobin 1998)—or what you could call mentalism (Badcock 2004). According to this hypothesis, genes that increase risk of schizophrenia have been subject to positive selection in the evolution of human beings, thanks to their key role in mentalistic cognition. Recently, Crespi, Summers, and Dorus evaluated this hypothesis by screening human and primate genes for evidence of positive selection (Crespi et al. 2007). They found statistically significant evidence for positive selection on 26 of 80 genes mediating liability to schizophrenia, including some which exhibit some of the best-supported functional and genetic links to this disorder. Previous studies indicated that recent positive selection in humans has driven the evolution of a suite of additional genes linked with schizophrenia risk, and variants of three genes associated with schizophrenia have recently been linked with measures of creativity. Taken together, the authors conclude that these findings provide evolutionary and genetic support for the hypothesis that schizophrenia represents “the illness that made us human” (Horrobin 1998)—or at least, half of what makes us human.
If mentalism gave us our mental culture, then there is good evidence for a contrasting form of cognition—what you might call mechanistic cognition. This is the mode of cognition that we have evolved to interact with the physical, nonhuman, natural environment, and it stands in contrast to mentalistic cognition, which evolved to facilitate social contact and cognition in relation to other people (Badcock 2004).
Significantly, autistics sometimes show remarkable compensations for their mentalistic deficits in mechanistic cognitive skills—something otherwise known as autistic savantism. Among the most common such skills are calendar calculation (such as knowing the date of Easter in any year you care to name), rote memorization, and math skills (Happé and Frith 2009). But as the diametric model of the mind might also suggest, psychotics show the contrary cognitive configuration: despite the mentalistic gifts mentioned above, they also reveal deficits in mechanistic skills (Toulopoulou et al. 2006). According to one authority, “Intellectual asymmetry with a relative superiority of verbal skills to spatial skills represents a putative endophenotype of schizophrenia” (Kravariti et al. 2006). Indeed, a recent finding confirms that visuospatial ability (and especially mental rotation) is impaired in schizophrenia patients when compared with healthy controls and implicates one particular gene (S100B) in accounting for this deficit (Zhai et al. 2011).
According to a survey of 919 families of children with ASD which listed occupations of parents, fathers of children with ASD were twice as often employed in engineering as were fathers in any of four control groups of children with Tourette’s or Down syndrome. This was also true of grandfathers: among the fathers of children with autism, the ratio of those working in engineering to those working in other fields was 6:1, whereas in the Tourette’s and Down cases, it was less than 3:1. The authors conclude that there seems to be a small but statistically significant link between autism and engineering and that their result might also help to explain why a condition like autism persists in the gene pool. They speculate that the very same genes that lead an individual to have a child with autism can lead to superior functioning in folk physics and observe that engineering and related folk physics skills have transformed the way in which our species lives. Indeed, they conclude that “without such skills, Homo sapiens would still be pre-industrial” (Baron-Cohen et al. 1997).
Part of the paradox of why severe mental illnesses like autism and schizophrenia have genetic causes may therefore lie in the fact that the very same genes that can produce these pathological conditions also underpin the twin cognitive systems on which human preeminence as a species relies: mentalistic and mechanistic cognition. One gave us our society, culture, language, and ability to empathize and interact with other people’s minds. The other gave us science, technology, and all the manual, mechanical, and technical skills on which our civilization depends. If this view is correct, autism and psychoses like schizophrenia are the price we pay for these critical cognitive adaptations (Badcock 2004).
7 Nurture via Nature: Environmental Epigenetics
A final and equally challenging and controversial implication of the imprinted brain theory is that it may be able to explain the seemingly non-genetic, environmental, and social factors in the incidence of mental illnesses like autism and schizophrenia mentioned at the beginning.
A possible explanation for the findings relating maternal starvation to schizophrenia mentioned earlier is that maternal starvation has the same effect as maternally active genes in restricting growth and, according to the hypothesis advanced here, also predisposes towards the risk of psychosis in later life: nurture—or the lack of it—via nature, so to speak. Furthermore, a study of IGF2 expression in children born during the Dutch wartime famine provided the first evidence that transient environmental conditions early in human gestation can affect the expression of such imprinted genes (Heijmansa et al. 2008). Although this effect was found among those with normal birth weight who were exposed to famine early in gestation but was not found among those with low birth weight unrelated to IGF2 expression exposed to famine late in gestation, the finding suggests that more direct effects cannot be ruled out in principle. On the contrary, it establishes a strong precedent for thinking that environmental factors could directly or indirectly affect gene expression in accordance with the theory set out here.
