
Abstract
Selected concepts and techniques of Information-Theory (IT) are summarized and their use in probing the molecular electronic structure is advocated. The electron redistributions accompanying formation of chemical bonds, relative to the (molecularly placed) free atoms of the corresponding “promolecule,” generate the associated displacements in alternative measures of the amount of information carried by electrons. The latter are shown to provide sensitive probes of information origins of the chemical bonds, allow the spatial localization of bonding regions in molecules, and generate attractive entropy/information descriptors of the system bond multiplicities. Information-theoretic descriptors of both the molecule as a whole and its diatomic fragments can be extracted. Displacements in the molecular Shannon entropy and entropy deficiency, relative to the promolecular reference, are investigated. Their densities provide efficient tools for detecting the presence of the direct chemical bonds and for monitoring the promotion/hybridization changes the bonded atoms undergo in a molecular environment. The nonadditive Fisher information density in the Atomic Orbital (AO) resolution is shown to generate an efficient Contra-Gradience (CG) probe for locating the bonding regions in molecules. Rudiments of the Orbital Communication Theory (OCT) of the chemical bond are introduced. In this approach molecules are treated as information systems propagating “signals” of electron allocations to basis functions, from AO “inputs” to AO “outputs.” The conditional probabilities defining such an information network are generated using the bond-projected superposition principle of quantum mechanics. They are proportional to squares of the corresponding elements of the first-order density matrix in AO representation. Therefore, they are related to Wiberg’s quadratic index of the chemical bond multiplicity. Such information propagation in molecules exhibits typical communication “noise” due to the electron delocalization via the system chemical bonds. In describing this scattering of electron probabilities throughout the network of chemical bonds, due to the system occupied Molecular Orbitals (MO), the OCT uses the standard entropy/information descriptors of communication devices. They include the average communication noise (IT covalency) and information flow (IT ionicity) quantities, reflected by the channel conditional entropy and mutual information characteristics, respectively. Recent examples of applying these novel tools in an exploration of the electronic structure and bonding patterns of representative molecules are summarized. This communication perspective also predicts the “indirect” (through-bridge) sources of chemical interactions, due to the “cascade” probability propagation realized via AO intermediates. It supplements the familiar through-space mechanism, due to the constructive interference between the interacting AO, which generates the “direct” communications between bonded atoms. Such bridge “bonds” effectively extend the range of chemical interactions in molecular systems. Representative examples of the π systems in benzene and butadiene are discussed in a more detail and recent applications of the information concepts in exploring the elementary reaction mechanisms are mentioned.
Graphical Abstract

Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Bond information probes
- Bond localization
- Chemical bonds
- Chemical reactivity
- Contra-gradience criterion
- Covalent/ionic bond components
- Direct/indirect bond multiplicities
- Entropic bond indices
- Fisher information
- Information theory
- Molecular information channels
- Orbital communications
1 Introduction
The Information Theory (IT) [1–8] is one of the youngest branches of the applied probability theory, in which the probability ideas have been introduced into the field of communication, control, and data processing. Its foundations have been laid in 1920s by Sir R. A. Fisher [1] in his classical measurement theory and in 1940s by C.E. Shannon [3] in his mathematical theory of communication. The quantum state of electrons in a molecule is determined by the system wave function, the amplitude of the particle probability distribution which carries the information. It is thus intriguing to explore the information content of electronic probability distributions in molecules and to extract from it the pattern of chemical bonds, reactivity trends and other molecular descriptors, e.g., the bond multiplicities (“orders”) and their covalent/ionic composition. In this survey we summarize recent applications of IT in probing chemical bonds in molecules. In particular, changes in the information content due to subtle electron redistributions accompanying the bond formation process will be examined. Elsewhere, e.g., [9–14], it has been amply demonstrated that many classical problems of theoretical chemistry can be approached afresh using such a novel IT perspective. For example, the displacements in the information distribution in molecules, relative to the promolecular reference consisting of the nonbonded constituent atoms, have been investigated [9–16] and the least-biased partition of the molecular electron distributions into subsystem contributions, e.g., densities of bonded Atoms-in-Molecules (AIM), have been investigated [9, 17–24]. These optimum density pieces have been derived from alternative global and local variational principles of IT. The IT approach has been shown to lead to the “stockholder” molecular fragments of Hirshfeld [25].
The spatial localization of specific bonds presents another challenging problem to be tackled by this novel treatment of molecular systems. Another diagnostic problem in the theory of molecular electronic structure deals with the shell structure and electron localization in atoms and molecules. The nonadditive Fisher information in the Atomic Orbital (AO) resolution has been recently used as the Contra-Gradience (CG) criterion for localizing the bonding regions in molecules [10–14, 26–28], while the related information density in the Molecular Orbital (MO) resolution has been shown [9, 29] to determine the vital ingredient of the Electron-Localization Function (ELF) [30–32].
The Communication Theory of the Chemical Bond (CTCB) has been developed using the basic entropy/information descriptors of molecular information (communication) channels in the AIM, orbital and local resolutions of the electron probability distributions [9–11, 33–48]. The same bond descriptors have been used to provide the information-scattering perspective on the intermediate stages in the electron redistribution processes [49], including the atom promotion via the orbital hybridization [50], and the communication theory for the excited electron configurations has been developed [51]. Moreover, a phenomenological treatment of equilibria in molecular subsystems has been proposed [9, 52–54], which formally resembles the ordinary thermodynamic description.
Entropic probes of the molecular electronic structure have provided attractive tools for describing the chemical bond phenomenon in information terms. It is the main purpose of this survey to summarize alternative local entropy/information probes of the molecular electronic structure [9–14, 21, 22] and to explore the information origins of the chemical bonds. It is also our goal to present recent developments in the AO formulation of CTCB, called the Orbital Communication Theory (OCT) [10, 11, 39, 48, 55–58]. The importance of the nonadditive effects in the chemical-bond phenomena will be emphasized and the information-cascade (bridge) propagation of electronic probabilities in molecular information systems, generating the indirect bond contributions due to the orbital intermediaries [59–63], will be examined. Throughout the article symbol A denotes a scalar quantity, A stands for the row-vector, and A represents a square or rectangular matrix. In the logarithmic measure of information the logarithm is taken to base 2, log = log2, which expresses the amount of information in bits (binary digits). Accordingly, selecting log = ln measures the information in nats (natural units): 1 nat = 1.44 bits.
2 Measures of Information Content
We begin with a short summary of selected IT concepts and techniques to be used in diagnosing the information content of electronic distributions in molecules and in probing their chemical bonds. The Shannon entropy [3, 4] in the (normalized) discrete probability vector p = {p i },
where the summation extends over labels of all elementary events determining the probability scheme in question, provides a measure of the average indeterminacy in p. This function also measures the average amount of information obtained when the uncertainty is removed by an appropriate measurement (experiment).
The Fisher information for locality [1, 2], called the intrinsic accuracy, historically predates the Shannon entropy by about 25 years, being proposed in about the same time when the final form of quantum mechanics was shaped. It emerges as an expected error in a “smart” measurement, in the context of an efficient estimator of a parameter. For the “locality” parameter, the Fisher measure of the information content in the continuous (normalized) probability density p(r) reads:
This Fisher functional can be simplified by expressing it as the functional of the associated classical (real) amplitude A(r) = [p(r)]−1/2 of the probability distribution:
The amplitude form is then naturally generalized into the domain of complex probability amplitudes, e.g., the wave functions encountered in quantum mechanics [2, 26]. For the simplest case of the spinless one-particle system, when p(r) = ψ *(r)ψ(r) = |ψ(r)|2,
The Fisher information is reminiscent of von Weizsäcker’s [64] inhomogeneity correction to the electronic kinetic energy in the Thomas–Fermi theory. It characterizes the compactness of the probability density p(r). For example, the Fisher information in the familiar normal distribution measures the inverse of its variance, called the invariance, while the complementary Shannon entropy is proportional to the logarithm of variance, thus monotonically increasing with the spread of the Gaussian distribution. Therefore, the Shannon entropy and intrinsic accuracy describe complementary facets of the probability density: the former reflects the distribution “spread,” providing a measure of its uncertainty (“disorder”), while the latter measures a “narrowness” (“order”) in the probability density.
An important generalization of Shannon’s entropy, called the relative (cross) entropy, also known as the entropy deficiency, missing information or the directed divergence, has been proposed by Kullback and Leibler [5, 6]. It measures the information “distance” between the two (normalized) probability distributions for the same set of events. For example, in the discrete probability scheme identified by events a = {a i } and their probabilities P(a) = {P(a i ) = p i } ≡ p, this discrimination information in p with respect to the reference distribution p 0 = {P 0(a i ) = p i 0} reads:
The entropy deficiency provides a measure of the information resemblance between the two compared probability schemes. The more the two distributions differ from one another, the larger the information distance. For individual events the logarithm of the probability ratio I i = log(p i /p i 0), called the surprisal, provides a measure of the event information in the current distribution relative to that in the reference distribution. The equality in the preceding equation takes place only for the vanishing surprisal for all events, i.e., when the two probability distributions are identical.
For two mutually dependent (discrete) probability vectors P(a) = {P(a i ) = p i } ≡ p and P(b) = {P(b j ) = q j } ≡ q, one decomposes the joint probabilities of the simultaneous events a∧b = {a i ∧b j } in the two schemes, P(a∧b) = {P(a i ∧b j ) = π i,j } ≡ π, as products of the “marginal” probabilities of events in one set, say a, and the corresponding conditional probabilities P(b|a) = {P(j|i)} of outcomes in another set b, given that events a have already occurred: {π i,j = p i P(j|i)}. Relevant normalization conditions for the joint and conditional probabilities then read:
The Shannon entropy of this “product” distribution,
has been expressed above as the sum of the average entropy S(p) in the marginal probability distribution, and the average conditional entropy in q given p:
The latter represents the extra amount of uncertainty about the occurrence of events b, given that events a are known to have occurred. In other words, the amount of information obtained as a result of simultaneously observing events a and b of the two discrete probability distributions equals to the amount of information in one set, say a, supplemented by the extra information provided by the occurrence of events in the other set b, when a are known to have occurred already. This is qualitatively illustrated in Fig. 1 [7, 8].

