
Abstract
The inflammatory bowel diseases (IBD); Crohn’s and Ulcerative colitis, result from an altered host response to intestinal flora. Recurrent inflammation with ulceration and tissue restitution confers an increased risk of cancer in both UC and Crohns, and genome wide searches have identified a number of disease susceptibility alleles. The carcinogenesis pathway in colitis-associated colorectal cancer (CACRC) is less clearly understood than it’s sporadic counterpart. Clonal ordering experiments have indicated the order and timing of chromosomal instability and common genetic mutations. Epigenetic changes such as DNA methylation and histone modification are thought to play an increasingly important role in inflammation induced carcinogenesis. Clonal expansion of procarcinogenic mutations can lead to large fields of mutant tissue from which colitis associated cancers can arise (field cancerisation). Endoscopic screening is the mainstay of surveillance in high-risk patients although the development of appropriate, clinically applicable biomarkers remains a research priority. Despite the expanding field of biological therapy in inflammatory bowel disease the ASA compounds remain the best-studied and most efficacious chemopreventive agents. Colitis associated CRC appears to have a different aetiology, carcinogenesis pathway and clinical course to its sporadic counterpart. Further research including long-term follow up of patient cohorts taking biological therapies will improve the detection and treatment of these important, inflammation-induced malignancies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Inflammatory Bowel Disease
- Ulcerative Colitis
- Primary Sclerosing Cholangitis
- Stem Cell Therapy
- Field Cancerisation
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Ulcerative colitis (UC) and Crohn’s disease (CD) both have a prevalence of two per 1,000 people in northern Europe, with an incidence of 10 and 6 per 100,000 people per year, respectively, in Western countries (Shivananda et al. 1996). Clinically the conditions are characterised by chronic, relapsing inflammation affecting the colon only in UC or any portion of the gut in CD. The aetiology of these conditions is unclear, but it is often presumed that they result from an altered host response to normal intestinal flora.
Inflammatory bowel disease (IBD) confers a high risk of development of a number of malignancies especially colorectal cancer, with a standardised incidence ratio of 2.4 (95% CI 0.6–6.0) in patients with extensive or pan UC. This risk is associated with longer disease duration, an earlier age of onset (Ekbom et al. 1990) the greater the severity of inflammation (Rutter et al. 2004), and the presence of concomitant inflammatory conditions such as primary sclerosing cholangitis (PSC). This suggests that the acquired cancer risk is a consequence of the inflammatory process which results in cycles of recurrent ulceration and tissue restitution. It is now accepted that the cancer risk in both Crohn’s disease and UC is approximately the same if similar disease patterns are compared (Gillen et al. 1994), and this is further evidence for similar inflammation-related tumour biology.
2 Genetic Epidemiology of IBD Aetiology
Genetic and environmental factors both appear to be important in the development of IBD. At least 13 genome-wide linkage studies have been completed since 1996, but the results have varied widely. The most frequently reported linkages were designated the IBD loci (Table 1).
A recent genome-wide association study (GWAS) of UC has recently identified susceptibility loci that provides the first genetic link between UC and colorectal cancer (Barrett et al. 2009). The strongest new association intervals include CDH1 on chromosome 16q22, which encodes E-cadherin, a transmembrane glycoprotein and one of the main components of the adherens junction. It is a key mediator of intercellular adhesion in the intestinal epithelium and also plays a key role in epithelial restitution and repair following mucosal damage. The observation of correlated association signals at the CDH1 locus in both colorectal cancer and UC is significant.
This is the first time that variants within genetic loci encoding epithelial barrier genes have shown association with IBD at rigorous genome-wide significant thresholds and provide further evidence for the re-emerging concept that altered epithelial barrier function and may be a strategic factor in UC pathogenesis. Additional fine mapping and functional studies are undoubtedly necessary to explore this association; however this study provides strong scientific rationalisation for the investigation of novel therapeutic targets pertinent to epithelial barrier function.
2.1 Genetic Variation in Inflammation-Related Genes
Associations between CRC and genetic variation in genes involved in inflammation-related pathways provide support to the mounting body of evidence that suggests inflammation-related pathways are important in the aetiology of CRC. Of potential importance are genes such as IL6, which is a critical regulator of inflammation signalling (Slattery et al. 2007). Polymorphisms in the IL6 gene promoter have been found to be associated with levels of circulating C-reactive protein, an important biomarker for pro-inflammatory status in several diseases (Ferrari et al. 2003). A study by Slattery et al. (2007) suggests that IL6 genotype may influence risk of CRC. Individuals with the C allele of the c.572G > C SNP and the GG genotype for the c.174G > C polymorphism were at a slightly reduced risk of colon cancer, but possibly at a slightly increased risk of rectal cancer. Associations were comparable for men and women and for all age groups and appeared to be modified by use of aspirin or non-steroidal anti-inflammatory drugs (NSAIDs): especially for colon cancer (Macarthur et al. 2005). In addition, if users had a C allele in either IL6 polymorphism, they had a greater reduction in risk of colon cancer (Slattery et al. 2007). Although these data are supportive of genetic involvement in an inflammation-related pathway, additional work is necessary that will encompass more genes in this pathway to obtain a better understanding of the associations between inflammation and genetic factors and CRC development.