The suggestion that severe deficits in nutrition like those associated with maternal starvation during pregnancy might have pathological consequences where development is concerned is hardly surprising. But if that were true, the theory proposed here would have the contrary, very counterintuitive implication. This is that environmental influences which enhanced growth might predispose towards ASD, perhaps by way of increasing the expression of genes like IGF2 or at the very least by mimicking their effects. This in itself might explain quite a lot of the so-called autism epidemic of recent years. Growth enhancement, thanks to higher standards of living in developed countries, could be predicted to predispose towards milder forms of ASD such as Asperger’s syndrome. Indeed, birth weights of newborn babies in Vienna rose an “unprecedented amount” (from a mean of 3 kg to a peak of 3.3 kg) during the 1920s (Ward 1993, p. 56), and perhaps this partly explains why Asperger was to discover the syndrome named after him during the next couple of decades.
Again, critics of Kanner, autism’s other independent discoverer, have pointed out that he portrayed it as an “upper class” disorder, but that later research, particularly in Sweden (the Gothenburg studies) contradicted this and found no clear link to social class (Gillberg 1992). However, it might simply be that during the 1940s the heavier-birth-weight effect was mainly seen among better-off people in the USA, but that by the 1980s it had spread to just about everyone in welfare-state Sweden—and today to most people in modern Western societies, where obesity, rather than undernourishment, has become the primary health problem related to food intake.
A controversial and counterintuitive prediction of the theory for which there is already much evidence is that if ASD has increased in modern societies with higher standards of living as it so spectacularly has done, then PSD should be falling. Interestingly in this respect, rates of admission for PSDs like schizophrenia have decreased by between 10 % and 57 % in England and Wales, Scotland, Denmark, Australia, and New Zealand. Indeed, even Bleuler, who coined the term schizophrenia, noticed a secular decline in his own lifetime (Der et al. 1990), and a recent Canadian study showed a 42 % decrease in the number of first-admission schizophrenia cases over 20 years. It found that annual inpatient prevalence rates decreased by 52 % between 1986 and 1996, with no corresponding change in outpatient rates, regardless of sex (Woogh 2001).
The exception is major depressive disorder (MDD); however, there are now reasons for thinking that MDD may be an immunoregulatory disorder with major psychiatric symptoms: in other words, the outcome of an immune system dysregulated by modern hygienic living conditions, rather than a dysregulated brain (Raison et al. 2010). Again, where schizophrenia is concerned, there are suspicions that the protozoan parasite Toxoplasma gondii may sometimes be a contributing factor to the development of the illness (Webster et al. 2006). But as I have argued elsewhere, T. gondii is known to attack the amygdala, a key component of what I have been calling the paternal brain, perhaps explaining its link with PSD by way of disturbing the balance of brain function in a maternal direction, as predicted by the theory (Badcock 2009). Indeed, given its known affinity for the limbic system, much the same might be said for the other suspected infectious cause of schizophrenia: Cytomegalovirus (CMV) (Yolken and Torrey 2008).
8 Summary and Conclusion
The imprinted brain theory proposes that development can be pushed to either mental extreme by any factors that affect gene expression either before or after birth (Fig. 5.1). Valproic acid is known to do this, as is thalidomide and other environmental causes of autism (Rodier 2000). Where purely genetic factors are concerned, we have seen that the theory proposes that increased expression of paternal genes like IGF2 will predispose to autism—and expression of that gene is now known to be enhanced in individuals with ASD. And because all fathers are male, the new theory can also be reconciled with the extreme male brain theory of autism, which persuasively argues that ASD can often be linked to increased testosterone exposure in utero, and to the more lateralized brain characteristic of males. But because all mothers are female, enhanced expression of maternal genes also goes with reduced fetal testosterone and the less lateralized brain typical of women. Moreover, in cases where an extra X chromosome is present, it results in brain features similar to those found in schizophrenia, along with a notably increased vulnerability to psychosis, just as the theory would predict.
Two recent discoveries both illustrate and corroborate the imprinted brain theory. Retinoic acid-related orphan receptor-alpha (RORA) is the first candidate gene for autism that has been found to be responsive to both male and female sex hormones. In research on twins, the expression of RORA and other candidate autism genes was shown to be affected by a key epigenetic factor, DNA methylation, which in turn might explain how one of a pair of monozygotic twins could be affected by ASD, but the other not (Nguyen et al. 2010). RORA is involved in several key processes implicated in autism, including Purkinje cell differentiation, muscle tone and development of the cerebellum, protection of neurons against oxidative stress, suppression of inflammation, and regulation of circadian rhythm. Testosterone acts on androgen receptors to reduce RORA, while estrogen acts to increase it. Aromatase (an enzyme responsible for the conversion of testosterone to estrogen) is also a target for RORA, and both RORA and aromatase are strongly correlated in brain tissue and relatively reduced in the frontal cortex of ASD subjects—a region of the brain known to be critically involved in mentalistic cognition (Sarachana et al. 2011). As such, this is a finding that beautifully endorses the imprinted brain theory because it demonstrates how male genetic influence predisposes to ASD and how the presence of female sex hormones such as estrogen is protective.
