
Abstract
Whenever the reference electrode compartment has to be separated by electrolyte bridges and diaphragms from the other parts of the electrochemical cell, the separation has to meet the following requirements:
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


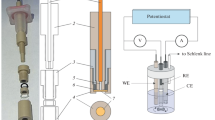
Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
4.1 Common Types of Salt Bridges and Diaphragms
4.1.1 Introduction
Whenever the reference electrode compartment has to be separated by electrolyte bridges and diaphragms from the other parts of the electrochemical cell, the separation has to meet the following requirements:
-
(i)
The electrolytic contact between the electrodes must be maintained.
-
(ii)
The ohmic resistance \( {R_{\Omega}} \) of the electrolyte bridge(s) and diaphragm(s) must be small enough so that the voltage drop \( i{R_{\Omega}} \) is minimal or, if possible, negligible. (Electronic \( i{R_{\Omega}} \) compensation will not be considered here.)
-
(iii)
The diffusion potentials at all places were different electrolyte solutions border, must be as small as possible, and they must be as stable (time invariant) as possible. It is the major task of a salt bridge to reduce the diffusion potential which would build up at the junction of the electrolytes A and A′ of the two electrode compartments by interposing the bridge electrolyte B.
-
(iv)
The electrolyte of the reference electrode should not be contaminated by the electrolyte (or generally the solution) of the working electrode and vice versa.
Strictly taken, these requirements can never be completely met, and at best some good compromise can be achieved. One fact facilitates these problems: to maintain a constant potential of the reference electrode, generally high-input impedance measuring instruments are used, so that the current flowing through the salt bridge and diaphragms is practically negligible.
The term salt bridge means a device containing an electrolyte solution and placed between two electrodes or electrode compartments in such way that the electrolyte solution serves as the ionic conductor between the two electrodes. Normally, a salt bridge is constructed of tubes containing the electrolyte solution. The ends of the tubes may have diaphragms (e.g., glass frits) to keep the solution as good as possible confined to the inner of the tubes. Another possibility is that one end of the tube is drawn out to a capillary, the orifice of which can be placed very near to the surface of the working electrode. This is called a Luggin capillary.Footnote 1 A very simple salt bridge is depicted in Fig. 4.1.1. Such salt bridge is filled by sucking the electrolyte solutions through the frits and glass tubes. It is possible to fill both sides of the bridge with different solutions, provided they are chemically compatible. If the electrolyte bridge is filled with only one solution, a two-way cock can be placed in the top glass tube. The sleeve of the cock should be free of any grease as the electrolyte film in the annular gab between the cone and the sleeve of the glass cock provides the ionic conductivity. When the three-way cock is closed and the bridge is in use, the cock acts as a ground-joint diaphragm. The cock should possess high-precision grounds of a large ground surface area. The high-precision grounding is important to minimize leakage by still guaranteeing low resistance of the electrolyte film. It was the important discovery of Olin Freeman Tower (1872–1945), an American student of F. W. Ostwald in Leipzig, that rather concentrated solutions of potassium chloride offer the great advantage that the diffusion potentials are very small because the effects of potassium cations and chloride anions almost cancel (for a detailed discussion, see Chap. 3). Still today, there is no better salt for the aqueous electrolyte bridges than KCl, if possible at saturation concentration. In case that the two solutions which need to be separated by a diaphragm are chemically incompatible (e.g., one solution containing K+ and the other \( \mathrm{ ClO}_4^{-} \) ions) one needs to use a salt bridge as depicted in Fig. 4.1.2: that bridge allows the tube between the two cocks to be filled with an electrolyte solution which is compatible with the two other electrolytes (e.g., NaCl in the above given example). Bates et al. reported in 1950 [1] a special salt bridge for high-accuracy measurements which allowed to form a very stable liquid|liquid electrolyte junction in two vertical glass tubes. That bridge, and also others, has two cocks, so that the two electrolytes of the electrodes can be separated by a third electrolyte. Bridges like that depicted in Fig. 4.1.1 always possess the property of a siphon, the flow of liquids being only impeded by the frits at the two ends and the cock. Nevertheless, it is important to arrange the hydrostatic pressure on both sides in such way that no pressure gradient will drive the liquids from one to the other side. Very small pore frits are also helpful to limit the contamination of solutions by a directed flow. Especially for reference electrodes, the described problem of pressure gradient can be easily solved by using a single glass tube with one frit at the lower end, and tightly fitting the reference electrode to the tube as shown in Fig. 4.1.3. Here the frit is a small stick of magnesia (MgO) melted in the glass. Several other materials can be also used as diaphragm. The construction shown in Fig. 4.1.3 has the advantage that normally no pressure can force the solution to flow in or out; however, a pressure gradient can build up when the bridge/electrode system is subjected to temperature changes as the thermal contraction or expansion of the inner solutions may lead to pressure gradients. When very tight frits (like a magnesia stick or Vycor® plate) are used, this is no issue in case of normal room temperature variations of some Kelvin. In any case, care must be taken to prevent any air bubbles inside the bridge and electrode, as gas has a much higher temperature coefficient of volume, and thus a gas bubble would create larger pressure gradients in response to temperature changes. Figure 4.1.4 depicts some common diaphragms used for salt bridges. Diaphragms for reference electrodes may be made of (1) sintered glass frits, (2) sintered ceramics (e.g., sintered magnesia (MgO), (3) asbestos fiber (not anymore used), (4) Vycor®, (5) organic polymer membranes, (6) stopper-in-sleeve constructions (glass), (7) stopper with thread (any material), (8) sintered platinum, and (9) they may also be simply a plug of a gel, like Agar. Vycor® (registered trademark of Corning, Inc.) is a porous glass possessing a sufficient conductivity and being very well suited to be used as diaphragm between aqueous and nonaqueous solutions. Thin plates of about 2 mm thickness are cut from a rod and fitted to the end of a glass tube with the help of a shrink tube or in another suitable way. Vycor® diaphragms cannot be used in strongly alkaline solutions as they will crack and deteriorate rapidly. All diaphragms have specific properties, of which the following are of special importance: (a) ohmic resistance, (b) chemical stability towards the used electrolyte solutions, and (c) leaking rate (permeability for solutions). Of course, thermal stability, pressure stability, etc. may be also important in some cases. Table 4.1.1 gives a compilation of properties of common diaphragms. Pore diaphragms (e.g., the Twin Pore Diaphragm of Metrohm AG, Switzerland) consist of open pores which connect the analyte solution and the reference electrode solution with a gel electrolyte. The gel practically does not interact with the analyte solution, so that interference by blocking (e.g., protein precipitation) is negligible. A platinum thread consisting of fine twinned Pt wires has similar advantages. Platinum threads, however, may suffer from wrong potentials in strongly redox buffered solutions.

Salt bridge constructed from glass tubes, glass frits, and a 3-way cock

Salt bridge constructed from glass tubes, glass frits, and two 3-way cocks for the purpose of separating chemically incompatible electrolyte solutions by a third solution which is compatible with the two terminal solutions

Construction of a silver/silver chloride reference electrode with a second salt bridge. Reproduced with permission from [20]

Common diaphragms for salt bridges. The stopper/sleeve shown in (e) is usually made in an inverted way, so that the stopper can be removed from the outside of the tube. Reproduced with permission from [21]
Diaphragms tend to fail because of (1) leakage of electrolyte leading to higher resistance, because the diaphragm may contain gas (air) instead of electrolyte solution and (2) precipitations of sparingly soluble salts, proteins, etc., leading also to higher resistance or even complete blocking. The leakage can be diminished by adding glycerol or similar liquids to the aqueous electrolyte solution, and or gelling agents. Adding viscous water-miscible solvents also diminishes the mobility of the ions [9] according to the Stokes–Einstein law and thus considerably contributes to a decrease of the junction potentials.
