
Abstract
TRIM5α protein blocks retroviral replication at early postentry stage reducing the accumulation of reverse transcriptase products. TRIM5α proteins of Old World primates restrict HIV-1 infection whereas TRIM5α proteins of most New World monkeys restrict SIVmac infection. TRIM5α protein has a RING domain, B-box 2 domain, coiled-coil domain, and PRYSPRY domain. The PRYSPRY domain of TRIM5α determines viral specificity and restriction potency by mediating recognition of the retroviral capsid. The coiled-coil domain is essential for TRIM5α oligomerization, which contributes to binding avidity for the viral capsid. The RING domain and B-box 2 domain are required for efficient restriction activity of TRIM5α protein but the mechanisms remain to be defined.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Following entry into the host cells, retroviruses must execute a series of processes including the uncoating of the viral core, reverse transcription, nuclear import, and integration of the viral DNA into the host genome before they establish a successful infection (Arts and Wainberg 1996; Whitcomb and Hughes 1992). The prevalence of retroviral DNAs in the genomes of all eukaryotes suggests that they have been present throughout the course of evolution, and about 8% of the human genome is made of the relics of past infections in the form of extinct endogenous retroviruses (Bannert and Kurth 2004). The integration of viral DNA and expression of viral proteins are potentially mutagenic or pathogenic. Therefore, the ability of an organism to limit or restrict retrovirus replication should have a selective edge over virus-susceptible counterparts. There are multiple genetic barriers to HIV-1 replication in most nonhuman primates hampering our efforts in developing a robust animal model for HIV-1 infection and pathogenesis. A major cellular factor responsible for blocking HIV-1 infection in Old World monkeys at early postentry step was identified as TRIM5α by Sodroski and colleagues in 2004 (Stremlau et al. 2004). Our efforts in understanding how TRIM5α interferes with the replication cycle of HIV-1 will hopefully allows us to manipulate this system to induce antiviral states in the near future. What follows is a brief review of the postentry restrictions of retroviruses, followed by highlights of these recent new developments on the antiretroviral activity of TRIM5α.
2 Postentry Restrictions
2.1 Fv1
Evidence of a specific, postentry restriction of retroviruses was first provided by studies of murine leukemia virus (MLV) replication in mouse cells of different genetic backgrounds regarding the Fv1 gene, which was first identified as a locus that controlled susceptibility to Friend leukemia virus disease (Lilly 1970; Pincus Hartley and Rowe 1971). The virus resistance induced by Fv1 was genetically dominant over susceptibility and was evident in cells cultured in vitro. Among a variety of Fv1 alleles, two alleles of Fv1 were shown to provide resistance to infection by particular MLV types. The Fv1 b allele, present in Balb/c mice, blocks infection of N-MLV, whereas the Fv1 n allele, present in NIH/swiss mice, blocks infection of B-MLV. This block occurs after reverse transcription but prior to integration, and targets the MLV capsid (DesGroseillers and Jolicoeur 1983; Jolicoeur and Rassart 1980; Ou et al. 1983; Pryciak and Varmus 1992; Sveda and Soeiro 1976). Indeed, a single amino acid residue at position 110 in the viral capsid can determine the susceptibility of the virus to the blocking effects of different Fv1 alleles (Kozak and Chakraborti 1996). Fv1 activity is saturated or titrated at high multiplicity of infection.
Fv1 gene arose from the germ-line integration of an endogenous retrovirus and encodes a Gag-like product (Best et al. 1996). An intact sequence corresponding to the major homology region (MHR), which is conserved in all retroviral capsids and contributes to capsid–capsid interactions, is important for Fv1 function (Bishop et al. 2001). However, the mechanism of the Fv1-mediated block is not understood. It is possible that Fv1 interferes with the trafficking of preintegration complex (PIC) or inhibits the integration of PIC into the host chromosome.
2.2 Ref1 and Lv1
In addition to governing the ability of retroviruses to infect particular mouse strains, early postentry restriction can also determine tropism at the species level. N-MLV, for example, inefficiently infects human cells and certain cell lines from African green monkeys (Besnier et al. 2003; Towers et al. 2000). The cellular factor restricting N-MLV in human cells has been referred to as Ref1 (restriction factor 1). As for Fv1 mediated restriction, the major determinant for virus susceptibility to Ref1 was the amino acid 110 of the capsid protein, and Ref1 blocked N-MLV in a saturable manner. However, Ref1 blocked infection at a slightly earlier stage than Fv1, before reverse transcription.