A second recent discovery (Ressler et al. 2011) illustrates epigenetic effects working as predicted by the theory in the opposite direction. Over a lifetime, posttraumatic stress disorder (PTSD) is diagnosed in as many as 40 % of individuals exposed to traumatic events. The classic triad of symptoms is as follows: waking or dreaming flashbacks or recurrent involuntary reactions to the trauma or things recalling it; avoidances, fears, and phobias associated with the trauma; and finally, hyperarousal, hypervigilance, and exaggerated startle response. As such, these symptoms clearly mark PTSD out as a hyper-mentalizing disorder on the psychotic side of the spectrum according to the diametric model of mental illness peculiar to the imprinted brain theory. Indeed, in severe or chronic cases, classic psychotic symptoms such as paranoid delusions and auditory hallucinations may be present. And as the imprinted brain theory would predict, females may be at twice the risk of PTSD compared to males (Breslau 2001).
A study of 1,200 highly traumatized subjects with and without PTSD matched for age, sex, race, and trauma history found that levels of pituitary adenylate cyclase-activating polypeptide (PACAP) correlated strongly with PTSD symptoms and diagnosis in female, but not in male subjects (Ressler et al. 2011). This hormone is involved in activation and growth of neurons and their connections, and in rodents an-order-of-magnitude higher concentration of it is found in a part of the brain involved with conditioned fear reactions (the amygdala and associated regions). A separate experiment on startle reflexes in 16 male and 11 female subjects revealed that only female participants with the high PACAP levels showed correlated conditioned fear responses. A presumed estrogen receptor for PACAP seems to explain the striking limitation of the effect to women just as in the previous case we saw that RORA’s links with androgens and androgen receptors might explain some of the male-biased incidence of autism. Furthermore, it is worth pointing out that the PACAP receptor gene is subject to differential methylation. This is the same epigenetic mechanism found in imprinted genes, in X-chromosome gene inactivation, and in RORA, and it is the methylation of the critical part of the PACAP receptor gene (ADCYAP1R1) which correlates with PTSD symptoms (Ressler et al. 2011). Such epigenetic mechanisms are known to be affected by environmental factors and insults during development, perhaps suggesting a further explanation for constitutional variations in susceptibility to PTSD.
In other words, PTSD and its associated receptor gene now seems to fit in as aptly on the psychotic side of the spectrum as RORA does on the autistic side. Both discoveries vindicate the twin predictions of the imprinted brain theory: namely, that mental illness is caused by epigenetic mechanisms affecting gene expression as well as by inheritance and that enhanced expression on the male/paternal side results in autistic spectrum disorders (as with RORA), while biased expression on the female/maternal side results in psychotic spectrum disorders—such as PTSD. Together, these two very recent discoveries suggest that as the genetic basis of mental disorders is discovered, more and more of them will be found to fit into the new paradigm proposed by the imprinted brain theory and its associated diametric model of the mind and of mental illness.
A further conclusion is that normality represents a more or less balanced expression of genes and environmental developmental influences; however, the sexes are likely to be slightly offset. This would fit with the finding that ASD afflicts more males than females and that men typically do worse on tests of mentalistic competence than do women. Women, on the other hand, would be symmetrically offset to the more mentalistic side of the spectrum, and this might explain why BPD is three times more common in women than in men and why rates of incidence of schizophrenia among family members of women with the disorder are higher than those among family members of men with schizophrenia. And although there is a slightly higher incidence of schizophrenia overall in men, erotomania appears to be a predominantly female pathology, with women suffering more paranoid delusions and hallucinations than men, particularly in late-onset cases.
The model appears to rule out anyone suffering from an ASD and a PSD simultaneously, and such comorbidity does appear to be rare—but is not unknown. There are cases of individuals diagnosed with bipolar disorder who also show unmistakable signs of ASD during their non-manic phases. Indeed, in my book I quote one who suffers from severe gaze-aversion, autistic deficits in a sense of self and social anxiety most of the time, but who becomes comfortable with other people during manic episodes when his sense of self hypertrophies into megalomania with the feeling that he is the returned Jesus Christ (Badcock 2009, pp. 96–7)! Furthermore, there is evidence of both ASD and PSD in Newton and Beethoven and also so in the Nobel Prize-winning mathematician John Nash. Here the theory predicts that the ASD must come first (typically in childhood) and leave a permanent savant-like basis later built on by hyper-mentalistic tendencies to produce an unusually broadened and dynamically balanced cognitive configuration: that of true genius.