4.1.2 Gels for Stabilizing Salt Bridges
To decrease the leakage of salt solutions from bridges, especially in case of reference electrodes, glass electrodes, etc., the viscosity of the salt solutions is increased with gelling agents. Gelling also contributes to decreased diffusion coefficients [10, 11] of the salt ions and thus to diminished liquid junction potentials. The decreased ion mobility in gels results from geometric obstructions, and in case of polyelectrolyte gels also from electrostatic obstruction [12]. A very convenient way to prepare gelled salt bridges for connecting a reference electrode compartment with an electrochemical cell housing the other electrode(s) is as follows: A thin polyethylene tube (e.g., 0.5–1 mm inner diameter) is filled with the liquid gel electrolyte solutions, and after gelling the tube can be cut in appropriate pieces to make the solution connections. Such tubes can be easily exchanged, or the end pieces can be cut and disposed off if contamination has occurred. The tubes may also be thrown out before filling, so that the cut can be performed at places of diminished radius, allowing to decrease the contact area at the orifice, i.e., at the gel solution junction. To fill a tube, e.g., with warm agar gel, a syringe has to be used. The tube can be kept very long times. Before use one end has to be cut and disposed, and then a part of appropriate length can be cut for use. Such tubes are especially useful to contact small electrochemical cells, as e.g., for in situ AFM/STM [13], or also flow-through cells [14]. Such tubes can be also used like Luggin capillaries to approach the surface of a working electrode.
4.1.2.1 Agar and Pure Agarose Gels
Agar (also called agar-agar) is a mixture of agarose and agaropectin. Agarose is a polymer build up of the repeating units of agarobiose. Agarobiose is a disaccharide from d-galactose and 3,6-anhydro-l-galactopyranose. Agaropectin is a heterogeneous mixture of smaller acidic molecules that gel poorly. Instead of using agar, one can purchase purified agarose and use it for the gelation of electrolyte solutions: typically the salt solution contains 2–5 % (w/v) agar (or agarose). Agarose gels have a concentration-dependent pore structure: the pore size varies from 100 to several hundred nanometers [15, 16]. The gel is prepared by gently heating the agar (or agarose) with the salt solution and the liquid is allowed to cool down in the appropriate tubes. Solidification occurs between 40 and 32 °C. Agar and agarose gels are probably most popular for self-made laboratory diaphragms, but they suffer from an easy drying out at air, and microbes like to settle and grow on the gel. Therefore commercial reference electrode systems use synthetic polymers for gel formation.
4.1.2.2 Cellulose Gel
Hydroxyethyl cellulose is the gelling ingredient of a gel commercially used in the reference gel electrolyte of Ionode Pty Ltd, Australia. It consists of KCl 26 %, triethylene glycol 21 %, hydroxyethyl cellulose 2 %, and water to 100 % [17]. This gel is a highly viscous liquid minimizing leakage.
In another commercial product, methyl cellulose is used as gelling agent, with 40 % KCl (KCl saturated) and a not specified amount of water [18].
4.1.2.3 Acrylate Hydrogels
Among the methacrylate hydrogels, the most common gelating polymer is poly-2-hydroxyethyl-methacrylate (p-HEMA) [19]. The monomer and the aqueous electrolyte solutions are mixed, degassed, and kept on ice when 100 μl of a 6 % solution of sodium bisulfite and 100 μl of a 12 % solution of ammonium persulfate are added per 1 ml of monomer. The mixture is left for solidification in an appropriate vessel (or tube) at 37 °C for 2 h. The polymerized material has to be dialyzed against the used electrolyte solution to remove non-polymerized monomers and catalyst. Generally, health hazard issues have to be severely observed in case of acrylates.
References
-
1.
Bates RG, Pinching GD, Smith ER (1950) J Res Natl Bur Stand 45:418–429
-
2.
Material safety data sheet (21 April 2009) 6.2308.040 Idrolyte. Metrohm Ltd., CH-9101 Herisau, Switzerland
-
3.
Smith TJ, Stevenson KJ (2007) Reference electrodes. In: Zoski CG (ed) Handbook of electrochemistry. Elsevier, Amsterdam, p 96
-
4.
Data sheet: VYCOR® Brand Porous Glass 7930, Corning Incorporated, 2001
-
5.
Gille W, Enke D, Janowski F (2002) J Porous Mater 9:221–230
-
6.
Technical speciation of pH glass electrode 6.0224.100 of Metrohm AG, Switzerland
-
7.
Technical speciation of pH glass electrode 6.0221.100 of Metrohm AG, Switzerland
-
8.
Technical speciations of various glass electrodes 6.01 – 6.02 of Metrohm AG, Switzerland
-
9.
Steel BJ, Stokes JM, Stokes RH (1958) J Phys Chem 62:1514–1516
-
10.
Fuji T, Thomas HC (1958) J Phys Chem 62:1566–1568
-
11.
Gokarn NA, Rajurkar NS (2006) J Solut Chem 35:1673–1685
-
12.
Darwish MIM, van der Maarel JRC, Zitha PLJ (2004) Macromolecules 37:2307–2312
-
13.
Hasse U, Scholz F (2005) Electrochem Commun 7:173–176
-
14.
Vahl K, Kahlert H, Scholz F (2010) Electroanalysis 22:2172–2178
-
15.
Narayanan J, Xiong J-Y, Liu X-Y (2006) J Phys Conf Ser 28:83–86
-
16.
Hasse U, Scholz F (2006) J Solid State Electrochem 10:380–382
-
17.
Material safety data sheet (April 2011, Version 3) Reference Gel Electrolyte RE45. Ionode Pty Ltd, 8/148 Tennyson Memorial Avenue, Tennyson Qld 4105, Australia
-
18.
Material safety data sheet (21 April 2009) 6.2308.030 Electrolyte KCl sat. gel. Metrohm Ltd., CH-9101 Herisau, Switzerland
-
19.
Kindler DD, Bergethon PR (1990) J Appl Physiol 69:371–375
-
20.
Kahlert H (2010) In: Scholz F (ed) Electroanalytical methods. Guide to experiments and applications, 2nd edn. Springer, Berlin, p 305
-
21.
Kahlert H (2010) In: Scholz F (ed) Electroanalytical methods. Guide to experiments and applications, 2nd edn. Springer, Berlin, p 304
4.2 Ionic Liquid Salt Bridge
4.2.1 Introduction
Ionic liquid salt bridge (ILSB) [22, 23] is a promising alternative to KCl-based salt bridge (KClSB) and is capable of solving many of the problems intrinsic to KClSB: unstable liquid junction potential in low-ionic-strength sample solutions, clogging of the junction plug in pH composite electrodes, frequent necessity of renewing internal solution, contamination of sample solutions with KCl, and the dependence of liquid junction potential on the type of the junction [23].
ILSB is not omnipotent, because the liquid junction potential is interfered by ions whose hydrophobicity is nearly comparable to or higher than that of ions constituting the ILSB, when the concentration of the ion is comparable to or higher than the solubility of ILSB-constituent ions in a sample solution [23]. Nevertheless, ILSB outperforms KClSB in certain important applications of electroanalytical chemistry, notably, precise pH measurements [24, 25]. The stable liquid junction potential that formed when an ILSB is in contact with a low-ionic-strength aqueous solution is a strong advantage of ILSB over KClSB in that the former enables the determination of single ion activities in aqueous solutions with the precision on the order of 1 mV or better [24, 26]. Unlike KClSB, ILSB is currently in the development phase in view of optimization of ionic liquids depending on the purposes. Nonetheless, ILSB has been shown to be a workable and superior alternative to KClSB in a few important applications and it seems worthwhile at the current stage to describe the principles and applications of ILSB.
4.2.2 Principles of ILSB
A hydrophobic ionic liquid (IL) forms in contact with water (W) a liquid–liquid two-phase system. A notable feature of this two-phase system is that the phase-boundary potential, that is, the potential drop across the interface that develops in the IL-W two-phase system, is mainly determined by the partitioning of the IL-constituent ions between ILSB and W. First, this principle is briefly introduced. Following sections summarize other factors that can influence the phase-boundary potential at the contact of an IL with W.
4.2.2.1 Distribution Potential
When an ionic liquid that consists of moderately hydrophobic cationic and anionic species is in contact with an aqueous solution, the phase-boundary potential, \( \Delta_{{_{\mathrm{ IL}}}}^{{^{\mathrm{ W}}}}\phi \), which is the inner potential of the W phase with respect to that of IL phase, is established across the interface at a distribution equilibrium, where the superscript W and subscript IL stand for the aqueous phase and the IL phase, respectively.