HIV-1 encounters a postentry block in Old World monkeys, whereas simian immunodeficiency virus (SIVmac) is blocked in most New World monkey cells (Himathongkham and Luciw 1996; Hofmann et al. 1999; Shibata et al. 1995). The cellular factor dictating the susceptibility of primate cells to the lentiviruses was referred to as Lv1 (lentiviral susceptibility factor 1) (Cowan et al. 2002). Lv1 restricted HIV-1 in a saturable manner (Hofmann et al. 1999; Towers et al. 2000). Like the restriction mediated by Ref1, the Lv1 restriction occurred early after entry, before reverse transcription, and the resistance was dominant over sensitivity (Cowan et al. 2002; Munk et al. 2002). As for Fv1 and Ref1, the determinant for virus susceptibility was the viral capsid protein (Cowan et al. 2002; Dorfman and Gottlinger 1996; Hatziioannou et al. 2003; Owens et al. 2003) and the block could be abrogated with wild-type HIV-1 (Kootstra et al. 2003) or with replication-defective particles lacking reverse transcriptase activity (Besnier et al. 2003; Cowan et al. 2002).
These species-specific, postentry restrictions mediated by Ref1 and Lv1 share common features: (1) the block occurs prior to reverse transcription (Cowan et al. 2002; Himathongkham and Luciw 1996; Munk et al. 2002; Shibata et al. 1995); (2) the viral determinant of the susceptibility to restriction is the capsid protein (Cowan et al. 2002; Kootstra et al. 2003; Owens et al. 2003; Towers et al. 2000); and (3) the host cell restricting factor can be competed by virus-like particles containing proteolytically-processed capsid proteins of the restricted viruses (Besnier Takeuchi and Towers 2002; Cowan et al. 2002; Hatziioannou et al. 2003; Owens et al. 2004).
3 TRIM5α
3.1 Identification of TRIM5α Restriction
While the search for the genes encoding Ref1 and Lv1 continued, the similarities in the nature and timing of the block imposed by the two loci raised the possibility that Ref1 and Lv1 might be human and monkey versions of the same gene. This possibility was further supported by a cross-abrogation experiment in African green monkey (AGM) cells which show a broad range of restriction against retroviruses including HIV-1, HIV-2, EIAV, and N-MLV. For example, the restriction of HIV-1 in AGM cells could be abrogated by EIAV, and the restriction of N-MLV could be abrogated by the lentiviruses (Besnier et al. 2002; Hatziioannou et al. 2003; Stoye 2002; Towers et al. 2000).
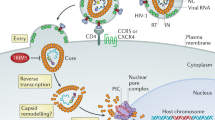
A major breakthrough in the field was accomplished with the identification of the gene responsible for Lv1 activity (Stremlau et al. 2004). TRIM5α was identified during a screen for cDNA clones derived from HIV-resistant, rhesus macaque lung fibroblasts that would protect human HeLa cells from infection by single-cycle GFP-expressing HIV-1 vector pseudotyped with VSV-G envelope when introduced into HIV-susceptible human HeLa cells. The promiscuous envelope protein VSV-G allows entry into most mammalian cell types and thus bypasses blocks related to cell-surface binding, fusion, and entry. This particular screen, therefore, specifically revealed the presence of barriers to the first half of the retroviral life cycle, including reverse transcription, integration, and expression. The expression of the rhesus cDNA was sufficient to restrict incoming HIV-1, whereas the human cDNA was not. Rhesus TRIM5α activity was specific for the HIV-1 capsid, as expected for Lv1 activity. In the cells expressing rhesus TRIM5α, the accumulation of reverse transcripts was significantly reduced indicating that TRIM5α blocks HIV-1 replication before or during early reverse transcription (Fig. 1). Furthermore, siRNA-mediated knockdown of endogenous TRIM5α expression in rhesus cells abrogated the early postentry restriction to HIV-1 infection indicating that TRIM5α was required for Lv1 activity. Work performed by several laboratories soon confirmed that Ref1 and Lv1 were indeed the human and monkey orthologues of TRIM5α (Hatziioannou et al. 2004b; Keckesova Ylinen and Towers 2004; Perron et al. 2004; Song et al. 2005c; Yap et al. 2004).