9 Future Prospects: The Epigenetic Revolution
Such a speculative theory as this can be expected to be controversial, and much remains to be done to work out its details. But the hypothesis does have one outstanding merit: it makes clear and counterintuitive predictions about which genes are likely to be involved, about how they should be expressed, and about what effects they should be found to have in the brain and on cognition. Rapid progress now being made in genomics and neuroscience should be able to disprove the basic concept quickly if it is indeed as wrong as some of its critics believe. Full corroboration may take some time and in practice would probably need something of a Human Imprintome Project (i.e., one focused on differential paternal/maternal methylation or other evidence of actual imprinting—something not currently provided by the Human Epigenome Project) (Crespi, 2010, personal communication). But were the theory to be proved even partly true, it would have a number of far-reaching and indeed revolutionary implications.
First and foremost, the new theory would place human epigenetics in its proper biological and evolutionary context and, if proved, would vindicate the kinship/conflict theory of imprinting in its most crucial application: the mind and brain. The study of human evolution would be put on a secure genetic basis, with genes and their expression, rather than phenotypes, being the new focus of research and explanation in accordance with the “selfish-gene” approach of modern Darwinism (Badcock 2009; Dawkins 1989).
Secondly, the diametric model and its basis in genetic conflict outlined above would provide a completely new paradigm for psychology. The twentieth century saw the emergence of two opposed psychological paradigms: psychoanalysis, which was guilty of hyper-mentalizing to an almost paranoid extent, and behaviorism, which was hypo-mentalistic in its autism-like denial of the mind (Badcock 2004). The new theory’s dualistic mentalistic/mechanistic model of cognition does full justice to the mind without going to either extreme and roots cognition in a sound biological foundation (Badcock 2009).
Finally, success for the theory would revolutionize psychiatry and psychotherapy as a whole. Mental disorders could be classified in terms of their position on the mentalistic continuum and diagnosis confirmed by epigenetic testing for the patterns of gene expression predicted by the theory. Prevention and therapy would also be revolutionized by such objective measures, along with new drugs and other interventions tailored to the individual’s epigenome. Indeed, individual epigenetic profiling might open up a completely new window on personal development, especially if psychological changes over a person’s lifetime could be linked to variations in the pattern of gene expression as they conceivably might (Abu-Akel, 2011, private communication). Psychotherapy could certainly be founded on a secure basis in epigenetics, and the diametric model would become the basis of a completely new rationale for intervention. Mentalistic skills training has already been shown to benefit autistics, but only the diametric model explains why mechanistic skills training appears to benefit psychotics (e.g., pitch discrimination, which is often perfect in autistics (Fisher et al. 2009)).
At the very least, the imprinted brain theory poses a challenging new perspective for thinking about epigenetics and its relationship to the brain, the mind, and mental illness. The next few years will certainly reveal how successful it is going to be in that respect.
Abbreviations
- ADCYAP1R1:
-
PACAP receptor
- AG:
-
Androgenetic
- AS:
-
Angelman syndrome
- ASD:
-
Autism spectrum disorder
- BPD:
-
Borderline personality disorder
- BWS:
-
Beckwith-Wiedemann syndrome
- CMV:
-
Cytomegalovirus
- CNV:
-
Copy number variation
- MDD:
-
Major depressive disorder
- PACAP:
-
Pituitary adenylate cyclase-activating polypeptide
- PG:
-
Parthenogenetic
- PSD:
-
Psychotic spectrum disorder
- PTSD:
-
Posttraumatic stress disorder
- PWS:
-
Prader-Willi syndrome
- RORA:
-
Retinoic acid-related orphan receptor-alpha
- SRS:
-
Silver-Russell syndrome
- TPJ:
-
Temporoparietal junction