We consider the simplest case when the ions in W phase are all hydrophilic and do not significantly partition into the IL phase and an ILSB, which is made of a 1–1 electrolyte. In such an expedient case, \( \Delta_{{_{\mathrm{ IL}}}}^{{^{\mathrm{ W}}}}\phi \) is determined by the partitioning of the IL-constituent ions, C+ and A−, and is given by [23]
where \( \Delta_{\mathrm{ IL}}^{\mathrm{ W}}\phi_{{{{\mathrm{ C}}^{+}}}}^{{\rlap{{-}}{\mathrm{ o}}}} \) and \( \Delta_{\mathrm{ IL}}^{\mathrm{ W}}\phi_{{{{\mathrm{ A}}^{-}}}}^{{\rlap{{-}}{\mathrm{ o}}}} \) are the standard ion transfer potentials of C+ and A−, which is defined in terms of the standard Gibbs energy of the transfer of ion, j, from IL to W, \( \Delta G_{\mathrm{ j}}^{{^{{\mathrm{ IL}\to \mathrm{ W},{{\rlap{{-}}{\mathrm{ o}}}}}}}} \), as \( \Delta_{\mathrm{ IL}}^{\mathrm{ W}}\phi_{\mathrm{ j}}^{{\rlap{{-}}{\mathrm{ o}}}} = -\Delta G_{\mathrm{ j}}{{^{{^{{\mathrm{ IL}\to \mathrm{ W},{{\rlap{{-}}{\mathrm{ o}}}}}}}}/({z_{{_{\mathrm{ j}}}}}F)\cdot \gamma_{{_{{{{\mathrm{ C}}^{+}}}}}}^{{^{\alpha }}}}} \) and \( \gamma_{{_{{{{\mathrm{ A}}^{-}}}}}}^{{^{\alpha }}} \) in Eq. (4.2.1) are the activity coefficients of C+ and A− in phase \( \alpha \) (α = W or IL) and \( {z_{\mathrm{ j}}} \) is the ionic charge on ion j in signed units of electronic charge.
Usually, \( \Delta_{\mathrm{ IL}}^{\mathrm{ W}}\phi_{{_{{{{\mathrm{ C}}^{+}}}}}}^{{\rlap{{-}}{\mathrm{ o}}}}\ne -\Delta_{\mathrm{ IL}}^{\mathrm{ W}}\phi_{{{{\mathrm{ A}}^{-}}}}^{{\rlap{{-}}{\mathrm{ o}}}} \) and, hence, \( \Delta_{\mathrm{ IL}}^{\mathrm{ W}}\phi \) is not null. However, in an IL sandwiched by two aqueous solutions, \( \Delta_{\mathrm{ IL}}^{\mathrm{ W}}\phi \) values on both sides of the IL phase have the same magnitudes but with opposite signs, and the second term on the right-hand side of Eq. (4.2.1) does not strongly depend on the electrolyte composition of the aqueous solution. Hence, the IL phase functions as an ILSB, because the liquid junction potential, which would develop if the ILSB is absent, is effectively canceled out by the IL phase inserted in between.
4.2.2.2 Contributions of Ion Transport to the Phase-Boundary Potential
In many cases of practical applications of ILSB, the distribution equilibrium throughout a two-phase system of an ILSB and a sample solution is seldom achieved. In the course of the dissolution of the IL into the sample solution, a quasi-distribution equilibrium is established within the diffusion layer in the sample solution side of the interface and the Nernst equation [27] for the distribution of an ionic species holds only for surface concentrations of the ions on both sides of the interface [28]. The phase-boundary potential at zero current is then characterized as a mixed potential [29] determined by the sum of the partial currents caused by the transfer of different ions across the interface [30].
To calculate partial currents, the Nernst–Planck equation may be used to describe the mass transfer of C+ and A− in ILSB. In W, the mode of mass transfer of C+ and A− depends on the electrolyte composition. In the simplest case when only C+ and A− transfer across a planar interface by diffusion and the aqueous phase contains sufficient amount of indifferent electrolytes, so that the diffusion is the mode of mass transfer of C+ and A− in W, the value of \( \Delta_{\mathrm{ IL}}^{\mathrm{ W}}\phi \) is determined by the potential at which the sum of two partial currents due to the transfer of C+ and A− across the phase boundary is null. Then, the mixed potential, \( \Delta_{\mathrm{ IL}}^{\mathrm{ W}}{\phi_{{_{\mathrm{ mix}}}}} \), is described by [31]:
where \( D_{{{{\mathrm{ C}}^{+}}}}^{\mathrm{ W}} \) and \( D_{{{{\mathrm{ A}}^{-}}}}^{\mathrm{ W}} \) are the diffusion coefficients of C+ and A− in W.
When the concentration of indifferent electrolytes in W is low, the mixed potential simply reduces to
However, in this case the diffusion potential on the aqueous solution side of the interface due to the difference in \( D_{{{{\mathrm{ C}}^{+}}}}^{\mathrm{ W}} \) and \( D_{{{{\mathrm{ A}}^{-}}}}^{\mathrm{ W}} \) may become significant [32]. When the aqueous phase contains a salt MX, where M+ and X− do not partition in the ILSB, the Henderson equation [33] for the diffusion potential takes the form
where \( \Delta \phi_{\mathrm{ diff}}^{\mathrm{ W}} \) is the diffusion potential in W referred to the electrostatic potential in W at the interface between the IL and W, \( c_{{_{\mathrm{ CA}}}}^{\mathrm{ W}} \) is the concentration of [C+][A−] in the aqueous solution side of the interface determined by the solubility of [C+][A−], \( c_{{_{\mathrm{ MX}}}}^{\mathrm{ W}} \) is the concentration of MX in W, and \( {u_{\mathrm{ i}}} \) is the mobility of ion i (i = M+, X−, \( {{\mathrm{ C}}^{+}} \), or \( {{\mathrm{ A}}^{-}} \)) in W. From Eq. (4.2.4) it is seen that \( \Delta \phi_{\mathrm{ diff}}^{\mathrm{ W}} \) becomes negligible when a sufficient amount of hydrophilic salt, MX, is in W.
Note the difference in the length scales between the distribution potential and the diffusion potential. In the latter, the region where the potential drop develops is a function of time and spans across the diffusion layer [34], whereas in the former the potential drop is limited within the diffuse part of the electrical double layer and is time-invariant. In the case when \( \Delta \phi_{\mathrm{ diff}}^{\mathrm{ W}} \) is not negligible, \( \Delta_{\mathrm{ IL}}^{\mathrm{ W}}\phi \) is thus given by the sum of the two quantities given in Eqs. (4.2.3) and (4.2.4). To minimize the deviation of the phase-boundary potential from that determined by the distribution potential of C+ and A−, the use of an ionic liquid that consists of cation and anion having similar mobility values in W is recommended [32, 35].
4.2.2.3 Experimental Examination of the Constancy of Liquid Junction Potential at ILSB
Although any phase-boundary potential is not thermodynamically accessible, it is possible to examine the constancy of the liquid junction potential at ILSB, for example, by use of the following cell:
I | II | III | IV | V | VI | VII | |
a mol dm−3 | x mol dm−3 | ||||||
Ag | AgX | Aqueous M1X | ILSB | Aqueous M2X′ | AgX′ | Ag | (A) |
solution | solution |
where M1X and M2X′ are salts of monovalent cations, such as H+ and Na+, with halogen ions, X− and X′−. By changing \( x \) in phase V while keeping \( a \) in phase III constant, the cell voltage, \( E \), should vary with the single ion activity of X′−, provided that the phase-boundary potential at the interface between IV and V, \( \Delta_{{_{\mathrm{ IL}}}}^{{^{\mathrm{ W}}}}{\phi_{{_{\mathrm{ SB}}}}}, \) and the Ag/AgX′ electrode on the right-hand side of cell (A) responds to the activity of X′− in phase V according to the Nernst equation; the constancy of the potential of the Ag/AgX electrode on the left-hand side at a given \( a \) value is naturally expected as long as the compositions of phases III and V are independent of each other.