Postentry restriction of retroviral infection by TRIM5α. The early steps of retrovirus replication cycle and the position of block generated by TRIM5α are shown. PIC Preintegration complex
3.2 TRIM5α
TRIM5α is a member of the large family of tripartite motif proteins (TRIM) (Reymond et al. 2001). TRIM proteins contain RING, B-box 2, and coiled-coil domains and thus have been referred to as RBCC proteins (Reymond et al. 2001) (Fig. 2). Human TRIM5 gene is located in chromosome 11p15 in a cluster with other TRIM genes including TRIM3, TRIM6, TRIM21, TRIM22, TRIM34, and TRIM68 (Fig. 2). Among these TRIM genes, TRIM5, TRIM6, TRIM22, and TRIM34 are located at adjacent loci. TRIM5α displays higher identities to adjacent TRIM proteins in RING and B-box domains, but lower identities in coiled-coil and PRYSPRY domains. Consistent with this observation, the carboxy-terminal PRYSPRY domain has recently been shown as a variable region that determines the species specificity of retroviral restriction in primates.

Chromosomal localization and domain structure of TRIM5α. (a) TRIM genes in the segment of 11p15, which is present in the distal region of human chromosome 11. (b) Schematic representation of TRIM5 gene. The coding sequences for the alpha isoform is spread across 7 exons, beginning with the RING domain in exon 2 and ending with the PRYSPRY domain in exon 8. (c) Schematic representation of the domain structure of human TRIM5α protein. Other isoforms such as TRIM5γ or TRIM5δ do not have the PRYSPRY domain and they do not restrict retroviruses. (d) Schematic representation of the domain structure of TRIMCyp fusion protein expressed in owl monkey cells. TRIMCyp fusion consists of the RBCC domain of TRIM5 fused to cyclophilin A due to the insertion of a CypA sequence between exons 7 and 8
Differential splicing of the TRIM5 primary transcript gives rise to the expression of several isoforms of the protein product (Reymond et al. 2001). The TRIM5α is the largest product (493 amino acid residues in humans) and contains the PRYSPRY domain. The other TRIM5 isoforms lack an intact PRYSPRY domain and are incapable of restricting HIV-1. Two TRIM5 isoforms, TRIM5δ and TRIM5α, are reported to have ubiquitin ligase activity typical of RING-containing proteins (Xu et al. 2003; Yamauchi et al. 2008).
TRIM proteins often self-associate and form nuclear or cytoplasmic bodies of undefined function (Diaz-Griffero et al. 2006; Reymond et al. 2001; Song et al. 2005a). Although TRIM proteins have been implicated in transcriptional regulation, cell division, antiviral activity, determination of cell polarity, and differentiation, the precise functions of most TRIM proteins remain to be determined (Meroni and Diez-Roux 2005; Nisole et al. 2005; Towers 2005). TRIM proteins arose with the metazoans and have expanded in number during vertebrate evolution (Reymond et al. 2001). To date, more than 70 TRIM proteins have been identified in the human genome; homologues exist in other species as well. Dysregulation and mutations of some TRIM family members have been linked to a variety of pathological conditions, including genetic diseases and oncogenesis. Many TRIM proteins, including TRIM19, TRIM21, TRIM22, TRIM34, and TRIM5α itself, can be upregulated by interferon, supporting their potential role as effectors in the antiviral cellular response (Asaoka et al. 2005; Chelbi-Alix et al. 1995; Gongora et al. 2000; Orimo et al. 2000; Tissot and Mechti 1995). Indeed, several primate TRIM proteins, including TRIM1, TRIM5α, TRIM19, TRIM22, TRIM32, and TRIM34 have been shown to have antiviral activity against different viruses (Nisole et al. 2005).
3.3 TRIMCyp
Cyclophilin A (CypA) is a highly conserved peptidyl prolyl isomerase that binds to HIV-1 capsid (CA) (Franke et al. 1994; Luban et al. 1993; Thali et al. 1994). HIV-1 directly interacts with the CypA activie site by virtue of residues in a loop between the fourth and fifth alpha helices of CA (4–5 loop) (Bukovsky et al. 1997; Gamble et al. 1996). A proline residue P90 in HIV-1 CA is required for CypA binding (Franke et al. 1994). Blocking the CypA-CA interaction by either mutation of the critical proline itself or nearby residues in CA, by mutation of the cyclophilin gene, or by addition of the drug cyclpsporin inhibited virus replication in human cells (Braaten et al. 1996; Braaten and Luban 2001; Dorfman and Gottlinger 1996; Franke et al. 1994; Thali et al. 1994). The affected step was early after entry, before reverse transcription, at the same time as the Ref1 and Lv1 blocks. Analysis of the retroviral replication cycle, using RNA interference to disrupt CypA in the virion producer cell or in the target cell, indicated that target cell CypA alone promotes HIV-1 infectivity (Kootstra et al. 2003; Towers et al. 2003).