References
Aitken KJ (2008) Intersubjectivity, affective neuroscience, and the neurobiology of autistic spectrum disorders: a systematic review. Keio J Med 57:15–36
Allen ND, Logan K, Lally G, Drage JD, Norris ML, Keverne B (1995) Distribution of parthenogenetic cells in the mouse brain and their influence on brain development and behavior. Proc Natl Acad Sci U S A 92:10782–10786
Anderson GM, Jacobs-Stannard A, Chawarska K, Volkmar FR, Kliman HJ (2007) Placental trophoblast inclusions in autism spectrum disorder. Biol Psychiatry 6:487–491
Angelman H (1965) ‘Puppet’ children: a report on three cases. Dev Med Child Neurol 7:681–688
Badcock CR (1995) Pre-printed gift tags. The Times Higher Education Supplement, p 30. 16 June
Badcock CR (1999) Genetic conflict: a new biological foundation for freud’s fundamental findings. Psychother Rev 1:116–122
Badcock CR (2000) Evolutionary psychology: a critical introduction. Polity Press, Cambridge
Badcock CR (2004) Mentalism and mechanism: the twin modes of human cognition. In: Crawford C, Salmon C (eds) Evolutionary psychology, public policy, and personal decisions. Lawrence Erlbaum Associates, Mahwah, pp 99–116
Badcock CR (2008) An evolutionary theory of mind and of mental illness: genetic conflict and the mentalistic continuum. In: Crawford C, Krebs D (eds) Foundations of evolutionary psychology. Lawrence Erlbaum Associates, New York, pp 431–450
Badcock CR (2009) The imprinted brain: how genes set the balance between autism and psychosis. Jessica Kingsley, London/Philadelphia
Badcock CR, Crespi B (2006) Imbalanced genomic imprinting in brain development: an evolutionary basis for the etiology of autism. J Evol Biol 19:1007–1032
Badcock CR, Crespi B (2008) Battle of the sexes may set the brain. Nature 454:1054–1055
Baird G, Simonoff E, Pickles A, Chandler S, Loucas T, Meldrum D, Charman T (2006) Prevalence of disorders of the autism spectrum in a population cohort of children in South Thames: the special needs and autism project (SNAP). Lancet 368:210–215
Bara BG, Ciaramidaro A, Walter H, Adenzato M (2011) Intentional minds: a philosophical analysis of intention tested through fMRI experiments involving people with schizophrenia, people with autism, and healthy individuals. Front Hum Neurosci 5. doi:10.3389/fnhum.2011.00007
Barlow D (1995) Gametic imprinting in mammals. Science 270:1610–1613
Baron-Cohen S (2002) The extreme male brain theory of autism. Trends Cogn Sci 6:248–254
Baron-Cohen S (2005) The empathizing system. In: Ellis BJ, Bjorklund DF (eds) Origins of the social mind. The Guilford Press, New York/London, pp 468–492
Baron-Cohen S, Wheelwright S, Stott C, Bolton P, Goodyer I (1997) Is there a link between engineering and autism? Autism 1:101–109
Baron-Cohen S, Knickmeyer RC, Belmonte MK (2005) Sex differences in the brain: implications for explaining autism. Science 310:819–823
Baron-Cohen S, Ashwin E, Ashwin C, Tavassoli T, Chakrabarti B (2009) Talent in autism: hyper-systemizing, hyper-attention to detail and sensory hypersensitivity. Philos Trans R Soc Lond B Biol Sci 364:1377–1383
Bonati MT, Russo S, Finelli P, Valsecchi MR, Cogliati F, Cavalleri F, Roberts W, Elia M, Larizza L (2007) Evaluation of autism traits in Angelman syndrome: a resource to unfold autism genes. Neurogenetics 8:169–178
Breslau N (2001) The epidemiology of posttraumatic stress disorder. J Clin Psychiatry 62:16–22
Brosnan M, Ashwin C, Walker I, Donaghue J (2010) Can an ‘extreme female brain’ be characterised in terms of psychosis? Pers Indiv Differ 49:738–742
Brown WM (2011) The parental antagonism theory of language evolution: preliminary evidence for the proposal. Hum Biol 83:213–245
Brown WM, Consedine NS (2004) Just how happy is the happy puppet? An emotion signaling and kinship theory perspective on the behavioral phenotype of children with Angelman syndrome. Med Hypotheses 63:377–385
Chorney MJ, Chorney K, Seese N, Owen MJ, Daniels J, McGiffin P, Thompson LA, Detterman DK, Benbow C, Lubinski D, Eley T, Plomin R (1998) A quantitative trait locus associated with cognitive ability in children. Psychol Sci 9:1–7
Crespi B (2007) Sly FOXP2: genomic conflict in the evolution of language. Trends Ecol Evol 22:174–175