3 Comparison of Carcinogenesis Pathways in Sporadic and Colitis-Associated Colorectal Cancer
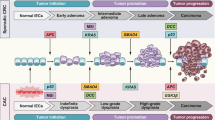
There are several distinguishing clinical features when comparing colitis-associated colorectal cancer (CACRC) to sporadic colorectal carcinoma (SCRC). Firstly, CACRC arises in a younger population, often from flat, not polyploid dysplasia and has a more proximal distribution. Furthermore, there is a greater frequency of mucinous or signet cell histology and a higher incidence of multiple synchronous lesions (Itzkowitz and Yio 2004). From a histological perspective, sporadic tumours tend to follow the adenoma-carcinoma sequence (Vogelstein et al. 1988), whereas CACRC progresses from no dysplasia to indefinite dysplasia, usually through low (LGD) and high-grade dysplasia (HGD) to carcinoma. The stepwise accumulation of genetic mutations in onco- and tumour suppressor genes that underpins the SCRC carcinogenesis pathway is well established and has significantly altered worldwide clinical practice (Vogelstein et al. 1988). The CACRC carcinogenesis pathway is less explored and significantly differs in the requirement and timing of genetic and epigenetic alterations (Fig. 1).

Comparison of Colitis Associated and Sporadic Colorectal Cancer Pathways. Both types of cancer show multistep development with sequential mutation in tumour suppressor and oncogenes. The main differences between the pathways are in the timing of these mutations. Abbreviations: APC, Adenomatous Polyposis Coli; DCC, Deleted in Colon Cancer; LOH, loss of heterozygosity; MSI, Microsatellite instability
3.1 Genetic Instability
3.1.1 Chromosomal Instability
In sporadic cancer carcinogenesis, chromosomal instability leading to aneuploidy, detectable by both image and flow cytometry, is rare in established precursor lesions before the development of high-grade dysplasia or cancer (Sieber et al. 2002). Yet, in ulcerative colitis, chromosomal instability (CIN) can be detected in histologically non-dysplastic tissue from high-risk patients (extensive disease distribution and long duration of disease), by comparative genomic hybridisation (Willenbucher et al. 1997), image (Keller et al. 2001) or flow cytometry and is thought to precede the development of dysplasia in these patients (Rubin et al. 1992; Lofberg et al. 1992; Befrits 1994). It has been suggested that CIN occurs as a consequence of the effect of inflammation and reactive oxygen species encouraging telomere shortening, permitting chromosomal end fusion. This results in cycles of chromatin bridge breakage and fusion, promoting the accumulation of chromosomal aberrations (O’Sullivan et al. 2002).
3.1.2 Initiating Genetic Mutations
In sporadic CRC carcinogenesis, mutations in APC are found in about 60% of sporadic adenomas and 80% of tumours (Powell et al. 1992) and are considered to be the gate-keeping, initiating mutations (Kinzler and Vogelstein 1996). It is now becoming clear that the inflammation and restitution processes that underly IBD, select for alternative initiating genetic mutations in CACRC. A recent clonal ordering study determining the spatial distribution of shared mutations in UC-associated neoplasia allowed insight into the timing of genetic mutations (Leedham et al. 2009). p53 was the most common single founding mutation with K-RAS mutations as the only other detected unique gate-keeping mutation. APC mutations were uncommon suggesting that APC is unlikely to have a gatekeeper function in colitis. This is consistent with other work. Point mutations in the p53 gene can be detected in non-dysplastic tissue from patients with UC preceding the development of aneuploidy and LOH, and appear to be linked to the presence of inflammation (Brentnall et al. 1994; Hussain et al. 2000). Additionally, LOH for p53 correlates with malignant progression, occurring in 6% of non-dysplastic biopsies, 33% of LGD, 63% of HGD and 85% of cancers (Burmer et al. 1992). The mutation spectrum in p53 is dominated by transition mutations (Yin et al. 1993; Hussain et al. 2000; Yoshida et al. 2003), and this is likely to reflect the effect of the inflammatory process causing oxidative DNA damage and deamination of 5-methylcytosine, promoting G:C to A:T transitions (Hussain et al. 2000; Seril et al. 2003). It is simple to comprehend why p53 may act as an initiating mutation in colitis. If underlying chromosomal instability throughout the colon is the main tumourigenic driving force in colitis (Chen et al. 2003, 2005), early p53 would be selected for, on the basis that disruption of a mitotic checkpoint would permit the survival and selection of clones with gross chromosomal changes.
3.2 Role of Inflammation in Cancer Epigenetics
One of the many potential processes by which inflammation can contribute to carcinogenesis includes alterations in epigenetic events and subsequent inappropriate gene expression.
