
Abstract
Boron neutron capture therapy (BNCT) strongly depends on the selective uptake of 10B in tumor cells and on the 10B distribution inside single cells. The chemical properties of boron and the need to discriminate different isotopes make the investigation of the concentration and distribution of 10B a challenging task. The most advanced techniques to measure the boron concentration and distribution in tissues and liquids are described in this chapter.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Positron Emission Tomography
- Inductively Couple Plasma Atomic Emission Spectroscopy
- Boron Concentration
- Inductively Couple Plasma Optical Emission Spectrometry
- Electron Energy Loss Spectroscopy
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Boron has two stable naturally occurring isotopes, 10B and 11B, with natural abundance of approximately 19 and 81 %, respectively. In biology, boron is an essential trace element for the growth of higher plants. Even in organisms in which boron is present but has not been established as essential (e.g., animals and humans), this element has been recognized to be beneficial.
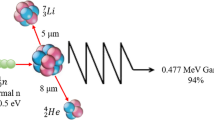
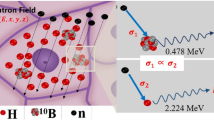
Boron neutron capture therapy (BNCT) exploits the ability of the isotope 10B to capture thermal neutrons with a very high probability, leading to the nuclear reaction 10B(n,α,γ)7Li. This reaction produces 478 keV gamma rays, He-4 particles, and Li-7 recoil ions, the latter two having high linear energy transfer (LET) properties and a high relative biological effectiveness (RBE) relative to photon irradiation. The range of these particles in tissue is limited to 8 and 4 μm, respectively, which restricts their effects to one cell diameter. Therefore, if the 10B can be selectively delivered to tumor cells, the short range of the high LET-charged particles offers the potential for a targeted irradiation of individual tumor cells [1, 2].
The energy released in matter by the α and 7Li particles is inhomogeneous. As the range of these particles is so small, the clinical efficacy of the therapy is only guaranteed if the boron is located inside a tumor cell and ideally close to the cell nucleus.
It is therefore a crucially important requirement to investigate the physiological and pharmacological behavior of the 10B in the patient. The methods used should be suitable for accurately measuring the bulk 10B concentration in tissue and fluids such as blood and urine as well as its microscopic spatial distribution at the cellular or subcellular level. Ideally, these methods should be noninvasive, allowing for in vivo measurements in the patient, and results should be obtained within a time period allowing for clinical decisions based on such results.
The following sections describe methods for measuring the quantitative distribution of boron and boron compounds on a microscopic and macroscopic level. All methods have been used in BNCT applications both in preclinical and clinical investigations and have partly been adapted for this special purpose. This chapter aims at describing and comparing these methods with emphasis on a short technical background, the specific endpoint, application possibilities complexity, and pitfalls. This chapter condenses a more detailed description especially of technical aspects as published in 2008 [2], which might be additionally consulted by the interested reader.
2 Description of Methods
2.1 Prompt Gamma-Ray Spectroscopy
Prompt gamma-ray analysis (PGRA) is a fast method for measuring the average 10B content in macroscopic samples [3–5], which has been used in BNCT studies since many years [6–10].
The principle of PGRA is based on gamma-ray spectroscopy following neutron capture in 10B. The recoiling 7Li nuclei from the 10B(n, α)7Li reaction decays to the ground state of 7Li by the emission of 478 keV photons. The emission rate of the photons is proportional to the reaction rate of the neutron capture reaction and therefore carries information of the 10B concentration. Among other possibilities, nuclear reactors provide the most usual sources of neutrons. Often, filtering systems are needed to produce a beam of neutrons with the appropriate quality. Other elements present in the sample also emit photons after neutron capture, the most relevant being the 2.2 MeV photon from the 1H(n, γ) reaction. The count rate of this hydrogen line can be used as a neutron fluence monitor [11].
Of note, exclusively the 10B isotope can be quantified with this method. PGRA is not applicable for 11B and only indirectly applicable for the naturally occurring B isotope if the 10B to 11B ratio is precisely known.
The content of the 10B line in PGRA for an unknown sample must be calibrated against 10B reference samples with known 10B concentrations. Intercalibration between PGRA and other methods for 10B quantitation, is mandatory for the detection of possible systematic errors.
Sample changing and 10B analysis can be fully automatized. Facilities operating 24 h a day are therefore suited to process a large number of samples. Sample preparation is quite simple. For liquid samples (e.g., blood, urine), a fixed volume is injected into a standard vial and then weighted. Tissue samples are just weighted. The vials are positioned in a sample changer, which allows for automatic measurements.
Present PGRA facilities provide fast (about 5 min/sample), accurate (standard deviation about 0.5 ppm), and nondestructive measurements of 10B concentrations down to 1 ppm, suited for macroscopic samples (0.4–1.0 ml) of tissue, blood, and urine.
The major limitation of this method, the relatively large sample size, can be further reduced by improvement of the shielding and the geometry of the facility to decrease the background in the gamma-ray spectra. PGRA measures the integral 10B concentration within a sample; therefore, it cannot show any inhomogeneities of the 10B concentration within a possibly inhomogeneous sample. Such inhomogeneities can however be relevant in BNCT as the tissue volume, which can be measured with PGRA, is far bigger than the volume irradiated with a boron neutron capture reaction.
The PGRA has been applied to quality control of the compounds BSH and BPA, boron uptake studies, pharmacokinetic studies, and blood-boron concentration measurements during BNCT of patients [2, 5]. The latter is an important tool for the patient treatment planning adjustment and dosimetry during BNCT: the measured 10B concentration in the blood of a patient can be used to adjust the calculated pharmacokinetic curve to the actual situation of an individual patient. This allows readjustment of the calculated beam exposure time during a BNCT treatment to reach the intended total absorbed dose. To make such adjustment during a BNCT treatment, a very quick analytical method is mandatory, as is the case for PGRA. As of today, PGRA is the only available tool for such purpose; therefore, the method plays an essential role for clinical BNCT [2].
Further application of PGRA for BNCT includes the possibility of in vivo gamma-ray spectroscopy of the patient during treatment [12, 13]. The gamma-ray telescope can provide in vivo dosimetry and measurement of 10B concentrations, averaged over a volume of several cm3 and over a time interval of about 2 min. This method, however, needs further improvement for implementation in a clinical routine [2].
2.2 Inductively Coupled Plasma Spectroscopy
Inductively coupled plasma atomic emission spectroscopy (ICP-AES), also referred to as inductively coupled plasma optical emission spectrometry (ICP-OES), is an emission spectrophotometric technique, exploiting the fact that excited electrons emit energy at a given wavelength as they return to ground state. The fundamental characteristic of this process is that each element emits energy at specific wavelengths peculiar to its chemical character. Thus, by determining which wavelengths are emitted by a sample and by determining their intensities, the elemental composition of the given sample relative to a reference standard can be quantified [2].
ICP-MS (inductively coupled plasma mass spectrometry) is a type of mass spectrometry that is highly sensitive and capable of the determination of a range of metals and several nonmetals at concentrations below one part in 10 × E12. It is based on coupling an inductively coupled plasma as a method of ionization with a mass spectrometer as a method of separating and detecting the ions. ICP-MS is also capable of monitoring isotopic speciation for the ions of choice such as the individual isotopes of boron with the atomic masses of 10 and 11. Hence, ICP-MS can quantify each isotope individually.
ICP-MS methods are applicable to quantify boron at ppb levels in serum, plasma, urine, saline, water, and tissue. Boron isotopes are determinable with better than 2 % relative standard deviation (RSD), which means very accurate data can be obtained using isotope dilution. While the sensitivity of ICP-MS for boron and other elements is excellent, their backgrounds, their tendency to adsorb onto glass, and their chemistry all have to be controlled for successful quantitative analysis in complex matrices such as biological fluids or tissues [2]. Therefore, a rugged, validated analytical procedure for sample preparation and analysis using ICP-MS is to be demanded for boron determination.
The principal concerns when using ICP-MS for isotopic determinations of boron are memory effects and mass discrimination. Special washing agents help preventing the memory effect [2]. Mass discrimination between 11B and 10B is accentuated due to the 10 % isotopic mass difference and arises during ion transport caused by instrumental adjustments or by the sample matrix [14]. The instrumental effect is corrected by analyzing a reference standard with a certified 11B/10B ratio [15]. The sample matrix effect on mass discrimination can be corrected using internal standards [16, 17].