As an example, the results when the ILSB is made of N-heptyl-N-methylpyrrolidinium bis(pentafluoroethanesulfonyl)amide, [C1,7pyrr+][C2C2N−], are shown in Figs. 4.2.1 and 4.2.2. The concentration of HBr in phase V (M2X′) was changed from 5 μmol kg−1 to 2 mol kg−1, whereas the composition of phase III was kept constant at 10 mmol dm−3 chloride salt of C1,7pyrr+ (M1X). The latter salt was chosen to lower the electrochemical polarizability of the interface between III and V [31] and fix the phase-boundary potential across the interface between III and IV at a value determined by the Nernst equation for the partition of C1,7pyrr+ between III and IV [22].

Time courses of \( E \) at 25 °C for [C1,7pyrr+][C2C2N−] ILSB at 11 different concentrations of HBr: 5 × 10−6, 1 × 10−5, 2 × 10−5, 5 × 10−5, 1 × 10−4, 2 × 10−4, 2 × 10−3, 2 × 10−2, 2 × 10−1, 5 × 10−1, and 2 mol kg−1 from top to bottom [37]

Figure 4.2.1 displays stable time courses of \( E \) for 15 min at 11 different concentrations of HBr. The constancy of \( E \) is seen at all concentrations. The stability of \( \Delta_{{_{\mathrm{ IL}}}}^{{^{\mathrm{ W}}}}{\phi_{{_{\mathrm{ SB}}}}} \) over 1 h has been published for a similar ILSB [36].
\( E \) values in Fig. 4.2.1 are plotted against the decadic logarithm of the mean activity of HBr, \( a_{{_{\mathrm{ HBr}}}}^{{^{\pm }}} \), in Fig. 4.2.2 (filled circles) [36]. The dashed line has a slope of 59.16 mV per decade expected for the Nernstian response of the \( \mathrm{ Ag}|\mathrm{ AgBr} \) electrode at 25 °C. Except at the low and high extremes of the HBr concentrations, the variation of \( E \) with \( \mathrm{ lo}{{\mathrm{ g}}_{{_{10 }}}}a_{{_{\mathrm{ HBr}}}}^{\pm } \) is well explained by the Nernstian slope, which suggests the constancy of \( \Delta_{{_{\mathrm{ IL}}}}^{{^{\mathrm{ W}}}}{\phi_{{_{\mathrm{ SB}}}}} \) over four orders of magnitude change in the HBr concentration.
Note that the phase-boundary potential at III and IV includes a contribution of the transfer Gibbs energy of C1,7pyrr+ between III and IV [22], which shifts \( E \) up to a positive value of a few tens mV.
The deviations at the low and high extremes probably have different origins. At the low ionic strength, the diffusion potential can be significant as described above. On the other hand, the deviation at the high extreme is likely to be due to the response of the \( \mathrm{ Ag}|\mathrm{ AgBr} \) electrode to the single ion activity of Br−, and not to the mean activity of HBr; the single ion activity of Br− is smaller than the mean activity of HBr [38] and the difference becomes greater with increasing concentration of HBr. The applicability of the ILSB then extends to higher concentrations of HBr to 1 mol dm−3 [39].
4.2.2.4 Interference by Other Ions
When ions dissolved in W are not hydrophilic enough, the partition of these ions into ILSB can participate in the distribution equilibrium and can shift \( \Delta_{\mathrm{ IL}}^{\mathrm{ W}}\phi \) from the value given by Eq. (4.2.1) in a way similar to the interference by foreign ions in ion-exchange-membrane-type ion-selective electrodes [30, 40]. To estimate the degree of interference, we need to know for a particular ionic liquid used for an ILSB the values of \( \Delta_{\mathrm{ IL}}^{\mathrm{ W}}\phi_{\mathrm{ i}}^{{\rlap{{-}}{\mathrm{ o}}}} \), which are thermodynamically not accessible in principle and their estimates for each IL based on an extrathermodynamic assumption are also not usually available. Because most of the ILs are similar to a polar aprotic solvent with regard to the partition of a number of substances [41], a good quantitative measure of the hydrophobicity of ions between an IL and W is the standard ion transfer potential of the ions between nitrobenzene (NB) and W, \( \Delta_{\mathrm{ NB}}^{\mathrm{ W}}\phi_{\mathrm{ i}}^{{\rlap{{-}}{\mathrm{ o}}}} \), whose values are available for ions relevant to ILs [42, 43].
Figure 4.2.3 shows apparent values of \( \Delta_{\mathrm{ NB}}^{\mathrm{ W}}\phi_{\mathrm{ i}}^{{\rlap{{-}}{\mathrm{ o}}}} \) for moderately hydrophobic ions that are relevant to ILSB, together with typical ions employed in electrochemistry of liquid–liquid interfaces.

Apparent standard ion transfer potentials of some hydrophobic ions across nitrobenzene and water interface for preparing moderately hydrophobic ionic liquids 25 °C [43] C n mim+: 1-alkyl-3-methylimidazolium (alkyl = butyl (n = 4), pentyl(n = 5), hexyl(n = 6), heptyl(n = 7), octyl(n = 8), decyl(n = 10), dodecyl(n = 12)); \( \mathrm{ Py}{{\mathrm{ C}}_1}\mathrm{ C}_n^{+} \): N-alkyl-N-methylpyrrolidinium (aklyl = butyl(n = 4), octyl(n = 8)); C n Py+: N-alkylpyridinium (alkyl hexyl (n = 6), octyl(n = 8), decyl(n = 10), dodecyl(n = 12)); C n Iq+: N-alkyllisoquinolinium (alkyl = butyl(n = 4), dodecyl(n = 12), hexadecyl(n = 14)); C m C n N− (bis(perfluoroalkanesulfonyl)amide: m and n stand for the number of difluoromethylene moiety). The value after each abbreviated name of ion indicates the value of \( \Delta_{\mathrm{ NB}}^{\mathrm{ W}}\phi_{\mathrm{ i}}^{{\rlap{\text{--}}{\mathrm{ o}}^{\prime}}} \) in V
As an example, the vertical upward arrow indicates the location of \( \Delta_{\mathrm{ NB}}^{\mathrm{ W}}\phi \) at −0.051 V when the NB phase contains 1-methyl-3-octylimidazolium (C8mim+) and bis(trifluoromethanesulfonyl)amide (C1C1N−), which suggests that \( \Delta_{\mathrm{ IL}}^{\mathrm{ W}}\phi \) between the ILSB made of these ions and an aqueous solution, \( \Delta_{\mathrm{ IL}}^{\mathrm{ W}}{\phi_{{_{\mathrm{ IL}\mathrm{ SB}}}}} \), would have a small negative value.
The degree of interference by a hydrophobic ion becomes significant when its surface concentration at \( \Delta_{\mathrm{ IL}}^{\mathrm{ W}}{\phi_{{_{\mathrm{ IL}\mathrm{ SB}}}}} \) is comparable to the solubility of the ILSB-constituent ions and, hence, depends on the relative magnitude of \( \Delta_{\mathrm{ IL}}^{\mathrm{ W}}\phi_{{_{\mathrm{ i}}}}^{{\rlap{{-}}{\mathrm{ o}}}} \) with respect to \( \Delta_{\mathrm{ IL}}^{\mathrm{ W}}{\phi_{{_{\mathrm{ IL}\mathrm{ SB}}}}} \), that is, \( |\Delta_{\mathrm{ IL}}^{\mathrm{ W}}\phi_{\mathrm{ i}}^{{\rlap{{-}}{\mathrm{ o}}}}-\Delta_{\mathrm{ IL}}^{\mathrm{ W}}{\phi_{{_{\mathrm{ IL}\mathrm{ SB}}}}}| \). This interference is basically the same as those seen in the case of ion-exchange-type liquid-membrane ion-selective electrodes [30].