Owl monkey cells restricted HIV-1 whereas most other New World monkey cells blocked SIVmac infection (Hofmann et al. 1999). In contrast to the positive effect of CypA-CA interaction on HIV-1 replication in human cells, Towers et al. showed that inhibiting CypA in owl monkey cells rescued HIV-1 restriction (Towers et al. 2003). This was later explained by the identification of TRIM5-cyclophilin A fusion protein (TRIMCyp) (Nisole et al. 2004; Sayah et al. 2004), which arose by retrotransposition of a complete CypA cDNA into TRIM5 intron 7 (Fig. 2). These findings raised a possibility that the CypA domain in the owl monkey TRIMCyp and the PRYSPRY domain in the rhesus TRIM5α could provide a binding domain to target the incoming HIV-1 core and the N-terminal domain(s) of TRIM5α and TRIMCyp could serve an effector function.
It was also shown that inhibiting CypA in Old World monkey cells reduced HIV-1’s sensitivity to TRIM5α (Berthoux et al. 2005; Keckesova et al. 2006; Kootstra et al. 2003; Stremlau et al. 2006b) and this was concluded to be due to CypA-mediated prolyl isomerization of CA residue P90 impacting on sensitivity to TRIM5α binding (Berthoux et al. 2005; Keckesova et al. 2006). It was also reported that TRIM5α does not have a role for CypA sensitivity of HIV-1 in human cells (Hatziioannou et al. 2005; Keckesova et al. 2006; Sokolskaja et al. 2006).
Initially, a TRIMCyp fusion gene was thought to exist only in the owl monkey, a New World monkey. However, recent studies reported that the TRIMCyp fusion gene is also found in Old World monkeys including rhesus macaque and pig-tailed macaque (Brennan et al. 2008; Liao et al. 2007; Newman et al. 2008; Virgen et al. 2008; Wilson et al. 2008). Rhesus TRIMCyp restricts infection of HIV-2 and FIV but not HIV-1 (Wilson et al. 2008) and pig-tailed TRIMCyp restricts FIV but not HIV-1 (Brennan et al. 2008; Virgen et al. 2008). TRIMCyp genes of Old World monkeys were proposed to be generated independently from that in owl monkeys as indicated by different position of CypA cDNA sequence, and these events constitute a remarkable example of convergent evolution.
4 Restriction Activity of TRIM5α
4.1 Interspecies Variation of TRIM5α and Retroviral Restriction
Sequence analysis revealed significant interspecies variability in the PRYSPRY domains of TRIM5α proteins of Old World and New World monkeys (Sawyer et al. 2005; Song et al. 2005b). These studies showed substitution patterns indicative of selection in the PRYSPRY domain and revealed lineage-specific expansion and sequential duplication in the PRYSPRY domain (Song et al. 2005b). For the sequences encoding the PRYSPRY domain, the Ka/Ks ratio was very high, indicative of selectively driven diversity. These results suggest that occasional, complex changes were incorporated into the TRIM5α PRYSPRY domain at discrete time points during the evolution of primates. Some of these time points correspond to periods during which primates were exposed to retroviral infections, based on the appearance of particular endogenous retroviruses in primate genomes.
Soon after the identification of TRIM5α as a restriction factor blocking HIV-1 replication at early postentry steps in rhesus monkey cells, several laboratories cloned the TRIM5α cDNAs from diverse primate species and tested their antiretroviral activities (Table 1) (Hatziioannou et al. 2004b; Newman et al. 2006; Ohkura et al. 2006; Perez-Caballero et al. 2005a; Sawyer et al. 2005; Song et al. 2005a,2005c; Stremlau et al. 2004; Yap et al. 2004). The ability of TRIM5α proteins from different primate species to restrict infection by various retroviruses (Table 1) and the evidence for positive evolutionary selection of TRIM5 genes support the possibility that a major natural function of TRIM5α is its antiviral activity.