Diagram of the conditional entropy and mutual information quantities for two dependent probability distributions p and q. Two circles enclose areas representing the entropies S(p) and S(q) of the separate probability vectors, while their common (overlap) area corresponds to the mutual information I(p:q) in two distributions. The remaining part of each circle represents the corresponding conditional entropy, S(p|q) or S(q|p), measuring the residual uncertainty about events in one set, when one has the full knowledge of the occurrence of events in the other set of outcomes. The area enclosed by the circle envelope then represents the entropy of the “product” (joint) distribution: \( S(\mathbf{\uppi} ) = S\left( {{\mathbf{P}}(\user2{a} \wedge \user2{b})} \right) = S(\user2{p}) + S(\user2{q}) - I(\user2{p}:\user2{q}) = S(\user2{p}) + S(\user2{q}|\user2{p}) = S(\user2{q}) + S(\user2{p}|\user2{q}). \)
The common amount of information in two events a i and b j , I(i:j), measuring the information about a i provided by the occurrence of b j , or the information about b j provided by the occurrence of a i , is called the mutual information in two events:
It may take on any real value, positive, negative, or zero. It vanishes when both events are independent, i.e., when the occurrence of one event does not influence (or condition) the probability of the occurrence of the other event, and it is negative when the occurrence of one event makes a nonoccurrence of the other event more likely. It also follows from the preceding equation that
where the self-information of the joint event I(i∧j) = −logπ i,j . Hence, the information in the joint occurrence of two events a i and b j is the information in the occurrence of a i plus that in the occurrence of b j minus the mutual information. For independent events, when π i,j = p i q j , I(i:j) = 0 and hence I(i, j) = I(i) + I(j).
The mutual information of an event with itself defines its self-information: I(i:i) ≡ I(i) = log[P(i|i)/p i ] = −logp i , since P(i|i) = 1. It vanishes when p i = 1, i.e., when there is no uncertainty about the occurrence of a i , so that the occurrence of this event removes no uncertainty and hence conveys no information. This quantity provides a measure of the uncertainty about the occurrence of the event itself, i.e., the information received when the event occurs. The Shannon entropy of Eq. (1) can be thus interpreted as the mean value of self-information in all individual events: S(p) = ∑ i p i I(i). One similarly defines the average mutual information in two probability distributions as the π-weighted mean value of the mutual information quantities for individual joint events:
where the equality holds only for the independent distributions, when π i,j = π i,j 0 = p i q j . Indeed, the amount of uncertainty in q can only decrease when p has been known beforehand, S(q) ≥ S(q|p) = S(q) − I(p:q), with the equality being observed only when the two sets of events are independent (nonoverlapping). These average entropy/information relations are also illustrated in Fig. 1.
The average mutual information is an example of the entropy deficiency, measuring the missing information between the joint probabilities P(a∧b) = π of the dependent events a and b, and the joint probabilities P ind(a∧b) = π 0 = p T q for the independent events: I(p:q) = ΔS(π|π 0). The average mutual information thus reflects a degree of dependence between events defining the compared probability schemes. A similar information-distance interpretation can be attributed to the conditional entropy: S(p|q) = S(p)−ΔS(π|π 0).
3 Communication Systems
We continue this short introduction to IT with the entropy/information descriptors of a transmission of signals in communication systems [3, 4, 7, 8] (Fig. 2). The signal emitted from n “inputs” a = (a 1, a 2, …, a n ) of the channel source A is characterized by the probability distribution P(a) = p = (p 1, p 2, …, p n ). It can be received at m “outputs” b = (b 1, b 2, …, b m ) of the system receiver B. A transmission of signals in the channel is randomly disturbed thus exhibiting the communication noise. Indeed, in general the signal sent at the given input can be received with a nonvanishing probability at several outputs. This feature of communication systems is described by the conditional probabilities of outputs-given-inputs, P(b|a) = {P(b j |a i ) = P(a i ∧b j )/P(a i ) ≡ P(j|i)}, where P(a i ∧b j ) ≡ π i,j stands for the probability of the joint occurrence of the specified pair of the input–output events. The distribution of the output signal among the detection “events” b is thus given by the output probability distribution P(b) = q = (q 1, q 2, …, q m ) = p P(b|a).

Schematic diagram of the communication system characterized by two probability vectors: P(a) = {P(a i )} = p = (p 1, …, p n ), of the channel “input” events a = (a 1, …, a n ) in the system source A, and P(b) = {P(b j )} = q = (q 1, …, q m ), of the “output” events b = (b 1, …, b m ) in the system receiver B. The transmission of signals in this communication channel is described by the (n × m)-matrix of the conditional probabilities P(b|a) = {P(b j |a i ) ≡ P(j|i)}, of observing different “outputs” (columns, j = 1, 2, …, m), given the specified “inputs” (rows, i = 1, 2, …, n). For clarity, only a single scattering a i → b j is shown in the diagram
The Shannon entropy S(p) of the input (source) probabilities p determines the channel a priori entropy. The average conditional entropy H(B|A) ≡ S(q|p) of the outputs-given-inputs, determined by the scattering probabilities P(b|a), then measures the average noise in the a → b transmission. The so-called a posteriori entropy H(A|B) ≡ S(p|q), of inputs-given-outputs, is similarly defined by the “reverse” conditional probabilities of the b → a transmission signals: P(a|b) = {P(a i |b j ) = P(i|j)}. It reflects the residual indeterminacy about the input signal, when the output signal has already been received. The average conditional entropy S(p|q) thus measures the indeterminacy of the source with respect to the receiver, while the conditional entropy S(q|p) reflects the uncertainty of the receiver relative to the source. Hence, an observation of the output signal provides on average the amount of information given by the difference between the a priori and a posteriori uncertainties, which defines the mutual information in the source and receiver: S(p) − S(p|q) = I(p:q). This quantity measures the net amount of information transmitted through the communication channel, while the conditional information S(p|q) reflects a fraction of S(p) transformed into “noise” as a result of the input signal being scattered in the channel. Accordingly, S(q|p) reflects the noise part of S(q) = S(q|p) + I(p:q) (see Fig. 1).
Consider as an illustrative example the familiar Binary Channel of Fig. 3 defined by the symmetric conditional probability matrix of observing its two outputs, given the two inputs,

The symmetric binary channel
The given input probabilities p = (x, 1 – x) characterize the way this communication device is used. Its input (a priori) entropy is determined by the Binary Entropy Function (BEF) S(p) = −xlogx − (1 − x)log(1 − x) ≡ H(x) shown in Fig. 4. The system output entropy is also determined by another BEF, S(q) = H(z(x, ω)), where z(x, ω) ≡ q 2 = xω + (1 − x)(1 − ω), while the channel conditional entropy S(q|p) = H(ω) measures its average communication noise. The mutual information between the system inputs and outputs, I(p:q) = S(q) − S(q|p) = H[z(x, ω)] − H(ω), thus reflects the system net information flow (see Fig. 4). Since z always lies between ω and 1 − ω, H(z) = H(1 − z) ≥ H(ω) = H(1 − ω). This demonstrates the nonnegative character of the mutual information, represented by the overlap areas between the two entropy circles in a qualitative diagram of Fig. 1.

The binary entropy function H(x) = −xlog2 x − (1 − x)log2(1 − x) and the geometric interpretation of the conditional entropy S(q|p) = H(ω) ≡ H(B|A) and mutual information I(p:q) = H(z) − H(ω) ≡ I(A:B) quantities for the information network of Fig. 3
The amount of information I(p:q) flowing through this transmission system thus depends on both the cross-over probability ω, which characterizes the communication channel itself, and on the input probability x, which determines the way the device is exploited (probed). For x = 0 (or 1) H(z) = H(ω) and thus I(p:q) = 0, i.e., there is no net flow of information from the source to the receiver. For x = ½ one finds H(z) = 1 bit, thus giving rise to the maximum value of the mutual information (transmission capacity): C(ω) ≡ max p I(p:q) = max x {H[z(x, ω)] − H(ω)} = 1 – H(ω). Hence, for ω = ½ the information capacity of SBC identically vanishes.
4 Information Displacements in Molecules
The separated atoms (in their ground states), when shifted to their molecular positions define the (isoelectronic) molecular prototype called the “promolecule” [9, 25]. Its overall electron density, given by the sum of the free-atom distributions {ρ i 0}, ρ 0 = ∑ i ρ i 0, defines the initial stage in the bond-formation process. It determines a natural reference for extracting changes due to formation of chemical bonds. They are embodied in the density-difference function Δρ = ρ − ρ 0, between the molecular ground-state density ρ = ∑ i ρ i , a collection of the AIM densities {ρ i }, and the promolecular electron distribution ρ 0. This deformation density has been widely used to probe the electronic structure of molecular systems. In this section we compare it against selected (local) IT probes, in order to explore the information content of the molecular ground-state electron distribution ρ(r), or its shape (probability) factor p(r) = ρ(r)/N [9–16], obtained from the Kohn–Sham (KS) [65] calculations in the Local Density Approximation (LDA) of the Density Functional Theory (DFT) [66].
Consider the density Δs(r) of the Kullback–Leibler functional for the information distance between the molecular and promolecular electron distributions,
where w(r) and I[w(r)] denote the density-enhancement and surprisal functions relative to promolecule, respectively. We shall also explore the density Δh ρ (r) of the molecular displacement in the Shannon entropy,
testing its performance as an alternative IT tool for diagnosing the presence of chemical bonds and monitoring the effective valence states of bonded atoms.
It should be observed that the molecular electron density ρ(r) = ρ 0(r) + Δρ(r) is only slightly modified relative to the promolecular distribution ρ 0(r): ρ(r) ≈ ρ 0(r) or w(r) ≈ 1. Indeed, the formation of chemical bonds involves only a minor reconstruction of the electronic structure, mainly in the valence shells of the constituent atoms, so that |Δρ(r)| ≡ |ρ(r) − ρ 0(r)| ≪ ρ(r) ≈ ρ 0(r). Hence, the ratio Δρ(r)/ρ(r) ≈ Δρ(r)/ρ 0(r) is generally small in the energetically important regions of large density values.
As explicitly shown in the first column of Fig. 5, the largest values of the density difference Δρ(r) are observed mainly in the bond region, between the nuclei of chemically bonded atoms. The accompanying reconstruction of atomic lone pairs is also seen to lead to an appreciable displacement in the molecular electron density. By expanding the logarithm of the molecular surprisal I[w(r)] around w(r) = 1, to the first-order in the relative displacement of the electron density, one obtains the following approximate relation between the local value of the molecular surprisal density and that of the difference function itself:

The contour diagrams of the molecular density difference function Δρ(r) = ρ(r) − ρ 0(r) (first column), the information-distance density Δs(r) = ρ(r)I[w(r)] (second column), and its approximate, first-order expansion Δs(r) ≅ Δρ(r)w(r) (third column), for selected diatomic and linear triatomic molecules: H2, HF, LiF, HCN, and HNC. The solid, pointed and broken lines denote the positive, zero, and negative values, respectively, of the equally spaced contours [15, 67]
It provides a semiquantitative IT interpretation of the relative density difference diagrams and links the local surprisal of IT to the density difference function of quantum chemistry. This equation also relates the integrand of the information-distance functional with the corresponding displacement in the electron density: Δs(r) = ρ(r)I[w(r)] ≅ Δρ(r)w(r) ≈ Δρ(r).
This approximate relation is numerically verified in Fig. 5, where the contour diagram of the directed-divergence density Δs(r) (second column) is compared with the corresponding map of its first-order approximation Δρ(r)w(r) (third column) and the density difference function itself (first column). A general similarity between the diagrams in each row of the figure confirms a semiquantitative character of this first-order expansion. A comparison between panels of the first two columns in the figure shows that the two displacement maps so strongly resemble one another that they are hardly distinguishable. This confirms a close relation between the local density and entropy-deficiency relaxation patterns, thus attributing to the former the complementary IT interpretation of the latter. The density displacement and the missing-information distribution can be thus viewed as practically equivalent probes of the system chemical bonds.
The density difference function Δρ(r) for representative linear diatomic and triatomic molecules reflects all typical reconstructions of free atoms in molecules, which accompany the formation of a single or multiple chemical bonds with varying degree of the covalency (electron sharing) and ionicity (charge transfer) components. For example, the single covalent bond in H2 gives rise to a relative accumulation of electrons in the bond region, between the two nuclei, at the expense of the outer, nonbonding regions of space. The triple-bond pattern for N2 reflects density accumulation in the bonding region, due to both the σ and π bonds, and the accompanying increase in the density of the lone pairs on both nitrogen atoms, due to their expected sp-hybridization. One also observes a decrease in the electron density in the vicinity of the nuclei and an outflow of electrons from the 2p π AO to their overlap area, a clear sign of their involvement in formation of the double π bond system. In triatomic molecules one identifies a strongly covalent pattern of the electron density displacements in the regions of the single N–H and C–H atoms. A typical buildup of the bond charge due to the multiple CN bonds in the two isomers HCN and HNC can be also observed. The increase in the lone-pair electron density on the terminal heavy atom, N in HCN and or C in HNC, can be also detected, thus confirming the expected sp-hybridization of these bonded atoms.
Both heteronuclear diatomics represent partially ionic bonds between the two atoms exhibiting a small and large difference in their electronegativity (chemical hardness) descriptors, respectively. A pattern of the density displacement in HF reflects a weakly ionic (strongly covalent) bond, while in LiF the two AIM are seen to be connected by the strongly ionic (weakly covalent) bond. Indeed, in HF one detects a common possession of the valence electrons by the two atoms, giving rise to the bond charge located between them, and hence a small H → F charge transfer. Accordingly, in LiF a substantial Li → F electron transfer is detected.
In Fig. 6 the contour maps of the entropy-displacement density Δh ρ (r) are compared with the corresponding density difference diagrams for representative linear molecules of Fig. 5. Again, Δρ and Δh ρ diagrams for H2 are seen to qualitatively resemble one another and the corresponding Δs map of Fig. 5. The main feature of Δh ρ plot, an increase in the electron uncertainty between nuclei, is due to the inflow of electrons to this region. Again this covalent charge/entropy accumulation reflects the electron-sharing effect and the delocalization of the AIM valence electrons, which effectively move in the field of both nuclei, towards the bond partner. The entropy difference function is seen to display typical features in the reconstruction of atomic electron distributions in a molecule, relative to the promolecule.

Next we examine the central-bond problem in small [1.1.1], [2.1.1], [2.2.1], and [2.2.2] propellanes shown in Fig. 7. The main purpose of this study [9, 16] was to examine the effect on the central C′–C′ bond between the “bridgehead” carbon atoms of an increase in the size of the carbon bridges. The contour maps of Fig. 8 compare Δρ, Δs, and Δh ρ plots in the planes of section shown in Fig. 7 generated using the DFT–LDA calculations in the extended (DZVP) basis set. They display a depletion of the electron density between the bridgehead carbons in [1.1.1] and [2.1.1] propellanes, while larger bridges in [2.2.1] and [2.2.2] systems generate a density buildup in this region. A similar conclusion follows from the entropy-displacement and entropy-deficiency plots of the figure. The two entropic diagrams are again seen to be qualitatively similar to the corresponding density difference plots. As before, this resemblance is seen to be particularly strong between the Δρ and Δs diagrams.

The propellane structures and the planes of sections containing the bridge and bridgehead (C′) carbon atoms, all identified by black circles

5 Contra-Gradience Probe of Bond Localization
Each density functional A[ρ] can be regarded as the corresponding multicomponent functional A[ρ] ≡ A total[ρ] of the pieces ρ = {ρ α } into which the electron distribution is decomposed:
Such functionals appear in the non-Born–Oppenheimer DFT of molecules [68], in partitioning the electron density into distributions assigned to AIM, and in the fragment embedding problems [69–72]. Each resolution of the molecular electron density also implies the associated division of the molecular (total) quantity A total[ρ] into its additive, A add.[ρ], and nonadditive, A nadd.[ρ], contributions:
For example, the Gordon–Kim [73] type division of the kinetic energy functional defines the nonadditive contribution which constitutes the basis of the DFT-embedding concept of Cortona and Wesołowski [69–72]. Similar partition can be used to resolve the information quantities themselves. In particular, the inverse of the nonadditive Fisher information in the MO resolution has been shown to define the IT-ELF concept [29], in the spirit of the original Becke and Edgecombe formulation [30], while the related quantity in the AO resolution of the SCF MO theory generates the CG criterion for localizing the chemical bonds [9–14, 26–28]. It has been argued elsewhere that the valence basins of the negative CG density identify the bonding (constructive) interference of AO in the direct bonding mechanism, while the positive values of this local IT probe similarly delineate the antibonding regions in molecules (see Fig. 9).

The circular contour passing through both nuclei of the vanishing CG integrand for two 1s orbitals on atoms A and B, i cg(r) = 0. It separates the bonding region (inside the circle), where i cg(r) < 0, from the region of positive contributions i cg(r) > 0 (outside the circle). One observes that the heavy arrows representing the negative gradients of the two basis functions are mutually perpendicular on the dividing (spherical) surface i cg(r) = 0
For the given pair of AO, say A(r) and B(r) originating from different atoms A and B, respectively, the negative contribution to the nonadditive Fisher information results, when the gradient of one AO exhibits the negative projection on the direction of the gradient of the other AO. This explains the name of the CG criterion itself. The zero contour, which encloses the bonding region and separates it from the antibonding environment, is thus identified by the equation \( {{i}^{\mathrm{ cg}}}(\user2{r}) \equiv \nabla A(\user2{r}) \cdot \nabla B(\user2{r}) = 0 \). As shown in the qualitative diagram of Fig. 9, for the two 1 s orbitals in H2 this dividing surface constitutes the sphere passing through both nuclei, on which the two AO gradients are mutually perpendicular. Integration of i cg over the whole space gives the associated CG integral,
where μ stands for the electronic mass, while limiting this integration to the valence (bonding) region of the negative i cg, inside the closed surface i cg(r) = 0 (see Fig. 9), provides a useful tool for indexing the chemical bonds [28]. As also indicated in the preceding equation, the CG integral measures the coupling (off-diagonal) element T A,B of the electronic kinetic energy operator \( \hat{\rm T} \). Thus, CG integral reflects the kinetic energy coupling between the two chemically interacting AO. Such integrals are routinely calculated in typical quantum-chemical packages for determining the electronic structure of molecular systems. This observation emphasizes the crucial role of the kinetic energy component in interpreting the IT origins of the chemical bonding [74–77].
Consider a general case of the AO basis set χ = (χ 1, χ 2, …, χ m ) used to describe the occupied MO in the N-electron system. In the ground-state electron configuration defined by the singly occupied subspace \( {\boldsymbol{\psi}} = \{ {{\psi}_k}\} \) of the N lowest spin-MO, with the spatial (MO) parts \( {\boldsymbol{\varphi}} = {\boldsymbol{\chi}} \mathbf{C} = \{ {{\varphi}_k},k = 1,2,\ldots, N\} \) generated by the Hartree–Fock (SCF MO) or KS calculations, the nonadditive Fisher information in AO resolution reads as follows:
Here, the elements of the Charge-and-Bond-Order (CBO), first-order density matrix,
provide the AO representation of the projection operator \( {{{\hat{\mathrm{ P}}}}_{\boldsymbol{\varphi} }} \) onto the occupied MO subspace. The average nonadditive information is thus proportional to the associated component T nadd.[χ] of the system average kinetic energy: T total[χ] = tr(γT) = T add.[χ] + T nadd.[χ], where the kinetic energy matrix in AO representation \( {\mathbf{T}} = \{ {{T}_k}_{{,l}} = \left\langle {{{\chi}_k}} \right|\hat{\rm T}\left| {{{\chi}_l}} \right\rangle \} \) and T add.[χ] = ∑ k γ k,k T k,k .
In this orbital approximation one uses the most extended (valence) basins of the negative CG density, f nadd.(r) < 0, enclosed by the associated f nadd.(r) = 0 surfaces, as locations of the system chemical bonds. This probe has been successfully validated in recent numerical calculations [28]. In the remaining part of this section, we shall present selected results of this study. They have been obtained using the standard SCF MO calculations (GAMESS software) in the minimum (STO-3 G) Gaussian basis set. The contour maps, for the optimized geometries, are reported in atomic units. In these plots the broken-line contours correspond to the negative CG values. For visualization purposes representative perspective views of the CG bonding regions are also presented.
The contour map of Fig. 10 confirms qualitative predictions of Fig. 9 for H2. In the minimum basis set one indeed observes a spherical CG bonding region between the two nuclei, with the accompanying increases of this quantity being observed in the nonbonding surroundings. A similar analysis for HF is presented in Fig. 11. It indicates existence of three basins of the negative CG density: a large valence region between the two atoms, and two small volumes in the inner shell of F. In the triple-bond case of N2 (Fig. 12) the bonding basin is now distinctly extended away from the bond axis, due to the presence of the two π bonds accompanying the central σ bond. Small core-polarization basins near each nucleus are again seen in the perspective plot. The sp-hybridization promotion of the nonbonding regions on both atoms is again much in evidence in the accompanying contour map, and the bonding region is seen to be “squeezed” between the two atomic cores. The positive values of CG density in transverse directions on each nucleus reflect the charge displacements accompanying the π-bond formation (see also the N2 diagrams in Figs. 5 and 6).