The ICP-MS is often the method of choice over ICP-OES and spectrophotometric methods for analysis of boron [18]. The advantages of ICP-MS over other methods are higher sensitivity, lower detection limits, and simultaneous measurement of 10B to 11B isotopic ratio and total B concentration in a sample. The ability of ICP-MS to measure B isotope ratios renders this instrument especially suitable for biological B tracer studies [19].
The detection limits are at the ppb level, e.g., 1 ppb [20] to 3 ppb [18] in biological materials, 0.15 ppb in saline waters [16], and 0.5 ppb in human serum [21]. The uniqueness of ICP-MS is also due to its capability to carry out B determination by the isotope dilution method which is considered the most precise for quantitative determination. Laakso et al. [22] compared both the ICP-MS as well as the ICP-AES (ICP-OES) method and found a strong correlation between results of the ICP-AES and ICP-MS (r = 0.994). They concluded, that their established ICP-AES method is also feasible, accurate, and one of the fastest for boron determination during BNCT. ICP-AES has been applied to quality control of the compounds and boron uptake studies as well as boron concentration measurements in blood of patients [23].
ICP-MS is a reliable technique for the determination of the mean concentration of total boron and mean isotopic ratios of boron in biological samples. It is possible to quantify boron in liquids and – after suitable disintegration – in tissue samples. In contrast to PGRA which measures the mean 10B concentration in a sample, an irradiation with neutrons is not necessary, and therefore the method is independent of a nuclear reactor site. ICP-MS however is destructive. It is not appropriate for imaging the distribution of boron in histological preparations [2].
2.3 High-Resolution Alpha Autoradiography, Alpha Spectroscopy, and Neutron Capture Radiography
2.3.1 High-Resolution Alpha Autoradiography
The first experiments in which a neutron-induced nuclear reaction was utilized to image a stable nuclide in a sample was reported by Ficq [24] and reported in the context of BNCT first by Edwards [25]. Subsequently, variants of Edwards’ approach that provide cellular level spatial resolution have been reported by Solares et al. [26, 27] and Yam et al. [28]. These approaches are based on the transient activation of the stable isotope 10B using thermal neutron activation to convert 10B to the unstable isotope 11B. 11B disintegrates into alpha and 7Li particles sharing 2.3–2.4 MeV of kinetic energy between them. These charged particles have corresponding ranges in tissue of 8 and 4 μm, respectively, making the mapping of these particles a useful surrogate measurement of the 10B distribution itself.
The high-resolution alpha-track autoradiography (HRAR) is possible in small tissue samples, which are surgically removed after 10B compound administration and immediately frozen. One to two μm thick histological frozen tissue sections are cut and mounted on top of submicrometer thick Ixan and Lexan films on top of a quartz glass slide. The quartz slide/tissue/Ixan/Lexan units are packed with dry ice and irradiated with thermal neutrons. Upon completion of irradiation, the tissue sections are histologically stained. The quartz glass slide is then remounted in contact with the tissue section on the opposing side of the unit thereby exposing the Lexan film. During neutron irradiation, the interaction of the alpha- and 7Li-charged particles with the Lexan film from the 10B neutron capture reactions in the frozen tissue causes molecular weakening of the Lexan along the paths of these particles. Chemically etching the exposed surface of the Lexan film yields optically visible 1–2-μm diameter tracks. The Ixan film located between the frozen tissue section and the Lexan film act as a chemical barrier ensuring that the corrosive etchant fluid seeping through the etched tracks does not damage the stained tissue. Finally, digital microphotographs are taken of the superimposed tracks and stained tissue sections. Correlation of the distribution of tracks with the anatomical features of the stained tissue provides maps of 10B distribution in the tissue. A calibration curve of track density in standard tissues vs. known concentration of boron provides the ability to relate the observed track densities in the experimental tissue samples to absolute boron concentrations. The HRAR calibration curve is highly linear over the entire range of boron concentrations of interest. Saturation effects at the high end of the calibration curve can be corrected by irradiation with lower thermal neutron fluence. This approach of track counting as opposed to film darkening, as utilized in boron neutron capture radiography, provides a more quantitative analysis of the boron distribution in tissue but does not provide the useful macroscopic visual depiction of boron distribution that is extremely useful for many applications in BNCT [2].
The method has been used to study the boron concentration in normal tissues and tumors BNCT animal models, e.g., rat tongue and brain [29, 30].
Another level of analysis using HRAR at the initial stage first described by Solares et al. [31] and Kiger et al. [30] uses a Monte Carlo code to simulate the trajectories of the alpha- and 7Li-charged particles within the tissue using the measured track locations from the HRAR images to determine the number of nuclear “hits” and other important microdosimetric quantities. This approach investigates the more realistic domain of the expected responses of normal and tumor tissues to BNCT [2].
2.3.2 Neutron Capture Radiography
Neutron capture radiography (NCR) can be combined with particle spectroscopy and histological analysis of neighboring tissue sections to measure the 10B concentration and its spatial distribution in relation to the histological information [32–36]. Unlike the HRAR technique, NCR provides intuitive visualization of the boron distribution, which may also be quantified on a macroscopic scale by densitometric analysis [37]. The 10B concentration is measured using the spectroscopy of charged particles [38] emitted in the boron neutron capture reaction and is then spatially correlated with stained tissue sections. Tissue sample of a few millimeter thickness and an area of approximately 0.5 cm2 is needed for the NCR technique. The tissue is cryofixed by immersion of the sample into liquid nitrogen to statically “freeze” the physiological distribution of the 10B-containing molecule. For each measurement, a minimum of three neighboring slices are cut from the tissue block using a cryostat operating at a temperature of −20 °C.
-
1.
The first section cut is deposited on a Mylar disk for charged particle spectroscopy.
-
2.
The second section is deposited on a glass slice for histological analysis by hematoxylin-eosin staining using a light microscope coupled with software for image analysis. It is optimized to gain information on the biological composition of the sample, especially to evaluate the percentage of tumor cells, healthy parenchyma, necrosis, fibrosis, mucus, blood vessels, and other tissues in a specific sample.
-
3.
The third slice is deposited on a solid state nuclear track detector for imaging of the macroscopic boron distribution by neutron capture radiography (NCR).
The third tissue slice is positioned on a nuclear detection film and exposed to thermal neutrons. At the locations in the tissue sample where a 10B atom is present, an α particle and a 7Li ion are emitted and cause damage to the nuclear detection film. After irradiation, the tissue sample is removed from the film, and the image of the 10B distribution is obtained by chemical etching in a dilute NaOH solution [39]. The distribution of the tracks in the detector is representative of the macroscopic distribution of the 10B atoms in the sample. Using the NCR technique, it is possible to digitally superimpose and compare the histological image with a map of the spatial 10B distribution and relative 10B concentration [2]. One example of such correlation between the macroscopic boron distribution and tissue histology is depicted in Fig. 9.1.

Comparison between (a) histology and (b) neutron capture radiography of a liver sample from a patient with liver metastases of colorectal adenocarcinoma after infusion of BPA. The tumor zone is above the green line while normal liver is below it. In the neutron capture radiography image the darker zone (higher boron concentration) corresponds to the tumor while the lighter one (lower boron concentration) corresponds to normal liver. The boron concentration ratio between tumor and liver is about 3×. The tissue sample is 7 mm × 10 mm × 10 μm thick (S. Altieri, Department of Nuclear and Theoretical Physics, University of Pavia, and National Institute of Nuclear Physics (INFN) – Pavia Section, Pavia, Italy) (Reprinted from Wittig et al. [2])
To determine the absolute 10B concentration using particle spectroscopy, section 1 is used. If exclusively normal tissue is present in the sample, the absolute 10B concentration can be directly determined by spectrometric analysis. If the sample contains both, normal and tumor tissue, the regional tumor tissue 10B concentration can be calculated by subtracting the amount of the boron contained in normal tissue (determined in a tumor-free location based on morphological appearance) from the total 10B content.
The first tissue sample section on Mylar disk is placed in front of a silicone solid state detector (Si detector) in a vacuum container and exposed to thermal neutron irradiation. Alpha particles and 7Li ions induced in the boron by the neutrons are detected in the Si detector and their energy distribution spectra are recorded by a multichannel analyzer.
The experimental error of this method is in the order of ±10 %. The lower limit of detection is approximately 0.5 ppm and is limited by the naturally occurring boron background concentration in tissue. The accuracy of the method depends on the number of counts, the irradiation time, and the number of boron atoms in the sample.