4.2.2.5 Solubility of IL in W and Electrochemical Polarizability at the ILSB∣W Interface
The solubility is another important factor in designing ILSB. It directly determines the degree of contamination of a sample solution by ILSB-constituent salts, and also the duration of life of an ILSB. The solubility product, \( K_{\mathrm{ s}}^{\mathrm{ W}} \), of an ILSB-constituent salt, [C+][A−], in water is quantitatively related to the difference between \( \Delta_{\mathrm{ IL}}^{\mathrm{ W}}\phi_{{{{\mathrm{ C}}^{+}}}}^{{\rlap{{-}}{\mathrm{ o}}}} \) and \( \Delta_{\mathrm{ IL}}^{\mathrm{ W}}\phi_{{{A^{-}}}}^{{\rlap{{-}}{\mathrm{ o}}}} \) through
The length of the horizontal bar in Fig. 4.2.3 is thus a direct measure of \( K_{\mathrm{ s}}^{\mathrm{ W}} \).
Importantly, the electrochemical polarizability of the interface is inversely proportional to the solubility of the ILSB-constituent salt in water [31]. The electrical resistance may not be a serious problem in conventional potentiometry with a high-input impedance electrometer or voltammetry with a potentiostat. Nonetheless, it is preferable to lower the polarizability of the ILSB∣W interface as low as possible to minimize the interference by other ions on \( \Delta_{\mathrm{ IL}}^{\mathrm{ W}}\phi \) determined by Eq. (4.2.3).
[C n mim+][C1C1N−]: 1-alkyl-3-methylimidazolium bis(trifluoromethanesulfonyl)amide (hexyl (n = 6), octyl(n = 8)); [C n mim+][C2C2N−]: 1-alkyl-3-methylimidazolium bis(pentafluoroethanesulfonyl)amide (hexyl (n = 6), octyl(n = 8)); [TBMOEP+][C2C2N−]: tributylmethoxyethylphosphonium bis(pentafluoroethanesulfonyl)amide; [C2,n pyrr+][C2C2N−]: N-alkyl-N-methylpyrrolidinium bis(pentafluoroethanesulfonyl)amide (hexyl (n = 6), octyl(n = 8)).
Table 4.2.1 lists the solubility of some moderately hydrophobic ILs suitable to ILSBs in water. A practical range of the solubility of ILSB-constituent ILs is between a few tenth and a few mmol dm−3. The lower end, ~0.1 mmol dm−3, is mainly for of ILSBs specialized for low-ionic-strength samples, and the higher end is of ILSBs for samples with high ionic strength or those that may contain a certain level of hydrophobic ions.
4.2.2.6 Dissolution of Water in ILSB
Usually, the more hydrophobic an IL, the higher the solubility of water in the IL is. The solubility of water in moderately hydrophobic ILs suitable to an ILSB is typically 0.5–1 weight percent, which corresponds to the mole fraction of water being ca. 0.2 at room temperature, and increases with temperature [44–46]. A high content of water in an ILSB is unwelcome. When the ambient temperature varies during use or storage of ILSB, tiny water droplets may appear as suspensions in the ILSB or may absorb more water from a sample solution than that at a normal operating temperature. Such variability of water content in ILSB can be deleterious for the reproducible liquid junction potential, and minimization of water dissolution is preferable. However, the use of more hydrophobic ILs than those exemplified in Table 4.2.1 is bound by the electrochemical polarizability and the vulnerability to interfering ions, as described above.
4.2.2.7 ILSB Based on Mixed ILs
A simple possibility to overcome the above dichotomy regarding the water dissolution in ILSB is the use of a mixture of two ionic liquids, a very hydrophobic and a moderately hydrophobic IL [47]. The former works simply as a hydrophobic medium to suppress water dissolution and the latter does as a potential determining salt [47].
A mixture of pentyltripropylammonium bis(pentafluoroethanesulfonyl)amide as a potential determining salt and heptadecafluorodecyltrioctylphosphonium tetrakis [3,5-bis (trifluoromethyl)phenyl]borate as a very hydrophobic liquid matrix shows a reproducibility of \( \pm 0.6\ \mathrm{ mV} \) (95 % confidence interval) for aqueous KCl solutions in the range between 0.5 mmol dm−3 and 0.5 mol dm−3; the average excursion of \( E \) over 1 h is within \( \pm 0.3\ \mathrm{ mV} \) [47]. Moreover, such a strategy of using mixed ILs for ILSB allows us to employ moderately hydrophobic salts having melting points higher than room temperature or target temperature for use as ILSBs because of the lower melting points of the mixtures than their component salts.
4.2.3 Preparation of ILSB
4.2.3.1 Selection of Ionic Liquids for ILSB
Guidelines in selecting ionic liquids for ILSB with regard to the value of \( \Delta_{\mathrm{ IL}}^{\mathrm{ W}}{\phi_{{_{\mathrm{ IL}\mathrm{ SB}}}}} \), the mutual solubility, and the diffusion potential are described in the preceding sections. In addition, there are several factors to be considered.
4.2.3.2 Chemical Stability and Purity of ILs
Moderately hydrophobic ILs for ILSB are preferably inert. However, ILs are not necessarily chemically “innocent” [48, 49], nor green [50, 51]. The electrochemical stability of cation and anions used for moderately hydrophobic ILs in its ionic liquid states has been studied extensively in the relevance with their applications to electrochemical devices. Aside from impurities, such as water, both ends of the polarization windows are limited by redox reactions of cations and anions [52, 53]. For our interest in ILSB aimed to be used in aqueous solutions, ILSB-constituent ions dissolved in aqueous sample solutions are electrochemically inactive within potential window available in aqueous solutions.
4.2.3.2.1 Anions
PF6, which has been used for preparing moderately hydrophobic ILs, is hydrolyzed in water, though its rate is slower than that of \( \mathrm{ BF}_4^{-} \) [54, 55]. A more hydrophobic tri(pentafluoroethyl)trifluorophosphate is more stable against hydrolysis in water, but the hydrolysis is discernible [54]. These anions are therefore not suitable for ILSB. Perfluoroalkyltrifluoroborates are stable in water and tuning of hydrophobicity is feasible by changing the length of perfluoroalkane moiety [56].
Bis(perfluoroalkanesulfonyl)amides are stable in water. The hydrophobicity can be tuned by changing the length of the perfluoroalkyl moieties. The complexation with metal ions in IL is probably weak, but the ion pair formation with transition metal ions, such as Cd2+, can be appreciable when the concentrations of metal ions are high. \( \Delta_{\mathrm{ IL}}^{\mathrm{ W}}{\phi_{{_{\mathrm{ IL}\mathrm{ SB}}}}} \) is then affected by such extraction of the ions in ILSB [57].
4.2.3.2.2 Cations
Quaternary ammonium and phosphonium, as well as dialkylimidazolium ions, are typical ions used for preparing moderately hydrophobic ILs. It is known that these ions decompose through Hofmann elimination or ylide formation in strongly alkaline environment, e.g., solid alkyltrimethylammonium hydroxide hydrate, even at room temperature and the decomposition rate increases with elevating temperature [58, 59]. However, these cations are stable in an aqueous alkaline solution [58, 60], and, hence, ILSBs made of these ions are stable when they are in contact with an aqueous alkaline solution, e.g., 0.1 mol dm−3 NaOH.
4.2.3.2.3 Purification
The purity of ILs is an important issue for characterization of the physicochemical properties of ILs. For example, in application of [TBMOEP+][C2C2N−] (see Table 4.2.1) to an ILSB, the removal of unreacted tributylphosphine and other impurities is essential to obtain a stable liquid junction potential [61].
For moderately hydrophobic ILs, washing of ILs with copious water repeatedly, typically 30 times, is highly recommended to remove extraneous ions and other hydrophilic impurities. Decolorization based on a silica-charcoal column [62] or a treatment with charcoal [63] is simple yet powerful, though the loss of the IL is considerable.
4.2.3.3 Type of Liquid Junction
The thermodynamic nature of the working mechanism of ILSB liberates us from concern about the shape of the liquid junction in designing ILSBs. This is a distinctive advantage of ILSB over KClSB.