3.2.1 DNA Methylation and Transcriptional Silencing
CpG island hypermethylation often starts in normal mucosa as a function of age and is markedly increased in cancer (Issa et al. 2001). Such silencing is clonal and is thought to be physiologically irreversible in somatic cells. Neoplastic cells often display aberrant promoter region methylation with epigenetic silencing of multiple genes including genes that regulate critical processes such as cell cycle control, DNA repair and angiogenesis. In the colon, CpG islands methylated in cancer have been divided into two groups: those that display cancer-restricted methylation (type C), and those that are methylated initially in aging normal epithelial cells (type A). It has been proposed that age-related methylation contributes to an acquired predisposition to colorectal neoplasia because methylation alters the physiology of aging cells and tissues (Issa et al. 2001). This hypothesis predicts that higher levels of age-related methylation are associated with a heightened susceptibility to developing colorectal cancer, and it may be present in conditions of rapid cell turnover that mimic premature aging such as IBD.
Issa et al. (2001), investigated the methylation status of 4 genes in patients with UC versus controls (ER, MYOD1, CSPG2 and p16). All four genes were highly methylated in dysplastic epithelium from patients with colitis-associated HGD or cancer. In addition, three of the four genes (ER, MYOD and p16) were also highly methylated in the normal appearing (non-dysplastic) epithelium from these same HGD/cancer patients, indicating that methylation precedes dysplasia and is widespread in these patients. These results are consistent with the hypothesis that age-related methylation marks (and may lead to) the field defect that reflects acquired predisposition to colorectal neoplasia. More recently, Kukitsu et al. (2008) identified hypermethylation and subsequently reduced p16 gene expression in aberrant crypt foci (ACF) in UC. These are the earliest detectable lesions in the CACRC pathway and suggest that aberrant methylation of tumour suppressor genes may be an early event in CACRC carcinogenesis.
3.2.2 Histone Modification
A well-proven epigenetic mechanism of gene expression control involves chromatin remodelling via histone modification. Transcriptional regulation of a variety of cancer-related genes are controlled by two contrasting classes of enzymes—histone deacetylase (HDAC) and histone acetyl transferases (HATs). The acetylation of lysine residues on the N-terminus of histones by HATs activates gene transcription, while removal of an acetyl group from lysine residues in histone tails by HDACs results in transcriptional repression. Therefore, HDACs and HATs, in general, act as transcriptional co-repressors and co-activators, respectively. In chronic inflammatory responses and carcinogenesis the inappropriate activation/inactivation of HDACs and HATs has been implicated. In a study by Cao et al. (2007), the exposure of human bronchial epithelial cells (BEAS-2B) to the diesel exhaust particulate matter induced the transcriptional activation of a representative pro-inflammatory gene cyclooxygenase-2 (COX-2) by promoting acetylation of histone-4 by degradation of HDAC-1. Similarly, the activation of NF-κB and expression and release of IL-8 and IL-6 in human alveolar (A549) cells by H2O2 were associated with augmented acetylation of histone-4 and diminished expression and activity of HDAC-2 (Cao et al. 2007). Thus histone modification and the ensuing upregulation of COX-2 and NF-κB demonstrates that inflammation induced modification in cellular epigenetic apparatus may also contribute to the genetic instability of cancer cells.
4 Mutator Phenotype versus Clonal Expansion
Multiple aneuploidy detection techniques have shown gross chromosomal changes occurring in non-dysplastic tissue in UC (see Sect. 3.1). Chen et al. (2003, 2005) used arbitrarily-primed (AP-PCR) and inter-simple-sequence repeat PCR (ISSR-PCR) genetic fingerprinting techniques to further analyse genomic instability in colitis. The identification of DNA fingerprint abnormalities throughout normal and dysplastic areas of the colon allowed the subdivision of patients with IBD into UC progressors: patients with identifiable genomic instability who are likely to progress to dysplasia or cancer, and UC non-progressors, patients with normal DNA fingerprints who are not (Chen et al. 2003). The authors proposed that this colon-wide genomic instability in UC progressors provides a field from which dysplasia develops, and is evidence of a mutator phenotype where mutations in genes maintaining genetic stability result in an increased mutation rate driving colitis-associated tumourigenesis (Chen et al. 2003, 2005; Loeb and Loeb 1999). This is a controversial subject and proponents of an evolutionary theory of carcinogenesis argue that the mutator phenotype theory underestimates the power of natural selection (Tomlinson and Bodmer 1999; Bodmer 2008). The recent identification of colitis-associated neoplasia clonality with p53 as the commonest initiating mutation (Leedham et al. 2009) lends weight to a Darwinian model where natural selection and clonal expansion are the dominant forces driving CACRC evolution—the somatic mutation theory of carcinogenesis. The close association between the cell cycle and DNA repair suggests that a number of genes involved in the cellular response to DNA damage, such as p53 may have a two-fold responsibility in controlling DNA repair and growth. Consequently mutations in these genes may provide both a selective growth advantage and an increased mutation rate driving selection and the mutator phenotype simultaneously, although evolutionary geneticists argue that the mutator phenotype component is a coincidental by-product of direct selection of mutation of these genes for their anti-apoptotic effects (Bodmer 2008). The debate continues!