The main advantage of the charged particle spectrometry is the ability to measure the 10B concentration as well as the 10B distribution in a two-dimensional slice of tissue and to directly correlate the spatial information at the macroscopic level to the histology of the specimen analyzed. The lateral resolution of the method is limited to approximately 100 μm.
The necessity to irradiate tissue samples with thermal neutrons limits this method as the other two techniques described in this section to very few specialized nuclear research centers. Moreover, it is not possible to directly measure liquid samples using charged particle spectroscopy as samples have to be measured under vacuum. To obtain absolute results, it is necessary to know the mass stopping power for each material to be analyzed or to use a set of reference standards with known boron concentrations representing typical biological tissue types [2].
2.4 Laser Post-ionization Secondary Neutral Mass Spectrometry
The range of the alpha particles from the boron capture reaction is approximately 10 μm. If the irradiated volume is to be investigated, then methods are necessary that have a similar resolution. Determination of boron distributions at the subcellular level is crucial to understanding the behavior of boron compounds in tissues and cells and its impact on radiation response.
Laser post-ionization secondary neutral mass spectrometry (laser-SNMS) and secondary ion mass spectrometry (SIMS) are alternative techniques for element- and molecule-specific imaging. SIMS analysis utilizes a technique in which the sample is bombarded with a focused, energetic ion beam that sputters atoms and molecules off the surface. A fraction of these particles are ejected as ions which are extracted and mass analyzed. Combining SIMS with a time-of-flight mass spectrometer (ToF-SIMS), all masses can be detected simultaneously on each primary ion pulse, and charge compensation for insulator analysis is then possible using pulsed low-energy electrons, which are introduced during the time interval between ion pulses. If a liquid metal ion gun is used, the ion beam can be focused from a few μm down to <200 nm in diameter, enabling analysis with nanometer-scale spatial resolution. Imaging is carried out either by scanning the ion beam over the sample (500 μm × 500 μm or smaller) or by moving the sample under a fixed ion beam position (up to 10 cm × 10 cm). By combining the imaging technique with an additional sputtering ion beam to remove successive atomic/molecular layers, a three-dimensional image of the chemical composition of the surface can be obtained [2].
Although SIMS has many advantages and has often been used in the dynamic SIMS mode for imaging boronated compounds in cell cultures and tissues [40–45], its application for boron imaging is often limited because of a lack of sensitivity and quantitation. Detection limit, efficiency, and quantitation, however, can be significantly improved by the use of laser-SNMS [46–48]. This technique, with the same technical features as ToF-SIMS, decouples the sputtering and ionization processes by applying laser beams to ionize the majority of neutral particles sputtered from the sample surface. The neutral particles yield is less affected by the chemical composition of the surface than the much smaller yield of secondary ions used for SIMS analysis, resulting in much greater sensitivity and accuracy of laser-SNMS than SIMS. Furthermore, laser-SNMS images are significantly less influenced by topographical effects than SIMS images.
Laser-SNMS has been used for examining the subcellular distribution of intrinsic elements and molecules as well as the potential of BSH and BPA to deliver 10B to tumor cells in vitro and in vivo to cells of experimental tumors and of normal tissues. The analysis of the samples is carried out with a combined gridless reflectron-based laser-SNMS/ToF-SIMS instrument. Ion-induced electron images (IIEI) can be obtained from all samples to select regions of interest for further analysis [2].
In order to be able to analyze the samples in the vacuum system, cryofixation, freeze-fracturing, and freeze-drying of the living cell while preserving its structural and chemical integrity must be carried out [49–51]. A dedicated cryofixation method with very high cooling rate must therefore be used.
Laser-SNMS was used in vitro and in animal experiments employing the described preparation technique to analyze normal tissues and different experimental tumors after treatment of mice with the compounds BPA and BSH. Laser-SNMS simultaneously delivers maps of the distribution of elements and molecules such as K, Na, Ca, CN, and C3H8N as well as a distinct boron distribution. Proof of a Na and K concentration and distribution as known from living cells serve to ascertain successful preparation of the tissue.
A challenging task is however correlation of elemental/molecular maps with histological information [2]. Such correlation is possible through analysis of distinct molecular fragments: earlier studies on L-α-dipalmitoylphosphatidylcholine (DPPC) membrane model systems have shown that C3H8N is a typical fragment of the choline headgroup of the DPPC [52, 53]. The CN signal contribution originates mainly from DNA (purine and pyrimidine) and proteins. In particular, the subcellular distribution of the membrane-characteristic C3H8N signal shows a distinct pattern. The signal almost disappears in the center of some cells. It may be presumed that, in these areas, the nucleic membrane was removed by the fracturing process, leaving behind an open nucleus. In the CN pattern, however, no decreases are observed at the positions where the C3H8N signal almost disappears. This can be explained by the fact that CN originates not only from fragments of membrane constituents and proteins but also from fragments of DNA constituents such as purine and pyrimidine rings. Therefore, at the site of the nucleus, the CN signal should not disappear.
By overlaying images of the distribution of different molecules with distinct distribution [2], e.g., CN and C3H8N, it is possible to denote cell nuclei and cell membranes and to relate the boron signal to such regions. With this method, it is possible to investigate the boron concentration and distribution within single cells. The laser-SNMS has proven 10B as delivered with BSH and BPA within tumor cells and even within the cell nucleus [2].
Figure 9.2 shows an example of laser-SNMS images of a kidney sample from a nude mouse treated with a combination of BPA and BSH. Intensive signals from several elements and molecules such as 10B, CN, and C3H8N were observed in the laser-SNMS images (top row). The 10B image (top left) shows a distinct boron distribution. The molecules (top middle and right) represent mainly lipids, proteins, and nucleic acids and also exhibit distinct features. The bottom images show overlay images, where the CN image is overlaid with the C3H8N image (bottom left) and 10B with the C3H8N image (bottom right). Here, the CN signal, corresponding to nucleus sites, is presented as red, the C3H8N as green, and 10B signal as blue in color. Individual kidney tubules and lumen can be identified in the overlay image (tubules surrounded by single cells). The high concentrations of boron are observed especially in the lumen of the tubule, which was cut along its longitudinal axis (bottom left part of the image), and in the basal membrane surrounding the tubules.

Laser-SNMS images of a kidney sample from a NMRI nude mouse treated with a combination of BPA and BSH (H.F. Arlinghaus, Physikalisches Institut, Westfälische Wilhelms-Universität Münster, Münster, Germany) (Reprinted from Arlinghaus et al. [49])
Figure 9.3 shows an image of a murine sarcoma grown in a mouse treated with BPA [2]. The top row shows the 10B, CN, and C3H8N images again showing distinct distributions. The bottom row demonstrates images where CN is overlaid with C3H8N (bottom left) and 10B is overlaid with C3H8N (bottom right). Here, the highest 10B signal is presented as blue and the highest CN signal, which again denotes the nuclei, as red.

Laser-SNMS images of a murine sarcoma tumor sample from a NMRI nude mouse treated with the compound BPA (H.F. Arlinghaus, Physikalisches Institut, Westfälische Wilhelms-Universität Münster, Münster, Germany) (Reprinted from Arlinghaus et al. [49])
In summary, the cryopreparation techniques described preserve the structural and chemical integrity of cell cultures and tissues and make cellular structures directly accessible for laser-SNMS analysis. In particular, where molecular images are available, different functional areas of the cells can be identified. Laser-SNMS is particularly well suited for identifying specific cell structures and for imaging ultratrace 10B concentrations in tissues with nanometer-scale lateral resolution and a detection limit in the upper ppb renage. These possibilities make laser-SNMS a very valuable tool in directly imaging and quantifying 10B and (simultaneously) other elements. Due to the necessity of the demanding preparation of cells or tissues and the challenging measurements, the method is reserved to special questions but not suitable as a routine method for direct clinical decisions [2].
2.5 Electron Energy Loss Spectroscopy
Regarding boron imaging, the best spatial resolution is obtained using methods based on electron microscopy. Two such methods exist: immunohistochemistry [54] and electron energy loss spectroscopy (EELS) [55, 56]. Immunochemistry is a very sensitive method, but it has its drawbacks: indirect detection leading to the possibility of artifacts (e.g., redistribution during the fixation process) and the need to prepare a specific antibody for each potential boron carrier molecule. EELS is based on the inelastic scattering of incoming electrons after interaction with the sample electrons. Inelastic scatter results in a characteristic energy loss depending on the atomic or molecular energy level involved. It has been well established that EELS is the most sensitive nanoanalytical method for detecting light elements such as carbon, boron, or even phosphorus and particularly in a biological tissue [57, 58].