It has been shown by using cell (A) that the ILSB made of [TBMOEP+][C2C2N−] gives a stable \( \Delta_{\mathrm{ IL}}^{\mathrm{ W}}{\phi_{{_{\mathrm{ IL}\mathrm{ SB}}}}} \) down to 1 μmol dm−3 LiI, NaI, KI, and HI [10, 14]. This stability of \( \Delta_{\mathrm{ IL}}^{\mathrm{ W}}{\phi_{{_{\mathrm{ IL}\mathrm{ SB}}}}} \) of the ILSB at low-ionic-strength solutions allows us to more reliably estimate single ion activities. For example, by inserting [TBMOEP+][C2C2N−] in a Harned cell,
I | II | III | IV | V | VI | |
Pt | H2 (g), aqueous HCl soln. | ILSB | Aqueous HCl soln. | AgCl | Ag | (B) |
it is possible to determine the activity coefficients of hydrogen ions and chloride ions, independently, by changing the concentration of HCl in phase IV, while keeping the solution composition in phase II, or vice versa [26]. The shape of the cell is shown in Fig. 4.2.4, in which the liquid ILSB is in the bottom of a J-shaped glass tube in the right-hand side compartment of the cell.

Illustration of cell configuration for a fluid ILSB inserted between a hydrogen electrode and a Ag∣AgCl electrode, corresponding to cell (B)
Another simple water-jacketed cell is illustrated in Fig. 4.2.5, in which a liquid ILSB is supported on a glass frit of ca. 4 mm thickness. The upper aqueous phase is conveniently changed, while the lower aqueous phase remains unchanged. It is important to avoid electrical leakage between upper and lower solutions through inner glass wall. A way to minimize this effect is the silanization of the inner glass wall where the IL is in contact.

Illustration of a cell configuration for a fluid ILSB supported by a glass frit
4.2.3.4 Gelation of ILSB
In most of practical applications of ILSB-equipped reference electrodes, it is preferable to have a solidified ILSB. For example, 1-methyl-3-octylimidazolium bis(trifluoromethanesulfonyl)amide, [C8mim+][C1C1N−], is gelled at room temperature with poly(vinylidenefluoride-co-hexafluoropropylene) [64, 65] (PVDF-HFP), when the weight percent of [C8mim+][C1C1N−] is below 80.
A 1:1 mixture of an IL and PVDF-HFP has been used for ILSB. There is no difference in performance of gelled and non-gelled ionic liquids for ILSB [23]. A gelled ILSB membrane can be mounted in a membrane holder, as the one used for liquid-membrane-type ion-selective electrodes [66].
Gelation of ionic liquid with PVDF-HFP apparently suppresses the intake of water into ILSB and improves the reproducibility of \( \Delta_{\mathrm{ IL}}^{\mathrm{ W}}\phi \). The IL gradually leaches out from the gelled ILSB into a bathing aqueous solution, which causes a gradual thinning of the membrane and eventual loss of its function in a long time use.
An example of a gelled ILSB assembled in a glass composite electrode for pH measurements is shown in Fig. 4.2.6 [67]. A torus-shaped ring of a gelled IL is held between the upper and lower plastic cylinders through two O-rings. The inner wall of the IL ring is in contact with an internal solution. Accurate determination of the pH of dilute sulfuric acid solutions has been successfully made with this composite electrode [67]. A smaller version of a glass composite pH electrode equipped with an ILSB is commercially available.

Structure of a glass composite electrode equipped with a gelled ILSB torus ring [67]. 1: electric cable; 2: a plastic cap; 3: cylindrical plastic body; 4: internal solution of reference electrode; 5: internal solution of glass electrode; 6 and 6′: Ag/AgCl electrodes; 7: gelled ILSB; 8 and 8′: silicon O-ring; 9: glass membrane; 10: stem glass tube
4.2.3.5 ILSB-Coated Reference Electrodes
The presence of an internal solution is the bottleneck in miniaturization of reference electrodes. A reference electrode with gelled ILSB coated on a Ag∣AgCl electrode has been proposed [68] in which a gelled ILSB layer in contact with AgCl is saturated with AgCl.
As an example of a miniature reference electrode, Fig. 4.2.7 illustrates a glass capillary, with 150 μm inner diameter at the tip, filled with an non-gelled [C8mim+][C1C1N−]. A AgCl-coated silver wire is inserted in the capillary.

Ag∣AgCl electrode inserted in [C8mim+][C1C1N−] filled with a glass capillary with 150 μm inner diameter at the tip [69]
Figure 4.2.8 shows cyclic voltammograms in the presence (solid line) and absence (dotted line) of 1 mmol dm−3 ferrocenedimethanol in an aqueous KCl solution. Voltammograms in Fig. 4.2.8a were recorded with the miniature [C8mim+][C1C1N−]-coated AgCl reference electrode, and those in Fig. 4.2.8b are obtained with a conventional Ag∣AgCl electrode.

Cyclic voltammograms for oxidation–reduction of ferrocenedimethanol in 0.1 mol dm−3 KCl at 15 μm diameter Pt disk electrode referred to Ag∣AgCl electrode inserted in [C8mim+][C1C1N−] filled with a glass capillary with 150 μm inner diameter at the tip (a) and referred to a conventional Ag∣AgCl electrode in a saturated KCl solution (b). Counterelectrode is a 6-mm diameter Pt electrode in both cases. Scan rate: 50 mV s−1 [69]
Although a calibration of potential is required to compare data obtained with a ILSB-coated electrodes with those obtained with traditional reference electrodes, such a small reference electrode will find certain applications, e.g., electrophysiology.
4.2.4 Some Remarks on ILSB
4.2.4.1 Difference and Similarity of KClSB and ILSB
Salt bridge is an electrochemical device interposed between two electrolyte solutions of different compositions to isolate two solutions from each other while keeping the electrical connection, and hence is intrinsically nonthermodynamic in nature. The reason is easily seen in the case of KClSB; a system of an internal solution, KClSB, and a sample solution eventually end up with a homogeneous solution phase, sooner or later. In the case of ILSB, because of finite solubility of any kind of ions into the ILSB, the compositions of the two solution phases eventually become equal at equilibrium. Both solution phases are saturated with ILSB-constituting ions, whereas the ILSB is saturated with the components originally contained in the sample solution. There is therefore no fundamental or thermodynamically distinctive difference between KClSB and ILSB, although the working principle of minimizing the liquid junction potential between two electrolyte solutions is conceptually different [23]. Both KClSB and ILSB are useful only before, well before, the system reaches a thermodynamic equilibrium. In this sense, the ILSB is based on a quasi-thermodynamic basis. However, even technically, the difference between them is substantial, and is of considerable significance inasmuch as only technically both find raison d’etre. Aside from technical advantages of ILSB over KClSB described in Introduction (see also [23]) originated with the quasi-thermodynamic nature of ILSB, there are some more points worth mentioning.
The liquid junction potential at the interface between an ILSB and an aqueous solution of a simple mineral acid or an alkali hydroxide is distinctively more stable and accountable than that in the case of KClSB with regard to a much wider concentration range of an electrolyte, more than five orders of magnitude, and much wider acidity range, about 14 pH units. Such stability of phase-boundary potential over the wide pH range at ILSB stems from the working principle based on the distributing potential, distinguished from the diffusion potential to which the difference in the mobility of H+ and OH− and that of other ions contributes significantly, and enables many experiments in chemistry of electrolyte solutions that have been not possible or not reliable enough with KClSB.
The magnitude of the contamination of a sample solution by dissolution of electrolytes from an SB is more than three orders of magnitude lower in ILSB. These constitute the basis of reliable estimates of single ion activities [24, 26], including pH measurements, and other subjects related to liquid junction potential.
4.2.4.2 Comparison of Electrode Potentials Obtained with KClSB and ILSB
A comparison of the cell voltage data obtained with an ILSB or the potential of a working electrode with respect to an ILSB-equipped reference electrode, such as half-wave potential in voltammetry, with those obtained with an KClSB is made without any difficulty. A cell voltage of the cell (B), \( E \), that is, the electrode potential of the working electrode
I | II | III | IV | V | VI | |
Ag | Ag X | a mol dm−3 MX | SB | Sample solution | Working electrode | (B) |
does not depend on the type of the salt bridge, as long as the salt bridge works ideally, that is, it nullifies the liquid junction potential otherwise formed at the direct contact of phases III and V without disturbing the compositions of II and V. Therefore, there is no conceptual difference in \( E \) obtained with ILSB and that obtained with KClSB.