The contour map of the CG density \( {{f}^{{{\mathrm{ nadd}}.}}}(\user2{r}) \) for H2

The perspective view of the negative CG basins and the contour map of \( {{f}^{{{\mathrm{ nadd}}.}}}(\user2{r}) \) for HF

The same as in Fig. 11 for N2
The chemical bonds in hydrocarbons are similarly probed in Figs. 13 and 14. These diagrams testify to the efficiency of the CG criterion in localizing both the C–C and C–H bonding regions in acetylene, butadiene, and benzene. In Fig. 13a the CG pattern of the triple bond between the carbon atoms in acetylene strongly resembles that observed in N2. It directly follows from the two perpendicular cuts of Fig. 13b that the π bond between the neighboring peripheral carbons in butadiene is stronger than its central counterpart. One thus concludes that the CG probe of chemical bonds indeed provides an efficient tool for locating the directly bonded regions in typical diatomic and polyatomic molecules.

The contour map of the CG density \( {{f}^{{{\mathrm{ nadd}}.}}}(\user2{r}) \) for acetylene (left diagram of panel a) and butadiene (remaining diagrams). The right diagram of panel a shows the contour map in the molecular plane of butadiene, reflecting only the σ-bonds, while panel b reports additional, perpendicular cuts for this molecule, passing through terminal (lower-left diagram) and central (lower-right diagram) C–C bonds, which additionally reflect the π-bonds

The contour maps of the CG density \( {{f}^{{{\mathrm{ nadd}}.}}}(\user2{r}) \) for benzene: in the molecular plane (left panel) and in the perpendicular plane passing through the C–C bond (right panel)
Finally, the bonding patterns in a series of four small propellanes of Fig. 7 are examined in the contour maps of Fig. 15. Each row of this figure is devoted to a different propellane in a series: [1.1.1], [2.1.1], [2.2.1], [2.2.2]. The left panels of each row correspond to the plane of section perpendicular to the central bond between the bridgehead carbons, at the bond midpoint, while the axial cuts of the right panels involve one of the carbon bridges. The main result of the density-difference and entropy-deficiency analysis of Fig. 8, of an apparent absence of the direct (through-space) bond between the carbon bridgeheads in the [1.1.1] and [2.1.1] systems and its full presence in the [2.2.1] and [2.2.2] propellanes, remains generally confirmed by the CG probe of Fig. 15. However, this transition is now seen to be less sharp, with very small bonding basins between bridgeheads being also observed in the two smallest molecules. This small bonding basin of [1.1.1] system is seen to gradually evolve into that attributed to the full bond in the [2.2.2] propellane. The bridge C–C and C–H chemical bonds are again seen to be perfectly localized by the closed valence surfaces of the vanishing CG density.

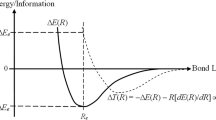
One of the primary goals of theoretical chemistry is to identify also the physical sources of the chemical bond. Most of existing theoretical interpretations of its origins emphasize, almost exclusively, the potential (interaction) aspect of this phenomenon, focusing on the mutual attraction between the accumulation of electrons between the two atoms (the negative “bond charge”) and the partially screened (positively charged) nuclei. This is indeed confirmed by the virial-theorem decomposition of the diatomic Born–Oppenheimer potentials. The latter indicates that for the equilibrium bond length it is the change in its potential component, relative to the dissociation limit, which is ultimately responsible for the net bonding effect of the atomic interaction.
The ELF and CG criteria adopt the complementary view, stressing the importance of the kinetic-energy bond component in locating the bonding regions in molecules. We recall that the associated displacement of the total kinetic energy of electrons, relative to that in the separated atom limit, is bonding only at an earlier stage of the mutual approach by both atoms. At this stage it is dominated by the longitudinal contribution associated with the gradient component along the bond axis. It then assumes the destabilizing character at the equilibrium internuclear separation, mainly due to its transverse contribution due to the gradient components in the directions perpendicular to the bond axis [74, 76, 77]. This virial theorem perspective also indicates that the kinetic energy constitutes a driving force of the bond-formation process. It follows from the classical analysis by Ruedenberg and coworkers [74, 76, 77] that a contraction of atomic electron distributions is possible in molecule due to this very lowering of the kinetic energy at large internuclear separation. The process of redistributing electrons in the chemical bond formation can be thus regarded as being “catalyzed” by the gradient effect of the kinetic energy. A similar conclusion follows from the theoretical analysis by Goddard and Wilson [75].
Therefore, the overall change in the kinetic energy (Fisher information) due to the chemical bond formation emphasizes a contraction of the electronic density in the presence of the remaining nuclear attractor in the molecule. It only blurs the subtle (interference) origins of chemical bonds. Indeed, the total kinetic energy component combines delicate, truly bonding (nonadditive) interatomic effects, originating from the stabilizing combinations of AO in the occupied MO, and the accompanying processes of the intraatomic polarization. In other words, the overall displacement in the kinetic energy contribution effectively hides a contribution due to minute changes in the system valence shell, which are associated by chemists with the chemical bond concept. Some partitioning of this overall energy component is thus called for to separate these subtle bonding phenomena from the associated nonbonding promotion of constituent atoms. Only by focusing on the nonadditive part of the electronic kinetic energy in CG criterion can one uncover the real information origins of the chemical bond and ultimately define efficient IT probe for its localization.
6 Orbital Communications and Information Bond Multiplicities
The molecular information system [9–13, 33, 39, 48, 55–58] represents the key concept of CTCB. It can be constructed at alternative levels defined by the underlying electron-localization “events,” which determine the channel inputs a = {a i } and outputs b = {b j }. In OCT the AO basis functions of the SCF MO calculations determine a natural resolution for discussing the information multiplicity (order) of the system chemical bonds: a = {χ i } and b = {χ j }. This AO network describes the probability/information propagation in the molecule. It can be described by the standard quantities developed in IT for real communication devices. The transmission of the AO-assignment “signals” becomes randomly disturbed, due to the electron delocalization throughout the network of chemical bonds, thus exhibiting typical communication “noise.” Indeed, an electron initially attributed to the given AO in the channel “input” can be later found with a nonvanishing probability at several AO in the molecular “output.”
This electron delocalization is embodied in the conditional probabilities of the “outputs-given-inputs,” P(b|a) = {P(χ j |χ i ) ≡ P(j|i)}, which define the “forward” channel of orbital communications. In OCT one constructs these probabilities [39, 48, 55–58, 78] using the bond-projected superposition principle of quantum mechanics [79]. This “physical” projection involves all occupied MO, which ultimately determine the entire network of chemical bonds in the molecular system of interest. Both the molecule as a whole and its constituent subsystems can be adequately described in terms of such IT bond multiplicities [9–13]. The off-diagonal orbital communications are related to the familiar Wiberg [80] contributions to the molecular bond orders or the related “quadratic” bond multiplicities [81–90] formulated in the MO theory.
The IT descriptors of the chemical bond pattern have been shown to account for the chemical intuition quite well providing the resolution of the diatomic bond multiplicities into the complementary IT-covalent and IT-ionic components [10, 11, 56]. The internal (intrafragment) and external (interfragment) indices of molecular subsystems (groups of AO) can be efficiently generated using the appropriate reduction of the molecular AO channel by combining the selected outputs into larger molecular fragments [9, 37, 42, 78].
In the SCF MO theory the bond network is determined by the occupied MO in the system ground state. For reasons of simplicity we assume the closed-shell (cs) configuration of N = 2n electrons in the standard (spin-restricted) Hartree–Fock (RHF) description, which involves n lowest (doubly occupied, orthonormal) MO. In the familiar LCAO MO approach they are expanded as linear combinations of the (Löwdin) orthogonalized AO χ = (χ 1, χ 2, …, χ m ) = {χ i } contributed by the system constituent atoms:
here, the rectangular matrix \( {\mathbf{C}} = \{ {{C}_i}_{{,s}}\} { } = \left\langle {\boldsymbol{\chi}} \right|\left. {\boldsymbol{\varphi}} \right\rangle \) groups the relevant LCAO MO coefficients to be determined using the iterative self-consistent-field procedure.
The molecular electron density,
and hence also the probability distribution p(r) = ρ(r)/N, the shape-factor of ρ, are both determined by the CBO matrix,
The latter constitutes the AO representation of the projection operator onto the subspace of all (doubly) occupied MO, \( {{\hat{\rm P}}_{\boldsymbol{\varphi} }} = |{\boldsymbol{\varphi}} \rangle \langle {\boldsymbol{\varphi}} | = {{\sum}_s}|{{\varphi}_s}\rangle \langle {{\varphi}_s}| \equiv {{\sum}_s}{{\hat{P}}_s} \), and satisfies the idempotency relation:
The CBO matrix reflects the promoted, valence state of AO in the molecule. Its diagonal elements reflect the effective electron occupations of the basis functions, {N i = γ i,i = Np i }, with the normalized probabilities p = {p i = γ i,i /N} of the basis functions occupancy in molecule: ∑ i p i = 1.
The orbital information system involves the AO events in the channel input a = {χ i } and output b = {χ j }. In this description the AO → AO communication network is determined by the conditional probabilities:
where the joint probabilities of simultaneously observing two AO in the system chemical bonds P(a∧b) = {P(i∧j)} exhibit the usual normalization relations:
These probabilities involve squares of corresponding elements of the CBO matrix [39, 48]:
where the constant R i = (2γ i,i )−1 satisfies the normalization condition of Eq. (24). These probabilities explore the dependencies between AO resulting from their participation in the framework of the occupied MO, i.e., their simultaneous involvement in the entire network of chemical bonds in the molecule. This orbital channel can be subsequently probed using both the promolecular (p 0 = {p i 0}), molecular (p = {p i }), or general (ensemble) input probabilities, in order to extract the desired aspects of the chemical bond pattern [10, 11, 56, 63].
The off-diagonal conditional probability of jth AO output given ith AO input is thus proportional to the squared element of the CBO matrix linking the two AO, γ j,i = γ i,j , thus being also proportional to the corresponding AO contribution M i,j = γ i,j 2 to Wiberg’s [80] index of the overall chemical bond order between two atoms in the molecule,
or to related quadratic descriptors of molecular bond multiplicities [81–90].
In OCT the entropy/information indices of the covalent/ionic components of chemical bonds represent complementary descriptors of the average communication noise and the amount of information flow in the molecular information channel [9–13, 33, 63]. One observes that the molecular input P(a) ≡ p generates the same distribution in the output of the molecular channel,
thus identifying p as the stationary probability vector of the molecular ground state. This purely molecular channel is devoid of any reference (history) of the chemical bond formation and generates the average-noise index of the molecular overall IT covalency. It is measured by the conditional entropy of the system outputs given inputs:
The AO channel with the promolecular input “signal,” P(a 0) = p 0, refers to the initial state in the bond formation process. It corresponds to the ground state (fractional) occupations of the AO contributed by the system constituent (free) atoms, before their mixing into MO. These input probabilities give rise to the average information flow descriptor of the bond IT ionicity, given by the mutual information in the channel inputs and outputs [63]:
In particlular, for the molecular input, when p 0 = p and hence the vanishing information distance ΔS(p|p 0) = 0, I(p:p) = S(p) − S ≡ I.
The sum of these two bond components,
measures the overall IT bond multiplicity of all bonds in the molecular system under consideration. For the molecular input this quantity preserves the Shannon entropy of the molecular input probabilities:
As an illustration consider the familiar problem of combining the two (Löwdin-orthogonalized) AO, A(r) and B(r), say, two 1s orbitals centered on nuclei A and B, respectively, which contribute a single electron each to form the chemical bond A–B. The two basis functions χ = (A, B) then form the bonding (ϕ b ) and antibonding (ϕ a ) MO combinations, ϕ = (ϕ b , ϕ a ) = χ C:
where the square matrix
groups the LCAO MO expansion coefficients expressed in terms of the complementary probabilities: P and Q = 1 − P. They represent the conditional probabilities of observing AO in MO:
In the ground-state configuration of the doubly occupied bonding MO ϕ b the CBO matrix γ = 2d, the double density matrix \( {\mathbf{d}} = \left\langle {\boldsymbol{\chi}} \right|\left. {{{\varphi}_b}} \right\rangle \left\langle {{{\varphi}_b}} \right.\left| {\boldsymbol{\chi}} \right\rangle \), reads:
It generates the following conditional probabilities of communications between AO in the molecular bond system,
which determine the χ → χ communication network shown in Fig. 16.

