
Abstract
The acidity and basicity of the oxides and metal oxide surfaces is one among the most peculiar properties of this group of inorganic products that have been exploited in catalysis from decades. The different types of oxides (single oxides, doped and modified oxides, supported oxides, mixed and complex oxides) are here described with emphasis on their acid-base functionalities in relation with the catalytic activity. The prediction of the acidity insurgence from the oxide composition is discussed. The possibility to have a deep knowledge of the amount of acid sites and of the acid strength and strength distribution at the oxide surface is here presented. Adsorption by suitable probes with basic/acidic characteristics can be realized in a microcalorimetry of adsorption giving information on the heterogeneity of the surface. Alternatively, probe desorption can be realized by a temperature increasing program to obtain the acid-site strength distribution. In any case, quantitative information on the acid oxide surfaces can be obtained with the relevant thermodynamic of kinetic parameters which determine the amount and strength of the acid sites.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
1 Introduction
Oxides are relevant products of the inorganic chemistry that are extensively used in many applied fields such as adsorption technology, pigment technology, ceramics, and heterogeneous catalysis. In any applied field, the oxide functionality is governed by surface interaction with the environment. In particular, the interactions of the oxide surfaces with gases and liquids, active in the fields of adsorption and catalysis, are mainly governed by the acid-base properties of the oxide. These properties are tightly connected to the catalytic functionality of several divided, wide surface area oxide surfaces. Acid oxide catalysts have great importance in petroleum chemistry (e.g., amorphous silica-alumina cracking catalyst) and in any reaction involving hydrocarbons and they have been extensively investigated, while basic oxide catalysts have been studied to a lesser extent. The nature of the acid and base sites on oxide surfaces is an intriguing and moot subject. Surface exposed unsaturated cations and hydroxyl groups give rise to Lewis and Brønsted acid sites, respectively, while basic sites could be associated with surface oxygen atoms able to interact attractively with a proton. Severe pretreatment conditions of the oxides are needed for generating active basic sites by removal of carbon dioxide, water, and in some cases, oxygen from the surface.
Among the various types of heterogeneous acid and basic catalysts that found real application in various reactions and catalytic processes (Table 8.1, [1–4]), various types of oxide structures are found. The surface properties, nature and structure of acid and base sites of oxides have been investigated for many decades by various approaches, comprising spectroscopic, thermal, and theoretical studies, and newly developed measurement methods using modern techniques are nowadays used for this purpose. Other studies are aimed at developing novel acid and base oxide formulations with improved functionality. In the following, the acidity-basicity of oxide materials of catalytic importance by adsorption calorimetry by conventional and novel methodologies of study is presented. Beside the conventional gas-solid adsorption calorimetry studies, liquid-solid adsorption calorimetry has been developed to disclose sound relations between acid-base properties and activity of catalysts working in liquid-solid heterogeneous processes.
2 The Surface Acido–Basicity of Metal Oxides
To introduce the concept of acidity and basicity of surface sites of any solid, it is necessary to start with the very general definition of acids and bases. Two different definitions are used today; they are the most relevant and accepted ones among all the others. Brønsted and Lowry in 1923 defined that an acid (HA) is any hydrogen-containing species able to release a proton and a base (B) is any species capable of combining with a proton. In this view, the acid-base interaction consists in an equilibrium exchange of a proton and the final product of the exchange is the conjugated base (\(\mathrm{A}^{-}\)) and acid (\(\mathrm{BH}^{+}\)), respectively. Alternatively, Lewis proposed in the same year (1923) a different view to define the acidity-basicity concept. An acid (A) is any species possessing an incomplete electronic grouping that can accept an electron pair to form a dative or coordination bond (\(\mathrm{A}^{{\delta }^{-}}\)). A base (B) is any species possessing a non-bonding electron pair that can be donated to form a dative or coordination bond (\(\mathrm{B}^{{\delta }+}\)). This second definition is more general than the first one and can be applied to any species, even if hydrogen is not concerned in the exchange. The Brønsted and Lewis acid and basic strength can be quantitatively determined. According to the Brønsted theory, \(\mathrm{p}K_{a}\) measures the position of the equilibrium in water solution or in gas-phase so allowing the quantitative determination of the relative strength of the acids and bases. The \(\mathrm{p}K_{a}\) of the various acids/bases is usually reported with respect to water, while in the gas-phase, where solvation effects are absent, the acidity strength scale of given acids can be greatly different. If HA bond is fully ionic, the equilibrium of the acid-base couple is modified and now the Hammett acidity function (\(H_{o})\) is introduced. \(H_{o}\) holds in concentrated solutions where the pH concept is no longer applicable. The Hammett function is determined by the extent to which an indicator (weak or very weak organic base) is protonated, by measuring the [BH\(^{+}\)]/[B] ratio by UV-vis spectroscopy. The acid strength concept of the Lewis acids is more difficult to be quantified. A simple scale of acid strength for Lewis acids is not feasible because in this case the acidity depends upon the nature of the base involved in the acid-base equilibrium. Only few quantitative measurements of acid strength are available compared to Brønsted acids.
Other definitions of the acidity concept have been proposed in the literature; the concept of “Hard and Soft Acids and Bases (HSAB)” initially proposed by R. Pearson in 1963 [5] and extended by G. Klopman merits to be mentioned. Pearson classified Lewis acids and Lewis bases as hard, borderline, or soft and G. Klopman attempted to quantify Pearson’s HSAB principle using frontier molecular orbital (FMO) theory [6]. Hard acids are defined as small-sized, highly positively charged, and not easily polarizable electron acceptors. Hard bases are species holding their electrons tightly as a consequence of large electronegativities, low polarizabilities, and difficulty of oxidation of their donor atoms. Hard acids prefer to associate with hard bases, while soft acids prefer to associate with soft bases. The formed complexes are thermodynamically more stable than mixed complexes and also form faster. According with this theory, protons and hydroxides and oxide ions are all hard species, as are most O- and N-terminated anions. C- and S- terminated anions, sulphides, phosphines, and aromatic hydrocarbons are soft bases.
It may be taken into consideration that the proton affinity, PA, of a anion or of a neutral atom or molecule is a measure of its gas-phase basicity. It is the energy released in the following reactions: \(\mathrm{A}^{-} + \mathrm{H}^{+} \rightarrow \) HA or B + \(\mathrm{H}^{+} \rightarrow \mathrm{BH}^{+}\). For any species, the higher the proton affinity, the stronger the base and the weaker the conjugate acid in the gas phase. Recently, the relationships between the proton affinity and atomic charge and with the transfer of charge have been reexamined [7].
When getting on to the acidity-basicity of solids, we face with a different problem. The quantitative concepts that hold in dilute and concentrated solutions (\(\mathrm{p}K_{\mathrm{a}}\) and \(H_{\mathrm{o}})\) cannot be extended to solid surfaces. In general terms, a solid acid may be viewed as a surface on which a base is chemically adsorbed and, if the base acts as an indicator, its color changes upon adsorption. Following both the Brønsted and Lewis definitions, a solid acid shows a tendency to donate protons or to accept electron pairs, respectively, while a solid base should tend to accept protons or to donate electron pairs. For a complete evaluation of the solid acidity/basicity, the knowledge of the nature, amount and strength of the acid/base sites is of crucial importance, besides the knowledge of the distribution and density of the sites in nature and strength. This last point is of great importance when the solid acts as catalyst since the catalyst activity mainly depends on the amount of active sites but the catalyst selectivity may be different as a function of the site nature and their distribution and density on the surface. The amount of acid/base on a solid is usually expressed as the number of moles of acid/base sites per unit mass or per unit surface area of the solid, and it is obtained by measuring the amount of a base which is adsorbed on it. The extent of the adsorption reaction can be studied with different analytical techniques, among these UV-vis spectroscopy, volumetric and gravimetric apparatus. Others techniques of study have to be selected to investigate on the nature, strength, and distribution of the acid/base sites. The need for integrated approach of study clearly emerges to successful face the solid acidity/basicity question.
Confining our interest on metal oxides, knowing that they are solid compounds of oxygen and metal or semimetal elements, their surfaces are constituted by positive and negative arrays of coordinated ions which are bound through ionic or covalent bonds, depending on the nature of the cations and structure of the oxide. The positive and negative centers can behave as Lewis acids and Lewis bases that con be described as a bidimensional organization of AB acid-base pairs \(\mathrm{(AB)}_{\mathrm{n}}\). Above that, hydroxyl groups variably cover any real oxide surfaces, as these groups are potential proton donors or acceptors, it is evident that Brønsted acidity and basicity should be considered to appear on such surfaces. Of course Lewis and Brønsted species are often simultaneously present on the same surface where they form densely populated patches. Consequently due to the complexity of any real metal oxide surface, the quantitative study of their acid-base properties is not a trivial matter and it deserves considerable work.
3 Acid, Basic, and Amphoteric Oxides
Before providing an overview of the acid-base properties of metal oxide surfaces, a brief systematic description of their bulk and structural properties and surface chemistry is needed. According to basic inorganic chemistry concepts, the oxides of non-metals as well as the oxides of the metals in very high oxidation states are defined as acidic oxides and anhydrides, respectively, the oxides of metals as basic oxides, and the oxides having both acidic and basic characters are denoted as amphoteric.
Basic oxides possess low-valency metals (oxidation number lower than or equal to \(+4\)) and they are typically ionic compounds obtained in crystalline forms. In such oxides the coordination of the cations (from 4 to 8) is generally higher than the valency (from 1 to 4) and the same occurs for the coordination of the oxide ions (from 3 to 6). The basic nature of the ionic oxides is associated with the strong polarization of the metal-oxygen bond and with the formation of basic products (metal hydroxides) formed from the reaction with water. Examples of relevance in catalysis are: \(\mathrm{Cu}_{2}\mathrm{O}, \mathrm{CuO}, \mathrm{Ag}_{2}\mathrm{O}, \text {MgO, BaO, MnO, FeO, La}_{2}\mathrm{O}_{3}\), etc. They found application in various base-catalyzed reactions (alcohol dehydrogenation, olefines, aromatic carboxylic acids hydrogenation, amination of primary and secondary amines, Meerwein-Ponndorf-Verley reduction, side-chain alkylation of aromatics, aldol addition and condensation, Tischenko reaction to produce esters by aldehydes dimerization, Michael addition, Knoevenagel condensation, etc.) [2].
Acidic oxides are oxides of non-metal elements with high electronegativity characterized by a nearly covalent bond between the non-metal and oxygen. In normal conditions, some of these oxide structures are gaseous or liquids while others are solids (e.g., \(\mathrm{Se}_{4}\mathrm{O}_{12}, \mathrm{P}_{4}\mathrm{O}_{10})\). Decreasing the electronegativity of the element, covalent network structures are formed (e.g., \(\mathrm{SiO}_{2}, \mathrm{B}_{2}\mathrm{O}_{3})\). In this case, the covalent nature of the non-metal oxides is evidenced by the coordination number of the non-metal element which corresponds to its valency or even lower and coordination of oxygen is two or one. They can form crystalline phases but they usually exist in very stable amorphous states that are utilized for wide applications (e.g., silica) and convert into stable crystalline phases after treatment at high temperatures. The acid nature of these oxides is associated with the acidity of the products of their reaction with water: oxo-acids. Different types of acid oxides, denoted as anhydrides, are constituted of metal elements in very high oxidation states (e.g., \(\mathrm{Mn}_{2}\mathrm{O}_{7}, \mathrm{CrO}_{3}, \mathrm{MoO}_{3}, \mathrm{WO}_{3}, \mathrm{Sb}_{2}\mathrm{O}_{5}, \mathrm{Nb}_{2}\mathrm{O}_{5}\), etc.). The very high electronegativity of the metal imparts them nearly the same properties of non metal oxides. Concerning the structure of these oxides, ii depends on the position of the metal in the period (from group 4 to group 7); network structure, as \(\mathrm{TiO}_{2}\), layered structure as \(\mathrm{V}_{2}\mathrm{O}_{5}\), linear polymeric structure, \(\mathrm{asCrO}_{3}\), and molecular structure, as \(\mathrm{Mn}_{2}\mathrm{O}_{7}\). These metal oxides react with water giving rise to acid and polyoxoacid products.

