
Abstract
Alternative splicing is one of the most powerful mechanisms for generating functionally distinct products from a single genetic loci and for fine-tuning gene activities at the post-transcriptional level. Alternative splicing plays important roles in regulating genes critical for cell death. These cell death genes encode death ligands, cell surface death receptors, intracellular death regulators, signal transduction molecules, and death executor enzymes such as caspases and nucleases. Alternative splicing of these genes often leads to the formation of functionally different products, some of which have antagonistic effects that are either cell death-promoting or cell death-preventing. Differential alternative splicing can affect expression, subcellular distribution, and functional activities of the gene products. Molecular defects in splicing regulation of cell death genes have been associated with cancer development and resistance to treatment. Studies using molecular, biochemical, and systems-based approaches have begun to reveal mechanisms underlying the regulation of alternative splicing of cell death genes. Systematic studies have begun to uncover the multi-level interconnected networks that regulate alternative splicing. A global picture of the complex mechanisms that regulate cell death genes at the pre-mRNA splicing level has thus begun to emerge.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
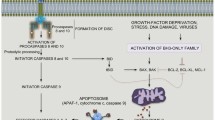
Although the major mechanism of programmed cell death (PCD) has been characterized by activation of caspases and packaging of cells into apoptotic bodies, several types of cell death have been described with distinct features: apoptosis, necrosis, pyroptosis, and autophagic cell death [15, 47]. Prominent morphological features of apoptosis include cell shrinkage, nuclear condensation, nuclear fragmentation, chromatin condensation, membrane blebbing, retention of intact organelles, vacuole formation, and DNA fragmentation [83, 189]. Necrosis, now recognized as an alternate form of programmed cell death, often involves prominent immune responses. Recent studies suggest that by initiating inflammatory and reparative responses, necrosis may also serve to maintain tissue homeostasis and organismal integrity (reviewed in [178, 203]). In addition, autophagy plays important roles in cell death signaling in both promotional and inhibitory manners (for reviews: [55, 176]). Cell death can also be induced by microbial and viral infection. Infection induced cell death, termed "pyroptosis", involves an inflammatory response with neighboring cells [15].
A common feature of cell death is the activation of proteases followed by DNA degradation. Similar to the activation of caspases during apoptosis, other types of PCD are triggered by specific proteases. Cells undergoing necrotic death activate cathepsins and calpains, whereas pyroptotic cells activate caspase-1 and caspase-7. Furthermore, many downstream DNases have been discovered, such as Caspase-activated DNase (CAD), EndoG, LEI/L-DNase II, and Granzyme A-activated DNase (GAAD). PCD is a complex phenomenon that engages more than 400 proteins with diverse functions ranging from PCD receptors to the execution proteins.
Regulated cell death processes are critical for the development and homeostasis of multi-cellular organisms. Disruption of cell death regulation plays an important role in the pathogenesis of a wide spectrum of human diseases. Cell death regulation occurs at multiple levels and involves many different PCD associated proteins (for recent reviews, [15, 33, 60, 176]). One important step of this regulation occurs during pre-mRNA splicing because many cell death genes are expressed as functionally distinct or even antagonistic isoforms as a result of alternative splicing. Genes implicated in autophagy have been reported to undergo alternative splicing, including autophagosome markers (reviewed in [188]), although the functional significance remains to be investigated.
In this chapter, we review recent molecular and biochemical studies on alternative splicing of genes involved in regulating PCD and illustrate the current PCD paradigms. These genes range from extracellular signals and death receptors to the intracellular components of the PCD machinery. It is possible that alternative splicing profoundly contributes to the life-or-death decision of cells. Because of the large number of different PCD genes that undergo alternative splicing, it is difficult to cover all relevant aspects of these genes within this short chapter. We will describe selected examples to illustrate the extreme complexity of alternative splicing in cell death regulation and its impact on cancer pathogenesis and treatment.
2 Pre-mRNA Splicing and Alternative Splicing Regulation
In mammals, nascent transcripts (messenger RNA precursor, or pre-mRNAs) produced from the vast majority of protein-encoding genes contain intervening sequences or introns, that must be accurately removed to form functional messenger RNAs. In addition, it is now estimated that more than 90 % of human protein-coding genes undergo alternative splicing to generate distinct transcripts [114, 142], providing a robust post-transcriptional mechanism for increasing genetic diversity. Mammalian gene regulation is a highly complex process, involving multiple interconnected networks of regulation [184]. Disruption of such a regulated process leads to a wide range of diseases including cancer [36, 165].
Alternative splicing is regulated by intricate interactions between cis-regulatory elements and splicing machinery. There are four types of cis-acting regulatory sequence elements: splicing enhancers (SE) in introns (ISE) or exons (ESE) and splicing silencers (SS) in introns (ISS) or exons (ESS) [177]. Splicing enhancers recruit splicing activators to the spliceosome to stimulate splicing, whereas splicing silencers block the interaction between the spliceosome and the corresponding splice site. Multiple families of splicing factors have been identified with characteristic sequences such as the RNA-recognition motifs (RRM) and serine/arginine-rich (SR) proteins among others [124].
3 Alternative Splicing Regulation of Cell Death Genes
Cell death is a tightly regulated process. The components of the cell death machinery that control and execute this process are under strict regulation as well. At the post-transcriptional level, alternative splicing is one of the most powerful and diverse mechanisms that regulate the expression and function of many PCD genes. A large number of players function together in a carefully orchestrated manner to control cell death.
3.1 Alternative Splicing Isoforms of Genes Encoding Caspases and Other PCD-Related Proteases
Caspases are a family of cysteine proteases that play important roles in cell death regulation and cytokine secretion. Activation of pro-apoptotic caspase (caspase 2, 3, 6, 7, 8, 9, and 10) is a central pathway for different death signals (for reviews; [104, 156]). Each caspase has different sets of isoforms as a result of alternative splicing ([74, 75, 76, 95, 128], for reviews see [185, 190]). Many caspase genes produce pro- and anti-PCD isoforms as a result of alternative splicing, further support that alternative splicing is a common mechanism for regulating caspase gene expression [74, 85].
Caspase-1 is also known as IL-1β converting enzyme (ICE) that processes pro-inflammatory cytokine precursors pro-IL-1β and pro-IL-18. Recent studies show that caspase-1 also activates pro-apoptotic caspase-7 by processing pro-caspase-7 ([3, 105], for review [55]). Caspase-1 is activated by various cell death stimuli, such as bacterial toxin, LPS, microbial/viral nucleic acids, viral infection, reactive oxygen species (ROS), and asbestos. Five splicing isoforms of caspase-1 (caspase 1α − 1ε) have been reported. Among these, caspase-1δ and -1ε display an anti-apoptotic effect [6, 52].
Alternative splicing of caspase-2 generates five splicing isoforms: caspase-2L, caspase-2S, caspase-2β, caspase-2L-Pro, and caspase-2S-Pro. Caspase-2β, a possible negative regulator of caspase-2 activity, contains a deletion downstream of the first potential aspartic proteolytic cleavage site between the large and small subunits [5]. In addition, caspase-2S has anti-apoptotic activity [14, 46]. Interestingly, caspase-2S is abundant in the developing brain, suggesting that caspase-2 alternative splicing is developmentally regulated [14].
In caspase-8, retention of intron 8 results in the formation of caspase-8L mRNA and the caspase-8L protein that lacks the C-terminal half of the proteolytic domain. Patients with systemic lupus erythematosus (SLE) also display alternative splicing of caspase-8 [76]. Overexpression of caspase-8L inhibits Fas-mediated apoptosis, suggesting dominant negative effects of caspase-8L [75].
In addition to caspase genes, other protease genes involved in cell death also undergo alternative splicing. Calpains are Ca2+-regulated proteases important for apoptotic, necrotic, and pyroptotic cell death. M- and μ-calpain are expressed ubiquitously and share the regulatory small subunit called calpain 4. Cells lacking calpain 4 exhibit resistance against certain PCD stimuli. In addition, an intrinsic calpain inhibitor, calpastatin, has at least eight splicing isoforms, some of which exhibit tissue-specific expression [66]. The role of alternative splicing in regulating calpains and calpastatin remains to be further characterized.
Cathepsins are a family of lysosomal proteases participating in the hydrolysis of macro-molecules in lysosomes. Cathepsins are released into the cytosol by various cell death signals and activate downstream PCD signaling. For example, Bid, a pro-apoptotic Bcl-2 family member, is cleaved by cathepsins released from lysosomes and translocates from the cytosol to the mitochondria, inducing mitochondrial death. Cathepsins activate other PCD associated DNases. Cathepsin leakage from lysosomes is observed during necrosis and pyroptosis as well [192].
The cathepsin B gene generates at least 3 splicing isoforms. Exon 2, which is an alternative cassette exon located at the 5′ untranslated region (UTR) of cathepsin B full-length mRNA, down-regulates cathepsin B expression [203]. On the other hand, alternative splicing also modulates the subcellular localization of cathepsin B. Exclusion of exons 2 and 3 results in a protein with a different N-terminus that serves as a mitochondrial targeting sequence [135]. This isoform contains a catalytic domain but does not have protease activity possibly because of misfolding. Overexpression of this isoform changes the mitochondrial morphology and induces cell death. Therefore, alternative splicing may trigger cell death by changing the cellular localization of cathepsin B.
3.2 Bcl-2 Superfamily
The Bcl-2 gene family of proteins mediates a complex network of interactions among different pro- and anti-apoptotic proteins as well as downstream molecules [99, 112]. They play important roles in controlling mitochondrial permeability, cytochrome C release, and caspase activation. The human Bcl-2 family has multiple members including pro- and anti-apoptotic genes. Prototypical anti-apoptotic members including Bcl-2, Bcl-w, Bfl-1/A1, Mcl-1, and Boo/DIVA usually contain four Bcl2-homology (BH) (BH1-4) domains. Pro-apoptotic members, on the other hand, contain two or more BH domains (BH1-3) (Bax, Bak, Bok/Mtd, Bcl-G, Boo/Diva, and Bfk) or only the BH3 domain (BH3-only protein family). The list of BH3-only protein family members has continuously grown (Bad, Bid, Bik, Bim, Blk, Hrk, Noxa, BNIP1, BNIP3, BNIP3L, Bmf, and Beclin-1). Furthermore, many proteins containing BH3-like domains have been reported (APO L1, APO L6, BRCC2, HER2, HER4, MAP-1, MULE, SPHK2, Rad9, and TGM2). These proteins have death inducing activity as well as the ability to interact with the anti-apoptotic Bcl-2 family as well as BH3-only proteins. Beclin-1, for example, is a critical effecter of autophagy. Bad and Bik can induce autophagic cell death.
The role of alternative splicing in the Bcl-2 family gene regulation has not yet been fully elucidated. Several genes of this superfamily including Bcl-x, Bak, Mcl-1, and Bid encode for anti-apoptotic (Bcl-xL, N-Bak, Mcl-1L, BidS) and pro-apoptotic (Bcl-xS, Bak, Mcl-1S, Bid) isoforms as a result of alternative splicing [10, 18, 154]. Bid is a prominent member of the BH3-only family that bridges the death receptor signaling pathway to the mitochondrial signaling pathway, which is mediated by other Bcl-2 family member proteins. Alternative splicing of Bid pre-mRNA generates isoforms lacking proteolytic cleavage sites (BidES, lacking all cleavage sites; BidS, lacking the granzyme B cleavage site) [110, 117]. In addition, some of Bcl-2 family proteins have a transmembrane domain that can be removed by alternative splicing, leading to differential subcellular distribution. Therefore, alternative splicing regulation of Bcl-2-related proteins has a considerable impact on cell death signaling pathway.
3.3 Death Ligands and Death Receptors
Death ligands and their receptors have been studied since the discovery of FAS, a member of the TNF (Tumor Necrosis Factor) superfamily. TNF-α, TNFSF1/Lynphotoxin-α, TNFSF3/Lynphotoxin-α, TRAIL, VEGI, TWEAK, and LIGHT proteins mediate apoptosis. TRAIL (TNF-related apoptosis-inducing ligand) has three splicing isoforms: TRAIL-α, β, and γ. TRAIL-α, the full-length form, contains five exons and promotes apoptosis. In contrast, apoptotic activity is not observed when exon 3 (TRAIL-β) or exons 2 and 3 (TRAIL-γ) are skipped [100]. In addition to TRAIL, splicing isoforms of TNFSF1 and VEGI are reported [26, 170]. In naïve T cell lymphocytes, TNF-α transcripts do not undergo splicing and are instead stored as pre-mRNA. TNF-α pre-mRNA splicing is initiated to produce TNF-α protein following the engagement of T cell receptors [195].