5 Field Cancerisation
The term field cancerisation was proposed by Slaughter et al. (1953) to explain the presence of multifocal head and neck cancers developing out of a field of precancerous change that had developed following carcinogen exposure. Braakhuis et al. (2003) expanded this theory and proposed that the field was actually a clonally expanded area of mutated cells. Clonally expanded mutated patches have been documented previously in dysplastic and phenotypically normal mucosa of colitis patients (Lyda et al. 1998, 2000). Leedham et al. (2009) identified field cancerisation in one interesting patient when they demonstrated that three left-sided tumours and some of the intervening chronically inflamed but phenotypically non-dysplastic mucosa shared the same founder mutation, suggesting widespread clonal expansion of a progenitor clone from which the three spatially independent tumours arose. Niche succession and crypt fission are likely to be the mechanisms behind clonal expansion in CACRC. Occasional symmetrical division of individual crypt stem cells results in the extinction or amplification of one cell lineage (Kim and Shibata 2002). This process will occur faster if the mutation provides a growth advantage. Crypt fission has been shown to be responsible for the spread of individual clones into daughter crypts in the colon (Greaves et al. 2006) and this process is a histological feature of colitis and dysplasia (Park et al. 1995). Chen et al. (2005) used a fluorescent in situ hybridisation technique to demonstrate the spread of p53 mutations into the daughter crypts of a crypt in the process of fission in UC. The suggestion of field cancerisation in this condition has possible clinical implications, raising questions about the use of molecular genetic analysis of non-dysplastic tissue in high-risk cancer patients to detect fields from which future tumours may arise.
6 Screening and Detection
6.1 Endoscopic Screening
Early detection and screening is the mainstay of reducing cancer morbidity and mortality in the IBD population. As the risk of CRC is influenced by the extent and duration of the disease current European guidelines suggest an initial assessment colonoscopy 8–10 years after the onset of symptoms in UC (Moum et al. 1999). The development of PSC is an independent risk factor and patients should be offered yearly surveillance as soon as PSC is diagnosed. Many dysplastic lesions in colitis are flat rather than polypoid (Allen et al. 1985). These are more difficult to detect endoscopically, which leads us to the question of how many random biopsies to take to maximise the chance of detecting dysplasia? Current recommendations suggest that four biopsies should be taken every 10 cm with additional biopsies in strictured, raised or other abnormal areas of the colon; however this is time consuming for patients, nurses, colonoscopists and histopathologists. There are gradual moves towards a more focused approach to obtain targeted biopsies aided by the use of chromendoscopy with indigo carmine or methylene blue. This has shown to give a superior yield in the detection of dysplasia (Biancone et al. 2008; Eaden and Mayberry 2002; Winawer et al. 2003). The role of other methods of targeting biopsies—such as trimodal, autofluoresence and narrow band imaging are also being studied and may feature in future recommendations (East et al. 2006; van den Broek et al. 2008; Dekker et al. 2007).
6.2 Biomarkers
A biomarker is an indicator of a pathological process that may be measured or used to assess the response to therapeutic intervention. At present the histological detection of dysplasia in a biopsy sample is the only marker that has entered widespread clinical practice, and the detection of high-grade dysplasia is an indication for endoscopic resection or colectomy. The discomfort, difficulty and expense of obtaining histological samples mean that the development of a biomarker detectable in stool is a research priority. As yet studies on calprotectin (von Roon et al. 2007) and SFRP2 hypermethylation (Huang et al. 2007) from stool samples have failed to show the sensitivity and specificity required for clinical applicability.
7 Chemoprevention
Prevention is the best strategy to minimise the impact of cancer and may be theoretically achieved by good disease control and reduction of modifiable risk factors. A number of pharmacological agents have been proposed to have a chemopreventive role and these include 5-amino salicylic acid (5-ASA) compounds. The efficacy of these agents may only be partially explained by anti-inflammatory effects of these drugs as other more potent anti-inflammatory agents such as glucocorticoids and immunomodulators such as azathioprine have a less significant cancer protective effect. Additional chemopreventative effects of 5-ASA compounds include; modulation of inflammatory cytokine production (Zimmerman and Jewell 1996), inhibition of cyclooxygenase (Allgayer 2003), inducible NO synthase (Hasko et al. 2001; Kennedy et al. 1999) and nuclear factor KB (Greten et al. 2004; Wahl et al. 1998) as well as activation of peroxisome proliferator activated receptor (PPAR) gamma (Dubuquoy et al. 2006; Rousseaux et al. 2005). In addition to this 5-ASA’s scavenge oxygen free radicals and have an antimicrobial action (Swidsinski et al. 2005). 5-ASA compounds can also act as an inhibitor of protein phosphatase 2A—which can reduce the activity of the Wnt pathway (Bos et al. 2006). Although, theoretically these mechanisms could help to prevent cancer, there are no prospective randomised controlled trials to confirm the protective effect of 5-ASA in cancer chemoprevention in colitis. The best evidence to support their use comes from the meta-analysis by Velayos et al. (2008) that revealed a reduced risk of the development of cancer or dysplasia in UC patients on regular 5-ASA (pooled odds ratio of 0.51 (95% CI 0.38–0.69)).