Heat evolved for given amounts of CO\(_{2}\) and NH\(_{3}\) adsorbed over a series of metal oxides (from Ref. [9]); top oxides of Al, Be, La, Mo, Nd, Pr, Si, Ta, Th, and Zr; bottom oxides of Cr, Ca, Ga, Mg, Nb, Mg, Ti, V, and W
Some oxides exhibit both acidic and basic properties, that is, they have amphoteric properties. In a strongly acidic environment, they act as bases while in a strongly basic environment they act as acids. In general the electropositive character of the oxide central atom determines whether the oxide will be acidic or basic. The more electropositive the metal is the more basic the oxide will be and the more electronegative the central atom is, the more acidic oxide will be. Electropositive character increases from right to left across the period and increases down the column for each period column. A resultant borderline between basic and acidic oxides occurs along the diagonal of the period including oxides of Zn, Al, Ga, Sb, etc. Alumina represents the most well-known example of amphoteric oxide able to react with acids and d bases:
Taking into account a given series of metal oxides of different acid-base properties, including \(\mathrm{Al}_{2}\mathrm{O}_{3}, \mathrm{BeO}, \mathrm{CaO}, \mathrm{Cr}_{2}\mathrm{O}_{3}, \mathrm{Ga}_{2}\mathrm{O}_{3}, \mathrm{La}_{2}\mathrm{O}_{3}, \mathrm{MgO}, \mathrm{MoO}_{3}, \mathrm{Nb}_{2}\mathrm{O}_{5}, \mathrm{Nd}_{2}\mathrm{O}_{3}, \mathrm{Pr}_{6}\mathrm{O}_{11}, \mathrm{SiO}_{2}, \mathrm{Ta}_{2}\mathrm{O}_{5}, \mathrm{ThO}_{2}, \mathrm{TiO}_{2}, \mathrm{V}_{2}\mathrm{O}_{5}, \mathrm{WO}_{3}, \mathrm{ZnO}, \mathrm{ZrO}_{2}\) [8, 9], those able to adsorb at their surface both carbon dioxide (an acid molecular probe) and ammonia (a basic molecular probe) with evolution of an appreciable amount of heat, typically in the range of chemisorbed reactions (from 50 to 200 kJ\({\cdot }\)mol\(^{-1})\) are classed as amphoteric oxides, while if only ammonia or carbon dioxide are retained on a given surface, the complete acidic or basic character of the oxide, respectively, can be evinced. Figure 8.1 shows a quantitative description of the acid base characteristics of the above cited series of oxides by using a three-dimensional picture in which each experimental point of each curve refers to three axes: the amount of adsorbed ammonia and the amount of adsorbed carbon dioxide (expressed per unit surface area of oxide sample), and the relevant evolved heat of adsorption (expressed by the differential heat, \(\mathrm{kJ}{\cdot }\mathrm{mol}^{-1})\). The pure acidic oxides are settled completely in the right hand side plane of the diagrams, the basic ones which reacted noticeably with carbon dioxide only stayed completely in the left hand side of the diagrams, and the amphoteric oxides can be found in the middle of the diagram planes with their experimental points in the forehead space. For instance, ZnO showed high acidic tendency for its strong sites and much more basic tendency for its weak sites (starting from 100 \(\mathrm{kJ}{\cdot }\mathrm{mol}^{-1}\)).
4 Heterogeneous Character of Oxides
The heterogeneous character of oxides is due to (i) irregularity in the surface crystallographic structure (different types of crystal planes, growth steps, crystal edges and corners, Fig. 8.2); (ii) presence of various functional groups (\({\text {M--O}}^{-}, \text {M--O--M, M=O}\), \(\text {(M)}_{x}{\text {--OH}}, {\text {M(--OH)}}_{2}\), M–[ ], etc. [8, 10]); and (iii) presence of impurities strongly bounded to the surface. Concerning the acid and basic functionalites of any oxide surface, different species can be distinguished: (i) exposed oxide species (mono-oxo or di-oxo surface species), potentially acting as basic oxides; (ii) exposed hydroxyl groups arising from water adsorption, potentially acting as acid Brønsted sites or basic sites; (iii) exposed cationic centers, potentially acting as Lewis acid sites; and (iv) cationic or anionic vacancies. The acid or basic character of the hydroxyl species is dependent on the nucleophilic character of the hydroxyl group that is governed by the M–OH bond strength; the proton acidity increases with the increase of the coordination number of the OH group (Scheme 8.1 [10, 11]).
Other than the intrinsic heterogeneous character of any surface, it is possible to induce surface heterogeneity by several actions as (i) the mode of formation of the solid (e.g., by precipitation from liquid phase, flocculation of colloidal suspension, sintering of powder, partial/total re-crystallization, sputtering/condensation from vapor phase, surface chemical reaction, etc.); (ii) the thermal treatment of the solid; in fact, heating causes roughness of initially smooth surface by atom displacement and migration to sites of higher energy on top of the surface; and (iii) the introduction of additional phase (active phase or promoter) that imparts functionality to the surface.

Schematic representation of a real oxide particle with irregularity in crystal planes, growth steps, edges, and corners which causes the presence of surface species with different coordinative unsaturation (from Ref. [12])

Hydroxyl groups of metal oxide surfaces [11]: Type 1, mono bridging hydroxyl group; Type II, doubly bridging hydroxyl group; Type III, triply bridging hydroxyl group
The complex picture of oxide surfaces can be studied from both qualitative and quantitative point of view by spectroscopic techniques, conventionally infrared (FT-IR) but also Raman and NMR spectroscopies [11, 13–15], spectrophotometric techniques, in particular XPS [16, 17], by directly observing the surface and by the help of suitable probe molecules adsorption [2, 4, 18–21] which give the possibility to study the molecular adduct formed between the acid/base site and the probe [2, 10, 11]. Reasons for the choice of one or another probe are essentially linked with their size (i.e., kinetic diameter) and basicity [10, 22].
The most used molecular probes are among different chemical families: cyclic amines (piperidine), alkyl amine (n-butylamine), heterocyclic amines (pyridine, 2-phenylethylamine), phosphines (trimethyl-phosphine), ammonia, carbon monoxide. The probe basicity can be expressed as \(\mathrm{p}K_{\mathrm{a}}\) of the conjugated acid and as proton affinity (PA). The former parameter, \(\mathrm{p}K_{\mathrm{a}}\), is relevant with respect to acid-base interactions occurring in water solution, also implying solvation with water of both the base and its conjugated acid. The latter parameter, PA, is more appropriate for gas-phase interactions. The two basic scales are not coincident, for example ammonia is associated with a higher \(\mathrm{p}K_{\mathrm{a}}\) and a lower PA than pyridine: This suggests that ammonia is more basic than pyridine in water but it is less basic than pyridine in gas phase. The inversion of the relative acidity in water with respect to the gas phase observed for the ammonia and pyridine probes is due to the greater solvation of ammonium ion in water compared with the pyridinium ion. At last, the choice of the probe is also dependent on the analytical technique and the method used to study the adsorbed molecular acid-base complex formed. For example, a high molecular mass probe is preferred when the acid-sites of a surface are determined by gravimetric analysis (by adsorption or thermal desorption), in this case 2-phenylethylamine is a better choice than pyridine [23, 24].
The quantitative evaluation of the amount of acid/base sites and relative acid/base strength of a surface can be more easily determined by gravimetric, volumetric, microcalorimetric, and thermal techniques (Temperature Programmed Desorption, TPD, among others) [8, 24–35]. Moreover, calorimetric and thermal techniques are able to provide quantitative information on the strength of interaction between the acid/base site and the probe, that is, the energy of the bond formed, through the direct determination of the heat of adsorption or desorption, depending on the experimental method adopted for the measurements. Such information let us to classify the acid/base sites of any solid following a scale of acid/base strength with important relationship with the solid reactivity [8, 36, 37]. When a heterogeneous surface is concerned, the acid/base site strength distribution can be determined by mathematical analysis of the experimental data obtained from adsorption and desorption thermal techniques [30–32, 34, 38]. In this context, the studies of adsorption calorimetry can give an estimation of the energy distribution of acid or base sites of heterogeneous surfaces by the direct inspection of the collected calorimetric profiles which show a more or less pronounced decreasing trend of the differential heat values as a function of the degree of coverage of the used probe (\(\mathrm{q}_{\mathrm{diff}}\) versus adsorbed moles). The decreasing trend reflects heterogeneity of the surface sites but also induced heterogeneity, due to lateral interactions between the adsorbed molecules [38, 39], leads to the observed decreasing trends, so making complicated the quantitative evaluation of the energy distribution of any surface. Following a common and simplified viewing, the different types of site are assumed to be covered by the probe successively starting from the strongest ones. This assumption can hold only if large differences among the adsorption constants are involved [40, 41].
Carniti and Gervasini [42] presented the possibility to study the heterogeneity of oxide surfaces by performing adsorption calorimetry experiments, by using a suitable base probe, at different adsorption temperatures. If on a same sample, adsorption calorimetry measurements are performed at different temperatures, different distributions of sites could be observed [42–44]. If no modification of the surface occurs in the temperature range considered, the different observed distributions can be due to either kinetic or thermodynamic effects. Regarding the kinetic effects, at low adsorption temperature, the strongest sites may not be activated or, even if sufficiently activated, may be covered by the probe at an adsorption rate too low to achieve equilibrium during the experiment. Moreover, surface diffusion could not be accomplished if enough time does not elapse in the equilibration. When the temperature and the time employed in the measurements ensure that the adsorption equilibrium has been obtained on all sites, the differences among the observed distributions have to be ascribed to the influence of temperature on the thermodynamic parameters of adsorption, particularly on the adsorption constants. Accurate adsorption calorimetric measurements carried out at different temperatures can give the true site distribution; besides, the thermodynamic parameters of adsorption can be obtained as functions of temperature. The true distribution can be different from the observed one obtained by using the above-mentioned oversimplification from a single adsorption calorimetric curve. Concerning the temperature range employed, temperatures should be sufficiently high that the adsorption process is activated on all types of site, but low enough that the adsorption equilibrium constants are favorable.
Once obtained the data (sets of adsorption and calorimetric isotherms collected at different temperatures, n and Q, adsorbed moles and evolved heat, respectively), a computation fitting procedure has to be applied [42]. In the model, the adsorption isotherms are interpreted as summations of single Langmuir isotherms, each of them relevant to the sites of a definite type (with \(\mathrm{n}_{\mathrm{max,i}}\) representing the maximum number of moles adsorbed on sites of \(i\)th type and having a defined adsorption enthalpy with the probe, \(\Delta _{\mathrm{a}}\mathrm{H}_{\mathrm{i}}\)). The temperature dependence of the Langmuir adsorption constant of each site-type (\(\mathrm{b}_{\mathrm{i}}\)) was obtained through the integration of the vant’Hoff differential equation, considering the adsorption enthalpy either independent or dependent on temperature by introducing the heat capacity of adsorption (\(\Delta _{\mathrm{a}}\mathrm{C}_{\mathrm{p,i}}\)). On the basis of the experimental values of n and Q obtained at the investigated temperatures as a function of equilibrium pressure and surface coverage, respectively, the characteristic parameters of adsorption (\(\mathrm{n}_{\mathrm{max,i}}, \Delta _{\mathrm{a}}\mathrm{H}_{\mathrm{i}}\), number of acid sites and adsorption enthalpy for each type of \(i\)th site, and any other parameters related to them) can be evaluated by a computational program including an optimization subroutine. When the vant’Hoff differential equation is considered,