Two experimentally different spectroscopic approaches have been derived from this theoretical basis: EELS and electron spectroscopic imaging (ESI), which is an imaging technique coupled with energy-filtering transmission electron microscopy (EFTEM). Furthermore, EELS is an analytical technique generally coupled with transmission or scanning transmission electron microscopy (TEM/STEM). It is important to keep in mind that despite significant experimental differences, these two approaches (EELS and ESI) are based on the same physical signal and that knowledge of the EELS spectrum is required in order to understand and to safely use the EFTEM images. An electron energy loss spectrum corresponds to the counting of the electrons passing through a defined area of the sample.
A critical point in the study of elemental distributions in tissues and cells is sample preparation. A cryopreparation method, avoiding any chemical treatments, has to be used in order to immobilize also small molecules and ions close to their positions in the natural state. It implies successive cryofixation, cryosectioning, and cryo-observation. A specific step in EELS is the need for ultrathin cryosections (100 nm or less), which are freeze-dried inside the microscope by raising the temperature to 193 K and finally investigated at a temperature below 110 K.
The boron signal in the EELS spectrum is very weak and superimposed over a strong noncharacteristic background, which requires a dedicated method based on digital filtering to extract the signal [59]. Moreover, the boron K edge located at 188 eV can be confused with the phosphorus L1 signal. Consequently, the experimental filtered spectrum has to be modeled as a sum of a boron-filtered reference and a phosphorus-filtered reference spectrum [56]. Neglecting the phosphorus signal can lead to erroneous images, where boron is systematically detected in phosphorus-rich regions. This problem has also to be resolved using ESI. The detection limits can be estimated to be around a few tens of ppm in, for a sample area of 0.1 μm2 (see Ref. [56] for details of the method for boron detection and quantification).
Boron imaging is performed using the spectrum-imaging acquisition mode in which a spectrum is collected from each pixel in a defined STEM image area. In order to reveal the boron signal, each spectrum is processed (digital filtering, multiple least-squares fitting). Measured signals can then be presented as elemental maps to be correlated with the STEM image (Fig. 9.4) [2].

(a) Dark-field STEM image (inverted contrast) of a freeze-dried cryosection of mouse kidney tissue. (b) Phosphorus map, (c) corrected boron map, (d) uncorrected boron map (J. Michel, Laboratoire de Microscopie Electronique Analytique, INSERM ERM 0203, Université de Reims Champagne-Ardenne, Reims, France) (Reprinted from Michel et al. [90])
Measurements using EELS, thanks to their high spatial resolution, are most complementary to sensitive measurements obtained by other methods, such as SIMS or a nuclear microprobe technique. In this way, EELS can be a very specific tool to detect small accumulation areas of boron or boron compounds and to localize them at the subcellular level. Furthermore, a lack of boron in the EELS measurement in samples where boron is detected by other methods on a larger scale implies a homogeneous distribution of boron at the subcellular level in the limits of the EELS detection sensitivity. Boron detection by is independent of the isotopic form of the element. The isotope 10B cannot be discerned from 11B in the measurements. 10B contents have to be deduced from the previously known relative abundance of the different isotopes which can increase uncertainties in the final quantitative values. Similar to SIMS, EELS is an important data source for BNCT; the complexity of sample preparation, measurement, and data processing however prevents using EELS as a routine method [2].
2.6 Ion Trap Mass Spectrometry and Proteomic Technologies
The analytical methods described in the previous sections are usually applied to detect the isotope 10B only, but cannot be used to analyze and characterize the specific molecule of the relevant 10B-containing compound under investigation, which is highly desirable in in vivo and in clinical investigations.
This is however possible with ion trap mass spectrometry and proteomic technologies. For this technique, a liquid sample is injected into the mass spectrometer by means of electrospray interface (ESI). Thereafter, fragmentation of each specific molecule is performed using the ion trap mass spectrometer (tandem mass spectrometry) that allows isolation and fragmentation in the same space [60]. Ion trap mass spectrometry monitors the entire MS/MS spectrum with the same sensitivity. Mauri [61] proposed an analytical method using flow injection coupled with tandem mass spectrometry (FI/ESI-MS/MS) in order to obtain quantitative data of the boron carrier molecules. For such analysis, a sample is flow injected (FI) through an HPLC injector (injected volume approximately 1 μl) into the ESI-MS/MS system [2].
BSH is detected in a negative mode, and the main ion is m/z 187.4, corresponding to [(10B12H11SH)Na]−, while the product of its MS/MS fragmentation is an ion with an m/z of 131.5, due to the loss of the [SNa] residue. The dimer (BSSB) of BSH has a molecular weight of m/z 395.7, and its fragment ion is m/z 391.9 [61]. The detection of BPA has been performed in a positive mode, and its molecular and fragment ions are m/z 209.1 and m/z 163.1, respectively.
The FI/ESI-MS/MS methodology results in typical fingerprinting mass spectra of boron derivatives and allows for their quantitative determination [62]. When extracting the specific fragment ions, a linear relationship between the peak area and the concentration of the 10B-containing compound is found in the range of 10–10,000 ng/ml.
The high sensitivity of the FI/ESI-MS/MS method allows for dilution of biological samples, which reduces the matrix effect occurring due to the high salt concentrations in urine and plasma. High salt concentration produces an overloaded response on the mass spectrometer and reduce the signal (ion suppression) related to the metabolites of interest. 10B-containing compounds could be detected by FI/ESI-MS/MS in biological samples (urine and plasma, diluted 10,000- and 1,000-fold, respectively) when injecting a small volume (1 μl) of the diluted samples [62].
In conclusion, the FI/ESI-MS/MS is a rapid and quantitative method for the analysis of 10B-containing compounds in pharmaceutical preparations and biological samples. The method is particularly suited for analyzing liquid samples. The assay of 10B compounds in tissue samples requires extraction by 50 % methanol [2].
2.6.1 Ion Trap and Proteomics
Ion trap mass spectrometry coupled to two-dimensional chromatography (2DC-MS/MS) has been applied to develop a shotgun approach for proteomic studies [63]. It permits the simultaneous characterization of each of many proteins without limits concerning molecular weight or isoelectric point. Quantitative analysis is also possible [64]. This approach, also known as MudPIT (multidimensional protein identification technology), is very useful in clinical proteomics for biomarker discovery and to identify molecular targets for new drugs. This translational research aspect can also be applied for BNCT [65]. The MudPIT proteomic approach can be used for characterizing protein profiles of biological samples (urine and tissues) after 10B application, investigating the accumulation of boron in cancer cells and combining proteomic with pharmacokinetic data. The use of the above methodologies means that transport, metabolism, and uptake of the different drugs used in BNCT can be investigated in great detail [2].
2.7 Nuclear Magnetic Resonance and Magnetic Resonance Imaging
Both the 10B and 11B nuclei are amenable for detection by nuclear magnetic resonance (NMR), and therefore their spatial distribution can be mapped using magnetic resonance imaging (MRI). MRI is a special type of the NMR method where the frequencies of the nuclear spins are encoded according to their locations in space. Moreover, NMR can distinguish between nuclei of the same type in different molecular species (typically through an interaction called “chemical shift”) [2].
The sample is placed in the magnetic field of the NMR spectrometer or MRI scanner. Signal excitation and detection is achieved through a properly tuned radio-frequency (RF) coil, which (depending on its geometry) either encompasses the sample or is placed adjacent to it. Energy is delivered by short pulses (10−3–10−6 s) and can be absorbed by the nuclear spins if it matches the gaps in energy levels corresponding to different spin states. The frequencies of these transitions are proportional to the strength of the magnetic field and are typically in the order of 106–108 Hz. The RF pulses perturb the Boltzmann equilibrium of the nuclear spin populations, which decay back to equilibrium after the pulses are turned off. During this return to equilibrium, the nuclei emit signals at their characteristic resonance frequencies, which are detected, digitized, and interpreted to create spectra or images. Immediately after the pulse, the signals from individual spins have the same phase and are therefore summed coherently, which is what makes the total signal detectable. The return to equilibrium proceeds exponentially at a rate characterized by a time constant T 1, which is rather short for 10B and 11B in the molecules used for BNCT. The short T 1 is an advantage for NMR detection because it permits to repeat successive signal excitations at a high rate, which is necessary to gain enough independent input for creating images, and achieving a sufficiently high signal-to-noise (S/N) ratio. The return to equilibrium also results in an irreversible, exponential loss of coherence, characterized by a time constant T 2, which is often significantly shorter than T 1. The short T 2 for 10B and 11B, especially in biological tissue, can be problematic. The reason is that, for mainly technical reasons, a finite delay between signal excitation and detection is inevitable, and unless this delay is much shorter than T 2 (which is often not the case), a significant part of the signal is lost, which not only decreases the S/N ratio and sensitivity, but also introduces uncertainties in the quantitation of the boron presence in the sample [2].