8 Future Perspectives
8.1 Biological Therapies and Cancer
With the advent of the use of biological therapies, we have seen the medical management of IBD patients who are refractory to steroids and revolutionised immunomodulators (Rutgeerts et al. 2009). As yet there is no evidence to suggest that biologics offer any cancer chemoprevention. In fact, data from the British Society of Rheumatology Biologics Registry show that patients with pre-existing cancer have an increased risk of recurrence with the use of biologics, and those without pre-existing cancer have no increased incidence except in two cohorts—teenagers and young adolescents (in particular with the risk of hepatosplenic T-cell lymphoma) (Rosh et al. 2007). There remains many unanswered questions about the mechanism of action, appropriate time to use biologics and their long-term safety profile and as more long-term data emerges, our understanding of these novel therapies will expand.
8.2 Stem Cell Therapy
It is now appreciated that bone marrow-derived stem cells have a dynamic role in inflammation and cancer throughout the body. Bone marrow-derived cells contribute to myofibroblast populations in the colon and small intestine of mice and humans (Brittan et al. 2002) as well as in mouse models of colitis where they also contribute to vascular lineages (Brittan et al. 2005). Not only this, but also in the IL-10 knock-out model, the colitis that develops can be ameliorated by transplantation of wild-type bone marrow (Bamba et al. 2006). Bone marrow has been shown to contribute to stromal cell populations in cancer (Direkze et al. 2004) and this may offer an alternative route to target therapies to control not only IBD itself but also CACRC, a finding that has been seen in mouse cancer models (Studeny et al. 2002; Nakamizo et al. 2005). In the human, case reports of amelioration of IBD in haematopoietic stem cell (HSC) transplant recipients for co-incident haematologic malignancy prompted interest in stem cell therapy for IBD (reviewed in (Lanzoni et al. 2008)). More recently adipose-derived mesenchymal stem cells have been successfully used in the treatment of refractory perianal fistulae (Garcia-Olmo et al. 2005) and a European-wide phase III trial on the effect of autologous stem cell transplantation in CD is underway (ASTIC trial). Whether the beneficial effect of stem cell therapy arises from concomitant immunosuppressive therapy or from a ‘resetting’ of the colonic stem cell niche remains to be seen.
There is increasing evidence that CACRC has a different aetiology, carcinogenesis pathway and clinical course to its sporadic counterpart. Genome-wide association studies have revealed new susceptibility loci and opened up new lines of investigation. The recognition that intestinal immune system dysregulation provokes chronic inflammation with resultant carcinogenesis has already shifted the focus of management of the IBD. The development of further biological treatments including stem cell therapy promises further tantalising insight into the pathogenesis of these, and other chronic inflammatory conditions.
References
Allen DC, Biggart JD, Pyper PC (1985) Large bowel mucosal dysplasia and carcinoma in ulcerative colitis. J Clin Pathol 38:30–43
Allgayer H (2003) Review article: mechanisms of action of mesalazine in preventing colorectal carcinoma in inflammatory bowel disease. Aliment Pharmacol Ther 18(suppl 2):10–14
Asano K, Matsushita T, Umeno J et al (2009) A genome-wide association study identifies three new susceptibility loci for ulcerative colitis in the Japanese population. Nat Genet 41:1325–1329
Bamba S, Lee CY, Brittan M et al (2006) Bone marrow transplantation ameliorates pathology in interleukin-10 knockout colitic mice. J Pathol 209:265–273
Barrett JC, Lee JC, Lees CW et al (2009) Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat Genet 41:1330–1334
Befrits R, Hammarberg C, Rubio C et al (1994) DNA aneuploidy and histologic dysplasia in long-standing ulcerative colitis. A 10-year follow-up study. Dis Colon Rectum 37:313–319 discussion 319-20
Biancone L, Michetti P, Travis S, Escher JC, Moser G, Forbes A, Hoffmann JC, Dignass A, Gionchetti P, Jantschek G, Kiesslich R, Kolacek S et al (2008) European evidence-based consensus on the management of ulcerative colitis: special situations. J Crohn’s Colitis 2:63–92
Bodmer W (2008) Genetic instability is not a requirement for tumor development. Cancer Res 68:3558–3560 discussion 3560-1
Bos CL, Diks SH, Hardwick JC et al (2006) Protein phosphatase 2A is required for mesalazine-dependent inhibition of Wnt/beta-catenin pathway activity. Carcinogenesis 27:2371–2382