Acid site energy distribution for several oxides of catalytic relevance (from Ref. [42])
the b\(_{0,\mathrm{i}}\) pre-exponential parameter has to be optimized too, in order to obtain the energy distribution of the surface, that is \(\mathrm{n}_{\mathrm{max,i}}\) versus \(\Delta _{\mathrm{a}}\mathrm{H}_{\mathrm{i}}\). The validity of the \(\mathrm{b}_{\mathrm{0,I}}\) parameter is difficult to evaluate because it does not have a simple physical meaning and the range of its numerical values is far from being clearly defined. A new parameter [45] related to b\(_{0,\mathrm{i}}\) and defined as half-coverage temperature at unit pressure, \(\mathrm{T}^{0}_{1/2,\mathrm{i}}\), can be optimized instead of \(\mathrm{b}_{\mathrm{0,i}}\). The new parameter corresponds to the temperature at which the adsorption constant, \(\mathrm{b}_{\mathrm{i}}\), becomes unity. In the case of a Langmuir isotherm, this condition corresponds to half-coverage; therefore the sites of \(i\)th type are half-covered at \(\mathrm{T} = \mathrm{T}^{0}_{1/2}\). Then \(\mathrm{b}_{\mathrm{0,I}}\) can be easily calculated from the optimized \(\mathrm{T}^{0}_{\mathrm{1/2,i}}\), values.
Following the approach above described, the energy distribution of several acidic simple metal oxides has been determined by using ammonia as probe [42]. The obtained total number of acid sites (\(\mathrm{n}_{\mathrm{max}}=\Sigma _{\mathrm{i}} \mathrm{n}_{\mathrm{max,i}}\)) allowed to disclose the following acidity scale: \(\mathrm{WO}_{3}\) (5.55 \(\upmu \mathrm{mol}/\mathrm{m}^{2}) > \mathrm{ZrO}_{2}\) (5.50 \(\upmu \mathrm{mol}/\mathrm{m}^{2}) > \mathrm{Nb}_{2}\mathrm{O}_{5}\) (4.76 \(\upmu \mathrm{mol}/\mathrm{m}^{2}) < \mathrm{BeO} (4.56\,{\upmu \mathrm{mol}/\mathrm{m}^{2}}) > \mathrm{TiO}_{2}\) (3.20 \(\upmu \mathrm{mol}/\mathrm{m}^{2}) > \mathrm{Al}_{2}\mathrm{O}_{3}\) (1.98 \(\upmu \mathrm{mol}/\mathrm{m}^{2})\) (Fig. 8.3). No more than three or four different types of site were found for each oxide. All the oxide surfaces showed a significant quantity of sites (70–80 %) associated with \(\Delta _{\mathrm{a}}\mathrm{H}_{\mathrm{i}}\) of \({-}40\) kJ/mol, corresponding to hydrogen-bonded ammonia (the weakest mode of interaction). The other 20–30 % of sites was distributed in a \(\Delta _{\mathrm{a}}\mathrm{H}_{\mathrm{i}}\) range of \(-160\) to \(-280\) kJ/mol, corresponding to more energetic interactions of ammonia with the sites.
5 Single Oxides, Doped and Modified Oxides, Supported Oxides, Mixed Oxides, and Complex Oxides
In catalysis, oxides with well defined acidic and basic properties are used in different forms that have found application in numerous catalytic applications in the gas-solid and liquid-solid heterogeneous catalysis [3, 46, 47]. Among the most used oxide materials in catalysis, we find: (i) bulk oxides (one component metal oxides); (ii) doped and modified oxides; (iii) supported metal oxides (dispersed active oxide component onto a support oxide component); (iv) bulk and supported binary metal oxides to quaternary metal oxides (mixed oxide compositions); (v) complex oxides (e.g., spinels, perovskites, hexa-aluminates, bulk and supported hydrotalcites, pillared clays, bulk and supported heteropolyacids, layered silicas, etc.).
One component metal oxides can crystallize with different morphologies (isotropic, anisoptropic, or remain amorphous) and local coordination. All one component metal oxide phases crystallize at high temperatures, providing detectable crystalline XRD spectra, and many phases can remain amorphous at moderate calcinations temperatures (\(\mathrm{SiO}_{2}, \mathrm{Al}_{2}\mathrm{O}_{3}, \mathrm{Nb}_{2}\mathrm{O}_{3}, \mathrm{MoO}_{3}\), etc.). Their surfaces terminate with various functionalities; for example, amorphous silica terminates with isolated Si–OH, hydroxylpairs of \({\text {(Si--OH)}}_{2}\), and \({\text {Si(--OH)}}_{2}\), bridging Si–O–Si bonds [48]; niobia terminates with Nb–OH, Nb–O–Nb, and Nb=O bonds [49, 50]. In general, surfaces terminating with M=O functionality typically possess \(\mathrm{M}^{+7}, \mathrm{M}^{+6}\), and \(\mathrm{M}^{+5}\) cations, while surfaces showing \(\mathrm{M}^{+4}, \mathrm{M}^{+3}, \mathrm{M}^{+2}\), and \(\mathrm{M}^{+1}\) cations do not possess enough electrons to form terminal bonds and typically terminate with M–OH or M–O–M bonds. The literature reports several investigations on acid-base properties of amorphous metal oxides by adsorption calorimetry in research and review articles [8, 9, 41].
The presence of the different functionalities on the various oxide surfaces leads to different reactivity properties. When molecules, metal salts, or metal complexes are adsorbed on the \(\mathrm{SiO}_{2}\) surface, for example, the anchoring sites are the more reactive isolated Si–OH groups rather than the less reactive bridged Si–O–Si groups. Acid and base sites can often be present simultaneously on oxide surfaces and they can work independently or in a concerted way. Alumina is the best known example of amphoteric oxide widespread used in catalysis (Scheme 8.2) [51].
In this case, multiple surface hydroxyl groups varying by the number of Al sites and Al coordination are present as well as oxygen vacancies of defects, such determining the characteristic surface chemistry of alumina [48]. As for \(\mathrm{SiO}_{2}\), the isolated Al–OH sites are the most reactive towards molecules, metal salts, and metal complexes that have to be adsorbed on the alumina surface, these sites are basic, for the most part, while the bridging Al–OH–Al sites are the most acidic ones, and the Al–[ ] defects possess Lewis acidic character.

Picture of acid and base sites creation on the alumina surface (from Ref. [51])
There are some other metal oxides crystallize with an anisotropic morphology, such as platelets [52]. Both \(\mathrm{MoO}_{3}\) and \(\mathrm{V}_{2}\mathrm{O}_{5}\) crystallize with platelet morphology. In these structures, the terminal M=O and bridging M–O–M groups are present on the basal planes and the terminal M–OH predominate on the edge plains. Concerning surface reactivity of such surfaces, it was demonstrated that the \(\mathrm{MoO}_{3}\) basal planes do not chemisorb methanol while the flat portions of surface containing Mo–OH sites on the edges react with methanol to form \({\text {Mo--OCH}}_{3}\) and water [53]. The same behavior was observed for \(\mathrm{V}_{2}\mathrm{O}_{5}\) and ZnO when exposed to methanol [54–56]. On such materials, the number of catalytically active sites, i.e., acid sites, are only a small fraction of the total exposed metal oxide sites and we assist to structure sensitivity of active sites to different crystalline planes.
The recent discovery of mesoporous silica or in general mesoporous molecular sieves (MMS) has attracted a great deal of attention [57, 58]. The adjustable porosity of silica-based MMS allows large reactant molecules to penetrate into the internal void space to be processed at the active-acid sites and then diffuse out freely as products. Because of the low acidity of the silanol groups of such materials non-silica mesoporous transition-metal oxide materials have been recently prepared for catalytic purposes.
Many early transition metal oxides possess high acidity, especially in sulfated and phosphated forms; titanium, niobium, and tantalum oxides demonstrated very high activity for large series of acid-catalytic reactions such as benzylation, alkilation, and isomerisation [57]. For example, sulfated mesoporous Nb oxide has excellent activity (ca. 200 times greater than that of sulfated bulk Nb oxide) in the benzylation of anisole or toluene with benzyl alcohol in liquid phase. The mesoporous structure is maintained even after acid treatment (both with sulfuric or phosphoric acids), Brønsted and Lewis acid sites coexist in a roughly 50:50 mixture on the surface of mesoporous Nb oxide while a strong dominance of Brønsted acid sites is present on the sulfated and phosphated materials [59]. The very high surface area of these mesoporous materials results in a great increase in the exposed acid sites per mass, so justifying the observed excellent catalytic activity.
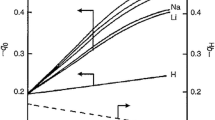
Doped and modified oxides are a wide family of samples which are synthesized with the aim of modifying some surface property of a given oxide. In particular, when an acid or base solid has to be used as catalyst, it can happen that the average acid/base strength of its surface active sites is not useful for the studied reaction. The acidic or basic sites can be either too strong causing some irreversible adsorption of the reactant species or too weakly acidic or basic to activate the reactants. Then, the oxide surface can be easily modified by incorporation of a second oxide component or by adding doping species that can regulate their acid-base strength by modification of the electronic and geometric characteristics of the acid or base sites. The effect of the addition of small amounts of various ionic species (\(\mathrm{Ca}^{2+}, \mathrm{Li}^{+}, \mathrm{Nd}^{3+}, \mathrm{Ni}^{2+}, \mathrm{Zr}^{4+}\), and \(\mathrm{SO}_{4}^{2-}\),) on the acid-base properties of \(\upgamma \)-alumina, silica, and magnesia surfaces was studied by ammonia and sulfur dioxide probe molecules, respectively, in an adsorption calorimeter [60].
The added metal ion concentration was in the range from 0.1 to 0.3 \(\upmu \mathrm{mol}{\cdot }\mathrm{m}^{2}\) corresponding to a surface coverage of about 0.5–1 % of moles of metal ion per mole of support. It was found that the modification of \(\upgamma {\text {-Al}}_{2}\mathrm{O}_{3}\) surface properties by the ion dopants only moderately changed its amphoteric properties, the surfaces of the doped alumina samples remained amphoteric with acid/base pairs independent of the additives. More substantial changes were observed on magnesia concerning its basic properties, formation of even more basic sites in the domain of medium weak sites, but not in the strong sites domain, were revealed (Fig. 8.4 upper). On the weakly acid silica sample, the number of acid-base centers was strongly affected by the introduction of the doping ions, as shown in Fig. 8.4 down.