NMR is quantitative as the signal is proportional to the total amount of the detected nucleus within the sample volume. For MRI, the image intensity is proportional to the amount of the detected nucleus in a voxel. Therefore, quantitation through comparison with the signal from calibration or reference samples should be straightforward. However, the accuracy of boron quantitation by NMR or MRI can be compromised by several factors, mainly due to the dependence of T 2 on molecular mobility. In practice, only boron in relatively mobile molecules in liquid or liquid-like environments can be properly detected. If the boron atoms themselves, or the molecules containing boron, are tightly bound or attached to entities with high molecular weight (e.g., membranes, proteins, nucleic acids), their NMR signal can either be completely missed or severely underestimated. This problem is usually worse for MRI and in vivo experiments than for “test-tube” spectroscopy, because in the latter case shorter detection “dead times” can be achieved. The quantification accuracy can be improved to some extent by measuring the T 2 decay rates and extrapolating the measured signal intensities to zero decay time [2].
Sensitivity and spatial resolution are inherently connected for MRI of boron compounds. The sensitivity, defined as the “minimum detectable concentration,” is inversely proportional to the volume of the image voxel.
One of the aspired applications of boron imaging for BNCT is the real-time monitoring of 10B-enriched compounds administered to patients, in and around the tumor, prior to irradiation with neutrons. This goal has not yet been achieved [66, 67], but due to the high potential, this method strongly deserved further development. Recently, Porcari et al. used (19)F MR Imaging ((19)F MRI) and spectroscopy ((19)F MRS) to evaluate the boron biodistribution and pharmacokinetics of 4-borono-2-fluorophenylalanine ((19)F-BPA) in vivo [68].
One of the most significant advantages of NMR or MRI is that there are relatively few restrictions on sample preparation. The analysis is noninvasive and nondestructive, and can be performed on homogeneous liquid samples but also on intact chunks of tissue, cells, etc. and most important on laboratory animals and human patients. Therefore, NMR has considerable potential to fulfill the desired function of noninvasive mapping of the spatial distribution of the administered 10B carrier in the course of the treatment. Another advantage is its ability to distinguish between different molecular species, thereby providing an opportunity for assessing metabolism for the investigated compound. The main disadvantage is the rather low sensitivity of the method, particularly for imaging (Fig. 9.5) [2].

The upper 1H images show axial (left) and sagital (right) views through the head of a volunteer, with a cylindrical vial attached close to the ear and the temple. The lower images are the 10B images at the corresponding orientations and geometric scale. The vial contained a solution of 28 mM BSH. The 10B images have a voxel size of 1.7 cc (1.2 × 1.2 × 1.2 cm) and are slices through a 3D matrix acquired in under 3 min (P. Bendel, Chemical Research Support Department, The Weizmann Institute of Science, Rehovot, Israel) (Reprinted from Bendel et al. [91])
2.8 Positron Emission Tomography
Positron emission tomography (PET) is a key imaging tool in clinical practice and biomedical research to quantify and study biochemical processes in vivo. PET is a tracer technique based on the use of labeled molecules carrying a short-lived positron-emitting radionuclide [69]. PET can yield quantitative measurements of the distribution of radioactivity that is present in the scanned field of view (FOV) after correction for physical parameters, such as subject self-attenuation, spurious events (scattered, random), and scanner efficiency calibration. PET data are conveniently displayed in the form of 2D and volumetric 3D images (Bq/voxel). PET images are analyzed by defining regions of interest (ROI) in correspondence with relevant organs or organ substructures to assess activity concentration in the form of peak or average values. PET imaging can also be dynamic, to detect activity changes over an observation period. In this case, time-activity curves (TAC) can be extracted for the selected region. Pharmacokinetic modeling may be applied to such values during image postprocessing to noninvasively assess the tracer kinetics in vivo. Arterial concentration of the tracer and PET tissue measurements are then used to calculate radiotracer regional kinetic parameters. Blood sampling may be necessary to calculate derived biochemical parameters and/or apply corrections for metabolites [2]. PET molecular imaging is thus a powerful pharmacological tool.
Labeling of biologically active molecules to assess their body distribution and regional concentrations has emerged as an outstanding application of PET imaging. The recent physical integration of PET and computed tomography (CT) in hybrid PET/CT scanners allows combining anatomical and functional imaging [70]. The high-resolution images produced by CT can be overlaid on to PET images, thus yielding anatomical reference to biochemical and metabolic measurements and reducing the lack in spatial resolution that has affected PET imaging (spatial resolution: 4–6 mm at best). This development has also prompted the use of PET/CT for treatment planning in radiotherapy, adding the metabolic information coming from PET imaging to the assessment of tumor volumes made by CT for the optimization of treatment on areas [2, 71, 72] (Fig. 9.6).

Head and neck tumor, 66 years old patient, 18F-FDG study performed with PET/CT model GE Discovery RX (L. Menichetti, C.N.R. Institute of Clinical Physiology, Pisa, Italy) (Reprinted from Wittig et al. [2])
The in vivo pharmacokinetics of a boron carrier for BNCT and the quantification of 10B in living tissue were already performed by PET in the late 1990s by W. Kabalka in the USA [73] and Y. Imahori in Japan [74, 75]. PET and PET/CT have been used to address the issue of pharmacokinetics, metabolism, and accumulation of BPA in target tissue. Imahori et al. [74] reported a method for the quantitative measurement of boronated drug uptake in patients with high-grade gliomas, based on the use of L-[18F]-BPA. A three-compartment model was used to analyze PET data and to assess the tumor pharmacokinetics. The concentration of boron in the tumor calculated by the model was found to be close to that measured in surgical specimens. The similarity in pharmacokinetics between L-BPA and the labeled analogue was confirmed using a segmental convolution method. The estimated values of the 10B concentration of BPA can be calculated from a four rate constants model applied to a dynamic study by PET using 18F-BPA as a tracer. With this approach, the 10B concentration was assessed during neutron irradiation of patients to be treated with BNCT after L-BPA administration. Following a similar approach, 18F-BPA has been used to measure its uptake in recurrent cancer of the oral cavity and cervical lymph node metastases [76]; malignant glioma [77]; low-grade brain tumors, such as schwannoma and meningioma [78]; head and neck malignancies [79]; and metastatic malignant melanomas [73]. These early clinical findings with 18F-BPA/PET led to study the transport and the net influx and accumulation of BPA and to show the capability of PET to screen the different types and different grades of tumor lesions as candidates for BNCT [2, 80].
The added value of the use of L-18F-BPA and PET/CT in BNCT is to provide key data on the tumor uptake of 10BPA and normal tissue and to predict the efficacy of the treatment of individual patients. Based on the assumption that the biochemical fate of the BPA is due to the molecular similarity to tyrosine [81–83], the PET of aromatic amino acid analogues might help in proving the pharmacokinetic rate constants (k 1, k 2, k 3, k 4) for the tumor and the input function for plasma L-BPA concentration (as previously demonstrated for L-BPA and L-18F-BPA).
One key role of PET/CT in BNCT is further to test the L-18F-BPA uptake in the tumor relative to surrounding normal tissues in individual patients and to use this information as a predictor for BNCT effectivity. Such data can be used as inclusion/exclusion criterion in clinical trials and might enormously help in patient selection to avoid treatment failures due to insufficient 10B uptake in tumors of individual patients [76, 79].
Feasibility studies are moreover conducted to use single-photon emission computed tomography (SPECT) during BNCT treatments to compute online boron dose maps without the large current uncertainties in the assessment of the boron concentration in different tissues [84, 85].