Many of half-wave potentials and formal potentials in electrochemistry have been compiled with respect to a calomel electrode or Ag∣AgCl electrode in contact with a concentrated KCl solution (phases III and IV in cell (B)), e.g., saturated or 3 mol dm−3 KCl. By replacing the KClSB with and ILSB in cell (B), however, we would not be able to directly compare the electrode potential because of a possible dissolution of K+ in ILSB at such high concentrations. It is recommended to examine \( E \) at a lower KCl concentration in phase III in cell (B) with an ILSB, e.g., \( a=0.01 \), and then convert \( E \) with a known difference in the electrode potential of Ag∣AgCl electrodes at 10 mmol dm−3 and saturated KCl.
4.2.4.3 Challenges in Future Studies
The reproducibility of \( \Delta_{\mathrm{ IL}}^{\mathrm{ W}}{\phi_{{_{\mathrm{ IL}\mathrm{ SB}}}}} \) of one type of ILSB prepared and tested on different days is 0.6 mV (95 % confidence interval) [47]. This is comparable or slightly less reproducible than that reported in the liquid junction potential of KClSB at the optimized conditions [70, 71]. Further studies seem to be required to experimentally establish the highly reproducible liquid junction potential at ILSB. On the one hand, the wide degree of freedom in selecting ILs for ILSB allows us to optimize the composition of ILSBs depending on particular purposes. It is preferable, on the other hand, to narrow them down to one or few ILs for the ILSB on which more extensive and intensive studies are to be made for standardizing this new salt bridge.
Temperature and pressure dependence of the performance of ILSB have not been elucidated. There is no conceivable obstacle in applying ILSB at wider temperature and pressure conditions.
The use of ILSB in organic solvents has not been challenged. Because ILs are considerably, if not completely, miscible with polar protic and aprotic solvents, the formation of phase-separated systems is not expected. Even in such cases, ILSBs described above presumably work as conventional salt bridges based on diffusion potential, as is the case of KClSB. As ILs are not completely miscible with nonpolar organic solvents, ILSB should work to correlate electrode potentials in such solvents with those in aqueous solutions.
References
-
22.
Kakiuchi T, Tsujioka N, Kurita S, Iwami Y (2003) Electrochem Commun 5(2):159–164
-
23.
Kakiuchi T, Yoshimatsu T (2006) Bull Chem Soc Jpn 79(7):1017–1024
-
24.
Shibata M, Sakaida H, Kakiuchi T (2011) Anal Chem 83(1):164–168
-
25.
Kakiuchi T (2011) J Solid State Electrochem 15(7–8):1661–1671
-
26.
Sakaida H, Kakiuchi T (2011) J Phys Chem B 115(45):13222–13226
-
27.
Nernst W (1889) Z Phys Chem 8:129–181
-
28.
Kakiuchi T (2001) J Electroanal Chem 496(1–2):137–142
-
29.
Kolthoff IM, Miller CS (1940) J Am Chem Soc 62:2171–2174
-
30.
Kakiuchi T, Senda M (1984) Bull Chem Soc Jpn 57(7):1801–1808
-
31.
Kakiuchi T, Tsujioka N (2007) J Electroanal Chem 599(2):209–212
-
32.
Yoshimatsu T, Kakiuchi T (2007) Anal Sci 23(9):1049–1052
-
33.
Henderson P (1907) Z Phys Chem 59(1):118–127
-
34.
Dickinson EJF, Freitag L, Compton RG (2010) J Phys Chem B 114(1):187–197
-
35.
Sakaida H, Kitazumi Y, Kakiuchi T (2010) Talanta 83(2):663–666
-
36.
Fujino Y, Kakiuchi T (2011) J Electroanal Chem 651(1):61–66
-
37.
Fujino Y, Kakiuchi T (2010) Unpublished work
-
38.
Fraenkel D (2011) J Phys Chem B 115:557–568
-
39.
Sakaida Y, Kakiuchi T, to be published
-
40.
Kakiuchi T, Obi I, Senda M (1985) Bull Chem Soc Jpn 58(6):1636–1641
-
41.
Abraham MH, Acree WE (2006) Green Chem 8(10):906–915
-
42.
Kakiuchi T (2007) Anal Chem 79(17):6442–6449
-
43.
Tanaka S, Matsuoka Y, Belkada F, Kitazumi Y, Suzuki A, Nishi N, Kakiuchi T, in preparation
-
44.
Freire MG, Carvalho PJ, Gardas RL, Marrucho IM, Santos LMNBF, Coutinho JAP (2008) J Phys Chem B 112(6):1604–1610
-
45.
Ferreira AR, Freire MG, Ribeiro JC, Lopes FM, Crespo JG, Coutinho JAP (2011) Ind Eng Chem Res 50(9):5279–5294
-
46.
Rehak K, Moravek P, Strejc M (2012) Fluid Phase Equil 316:17–25
-
47.
Zhang LM, Miyazawa T, Kitazumi Y, Kakiuchi T (2012) Anal Chem 84(7):3461–3464
-
48.
Dupont J, Spencer J (2004) Angew Chem Int Ed 43(40):5296–5297
-
49.
Chowdhury S, Mohan RS, Scott JL (2007) Tetrahedron 63(11):2363–2389
-
50.
Frade RFM, Afonso CAM (2010) Hum Exp Toxicol 29(12):1038–1054
-
51.
Petkovic M, Seddon KR, Rebelo LPN, Pereira CS (2011) Chem Soc Rev 40(3):1383–1403
-
52.
Opallo M, Lesniewski A (2011) J Electroanal Chem 656(1–2):2–16
-
53.
Silvester DS (2011) Analyst 136(23):4871–4882
-
54.
Schmidt M, Heider U, Kuehner A, Oesten R, Jungnitz M, Ignat’ev N, Sartori P (2001) J Power Sources 97–98:557–560
-
55.
Freire MG, Neves CMSS, Marrucho IM, Coutinho JAP, Fernandes AM (2010) J Phys Chem A 114(11):3744–3749
-
56.
Nishi N, Suzuki A, Kakiuchi T (2009) Bull Chem Soc Jpn 82(1):86–92
-
57.
Yoshimatsu T (2007) Thesis of the master’s degree, Department of Energy and Hydrocarbon Chemistry, Graduate School of Engineering, Kyoto University
-
58.
Macomber CS, Boncella JM, Pivovar BS, Rau JA (2008) J Therm Anal Calorim 93(1):225–229
-
59.
Edson JB, Macomber CS, Pivovar BS, Boncella JM (2012) J Membr Sci 399:49–59
-
60.
Ye YS, Elabd YA (2011) Macromolecules 44(21):8494–8503
-
61.
Sakaida H (2010) Master’s Thesis of Graduate School of Engineering, Department of Energy and Hydrocarbon Chemistry, Kyoto University
-
62.
Earle MJ, Gordon CM, Plechkova NV, Seddon KR, Welton T (2007) Anal Chem 79(2):758–764
-
63.
Stark A, Behrend P, Braun O, Muller A, Ranke J, Ondruschka B, Jastorff B (2008) Green Chem 10(11):1152–1161
-
64.
Fuller J, Breda AC, Carlin RT (1997) J Electrochem Soc 144(4):L67–L70
-
65.
Fuller J, Breda AC, Carlin RT (1998) J Electroanal Chem 459(1):29–34
-
66.
Shibata M, Yamanuki M, Iwamoto Y, Nomura S, Kakiuchi T (2010) Anal Sci 26(11):1203–1206
-
67.
Shibata M, Kato M, Iwamoto Y, Nomura S, Kakiuchi T, submitted for publication
-
68.
Kakiuchi T, Yoshimatsu T, Nishi N (2007) Anal Chem 79(18):7187–7191
-
69.
Kitazumi Y, Kakiuchi T (2012) Unpublished
-
70.
Covington AK, Rebelo MJF (1987) Anal Chim Acta 200(1):245–260
-
71.
Davison W, Covington AK, Whalley PD (1989) Anal Chim Acta 223(2):441–447
Notes
- 1.
This kind of capillary salt bridge was suggested by Hans Luggin, an Austrian scientist (1863–1899), to Fritz Haber (1868–1934) when both worked in Karlsruhe, Germany.