In addition to the TNF superfamily, other ligands, such as growth factors, neurotrophic factors, and cytokines participate in cell death regulation. Several neurotrophic factor genes of the TGF-β superfamily contain introns in their prodomains, including persephin, neurturin (NTN), and glial cell line–derived neurotrophic factor (GDNF). The GDNF gene produces a short isoform by removing 78 bases at the end of exon 1. The short isoform GDNF∆78 accumulates in the Golgi apparatus and secretion of GDNF∆78 is repressed [182]. It is conceivable that activation of this splicing event occurs in response to certain stimuli, providing an efficient post-transcriptional mechanism controlling the production of functional mRNAs for trophic factors such as NTN and GDNF.
Alternative splicing of transmembrane receptors may lead to the formation and secretion of the soluble receptors of cell death related ligands. In fact, many transmembrane receptors have splicing isoforms that encode soluble proteins capable of modifying the downstream ligand effects in antagonistic or agonistic manners [108]. For example, FAS has multiple alternative splicing isoforms lacking the transmembrane domain [25, 145]. Alternative splicing of Fas exon 6 produces a soluble (lacking exon 6) or a membrane-bound (containing exon 6) protein product. Soluble FAS proteins suppress PCD induced by FAS ligand and participate in T cell regulation, immune diseases, and cancer development. A membrane-bound form of the FAS receptor found in thymocytes lacks exon 7 and is missing the death domain (DD) due to the frame shift caused by exon 6 retention. This FAS alternative splicing isoform acts as a FAS decoy receptor (FDR) because it blocks PCD induced by FAS ligands as well as soluble FAS.
Other death receptors have multiple splicing variants: tumor necrosis factor receptor 1 (TNF-R1), lymphocyte-associated receptor of death (LARD, also named as DR3, Apo-3 or TRAMP), death receptor-4 (DR4), and TRAIL receptor inducer of cell killing-2 (TRICK-2) [101]. Additionally, many cytokine receptor genes are alternatively spliced to generate functionally diverse receptor isoforms. For example, alternative splicing generates truncated interleukin 7 (IL-7) receptor isoforms associated with leukemia [180]. Therefore, alternative splicing modulates the expression and function of genes encoding both death ligands and death receptors.
PCD signals from extracellular ligands are often transduced by proteins containing death domains (DDs) and death effector domains (DEDs). DD and DED are homophilic protein interaction motifs that allow association between different proteins containing DDs or DEDs. For example, FAS recruits FADD through an interaction between their respective DDs. The DED of FADD in turn recruits caspase 8 via interaction between DEDs of the FADD and caspase 8. In addition, some DD and/or DED containing proteins modulate cell death signaling. Two DEDs are located at N terminus of caspase 8 and 10. Viral proteins named v-FLIPs have been found in equine herpesvirus-2, bovine herpesvirus-4, herpesvirus saimili human herpesvirus-8, and human poxvirus. These v-FLIP proteins show homology to the N-terminus of caspase 8/10 but lack the cysteine protease domain. Viral-FLIP proteins bind to FADD and/or caspase 8/10 and interfere with their recruitment to Fas-FADD receptor complex. Cellular FLIP (c-FLIP, also known as Casper/I-FLICE/FLAM/CASH/CLARP/MRIT/usurpin) was discovered based on viral protein sequence information. The c-FLIP gene generates three splicing isoforms—FLIPL, FLIPS, and FLIPR by alternative splicing [44, 65]. The sequence of FLIPL protein is similar to caspase 8, including the two aspartic acid residues at the auto-cleavage site, although it lacks caspase enzymatic activity. FLIPS and FLIPR lack a cysteine protease-like region, similar to v-FLIP. FLIPL is expressed in many tissues, whereas FLIPS is detectable mainly in T lymphocytes. FLIPS expression in T cells activated by CD3 and CD28 has been shown to correlate with the cellular resistance against FAS-mediated apoptosis [94]. On the other hand, IL-2 upregulates FAS expression and represses the transcription and expression of FLIPS in T cells, sensitizing T cells to apoptosis [152]. These results suggest important roles of FLIPS in activation-induced cell death (AICD) and self-tolerance. A similar expression pattern of FLIPR and FLIPS in CD3 and CD28-activated T cells has been reported [65]. Although the anti-apoptotic effects of FLIPL and FLIPS are reported, FLIPL also stimulates apoptosis under certain conditions. FLIPL can activate caspase 8 after the formation of a heterodimer on FADD and death receptor complexes. It has been proposed that activation of caspase 8 by FLIPL results in partial autocleavage of caspase 8 into the p43/41 and p12 subunits. This mechanism tethers caspase 8 on the death-induced signaling complex (DISC) by FADD and processes receptor-interacting kinase (RIP) within the FAS-signaling complex [125]. FLIPL may generate proliferation signals from DISC by processing DISC-proximal substrates, as a result of activation of ERK and NFkB by FLIPL in T cells [91].
The caspase activation and recruitment domain/caspase recruitment domain (CARD) is engaged in homophilic interactions with CARD containing caspases. TUCAN/CARD8/CARDINAL, cloned as an anti-apoptotic CARD containing protein, is overexpressed in colon cancer [147]. TUCAN can interact with caspase-9 via the CARD domains and interfere with the binding between Apaf-α and caspase-9. Interestingly, TUCAN also induces cell death when overexpressed in MCF7 and VERO cells. A TUCAN splicing isoform, TUCAN-54, has been recently reported as a cancer-specific protein expressed in gastric, colon, and breast cancer tissues. TUCAN-54 exhibits anti-apoptotic activity by inhibiting caspase-9 and caspase-8 activation. TUCAN along with caspase-9 and DRAL (a LIM domain protein) are recruited by the Shh receptor, Patched, to enhance cell death [127]. Therefore, TUCAN alternative splicing regulation may be important both in normal development and in cancer pathogenesis.
MAPK-activating death domain-containing protein (MADD) is a Rab3 GTP/GDP exchange protein containing a DD motif. There are six reported isoforms: IG20pa, MADD, IG20-SV2, DENN-SV, KIAA0358, and IG20-SV4 [111]. IG20pa render cells more susceptible to PCD by TNF-α, TRAIL, γ-irradiation, and cancer drugs; however, DENN-SV has an opposite effect, promoting cancer cell suvival [7, 48, 49, 102, 136, 137].
3.4 Intrinsic Cell Death Signals
The death domain mediates not only receptor-mediated cell death signals, but also intrinsic cell death signals. PIDD (P53-Induced Protein with Death Domain), which is induced by p53, interacts with caspase 2 by RAIDD bridging, and activates caspase-2, forming a protein complex known as PIDDosome. Binding between PIDD and RAIDD is mediated by a homophilic interaction of their DDs. Caspase 2 is recruited via an interaction between the CARDs of caspase 2 and RAIDD. Among the five reported splicing isoforms of PIDD, isoform 2 (also known as LRDD) counteracts the pro-apoptotic activity of full-length PIDD isoform 1 [35]. Similarly, Apaf-1 recruits caspase 9 with cytochrome C and dATP and forms apoptosome. Apaf-1 gene also generates pro- and anti- apoptotic splicing isoforms [140].
Similar to PIDDosome, NALP1 (NACHT leucine-rich-repeat protein 1) interacts with caspase 1 via ASC (Apoptosis-associated speck-like protein containing a CARD). The PYD motifs of NALP1 and ASC and the CARD motifs of ASC and caspase 1 interact together to comprise inflammasomes. The NALP family is composed of at least 14 genes (NALP1-14). NALP proteins consist of PYD, NACHT domain, and leucine-rich repeats (LRRs). The NACHT domain has a predicted P-loop NTPase sequence and is extracted across different proteins: NAIP (Neural Apoptosis Inhibitory Peptide), CIITA (class II transactivator), HET-E (incompatibility locus protein from Podospora anserine), and TP1 (telomerase-associated protein). Interestingly, NACHT domain shares homology with the dATP binding domain of Apaf-1. The LRRs of NALPs are thought to be able to sense various microbial molecules in intracellular space to form inflammasomes. Many members of NALP family have multiple alternative splicing isoforms (JYW, unpublished data).
PYD (pyrin domain) containing proteins and caspase 1 are involved in processing pro-IL-1/18 and pro-apoptotic caspase 7. PYD is a homophilic adapter domain as well as for DD and DED. Familial Mediterranean fever (MEFV) encodes PYD containing proteins interacting with ASC. The human MEFV gene produces multiple isoforms including full-length (MEFV-fl) and MEFV-d2 [42]. MEFV-fl encodes a cytoplasmic protein, whereas MEFV-d2 protein is concentrated in the nucleus [144]. Multiple components of inflammasome, including TUCAN, have distinct alternative splicing isoforms [2]. Alternative splicing may regulate the pyroptotic cell death signaling.
3.5 Inhibitor of Apoptosis Proteins (IAPs)
IAPs, first identified in baculovirus, contain 1–3 BIR motifs and one RING domain. BIR domains bind caspases and inhibit protease activity and RING domains recruit E2 ubiquitin conjugating enzyme and transfer ubiquitins onto itself or binding partners. Survivin, a member of the IAP family, contains a BIR domain but lacks a RING domain. Interestingly, Survivin still retains anti-apoptotic activity (for reviews, [141]). The Survivin gene generates several splicing isoforms. Among them, Survivin 2α and 2β uniquely display pro-apoptotic activity. Survivin 2β splicing is upregulated by Wilms Tumor 1 associated protein (WTAP) [169]. The alternative splicing pattern of Survivin pre-mRNA changes dynamically in cancer. Survivin alternative splicing has been considered as a diagnostic marker and therapeutic target (reviewed in [158]).
Two different splicing isoforms of livin, another member of the IAP family, livin α and β, have different tissue expression patterns and show distinct properties in inhibiting apoptosis induced by different signals [9].
3.6 Cell Death-Related DNases and Their Regulators
Caspase-activated DNases (CAD) are critical nucleases in PCD. Other notable DNAses are Endo G, LEI/L-DNase II, and GAAD. ICAD/DFF45 is a chaperonin for Caspase-activated DNase (CAD/DFF40) and inhibits the CAD/DFF40 activity. ICAD/DFF45 is one of substrates for capsases. Cleavage of ICAD/DFF45 by caspases releases CAD/DFF40 from the ICAD/DFF45-CAD/DFF40 complex and activates CAD/DFF40 DNase activity. There are at least two splicing isoforms encoded by the ICAD/DFF45 gene. The short isoform is generated by the retention of intron 5 which contains an in-frame stop codon. Although ICAD/DFF45 (ICAD-L) is necessary for CAD/DFF40 peptide to be folded properly, the short isoform ICAD-S/DFF35 does not have the ability to function as a chaperonin. However, ICAD-S binds to CAD/DFF40 more strongly than ICAD/DFF45 and can inhibit the DNase activity of CAD/DFF40. ICAD/DFF45 contains a nuclear localization signal and is expressed in nuclei whereas ICAD-S/DFF35 is also distributed to the cytosol. Aberrant splicing products of CAD/DFF40 have been detected in human hepatoma cells, but biological functions of those isoforms have not yet been characterized [78].
Endonuclease G (Endo G) is another DNase that is released from the mitochondria during PCD. Because the yeast orthologue of Endo G participates in caspase independent PCD, Endo G is thought to contribute to caspase independent PCD in mammalian cells [23, 34]. LEI/L-DNase II and GAAD also contribute to caspase independent PCD. There are many predicted alternative splicing isoforms for Endo G, LEI/L-DNase II, and GAAD isoforms; however, their biological roles and splicing regulation are still unclear.
3.7 Mitochondrial Cell Death Proteins
Since the discovery of the role of cytochrome C in apoptosis, many other mitochondrial factors have been reported to regulate cell death, such as Smac, Omi, and AIF.