One main challenge using PET for BNCT however is the need to prepare a radioactively labeled molecule for each potential boron carrier molecule, which has imaging qualities. To date, this was exclusively successful for the compound BPA. For BSH, the second compound used clinically, and even for newly designed compounds, such radioactively labeled compound is not available. New compounds, which come to the state of clinical testing, must undergo all legal testing requirements applicable for new drugs which will be a very cost and time-intensive process [2].
3 Conclusions
One crucially important requirement at every stage in the development of BNCT has been the ability to measure the concentration and image the spatial distribution of boron in biological samples. Without such capabilities, new boron compounds cannot be designed or evaluated and prospective controlled clinical trials cannot be developed. Furthermore, radiation dosimetry remains unacceptably basic, while the radiobiology of the 10B neutron capture reaction leaves many open questions. Boron analysis and boron imaging is therefore one of the scientific pillars upon which the success or failure of this modality rests.
Boron analysis and boron imaging is a very complex task. After decades of research, presently, numerous methods are available with distinct endpoints and advantages/disadvantages. Therefore, it is of importance to carefully choose the appropriate technique which can correctly answer the clinical or experimental question addressed:
PGRA, ICP-AES, and ICP-MS are rapid, reliable methods for quantitation of the 10B concentration in macroscopy. PGRA is suited for “online” measurements of the 10B concentration in blood of patients during a BNCT treatment. However, it is of the utmost importance to realize that the 10B compounds are inhomogeneously distributed on the microscopic level. However, measurements from PGRA, ICP-AES, and ICP-MS integrate the 10B concentration over a volume, which is always larger than the volume irradiated by the BNC reaction. Hence, such measurements cannot be used to directly predict the radiation effect. Some research groups have proposed methods to overcome this shortcoming by analyzing the inhomogeneities of the sample, e.g., percentage of tumor cells, normal tissue, and necrosis in the analyzed volume and correcting the measured data by a factor derived from the analysis [86, 87]. Such factors can complicate the comparison of data measured by different groups especially if methodology and factors induced are not reported in detail [2].
Techniques that might help to solve this problem and are able to image the 10B distribution in tissue with a lateral resolution similar that to histomorphological images are high-resolution alpha-track autoradiography and neutron capture radiography. Quantitative evaluations can be included to be able to consider the inhomogeneous 10B distribution in tissues [88].
Laser microprobe mass analysis (LMMA) was the first technique to reach a subcellular resolution in BNCT [89]. Further developments using TOF-SIMS and especially laser-SNMS have the capability to quantitatively map the spatial boron distribution in biological matrices with a detection limit in the sub-ppm range, extremely high selectivity, and high spatial resolution. Methods based on electron microscopy (i.e., EELS) are less sensitive but have a much higher spatial resolution that readily satisfies the needs of BNCT for calculating radiation response. However, laser-SNMS and EELS are so time-, labor-, and cost-intensive that the small amount of information available until now does not yet satisfy the statistics necessary to evaluate biological phenomena [2].
Another important aspect that has been investigated to a little extent only is the metabolism of the compounds BPA and BSH that are actually used in clinical trials for BNCT. Ion trap mass spectrometry in combination with proteomic technologies offers a possibility to investigate metabolites and transport of target molecules. Such information is mandatory to optimize the application of these drugs on a scientifically solid basis.
For clinical purposes, noninvasive in vivo techniques to follow the pharmacological and chemical behavior of the boron carrier are needed. The labeling of BPA with 18F and the use of PET to assess the molecule in a patient and the detection of boron compounds by MRI are feasible but not yet available for routine clinical use. PET with 18F-labeled BPA is already used for patient selection in the recent clinical trials in Japan [78]. The expensive procedure to introduce a new radiopharmaceutical in clinical practice has limited this approach in Europe and the USA. In Europe, 18F-BPA is available to patients in Finland only.
Available methods offer powerful tools to the clinical scientist to further investigate BNCT. However, none of these methods satisfies all needs at any time in each situation. Often, a complementary use of different techniques will become necessary. The complementary use of different methods, which are not accessible everywhere and which need special knowledge and trained personnel, is only feasible in the framework of a well-organized and structured network. Important tasks for such collaboration will be not only to establish a standardized system for reporting the 10B concentration, especially in the clinical situation, but also to be able to provide answers to questions in basic science. Moreover, the design of standardized samples with known boron concentration and distribution should be developed, such that cross calibration and comparison of different analytical approaches can be made [2].
References
Sauerwein W (1993) Principles and history of neutron capture therapy. Strahlenther Onkol 169(1):1–6
Wittig A, Michel J, Moss RL, Stecher-Rasmussen F, Arlinghaus HF, Bendel P et al (2008) Boron analysis and boron imaging in biological materials for boron neutron capture therapy (BNCT). Crit Rev Oncol Hematol 68(1):66–90
Kobayashi T, Kanda K (1983) Microanalysis system of ppm order B-10 concentrations in tissue for neutron capture therapy by prompt gamma-ray spectrometry. Nucl Instrum Methods Phys Res 204:525–531
Konijnenberg MW, Raaijmakers CPJ, Constantine G, Dewit LGH, Mijnheer BJ, Moss RL et al (1993) Prompt gamma-ray analysis to determine 10B-concentrations. In: Soloway AH (ed) Advances in neutron capture therapy. Plenum Press, New York, pp 419–422
Raaijmakers CPJ, Konijnenberg MW, Dewit L, Haritz D, Huiskamp R, Philipp K et al (1995) Monitoring of blood-10B concentration for boron neutron capture therapy using prompt gamma-ray analysis. Acta Oncol 34(4):517–523
Fairchild RG, Gabel D, Laster BH, Greenberg D, Kiszenick W, Micca PL (1986) Microanalytical techniques for boron analysis using the 10B(n, alpha)7Li reaction. Med Phys 13(1):50–56
Matsumoto T, Aoki M, Aizawa O (1991) Phantom experiment and calculation for in vivo 10boron analysis by prompt gamma ray spectroscopy. Phys Med Biol 36(3):329–338
Mukai K, Nakagawa Y, Matsumoto K (1995) Prompt gamma ray spectrometry for in vivo measurement of boron-10 concentration in rabbit brain tissue. Neurol Med Chir (Tokyo) 35:855–860
Wittig A, Huiskamp R, Moss RL, Bet P, Kriegeskotte C, Scherag A et al (2009) Biodistribution of 10B for boron neutron capture therapy (BNCT) in a mouse model after injection of sodium mercaptoundecahydro-closo-dodecaborate and l-para-boronophenylalanine. Radiat Res 172(4):493–499
Kashino G, Fukutani S, Suzuki M, Liu Y, Nagata K, Masunaga S et al (2009) A simple and rapid method for measurement of 10B-para-boronophenylalanine in the blood for boron neutron capture therapy using fluorescence spectrophotometry. J Radiat Res 50(4):377–382
Vega-Carrillo HR, Manzanares-Acuna E, Hernandez-Davila VM, Chacon-Ruiz A, Gallego E, Lorente A (2007) Neutron fluence rate measurement using prompt gamma rays. Radiat Prot Dosimetry 126(1–4):265–268