Heat of adsorption for \(\mathrm{NH}_{3}\) and \(\mathrm{SO}_{2}\) probes on doped magnesia samples (upper) and on doped silica samples (bottom) (from Ref. [60])
Concerning the relationships between the surface acid-base properties and more general intrinsic properties of the ions, such as electronegativity, ionicity character of the cation-oxygen bond, etc., it was found that the acidity of the samples correlated with the charge/radius ratio of the ionic species and with the electronegativity of the doping ions. The basicity correlated well with the partial oxygen charge associated with the cationic dopant [60]. Cardona-Martíinez and Dumesic [61] arrived at the same conclusion studying the acid properties of a series of doped silicas with small amounts (0.2–0.3 \(\upmu \mathrm{mol}{\cdot }\mathrm{m}^{2})\) of \(\mathrm{Mg}^{2+}, \mathrm{Sc}^{3+}, \mathrm{Fe}^{2+}, \mathrm{Fe}^{3+}, \mathrm{Al}^{3+}, \mathrm{Zn}^{2+}\), and \(\mathrm{Ga}^{3+}\) ions by adsorption microcalorimetry using pyridine as probe. The new acidity created by the ion introduction could be correlated with the Sanderson electronegativity of the corresponding oxide formed on the surface (Fig. 8.5). In particular, the Ga-, Al-, and Sc-silica samples were found to have both Brønsted and Lewis acidity while all the other samples showed only Lewis acidity.

Integral heat of pyridine adsorption as a function of the Sanderson electronegativity of the cations introduced on silica surface (from Ref. [61])
Besides the doped oxides, modified oxides are gained a prominent role in the catalytic scenario. Many different oxide materials were synthesized chemically modifying in particular, but not exclusively, silica structures with alumina, titanium, niobium, tantalum, and zirconium giving rise to many successful materials used in catalysis as active phases or support phase [23, 24, 34, 62–67].

Integral (upper) and differential (bottom) heats of ammonia adsorption versus coverage for a series of modified silicas with alumina, SA, titania, ST, and zirconia, SZ; for comparison the curves for silica, S, and alumina, A, are reported (from Ref. [34])
The possibility of modifying the acid properties of the silica surface by chemical modification, for example, by covalent anchoring of acid groups or by introducing elements of other valence [68–71] which create a defect of charge causing the formation of a Brønsted site to balance it, is well known from long time and it has been well exploited in catalysis to synthesize solid acids with modulated acidity strength. The integral and differential heats evolved from ammonia adsorption on a series of oxides comprising silica, alumina, and three modified silicas with amounts of Al, Ti, and Zr, are shown in Fig. 8.6. The curves for alumina (A), silica-alumina (SA), and silica-zirconia (SZ) run together while that for silica-titania (ST) lays well below those of the other samples and the curves for all the samples are very higher than that of silica. This suggests that by chemical modification of the silica structure it is possible to enhance the surface acidity of the resultant samples and in particular that alumina, silica-alumina, and silica-zirconia samples have surfaces with the highest acidity in terms of number and strength of sites with high heterogeneity of the surfaces as indicated by the continuous decreasing heat values as a function of coverage [34].
Among these kinds of oxides, the most important example is silica-alumina that gained success as substitute for acidic zeolites in many catalytic processes of petrol chemistry and refining and in general of hydrocarbon chemistry [72]. Synthetic silica-aluminas are amorphous materials with structure consisting of a random array of silica and alumina tetrahedral interconnected over three dimensions. In order to maximize the number of acid sites in a silica-alumina it is important to prepare samples with the maximum amount of tetrahedrally coordinated Al; in this way, one acidic proton would be generated by each Al atom, while formation of Al–O–Al should be avoided. Figure 8.7 reports a comparative view of the acidity of some zeolite and silica-alumina samples [33]. In the case of a zeolite, a typical differential heat plot shows three regions; the sharp decrease of adsorption heats at low coverage indicates the presence of small concentration of very strong Lewis type-acid sites. The plateau of constant heats of adsorption reveals the presence of Brønsted type-acid sites. The differential heat versus coverage curves for silica-aluminas reveal a more heterogeneity of the acid sites with a more balanced presence of Brønsted and Lewis acid sites than zeolite materials that have a predominant Brønsted acidity.

Differential heat of ammonia adsorption versus coverage for a series of zeolites (H-ZSM-5, H-BETA, and H-MCM-22) and two silica-aluminas (SAH and SAG) (from Ref. [33])
Not only the number and strength of the acid sites but also the accessibility to the reactant molecules is a target in the case of the solid acids when reaction forecasts large reactants to transform. A new family of mesoporous aluminosilicates with a regular array of uniform pores of 20–100 Å diameter has been discovered [73]. These MCM-41 materials can be synthesized over a large range of framework Si/Al ratios developing acidity properties [74]. The presence of these very large uniform pores combined with acidic properties opened new possibilities for cracking heavier feeds and other hydrocarbon transformation.
Supported metal oxides obtained by spreading an active metal oxide component over an oxide support, in prevalence of ceramic type (\(\mathrm{Al}_{2}\mathrm{OI}_{3}, \mathrm{SiO}_{2}, \mathrm{ZrO}_{2}, \mathrm{TiO}_{2}\), etc.), represent a very large family of oxides widely used in heterogeneous catalysis. The driving force for this surface wetting of metal oxides is the lower surface free energy of the final supported metal oxide system. The hydroxylated oxide support surface possesses a much higher surface free energy than the terminal M=O bonds formed in the surface metal oxide monolayer. The electronic and molecular structures of the surface metal oxide species dispersed on oxide supports have received enormous attention over the past decades because (i) the industrial significance as catalysts for numerous applications and (ii) the ability to serve as model mixed metal oxides systems due to the essentially completed dispersed state.
Depending on the mutual characteristics and nature of the supported oxide and oxide components, supported systems with different properties and stability can be formed [10]. As a general trend, it is possible to support ionic oxides on ionic oxides even if the resultant compositions are quite unstable due to the ability of the two oxides to react with each other giving rise to solid solutions or ternary phases. This occurs, for example, for CuO on alumina which forms \(\mathrm{CuAl}_{2}\mathrm{O}_{4}\) spinel. Only when the supported oxide is formed by big cations (e.g., \(\mathrm{K}_{2}\mathrm{O}, \mathrm{CaO}, \mathrm{La}_{2}\mathrm{O}_{3}\), etc.) unable to enter the close packing of the oxide ions forming the support (e.g., \(\mathrm{Al}_{2}\mathrm{O}_{3}, \mathrm{TiO}_{2}\), etc.), fairly stable oxide-on-oxide structures can be formed. Systems based on covalent oxides deposited on covalent oxides are frequently used in catalysis (e.g., \(\mathrm{MoO}_{3}/\mathrm{SiO}_{2}, \mathrm{WO}_{3}/\mathrm{SiO}_{2}\), etc.). They are generally viewed as constituted of small particles of the supported phase weakly interacting with the support surface. Also covalent oxides supported on ionic oxides are frequently used in catalysis. Oxides such as vanadia, tungsta, niobia, rhenia can be usefully supported on oxides as alumina, titania, and zirconia. This possibility gives rise to monolayer-like supported phases which are quite stable because of the slow reactivity of the two components. If, however, the ionic oxide support has a too pronounced basic feature the reactivity between the two components is high and a salt is easily produced (e.g., vanadia on magnesia gives rise to Mg vanadate). Ionic oxides supported on covalent oxides are sometimes used in catalysis. The covalent support is almost amorphous silica, on which many ionic oxides can be supported. The stability of such compositions appears to be quite limited, due to salt formation.
The acidic metal oxides (e.g., \(\mathrm{CrO}_{\mathrm{x}}, \mathrm{MoO}_{\mathrm{x}}, \mathrm{WO}_{\mathrm{x}}, \mathrm{VO}_{\mathrm{x}}, \mathrm{NbO}_{\mathrm{x}}, \mathrm{TaO}_{\mathrm{x}}\), etc.) usually anchor to the oxide substrate by preferentially titrating the basic surface hydroxyls of the support surfaces [47]. The active basic metal oxides (e.g., \(\mathrm{NiO}_{\mathrm{x}}, \mathrm{CoO}_{\mathrm{x}}, \mathrm{InO}_{\mathrm{x}}\), CuO, etc.) usually anchor to the oxide substrate by preferentially titrating the surface Lewis acid sites of the oxide supports [47, 75]. For many supported metal oxide systems, the active component can be present as a 100 % dispersion (typically when \(\mathrm{Al}_{2}\mathrm{O}_{3}, \mathrm{TiO}_{2}, \mathrm{ZrO}_{2}\) are used as oxide supports) when its loading on the support is not high and the support coverage is below the monolayer. Less than 100 % dispersion is usually obtained for metal oxides at high loading and when the support surface presents lower reactivity of the surface hydroxyls. In addition, it is known that some active basic metal oxides do not interact strongly with the different oxide functionalities of the oxide supports and, consequently, do not disperse to form well-dispersed phases (e.g., \(\mathrm{MnO}_{\mathrm{x}}, \mathrm{CeO}_{\mathrm{x}}\), [47, 76]).
Another important distinction has to be made based on the strength of the interaction between the support and the supported phase, designated as active phase, in view of its catalytic role [77]. By varying the support and the active oxide nature, different situations can be met which cover a continuous range of interaction strength between the two phases (Fig. 8.8). When weak interaction strength occurs (Fig. 8.8a), the supported phase gives rise to isolated crystallites deposited on, but not necessarily covering, the support phase. For medium interaction strength (Fig. 8.8b), the active phase tends to spread over the support surface if its loading is low, while, for increasing concentrations, multiple layers, and even distinct crystallites, of active oxide are built. For strong interaction strength (Fig. 8.8c), a spreading of the supported phase id favored, but a strong interaction can also lead to a new surface compound (Fig. 8.8c, \(\upalpha )\) or to bulk compound (Fig. 8.8c, \(\upbeta )\) or to solid solution (Fig. 8.8c, \(\upgamma )\). Figure 8.8d–f illustrate other situations with formation of a gradient composition compound, the layered support with adsorbed particles, and a diamond-layered model, respectively.