Initially identified as an inhibitor of IAPs, Smac/DIABLO has a number of splicing isoforms including Smac-β (Smac S), γ, δ, and Smac 3 [56, 172]. As a result of alternative splicing, Smac-β lacks the mitochondrial targeting sequence at its N terminus and displays a distinct distribution in cells, rather than the mitochondrial localization. Although Smac-β does not interact with IAPs, Smac-β is pro-apoptotic. Smac 3 is missing exon 4, but still contains the mitochondria targeting sequence and IAPs binding domain. Smac 3 disrupts caspase 9 binding to XIAP, promotes caspase 3 activation, and accelerates the auto-ubiquitination of XIAP, whereas the full-length Smac does not accelerate XIAP auto-ubiquitination. Smac 3 localizes to the mitochondria and is released into the cytosol following apoptosis signaling [56].
AIF induces cell death after being released from mitochondria. However, AIF induced cell death does not show oligonucleosome fragmentation, a hallmark of caspase-dependent cell death induced by CAD activation. In fact, cathepsins and calpains can release AIF from the mitochondria to trigger necrotic cell death [37]. Human AIF gene generates multiple alternative splicing isoforms: AIF, AIF-exB, AIFsh, AIFsh2, and AIFsh3 [38, 39]. AIFsh retains pro-apoptotic activity and is expressed in the cytosol due to a missing mitochondria targeting sequence caused by alternative splicing. Therefore, alternative splicing may induce cell death by producing cytosolic AIFsh.
3.8 Autophagy, Cell Death, and Alternative Splicing of Autophagy Regulatory Genes
Autophagy is an essential cellular process mediating the degradation of damaged or degenerated cellular materials. During this process, an isolated membrane sequesters unwanted macromolecules and organelles, such as aggregation-prone proteins and malfunctioning mitochondria. The formed double-membraned vacuoles are called autophagosomes, which in turn fuse with lysosomes forming autolysosomes to degrade their contents [96, 107]. The autophagy pathway is highly conserved from yeast to humans, playing important roles in cellular homeostasis [11, 153]. Under physiological conditions, autophagy degrades altered proteins and organelles, eliminating from the cell malfunctioning components and simultaneously recycling molecular components for the regeneration of new organelles. During nutrient deprivation, autophagy plays critical roles in the adaptation of organisms to new environmental conditions, providing nutrients from degraded cellular contents to maintain cellular metabolism [129].
In addition to cell survival, autophagy mediates cell death, although the underlying molecular mechanisms remain to be elucidated. The phenomenon of autophagic cell death (also known as type II programmed cell death) was observed in the 1960s. At the ultrastructural level, autophagic cell death is characterized primarily by the formation of numerous autophagic vacuoles in dying cells [27, 160, 161]. Autophagy genes Atg7 and Beclin1 are required for cell death in certain types of cells [197, 198]. However, the role of autophagy in regulating cell death remains unclear.
The mammalian target of rapamycin (mTOR) is a kinase that plays important roles in cellular metabolism, cell growth, cell proliferation, and autophagy [41, 43, 71, 188]. mTOR is initially inhibited during starvation, which triggers autophagy. A recent study shows that the mTOR signaling can be reactivated by prolonged starvation, which in turn forms an evolutionarily conserved cycle that maintains energetic homeostasis during cellular starvation [199]. Two splicing isoforms of mTOR have been identified: mTORα (the full-length protein) and mTORβ. mTORβ is capable of regulating cell cycle and cell proliferation. Notably, mTORβ may act as a proto-oncogene, because overexpression of mTORβ leads to immortalization of cells and is tumorigenic in nude mice [143]. However, the role of mTORβ in autophagy remains to be investigated.
Microtubule-associated protein light chain 3 (LC3), a mammalian homologue of yeast Atg8, is another essential component of autophagy [88]. LC3-I can be converted to LC3-II and then processed to bind tightly to the autophagosomal membrane. In rats, two alternative splicing isoforms of LC3 are produced, LC3A and LC3B, which generate proteolytic II forms from precursor I forms and colocalize with LC3. In different rat tissues LC3A, LC3B, and LC3 show different expression patterns, suggesting possible regulation of LC3 by alternative splicing [186].
Tumor protein 53-induced nuclear protein 1, TP53INP1, shows sequence similarity to TP53INP2, a protein essential for autophagy in mammalian cells by interacting with VMP1 and recruiting LC3 and/or LC3-related proteins to initiate the autophagosome [139]. Alternative splicing of TP53INP2 appears to be important for cell invasion, although its role in autophagy remains unclear [134].
Many other autophagy regulatory genes have splicing isoforms. For instance, ULK (uncoordinated-51 like kinase) protein is the mammalian counterpart of yeast Atg1, a Ser/Thr protein kinase involved in the initial step of autophagosome formation in collaboration with its regulators, Atg13, Atg17, Atg29, and Atg31 [61, 86]. Human ULK2 has two alternatively spliced transcript variants that differ in the 3′ UTR that may have different mRNA stability. However, the functional difference among the splicing variants is still unknown. Further studies are necessary to understand the role of alternative splicing in regulating autophagy.
4 Alternative Splicing: A Versatile Mechanism for Regulating Expression and Function of Cell Death Genes
Emerging evidence, some of which is summarized above, supports that a wide range of cell death genes undergo alternative splicing that impact their activities in regulating programmed cell death. Many of these PCD genes express alternative splicing isoforms in a tissue- or development stage-specific manner. In naïve T lymphocytes, TNF-α transcripts do not undergo splicing and are stored as pre-mRNA. TNF-α pre-mRNA splicing is only initiated to produce TNF-α protein following the engagement of T cell receptors [194]. In na B and T cells, LARD-1 (lymphocyte-associated receptor of death 1) is expressed as alternative isoforms with very little full-length mRNA [163]. The full-length LARD-1 becomes the predominant product after T cell activation.
Bcl-x pre-mRNA is alternatively spliced to produce two isoforms with opposing functions. The two isoforms are produced by using different 5′ splice sites (ss) in exon 2. The use of the upstream 5′ ss produces a shorter product, Bcl-xS and the downstream site gives rise to Bcl-xL. Bcl-xS is pro-apoptotic while the longer form is anti-apoptotic. Adult neurons predominantly express the Bcl-xL mRNA; whereas immature thymocytes express a relatively high level of Bcl-xS transcript [18]. Adding further to the complexity, several critical PCD regulators have been found to use alternative splicing to generate gene products that have antagonistic activities in cell death. The mechanism by which alternative splicing may regulate function of these PCD genes can be summarized in at least three aspects:
-
regulating subcellular localization of PCD gene products
-
modulating functional activity of PCD gene products
-
influencing stability of mRNA and/or translational control
4.1 Regulation of Subcellular Localization
Many cell death genes, including the death receptor family, Bcl-2 superfamily, and cell death associated proteases, have both membrane associated as well as soluble isoforms (see review [185]). Alternative splicing of these cell death genes produce proteins that contain or lack their transmembrane domains, thus generating gene products with different subcellular localization. These different isoforms may have distinct functions in PCD. For example, several Fas splicing variant mRNAs encode soluble proteins that block Fas-mediated apoptosis induced by the agonistic antibody [25] and by the Fas ligand [145]. Another Fas isoform, FasEX08Del, is generated by exon 8 skipping. This isoform contains a premature termination codon and thus lacks the entire intracytoplasmic death domain. FasEX08Del is only expressed in resistant tumor clones [25]. It is likely that different alternative splicing products of the death receptor may have distinct roles in the propagation of the cell death or survival signals.
Genes from the Bcl-2 superfamily undergo alternative splicing to generate different isoforms with distinct intracellular localization (reviewed in [4]). For example, both membrane-bound and soluble isoforms of Bcl-2 gene members are produced as a result of alternative splicing. As described previously, different splicing isoforms of mitochondrial cell death proteins lacking or containing their mitochondrial targeting sequences may have distinct function in cell death. Finally, alternative splicing may regulate the terminal events in cell death such as chromosome fragmentation and DNA degradation. For example, the nuclear localized ICAD-L and cytoplasmic ICAD-S are generated by alternative splicing which removes the nuclear localization signal in ICAD-L to form ICAD-S [157]. ICAD-L and ICAD-S may have different regulatory activities in cell death.
4.2 Modulating Functional Activities
Interestingly, many PCD associated genes generate splicing isoforms that have antagonistic activities, such as caspase-2L versus caspase-2S [31, 59]. Recent analyses indicate that a majority of the known caspase family members have alternative splicing at or near the regions encoding peptide sequences contributing to their active sites for enzyme activity [74]. Alternative splicing events that generate truncated peptides provide an additional mechanism to quantitatively regulate gene expression. The delicate balance of different pro-apoptotic and anti-apoptotic products of these PCD genes likely plays a critical role in determining cellular susceptibility to death signals. Alternative splicing can regulate each step of cell death induction or execution by generating distinct gene products with different or even antagonistic activities in cell death.
4.3 Altering mRNA Stability or Translational Efficiency
Alternative splicing can generate mRNAs with differential turnover rates or with differential properties in translational control (reviewed in [79]). A large number of alternative splicing events occur at 5′ or 3′ untranslated regions of cell death genes. For example, exon 2 of cathepsin B is an alternative exon and encodes the 5′ UTR regulating the protein expression level [135]. Caspase-2 mRNA undergoes nonsense-mediated decay (NMD) under certain conditions, and its protein expression may be tightly regulated by combination of alternative splicing and NMD [29]. A conserved AU-rich element has been identified in the 3′ UTR of Bcl-2 mRNA. This element interacts with AU-rich binding proteins and is associated with bcl-2-down-regulation during apoptosis [45, 159]. IL-1RI-associated kinase-1 (IRAK1) is a serine-threonine kinase important for IL-1 signaling and has different splicing isoforms. The IRAK1b isoform is kinase inactive and more stable than IRAK1 isoform. It is conceivable that different splicing isoforms of critical cell death genes that contain distinct elements for controlling their mRNA or protein stability could have a significant impact on the expression and function of these cell death genes.
5 Molecular Mechanisms Regulating Alternative Splicing of Cell Death Genes
5.1 Splicing Signals, Splicing Machinery, and Alternative Splicing Regulators
With combined approaches, significant progress has been made in identifying the molecular components of splicing machinery and in understanding the mechanisms that control alternative splicing [17, 24, 73, 123, 132, 133, 166]. Through highly dynamic RNA–RNA, protein-RNA, and protein–protein interactions, components of the splicing machinery assemble onto the pre-mRNA and form the catalytically active spliceosome in which biochemical reactions of splicing take place.
The basic splicing signals include the 5′ splice site, branch site, and polypyrimidine track-AG at the 3′ splice site. These signals are initially recognized by the U1 snRNP, U2 snRNP, and U2 snRNP Auxiliary Factor (U2AF), respectively. In mammals, these basic splicing signals tend to be degenerate and are not sufficient by themselves to confer the specificity required to achieve accurate splice site selection. A number of other factors also contribute to splice site recognition and influence splicing efficiency. These include the distance between two splice sites, exon size, as well as local secondary structures in the pre-mRNAs. In addition, various types of exonic and intronic elements have been identified that modulate the usage of nearby splice sites. For example, exonic or intronic splicing enhancers (ESEs or ISEs) and exonic or intronic splicing silencers (ESEs or ISEs and ESSs or ISSs respectively) in different genes have been described (for reviews see Hertel et al. 2008; [70, 183]). These splicing enhancers or silencers can promote or inhibit the use of either upstream 3′ splice sites or downstream 5′ splice sites. Many splicing enhancer elements function by interacting with different members or combinations of SR proteins and hnRNP proteins. A general theme has begun to emerge that the alternative splicing events of a given gene are regulated by a number of different splicing activators and repressors. In most cases, precise molecular mechanisms underlying the splicing inhibition remain to be elucidated.
The specific recognition of splice sites and proper association between authentic 5′ and 3′ splice sites is the central issue for pre-mRNA splicing and alternative splicing regulation. Spliceosomal snRNPs including U1 (or U11), U2 (or U12), U4/U6 (or U4atac/U6atac), and U5 play important roles in splice site recognition and association. In addition to the snRNPs, several families of accessory proteins are also important in regulating alternative splicing. These factors include heteronuclear ribonucleoproteins family (hnRNP proteins), proteins containing serine-arginine-rich sequences (SR proteins) and other RNA-binding proteins ([16, 187]; see Chap. 3). In many cases, SR proteins function as splicing activators by binding enhancer sequences, whereas hnRNP proteins often function as splicing respressors by binding splicing silencer sequences [113]. Other splicing regulators include KH-domain-containing proteins, CUGBP proteins, and proteins containing other sequence motifs (for example, helicase, RGG, zinc finger) [87]. There are also a number of proteins that can act as either splicing activators or splicing repressors depending on the splicing substrates and their binding sites [186, 187].