Communication channel of the 2-AO model of the chemical bond and its entropy/information descriptors in bits
This nonsymmetrical binary channel adopts the molecular input signal p = (P, Q) to extract the bond IT covalency, measuring the channel average communication noise, and the promolecular input signal p 0 = (½, ½), in which the two basis functions contribute a single electron each to form the chemical bond, in order to determine the model IT-ionicity index measuring the channel information capacity. The bond IT covalency S(P) is determined by the binary entropy function H(P) reaching the maximum value H(P = ½) = 1 bit for the symmetric bond P = Q = ½, e.g., the σ bond in H2 or π-bond in ethylene (see also Figs. 4 and 17). It vanishes for the lone-pair molecular configurations, when P = (0, 1), H(P = 0) = H(P = 1) = 0, marking the alternative ion-pair configurations A+B− and A−B+, respectively, relative to the initial AO occupations N 0 = (1, 1) in the assumed promolecular reference, in which both atoms contribute a single electron each to form the chemical bond.

Conservation of the overall entropic bond multiplicity V 0(P) = 1 bit in the 2-AO model of the chemical bond. It combines the conditional entropy (average noise, bond covalency) S(P) = H(P) and the mutual information (information capacity, bond ionicity) I 0(P) = 1 − H(P). In MO theory the direct bond order of Wiberg is represented by (broken-line) parabola M A,B(P) = 4P(1 − P) ≡ 4PQ
The complementary descriptor I 0(P) = 1 − H(P) of the bond IT ionicity (see Fig. 17), which determines the channel mutual information relative to the promolecular input, reaches the highest value for the two limiting (electron-transfer) configurations: I 0(P = 0) = I 0(P = 1) = H(½) = 1 bit, and identically vanishes for the purely covalent, symmetric bond, I 0(P = ½) = 0. As explicitly shown in Fig. 17, these two components of the chemical bond multiplicity compete with one another, yielding the conserved overall IT bond index V 0(P) = S(P) + I 0(P) = 1 bit, marking a single bond in OCT in the whole range of admissible bond polarizations P ∈ [0, 1]. This simple model thus properly accounts for the competition between the bond covalency and ionicity, while preserving the single IT bond order measure reflected by the conserved overall multiplicity of the chemical bond.
Similar effects transpire from the quadratic bond indices formulated in the MO theory [81, 82, 87]. The corresponding plot of the Wiberg bond order for this model is given by the parabola (see Fig. 17):
which closely resembles the IT-covalent plot S(P) = H(P) in the figure.
7 Localized Bonds in Diatomic Fragments
The bond components of Fig. 17 can be also decomposed into the corresponding atomic contributions [9]. In communication theory this partition is accomplished by using the partial (row) subchannels of Fig. 18, each determining communications originating from the specified atomic input. The partial entropy covalencies of the given atomic channel, calculated for the full (unit) probability of its input, again recover the binary entropy estimate of Fig. 17. However, the partial mutual information indices of the bond IT iconicity (charge transfer) have to be calculated for the full list of inputs, thus being equal to the promolecular mutual information index of Fig. 16.

The elementary partial (row) subchannels due to atomic inputs A (solid lines) and B (broken lines) in the 2-AO model of the chemical bond and their IT covalency/ionicity components
In typical SCF LCAO MO calculations, the lone pairs of the valence- and/or inner-shell electrons can strongly affect the overall IT descriptors of the chemical bonds. Elimination of such lone-pair contributions in the resultant IT bond indices of diatomic fragments of molecules requires an ensemble (flexible input) approach [10, 11, 56]. In this generalized procedure the input probabilities are derived from the joint (bond) probabilities of two AO centered on different atoms, which reflect the actual simultaneous participation of the given pair of basis functions in chemical bonds. Such an approach effectively projects out the spurious contributions due to the inner- and outer-shell AO, which are excluded from mixing into the delocalized, bonding MO combinations. This probability-weighting procedure is capable of reproducing the Wiberg bond orders in diatomics, at the same time providing the IT-covalent/ionic resolution of these overall bond indices.
Let us illustrate this weighting procedure in the 2-AO model of the preceding section. In the bond-weighted approach, one uses the elementary subchannels of Fig. 18 and their partial entropy/information descriptors {S(χ|i)}, I 0(i:χ) = I 0(χ:χ); {i = A, B}, which are also listed in the diagram. In this particular case they are equal to the corresponding molecular conditional-entropy and mutual-information quantities of Fig. 16. Since these row descriptors represent the IT indices per electron in the diatomic fragment, these contributions have to be multiplied by N AB = N = 2 in the corresponding resultant IT components and the overall measure of multiplicity of the effective diatomic bond. Using the off-diagonal joint probability P(A∧B) = P(B∧A) = PQ = γ A,B γ B,A /4 as the ensemble probability for both AO inputs gives the following average quantities for this model diatomic system (see Fig. 19):