Examples of different supported oxide systems concerning the two oxide component interaction (from Ref. [77]): a weak interaction; b medium interaction; c strong interaction, with formation of \(\upalpha \)-surface compound, \(\upbeta \)-bulk compound, and \(\upgamma \)-solid solution; d composition with gradient-concentration; e layered support with adsorbed particles, f diamond layered structure
The electronic and molecular structures of surface metal oxide species present on oxide supports have received enormous attention over the past three decades because of their industrial significance as catalysts for numerous applications and their ability to serve as model mixed metal oxide catalytic systems due to the quite completely dispersed state. Here below, some examples of supported oxide catalysts are illustrated with emphasis on the possibility to judiciously develop surfaces with defined acid-base properties.
The modification of the acid-base properties of oxides following the deposition of variable amounts (from 1 to 50 % of the support surface coverage) of \(\mathrm{Li}^{+}, \mathrm{Ni}^{2+}\), and \(\mathrm{SO}_{4}^{2-}\) species on supports like alumina, magnesia, and silica have been studied [78]. Once again the adsorption microcalorimetric experiments using a basic and an acidic probes, \(\mathrm{NH}_{3}\) and \(\mathrm{SO}_{2}\), respectively, was used to study the acid properties of the surfaces. It has been found that any linear changes in the amount and in strength of acid-base sites with the increasing addition of lithium, nickel, or sulfate ions to alumina, silica, or magnesia were found. The addition of small amounts of additives to alumina has very slight impact on the heat of adsorption and density of acid/base sites until the attainment of a sudden change when half of its surface was covered. The possibility to adjust the acid-base properties of bulk oxides by a second oxide phase deposited on them was more effective on silica owing to its very weak intrinsic acid character.
Niobia is a fascinating oxide phase used in catalysis because it is a typical strong-metal-support-interaction (SMSI) oxide; for this property it is used as a support phase for metal and metal oxides [50, 79]. The strong interaction between niobia surface and the supported metal species enhances both the redox of the reducible metal oxides species and the acid properties of the supported phases. Such modifications could be favorable for given catalytic processes; for example, some group III oxides (\(\mathrm{B}_{2}\mathrm{O}_{3}, \mathrm{Al}_{2}\mathrm{O}_{3}, \mathrm{Ga}_{2}\mathrm{O}_{3}\), and \(\mathrm{In}_{2}\mathrm{O}_{3})\) are frequently studied as semiconductor materials used as sensor devices for the detection of \(\mathrm{NO}_{\mathrm{x}}, \mathrm{O}_{2}, \mathrm{H}_{2}\mathrm{O}\), CO [80–82], and more recently as de-NO\(_{x}\) catalysts [83, 84], or as catalysts for the dehydrogenation or aromatization of light alkanes [85] in the form of supported phases. When niobia is concerned as support phase for \(\mathrm{Al}_{2}\mathrm{O}_{3}\) (Al/Nb), \(\mathrm{Ga}_{2}\mathrm{O}_{3}\) (Ga/Nb), and \(\mathrm{In}_{2}\mathrm{O}_{3}\) (In/Nb) oxides, amphoteric surfaces are obtained giving interesting catalytic properties in the reaction of dehydrogenation of propene [86]. The Ga/Nb and In/Nb samples presented differential heat curves of ammonia adsorption clearly different from that of niobia. Up to a certain adsorbed amount (ca. 25 \(\upmu \mathrm{mol}{\cdot }\mathrm{g}^{-1})\) the samples have adsorption heat values higher than those of niobia, attributed to newly created centers of guest oxide; then the adsorption heats become lower than those of the bare support (Fig. 8.9, left). This means that gallium and indium oxides are preferentially deposited on the acid sites of niobia. Concerning the basicity of the surfaces, as determined by \(\mathrm{SO}_{2}\) adsorption, the differential heat curves obtained (Fig. 8.9, right) do not reflect the basicity order of the group III bulk oxides. Despite the well assessed basic properties of \(\mathrm{In}_{2}\mathrm{O}_{3}\), the poor In-dispersion on niobia (In/Nb sample) was responsible for the very lower adsorbed amount of \(\mathrm{SO}_{2}\) than on Ga/Nb and, in particular on Al/Nb (possessing the highest surface area). At this subject, the influence of the support nature in promoting the dispersion of the supported phase is of fundamental importance. Still considering the dispersed \(\mathrm{In}_{2}\mathrm{O}_{3}\) active phase, it was shown [84] that among a series of oxide supports (\(\mathrm{Al}_{2}\mathrm{O}_{3}, \mathrm{Nb}_{2}\mathrm{O}_{5}, \mathrm{SiO}_{2}\), and \(\mathrm{TiO}_{2}), \mathrm{Al}_{2}\mathrm{O}_{3}\) and, to a lower extent, \(\mathrm{TiO}_{2}\) were found to be the best ones for obtaining active de-NO\(_{x}\) catalysts, since the good In-dispersion and high active surface area.

Sulfur dioxide adsorption (left) and ammonia adsorption (right) on some supported group III oxides (\(\mathrm{Al}_{2}\mathrm{O}_{3}\), Al/Nb, \(\mathrm{Ga}_{2}\mathrm{O}_{3}\), Ga/Nb, and \(\mathrm{In}_{2}\mathrm{O}_{3}\), In/Nb) on niobia. Bulk niobia does not show any basicity [84]
A peculiar characteristic of the highly dispersed oxides in nanometer size is that their local structure and electronic properties vary with domain size [87]. For example, in the catalysis applications, turnover rates vary as oxide domains evolve from isolated monomers to two-dimensional oligomers, and into three-dimensional clusters with bulk-like properties. Many examples on dispersed metal oxide domains in relation to acid catalyzed reactions can be recalled. For example, \(\mathrm{WO}_{\mathrm{x}}\) domains dispersed on \(\mathrm{ZrO}_{2}\) constitute an excellent acid catalyst for xylene isomerization at low temperatures [87]. The maximum rates of reaction, expressed per W-atom, were observed at \(\mathrm{WO}_{3}\) surface densities of ca. 10 \(\mathrm{atom}_{\mathrm{{W}}}{\cdot }\mathrm{nm}^{-2}\), corresponding to the two-dimensional polytungstate monolayer formation. Raman evidences suggested that W–O–W connectivity (vibrational mode at 807 \(\mathrm{cm}^{-1})\) which predominate over the terminal W=O groups (vibrational mode at 1019 \(\mathrm{cm}^{-1})\) in two-dimensional and three-dimensional extended \(\mathrm{WO}_{\mathrm{x}}\) oligomers constitute the active acid sites, they could stabilize the cationic transition state involved in the xylene isomerization. Figure 8.10 reports the evolution of \(\mathrm{WO}_{\mathrm{x}}\) species on zirconia surface from the smallest to most wide aggregates.

Representation of the evolution of supported \(\mathrm{WO}_{\mathrm{x}}\) domains from isolated mono tungstates to two-dimensional polytungstates and three-dimensional \(\mathrm{WO}_{3}\) clusters. The increasing W–O–W connectivity with increasing domain size is related to the extent of oligomerization (from Ref. [87])
Bulk and supported mixed oxide compositions, from binary metal oxides to quaternary metal oxides, consist in general of large crystalline phases possessing low surface area values (typically from 1 to 10 \(\mathrm{m}^{2}{\cdot }\mathrm{g}^{-1})\). Examples of oxides of this type of catalytic relevance are V–Nb–O, Mo–Nb–O, Co–Ti–O, Ni–Ti–O, etc. The acid-base properties of mixed metal oxides have been found to change with the nature of the constituents and their relative concentrations, preparation (co-precipitation and sol-gel synthesis among are the most popular methods used), and pre-treatments procedures. Appropriately choosing the mentioned variables, mixed oxides can be used to prepare catalysts with the desired-acid-base characteristics.
The example of binary mixed oxides constituted of silica and a second component like alumina, titania, and zirconia has been above reported. The so constituted surfaces have different acidity properties in terms of number of sites and site strength distribution, depending on the nature of the second component (Al, Ti, or Zr), the Si/Al, Si/Ti, and Si/Zr ratios, and the final sample structure [23, 34]. In catalysis such oxides can be used as support phases as well as acid catalysts. When they are used as support phases, the acid sites serve as anchoring points for the supported phase stabilizing it in the dispersed state, even when the samples operate under high temperature conditions. It was demonstrated by XPS spectrophotometry [23] that for silica-zirconia compositions, Si/Zr, it was possible to regularly increase the amount of surface acid sites by tuning the amount of Zr during preparation (\(\mathrm{Si}_{\mathrm{x}}\mathrm{Zr}_{\mathrm{1-x}}\mathrm{O}_{2}\) with 0.715 \(<\) x \(<\) 1, corresponding to \(\mathrm{ZrO}_{2}\) from 5 to 45 wt% in the silica) (Fig. 8.11). On the different synthesized Si/Zr acidic surfaces, the iron oxide dispersed phase was supported and new acidic surfaces were created, \(\text {Fe/Si}_{\mathrm{x}}\mathrm{Zr}_{\mathrm{1-x}}\mathrm{O}_{2}\), differing from the relevant bare supports for the nature, strength, and amount of the acid sites.
The average acid strength of the Fe-samples was higher than that of \(\mathrm{Si}_{\mathrm{x}}\mathrm{Zr}_{\mathrm{1-x}}\mathrm{O}_{2}\) supports and tuned towards a prominent Lewis acidity. Concerning the amount of acid sites of the Fe-catalysts, they were higher or lesser compared with that of the relevant supports depending on the acidity of the bare support. When a portion of highly acidic surface of support (Si/Zr samples at high Zr content) was covered by iron oxide having lower surface area than its support, a decrease of the number of surface acid sites was observed (Fig. 8.12, see the SZ-15, SZ-30, and SZ-45 supports and relevant Fe-catalysts) while when low acidic surfaces, as pure silica or low containing zirconium samples were covered by iron oxide an increase of the amount of acid sites was observed (Fig. 8.12, see the S and SZ-5 supports and relevant Fe-catalysts). It is then possible to tune not only the amount of the acid sites but also the Brønsted and Lewis nature and the acid strength of a surface by a judicious presence of different components at the surface.