Aberrant pre-mRNA splicing has been implicated in human diseases associated with either excessive or insufficient cell death, although underlying molecular mechanisms remain to be elucidated (for reviews, [17, 89, 123, 149, 168, 183].
5.2 Mechanisms Underlying Alternative Splicing Regulation of PCD Genes
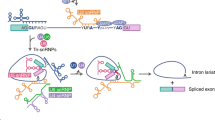
In recent years, a number of cell death genes have been characterized in detail for mechanisms underlying their alternative splicing regulation. Only a few examples are shown here, including FAS, Bcl-x, and caspase-2. As discussed before, alternative splicing of FAS exon 6 leads to the formation of the membrane-bound full-length FAS-L isoform and exon 6-skipped soluble FAS-s isoform. Two related splicing regulators TIA1 and TIAR stimulate exon 6 inclusion by binding to a U-rich sequence downstream of the 5′ splice site of exon 6 and promoting U1 snRNP interaction with this 5′ splice site [53, 54]. A protein kinase, Fas-activated serine/threonine kinase (FAST K) synergizes with TIA1 and TIAR to enhance FAS exon 6 inclusion [83]. On the other hand, Polypyrimidine Tract Binding protein (PTB), promotes exon 6 skipping by binding to an exonic splicing silencer [82]. RNA binding protein SPF45, which has a U2AF homology motif (UHM), interacts with the SF3b155 ULM (UHM-ligand motif) domain to enhance inclusion of Fas exon 6. This binding plays a critical role in FAS exon 6 inclusion [30]. During the early stages of apoptosis, U2AF65 is cleaved and the N-terminal fragment of U2AF65 has a dominant negative effect on FAS exon 6 splicing [81] thus leading to formation of the Fas-s isoform. Hu antigen R (HuR) binds to the ESS on FAS pre-mRNA and inhibits the association of U2AF65 with the 3′ss [84]. RBM5 excludes exon 6 of FAS pre-mRNA. Interestingly, RBM5 does not affect U1 and U2 snRNP assembly on FAS pre-mRNA but inhibits the transition from pre-spliceosome to mature spliceosome via interaction between U4/5/6 trisnRNP and the OCRE domain of RBM5 [20].
The production of anti- and pro-apoptotic isoforms of Bcl-x involves alternative selection of two competing 5′ splice sites. Both exonic and intronic elements have been identified in Bcl-x that regulate its alternative splicing [109, 120]. Trans-acting factors involved include SAP155, Sam68, hnRNA A1, hnRNP F/H, SRp30c, and RBM25 [28, 62, 146, 201]. An apoptotic agent Ro-31-8220 inhibits PKC and activates JNK, and concomitantly, Bcl-xL splicing is inhibited. This effect is repressed by okadaic acid, an inhibitor against PP1 and PP2A. Okadaic acid splicing regulation is mediated via a 16-nt G-tract element (Gt16) on pre-mRNAs [69]. Emetine, a protein synthesis inhibitor, also upregulates Bcl-xS splicing. However, emetine upregulation is blocked by calyculin A by inhibiting PP1 and PP2A [21]. Various stimuli are reported to change Bcl-xL/Bcl-xS ratio. For example, IL-6, GM-CSF, and TPA all upregulate Bcl-xL splicing in erythroleukemia and glioma. The Bcl-xS isoform is induced by S-adenosylmethionine (SAMe), whose synthesis is impaired by liver injury. β-adrenergic receptor activation upregulates Bcl-xS and induces cell death of cardiomyocytes. Consistently, β-blockers inhibit Bcl-xS induction. Ceramide responsive elements CRCE1 and 2 have been identified in Bcl-x pre-mRNA. Ceramide appears to regulate Bcl-x splicing via SAP155 which binds to CRCE1 [121, 122]. Although Bcl-x splicing responds to a number of stimuli, it still remains unclear how these signals transduce to the splicing machinery. A recent study using genomic siRNA screening for Bcl-x splicing regulators has uncovered a complex network of splicing regulators that link cell cycle and cell death controls [133].
In the case of caspase-2, several splicing factors have been identified to regulate the formation of anti-apoptotic (caspase-2S) and pro-apoptotic (caspase-2L) splicing isoforms. Interestingly, SR proteins including SC35 and ASF/SF2 promote exon skipping to produce caspase-2L, whereas, hnRNPA1 facilitates exon inclusion to produce caspase-2S [85]. The effects of these SR proteins and of hnRNP A1 on caspase-2 splicing are opposite to their effects on other model splicing substrates. A caspase-2 mini-gene model system has been used to dissect the cis-elements and trans-acting factors involved in caspase-2 alternative splicing ([85, 32]). An evolutionarily conserved 100 nt intronic element, In100, has been identified as an intronic splicing silencer element responsible for exon 9 exclusion between exon 9 and 10. The In100 element contains two domains: an upstream decoy 3′ splice site and a downstream PTB binding domain 32. The decoy 3′ splice site engages the 5′ splice site of the alternative exon 9 in a non-productive manner, effectively suppressing the use of this 5′ splice site without reducing U1 snRNP binding. PTB plays a role in regulating alternative splicing of a number of other genes (reviewed in [16, 171, 181]). The regulatory mechanism of PTB in caspase-2 alternative splicing appears to be distinct from that involved in other genes. Furthermore, our recent survey of human genome suggests that In100-like (Intron 100-like) intronic elements (i.e., decoy 3′ splice site juxtaposed to PTB binding domains) may represent a general intronic splicing regulatory motif and that such elements may play a role in the regulation alternative splicing of other cell death genes [74]. Recently, we reported that caspase 2 splicing is regulated by RBM5, which is frequently deleted in lung cancer [59]. RBM5 binds to the U/C-rich region immediately upstream of In100. These results suggest that splicing regulation plays an important role in cancer development via PCD-related gene products. However, crucial questions still remain unanswered. How does splicing machinery differentially recognize the decoy 3′ splice site inside In100 as a regulatory element as opposed to a true 3′ splice site? How are splicing regulators and cis-elements coordinated to regulate alternative splicing? Molecular dissection of cis-acting elements and trans-acting splicing regulators involved in caspase-2 alternative splicing has provided a good beginning point to understand the mechanisms underlying the complex regulation of alternative splicing of important PCD genes. The involvement of such a pseudo- or decoy splice site in alternative splicing regulation may provide an explanation for the phylogenic conservation of sequences containing pseudo-splicing signals in mammalian introns. Further studies are necessary to test whether pseudo-splice sites proximal to splicing repressor binding sites represent general splicing regulatory motifs.
5.3 Complex Networks Linking Alternative Splicing, Cell Death, and Other Processes
Recent efforts using systems biology approaches have begun to reveal the complex networks that link splicing regulation, cell death, and other important cellular processes [133]. Genome-wide analyses of splicing patterns can help identify specific gene products that are tumorigenic as well as involved in other cellular pathways. Gene regulation by alternative splicing plays critical roles in cellular differentiation, cell proliferation, and cell death. Therefore, imbalance in the splicing pathway can lead to tumorigenic events [63]. High-throughput microarray analyses and next-generation sequencing assays have been used to identify alternative splicing events or factors involved in specific pathways [191]. Such studies will help to decipher the splicing codes that dictate normal cell development and subsequently, how mutations affect these events to give rise to cancer. Given the importance of alternative splicing in generating antagonistic isoforms of pro- or anti-apoptotic proteins, it is hopeful to target alternative splicing machinery for future cancer therapeutics.
6 Cell Death Regulation, Pre-mRNA Splicing, and Cancer
Mounting evidence supports that alternative splicing of genes involved in cell cycle control, cell proliferation, apoptosis, angiogenesis, motility, and invasion are associated with tumor progression and metastasis [116, 200]. In cancer, aberrant alternative splicing has been associated with mutations affecting cis-elements that regulate splicing. Such mutations may alter the abundance, localization, or post-translationally modify trans-acting factors that determine splice site selection [123]. The precise mechanisms by which alternative splicing controls expression of genes related to cancer remain poorly understood.
6.1 Splicing Factors, Splicing Variants, and Cancer
A number of factors regulating alternative splicing of cell death genes show oncogenic properties [68]. Splicing factors, such as hnRNP A1 [22], hnRNP A2, hnRNP B1, polypyrimidine tract binding protein (PTB), [148] and HuR, are frequently overexpressed in tumors. Splicing factor overexpression can trigger malignant transformation.
In addition, cancer-related splicing factor isoforms could alter function of these splicing factors, resulting in aberrant alternative splicing. Depletion of splicing factors prevents the generation of cancer-associated isoforms, suggesting splicing factors as potential therapeutic targets for cancer therapies. Tumor-specific variations in splicing may also generate new epitopes that can serve as anticancer agents.
6.2 Death Receptors and Cancer
Fas (Apo-1/CD95), a transmembrane death receptor, has a soluble isoform (sFas) generated by alternative splicing of Fas mRNA. sFas lacks the transmembrane domain and antagonizes cell-surface Fas function. sFas is detected in patients with different types of leukemia and solid tumors [90, 126], such as adult T cell leukemia (ATL), large granular lymphocyte (LGL), leukemia and renal cell carcinoma.
TNF-related apoptosis-inducing ligand (TRAIL) and its receptors, namely DR4, DR5, DcR1, and DcR2 have become attractive targets for anti-cancer therapies, because they seem to trigger apoptosis selectively in cancer cells but not in normal cells. Thus far, several compounds and biologics (such as agonistic TRAIL antibodies) have gained attention due to their anti-tumor efficacy [131, 151, 196].
In addition, c-FLIP modulates the activation of procaspase-8 and thereby prevents induction of apoptosis mediated by death receptors. There is evidence for increased c-FLIP expression in various types of tumor cells, including colorectal cancer [97], gastric cancer [138], Hodgkin’s lymphoma [138], and ovarian cancer [1]. Thus, downregulating c-FLIP induced by pharmacological agents, such as proteasome inhibitors, protein or RNA synthesis inhibitors [57], or chemotherapeutic agents [103], may have therapeutic value for these types of cancer.
6.3 BCL-2 Family and Cancer
The Bcl-2 family of proteins are crucial players in regulation of apoptosis. Aberrant expression of members of this family has been associated with different cancers. Over-expression of Bcl-2 was originally observed in B cell lymphomas ([130]; reviewed in [98]). Bcl-2 overexpression has since been detected in other solid tumors in the lung, kidney, stomach, and brain [67, 119]. Interestingly, the relationship between Bcl-2 expression levels and cancer prognosis is cell type-dependent. For example, high levels of Bcl-2 were correlated with poor prognosis in certain lymphomas; however, low Bcl-2 expression as correlated with a poor prognosis in breast cancer. Experiments using knockout mice have advanced our understanding of the role of Bcl-2 family members in tumorigenesis. Bad-knockout mice develop B cell lymphoblastic leukemia/lymphoma when exposed to sub-lethal doses of γ-irradiation, whereas Bid-knockout mice show chromosomal aberrations and develop leukemia [202].
Furthermore, the imbalances between apoptosis-promoting and apoptosis-inhibiting members of the Bcl-2 family are also common in various human cancers. Myeloid cell leukemia-1 (MCL-1) has three splicing variants: anti-apoptotic MCL-1L and pro-apoptotic MCL-1S and MCL-1ES. There is an imbalance between the expression levels of MCL-1L, MCL-1S, and MCL-1ES in the skin basal cell carcinoma (BCC) cell line [167] and renal cancer [92]. Bcl-x mRNAs encoding a long isoform, Bcl-xL, predominates in various types of malignant lymphomas and may be involved in lymphomagenesis [190]. Bcl-xL was also expressed in human hepatoma cell lines at high levels and its down-regulation activated apoptosis [173]. Another Bcl-x alternative splicing product is Bcl-xAK, which contains the Bcl-2 homology domains, BH2 and BH4, as well as the transmembrane domain but lacks BH1 and BH3. Bcl-xAK is expressed in melanoma and other tumor cells and its overexpression results in significant induction of apoptosis in melanoma cells [77]. Another Bcl-2 family member, Bfk undergoes alternative splicing to produce four isoforms, out of which two are pro-apoptotic. In the transition from normal tissue to tumor, pro-apoptotic Bfk isoform expression is substantially reduced in tumors isolated from the human gastrointestinal tract [40].