Braakhuis BJ, Tabor MP, Kummer JA et al (2003) A genetic explanation of Slaughter’s concept of field cancerization: evidence and clinical implications. Cancer Res 63:1727–1730
Brentnall TA, Crispin DA, Rabinovitch PS et al (1994) Mutations in the p53 gene: an early marker of neoplastic progression in ulcerative colitis. Gastroenterology 107:369–378
Brittan M, Hunt T, Jeffery R et al (2002) Bone marrow derivation of pericryptal myofibroblasts in the mouse and human small intestine and colon. Gut 50:752–757
Brittan M, Chance V, Elia G et al (2005) A regenerative role for bone marrow following experimental colitis: contribution to neovasculogenesis and myofibroblasts. Gastroenterology 128:1984–1995
Burmer GC, Rabinovitch PS, Haggitt RC et al (1992) Neoplastic progression in ulcerative colitis: histology, DNA content, and loss of a p53 allele. Gastroenterology 103:1602–1610
Cao D, Bromberg PA, Samet JM (2007) COX-2 expression induced by diesel particles involves chromatin modification and degradation of HDAC1. Am J Respir Cell Mol Biol 37:232–239
Chen R, Rabinovitch PS, Crispin DA et al (2003) DNA fingerprinting abnormalities can distinguish ulcerative colitis patients with dysplasia and cancer from those who are dysplasia/cancer-free. Am J Pathol 162:665–672
Chen R, Bronner MP, Crispin DA et al (2005a) Characterization of genomic instability in ulcerative colitis neoplasia leads to discovery of putative tumor suppressor regions. Cancer Genet Cytogenet 162:99–106
Chen R, Rabinovitch PS, Crispin DA et al (2005b) The initiation of colon cancer in a chronic inflammatory setting. Carcinogenesis 26:1513–1519
Cho JH, Nicolae DL, Gold LH et al (1998) Identification of novel susceptibility loci for inflammatory bowel disease on chromosomes 1p, 3q, and 4q: evidence for epistasis between 1p and IBD1. Proc Natl Acad Sci USA 95:7502–7507
Dekker E, van den Broek FJ, Reitsma JB et al (2007) Narrow-band imaging compared with conventional colonoscopy for the detection of dysplasia in patients with longstanding ulcerative colitis. Endoscopy 39:216–221
Direkze NC, Hodivala-Dilke K, Jeffery R et al (2004) Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res 64:8492–8495
Dubuquoy L, Rousseaux C, Thuru X et al (2006) PPARgamma as a new therapeutic target in inflammatory bowel diseases. Gut 55:1341–1349
Duerr RH, Barmada MM, Zhang L et al (2000) High-density genome scan in Crohn disease shows confirmed linkage to chromosome 14q11–12. Am J Hum Genet 66:1857–1862
Eaden JA, Mayberry JF (2002) Guidelines for screening and surveillance of asymptomatic colorectal cancer in patients with inflammatory bowel disease. Gut 51(Suppl 5):V10–V12
East JE, Suzuki N, von Herbay A et al (2006) Narrow band imaging with magnification for dysplasia detection and pit pattern assessment in ulcerative colitis surveillance: a case with multiple dysplasia associated lesions or masses. Gut 55:1432–1435
Ekbom A, Helmick C, Zack M et al (1990) Ulcerative colitis and colorectal cancer. A population-based study. N Engl J Med 323:1228–1233
Fellermann K, Stange DE, Schaeffeler E et al (2006) A chromosome 8 gene-cluster polymorphism with low human beta-defensin 2 gene copy number predisposes to Crohn disease of the colon. Am J Hum Genet 79:439–448
Ferrari SL, Ahn-Luong L, Garnero P et al (2003) Two promoter polymorphisms regulating interleukin-6 gene expression are associated with circulating levels of C-reactive protein and markers of bone resorption in postmenopausal women. J Clin Endocrinol Metab 88:255–259
Fisher SA, Tremelling M, Anderson CA et al (2008) Genetic determinants of ulcerative colitis include the ECM1 locus and five loci implicated in Crohn’s disease. Nat Genet 40:710–712
Franke A, Balschun T, Karlsen TH et al (2008) Sequence variants in IL10, ARPC2 and multiple other loci contribute to ulcerative colitis susceptibility. Nat Genet 40:1319–1323
Garcia-Olmo D, Garcia-Arranz M, Herreros D et al (2005) A phase I clinical trial of the treatment of Crohn’s fistula by adipose mesenchymal stem cell transplantation. Dis Colon Rectum 48:1416–1423
Gillen CD, Walmsley RS, Prior P et al (1994) Ulcerative colitis and Crohn’s disease: a comparison of the colorectal cancer risk in extensive colitis. Gut 35:1590–1592
Greaves LC, Preston SL, Tadrous PJ et al (2006) Mitochondrial DNA mutations are established in human colonic stem cells, and mutated clones expand by crypt fission. Proc Natl Acad Sci USA 103:714–719
Greten FR, Eckmann L, Greten TF et al (2004) IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 118:285–296