Relation between the amount of acid sites of \(\mathrm{Si}_{\mathrm{x}}\mathrm{Zr}_{\mathrm{1-x}}\mathrm{O}_{2}\) oxides determined by 2-phenylethylamine probe and the amount of surface Zr concentration determined by XPS (from Ref. [23])
Silica-alumina has been used to support the CuO coupled with \(\mathrm{Ga}_{2}\mathrm{O}_{3}\), and \(\mathrm{SnO}_{2}\) dispersed phases to enhance the catalytic properties of CuO-based catalysts in reactions of environmental importance (hydrocarbon combustion, NO and \(\mathrm{N}_{2}\mathrm{O}\) decomposition and reduction [88]). The acidic properties of such oxide systems were studied from a qualitative (nature of the acid sites) and quantitative (number, acid strength, and strength distribution of acid sites) points of view through the adsorption and desorption of two basic probes (ammonia and pyridine) by coupled volumetric-calorimetric technique and XPS and FT-IR spectroscopy.

Amount of acid sites of the \(\mathrm{Si}_{\mathrm{x}}\mathrm{Zr}_{\mathrm{1-x}}\mathrm{O}_{2 }\)supports in comparison with the Fe oxide covered surfaces, determined by 2-phenylethylamine probe (S, pure silica, and SZ-X, silica-zirconia oxides with 5 \(<\) x \(<\) 45 zirconia mass concentration) (from Ref. [23])
The Brønsted acidity of the silica-alumina support was converted into predominantly Lewis acidity upon metal oxide deposition, in particular that of the CuO supported phase. The highest amount of Lewis acid centers were formed on the CuO sample, the partial coverage of the Cu phase by Ga-phase caused a marked decrease of the amount of Lewis acid centers of the Cu/Ga surfaces. The Lewis to Brønsted acid site ratio of the Cu/Sn samples was higher compared to that of Cu/Ga samples, accounting for a higher surface Sn-dispersion. Concerning the acid strength of the surfaces, moderate acidity was associated with the Cu sites (100 \(\mathrm{kJ}{\cdot }\mathrm{mol}^{-1} < \mathrm{q}_{\mathrm{diff}} < 150\,\mathrm{kJ}{\cdot }\mathrm{mol}^{-1})\) whereas the most acidic fraction of the sites (\(\mathrm{q}_{\mathrm{diff}} > 150\,\mathrm{kJ}{\cdot }\mathrm{mol}^{-1})\) increased with the presence of Ga and Sn (Fig. 8.13). The results have been discussed in the light of the intrinsic acidity of the Cu, Ga, and Sn metal ions derived from electronic parameters (in particular electronegativity).

Acid site strength percent distributions of the Cu/Ga (a) and Cu/Sn (b) series samples supported on silica-alumina (SA) as obtained from ammonia adsorption by calorimetric measurements (the Ga/Cu and Sn/Cu atomic ratios, 25, 50, or 75, are indicated) (from Ref. [88])
Complex oxides comprise a large variety of structures which gained importance in the catalysis field due to their acidity or basicity properties (e.g., spinels, perovskites, hexa-aluminates, bulk and supported hydrotalcites, pillared clays, bulk and supported heteropolyacids, etc.).
Hydrotalcites are gained an important position, in particular, in basic catalysis. They are layered double hydroxides containing a divalent ion (e.g., \(\mathrm{Mg}^{2+}, \mathrm{Ca}^{2+}, \mathrm{Zn}^{2+}\), or \(\mathrm{Ni}^{2+})\) a trivalent ion (e.g., \(\mathrm{Al}^{3+}, \mathrm{Fe}^{3+}, \mathrm{Cr}^{3+})\) and a charge compensating anion (e.g., \(\mathrm{OH}^{-}, \mathrm{Cl}^{-}, \mathrm{NO}_{3}^{-}, \mathrm{CO}_{3}^{2-}\), or \(\mathrm{SO}_{4}^{2-})\). Thermal treatments cause dehydration and dehydroxylation and loss of anions giving rise to mixed oxide with MgO-type structures. The acid-base properties of Mg–Al oxides are governed by the Mg to Al molar ratio, besides the calcination temperature and preparation procedure. Increasing the Mg:Al ratio of the hydrotalcites, increase of the total number of basic sites is observed. The thermal decomposition of hydrotalcites gives rise to mixed oxides with basic properties and they can act as precursors for the preparation of basic oxides with catalytic activity [89]. Several reviews have been reported recently concerning the application of Mg–Al or calcined hydrotalcites as basic materials in particular in the field of fine organic chemistry [90–92].
Among the mixed oxides, those with perovskite-like structure are among the most widely studied. They have general formula \(\mathrm{ABO}_{3}\) where A is usually a large-radius metallic cation (e.g., lanthanide, alkali-earth metal ion) and B is a lower size transition metal ions. The principal advantages of this oxide family are the high versatility of their composition that can give compounds with widely different properties and their exceptional stability of the structures. The crystal field force of the perovskite structure can force some ions to assume unusual oxidation states. A typical example is Cu in Ba-substituted lanthanum cuprate, in which Cu is present in both \(2{+}\) and \(3{+}\) oxidation states [93]. Another interesting property of perovskites is their nonstoichimetry. As the whole structure has to be neutral, so cation charge have to be distributed to give an overall \({+}6\) sum: \(\mathrm{A}^{1+}\mathrm{B}^{5+}\mathrm{O}_{3}\), or \(\mathrm{A}^{2+}\mathrm{B}^{4+}\mathrm{O}^{3}\), or \(\mathrm{A}^{3+}\mathrm{B}^{3+}\mathrm{O}_{3}\). The presence of defects, like anionic or cationic vacancies, is very common in real structures. Anionic vacancies are the most frequent and they can extend up to a whole oxygen layer, leading to a different family of compounds, the brownmillerites [94], like \(\mathrm{Ca}_{2}\mathrm{Fe}_{2}\mathrm{O}_{5}\) and \(\mathrm{La}_{2}\mathrm{Ni}_{2}\mathrm{O}_{5}\) in which one sixth of the oxygen atoms are vacant. In other cases, an excess oxygen can be found; this is the case of \(\text {LaMnO}_{3+{\delta }}\).which shows very good catalytic activity for total oxidation reactions. This defect or excess of charge is connected with acid or basic properties of these structures which found wide application in the hydrocarbon catalytic combustion processes rather than in the acid-base catalytic reactions, due to their excellent thermal and chemical stability.
Another interesting family of oxide compounds are the heteropolycompounds possessing Keggin-type structure. They consist of heteropolyanions and counter-cations such as \(\mathrm{NH}^{4+}, \mathrm{Cs}^{+}, \mathrm{H}^{+}\). When the counter-cations are protons, the compounds are called heteropolyacids, they can have very strong Brønsted acid sites, like \(\mathrm{H}_{3}\mathrm{PW}_{12}\mathrm{O}_{40}\). They can have different acid strength depending on the nature of the compounds but always presents a plateau of sites of the same strength. For the catalytic application, they are often supported on high-surface area oxides or activated carbons to increase the surface contact with the reactants of the fluid phase. They are used in several acid-catalyzed reactions, like the hydration of alkanes, synthesis of methacrolein, isobuturric acid, etc. Various studies are reported in the literature [95–97] concerning the acid strength of heteropolyacids, influence of the support on acidity, influence of the acid site distribution on the substitution of protons with cationic species, change in acidity upon heat treatment, etc.
6 Acidity Prediction from Composition
From practical and theoretical points of view concerning binary metal oxides, it is interesting to find oxide combinations having well defined and tunable acid or basic properties. On a catalytic oxide surface, the acid or basic sites can be either too strong causing some irreversible adsorption of the substrate species or the sites can be too weak to activate the substrate species. Therefore, the possibility to regulate the acid-base strength, besides the acid site amount of the oxide surfaces, appears a necessary tool for catalytic purposes.
The acid/base strength of an oxide surface may be enhanced or decreased by the addition of a secondary component which modifies the electronic and/or geometric characteristics of the parent acid/base sites or creates new acid/base centers. The oxides belonging to the silica-alumina system, wherein the Al introduction generates new Brønsted or Lewis acid sites, are representative examples of acid binary oxides. Several authors have found generation of acidity also for other mixed oxide systems (silica-zirconia, silica-niobia, silica-titania, [23, 51, 98]). Then, several models appeared in the literature (Thomas [99], Tanaka [100], Tanabe [101], Seiyama [102], Dumesic [103], and Gervasini [60]) which attempted to generalize the experimental observations picturing acidity generation mechanisms.
The Tanabe model for acidity prediction in mixed oxide compositions is based on the interaction of the oxides at molecular level, the interaction generates an excess of negative or positive charge in the mixed composition localized around the guest element. According to this model, the substitution/introduction of a metal ion into the structure of a host oxide, whether the charge is an excess or not, and whether it is positive or negative, may generate acid sites. They are determined by the coordination numbers, C, and valences, V, of the positive and negative elements in the model oxide structure pictured according to two postulates: (i) the coordination numbers of the positive elements of the metal oxides are maintained even when mixed; (ii) the coordination number of oxygen of the major component oxide is retained for all the oxygen atoms in the mixed composition. It is then possible to explain the mechanisms of the acidity generation of mixed oxides and to predict whether the generated acid sites will be of the Brønsted or the Lewis type.
where \(\Delta \) is the excess/ of charge, V/NC is the ionic valence to coordination number ratio, and the A and O subscripts concern the positive element of the added ion and the negative element (oxygen) of the major component oxide, respectively. In any case, Lewis acidity is assumed to appear upon the presence of an excess of positive charge and Brønsted acidity for an excess of negative charge.
Some model oxide structures pictured according to Tanabe’s model are here below reported.
Silica-titania, with silica the major component oxide:
\(\Delta \quad = (+4/6 - 2/2)\cdot 2 = -2\)

Titania-silica, with titania the major component oxide:
\(\Delta \quad = (+4/4 - 2/3)\cdot 4 = +4/3\)

Silica-niobia with Nb tetrahedral coordination (NbO\(_{4})\):
\(\Delta \quad = (+5/4 - 2/2)\cdot 4 = +1\)

Silica-niobia with Nb octahedral coordination (NbO\(_{6})\):
\(\Delta \quad = (+5/6 - 2/2)\cdot 6 = -1\)

Silica-zirconia wirh 8-fold coordination of Zr (fluorite-like structure):
\(\Delta \quad = (+4/8 - 2/2)\cdot 8 = -4\)