6.4 Caspase Alternative Splicing and Cell Death Regulation in Cancer
Caspases play important roles in the regulation of physiological cell death, therefore, the disturbance of the caspases expression or function may contribute to the cancer formation. Caspase-9 initiates apoptosis and has two distinct protein isoforms generated as a result of alternative pre-mRNA splicing: pro-apoptotic caspase-9a and anti-apoptotic caspase-9b. A recent study demonstrated that hnRNP family member L (hnRNP L) is specifically phosphorylated in non–small cell lung cancer cells (NSCLC cells) and also associated with caspase-9 pre-mRNA. The interaction of hnRNP L with caspase-9 pre-mRNA in NSCLC cells promotes preferential expression of the 9b isoform of caspase-9, which is antiapoptotic, and promotes tumor growth [64].
Another example is the caspase-2 tumor suppressor, which is alternatively spliced to generate multiple isoforms (discussed in Sect. 3.1). RBM5, by virtue of binding to the U/C-rich region in In100 splicing repressor element (see Sect. 5.2), promotes production of the proapoptotic caspase-2L isoform and regulates the ratio of caspase-2 isoforms in HeLa cells [59, 155]. Fas can lead to cell death and also be alternatively spliced to produce shorter isoforms. Exclusion of exon 6 in Fas pre-mRNA generates FasDelE6 which can inhibit Fas-mediated cell death. Recently, RBM5 [20] and HuR [81] have been identified to play an important role in Fas exon 6 inclusion.
Caspase-8L, generated by alternative splicing of caspase-8, can suppresses caspase 8-dependent apoptosis. The imbalanced expression of the caspase-8L splicing isoform has been associated with cancer. Suppressing the formation of caspase-8L splice variant renders cells more sensitive to apoptosis-induced neuroblastoma cell death [128]. In addition, a splice variant of IG20 gene regulating the activation of caspase-8 was implicated during tumorigenesis [111, 137].
6.5 IAPS and Cancer
Survivin, a member of the IAPS (inhibitor of apoptosis proteins) family, functions as a key regulator of mitosis and programmed cell death. It regulates cell death by interrupting multiple cell cycle-related proteins, such as INCENP and Aurora B kinase. Studies have shown survivin overexpression results from several polymorphisms in the survivin gene promoter [19, 150], which also correlate with tumorigenesis and prognosis [115, 172]. Therefore, survivin has become a target for cancer therapeutics [174].
Livin, an IAPS-related protein, has two different functional splicing variants that are characterized as anti-apoptotic [9]. Livin interacts with downstream caspases, such as caspase-3, caspase-7, and caspase-9, leading to their inactivation and degradation. Aberrant expression of Livin has been reported to be associated with tumorigenesis in many different cancer types including melanoma [179], breast cancer [193], and lung cancer [72]. Therefore, Livin is believed to be a new target for immunotherapy and gene therapy for treatment of cancer.
6.6 Cell Death-Related DNases and Their Regulators
Previous studies have demonstrated that the activity of alkaline (DNase I; EC 3.1.21.1) and acidic DNases (DNase II; EC 3.1.22.1) was inhibited in non-necrotic cells in malignant tumors. In subsequent studies, vitamin C and K3 administration reactivated these DNases to induce cancer cell death [175].
Endonuclease G (EndoG) cleaves chromatin DNA into nucleosomal fragments in the nucleus and participates in the caspase independent apoptotic pathway [80]. As a pro-apoptotic protein, decreased expression of EndoG has been found in several cancers, such as hepatocellular carcinomas and breast cancer [12].
6.7 Mitochondrial Cell Death Proteins and Cancer
Apoptosis can be activated through two pathways: the extrinsic pathway (mediated by death receptors) or the intrinsic pathway (mediated by mitochondria). As mentioned previously, mitochondrial factors such as Smac and AIF can adhere to IAPs and inhibit their caspase-binding activity, thereby regulating cell death and tumorigenesis. For example, in a malignant glioma xenograft mice model, co-administration of Smac/DIABLO peptides and TRAIL sensitized glioma cells lead to apoptotic death and induced malignant glioma regression [58]. Furthermore, several Smad analogs can induce cancer cell death and have shown potential as cancer therapies [106, 164].
6.8 Defective Autophagy and Cancer
Autophagy is an evolutionarily conserved mechanism for protein degradation and maintains homeostasis. Studies have implicated autophagy in tumorigenesis and tumor progression. Autophagy deficiency predisposes cells to tumor development. To this end, haploinsufficiency of autophagy genes increased tumor formation in mouse [118]. Conversely, once tumors were established, autophagy may enable cancer cell survival. For example, autophagy increases cancer drug resistance to Imatinib in chronic myeloid leukemia [13] and facilitates resistance trastuzumab for HER2 positive breast cancer cells [178]. Conversely, autophagy abrogation by autophagy inhibitors re-sensitizes the resistant cancer cells to the chemotherapy or radiation [8, 13].
7 Concluding Remarks
Many genes involved in PCD undergo alternative splicing to produce multiple isoforms with distinct functional activities. Alternative splicing not only generates products with different subcellular localization (membrane associated versus soluble proteins; nuclear versus cytoplasmic) but also produces proteins with different, and often antagonistic functional activities. Molecular analyses of these cell death genes suggest fundamental importance of alternative splicing in regulating PCD. However, molecular mechanisms controlling the alternative splicing of these PCD genes remain unclear. Recent studies using model systems have initiated molecular dissection of the link between alternative splicing and PCD regulation. These studies are only the beginning, given the wide variety of functionally distinct proteins generated by alternative splicing. A global landscape of alternative splicing patterns as well as molecular mechanisms involved in cell death regulation await further investigation using multidimensional and combinatorial approaches. Due to the complex regulatory networks that work in harmony to control cell fate and cell differentiation, mutations affecting one pathway can have far reaching consequences at the cellular, multicellular, and tissue levels. Therefore, elucidating regulatory mechanisms underlying functionally important alternative splicing events will not only help us understand pathogenetic mechanisms of human diseases caused by splicing defects but also provide molecular insights into designing new cancer therapies by targeting aberrant or defective splicing.
References
Abedini MR, Qiu Q, Yan X, Tsang BK (2004) Possible role of FLICE-like inhibitory protein (FLIP) in chemoresistant ovarian cancer cells in vitro. Oncogene 23(42):6997–7004
Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J (2004) NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 20(3):319–325
Akhter A, Gavrilin MA, Frantz L, Washington S, Ditty C, Limoli D, Day C, Sarkar A, Newland C, Butchar J, Marsh CB, Wewers MD, Tridandapani S, Kanneganti TD, Amer AO (2009) Caspase-7 activation by the Nlrc4/Ipaf inflammasome restricts Legionella pneumophila infection. PLoS Pathog 5(4):e1000361
Akgul C, Moulding DA, Edwards SW (2004) Alternative splicing of Bcl-2-related genes: functional consequences and potential therapeutic applications. Cell Mol Life Sci 61(17):2189–2199
Alnemri ES (1997) Mammalian cell death proteases: a family of highly conserved aspartate specific cysteine proteases. J Cell Biochem 64:33–42
Alnemri ES, Fernandes-Alnemri T, Litwack G (1995) Cloning and expression of four novel isoforms of human interleukin-1 beta converting enzyme with different apoptotic activities. J Biol Chem 270(9):4312–4317
Al-Zoubi AM, Efimova EV, Kaithamana S, Martinez O, El-Idrissi M-A, Dogan RE, Prabhakar BS (2001) Contrasting effects of IG20 and its splice isoforms, MADD and DENN-SV, on tumor necrosis factor alpha-induced apoptosis and activation of caspase-8 and -3. J Biol Chem 276(50):47202–47211
Apel A, Herr I, Schwarz H, Rodemann HP, Mayer A (2008) Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res 68(5):1485–1494
Ashhab Y, Alian A, Polliack A, Panet A, Ben Yehuda D (2001) Two splicing variants of a new inhibitor of apoptosis gene with different biological properties and tissue distribution pattern. FEBS Lett 495(1–2):56–60
Bae J, Leo CP, Hsu SY, Hsueh AJ (2000) MCL-1S, a splicing variant of the antiapoptotic BCL-2 family member MCL-1, encodes a proapoptotic protein possessing only the BH3 domain. J Biol Chem 275(33):25255–25261
Baehrecke EH (2002) How death shapes life during development. Nat Rev Mol Cell Biol 3:779–787
Basnakian AG, Apostolov EO, Yin X, Abiri SO, Stewart AG, Singh AB, Shah SV (2006) Endonuclease G promotes cell death of non-invasive human breast cancer cells. Exp Cell Res 312(20):4139–4149
Bellodi C, Lidonnici MR, Hamilton A, Helgason GV, Soliera AR, Ronchetti M, Galavotti S, Young KW, Selmi T, Yacobi R, Van Etten RA, Donato N, Hunter A, Dinsdale D, Tirrò E, Vigneri P, Nicotera P, Dyer MJ, Holyoake T, Salomoni P, Calabretta B (2009) Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J Clin Invest 119(5):1109–1123
Bergeron L, Perez GI, Macdonald G, Shi L, Sun Y, Jurisicova A, Varmuza S, Latham KE, Flaws JA, Salter JC, Hara H, Moskowitz MA, Li E, Greenberg A, Tilly JL, Yuan J (1998) Defects in regulation of apoptosis in caspase-2-deficient mice. Genes Dev 12(9):1304–1314
Bergsbaken T, Fink SL, Cookson BT (2009) Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 7(2):99–109
Black DL (2003) Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem 72:291–336
Blencowe BJ (2006) Alternative splicing: new insights from global analyses. Cell 126:37–47
Boise LH, González-García M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Nuñez G, Thompson CB (1993) bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 74(4):597–608
Borbély AA, Murvai M, Szarka K, Kónya J, Gergely L, Hernádi Z, Veress G (2007) Survivin promoter polymorphism and cervical carcinogenesis. J Clin Pathol 60(3):303–306
Bonnal S, Martinez C, Forch P, Bachi A, Wilm M, Valcarcel J (2008) RBM5/Luca-15/H37 regulates Fas alternative splice site pairing after exon definition. Mol Cell 32:81–95
Boon-Unge K, Yu Q, Zou T, Zhou A, Govitrapong P, Zhou J (2007) Emetine regulates the alternative splicing of Bcl-x through a protein phosphatase 1-dependent mechanism. Chem Biol 14(12):1386–1392
Boukakis G, Patrinou-Georgoula M, Lekarakou M, Valavanis C, Guialis A (2010) Deregulated expression of hnRNP A/B proteins in human non-small cell lung cancer: parallel assessment of protein and mRNA levels in paired tumour/non-tumour tissues. BMC Cancer 10:434
Büttner S, Eisenberg T, Carmona-Gutierrez D, Ruli D, Knauer H, Ruckenstuhl C, Sigrist C, Wissing S, Kollroser M, Fröhlich KU, Sigrist S, Madeo F (2007) Endonuclease G regulates budding yeast life and death. Mol Cell 25(2):233–246