Recent studies identified TRIM5 genes with restriction activity against divergent retrovirus in cows (Si et al. 2006; Ylinen et al. 2006) and rabbits (Schaller et al. 2007) and revealed several TRIM5-like genes in rodents (Tareen et al. 2009), and phylogenetic analysis of these TRIM genes suggest that these factors have evolved from a common ancestor with antiretroviral properties and have undergone independent evolutionary expansions within species.
4.2 Role of TRIM5α Domains
The RING domain, a cycteine-rich zinc binding sequence, found at the N-terminus of TRIM5α is involved in specific protein–protein interactions and often associated with E3 ubiquitin ligase activity (Freemont 2000; Pickart 2001). Indeed, ubiquitination of TRIM5α (Diaz-Griffero et al. 2006; Yamauchi et al. 2008) and TRIM5δ (Xu et al. 2003) has been demonstrated. It was shown that TRIM5 is able to ubiquitinate itself in a RING domain-dependent manner (Xu et al. 2003; Yamauchi et al. 2008). Deletion of the RING domain as well as point mutations affecting residues known to be critical for ubiquitin ligase activity (C15 and C18) significantly reduced the HIV-1 restriction activity of rhesus TRIM5α (Stremlau et al. 2004).
The B-box is a distinct zinc binding sequence present on a number of developmentally important proteins (Torok and Etkin 2001) but the exact function of the B-box is unknown. The deletion or disruption of the B-box domain completely abolished HIV-1 restriction activity of rhesus TRIM5α, suggesting that this domain is essential for restriction activity (Javanbakht et al. 2005; Li et al. 2006a; Perez-Caballero et al. 2005a; Stremlau et al. 2004). Alteration of arginine 119 of human TRIM5α or the corresponding arginine 121 of rhesus TRIM5α diminished the abilities of the proteins to restrict retroviral infection and removal of the positively charged side chain from the B-box 2 arginines 119/121 resulted in diminished proteasome-independent turnover of TRIM5α (Diaz-Griffero et al. 2007b). A recent study by Sodroski’s group suggests that the B-box 2 domain of TRIM5α promotes cooperative binding to the retroviral capsid by mediating higher-order self-association (Li and Sodroski 2008). Thus, the B-box domain of TRIM5α modulates capsid binding and retroviral restriction.
The coiled-coil region is composed of multiple alpha-helices involved in protein-protein interactions that may result in homo- or hetero-multimers (Meroni and Diez-Roux 2005; Nisole et al. 2005; Reymond et al. 2001). In vitro cross-linking studies demonstrate that the coiled-coil domain plays a critical role in oligomer formation of TRIM5α protein (Javanbakht et al. 2006b; Mische et al. 2005). It is proposed that oligomer formation allows the B30.2 domain of TRIM5α protein to be better positioned for binding to the target capsid. Consistent with this hypothesis, TRIM5α mutants lacking the coiled-coil domain fail to restrict viral infection (Javanbakht et al. 2006b; Perez-Caballero et al. 2005a).
The PRYSPRY domain, located at the C-terminus of TRIM5α protein, has a core composed of two β-sheets sandwiched together to form a central hydrophobic core and loops of variable length and containing non-conserved residues that protrude out from the core structure, based on comparisons with the molecular structures of related proteins (Grutter et al. 2006; Masters et al. 2006; Ohkura et al. 2006; Woo et al. 2006). PRYSPRY domain was shown to be the determinant of the specificity of restriction (Nakayama et al. 2005; Ohkura et al. 2006; Perez-Caballero et al. 2005a; Stremlau et al. 2005; Yap et al. 2005). Substitution of R332 of human TRIM5α with a negatively-charged or non-charged amino acid is sufficient to allow restriction of HIV-1 without altering its ability to restrict N-MLV (Li et al. 2006b; Yap, et al. 2005). The regions of variability among TRIM5α PRYSPRY domains for different species are located on the protruding variable loops (Ohkura et al. 2006; Song et al. 2005b; Woo et al. 2006). Initial studies using chemical crosslinking suggested that TRIM5α may function as a trimer, but recent studies using purified recombinant TRIM5-21R, which contains the RING domain of TRIM21 in the backbone of TRIM5, by Sodroski’s and Sundquist’s groups suggest that TRIM5α forms stable dimers and recognizes retroviral capsids through direct interactions mediated by the PRYSPRY domain (Kar et al. 2008; Langelier et al. 2008).