Seiyama has presented a different model in which the oxygen bridging between the two different metal ions develops a positive or negative charge as a consequence of the different coordination of the two cations. In this case too, it is possible to calculate the effective charge of oxygen as the sum of the boundary charges of the two oxides:
where the S subscript concerns the positive element of the major component oxide (support oxide).
There is not very often accordance among the different models for the acidity prediction of a given oxide structure. Moreover, it is also hard to justify the predicted acid properties with the catalytic activity of the oxide composition; this is the case of titania-silica system [67, 104]. \(\mathrm{TiO}_{2}\mathrm{-SiO}_{2}\) mixed oxide is a very important industrial material and catalyst in both the amorphous and crystalline phases which found several industrial applications (e.g., isomerization of olefins, epoxidation of olefins with hydroperoxides, selective oxidation of a number of organic compounds, etc.). The \(\mathrm{TiO}_{2}\mathrm{-SiO}_{2}\) activity in the 1-butene isomerization, phenol ammination, and ethane hydration was attributed to the formation of Brønsted acidity [105]. While for the oxidation reactions there is a general consensus that the active sites are tetrahedrally coordinated \(\mathrm{Ti}^{4+}\) isolated in \(\mathrm{SiO}_{2}\) matrix, less clear is the source of the activity in the other reactions which demand acid sites. Because the catalytic activity could not be explained by the acid properties of the two pure oxides: in fact \(\mathrm{TiO}_{2}\) has only Lewis acid sites and the silanol groups of \(\mathrm{SiO}_{2}\) are too weakly acidic for any reaction requiring an acid catalysis. The performances of \(\mathrm{TiO}_{2}\mathrm{-SiO}_{2}\) as acid catalyst were at first explained by Tanabe on the basis of his theory of acidity generation in mixed compositions. From his first hypothesis, many authors studied the \(\mathrm{TiO}_{2}\mathrm{-SiO}_{2}\) system and support the Tanabe hypothesis. Notari et al. [67] did not agree with the literature evidences on the acidity of \(\mathrm{TiO}_{2}\mathrm{-SiO}_{2}\) and confuted them by selected experiments. In particular, he proved that highly divided \(\mathrm{TiO}_{2}\) operated the same acid reactions that \(\mathrm{TiO}_{2}\mathrm{-SiO}_{2}\) did with a radical or anion mechanism.
In some other different examples, acidity prediction of an oxide system correlates well with its activity. This is the case of \(\mathrm{TiO}_{2}\mathrm{-SnO}_{2}\) which generated strong new acid sites. The number of acid sites become maximum at the composition \(\mathrm{TiO}_{2}\) 50 %. At this composition, the oxide shows the maximum catalytic activity in the butane isomerization reaction [106].
7 Intrinsic and Effective Acidity of Oxide Surfaces
The distinction between the intrinsic acidity of a solid and the effective acidity displayed when the surface acidic groups are screened by interaction with solvent molecules becomes a topic of prominent importance when the solid has to work in contact with liquids for its practical uses. This is the case of liquid-solid heterogeneous catalysts in which the activity of the catalyst can be modified by the presence of solvent which may establish physical or chemical interactions with the acid sites of the surface. For reactions carried out in liquid phase, the knowledge of the effective acidity (in terms of number and strength of the sites) of the catalyst in given liquids allows determining sound relationships between the catalytic activity and surface acidity.
Adsorption calorimetry in liquid is a complex matter of study because many variables are simultaneously present: solids with different acidity in terms of distributions of acid sites (nature and acid strength); liquids of different polarity, proticity, and solvating ability; and probes with different basicity (pKa scale). Therefore, results permit comparing for a given solid and probe couple, the influence of the solvent; for a given solid and liquid couple, the influence of the probe; and for a given liquid and probe couple, the properties of different solids.
One of the main goals of the calorimetric experiments of acid-base titrations in liquid is to evaluate the effective acid strengths of the surfaces when suspended in liquids of different nature. As for the gas-solid acid-base titrations, solutions of base probe in liquids of various natures are used for titrating the acid sites of the solid which is placed in a reaction calorimeter equipped with a stirring system. Successive pulse injections of dosed amounts of the probe solution are sent onto the sample maintained at constant temperature up to surface saturation with the probe. In this case, the evolved heat from the adsorption reaction derives from two different contributions: the exothermic enthalpy of adsorption, \(\Delta _{\mathrm{ad}}\mathrm{H}_{\mathrm{probe}}\), and the endothermic enthalpy of displacement of the liquid, \(\Delta _{\mathrm{dpl}}\mathrm{H}_{\mathrm{liquid}}\), the enthalpy effects describing dilution and mixing phenomena can be neglected depending on the differential design and preheating of the probe solution.
Various papers have recently been published concerning the liquid-phase titration of acid solids, such as acidic polymeric resins [107–109]. Example is here presented on the study of the intrinsic and effective acidities of two catalysts based on niobium: niobium oxide (NBO) and niobium phosphate (NBP) [110] which found application in reaction of acid transformation of monosaccharides (fructose, glucose, in particular) to useful chemicals, like 5-hydroxymethyl-2-furaldehyde (HMF) [111, 112].
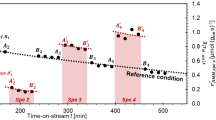
When a strongly basic probe, like 2-phenylethylamine (PEA), in the decane solvent is used for titrating the NBO and NBP surfaces (Fig. 8.14), the information obtained on the two acid surfaces support the conclusions obtained from the more classical determinations of acidity using gas-solid calorimetric adsorption and thermal desorption approaches [110]. The calorimetric curve of acid-base titration in decane for NBP always lies above that of NBO, and the difference between the two curves increases as the titration progresses further. This seems confirming that the main difference between the NBO and NBP surfaces has to be ascribed to the medium and weak strength acid sites.

Evolved heat for each dose of PEA solution injected as a function of the total amount of PEA injected in the liquid-solid adsorption experiments performed at 40 \(^\circ \)C in decane (from Ref. [110])