References
References to Sect. 4.1
Bates RG, Pinching GD, Smith ER (1950) J Res Natl Bur Stand 45:418–429
Material safety data sheet (21 April 2009) 6.2308.040 Idrolyte. Metrohm Ltd., CH-9101 Herisau, Switzerland
Smith TJ, Stevenson KJ (2007) Reference electrodes. In: Zoski CG (ed) Handbook of electrochemistry. Elsevier, Amsterdam, p 96
Data sheet: Vycor® Brand Porous Glass 7930, Corning Incorporated, 2001
Gille W, Enke D, Janowski F (2002) J Porous Mater 9:221–230
Technical speciation of pH glass electrode 6.0224.100 of Metrohm AG, Switzerland
Technical speciation of pH glass electrode 6.0221.100 of Metrohm AG, Switzerland
Technical speciations of various glass electrodes 6.01 – 6.02 of Metrohm AG, Switzerland
Steel BJ, Stokes JM, Stokes RH (1958) J Phys Chem 62:1514–1516
Fuji T, Thomas HC (1958) J Phys Chem 62:1566–1568
Gokarn NA, Rajurkar NS (2006) J Solut Chem 35:1673–1685
Darwish MIM, van der Maarel JRC, Zitha PLJ (2004) Macromolecules 37:2307–2312
Hasse U, Scholz F (2005) Electrochem Commun 7:173–176
Vahl K, Kahlert H, Scholz F (2010) Electroanalysis 22:2172–2178
Narayanan J, Xiong J-Y, Liu X-Y (2006) J Phys Conf Ser 28:83–86
Hasse U, Scholz F (2006) J Solid State Electrochem 10:380–382
Material safety data sheet (April 2011, Version 3) Reference Gel Electrolyte RE45. Ionode Pty Ltd, 8/148 Tennyson Memorial Avenue, Tennyson Qld 4105, Australia
Material safety data sheet (21 April 2009) 6.2308.030 Electrolyte KCl sat. gel. Metrohm Ltd., CH-9101 Herisau, Switzerland
Kindler DD, Bergethon PR (1990) J Appl Physiol 69:371–375
Kahlert H (2010) In: Scholz F (ed) Electroanalytical methods. Guide to experiments and applications, 2nd edn. Springer, Berlin, p 305
Kahlert H (2010) In: Scholz F (ed) Electroanalytical methods. Guide to experiments and applications, 2nd edn. Springer, Berlin, p 304
References to Sect. 4.2
Kakiuchi T, Tsujioka N, Kurita S, Iwami Y (2003) Electrochem Commun 5(2):159–164
Kakiuchi T, Yoshimatsu T (2006) Bull Chem Soc Jpn 79(7):1017–1024
Shibata M, Sakaida H, Kakiuchi T (2011) Anal Chem 83(1):164–168
Kakiuchi T (2011) J Solid State Electrochem 15(7–8):1661–1671
Sakaida H, Kakiuchi T (2011) J Phys Chem B 115(45):13222–13226
Nernst W (1889) Z Phys Chem 8:129–181
Kakiuchi T (2001) J Electroanal Chem 496(1–2):137–142
Kolthoff IM, Miller CS (1940) J Am Chem Soc 62:2171–2174
Kakiuchi T, Senda M (1984) Bull Chem Soc Jpn 57(7):1801–1808
Kakiuchi T, Tsujioka N (2007) J Electroanal Chem 599(2):209–212
Yoshimatsu T, Kakiuchi T (2007) Anal Sci 23(9):1049–1052
Henderson P (1907) Z Phys Chem 59(1):118–127
Dickinson EJF, Freitag L, Compton RG (2010) J Phys Chem B 114(1):187–197
Sakaida H, Kitazumi Y, Kakiuchi T (2010) Talanta 83(2):663–666
Fujino Y, Kakiuchi T (2011) J Electroanal Chem 651(1):61–66
Fujino Y, Kakiuchi T (2010) Unpublished work
Fraenkel D (2011) J Phys Chem B 115:557–568
Sakaida Y, Kakiuchi T, to be published
Kakiuchi T, Obi I, Senda M (1985) Bull Chem Soc Jpn 58(6):1636–1641
Abraham MH, Acree WE (2006) Green Chem 8(10):906–915
Kakiuchi T (2007) Anal Chem 79(17):6442–6449
Tanaka S, Matsuoka Y, Belkada F, Kitazumi Y, Suzuki A, Nishi N, Kakiuchi T, in preparation
Freire MG, Carvalho PJ, Gardas RL, Marrucho IM, Santos LMNBF, Coutinho JAP (2008) J Phys Chem B 112(6):1604–1610
Ferreira AR, Freire MG, Ribeiro JC, Lopes FM, Crespo JG, Coutinho JAP (2011) Ind Eng Chem Res 50(9):5279–5294
Rehak K, Moravek P, Strejc M (2012) Fluid Phase Equil 316:17–25
Zhang LM, Miyazawa T, Kitazumi Y, Kakiuchi T (2012) Anal Chem 84(7):3461–3464
Dupont J, Spencer J (2004) Angew Chem Int Ed 43(40):5296–5297
Chowdhury S, Mohan RS, Scott JL (2007) Tetrahedron 63(11):2363–2389
Frade RFM, Afonso CAM (2010) Hum Exp Toxicol 29(12):1038–1054
Petkovic M, Seddon KR, Rebelo LPN, Pereira CS (2011) Chem Soc Rev 40(3):1383–1403
Opallo M, Lesniewski A (2011) J Electroanal Chem 656(1–2):2–16
Silvester DS (2011) Analyst 136(23):4871–4882
Schmidt M, Heider U, Kuehner A, Oesten R, Jungnitz M, Ignat’ev N, Sartori P (2001) J Power Sources 97–98:557–560
Freire MG, Neves CMSS, Marrucho IM, Coutinho JAP, Fernandes AM (2010) J Phys Chem A 114(11):3744–3749
Nishi N, Suzuki A, Kakiuchi T (2009) Bull Chem Soc Jpn 82(1):86–92
Yoshimatsu T (2007) Thesis of the master’s degree, Department of Energy and Hydrocarbon Chemistry, Graduate School of Engineering, Kyoto University
Macomber CS, Boncella JM, Pivovar BS, Rau JA (2008) J Therm Anal Calorim 93(1):225–229
Edson JB, Macomber CS, Pivovar BS, Boncella JM (2012) J Membr Sci 399:49–59
Ye YS, Elabd YA (2011) Macromolecules 44(21):8494–8503
Sakaida H (2010) Master’s Thesis of Graduate School of Engineering, Department of Energy and Hydrocarbon Chemistry, Kyoto University
Earle MJ, Gordon CM, Plechkova NV, Seddon KR, Welton T (2007) Anal Chem 79(2):758–764
Stark A, Behrend P, Braun O, Muller A, Ranke J, Ondruschka B, Jastorff B (2008) Green Chem 10(11):1152–1161
Fuller J, Breda AC, Carlin RT (1997) J Electrochem Soc 144(4):L67–L70
Fuller J, Breda AC, Carlin RT (1998) J Electroanal Chem 459(1):29–34
Shibata M, Yamanuki M, Iwamoto Y, Nomura S, Kakiuchi T (2010) Anal Sci 26(11):1203–1206
Shibata M, Kato M, Iwamoto Y, Nomura S, Kakiuchi T, in preparation
Kakiuchi T, Yoshimatsu T, Nishi N (2007) Anal Chem 79(18):7187–7191
Kitazumi Y, Kakiuchi T (2012) Unpublished
Covington AK, Rebelo MJF (1987) Anal Chim Acta 200(1):245–260
Davison W, Covington AK, Whalley PD (1989) Anal Chim Acta 223(2):441–447
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Scholz, F., Kakiuchi, T. (2013). Salt Bridges and Diaphragms. In: Inzelt, G., Lewenstam, A., Scholz, F. (eds) Handbook of Reference Electrodes. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-36188-3_4
Download citation
DOI: https://doi.org/10.1007/978-3-642-36188-3_4
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-36187-6
Online ISBN: 978-3-642-36188-3
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)