Hampe J, Schreiber S, Shaw SH et al (1999) A genomewide analysis provides evidence for novel linkages in inflammatory bowel disease in a large European cohort. Am J Hum Genet 64:808–816
Hasko G, Szabo C, Nemeth ZH et al (2001) Sulphasalazine inhibits macrophage activation: inhibitory effects on inducible nitric oxide synthase expression, interleukin-12 production and major histocompatibility complex II expression. Immunology 103:473–478
Huang Z, Li L, Wang J (2007) Hypermethylation of SFRP2 as a potential marker for stool-based detection of colorectal cancer and precancerous lesions. Dig Dis Sci 52:2287–2291
Hugot JP, Laurent-Puig P, Gower-Rousseau C et al (1996) Mapping of a susceptibility locus for Crohn’s disease on chromosome 16. Nature 379:821–823
Hussain SP, Amstad P, Raja K et al (2000) Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: a cancer-prone chronic inflammatory disease. Cancer Res 60:3333–3337
Imielinski M, Baldassano RN, Griffiths A et al (2009) Common variants at five new loci associated with early-onset inflammatory bowel disease. Nat Genet 41:1335–1340
Issa JP, Ahuja N, Toyota M et al (2001) Accelerated age-related CpG island methylation in ulcerative colitis. Cancer Res 61:3573–3577
Itzkowitz SH, Yio X (2004) Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: the role of inflammation. Am J Physiol Gastrointest Liver Physiol 287:G7–G17
Keller R, Foerster EC, Kohler A et al (2001) Diagnostic value of DNA image cytometry in ulcerative colitis. Dig Dis Sci 46:870–878
Kennedy M, Wilson L, Szabo C et al (1999) 5-aminosalicylic acid inhibits iNOS transcription in human intestinal epithelial cells. Int J Mol Med 4:437–443
Kim KM, Shibata D (2002) Methylation reveals a niche: stem cell succession in human colon crypts. Oncogene 21:5441–5449
Kinzler KW, Vogelstein B (1996) Lessons from hereditary colorectal cancer. Cell 87:159–170
Kukitsu T, Takayama T, Miyanishi K et al (2008) Aberrant crypt foci as precursors of the dysplasia-carcinoma sequence in patients with ulcerative colitis. Clin Cancer Res 14:48–54
Lanzoni G, Roda G, Belluzzi A et al (2008) Inflammatory bowel disease: moving toward a stem cell-based therapy. World J Gastroenterol 14:4616–4626
Leedham SJ, Graham TA, Oukrif D et al (2009) Clonality, founder mutations, and field cancerization in human ulcerative colitis-associated neoplasia. Gastroenterology 136:542–506
Loeb KR, Loeb LA (1999) Genetic instability and the mutator phenotype. Studies in ulcerative colitis. Am J Pathol 154:1621–1626
Lofberg R, Brostrom O, Karlen P et al (1992) DNA aneuploidy in ulcerative colitis: reproducibility, topographic distribution, and relation to dysplasia. Gastroenterology 102:1149–1154
Lyda MH, Noffsinger A, Belli J et al (1998) Multifocal neoplasia involving the colon and appendix in ulcerative colitis: pathological and molecular features. Gastroenterology 115:1566–1573
Lyda MH, Noffsinger A, Belli J et al (2000) Microsatellite instability and K-ras mutations in patients with ulcerative colitis. Hum Pathol 31:665–671
Ma Y, Ohmen JD, Li Z et al (1999) A genome-wide search identifies potential new susceptibility loci for Crohn’s disease. Inflamm Bowel Dis 5:271–278
Macarthur M, Sharp L, Hold GL et al (2005) The role of cytokine gene polymorphisms in colorectal cancer and their interaction with aspirin use in the northeast of Scotland. Cancer Epidemiol Biomarkers Prev 14:1613–1618
Moum B, Ekbom A, Vatn MH et al (1999) Change in the extent of colonoscopic and histological involvement in ulcerative colitis over time. Am J Gastroenterol 94:1564–1569
Nakamizo A, Marini F, Amano T et al (2005) Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res 65:3307–3318
O’Sullivan JN, Bronner MP, Brentnall TA et al (2002) Chromosomal instability in ulcerative colitis is related to telomere shortening. Nat Genet 32:280–284
Park HS, Goodlad RA, Wright NA (1995) Crypt fission in the small intestine and colon. A mechanism for the emergence of G6PD locus-mutated crypts after treatment with mutagens. Am J Pathol 147:1416–1427
Powell SM, Zilz N, Beazer-Barclay Y et al (1992) APC mutations occur early during colorectal tumorigenesis. Nature 359:235–237
Rioux JD, Silverberg MS, Daly MJ et al (2000) Genomewide search in Canadian families with inflammatory bowel disease reveals two novel susceptibility loci. Am J Hum Genet 66:1863–1870
Rosh JR, Gross T, Mamula P et al (2007) Hepatosplenic T-cell lymphoma in adolescents and young adults with Crohn’s disease: a cautionary tale? Inflamm Bowel Dis 13:1024–1030