Variations of the IT-covalent [S AB(P)] and IT-ionic [I 0AB (P)] bond components [in the M A,B(P) units] of the 2-AO model with changing MO polarization P and conservation of the relative total bond order V 0AB (P)/M A,B(P) = 1
We have thus recovered the Wiberg index as the overall IT descriptor of the chemical bond in the 2-AO model, V 0AB = M A,B, at the same time establishing its covalent, S AB, and ionic, I 0AB , contributions. Again, these IT-covalency and IT-ionicity components compete with one another, while conserving the Wiberg bond order as the overall information measure of the bond multiplicity (in bits). This procedure can be generalized into the SCF MO calculations for general polyatomic systems and basis sets [10, 11, 56]. Illustrative RHF bond orders in diatomic fragments of representative molecules, for their equilibrium geometries in the minimum (STO-3G) basis set, are compared in Table 1. It follows from this numerical analysis that in polyatomics this weighting procedure gives rise to an excellent agreement with both the Wiberg bond orders and the accepted chemical intuition.
This ensemble averaging for the localized bond descriptors reproduces exactly the Wiberg bond order in diatomic molecules [56]. In a series of related compounds, e.g., in hydrides or halides, the trends exhibited by the entropic covalent and ionic components of a roughly conserved overall bond order also agree with intuitive expectations. For example, the single chemical bond between two “hard” atoms in HF appears predominantly covalent, while a substantial ionicity is detected for LiF, for which both Wiberg and information-theoretic results predicts roughly 3/2 bond, consisting of approximately 1 covalent and ½ ionic contributions. One also observes that all carbon–carbon interactions in the benzene ring are properly differentiated. The chemical orders of the multiple bonds in ethane, ethylene, and acetylene are properly reproduced and the triple bond in CO is correctly accounted for. As intuitively expected, the C–H bonds are seen to slightly increase their information ionicity when the number of these terminal bonds increases in a series: acetylene, ethylene, ethane.
8 Through-Space and Through-Bridge Bond Components
In OCT the direct communication χ i → χ j between the given pair of AO, manifested by the nonvanishing conditional probability P(j|i) > 0, reflects a “dialogue” between these basis functions in the molecular ground state. It indicates the existence of the direct (through-space) chemical bonding between these orbitals, due to their nonvanishing overlap/interaction giving rise to their constructive interference in the bonding MO combination. As we have demonstrated in the preceding section, the Wiberg bond-order contribution [see also Eqs. (20) and (22)],
provides a useful measure of the multiplicity of this explicit interaction. In this equation we have used the idempotency property of the projector \( {{{\hat{\mathrm{ P}}}}_{\boldsymbol{\varphi} }} \) onto the bonding (doubly occupied) subspace of MO, \( {\hat{\mathrm{ P}}}_{\boldsymbol{\varphi}}^2 = {{{\hat{\mathrm{ P}}}}_{\boldsymbol{\varphi} }} \). This operator determines the associated nonorthogonal projections of AO:
The direct “bond order” measures the magnitude of the overlap d i,j between bond projections of the interacting basis functions.
However, communications between χ i and χ j can be also realized indirectly via a cascade involving other orbitals, i.e., as a “gossip” spread through the remaining AO, χ′ = {χ k≠(i,j)}. For example, this implicit scattering process may involve a single AO intermediates, in the single-step “cascade” propagation: χ i → χ′ → χ j . However, since in the molecular channel each AO both emits and receives the signal to/from remaining basis functions, this process may also involve any admissible multistep cascade, χ i → {χ′→ χ′→ …. → χ′} → χ j , in which the consecutive sets of AO intermediaries {χ′ → χ′ → …. → χ’} form an effective multistep “bridge” for the information scattering. The OCT formalism has been recently extended to tackle such indirect chemical communications [59–62]. The corresponding Wiberg-type bond multiplicities and the associated IT bond descriptors for such cascade information channels have been proposed, capable of describing these through-bridge chemical interactions.
In MO theory the chemical coupling between, say, two (valence) AO or general basis functions originating from different atoms is strongly influenced by their direct overlap and interaction. Together these two factors condition the bonding effect experienced by electrons occupying their bonding combination in the molecule, compared to the nonbonding reference of electrons on the separated AO. This through-space bonding mechanism is generally associated with an accumulation of the valence electrons in the region between the two nuclei, due to the constructive interference in the bonding MO. Indeed, such “shared” bond charge is customarily regarded as the prerequisite for the bond covalency in the direct interaction between the two AO. It is also reflected by the familiar covalent Valence-Bond (VB) structure. In Sects. 4 and 5 a similar effect of the bonding accumulation of the information densities relative to the promolecular distribution has been detected. The complementary, ionicity aspect of the direct chemical bonding is manifested by the MO polarization, reflecting the charge transfer effects, or by the participation of the orthogonal part of the ionic structure in the ground-state wave function in VB theory. In OCT the bond ionicity descriptor reflects a degree of “localization” (determinicity) in direct communications between AO, while the complementary bond covalency index measures the “delocalization” (noise) aspect of the direct orbital channel.
To summarize, the direct (“through-space”) bonding interaction between neighboring atoms is in general associated with the presence of the bond charge between the two nuclei. However, for more distant atomic partners such an accumulation of valence electrons can be absent, e.g., in the cross-ring π-interactions in benzene or between the bridgehead carbon atoms in small propellanes [59]. Nonetheless, the bonding interaction lacking this accumulation of the charge (information) can be still realized indirectly, through the neighboring AO intermediaries forming a “bridge” for an effective interaction between more distant (terminal) AO, e.g., in the cross-ring interactions between two meta- or para-carbons in benzene, two bridgehead carbons in small propellanes [59], or higher order neighbors in the polymer chain [61, 62]. This indirect mechanism was shown to reflect the implicit dependencies between the AO bond projections χ b [60], which reflect the resultant AO participations in all chemical bonds in the molecular system under consideration. Indeed, the orthonormality constraints imposed on the occupied MO imply the implicit dependencies between the (nonorthogonal) bond projections of AO on different atoms, due to real chemical bridges in molecules (see Fig. 20).