4.3 How TRIM5α Works?
The viral determinant of susceptibility to TRIM5α-mediated restriction is the capsid protein (Cowan et al. 2002; Hatziioannou et al. 2004a; Owens et al. 2004; Owens et al. 2003; Towers et al. 2000). Restriction of retroviral infection by TRIM5α is saturated at high levels of input virions or virus-like particles and only the properly processed and assembled form of a condensed viral core is able to abrogate the restriction (Besnier et al. 2002; Cowan et al. 2002; Dodding et al. 2005; Forshey et al. 2005; Munk et al. 2002; Owens et al. 2004). Studies from several laboratories suggest that the PRYSPRY domain of TRIM5α is responsible for recognizing a conformational ligand on the viral capsid and that the RBCC domains provide an effector function by unknown mechanism (Javanbakht et al. 2006b; Li et al. 2006b; Owens et al. 2004; Perron et al. 2006, 2007; Sayah et al. 2004; Sebastian and Luban 2005; Stremlau et al. 2006a). Several possibilities can be envisioned for the possible mechanisms for TRIM5α restriction: it may bind and sequester the incoming virion core in a subcellular compartment; it may modify the virion core and target for degradation; it may interfere with normal uncoating; or it may inhibit trafficking of preintegration complex. Multiple mechanisms or pathways might be involved in TRIM5α restriction.
4.3.1 Inhibition of Normal Uncoating
In most instances, TRIM5α proteins impair retroviral infection early after entry into target cells, reducing the efficiency of reverse transcription (Keckesova et al. 2004; Perez-Caballero et al. 2005b; Stremlau et al. 2004), raising the possibility that TRIM5α might interfere with normal uncoating of the incoming viral cores. Recently, it has been shown that TRIM5α binds to the restriction-sensitive retroviral capsid (Sebastian and Luban 2005; Stremlau et al. 2006a) and causes an accelerated uncoating (Stremlau et al. 2006a). Uncoating is a poorly understood process, and it is still uncertain whether it is an active process requiring energy and/or specific host cell components, or whether it occurs passively (Greber et al. 1994; Narayan and Young 2004). HIV-1 cores are relatively unstable in vitro and, in the infected cell the capsid protein is thought to undergo disassembly soon after virus entry (Forshey et al. 2002; Grewe et al. 1990). Analyses of the viral components of the HIV-1 reverse transcription or preintegration complexes failed to detect significant amounts of the capsid protein (Bukrinsky et al. 1993; Farnet and Haseltine 1991; Fassati and Goff 2001; Karageorgos et al. 1993; Miller et al. 1997). The analysis of HIV-1 Gag mutants suggests that capsid disassembly demonstrates precise requirements; both increases and decreases in capsid stability resulted in decreased HIV-1 replication ability (Forshey et al. 2002). Therefore, an accelerated or premature uncoating could account for one of the modes of TRIM5α-mediated HIV-1 restriction.
4.3.2 Ubiquitin Ligase Activity of TRIM5α
Protein ubiquitination and the subsequent degradation of ubiquitinated proteins by the proteasomal pathway are essential for a wide range of cellular functions (Freemont 2000). Proteasome-independent functions of protein ubiquitination are also involved in regulating a variety of protein functions including transport and processing (Schnell and Hicke 2003). Recently, a RING domain-dependent, auto-ubiquitination activity of TRIM5 protein has been demonstrated in vitro (Xu et al. 2003; Yamauchi et al. 2008). Then, an interesting question arises: what is the substrate of the TRIM5α enzyme other than itself? The identification of interaction partner or cofactor of TRIM5α will hopefully answer the question. Ubiquitination of the incoming HIV-1 cores could potentially lead to the degradation of the modified viral cores by proteasome system or could interfere with the trafficking of the modified viral cores. However, TRIM5α-mediated ubiquitination of HIV-1 cores has not so far been demonstrated.
4.3.3 Proteasome
It has been shown that disrupting proteasome function relieves rhesus TRIM5α restriction of HIV-1 late RT products even though 2-LTR circle production and viral infection remained blocked, suggesting some contribution of proteasome activity to TRIM5α activity (Anderson et al. 2006; Wu et al. 2006). Therefore, a two step process of TRIM5α restriction was proposed: TRIM5α acts prior to complete reverse transcription of viral RNA and may also inhibit trafficking of the preintegration complex. A recent study showed that treatment of cells with proteasome inhibitors prevented TRIM5α-dependent loss of particulate CA protein (Diaz-Griffero et al. 2007a), indicating the potential involvement of proteasome activity in TRIM5α-induced virus uncoating. How much proteasome contributes to the restriction activity mediated by TRIM5α and its mechanism remain to be defined.