Rousseaux C, Lefebvre B, Dubuquoy L et al (2005) Intestinal antiinflammatory effect of 5-aminosalicylic acid is dependent on peroxisome proliferator-activated receptor-gamma. J Exp Med 201:1205–1215
Rubin CE, Haggitt RC, Burmer GC et al (1992) DNA aneuploidy in colonic biopsies predicts future development of dysplasia in ulcerative colitis. Gastroenterology 103:1611–1620
Rutgeerts P, Vermeire S, Van Assche G (2009) Biological therapies for inflammatory bowel diseases. Gastroenterology 136:1182–1197
Rutter M, Saunders B, Wilkinson K et al (2004) Severity of inflammation is a risk factor for colorectal neoplasia in ulcerative colitis. Gastroenterology 126:451–459
Satsangi J, Parkes M, Louis E et al (1996) Two stage genome-wide search in inflammatory bowel disease provides evidence for susceptibility loci on chromosomes 3, 7 and 12. Nat Genet 14:199–202
Seril DN, Liao J, Yang GY et al (2003) Oxidative stress and ulcerative colitis-associated carcinogenesis: studies in humans and animal models. Carcinogenesis 24:353–362
Shivananda S, Lennard-Jones J, Logan R et al (1996) Incidence of inflammatory bowel disease across Europe: is there a difference between north and south? Results of the European Collaborative Study on Inflammatory Bowel Disease (EC-IBD). Gut 39:690–697
Sieber OM, Heinimann K, Gorman P et al (2002) Analysis of chromosomal instability in human colorectal adenomas with two mutational hits at APC. Proc Natl Acad Sci USA 99:16910–16915
Slattery ML, Wolff RK, Herrick JS et al (2007) IL6 genotypes and colon and rectal cancer. Cancer Causes Control 18:1095–1105
Slaughter DP, Southwick HW, Smejkal W (1953) Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer 6:963–968
Studeny M, Marini FC, Champlin RE et al (2002) Bone marrow-derived mesenchymal stem cells as vehicles for interferon-beta delivery into tumors. Cancer Res 62:3603–3608
Swidsinski A, Weber J, Loening-Baucke V et al (2005) Spatial organization and composition of the mucosal flora in patients with inflammatory bowel disease. J Clin Microbiol 43:3380–3389
Tomlinson I, Bodmer W (1999) Selection, the mutation rate and cancer: ensuring that the tail does not wag the dog. Nat Med 5:11–12
van den Broek FJ, Fockens P, van Eeden S et al (2008) Endoscopic tri-modal imaging for surveillance in ulcerative colitis: randomised comparison of high-resolution endoscopy and autofluorescence imaging for neoplasia detection; and evaluation of narrow-band imaging for classification of lesions. Gut 57:1083–1089
Velayos F (2008) Colon cancer surveillance in inflammatory bowel disease patients: current and emerging practices. Expert Rev Gastroenterol Hepatol 2:817–825
Vogelstein B, Fearon ER, Hamilton SR et al (1988) Genetic alterations during colorectal-tumor development. N Engl J Med 319:525–532
von Roon AC, Karamountzos L, Purkayastha S et al (2007) Diagnostic precision of fecal calprotectin for inflammatory bowel disease and colorectal malignancy. Am J Gastroenterol 102:803–813
Wahl C, Liptay S, Adler G et al (1998) Sulfasalazine: a potent and specific inhibitor of nuclear factor kappa B. J Clin Invest 101:1163–1174
Willenbucher RF, Zelman SJ, Ferrell LD et al (1997) Chromosomal alterations in ulcerative colitis-related neoplastic progression. Gastroenterology 113:791–801
Winawer S, Fletcher R, Rex D et al (2003) Colorectal cancer screening and surveillance: clinical guidelines and rationale-update based on new evidence. Gastroenterology 124:544–560
Yin J, Harpaz N, Tong Y et al (1993) p53 point mutations in dysplastic and cancerous ulcerative colitis lesions. Gastroenterology 104:1633–1639
Yoshida T, Mikami T, Mitomi H et al (2003) Diverse p53 alterations in ulcerative colitis-associated low-grade dysplasia: full-length gene sequencing in microdissected single crypts. J Pathol 199:166–175
Zimmerman MJ, Jewell DP (1996) Cytokines and mechanisms of action of glucocorticoids and aminosalicylates in the treatment of ulcerative colitis and Crohn’s disease. Aliment Pharmacol Ther 10 (suppl 2):93–98 discussion 99
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2011 Springer-Velag Berlin Heidelberg
About this chapter
Cite this chapter
Jawad, N., Direkze, N., Leedham, S.J. (2011). Inflammatory Bowel Disease and Colon Cancer. In: Jankowski, J. (eds) Inflammation and Gastrointestinal Cancers. Recent Results in Cancer Research, vol 185. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-03503-6_6
Download citation
DOI: https://doi.org/10.1007/978-3-642-03503-6_6
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-03502-9
Online ISBN: 978-3-642-03503-6
eBook Packages: MedicineMedicine (R0)