Crespi B (2008) Genomic imprinting in the development and evolution of psychotic spectrum conditions. Biol Rev Camb Philos Soc 83:441–493
Crespi B, Badcock C (2008) Psychosis and autism as diametrical disorders of the social brain. Behav Brain Sci 31:241–261
Crespi B, Summers K, Dorus S (2007) Adaptive evolution of genes underlying schizophrenia. P Roy Soc Lond B Bio Sci 274:2801–2810
Crespi B, Stead P, Elliot M (2009a) Comparative genomics of autism and schizophrenia. Proc Natl Acad Sci U S A 107(Suppl 1):1736–1741
Crespi B, Summers K, Dorus S (2009b) Genomic sister-disorders of neurodevelopment: an evolutionary approach. Evol Appl 2:81–100
Crow TJ (1997) Is schizophrenia the price that homo sapiens pays for language? Schizophr Res 28:127–141
Dawkins R (1989) The selfish gene, 2nd edn. Oxford University Press, Oxford
De Chiara TM, Robertson EJ, Efstratiadis A (1991) Parental imprinting of the mouse insulin-like growth factor II gene. Cell 64:849–859
Der G, Gupta S, Murray RM (1990) Is schizophrenia disappearing? Lancet 335:513–516
Eggermann T, Schönherr N, Meyer E, Obermann C, Mavany M, Eggermann K, Rank MB, Wollmann HA (2005) Epigenetic mutations in 11p15 in Silver-Russell syndrome are restricted to the telomeric imprinting domain. J Med Genet 43:615–616
Fertuck EA, Jekal A, Song I, Wyman B, Morris MC, Wilson ST, Brodsky BS, Stanley B (2009) Enhanced ‘reading the mind in the eyes’ in borderline personality disorder compared to healthy controls. Psychol Med 39:1979–1988
Fisher M, Holland C, Subramaniam K, Vinogradov S (2009) Neuroplasticity-based cognitive training in schizophrenia: an interim report on the effects 6 months later. Schizophr Bull 36:869–879
Franzen N, Hagenhoff M, Baer N, Schmidt A, Mier D, Sammer G, Gallhofer B, Kirsch P, Lis S (2010) Superior ‘theory of mind’ in borderline personality disorder: an analysis of interaction behavior in a virtual trust game. Psychiatry Res 187:224–233
Gillberg CL (1992) The Emanuel Miller Memorial Lecture 1991. Autism and autistic-like conditions: subclasses among disorders of empathy. J Child Psychol Psychiatry 33:813–842
Goldstein JM, Seidman LJ, Horton NJ, Makris N, Kennedy DN (2001) Normal sexual dimorphism of the adult human brain assessed by in vivo magnetic resonance imaging. Cereb Cortex 11:490–497
Goos LM, Silverman I (2006) The inheritance of cognitive skills: does genomic imprinting play a role? J Neurogenet 20:19–40
Gregg C, Zhang J, Weissbourd B, Luo S, Schroth GP, Haig D, Dulac C (2010) High-resolution analysis of parent-of-origin allelic expression in the mouse brain. Science 329:643–648
Gur RC, Gunning-Dixon F, Bilker WB, Gur RE (2002) Sex difference in temporo-limbic and frontal brain volumes of healthy adults. Cereb Cortex 12:998–1003
Gur RE, Kohler C, Turetsky BI, Siegel SJ, Kanes SJ, Bilker WB, Brennan AR, Gur RC (2004) A sexually dimorphic ratio of orbitofrontal to amygdala volume is altered in schizophrenia. Biol Psychiatry 55:512–517
Haig D (2002) Genomic imprinting and kinship. Rutgers University Press, New Brunswick
Haig D (2004) Genomic imprinting and kinship: how good is the evidence? Annu Rev Genet 38:553–585
Haig D (2006) Intragenomic politics. Cytogenet Genome Res 113:68–74
Haig D, Graham C (1991) Genomic imprinting and the strange case of the insulin-like growth factor II receptor. Cell 64:1045–1046
Hamilton WD (1996) Narrow roads of gene land: evolution of social behaviour. WH Freeman/Spektrum, Oxford
Hannah J, Hayward BE, Sheridan E, Bonthron DT (2002) A global disorder of imprinting in the human female germ line. Nature 416:539–542
Happé F, Frith U (2009) Autism and talent. Philos Trans R Soc Lond B Biol Sci 364:1345–1480
Heijmansa BT, Tobia EW, Stein AD, Putter H, Blauwd GJ, Sussere ES, Slagbooma PE, Lumeye LH (2008) Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A 105:17046–17049
Holm VA, Cassidy SB, Butler MG, Hanchett JM, Greensway LR, Whitman BY, Greenbergy F (1993) Prader-Willi syndrome: consensus diagnostic criteria. Pediatrics 91:398–402
Horrobin DF (1998) Schizophrenia: the illness that made us human. Med Hypotheses 50:269–288
Ingudomnukul E, Baron-Cohen S, Wheelwright S, Knickmeyer R (2007) Elevated rates of testosterone-related disorders in women with autism spectrum conditions. Horm Behav 51:597–604