Direct (through-space) chemical interaction (broken line) between orbitals χ i and χ j contributed by atoms A and B, respectively, and the indirect (through-bridge) interaction (solid/pointed lines), through t AO-intermediaries (χ k , χ l , …, χ m , χ n ) = {χ r , r = 1, 2, …, t} contributed by the neighboring bonded atoms (C, D, …, F, G), respectively. The strength of each partial (direct) interaction is reflected by the magnitude of the corresponding elements of the density matrix \( {\mathbf{d}} = \langle {{{\boldsymbol{\chi}}}^b}|{{{\boldsymbol{\chi}}}^b}\rangle = \left\{ {{{d}_i}_{{,j}}} \right\} \), which measure the overlaps between the bond projections χ b of AO
In this generalized outlook on the chemical bond-index concept, emerging from both the Wiberg, quadratic measure of MO theory and the IT bond multiplicities of OCT, one thus identifies the chemical bond “order” as a measure of a “dependence” (nonadditivity) between orbitals on different atomic centers. On one hand, this dependence can be realized directly (through space), by the constructive interference of orbitals (probability amplitudes) on two atoms, which generally increases the electron density between them. On the other hand, it can also have an indirect origin, through the dependence on orbitals of the remaining AIM used to construct the occupied MO subspace ϕ o = χCo. These implicit (“geometrical”) dependencies are embodied in the (idempotent) density matrix:
Each pair of AO on different atoms is thus capable of exhibiting a partial through-space and through-bridge bond components. The order of the former quickly vanishes with an increase of the inter-atomic separation, and when the interacting AO are heavily engaged in forming chemical bonds with other atoms. At these separations the indirect bond orders can still assume appreciable values, when the remaining atoms form an effective bridge of the neighboring, chemically interacting atoms, which links the specified pair of terminal AO. The bridging atoms must be mutually bonded to generate the appreciable through bridge overlap of the interacting AO, so that a variety of significant bridges is practically limited to real chemical bridges of atoms in the molecular structural formula.
The representative indirect bond overlap through the t-bridge of Fig. 20, S i,j (k, l,.., m, n) = S i,j ({r}), constitutes a natural generalization of its direct, through-space analog by additionally including the product of bond projectors onto the indicated intermediate AO,
For specific single- or multi-step bridges realized by the indicated AO intermediates, this indirect bond overlap is given by the relevant products of direct overlaps in the bridge:
Hence, for a general t-bridge of Fig. 20 one finds
The square of this generalized bond overlap then defines the associated Wiberg-type bond order for such implicit interaction between orbitals χ i and χ j via the specified t-bridge:
The sum of contributions due to the most important (chemical) bridges {α}:
then determines the overall indirect Wiberg-type bond order, which supplements the direct component M i,j in the full quadratic bond-multiplicitiy index, between terminal orbitals χ i and χ j in presence of remaining AO:
As an illustration let us examine the indirect π-bonds between carbon atoms in benzene and butadiene, in the familiar Hückel approximation [59]. For the consecutive numbering of carbons in the ring/chain, the relevant CBO matrix elements in benzene read
while the off-diagonal part of the CBO matrix in butadiene is fully characterized by the following elements:
They generate the associated direct (through-space) bond multiplicities:
These direct bond orders are complemented by the following estimates of the resultant multiplicities of the indirect π-interactions due to chemical bridges:
Together these contributions give rise to the following resultant bond orders of Eq. (45):
Of interest also is a comparison of the bond-order contributions in benzene realized through the ring bridges of increasing length:
Thus, the longer the bridge, the smaller indirect bond order it contributes. The model and Hartree-Fock calculations for representative polymers [61, 62] indicate that the range of bridge interactions is effectively extended to third-order neighbors in the chain, for which the direct interactions practically vanish.
The artificial distinction in Wiberg’s multiplicity scheme of the π-interactions with the vanishing direct CBO matrix element as nonbonding is thus effectively removed, when the through-bride contributions are also taken into account. One observes the differences in composition of the resultant indices for the cross-ring interactions in benzene: the para interactions exhibit comparable through-space and through-bridge components, the meta multiplicities are realized exclusively through bridges, while the strongest ortho bonds have practically direct, through-space origin. A similar pattern can be also observed in butadiene.
The conditional probabilities of Eq. (26) determine the molecular information channel for the mutual communications between AO, which generate the associated covalency (noise) and ionicity (information flow) descriptors of the direct chemical bonds. One similarly derives the corresponding entropy/information multiplicities for the indirect interactions between the specified (terminal) orbitals χ i and χ j , generated from descriptors of the associated AO information cascades for the most important (chemical) bridges [11, 59, 61]. The resulting IT indices of specific bridge interactions have been shown to compare favorably with the generalized Wiberg measures of Eq. (43). For an attempt to separate the direct and indirect energy contributions, the reader is referred to [91].
9 Information Probes of Elementary Reaction Mechanisms
Interesting new results in the IT studies of the elementary reaction mechanisms have recently been obtained in the Granada group [92–94]. Both the global (Shannon) and local (Fisher) information measures have been used in these investigations. The course of two representative reactions has been examined: of the radical abstraction of hydrogen (two-step mechanism),
which requires extra energy to proceed, and of the nuclephilic substitution in the hydride exchange (SN2, one-step mechanism):
The abstraction process proceeds by homolysis and is kinetically of the first-order (SN1-like). It involves two steps: formation of new radicals, via the homolytic cleavage of the nonpolar, perfectly covalent bond in H2, in absence of any electrophile or nucleophile to initiate the heterolytic pattern, and the subsequent recombination of a new radical with another radical species. The hydride exchange is an example of the kinetically second-order, the first-order in both the incoming (nucleophile) and leaving (nucleofuge) hydride groups. It proceeds via the familiar Walden-inversion Transition State (TS) in a single, concerted reaction.
The central quantities of these IT studies are the Shannon entropies in both the position (r) and momentum (p) spaces,
and the related Fisher information measures:
Here, η(p) stands for the momentum density of electrons, efficiently generated from the Fourier transforms of the known position-space MO.
The reaction profiles of these information probes have discovered the presence of additional features of the two reaction mechanisms by revealing the chemically important regions where the bond forming and bond breaking actually occur. These additional features cannot be directly identified from the energy profile alone and from the structure of the TS densities involved. Consistency of predictions resulting from the global (Shannon) [92] and local (Fisher) [93, 94] information measures has additionally confirmed a more universal and unbiased character of these findings.
Indeed, either of the two complementary Shannon entropies for the model radical abstraction reaction displays a richer structure than the associated energy profile, which only exhibits one maximum at the TS point along the reaction coordinate. The position entropy S r displays a local maximum at this TS structure and two minima in its close proximity, whereas the momentum entropy S p exhibits the global minimum at TS complex and two maxima at points slightly more distant from TS than the corresponding positions of the S r minima. It thus follows from these entropy curves that the approach of the hydrogen molecule by the incoming hydrogen in the proximity of TS first localizes ρ in preparation for the bond rupture, which also implies an associated increase in the kinetic energy (delocalization of η). This preparatory stage is identified by the local minima of S r (maxima of S p ). Next, when the system relaxes and the new bond is formed at TS, the position (momentum) densities become more delocalized (localized). This is indeed manifested by the corresponding maximum (minimum) features of S r (S p ). The bond-breaking process requires energy, as indeed witnessed by an earlier maximum of S p in the entrance valley of the reaction Potential Energy Surface (PES), which is subsequently dissipated by relaxing the structure at TS. In other words, the reaction complex first gains the energy required for the bond dissociation, and then the position-space density gets localized to facilitate the bond cleavage, which in turn induces the energy/density relaxation towards the TS structure.
Therefore, the entropy representation of the reaction mechanism reveals the whole complexity of this transformation, while the associated Minimum Energy Path (MEP) profile only localizes the transition state on PES, missing the crucial transitory localization/delocalization and relaxational phenomena involved in this two-step process.
The corresponding S r (S p ) plot for the hydride-exchange SN2 process again exhibits the maximum (minimum) at TS, with two additional minima (maxima) in its vicinity, where the bond breaking is supposed to occur. These additional, pre- and post-TS features are symmetrically placed in the entrance and exit valleys, relative to TS structure, but now they appear at roughly the same values of the Intrinsic Reaction Coordinate (IRC) in both the position and momentum representations. This is in contrast to the two-stage abstraction mechanism, where in the entrance valley the p-space maximum of the Shannon entropy has preceded the associated minimum observed in the r-space. This simultaneous r-localization (p-delocalization) may be indicative of the single-step mechanism in which the approach of the nucleophile is perfectly synchronized with the concomitant departure of the nucleofuge, so that the bond forming and bond breaking occur in a concerted manner.
It should be observed that both these displacements increase the system energy. First, as the nucleophile approaches, this energy is required to overcome the repulsion between reactants and create the position localization (momentum delocalization) facilitating the bond weakening. As the reaction progresses forward, the energy continues to grow towards the maximum at TS, when the sufficient threshold of the new chemical bond has already been reached to start the structure relaxation inducing the reverse r-delocalization (p-localization) processes leading to the S r (S p ) maximum (minimum) at TS. This synchronous transformation picture is indeed customarily associated with this particular reaction.
The same sequences of the chemical events are seen in the complementary Fisher-information analysis of the structural features of distributions in both spaces. For the hydrogen abstraction reaction one observes with the progress of IRC towards TS that, relative to the separated reactants reference, both I r and I p at first decrease their values, thus marking a lower average gradient content of the associated probability amplitudes (wave functions) in both spaces, i.e., a more regular/uniform distribution (less structure, “order”). The I p profile is seen to exhibit a faster decay towards the local minimum preceding TS, where I p reaches the maximum value. These more uniform momentum densities also correspond to the local maxima of the system chemical hardness, with TS marking the local minimum of the latter. The I r monotonically decreases towards the minimum value at TS, thus missing the additional extrema observed in the S r plot, which have been previously associated with the bond homolysis. Thus, the I r is not capable of describing the bond breaking/forming processes, which is clearly uncovered by I p .
The disconcerted manner of the elementary bond forming and bond breaking is directly seen in the corresponding bond-length plots: the breaking of the bond occurs first, and then the system stabilizes by forming the TS structure. Additional insight into the density reconstruction in this homolytic bond rupture comes from examining the corresponding plots of the system dipole moment, reflecting the charge distortion during reaction progress. The observed behavior of these functions is opposite to I p . Therefore, in regions of the minimum I p the dipole moment reaches the maximum value and vice versa. As intuitively expected, the dipole moment identically vanishes at both TS and in separated reactants/products.
Finally, we turn to the Fisher-information analysis of the hydride-exchange reaction involving the heterolytic bond cleavage, with an accompanying exchange of charge between reactants. The corresponding I r and I p functions of the reaction coordinate now display similar behavior, both exhibiting maxima at TS, where the Shannon entropies have indicated a more delocalized position density and relatively localized momentum density. One also observes two minima of I r and I p in the proximity of TS. These IRC values coincide with the bond breaking/forming regions, and the change in the curvature of the bond-elongation curves for the incoming and outgoing nucleophile marks the start of an increase in the gradient content of the momentum density, towards the maximum value at TS structure.
10 Conclusion