4.3.4 TRIM5α Turnover
Human and rhesus TRIM5α proteins stably expressed in HeLa cells were shown to be rapidly turned over, with half-lives of 50–60 min (Diaz-Griffero et al. 2006). The high rate of TRIM5α turnover creates opportunities for a rapid regulation of the levels of these proteins in response to viral infection or other stimuli. Both proteasome-dependent and proteasome-independent modes for the turnover of TRIM5α protein has been proposed (Diaz-Griffero et al. 2006, 2007b ).
Recently, it was shown that TRIM5α is targeted for degradation by a proteasome-dependent mechanism following encounter of a restriction-sensitive retroviral core (Rold and Aiken 2008). This study proposed two potential outcomes of TRIM5α-CA interaction: (1) proteasomal degradation of a TRIM5α-CA complex, resulting in functional decapsidation of the viral core and a premature uncoating, and (2) dissociation of CA from the core followed by its release from TRIM5α, leading to destruction of the restriction factor and decapsidation of the core but not necessarily degradation of CA. It will be interesting to determine whether HIV-1-induced degradation of TRIM5α is dependent on the self-ubiquitination activity of TRIM5α or dependent on other host cell ubiquitin ligases.
4.3.5 TRIM5α dynamics and trafficking
It was shown that TRIM5α cytoplasmic bodies are highly mobile and use the microtubule network to navigate throughout the cytoplasm, and that TRIM5α proteins are dynamically exchanged between the cytoplasmic bodies and the diffuse cytoplasmic population (Campbell et al. 2007), suggesting a more active role of TRIM5α in antiviral activity. Furthermore, it was reported that there is a dynamic interaction between rhesus TRIM5α and HIV-1 viral complexes, including the de novo formation of TRIM5α cytoplasmic body-like structures around viral complexes (Campbell et al. 2008).
A previous study showed that heat shock proteins Hsp70 and Hsp90 colocalize with TRIM5α cytoplasmic bodies (Diaz-Griffero et al. 2006). Hsp70 and Hsp90 proteins are the components of molecular chaperones which play a critical function in protein folding by promoting and maintaining the native conformation of cellular proteins and in some cases in protein sorting (Young et al. 2004). It remains to be determined whether these molecular chaperones directly interact with TRIM5α and contribute to the turnover, trafficking, or the restriction activity of TRIM5α protein. Our understanding of the mechanism of HIV-1-restricting activity of TRIM5α may depend on the complete understanding of the components and function of the TRIM5α complexes and the dissection of the interaction partners of TRIM5α.
4.3.6 Cyclophilin A
The CypA-CA interaction has been shown to assist HIV-1 replication in some human cells (Franke and Luban 1996; Hatziioannou et al. 2005; Sokolskaja et al. 2004; Thali et al. 1994). Initially, it was thought that CypA in producer cells plays an important role in HIV-1 replication. However, recent findings support that CypA is more important in target cells than in producer cells for HIV-1 replication (Kootstra et al. 2003; Towers et al. 2003). It has been hypothesized that human encode an unknown factor that can negatively affect HIV-1 replication, and CypA binding to HIV-1 CA can protect HIV-1 from this unknown factor (Sokolskaja, et al. 2006; Towers et al. 2003). In contrast to the positive effects of CypA on HIV-1 replication in human cells, CypA exerts negative effects on HIV-1 replication in Old World monkey cells because CypA-CA interactions sensitize HIV-1 to the restriction from Old World monkey TRIM5α proteins (Berthoux et al. 2005; Keckesova et al. 2006; Stremlau et al. 2006b).