Munck af Rosenschold PM, Verbakel WF, Ceberg CP, Stecher-Rasmussen F, Persson BR (2001) Toward clinical application of prompt gamma spectroscopy for in vivo monitoring of boron uptake in boron neutron capture therapy. Med Phys 28(5):787–795
Verbakel WF, Sauerwein W, Hideghety K, Stecher-Rasmussen F (2003) Boron concentrations in brain during boron neutron capture therapy: in vivo measurements from the phase I trial EORTC 11961 using a gamma-ray telescope. Int J Radiat Oncol Biol Phys 55(3):743–756
Evans EH, Giglio JJ (1993) Interferences in inductively coupled plasma mass spectrometry – a review. J Anal Atomic Spectrom 8:1–18
Gregoire DC (1987) Determination of boron isotope ratios in geological materials by inductively coupled plasma mass spectrometry. Anal Chem 59:2479–2484
Gregoire DC (1990) Determination of boron in fresh and saline waters by inductively coupled plasma mass spectrometry. J Anal Atomic Spectrom 5:623–626
Al-Ammar A, Reitznerová E, Barnes RM (2000) Improving boron isotope ratio measurement precision with quadrupole inductively coupled plasma-mass spectrometry. Spectrochim Acta Part B 55:1861–1867
Evans S, Krahenbuhl U (1994) Boron analysis in biological material: microwave digestion procedure and determination by different methods. Fresenius Z Anal Chem 349:454–459
Brown PH, Hu H (1996) Phloem mobility of boron is species dependent: evidence for phloem mobility in sorbitol-rich species. Ann Bot 77:497–505
Smith F, Wiederin DR, Houk RS, Egan CB, Serfass RE (1991) Measurement of boron concentration and isotope ratios in biological samples by inductively coupled plasma mass spectrometry with direct injection nebulisation. Anal Chim Acta 248:229–234
Vanhoe H, Dams R, Vandecasteele C, Versieck J (1993) Determination of boron in human serum by inductively coupled plasma mass spectrometry after a simple dilution of the sample. Anal Chim Acta 281:401–411
Laakso J, Kulvik M, Ruokonen I, Vahatalo J, Zilliacus R, Farkkila M et al (2001) Atomic emission method for total boron in blood during neutron-capture therapy. Clin Chem 47(10):1796–1803
Heber EM, Kueffer PJ, Lee MW Jr, Hawthorne MF, Garabalino MA, Molinari AJ et al (2012) Boron delivery with liposomes for boron neutron capture therapy (BNCT): biodistribution studies in an experimental model of oral cancer demonstrating therapeutic potential. Radiat Environ Biophys 51(2):195–204
Ficq A (1951) Autoradiographie par neutrons: dosage du lithium dans les embryons d’amphibiens. C R Acad Sci 233:1684–1685
Edwards LC (1956) Autoradiography by neutron activation: the cellular distribution of 10B in the transplanted mouse brain tumor. Int J Appl Radiat Isot 1:184–190
Solares G, Zamenhof R, Saris S, Walzer D, Kerley S, Joyce M et al (1992) Biodistribution and Pharmacokinetics of p-Borono-phenylalanine in C57BL/6 Mice with GL261 Intracerebral Tumours, and Survival Following Neutron Capture Therapy for Cancer. In: Allen BJ, Harrington BV, Moore DE (eds) Progress in neutron capture therapy for cancer. Plenum Press, New York, London, pp 475–478
Solares GR, Zamenhof RG (1995) A novel approach to the microdosimetry of neutron capture therapy. Part I. High-resolution quantitative autoradiography applied to microdosimetry in neutron capture therapy. Radiat Res 144:50–58
Yam CS, Solares GR, Zamenhof RG (1994) Validation of the HR microdosimetry. Trans Am Nucl Soc 71:142–144
Goodarzi S, Pazirandeh A, Jameie SB, Baghban Khojasteh N (2012) Differentiation in boron distribution in adult male and female rats’ normal brain: a BNCT approach. Appl Radiat Isot 70(6):952–956
Kiger WS 3rd, Micca PL, Morris GM, Coderre JA (2002) Boron microquantification in oral muscosa and skin following administration of a neutron capture therapy agent. Radiat Prot Dosimetry 99(1–4):409–412
Solares GR, Zamenhof RG, Cano G (eds) (1993) Microdosimetry and compound factors for neutron capture therapy. Plenum Press, New York
Alfassi ZB, Probst TU (1999) On the calibration curve for determination of boron in tissue by quantitative neutron capture radiography. NIM A 428:502–507
Pugliesi R, Pereira MAS (2002) Study of the neutron radiography characteristics for the solid state nuclear track detector makrofol-de. NIM A 484:613–618
Roveda L, Prati U, Bakeine J, Trotta F, Marotta P, Valsecchi P (2004) How to study boron biodistribution in liver metastases from colorectal cancer. J Chemother 16(Suppl 5):5–8
Altieri S, Bortolussi S, Bruschi P, Chiari P, Fossati F, Stella S et al (2008) Neutron autoradiography imaging of selective boron uptake in human metastatic tumours. Appl Radiat Isot 66(12):1850–1855
Schutz C, Brochhausen C, Altieri S, Bartholomew K, Bortolussi S, Enzmann F et al (2011) Boron determination in liver tissue by combining quantitative neutron capture radiography (QNCR) and histological analysis for BNCT treatment planning at the TRIGA Mainz. Radiat Res 176(3):388–396
Nano R, Barni S, Chiari P, Pinelli T, Fossati F, Altieri S et al (2004) Efficacy of boron neutron capture therapy on liver metastases of colon adenocarcinoma: optical and ultrastructural study in the rat. Oncol Rep 11(1):149–153
Chiaraviglio D, De Grazia F, Zonta A, Altieri S, Braghieri A, Fossati F et al (1989) Evaluation of selective boron absorption in liver tumors. Strahlenther Onkol 1989(2/3):170–172
Enge W, Grabisch K, Beaujean R, Bartholoma KP (1974) Etching behaviour of cellulose nitrate plastic detector under various etching conditions. NIM 115:263–270
Bennett BD, Zha X, Gay I, Morrison GH (1992) Intracellular boron localization and uptake in cell cultures using imaging secondary ion mass spectrometry (ion microscopy) for neutron capture therapy for cancer. Biol Cell 74(1):105–108
Chandra S, Morrison GM (1992) Sample preparation of animal tissues and cell cultures for secondary ion mass spectrometry (SIMS) microscopy. Biol Cell 74:31–42
Chandra S, Smith DR, Morrison GH (2000) Subcellular imaging by dynamic SIMS ion microscopy. Anal Chem 72(3):104A–114A
Chandra S, Lorey ID, Smith DR (2002) Quantitative subcellular secondary ion mass spectrometry (SIMS) imaging of boron-10 and boron-11 isotopes in the same cell delivered by two combined BNCT drugs: in vitro studies on human glioblastoma T98G cells. Radiat Res 157(6):700–710
Smith DR, Chandra S, Barth RF, Yang W, Joel DD, Coderre JA (2001) Quantitative imaging and microlocalization of boron-10 in brain tumors and infiltrating tumor cells by SIMS ion microscopy: relevance to neutron capture therapy. Cancer Res 61(22):8179–8187
Yokoyama K, Miyatake S, Kajimoto Y, Kawabata S, Doi A et al (2007) Analysis of boron distribution in vivo for boron neutron capture therapy using two different boron compounds by secondary ion mass spectrometry. Radiat Res 67(1):102–109
Arlinghaus HF, Spaar MT, Switzer RC, Kabalka GW (1997) Imaging of boron in tissue at the cellular level for boron neutron capture therapy. Anal Chem 69(16):3169–3176
Fartmann M, Kriegeskotte C, Dambach S, Wittig A, Sauerwein W, Arlinghaus HF (2004) Quantitative imaging of atomic and molecular species in cancer cultures with TOF-SIMS and Laser-SNMS. Appl Surf Sci 231(2(SI)):428–431
Arlinghaus HF (ed) (2002) Laser Secondary Neutral Mass Spectrometry (Laser-SNMS). Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
Arlinghaus HF, Kriegeskotte C, Fartmann M, Wittig A, Sauerwein W, Lipinsky D (2006) Mass spectrometric characterization of elements and molecules in cell cultures and tissues. Appl Surf Sci 252:6941–6948
Fartmann M, Dambach S, Kriegeskotte C, Lipinsky D, Wiesmann HP, Wittig A et al (2003) Subcellular imaging of freeze-fractured cell cultures by TOF-SIMS and Laser SNMS. Appl Surf Sci 203–204:726–729
Wittig A, Wiemann M, Fartmann M, Kriegeskotte C, Arlinghaus HF, Zierold K et al (2005) Preparation of cells cultured on silicon wafers for mass spectrometry analysis. Microsc Res Tech 66(5):248–258
Arlinghaus HF, Fartmann M, Kriegeskotte C, Dambach S, Wittig A, Sauerwein W et al (2004) Subcellular imaging of cell cultures and tissue for boron localization with laser-SNMS. Surf Interface Anal 36(8):698–701