Jablonka E, Lamb M (1995) Epigenetic inheritance and evolution. Oxford University Press, Oxford
Jones D (2008) Killer instincts. Nature 451:512–515
Kaminsky ZA, Tang T, Wang S-C, Ptak C, Oh GHT, Wong AHC, Feldcamp LA, Virtanen C, Halfvarson J, Tysk C, McRae AF, Visscher PM, Montgomery GW, Gottesman II, Martin NG, Petronis A (2009) DNA methylation profiles in monozygotic and dizygotic twins. Nat Genet 41:240–245
Kent L, Bowdin S, Kirby GA, Cooper WN, Maher ER (2008) Beckwith-Wiedemann syndrome: a behavioral phenotype-genotype study. Am J Med Genet B Neuropsychiatr Genet 147B:1295–1297
Keverne EB, Fundele R, Narasimha M, Barton SC, Surani MA (1996) Genomic imprinting and the differential roles of parental genomes in brain development. Brain Res Dev Brain Res 92:91–100
Killian JK, Nolan CM, Wylie AA, Li T, Vu TH, Hoffman AR, Jirtle RL (2001) Divergent evolution in M6P/IGF2R imprinting from the Jurassic to the Quaternary. Hum Mol Genet 10:1721–1728
Knickmeyer R, Baron-Cohen S, Raggatt P, Taylor K (2004) Foetal testosterone, social relationships, and restricted interests in children. J Child Psychol Psychiatry 46:198–210
Kravariti E, Toulopoulou T, Mapua-Filbey F, Shultze W, Sham P, Murray RM, McDonald C (2006) Intellectual asymmetry and genetic liability in first-degree relatives of probands with schizophrenia. Br J Psychiatry 188:186–187
Lathe R (2006) Autism, brain, and environment. Jessica Kingsley Publishers, London/Philadelphia
LeDoux J (1996) The emotional brain: the mysterious underpinnings of emotional life. Simon & Schuster, New York
MacLean PD (1996) Limbic system. In: Beaumont JG, Kenealy P, Rogers MCJ (eds) The Blackwell dictionary of neuropsychology. Blackwell, Oxford
Maestro S, Muratori F, Cavallaro MC, Pei F, Stern D, Golse B, Palacio-Espasa F (2002) Attentional skills during the first 6 months of age in autism spectrum disorder. J Am Acad Child Adolesc Psychiatry 41:1239–1245
McNamara P, McLaren D, Smith D, Brown A, Stickgold R (2005) A “Jekyll and Hyde” within: aggressive versus friendly interactions in REM and Non-REM dreams. Psychol Sci 16:130–136
Mendrek A (2007) Reversal of normal cerebral sexual dimorphism in schizophrenia: evidence and speculations. Med Hypotheses 69:896–902
Mills JL, Hediger ML, Molloy CA, Chrousos GP, Manning-Courtney P, Yu KF, Brasington M, England LJ (2007) Elevated levels of growth-related hormones in autism and autism spectrum disorder. Clin Endocrinol 67:230–237
Milner KM, Craig EE, Thompson RJ, Veltman MW, Thomas NS, Roberts S, Bellamy M, Curran SR, Sporikou CM, Bolton PF (2005) Prader-Will syndrome: intellectual abilities and behavioural features by genetic subtype. J Child Psychol Psychiatry 46:1089–1096
Newton G (2001) The case of the biparental mole. Wellcome News, pp 18–19
Nguyen A, Rauch TA, Pfeifer GP, Hu VW (2010) Global methylation profiling of lymphoblastoid cell lines reveals epigenetic contributions to autism spectrum disorders and a novel autism candidate gene, RORA, whose protein product is reduced in autistic brain. FASEB J 24:3036–3051
Nicholls RD, Saitoh S, Horsthemke B (1998) Imprinting in Prader-Willi and Angelman syndromes. Trends Genet 14:194–200
Raison CL, Lowry CA, Rook GA (2010) Inflammation, sanitation, and consternation: loss of contact with coevolved, tolerogenic microorganisms and the pathophysiology and treatment of major depression. Arch Gen Psychiatry 67:1211–1224
Reik W, Surani A (1997) Genomic imprinting. IRL Press, Oxford
Ressler KJ, Mercer KB, Bradley B, Jovanovic T, Mahan A, Kerley K, Norrholm SD, Kilaru V, Smith AK, Myers AJ, Ramirez M, Engel A, Hammack SE, Toufexis D, Braas KM, Binder EB, May V (2011) Post-traumatic stress disorder is associated with PACAP and the PAC1 receptor. Nature 470:492–497
Rodier P (2000) The early origins of autism. Sci Am 282:56–63
Russell-Smith SN, Maybery MT, Bayliss DM (2010) Are the autism and positive schizotypy spectra diametrically opposed in local versus global processing? J Autism Dev Disord 40:968–977
Sarachana T, Xu M, Wu R-C, Hu VW (2011) Sex hormones in autism: androgens and estrogens differentially and reciprocally regulate rora, a novel candidate gene for autism. PLoS One 6:e17116