Cartegni L, Chew SL and Krainer AR (2002) Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet 3:285–298
Cascino I, Papoff G, Eramo A, Ruberti G (1996) Soluble Fas/Apo-1 splicing variants and apoptosis. Front Biosci 1:d12–d18
Chew LJ, Pan H, Yu J, Tian S, Huang WQ, Zhang JY, Pang S, Li LY (2002) A novel secreted splice variant of vascular endothelial cell growth inhibitor. FASEB J 16(7):742–744
Clarke PG (1990) Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol (Berl) 181:195–213
Cloutier P, Toutant J, Shkreta L, Goekjian S, Revil T, Chabot B (2008) Antagonistic effects of the SRp30c protein and cryptic 5′ splice sites on the alternative splicing of the apoptotic regulator Bcl-x. J Biol Chem 283(31):21315–21324
Corcos L, Solier S (2005) Alternative mRNA splicing, pathology and molecular therapeutics. Med Sci (Paris) 21(3):253–260
Corsini L, Bonnal S, Basquin J, Hothorn M, Scheffzek K, Valcárcel J, Sattler M (2007) U2AF-homology motif interactions are required for alternative splicing regulation by SPF45. Nat Struct Mol Biol 14(7):620–629
Côté J, Dupuis S and Wu JY (2001) Polypyrimidine track-binding protein binding downstream of caspase- 2 alternative exon 9 represses its inclusion. J Biol Chem 276:8535–8543
Coté J, Dupuis S, Jiang Z, and Wu JY (2001) Caspase-2 pre-mRNA alternative splicing: Identification of an intronic element containing a decoy 3’ acceptor site. Proc Nat Acad Sci USA 98:938–943
Cotter TG (2009) Apoptosis and cancer: the genesis of a research field. Nat Rev Cancer 9(7):501–507
Cregan SP, Dawson VL, Slack RS (2004) Role of AIF in caspase-dependent and caspase-independent cell death. Oncogene 23(16):2785–2796
Cuenin S, Tinel A, Janssens S, Tschopp J (2008) p53-induced protein with a death domain (PIDD) isoforms differentially activate nuclear factor-kappaB and caspase-2 in response to genotoxic stress. Oncogene 27(3):387–396
David CJ, Manley JL (2010) Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev 24:2343–2364
Delavallée L, Cabon L, Galán-Malo P, Lorenzo HK, Susin SA (2011) AIF-mediated caspase-independent necroptosis: a new chance for targeted therapeutics. IUBMB Life 63(4):221–232
Delettre C, Yuste VJ, Moubarak RS, Bras M, Lesbordes-Brion JC, Petres S, Bellalou J, Susin SA (2006) AIFsh, a novel apoptosis-inducing factor (AIF) pro-apoptotic isoform with potential pathological relevance in human cancer. J Biol Chem 281(10):6413–6427
Delettre C, Yuste VJ, Moubarak RS, Bras M, Robert N, Susin SA (2006) Identification and characterization of AIFsh2, a mitochondrial apoptosis-inducing factor (AIF) isoform with NADH oxidase activity. J Biol Chem 281(27):18507–18518
Dempsey CE, Dive C, Fletcher DJ, Barnes FA, Lobo A, Bingle CD, Whyte MK, Renshaw SA (2005) Expression of pro-apoptotic Bfk isoforms reduces during malignant transformation in the human gastrointestinal tract. FEBS Lett 579(17):3646–3650
Dennis PB, Jaeschke A, Saitoh M, Fowler B, Kozma SC, Thomas G (2001) Mammalian TOR: a homeostatic ATP sensor. Science 294:1102–1105
Diaz A, Hu C, Kastner DL, Schaner P, Reginato AM, Richards N, Gumucio DL (2004) Lipopolysaccharide-induced expression of multiple alternatively spliced MEFV transcripts in human synovial fibroblasts: a prominent splice isoform lacks the C-terminal domain that is highly mutated in familial Mediterranean fever. Arthritis Rheum 50(11):3679–3689
Diaz-Troya S, Perez–Perez ME, Florencio FJ, Crespo JL (2008) The role of TOR in autophagy regulation from yeast to plants and mammals. Autophagy 4:851–865
Djerbi M, Darreh-Shori T, Zhivotovsky B, Grandien A (2001) Characterization of the human FLICE-inhibitory protein locus and comparison of the anti-apoptotic activity of four different flip isoforms. Scand J Immunol 54(1–2):180–189
Donnini M, Lapucci A, Papucci L, Witort E, Tempestini A, Brewer G, Bevilacqua A, Nicolin A, Capaccioli S, Schiavone N (2001) Apoptosis is associated with modifications of bcl-2 mRNA AU-binding proteins. Biochem Biophys Res Commun 287(5):1063–1069
Droin N, Beauchemin M, Solary E, Bertrand R (2000) Identification of a caspase-2 isoform that behaves as an endogenous inhibitor of the caspase cascade. Cancer Res 60(24):7039–7047
Edinger AL, Thompson CB (2004) Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol 16(6):663–669
Efimova E, Martinez O, Lokshin A, Arima T, Prabhakar BS (2003) IG20, a MADD splice variant, increases cell susceptibility to gamma-irradiation and induces soluble mediators that suppress tumor cell growth. Cancer Res 63(24):8768–8776
Efimova EV, Al-Zoubi AM, Martinez O, Kaithamana S, Lu S, Arima T, Prabhakar BS (2004) IG20, in contrast to DENN-SV, (MADD splice variants) suppresses tumor cell survival, and enhances their susceptibility to apoptosis and cancer drugs. Oncogene 23(5):1076–1087
Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A (2009a) Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ 16(7):966–975
Eisenberg-Lerner A, Kimchi A (2009b) The paradox of autophagy and its implication in cancer etiology and therapy. Apoptosis 14(4):376–391
Feng Q, Li P, Leung PC, Auersperg N (2004) Caspase-1zeta, a new splice variant of the caspase-1 gene. Genomics 84(3):587–591
Forch P, Puig O, Kedersha N, Martinez C, Granneman S, Seraphin B, Anderson P, Valcarcel J (2000) The apoptosis-promoting factor TIA-1 is a regulator of alternative pre-mRNA splicing. Mol Cell 6(5):1089–1098
Forch P, Valcarcel J (2001) Molecular mechanisms of gene expression regulation by the apoptosis-promoting protein TIA-1. Apoptosis 6(6):463–468
Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G (2009) The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol 10(3):241–247
Fu J, Jin Y, Arend LJ (2003) Smac3, a novel Smac/DIABLO splicing variant, attenuates the stability and apoptosis-inhibiting activity of X-linked inhibitor of apoptosis protein. J Biol Chem 278(52):52660–52672
Fulda S, Meyer E, Debatin K-M (2000) Metabolic Inhibitors Sensitize for CD95 (APO-1/Fas)-induced Apoptosis by Down-Regulating Fas-associated Death Domain-like Interleukin 1-Converting Enzyme Inhibitory Protein Expression. Cancer Res 60:3947–3956
Fulda S, Wick W, Weller M, Debatin KM (2002) Smac agonists sensitize for Apo2L/TRAIL- or anticancer drug-induced apoptosis and induce regression of malignant glioma in vivo. Nat Med 8(8):808–815
Fushimi K, Ray P, Kar A, Wang L, Sutherland LC, Wu JY (2008) Up-regulation of the proapoptotic caspase 2 splicing isoform by a candidate tumor suppressor, RBM5. Proc Natl Acad Sci U S A 105:15708–15713
Galluzzi L, Blomgren K, Kroemer G (2009) Mitochondrial membrane permeabilization in neuronal injury. Nat Rev Neurosci 10(7):481–494
Ganley IG, du Lam H, Wang J, Ding X, Chen S, Jiang X (2009) ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 284:12297–12305
Garneau D, Revil T, Fisette JF, Chabot B (2005) Heterogeneous nuclear ribonucleoprotein F/H proteins modulate the alternative splicing of the apoptotic mediator Bcl-x. J Biol Chem 280(24):22641–22650
Germann S, Gratadou L, Dutertre M, Auboeuf D (2012) Splicing programs and cancer. J Nucleic Acids 2012:269570
Goehe RW, Shultz JC, Murudkar C, Usanovic S, Lamour NF, Massey DH, Zhang L, Camidge DR, Shay JW, Minna JD et al (2010) hnRNP L regulates the tumorigenic capacity of lung cancer xenografts in mice via caspase-9 pre-mRNA processing. J Clin Invest 120:3923–3939
Golks A, Brenner D, Fritsch C, Krammer PH, Lavrik IN (2005) c-FLIPR, a new regulator of death receptor-induced apoptosis. J Biol Chem 280(15):14507–14513
Goll DE, Thompson VF, Li H, Wei W, Cong J (2003) The calpain system. Physiol Rev 83(3):731–801
Groeger AM, Esposito V, De Luca A, Cassandro R, Tonini G, Ambrogi V, Baldi F, Goldfarb R, Mineo TC, Baldi A, et al. (2004) Prognostic value of immunohistochemical expression of p53, bax, Bcl-2 and Bcl-xL in resected non-small-cell lung cancers. Histopathology 44:54–63
Grosso AR, Martins S, Carmo-Fonseca M (2008) The emerging role of splicing factors in cancer. EMBO Rep 9:1087–1093
Hai Y, Cao W, Liu G, Hong SP, Elela SA, Klinck R, Chu J, Xie J (2008) A G-tract element in apoptotic agents-induced alternative splicing. Nucleic Acids Res 36(10):3320–3331
Han J, Xiong J, Wang D, Fu XD (2011) Pre-mRNA splicing: where and when in the nucleus. Trends Cell Biol 21(6):336–343
Harris TE, Lawrence JC Jr (2003) TOR signaling. Sci STKE 2003, re15
Hariu H, Hirohashi Y, Torigoe T, Asanuma H, Hariu M, Tamura Y, Aketa K, Nabeta C, Nakanishi K, Kamiguchi K, Mano Y, Kitamura H, Kobayashi J, Tsukahara T, Shijubo N, Sato N (2005) Aberrant expression and potency as a cancer immunotherapy target of inhibitor of apoptosis protein family, Livin/ML-IAP in lung cancer. Clin Cancer Res 11(3):1000–1009
Hastings ML, Krainer AR (2001) Pre-mRNA splicing in the new millennium. Curr Opin Cell Biol 13:302–309
Havlioglu N, Wang J, Fushimi K, Vibranovski MD, Kan Z, Gish W, Fedorov A, Long M, Wu JY (2007) An intronic signal for alternative splicing in the human genome. PLoS One 2(11):e1246
Himeji D, Horiuchi T, Tsukamoto H, Hayashi K, Watanabe T, Harada M (2002) Characterization of caspase-8L: a novel isoform of caspase-8 that behaves as an inhibitor of the caspase cascade. Blood 99(11):4070–4078
Horiuchi T, Himeji D, Tsukamoto H, Harashima S, Hashimura C, Hayashi K (2000) Dominant expression of a novel splice variant of caspase-8 in human peripheral blood lymphocytes. Biochem Biophys Res Commun 272(3):877–881
Hossini AM, Geilen CC, Fecker LF, Daniel PT, Eberle J (2006) A novel Bcl-x splice product, Bcl-xAK, triggers apoptosis in human melanoma cells without BH3 domain. Oncogene 25(15):2160–2169
Hsieh SY, Liaw SF, Lee SN, Hsieh PS, Lin KH, Chu CM, Liaw YF (2003) Aberrant caspase-activated DNase (CAD) transcripts in human hepatoma cells. Br J Cancer 88(2):210–216
Hughes TA (2006) Regulation of gene expression by alternative untranslated regions. Trends Genet 22(3):119–122
Ishihara Y, Shimamoto N (2006) Involvement of endonuclease G in nucleosomal DNA fragmentation under sustained endogenous oxidative stress. J Biol Chem 281(10):6726–6733
Izquierdo JM (2008) Hu antigen R (HuR) functions as an alternative pre-mRNA splicing regulator of Fas apoptosis-promoting receptor on exon definition. J Biol Chem 283:19077–19084
Izquierdo JM, Majós N, Bonnal S, Martínez C, Castelo R, Guigó R, Bilbao D, Valcárcel J (2005) Regulation of Fas alternative splicing by antagonistic effects of TIA-1 and PTB on exon definition. Mol Cell 19(4):475–484
Izquierdo JM, Valcárcel J (2007) Fas-activated serine/threonine kinase (FAST K) synergizes with TIA-1/TIAR proteins to regulate Fas alternative splicing. J Biol Chem 282(3):1539–1543
Izquierdo JM (2008) Fas splicing regulation during early apoptosis is linked to caspase-mediated cleavage of U2AF65. Mol Biol Cell 19(8):3299–3307
Jiang ZH, Zhang WJ, Rao Y, Wu JY (1998) Regulation of Ich-1 pre-mRNA alternative splicing and apoptosis by mammalian splicing factors. Proc Natl Acad Sci USA 95:9155–9160
Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH (2009) ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 20:1992–2003
Jurica MS, Moore MJ (2003) Pre-mRNA splicing: awash in a sea of proteins. Mol Cell 12(1):5–14
Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19:5720–5728
Kalsotra A, Cooper TA (2011) Functional consequences of developmentally regulated alternative splicing. Nat Rev Genet 12(10):715–729
Kamihira S, Yamada Y, Tomonaga M, Sugahara K, Tsuruda K (1999) Discrepant expression of membrane and soluble isoforms of Fas (CD95/APO-1) in adult T-cell leukaemia: soluble Fas isoform is an independent risk factor for prognosis. Br J Haematol 107(4):851–860
Kataoka T, Tschopp J (2004) N-terminal fragment of c-FLIP(L) processed by caspase 8 specifically interacts with TRAF2 and induces activation of the NF-kappaB signaling pathway. Mol Cell Biol 24(7):2627–2636
Kempkensteffen C, Hinz S, Johannsen M, Krause H, Magheli A, Christoph F, Köllermann J, Schrader M, Schostak M, Miller K, Weikert S (2009) Expression of Mcl-1 splicing variants in clear-cell renal cancer and their correlation with histopathological parameters and prognosis. Tumour Biol 30(2):73–79
Kerr JF, Wyllie AH, Currie AR (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26(4):239–257
Kirchhoff S, Müller WW, Krueger A, Schmitz I, Krammer PH (2000) TCR-mediated up-regulation of c-FLIPshort correlates with resistance toward CD95-mediated apoptosis by blocking death-inducing signaling complex activity. J Immunol 165(11):6293–6300
Kisenge RR, Toyoda H, Kang J, Tanaka S, Yamamoto H, Azuma E, Komada Y (2003) Expression of short-form caspase 8 correlates with decreased sensitivity to Fas-mediated apoptosis in neuroblastoma cells. Cancer Sci 94(7):598–605
Klionsky DJ, Emr SD (2000) Autophagy as a regulated pathway of cellular degradation. Science 290:1717–1721
Korkolopoulou P, Saetta AA, Levidou G, Gigelou F, Lazaris A, Thymara I, Scliri M, Bousboukea K, Michalopoulos NV, Apostolikas N, Konstantinidou A, Tzivras M, Patsouris E (2007) c-FLIP expression in colorectal carcinomas: association with Fas/FasL expression and prognostic implications. Histopathology 51(2):150–156
Korsmeyer SJ (1999) Programmed cell death and the regulation of homeostasis. Harvey Lect 1999–2000(95):21–41
Korsmeyer SJ, Gross A, Harada H, Zha J, Wang K, Yin XM, Wei M, Zinkel S (1999) Death and survival signals determine active/inactive conformations of pro-apoptotic BAX, BAD, and BID molecules. Cold Spring Harb Symp Quant Biol 64:343–50
Krieg A, Krieg T, Wenzel M, Schmitt M, Ramp U, Fang B, Gabbert HE, Gerharz CD, Mahotka C (2003) TRAIL-beta and TRAIL-gamma: two novel splice variants of the human TNF-related apoptosis-inducing ligand (TRAIL) without apoptotic potential. Br J Cancer 88(6):918–927
Krieg A, Schulte am Esch J 2nd, Ramp U, Hosch SB, Knoefel WT, Gabbert HE, Mahotka C. (2006) TRAIL-R4-beta: a new splice variant of TRAIL-receptor 4 lacking the cysteine rich domain 1. Biochem Biophys Res Commun.349(1):115–121
Kurada BR, Li LC, Mulherkar N, Subramanian M, Prasad KV, Prabhakar BS (2009) MADD, a splice variant of IG20, is indispensable for MAPK activation and protection against apoptosis upon tumor necrosis factor-alpha treatment. J Biol Chem 284(20):13533–13541
Lacour S, Micheau O, Hammann A, Drouineaud V, Tschopp J, Solary E, Dimanche-Boitrel MT (2003) Chemotherapy enhances TNF-related apoptosis-inducing ligand DISC assembly in HT29 human colon cancer cells. Oncogene 22(12):1807–1816
Lamkanfi M, Festjens N, Declercq W, Vanden Berghe T, Vandenabeele P (2007) Caspases in cell survival, proliferation and differentiation. Cell Death Differ 14(1):44–55
Lamkanfi M, Kanneganti TD, Van Damme P, Vanden Berghe T, Vanoverberghe I, Vandekerckhove J, Vandenabeele P, Gevaert K, Núñez G.(2008). Targeted peptidecentric proteomics reveals caspase-7 as a substrate of the caspase-1 inflammasomes. Mol Cell Proteomics 7(12):2350–2363
Lecis D, Drago C, Manzoni L, Seneci P, Scolastico C, Mastrangelo E, Bolognesi M, Anichini A, Kashkar H, Walczak H, Delia D (2010) Novel SMAC-mimetics synergistically stimulate melanoma cell death in combination with TRAIL and Bortezomib. Br J Cancer 102(12):1707–1716
Levine B, Klionsky DJ (2004) Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 6:463–477
Levine SJ (2008) Molecular mechanisms of soluble cytokine receptor generation. J Biol Chem 283(21):14177–14181
Li CY, Chu JY, Yu JK, Huang XQ, Liu XJ, Shi L, Che Y C, Xie J Y (2004) Regulation of alternative splicing of Bcl-x by IL-6, GM-CSF and TPA. Cell Res 14:473–479
Li H, Zhu H, Xu CJ, Yuan J (1998) Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94(4):491–501
Li LC, Sheng JR, Mulherkar N, Prabhakar BS, Meriggioli MN (2008) Regulation of apoptosis and caspase-8 expression in neuroblastoma cells by isoforms of the IG20 gene. Cancer Res 68(18):7352–7361
Lomonosova E, Chinnadurai G (2008) BH3-only proteins in apoptosis and beyond: an overview. Oncogene Suppl 1:S2–S19
Long JC, Caceres JF (2009) The SR protein family of splicing factors: master regulators of gene expression. Biochem J 417(1):15–27
Lopez-Bigas N, Audit B, Ouzounis C, Parra G, Guigo R (2005) Are splicing mutations the most frequent cause of hereditary disease? FEBS Lett 579:1900–1903
Lu H, Gan M, Zhang G, Zhou T, Yan M, Wang S (2010) Expression of survivin, caspase-3 and p53 in cervical cancer assessed by tissue microarray: correlation with clinicopathology and prognosis. Eur J Gynaecol Oncol 31(6):662–666
Luan XY, Liu XB (2010) Comparison the inhibitory effects of human bone marrow mesenchymal stem cells and human placenta mesenchymal stem cells on T cell proliferation. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 26: 849–851
Luo X, Budihardjo I, Zou H, Slaughter C, Wang X (1998) Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94(4):481–490
Mariño G, Salvador-Montoliu N, Fueyo A, Knecht E, Mizushima N, López-Otín C (2007) Tissuespecific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J Biol Chem 282:18573–18583
Martin S, Toquet C, Oliver L, Cartron PF, Perrin P, Meflah K, Cuillère P, Vallette FM (2001) Expression of bcl-2, bax and bcl-xl in human gliomas: a re-appraisal. J Neurooncol 52:129– 139
Massiello A, Salas A, Pinkerman RL, Roddy P, Roesser JR, Chalfant CE (2004) Identification of two RNA cis-elements that function to regulate the 5’ splice site selection of Bcl-x pre-mRNA in response to ceramide. J Biol Chem 279:15799–15804
Massiello A, Chalfant CE (2006) SRp30a (ASF/SF2) regulates the alternative splicing of caspase-9 pre-mRNA and is required for ceramide-responsiveness. J Lipid Res 47:892–897
Massiello A, Roesser JR, Chalfant CE (2006) SAP155 Binds to ceramide-responsive RNA cis-element 1 and regulates the alternative 5′ splice site selection of Bcl-x pre-mRNA. FASEB J 20(10):1680–1682
Matlin AJ, Clark F, Smith CW (2005) Understanding alternative splicing: towards a cellular code. Nat Rev Mol Cell Biol 6:386–398
Matlin AJ, Moore MJ (2007) Spliceosome assembly and composition. Adv Exp Med Biol 623:14–35
Micheau O, Thome M, Schneider P, Holler N, Tschopp J, Nicholson DW, Briand C, Grütter MG (2002) The long form of FLIP is an activator of caspase-8 at the Fas death-inducing signaling complex. J Biol Chem 277(47):45162–45171
Midis GP, Shen Y, Owen-Schaub LB (1996) Elevated soluble Fas (sFas) levels in nonhematopoietic human malignancy. Cancer Res 56(17):3870–3874
Mille F, Thibert C, Fombonne J, Rama N, Guix C, Hayashi H, Corset V, Reed JC, Mehlen P (2009) The Patched dependence receptor triggers apoptosis through a DRAL-caspase-9 complex. Nat Cell Biol 11(6):739–746
Miller MA, Karacay B, Zhu X, O’Dorisio MS, Sandler AD (2006) Caspase 8L, a novel inhibitory isoform of caspase 8, is associated with undifferentiated neuroblastoma. Apoptosis 11(1):15–24
Mizushima N (2007) Autophagy: process and function. Genes Dev 21:2861–2873
Monni O, Joensuu H, Franssila K, Klefstrom J, Alitalo K, Knuutila S (1997) BCL2 overexpression associated with chromosomal amplification in diffuse large B-cell lymphoma. Blood 90(3):1168–1174
Moon DO, Park SY, Choi YH, Ahn JS, Kim GY (2011) Guggulsterone sensitizes hepatoma cells to TRAIL-induced apoptosis through the induction of CHOP-dependent DR5: involvement of ROS-dependent ER-stress. Biochem Pharmacol 82(11):1641–1650
Moore MJ, Silver PA (2008) Global analysis of mRNA splicing. RNA (New York, N.Y.) 14(2):197–203
Moore MJ, Wang Q, Kennedy CJ, Silver PA (2010) An alternative splicing network links cell-cycle control to apoptosis. Cell 142:625–636
Moran-Jones K, Grindlay J, Jones M, Smith R, Norman JC (2009) hnRNP A2 regulates alternative mRNA splicing of TP53INP2 to control invasive cell migration. Cancer Res 69:9219–9227
Müntener K, Zwicky R, Csucs G, Baici A (2003) The alternative use of exons 2 and 3 in cathepsin B mRNA controls enzyme trafficking and triggers nuclear fragmentation in human cells. Histochem Cell Biol 119(2):93–101
Mulherkar N, Ramaswamy M, Mordi DC, Prabhakar BS (2006) MADD/DENN splice variant of the IG20 gene is necessary and sufficient for cancer cell survival. Oncogene 25(47):6252–6261
Mulherkar N, Prasad KV, Prabhakar BS (2007) MADD/DENN splice variant of the IG20 gene is a negative regulator of caspase-8 activation. Knockdown enhances TRAIL-induced apoptosis of cancer cells. J Biol Chem 282(16):11715–11721
Nam SY, Jung GA, Hur GC, Chung HY, Kim WH, Seol DW, Lee BL (2003) Upregulation of FLIP(S) by Akt, a possible inhibition mechanism of TRAIL-induced apoptosis in human gastric cancers. Cancer Sci 94(12):1066–1073
Nowak J, Archange C, Tardivel-Lacombe J, Pontarotti P, Pebusque MJ, Vaccaro MI, Velasco G, Dagorn JC, Iovanna JL (2009) The TP53INP2 protein is required for autophagy in mammalian cells. Mol Biol Cell 20:870–881
Ogawa T, Shiga K, Hashimoto S, Kobayashi T, Horii A, Furukawa T (2003) APAF-1-ALT, a novel alternative splicing form of APAF-1, potentially causes impeded ability of undergoing DNA damage-induced apoptosis in the LNCaP human prostate cancer cell line. Biochem Biophys Res Commun 306(2):537–543
Ouhtit A, Matrougui K, Bengrine A, Koochekpour S, Zerfaoui M, Yousief Z (2007) Survivin is not only a death encounter but also a survival protein for invading tumor cells. Front Biosci 12:1260–1270
Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ (2008) Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 40:1413–1415
Panasyuk G, Nemazanyy I, Zhyvoloup A, Filonenko V, Davies D, Robson M, Pedley RB, Waterfield M, Gout I (2009) mTORbeta splicing isoform promotes cell proliferation and tumorigenesis. J Biol Chem 284:30807–30814
Papin S, Duquesnoy P, Cazeneuve C, Pantel J, Coppey-Moisan M, Dargemont C, Amselem S (2000) Alternative splicing at the MEFV locus involved in familial Mediterranean fever regulates translocation of the marenostrin/pyrin protein to the nucleus. Hum Mol Genet 9(20):3001–3009
Papoff G, Cascino I, Eramo A, Starace G, Lynch DH, Ruberti G (1996) An N-terminal domain shared by Fas/Apo-1 (CD95) soluble variants prevents cell death in vitro. J Immunol 156:4622–4630