Evolved heat for each dose of aniline solution injected as a function of the total amount of aniline injected during the liquid-solid adsorption experiments performed at 40 \(^\circ \)C in various liquids: NBO (a) and NBP (b) (from Ref. [110])
The different effective acid strengths of the surfaces of NBO and NBP can be evidenced by the comparative experimental results summarized in Fig. 8.15a,b, respectively, obtained by using aniline as base probe. In these figures, the areas of the heat flow peaks obtained during aniline titration in cyclohexane, decane, toluene, and isopropanol are plotted as functions of the amount of aniline introduced. The higher acidity of NBP compared to that of NBO, in terms of both the number and strength of the sites, is clearly evident. The total heat evolved following the introduction of a definite amount of aniline, and the amount of aniline necessary to complete the adsorption reaction, are in all cases higher for NBP than for NBO, independently of the nature of the liquid. Since the evolved heat is a sum of two contributions with opposite signs (\(\Delta _{\mathrm{ad}}\mathrm{H}_{\mathrm{probe}}\), adsorption enthalpy, exothermic, and \(\Delta _{\mathrm{dpl}}\mathrm{H}_{\mathrm{liquid}}\), enthalpy of displacement of the liquid, endothermic), the higher the endothermic contribution, the lower the experimental heat flow measured. For a given solid and probe, \(\Delta _{\mathrm{ad}}\mathrm{H}_{\mathrm{probe}}\) depends only on the solid acidity, while \(\Delta _{\mathrm{dpl}}\mathrm{H}_{\mathrm{liquid}}\) can be different, depending on solid-solvent interactions. It may then be argued that the surface acid sites of NBO (Fig. 8.15a) present a stronger interaction with cyclohexane and decane than with toluene; the \(\Delta _{\mathrm{dpl}}\mathrm{H}_{\mathrm{liquid}}\) contribution is then higher in cyclohexane and decane, which results in lower measured heats than in toluene.
A completely different picture emerges when aniline titration is carried out in isopropanol, a protic solvent with high polarity. On both NBO and NBP, very low heats evolved have been measured; the initial value of evolved heat obtained for NBP is much higher than that for NBO (Fig. 8.15a,b). This confirms the ability of NBP to maintain a highly acidic character in solvents of high polarity, like alcohols or water.
The acid performances of the NBO and NBP catalysts were compared in the dehydration of fructose to 5-hydroxymethyl-2-furaldehyde (HMF). The reaction runs with high conversion and good selectivity to HMF in organic solvents but in agreement with a green chemistry process development, friendly solvents have to be used, like water or alcohol mixtures. In water, NBP shows higher fructose conversion and HMF selectivity than NBO, so justifying its higher effective acidity in polar and protic liquids [110].
The calorimetric titration of the acid sites in liquid phase makes it possible to discriminate the acid site strength more accurately than the more conventional gas-solid phase titration in order to understand the effective acidity of the solids measured in various liquids of very different polarities and proticities. The obtained calorimetric results are of not easy interpretation since solute-solvent and solid-solvent interactions have to be taken into account, besides the solute-site interaction.
References
G. Busca, Chem. Rev. 107, 5366 (2007)
H. Hattori, Chem. Rev. 95, 537 (1995)
K. Tanabe, W.F. Hölderich, Appl. Catal. A 181, 399 (1999)
G. Busca, Chem. Rev. 110, 2217 (2010)
R.G. Pearson, J. Am. Chem. Soc. 85, 3533 (1963)
G. Klopman, J. Am. Chem. Soc. 90, 223 (1968)
J.L. Reed, J. Phys. Chem. 98, 10477 (1994)
A. Auroux, A. Gervasini, J. Phys. Chem. 94, 6371 (1990)
A. Gervasini, A. Auroux, J. Therm. Anal. 37, 1737 (1991)
G. Busca, Phys. Chem. Chem. Phys. 1, 723 (1999)
J.C. Lavalley, Catal. Today 27, 377 (1996)
S. Coluccia, A.J. Tench, Proceedings of the 7th International Congress on Catalysis, Tokyo, Japan, Kodansha, 1980
I.E. Wachs, Catal. Today 27, 437 (1996)
G. Busca, Catal. Today 41, 191 (1998)
F. Haw, I.S. Chuang, B.L. Hawkins, G.E. Maciel, J. Am. Chem. Soc. 105, 7206 (1983)
M. Johansson, K. Klier, Top. Catal. 4, 99 (1997)
R. Borade, A. Sayari, A. Adnot, S. Kaliaguine, J. Phys. Chem. 94, 5989 (1990)
D. Barthomeuf, G. Coudurier, J.C. Vedrine, Mater. Chem. Phys. 18, 553 (1988)
R.J. Gorte, D. White, Top. Catal. 4, 57 (1997)
A. Boréave, A. Auroux, C. Guimon, Microporous Mater. 11, 275 (1997)
J. Kotrla, L. Kubelková, C.-C. Lee, R.J. Gorte, J. Phys. Chem. B 102, 1437 (1998)
G. Busca, in Metal Oxide Catalysis, ed. by S.D. Jackson, J.S.J. Hargreaves. The Use of Infrared Spectroscopy Methods in the Field of Heterogeneous Catalysis by Metal Oxide, vol 1 (Wiley-VCH, Weinheim, 2009), p. 95
A. Gervasini, C. Messi, D. Flahaut, C. Guimon, Appl. Catal. A 367, 113 (2009)
P. Carniti, A. Gervasini, S. Bennici, J. Phys. Chem. B 109, 1528 (2005)
V. Solinas, I. Ferino, Catal. Today 41, 179 (1998)
A. Auroux, Mol. Sieves 6, 45 (2008)
A. Auroux, Top. Catal. 19, 205 (2002)
A. Auroux, in Catalyst characterization: physical øøøtechniques for solid materials, ed. by B. Imelik, J.C. Vedriine (Plenum Press, New York, 1994), p. 611
M. Fadoni, L. Lucarelli, in Adsorption and its Applications in Industry and Environmental Protection, ed. by A. Dabrowski. Applications in Industry, vol 1 (Stud. Surf. Sci. Catal. 120A) (1999)
H.G. Karge, V. Dondur, J. Phys. Chem. 94, 765 (1990)
H.G. Karge, V. Dondur, J. Weitkamp, J. Phys. Chem. 95, 283 (1991)
F. Arena, R. Dario, A. Parmaliana, Appl. Catal. A 170, 127 (1998)
B. Dragoi, A. Gervasini, E. Dumitriu, A. Auroux, Thermochim. Acta 420, 127 (2004)
A. Gervasini, P. Carniti, A. Auroux, Thermochim. Acta 434, 42 (2005)
S. Bennici, A. Auroux, in Metal Oxide Catalysis, ed. by S.D. Jackson, J.S.J. Hargreaves. Thermal Analysis and Calorimetric Methods, vol 1 (Wiley-VCH, Weinheim, 2009), p. 391
A. Auroux, Top. Catal. 4, 71 (1997)
A. Auroux, A. Gervasini, L. Nemeth, G. Gati, I.S. Pap, G. Mink, Surf. Interface Anal. 19, 529 (1992)
W. Rudzinski, T. Borowiecki, T. Panczyk, A. Dominko, Langmuir 16, 8037 (2000)
M.V. Gargiulo, J.L. Sales, M. Ciacera, G. Zgrablich, Surf. Sci. 501, 282 (2002)
W. Rudzinski, D.H. Everett, Adsorption of Gases on Heterogeneous Surfaces (Academic Press, London, 1992)
N. Cardona-Martinez, J.A. Dumesic, Adv. Catal. 38, 149 (1992)
P. Carniti, A. Gervasini, A. Auroux, J. Catal. 150, 274 (1994)
K. Tsutsumi, Y. Mitani, H. Takahashi, Bull. Chem. Soc. Jpn. 56, 1912 (1983)
N. Cardona-Martinez, J.A. Dumesic, J. Catal. 125, 427 (1990)
P. Carniti, A. Gervasini, React. Kinet. Catal. Lett. 52, 285 (1994)
S.D. Jackson, J.S.J. Hargreaves (eds.), Wiley-VCH, Weinheim, vol I and II, 2009 (ISBN 978-3-527-31815-5).
I.E. Wachs, Catal. Today 100, 79 (2005)
H.-P. Boehnm, H. Knozinger, in Nature and Estimation of Functional Groups on Solid Surfaces, ed. by J.R. Anderson, M. Boudart. Catalysis, Science and Technology, vol 4 (Springer, Berlin, 1984)
I. Nowak, M. Ziolek, Chem. Rev. 99, 3603 (1999)
M. Ziolek, Catal. Today 78, 47 (2003)
K. Tanabe, in Solid Acid and Base Catalysts, ed. by J.R. Anderson, M. Boudart. Catalysis, Science and Technology, vol 5 (Springer, Berlin, 1987), p. 232
J. Haber, in Crystallography of Catalyst Types, ed. by M. Boudart, J.R. Anderson. Catalysis, vol 2 (Springer, Berlin, 1984)
W.E. Farneth, F. Ohuchi, R.H. Staley, U. Chowdhry, A.W. Sleight, J. Phys. Chem. 89, 2493 (1985)
M. Badlani, I.E. Wachs, Catal. Lett. 75, 137 (2001)
I.E. Wachs, Y. Chen, J.-M. Jehng, L.E. Briand, T. Tanaka, Catal. Today 78, 13 (2003)
L.E. Briand, J.-M. Jehng, L. Cornaglia, A.M. Hirt, I.E. Wachs, Catal. Today 78, 257 (2003)
Y. Rao, D.M. Antonelli, J. Mater. Chem. 19, 1937 (2009)
Y. Rao, J. Kang, M. Trudeau, D.M. Antonelli, J. Catal. 266, 1 (2009)
Y. Rao, M.L. Trudeau, D.M. Antonelli, J. Am. Chem. Soc. 128, 13996 (2006)
A. Gervasini, G. Bellussi, J. Fenyvesi, A. Auroux, J. Phys. Chem. 99, 5117 (1995)
N. Cardona-Martínez, J.A. Dumesic, J. Catal. 127, 706 (1991)
E. Cano-Serrano, G. Blanco-Brieva, J.M. Campos-Martin, J.L.G. Fierro, Langmuir 19, 7621 (2003)
A. Gervasini, C. Messi, A. Ponti, S. Cenedese, N. Ravasio, J. Phys. Chem. C 112, 4635 (2008)
C. Flego, L. Carluccio, C. Rizzo, C. Perego, Catal. Commun. 2, 43 (2001)
T. Klimova, M.-L. Rojas, P. Castello, R. Cuevas, J. Ramírez, Microporous Mesoporous Mater. 20, 293 (1998)
G. Guiu, P. Grange, J. Catal. 156, 132 (1995)
B. Notari, R.J. Willey, M. Panizza, G. Busca, Catal. Today 116, 99 (2006)
A. Desmartin-Chomel, J.L. Flores, A. Bourane, J.M. Clacens, F. Figueras, G. Delahay, A. Giroir-Fendler, C. Lehaut-Burnouf, J. Phys. Chem. 110, 858 (2006)
S.Y. Kim, J.G. Goodwin, S. Hammache, A. Auroux, D. Galloway, J. Catal. 201, 1 (2001)
M. Occelli, D.A. Schiraldi, A. Auroux, K.A. Keogh, B.H. Davis, Appl. Catal. A 209, 165 (2001)
D. Deutsch, V. Quaschning, E. Kemnitz, A. Auroux, H. Ehwald, H. Lieske, Top. Catal. 13, 281 (2000)
A. Corma, Chem. Rev. 95, 559 (1995)
J.S. Beck, C.W. Chu, I.D. Johnson, C.T. Kresge, M.E. Leonowicz, W.J. Roth, J.C. Vartuli, JWO 91/11390 (1991)
A. Corma, V. Fornés, M.T. Navarro, J. Pérez-Pariente, J. Catal. 148, 569 (1994)
M.A. Vuurman, D.J. Stufkens, A. Oskam, G. Deo, I.E. Wachs, J. Chem. Soc. Faraday Trans. 92, 3259 (1996)
F. Kapteijn, A.D. Vanlangeveld, J.A. Moulijn, A. Andreini, M.A. Vuurman, A.M. Turek, J.-M. Jehng, I.E. Wachs, J. Catal. 150, 94 (1994)
A. Cimino, D. Gazzoli, M. Valigi, J. Electron Spectrosc, Relat. Phenom. 104, 1 (1999)
A. Gervasini, J. Fenyvesi, A. Auroux, Langmuir 12, 5356 (1996)
T. Uchijima, Catal. Today 28, 105 (1996)
C. Rizzo, A. Carati, M. Tagliabue, C. Perego, Stud. Surf. Sci. Catal. 128, 137 (2000)
R. Pohle, M. Fleischer, H. Meixner, Sens. Actuators B 68, 151 (2000)
M. Kudo, T. Kosaka, Y. Takahashi, H. Kokusen, N. Sotani, S. Hasegawa, Sens. Actuators B 69, 10 (2000)
J.A. Perdigón-Melón, A. Gervasini, A. Auroux, J. Catal. 234, 421 (2005)
A. Gervasini, J.A. Perdigón-Melón, C. Guimon, A. Auroux, J. Phys. Chem. B 110, 240 (2006)
K. Nakagawa, C. Kajita, Y. Ide, M. Okamura, S. Kato, H. Kasuya, N. Ikenaga, T. Kobayashi, T. Suzuki, Catal. Lett. 64, 215 (2000)
A.L. Petre, J.A. Perdigón-Melón, A. Gervasini, A. Auroux, Catal. Today 78, 377 (2003)
J. Macht, E. Iglesia, Phys. Chem. Chem. Phys. 10, 5331 (2008)
S. Bennici, A. Auroux, C. Guimon, A. Gervasini, Chem. Mater. 18, 3641 (2006)
D. Tichit, M.H. Lhouty, A. Guida, B.H. Chiche, F. Figueras, A. Auroux, D. Bartolani, E. Garrone, J. Catal. 151, 50 (1995)
A. Vaccari, Appl. Clay Sci. 14, 161 (1999)
D. Tichit, B. Coq, CATTECH 7, 206 (2003)
A. Corma, S. Iborra, Adv. Catal. 49, 239 (2006)
M.A. Peña, J.L.G. Fierro, Chem. Rev. 101, 1981 (2001)
M.J. Sayagues, M. Vallet-Regi, A. Caneiro, J.M. Gonzales Calbet, J. Solid State Chem. 110, 295 (1994)
F.X. Liu-Cai, B. Sahut, E. Faydi, A. Auroux, G. Hervé Appl, Catal. A 185, 75 (1999)
L. Damjanovic, V. Rakic, U.B. Mioc, A. Auroux, Thermochim. Acta 434, 81 (2005)
T. Nakato, M. Kimura, S. Nakata, T. Okuhara, Langmuir 14, 319 (1998)
P. Carniti, A. Gervasini, M. Marzo, J. Phys. Chem. C 112, 14064 (2008)
C.L. Thomas, Ind. Eng. Chem. 41, 2564 (1949)
K.I. Tanaka, A. Ozaki, J. Catal. 8, 1 (1967)
K. Tanabe, T. Sumiyoshi, K. Shibata, T. Kiyoura, J. Kitagawa, Bull. Chem. Soc. Japan 47, 1064 (1974)
T. Seiyama, Metal Oxides and Their Catalytic Actions (Kodansha, Tokyo, 1978)
G. Connell, J.A. Dumesic, J. Catal. 102, 216 (1986)
B. Notari, Adv. Catal. 41, 253 (1996)
M. Itoh, H. Hattori, K. Tanabe, J. Catal. 35, 225 (1974)
M. Itoh, H. Hattori, K. Tanabe, J. Catal. 43, 192 (1976)
M. Hart, G. Fuller, D.R. Brown, J.A. Dale, S. Plant, J. Mol. Catal. A: Chem. 182–183, 439 (2002)
S. Koujout, B.M. Kiernan, D.R. Brown, H.G.M. Edwards, J.A. Dale, S. Plant, Catal. Lett. 85, 33 (2003)
S. Koujout, D.R. Brown, Catal. Lett. 98, 195 (2004)
P. Carniti, A. Gervasini, S. Biella, A. Auroux, Chem. Mater. 17, 6128 (2005)
P. Carniti, A. Gervasini, S. Biella, A. Auroux, Catal. Today 118, 373 (2006)
P. Carniti, A. Gervasini, M. Marzo, Catal. Today 152, 42 (2010)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Gervasini, A. (2013). Characterization of Acid–Base Sites in Oxides. In: Auroux, A. (eds) Calorimetry and Thermal Methods in Catalysis. Springer Series in Materials Science, vol 154. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-11954-5_8
Download citation
DOI: https://doi.org/10.1007/978-3-642-11954-5_8
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-11953-8
Online ISBN: 978-3-642-11954-5
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)