Shinawi M, Liu P, Kang SH, Shen J, Belmont JW, Scott DA, Probst FJ, Craigen WJ, Graham BH, Pursley A, Clark G, Lee J, Proud M, Stocco A, Rodriguez DL, Kozel BA, Sparagana S, Roeder ER, McGrew SG, Kurczynski TW, Allison LJ, Amato S, Savage S, Patel A, Stankiewicz P, Beaudet AL, Cheung SW, Lupski JR (2010) Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J Med Genet 47:332–341
St Clair D, Xu M, Wang P, Yu Y, Fang Y, Zhang F, Zheng X, Gu N, Feng G, Sham P, He L (2005) Rates of adult schizophrenia following prenatal exposure to the Chinese famine of 1959–1961. JAMA 294:557–562
Steffenburg S, Gillberg CL, Steffenburg U, Kyllerman M (1996) Autism in Angelman syndrome: a population-based study. Pediatr Neurol 14:131–136
Sugie Y, Sugie H, Fukuda T, Ito M (2005) Neonatal factors in infants with autistic disorder and typically developing infants. Autism 9:487–494
Susser E, St. Clair D, He L (2008) Latent effects of prenatal malnutrition on adult health: the example of schizophrenia. Ann NY Acad Sci 1136:185–192
Tamminga CA, Holcomb HH (2005) Phenotype of schizophrenia: a review and formulation. Mol Psychiatry 10:27–39
Toulopoulou T, Mapua-Filbey F, Quraishi S, Kravariti E, Morris RG, McDonald C, Walshe M, Bramon E, Murray RM (2006) Cognitive performance in presumed obligate carriers for psychosis. Br J Psychiatry 187:284–285
Trivers RL (1972) Parental investment and sexual selection. In: Campbell B (ed) Sexual selection and the descent of man 1871–1971. Heinemann, London, pp 136–179
Trivers R (1974) Parent-offspring conflict. Amer Zool 14:249–264
Veltman MW, Thompson RJ, Roberts SE, Thomas NS, Whittington J, Bolton PF (2004) Prader-Willi syndrome – a study comparing deletion and uniparental disomy cases with reference to autism spectrum disorders. Eur Child Adolesc Psychiatry 13:42–50
Veltman MWM, Craig EE, Bolton PF (2005) Autism spectrum disorders in Prader-Willi and Angelman syndromes: a systematic review. Psychiatr Genet 15:243–254
Walter H, Ciaramidaro A, Adenzato M, Vasic N, Ardito RB, Erk S, Bara BG (2009) Dysfunction of the social brain in schizophrenia is modulated by intention type: an fMRI study. Soc Cogn Affect Neurosci 4:166–176
Ward PW (1993) Birth weight and economic growth. University of Chicago Press, Chicago/London
Webster JP, Lamberton PHL, Donnelly CA, Torrey EF (2006) Parasites as causative agents of human affective disorders? The impact of anti-psychotic, mood-stabilizer and anti-parasite medication on Toxoplasma gondii’s ability to alter host behaviour. P Roy Soc Lond B Bio Sci 273:1023–1030
Whittington J, Holland T (2004) Prader-Willi syndrome: development and manifestations. Cambridge University Press, Cambridge
Wicks S, Hjern A, Gunnell D, Lewis G, Dalman C (2005) Social adversity in childhood and the risk of developing psychosis: a national cohort study. Am J Psychiatry 162:1652–1657
Woogh C (2001) Is schizophrenia on the decline in Canada? Can J Psychiatry 46:61–67
Yolken RH, Torrey EF (2008) Are some cases of psychosis caused by microbial agents? A review of the evidence. Mol Psychiatry 13:470–479
Zhai J, Zhang Q, Cheng L, Chen M, Wang K, Liu Y, Deng X, Chen X, Shen Q, Xu Z, Ji F, Liu C, Dong Q, Chen C, Li J (2011) Risk variants in the S100B gene, associated with elevated S100B levels, are also associated with visuospatial disability of schizophrenia. Behav Brain Res 217:363–368
Zwaigenbaum L, Bryson S, Rogers T, Roberts W, Brian J, Szatmari P (2005) Behavioral manifestations of autism in the first year of life. Int J Dev Neurosci 23:143–152
Acknowledgments
With thanks and acknowledgements to Ahmad Abu-Akel, Will Brown, Bernard Crespi, David Haig, and Randy Jirtle.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Badcock, C. (2013). The Imprinted Brain: How Genes Set the Balance Between Autism and Psychosis. In: Jirtle, R., Tyson, F. (eds) Environmental Epigenomics in Health and Disease. Epigenetics and Human Health. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-36827-1_5
Download citation
DOI: https://doi.org/10.1007/978-3-642-36827-1_5
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-36826-4
Online ISBN: 978-3-642-36827-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)