Paronetto MP, Achsel T, Massiello A, Chalfant CE, Sette C (2007) The RNA-binding protein Sam68 modulates the alternative splicing of Bcl-x. J Cell Biol 176(7):929–939
Pathan N, Marusawa H, Krajewska M, Matsuzawa S, Kim H, Okada K, Torii S, Kitada S, Krajewski S, Welsh K, Pio F, Godzik A, Reed JC (2001) TUCAN, an antiapoptotic caspase-associated recruitment domain family protein overexpressed in cancer. J Biol Chem 276(34):32220–32229
Pautz A, Linker K, Hubrich T, Korhonen R, Altenhofer S, Kleinert H (2006) The polypyrimidine tract-binding protein (PTB) is involved in the post-transcriptional regulation of human inducible nitric oxide synthase expression. J Biol Chem 281:32294–32302
Pio R, Blanco D, Pajares MJ, Aibar E, Durany O, Ezponda T, Agorreta J, Gomez-Roman J, Anton MA, Rubio A et al (2010) Development of a novel splice array platform and its application in the identification of alternative splice variants in lung cancer. BMC Genomics 11:352
Porebska I, Sobańska E, Kosacka M, Jankowska R (2010) Apoptotic regulators: P53 and survivin expression in non-small cell lung cancer. Cancer Genomics Proteomics 7(6):331–335
Prasad S, Yadav VR, Kannappan R, Aggarwal BB (2011) Ursolic acid, a pentacyclin triterpene, potentiates TRAIL-induced apoptosis through p53-independent up-regulation of death receptors: evidence for the role of reactive oxygen species and JNK. J Biol Chem 286(7):5546–5557
Refaeli Y, Van Parijs L, London CA, Tschopp J, Abbas AK (1998) Biochemical mechanisms of IL-2-regulated Fas-mediated T cell apoptosis. Immunity 8(5):615–623
Reggiori F, Klionsky DJ (2002) Autophagy in the eukaryotic cell. Eukaryot Cell 1:11–21
Renshaw SA, Dempsey CE, Barnes FA, Bagstaff SM, Dower SK, Bingle CD, Whyte MK (2004) Three novel Bid proteins generated by alternative splicing of the human Bid gene. J Biol Chem 279(4):2846–2855
Rintala-Maki ND, Sutherland LC (2004) LUCA-15/RBM5, a putative tumour suppressor, enhances multiple receptor-initiated death signals. Apoptosis 9(4):475–484
Salvesen GS, Abrams JM (2004) Caspase activation—stepping on the gas or releasing the brakes? Lessons from humans and flies Oncogene 23:2774–2784
Samejima K, Earnshaw WC (2000) Differential localization of ICAD-L and ICAD-S in cells due to removal of a C-terminal NLS from ICAD-L by alternative splicing. Exp Cell Res 255:314– 320
Sampath J, Pelus LM (2007) Alternative splice variants of survivin as potential targets in cancer. Curr Drug Discov Technol 4(3):174–191
Schiavone N, Rosini P, Quattrone A, Donnini M, Lapucci A, Citti L, Bevilacqua A, Nicolin A, Capaccioli S (2000) A conserved AU-rich element in the 3′ untranslated region of bcl-2 mRNA is endowed with a destabilizing function that is involved in bcl-2 down-regulation during apoptosis. FASEB J 14(1):174–184
Schin KS, Clever U (1965) Lysosomal and free acid phosphatase in salivary glands of chironomus tentans. Science 150:1053–1055
Schin KS, Clever U (1968) Ultrastructural and cytochemical studies of salivary gland regression in Chironomus tentans. Z Zellforsch Mikrosk Anat 86:262–279
Schwerk C, Schulze-Osthoff K (2005) Regulation of apoptosis by alternative pre-mRNA splicing.Mol Cell 19(1):1–13
Screaton GR, Xu XN, Olsen AL, Cowper AE, Tan R, McMichael AJ, Bell JI (1997) LARD: a new lymphoid-specific death domain containing receptor regulated by alternative pre-mRNA splicing. Proc Nat Acad Sci USA 94:4615–4619
Servida F, Lecis D, Scavullo C, Drago C, Seneci P, Carlo-Stella C, Manzoni L, Polli E, Lambertenghi Deliliers G, Delia D, Onida F (2011) Novel second mitochondria-derived activator of caspases (Smac) mimetic compounds sensitize human leukemic cell lines to conventional chemotherapeutic drug-induced and death receptor-mediated apoptosis. Invest New Drugs 29(6):1264–1275
Shankarling G, Lynch KW (2010) Living or dying by RNA processing: caspase expression in NSCLC. J Clin Invest 120:3798–3801
Sharma S, Black DL (2006) Maps, codes, and sequence elements: can we predict the protein output from an alternatively spliced locus? Neuron 52:574–576
Shieh JJ, Liu KT, Huang SW, Chen YJ, Hsieh TY (2009) Modification of alternative splicing of Mcl-1 pre-mRNA using antisense morpholino oligonucleotides induces apoptosis in basal cell carcinoma cells. J Invest Dermatol 129(10):2497–2506
Singh RK, Cooper TA (2012) Pre-mRNA splicing in disease and therapeutics. Trends Mol Med 18(8):472–482
Smirnova AS, Ferreira-Silva KC, Mine KL, Andrade-Oliveira V, Shulzhenko N, Gerbase-DeLima M, Morgun A (2008) Differential expression of new LTA splice variants upon lymphocyte activation. Mol Immunol 45(1):295–300
Solier S, Logette E, Desoche L, Solary E, Corcos L (2005) Nonsense-mediated mRNA decay among human caspases: the caspase-2S putative protein is encoded by an extremely short-lived mRNA. Cell Death Differ 12(6):687–689
Spellman R, Rideau A, Matlin A, Gooding C, Robinson F, McGlincy N, Grellscheid SN, Southby J, Wollerton M, Smith CWJ (2005) Regulation of alternative splicing by PTB and associated factors. Biochem Soc Trans 33:457–460
Stenner M, Weinell A, Ponert T, Hardt A, Hahn M, Preuss SF, Guntinas-Lichius O, Klussmann JP (2010) Cytoplasmic expression of survivin is an independent predictor of poor prognosis in patients with salivary gland cancer. Histopathology 57(5):699–706
Takehara T, Liu X, Fujimoto J, Friedman SL, Takahashi H (2001) Expression and role of Bcl-xL in human hepatocellular carcinomas. Hepatology 34(1):55–61
Talbot DC, Ranson M, Davies J, Lahn M, Callies S, André V, Kadam S, Burgess M, Slapak C, Olsen AL, McHugh PJ, de Bono JS, Matthews J, Saleem A, Price P (2010) Tumor survivin is downregulated by the antisense oligonucleotide LY2181308: a proof-of-concept, first-in-human dose study. Clin Cancer Res 16(24):6150–6158
Taper HS, Jamison JM, Gilloteaux J, Summers JL, Calderon PB (2004) Inhibition of the development of metastases by dietary vitamin C:K3 combination. Life Sci 75(8):955–967
Tsuchihara K, Fujii S, Esumi H (2009) Autophagy and cancer: dynamism of the metabolism of tumor cells and tissues. Cancer Lett 278(2):130–138
van Alphen RJ, Wiemer EA, Burger H, Eskens FA (2009) The spliceosome as target for anticancer treatment. Br J Cancer 100:228–232
Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA (2009) Autophagy facilitates the development of breast cancer resistance to the anti-HER2 monoclonal antibody trastuzumab. PLoS One 4(7):e6251
Vucic D, Stennicke HR, Pisabarro MT, Salvesen GS, Dixit VM (2000) ML-IAP, a novel inhibitor of apoptosis that is preferentially expressed in human melanomas. Curr Biol 10(21):1359–1366
Vudattu NK, Magalhaes I, Hoehn H, Pan D, Maeurer MJ (2009) Expression analysis and functional activity of interleukin-7 splice variants. Genes Immun 10(2):132–140
Wagner EJ, Garcia-Blanco MA (2001) Polypyrimidine tract binding protein antagonizes exon definition. Mol Cell Biol 21:3281–3288
Wang Y, Geng Z, Zhao L, Huang SH, Sheng AL, Chen ZY (2008) GDNF isoform affects intracellular trafficking and secretion of GDNF in neuronal cells. Brain Res 1226:1–7
Wang Z, Burge CB (2008) Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA 14(5):802–813
Ward AJ, Cooper TA (2010) The pathobiology of splicing. J Pathol 220:152–163
Wu JY, Tang H, avlioglu N (2003) Alternative pre-mRNA splicing and regulation of programmed cell death. Prog Mol Subcell Biol 31:153–185
Wu J, Dang Y, Su W, Liu C, Ma H, Shan Y, Pei Y, Wan B, Guo J, Yu L (2006) Molecular cloning and characterization of rat LC3A and LC3B–two novel markers of autophagosome. Biochem Biophys Res Commun 339:437–442
Wu JY, Yuan L, Havlioglu N (2004) Alternatively spliced genes. In Meyers RA (ed.) Encyclopedia of Molecular Cell Biology and Molecular Medicine. Wiley-VCH, pp. 125–177
Wullschleger S, Loewith R, Hall MN (2006) TOR signaling in growth and metabolism. Cell 124:471–484
Wyllie AH, Kerr JF, Currie AR (1980) Cell death: the significance of apoptosis. Int Rev Cytol 68:251–306
Xerri L, Parc P, Brousset P, Schlaifer D, Hassoun J, Reed JC, Krajewski S, Birnbaum D (1996) Predominant expression of the long isoform of Bcl-x (Bcl-xL) in human lymphomas. Brit J Haematol 92:900–906
Xiao X, Lee JH (2010) Systems analysis of alternative splicing and its regulation. Wiley Interdiscip Rev Syst Biol Med 2(5):550–556
Yamashima T, Oikawa S (2009) The role of lysosomal rupture in neuronal death. Prog Neurobiol 89(4):343–358
Yagihashi A, Ohmura T, Asanuma K, Kobayashi D, Tsuji N, Torigoe T, Sato N, Hirata K, Watanabe N (2005) Detection of autoantibodies to survivin and livin in sera from patients with breast cancer. Clinica chimica acta; Int J Clin Chem 362:125–130
Yang Y, Chang JF, Parnes JR, Fathman CG (1998) T cell receptor (TCR) engagement leads to activation-induced splicing of tumor necrosis factor (TNF) nuclear pre-mRNA. J Exp Med 188(2):247–254
Yang G, Huang SC, Wu JY, Benz EJ Jr (2005) An erythroid differentiation-specific splicing switch in protein 4.1R mediated by the interaction of SF2/ASF with an exonic splicing enhancer. Blood 105:2146–2153
Yano K, Horinaka M, Yoshida T, Yasuda T, Taniguchi H, Goda AE, Wakada M, Yoshikawa S, Nakamura T, Kawauchi A, Miki T, Sakai T (2011) Chetomin induces degradation of XIAP and enhances TRAIL sensitivity in urogenital cancer cells. Int J Oncol 38(2):365–374
Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ (2004) Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 304:1500–1502
Yu L, Lenardo MJ, Baehrecke EH (2004) Autophagy and caspases: a new cell death program. Cell Cycle 3:1124–1126
Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F et al (2010) Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465:942–946
Zhang T, Huang XH, Dong L, Hu D, Ge C, Zhan YQ, Xu WX, Yu M, Li W, Wang X et al (2010) PCBP-1 regulates alternative splicing of the CD44 gene and inhibits invasion in human hepatoma cell line HepG2 cells. Mol Cancer 9:72
Zhou A, Ou AC, Cho A, Benz EJ Jr, Huang SC (2008) Novel splicing factor RBM25 modulates Bcl-x pre-mRNA 5′ splice site selection. Mol Cell Biol 28:5924–5936
Zinkel SS, Hurov KE, Ong C, Abtahi FM, Gross A, Korsmeyer SJ (2005) A role for proapoptotic BID in the DNA-damage response. Cell 122(4):579–591
Zwicky R, Müntener K, Csucs G, Goldring MB, Baici A (2003) Exploring the role of 5′ alternative splicing and of the 3′-untranslated region of cathepsin B mRNA. Biol Chem 384(7):1007–1018
Acknowledgments
We apologize to our colleagues for not being able to cite their original research articles due to the space limit. The authors’ work was supported by grants from the NIH to Wu JY, a Scholar Award from the Leukemia Society of America. We would like to thank members of Wu lab for critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Ruirui, K. et al. (2013). Alternative Pre-mRNA Splicing, Cell Death, and Cancer. In: Wu, J. (eds) RNA and Cancer. Cancer Treatment and Research, vol 158. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-31659-3_8
Download citation
DOI: https://doi.org/10.1007/978-3-642-31659-3_8
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-31658-6
Online ISBN: 978-3-642-31659-3
eBook Packages: MedicineMedicine (R0)