Bourdos N, Kollmer F, Benninghoven A, Sieber M, Galla HJ (2000) Imaging of domain structures in a one-component lipid monolayer by time-of-flight secondary ion mass spectrometry. Langmuir 16(4):1481–1484
Neumann M, Kunz U, Lehmann H, Gabel D (2002) Determination of the subcellular distribution of mercaptoundecahydro-closo-dodecaborate (BSH) in human glioblastoma multiforme by electron microscopy. J Neurooncol 57(2):97–104
Zhu Y, Egerton RF, Malac M (2001) Concentration limits for the measurement of boron by electron energy loss spectroscopy and electron-spectroscopic imaging. Ultramicroscopy 87:135–145
Michel J, Sauerwein W, Wittig A, Balossier G, Zierold K (2003) Subcellular localization of boron in cultured melanoma cells by electron energy-loss spectroscopy of freeze-dried cryosections. J Microsc 210(Pt 1):25–34
Isaacson I, Johnson D (1975) The microanalysis of light elements using transmitted energy-loss electrons. Ultramicroscopy 1:33–52
Leapman RD, Kocsis E, Zhang G, Talbot TL, Laquerriere P (2004) Three dimensional distribution of elements in biological samples by energy filtered electron tomography. Ultramicroscopy 100:115–125
Michel J, Bonnet N (2001) Optimization of digital filters for the detection of trace elements in electron energy loss spectroscopy. Gaussian, homomorphic and adaptive filters. Ultramicroscopy 88:231–242
March RE (1997) An introduction to quadrupole ion trap mass spectrometry. J Mass Spectrom 32:351–369
Mauri PL, Basilico F, Pietta PG, Pasini E, Monti D, Sauerwein W (2003) New approach for the detection of BSH and its metabolites using capillary electrophoresis and electrospray ionization mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 788(1):9–16
Basilico F, Sauerwein W, Pozzi F, Wittig A, Moss R, Mauri PL (2005) Analysis of 10B antitumoral compounds by means of flow-injection into ESI-MS/MS. J Mass Spectrom 40(12):1546–1549
Washburn MP, Wolters D, Yates JRI (2001) Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol 19:242–247
Mauri P, Scarpa A, Nascimbeni AC, Benazzi L, Parmagnani E, Mafficini A (2005) Identification of proteins released by pancreatic cancer cells by multidimensional protein identification technology: A strategy for identification of novel cancer markers. FASEB J 19:1125–1127
Beretta L (2007) Proteomics from the clinical perspective: many hopes and much debate. Nat Methods 4:787–796
Bendel P (2005) Biomedical applications of 10B and 11B NMR. NMR Biomed 18(2):74–82
Bendel P, Koudinova N, Salomon Y (2001) In vivo imaging of the neutron capture therapy agent BSH in mice using 10B MRI. Magn Reson Med 46:13–17
Porcari P, Capuani S, D’Amore E, Lecce M, La Bella A, Fasano F et al (2009) In vivo 19F MR imaging and spectroscopy for the BNCT optimization. Appl Radiat Isot 67(7–8 Suppl):S365–S368
Martínez MJ, Ziegler SI, Beyer T (2008) PET and PET/CT: basic principles and instrumentation. Recent Results Cancer Res 170:1–23
Schöder H, Erdi YE, Larson SM, Yeung HW (2003) PET/CT: a new imaging technology in nuclear medicine. Eur J Nucl Med Mol Imaging 30:1419–1437
Lecchi M, Fossati P, Elisei F, Orecchia R, Lucignani G (2008) Current concepts on imaging in radiotherapy. Eur J Nucl Med Mol Imaging 35(4):821–837
Grosu AL, Piert M, Weber WA, Jeremic B, Picchio M, Schratzenstaller U et al (2005) Positron emission tomography for radiation treatment planning. Strahlenther Onkol 181(8):483–499
Kabalka GW, Nichols TL, Smith GT, Miller LF, Khan MK, Busse PM (2003) The use of positron emission tomography to develop boron neutron capture therapy treatment plans for metastatic malignant melanoma. J Neurooncol 62(1–2):187–195
Imahori Y, Ueda S, Ohmori Y, Kusuki T, Ono K, Fujii R et al (1998) Fluorine-18-labeled fluoroboronophenylalanine PET in patients with glioma. J Nucl Med 39(2):325–333
Imahori Y, Ueda S, Ohmori Y, Sakae K, Kusuki T, Kobayashi T et al (1998) Positron emission tomography-based boron neutron capture therapy using boronophenylalanine for high-grade gliomas: part II. Clin Cancer Res 4(8):1833–1841
Ariyoshi Y, Miyatake S, Kimura Y, Shimahara T, Kawabata S, Nagata K et al (2007) Boron neuron capture therapy using epithermal neutrons for recurrent cancer in the oral cavity and cervical lymph node metastasis. Oncol Rep 18(4):861–866
Nariai T, Ishiwata K, Kimura Y, Inaji M, Momose T, Yamamoto T et al (2009) PET pharmacokinetic analysis to estimate boron concentration in tumor and brain as a guide to plan BNCT for malignant cerebral glioma. Appl Radiat Isot 67(7–8 Suppl):S348–S350
Havu-Auren K, Kiiski J, Lehtio K, Eskola O, Kulvik M, Vuorinen V et al (2007) Uptake of 4-borono-2-[18F]fluoro-L-phenylalanine in sporadic and neurofibromatosis 2-related schwannoma and meningioma studied with PET. Eur J Nucl Med Mol Imaging 34(1):87–94
Aihara T, Hiratsuka J, Morita N, Uno M, Sakurai Y, Maruhashi A et al (2006) First clinical case of boron neutron capture therapy for head and neck malignancies using 18F-BPA PET. Head Neck 28(9):850–855
Takahashi Y, Imahori Y, Mineura K (2003) Prognostic and therapeutic indicator of fluoroboronophenylalanine positron emission tomography in patients with gliomas. Clin Cancer Res 9(16 Pt 1):5888–5895
Wyss MT, Hofer S, Hefti M, Bartschi E, Uhlmann C, Treyer V et al (2007) Spatial heterogeneity of low-grade gliomas at the capillary level: a PET study on tumor blood flow and amino acid uptake. J Nucl Med 48(7):1047–1052
Wang HE, Wu SY, Chang CW, Liu RS, Hwang LC, Lee TW et al (2005) Evaluation of F-18-labeled amino acid derivatives and [18F]FDG as PET probes in a brain tumor-bearing animal model. Nucl Med Biol 32(4):367–375
Ishiwata K, Kawamura K, Wang WF, Furumoto S, Kubota K, Pascali C et al (2004) Evaluation of O-[11C]methyl-L-tyrosine and O-[18F]fluoromethyl-L-tyrosine as tumor imaging tracers by PET. Nucl Med Biol 31(2):191–198
Minsky DM, Valda AA, Kreiner AJ, Green S, Wojnecki C, Ghani Z (2011) First tomographic image of neutron capture rate in a BNCT facility. Appl Radiat Isot 69(12):1858–1861
Murata I, Mukai T, Nakamura S, Miyamaru H, Kato I (2011) Development of a thick CdTe detector for BNCT-SPECT. Appl Radiat Isot 69(12):1706–1709
Wittig A, Malago M, Collette L, Huiskamp R, Buhrmann S, Nievaart V et al (2008) Uptake of two 10B-compounds in liver metastases of colorectal adenocarcinoma for extracorporeal irradiation with boron neutron capture therapy (EORTC Trial 11001). Int J Cancer 122(5):1164–1171
Coderre JA, Chanana AD, Joel DD, Elowitz EH, Micca PL, Nawrocky MM et al (1998) Biodistribution of boronophenylalanine in patients with glioblastoma multiforme: boron concentration correlates with tumor cellularity. Radiat Res 149(2):163–170
Thellier M, Hennequin E, Heurteaux C, Martini F, Pettersson M, Fernandez T et al (1988) Quantitative estimations in neutron capture radiography. Nucl Instrum Methods Phys Res B 30:567–579
Haselsberger K, Radner H, Gössler W, Schagenhaufen C, Pendl G (1994) Subcellular boron-10 localization in glioblastoma for boron neutron capture therapy with Na2B12H11SH. J Neurosurg 81:741–744
Michel J, Balossier G, Wittig A, Sauerwein W, Zierold K (2005) EELS Sprctrum-Imaging for boron detection in biological cryofixed tissues. Instrumentation Sciences and Technology 33:632–644
Bendel P, Koudinova N, Salomon Y, Hideghéty K, Sauerwein W (2002) Imaging of BSH by 10B MRI. In: Sauerwein W, Moss R, Wittig A, editors. Research and Development in Neutron Capture Therapy, Bologna: Monduzzi Editore, Bologna 877–880
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Wittig, A., Sauerwein, W.A.G. (2012). Boron Analysis and Boron Imaging in BNCT. In: Sauerwein, W., Wittig, A., Moss, R., Nakagawa, Y. (eds) Neutron Capture Therapy. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-31334-9_9
Download citation
DOI: https://doi.org/10.1007/978-3-642-31334-9_9
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-31333-2
Online ISBN: 978-3-642-31334-9
eBook Packages: MedicineMedicine (R0)