Information theory has been shown to provide a novel and attractive perspective on the entropic origins of the chemical bond. It also offers a complementary outlook on the transformation of the electronic information content in the elementary chemical reactions. In this short overview, we have first introduced the key IT concepts and techniques to be used in such a complementary analysis of electron distributions in molecular systems. They have been subsequently applied to explore the bonding pattern in typical molecules: in terms of the information distribution, the bond localization/multiplicity, and its ionic/covalent composition. The use of the information densities as local probes of electronic distributions in molecules has been advocated and the importance of the nonadditive entropy/information measures in extracting subtle changes due to the bond formation has been stressed. The use of the CG density, of the nonadditive Fisher information (electronic kinetic energy) in the AO resolution, as an efficient localization probe of the direct chemical bonds has been validated.
The OCT, in which molecules are regarded as information systems, has been shown to be capable of tackling several classical issues in the theory of bond multiplicities and their covalent/ionic components. The off-diagonal (inter-orbital) conditional probabilities it generates are proportional to the quadratic bond indices formulated in MO theory, and hence the strong inter-orbital communications correspond to strong Wiberg bond orders. The IT “fingerprints” of typical elementary mechanisms of chemical reactions have also revealed new stages in the bond-breaking–bond-forming processes involved. These illustrative results demonstrate that the alternative information probes increase our understanding of the complex chemical bond phenomena, with the communication-noise (covalency) and information-flow (ionic) measures of the IT bond orders adequately reflecting the accepted chemical intuition. This development extends our understanding of the chemical bond from the complementary IT viewpoint.
The CG criterion explores the nonadditive part of the Fisher information in AO resolution, which reflects the electron delocalization phenomena due to the direct chemical bonds. It has been also demonstrated elsewhere [29] that the key conditional probability ingredient of the original ELF concept [30–32] is also related to the nonadditive Fisher information in MO resolution. The inverse of the latter thus quantifies a degree of a localization of the system electrons in a molecule, in accordance with an earlier Fermi-hole analysis by Luken et al. [95, 96]. The spatial organization of the electronic structure can be also probed stochastically, by combining the path integral Markovian pair-conditional probability density with the basic concepts of the catastrophe theory [97], in a way consistent with the Heisenberg and Pauli principles of quantum mechanics. This analysis provides the Markovian derivation of the original ELF and extends this localization concept to new Markovian-type ELF classes.
The entropy/information descriptors can be generated for both the molecule as a whole and its localized fragments. The relevant OCT treatment of diatomic subsystems has been described. This approach reproduces the Wiberg bond multiplicity in diatomic molecules and closely approximates this index in polyatomic molecules. The extra computational effort of such an IT analysis of the molecular bonding patterns is negligible, compared to the cost of standard SCF LCAO MO calculations of the molecular electronic structure, since practically all computations using orbital approximation already determine the CBO and kinetic energy data required in the OCT probe of IT bond multiplicities and the CG localization of direct chemical bonds.
It has also been stressed that chemical interactions between the specified pair of AO have both the direct (through-space) and, hitherto neglected, indirect (through-bridge) components. The former reflects the direct interference between bonded atoms, while the latter is realized indirectly, through the remaining AIM which form effective bridges for the chemical coupling between more distant atoms. The most efficient bridges are the real (chemical) bridges of bonded atoms connecting such “terminal” atoms in question. Therefore, the bonded status of the given pair of atoms can be felt also at larger separations provided there exist real bridge of the direct chemical bonds connecting them. This implicit coupling of basis functions in molecular states has been first conjectured to explain the central bond in small propellanes, lacking the charge accumulation between the bridgehead carbons. This difficulty has also prompted the alternative proposition of the VB-inspired charge-shift [98] mechanism. The latter attributes this indirect bond to the instantaneous charge fluctuations between the bridgehead carbons.
We have also discussed the indirect Wiberg-type indices for this “through-bridge” interaction in molecular systems, which complement the familiar Wiberg bond orders of the “through-space” bonding. The bridge interactions have been shown to have important implications for the resultant multiplicities of π-interactions between more distant carbon atoms in hydrocarbons. For example, in benzene the ortho-carbons exhibit a strong Wiberg bond multiplicity, of almost exclusively through-space origin, while the cross-ring interactions between the meta- and para-carbons were shown to be described by much smaller, but practically equalized overall bond orders. The latter can be distinguished by their direct/indirect composition: the meta bonds have been shown to be realized exclusively through bridges, while the para bonds exhibit comparable direct and indirect contributions.
In OCT the “explicit” (through-space) bond component contributed by the specified pair of interacting AO originates from their mutual probability scattering. Its covalency is thus generated by finite conditional probabilities of their direct communications in the molecule, related to the square of the corresponding element of the system density matrix and hence also to the associated Wiberg index. These direct AO communications are thus determined by the electron delocalization throughout the system occupied (bonding) subspace of MO. The “implicit” (through-bridge) bond contributions can be similarly viewed as resulting from the indirect (cascade) information propagation via the bridging AO. In a sense, the through-space bonding can be regarded as reflecting the direct “conversation” between AO, while the through-bridge channel(s) can be compared to a chatty talk reporting “hearsay,” the “rumor” spread between the terminal AO via the connecting chain of the AO intermediaries of the bridge under consideration. Together the direct (“dialogue”) and indirect (“gossip”) contributions to the chemical bond order determine the resultant bond multiplicity between the specified AO in molecular environment. Each pair of atoms thus exhibits partial through-space and through-bridge bond components. The “order” of the former quickly vanishes with the increasing inter-atomic separation. The overall bond order between more distant atomic partners can still assume appreciable values, when some of the remaining atoms form an effective bridge of the neighboring, chemically bonded atoms, which links the two AIMs in question.
Therefore, the requirement for a nonvanishing density-matrix element coupling the two AO in the molecule, which in MO theory reflects their directly bonding status, is not essential for an existence of their effective through-bridge interaction, provided the two AO chemically couple to the bonded chain of orbitals connecting them.
This novel perspective on the entropic origins of chemical bonds is very much in the spirit of the Eugene Wigner’s observation, often quoted by Walter Kohn, that the understanding in science requires insights from several different points of view. The IT probes of molecular systems and chemical reactions generate such an additional perspective on the genesis of chemical bonds and the course of elementary reaction mechanisms. It complements the familiar MO interpretations of quantum chemistry and gives rise to a deeper understanding of these complex phenomena.
References
Fisher RA (1925) Proc Cambridge Philos Soc 22:700
Frieden BR (2004) Physics from the Fisher information—a unification, 2nd edn. Cambridge University, Cambridge
Shannon CE (1948) Bell System Tech J 27:379, 623
Shannon CE, Weaver W (1949) The mathematical theory of communication. University of Illinois, Urbana
Kullback S, Leibler RA (1951) Ann Math Stat 22: 79
Kullback S (1959) Information theory and statistics. Wiley, New York
Abramson N (1963) Information theory and coding. McGraw-Hill, New York
Pfeifer PE (1978) Concepts of probability theory, 2nd edn. Dover, New York
Nalewajski RF (2006) Information theory of molecular systems. Elsevier, Amsterdam
Nalewajski RF (2010) Information origins of the chemical bond. Nova, New York
Nalewajski RF (2012) Perspectives in electronic structure theory. Springer, Heidelberg (in press)
Nalewajski RF (2011) In: Hong WI (ed) Mathematical chemistry. Nova, New York, p 247
Nalewajski RF (2011) In: Putz MV (ed) Chemical information and computation challenges in 21st century. Nova Science Publishers, New York (in press)
Nalewajski RF, deSilva P, Mrozek J (2011) In: Roy AK (ed) Theoretical and computational developments in modern density functional theory. Nova Science Publishers, New York (in press)
Nalewajski RF, Świtka E, Michalak A (2002) Int J Quantum Chem 87:198
Nalewajski RF, Broniatowska E (2003) J Phys Chem A 107:6270
Nalewajski RF, Parr RG (2000) Proc Natl Acad Sci USA 97:8879
Nalewajski RF, Loska R (2001) Theoret Chem Acc 105:374
Nalewajski RF (2002) Phys Chem Chem Phys 4:1710
Nalewajski RF (2003) Chem Phys Lett 372:28
Parr RG, Ayers PW, Nalewajski RF (2005) J Phys Chem A 109:3957
Nalewajski RF (2003) Adv Quant Chem 43:119
Nalewajski RF, Broniatowska E (2007) Theoret Chem Acc 117:7
Nalewajski RF, Parr RG (2001) J Phys Chem A 105:7391
Hirshfeld FL (1977) Theoret Chim Acta (Berl) 44:129
Nalewajski RF (2008) Int J Quantum Chem 108:2230
Nalewajski RF (2010) J Math Chem 47:667
Nalewajski RF, deSilva P, Mrozek J (2010) J Mol Struct Theochem 954:57
Nalewajski RF, Köster AM, Escalante S (2005) J Phys Chem A 109:10038
Becke AD, Edgecombe KE (1990) J Chem Phys 92:5397
Silvi B, Savin A (1994) Nature 371:683
Savin A, Nesper R, Wengert S, Fässler TF (1997) Angew Chem Int Ed Engl 36:1808
Nalewajski RF (2000) J Phys Chem A 104:11940
Nalewajski RF, Jug K (2002) In: Sen KD (ed) Reviews of modern quantum chemistry: a celebration of the contributions of Robert G. Parr, Vol. I. World Scientific, Singapore, p. 148
Nalewajski RF (2004) Struct Chem 15:391
Nalewajski RF (2004) Mol Phys 102:531, 547
Nalewajski RF (2005) Theoret Chem Acc 114:4
Nalewajski RF (2008) J Math Chem 43:265
Nalewajski RF (2009) Int J Quantum Chem 109:425
Nalewajski RF (2005) Mol Phys 103:451
Nalewajski RF (2006) Mol Phys 104:365, 493, 1977, 2533
Nalewajski RF (2005) J Math Chem 38:43
Nalewajski RF (2008) J Math Chem 44:414
Nalewajski RF (2009) J Math Chem 45:607
Nalewajski RF (2009) J Math Chem 45:709
Nalewajski RF (2009) J Math Chem 45:776
Nalewajski RF (2009) J Math Chem 45:1041
Nalewajski RF (2010) J Math Chem 47:709
Nalewajski RF (2008) J Math Chem 43:780
Nalewajski RF (2007) J Phys Chem A 111:4855
Nalewajski RF (2006) Mol Phys 104:3339
Nalewajski RF (2003) J Phys Chem A 107:3792
Nalewajski RF (2006) Mol Phys 104:255
Nalewajski RF (2004) Ann Phys (Leipzig) 13:201
Nalewajski RF (2009) Adv Quant Chem 56:217
Nalewajski RF, Szczepanik D, Mrozek J (2011) Adv Quant Chem 61:1
Nalewajski RF (2010) J Math Chem 47:692, 709, 808
Nalewajski RF (2011) 49:592
Nalewajski RF (2011) J Math Chem 49:371, 546, 806
Nalewajski RF, Gurdek P (2011) J Math Chem 49:1226
Nalewajski RF (2012) Int J Quantum Chem 112:2355
Nalewajski RF, Gurdek P (2011) Struct Chem (M. Witko issue) 23:1383
Nalewajski RF (2011) J Math Chem 49:2308
vonWeizsäcker CF (1935) Z Phys 96:431
Kohn W, Sham LJ (1965) Phys Rev 140A:1133
Hohenberg P, Kohn W (1964) Phys Rev 136B:864
Nalewajski RF, Świtka E (2002) Phys Chem Chem Phys 4:4952
Capitani JF, Nalewajski RF, Parr RG (1982) J Chem Phys 76:568
Cortona P (1991) Phys Rev B 44:8454
Wesołowski T, Warshel A (1993) J Phys Chem 97:8050
Wesołowski T (2004) J Am Chem Soc 126:11444
Wesołowski T (2004) Chimia 58:311
Gordon RG, Kim YS (1972) J Chem Phys 56:3122
Feinberg MJ, Ruedenberg K, Mehler EL (1970) Adv Quant Chem 5:28
Goddard WA, Wilson CW (1972) Theoret Chim Acta (Berl) 26:195, 211
Feinberg MJ, Ruedenberg K (1971) J Chem Phys 54:1495
Feinberg MJ, Ruedenberg K (1971) J Chem Phys 55:5804
Nalewajski RF (2009) Int J Quantum Chem 109:245
Dirac PAM (1958) The principles of quantum mechanics, 4th edn. Clarendon, Oxford
Wiberg KA (1968) Tetrahedron 24:1083
Nalewajski RF, Köster AM, Jug K (1993) Theoret Chim Acta (Berl) 85:463
Nalewajski RF, Mrozek J (1994) Int J Quantum Chem 51:187
Nalewajski RF, Mrozek J, Mazur G (1996) Can J Chem 100:1121
Nalewajski RF, Mrozek J, Michalak A (1997) Int J Quantum Chem 61:589
Nalewajski RF (2004) Chem Phys Lett 386:265
Gopinathan MS, Jug K (1983) Theor Chim Acta (Berl) 63:497
Nalewajski RF, Formosinho SJ, Varandas AJC, Mrozek J (1994) Int J Quantum Chem 52:1153
Mrozek J, Nalewajski RF, Michalak A (1998) Polish J Chem 72:1779
Jug K, Gopinathan MS (1990) In: Maksić ZB (ed) Theoretical models of chemical bonding, vol II. Springer, Heidelberg, p 77
Mayer I (1983) Chem Phys Lett 97:270
Nalewajski RF (2012) Int J. Quantum Chem (in press)
Esquivel RO, Flores-Gallegos N, Iuga C, Carrera E, Angulo JC, Antolín J (2009) Theor Chem Acc 124:445
López-Rosa S (2010) Information-theoretic measures of atomic and molecular systems, PhD Thesis, University of Granada
López-Rosa S, Esquivel RO, Angulo JC, Antolín J, Dehesa JS, Flores-Gallegos N (2010) J Chem Theory Comput 6:145
Luken WL, Beratan DN (1984) Theoret Chim Acta (Berl) 61:265
Luken WL, Culberson JC (1984) Theoret Chim Acta (Berl) 66:279
Putz MV (2005) Int J Quantum Chem 105:1
Shaik S, Danovich D, Wu W, Hiberty PC (2009) Nat Chem 1:443
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Nalewajski, R.F. (2012). Information Theory Insights into Molecular Electronic Structure and Reactivity. In: Putz, M., Mingos, D. (eds) Applications of Density Functional Theory to Chemical Reactivity. Structure and Bonding, vol 149. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-32753-7_2
Download citation
DOI: https://doi.org/10.1007/978-3-642-32753-7_2
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-32752-0
Online ISBN: 978-3-642-32753-7
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)