CypA interacts with diverse lentiviral capsids including HIV-1, SIVcpz, SIVagmTAN, and FIV (Lin and Emerman 2006). It was proposed that CypA binding to HIV-1 CA induces the conformational change of viral core and renders HIV-1 CA more recognizable by the PRYSPRY domain of TRIM5α (Berthoux et al. 2005; Keckesova et al. 2006). However, HIV-1 variant (e.g., G89V), which does not bind CypA, is still susceptible to the TRIM5α restriction in a CypA-independent manner (Lin and Emerman 2008; Stremlau et al. 2006b). These findings support the idea that TRIM5α restriction of HIV-1 is composed of both CypA-dependent and CypA-independent components (Keckesova et al. 2006; Lin and Emerman 2008; Stremlau et al. 2006b). The ridge formed by helices 3 and 6 on CA has been reported to determine viral susceptibility to the TRIM5α restriction (Owens et al. 2004). A recent study showed that two loops on the HIV-1 capsid, one between the 4th and 5th helices (4–5 loop) and the other between the 6th and 7th helices (6–7 loop), are responsible for the HIV-1 susceptibility to the CypA-dependent TRIM5α restriction (Lin and Emerman 2008).
4.4 Polymorphism
An analysis of sequence data collected from HIV/AIDS cohorts, human genomic DNA diversity collections, and human SNP databases revealed polymorphism in TRIM5 (Goldschmidt et al. 2006; Javanbakht et al. 2006a; Sawyer et al. 2006; Speelmon et al. 2006; van Manen et al. 2008). These include residues in the RING domain (H43Y), in the B-box 2 domain (V112F), in or near the coiled-coil domain (R136Q, R238W, G249D), and in the PRYSPRY domain (H419Y). Two polymorphisms in the TRIM5 gene (H43Y and R136Q) were shown to affect the antiviral activity of TRIM5α in vitro. For example, human TRIM5α with the H-to-Y change at position 43 showed a reduced ability to restrict N-MLV in tissue culture-based assays (Goldschmidt et al. 2006; Javanbakht et al. 2006a; Sawyer et al. 2006). For the residue at position 136, one study reported that R-to-Q change at position 136 rendered a slightly more effective restriction of HIV-1 (Javanbakht et al. 2006a), although a different study did not detect a difference (Goldschmidt et al. 2006). A recent study reported that an accelerated disease progression was observed for individuals who were homozygous for the 43Y genotype as compared to individuals who were heterozygous or homozygous for the 43H genotype (van Manen et al. 2008), suggesting that polymorphisms in the TRIM5 gene may influence the clinical course of HIV-1 infection.
5 Conclusion
The recent discovery of TRIM5α has revealed a complex interaction between the incoming virion and the host factor, influencing the postentry replication steps in the retroviral life cycle. TRIM5α reduces the accumulation of reverse transcriptase products possibly by interfering with the normal uncoating process. Little is known about the uncoating process, that occurs shortly after a retrovirus fuses with the cell membrane. There are many questions to be addressed to understand the mechanisms of TRIM5α restriction. Defining the capsid uncoating process and the cellular factors involved will be critical to understanding the mechanisms of TRIM5α restriction. The role of ubiquitin and TRIM5α E3 ubiquitin ligase activity needs to be determined. The availability of methods for producing and purifying TRIM5 derivatives should expedite studies of their structure and mechanism of action in restricting retroviral infection. Equally important is identifying TRIM5α-binding proteins or cofactors and understanding the normal function of TRIM5α. The elucidation of TRIM5 mechanism may facilitate pharmacological and genetic intervention to induce currently nonrestrictive human TRIM genes to target and restrict HIV-1.
References
Anderson JL, Campbell EM, Wu X, Vandegraaff N, Engelman A, Hope TJ (2006) Proteasome inhibition reveals that a functional preintegration complex intermediate can be generated during restriction by diverse TRIM5 proteins. J Virol 80(19):9754–9760
Arts EJ, Wainberg MA (1996) Human immunodeficiency virus type 1 reverse transcriptase and early events in reverse transcription. Adv Virus Res 46:97–163
Asaoka K, Ikeda K, Hishinuma T, Horie-Inoue K, Takeda S, Inoue S (2005) A retrovirus restriction factor TRIM5alpha is transcriptionally regulated by interferons. Biochem Biophys Res Commun 338(4):1950–1956
Bannert N, Kurth R (2004) Retroelements and the human genome: new perspectives on an old relation. Proc Natl Acad Sci USA 101(Suppl 2):14572–14579
Berthoux L, Sebastian S, Sokolskaja E, Luban J (2005) Cyclophilin A is required for TRIM5{alpha}-mediated resistance to HIV-1 in Old World monkey cells. Proc Natl Acad Sci USA 102(41):14849–14853
Besnier C, Takeuchi Y, Towers G (2002) Restriction of lentivirus in monkeys. Proc Natl Acad Sci U S A 99(18):11920–11925
Besnier C, Ylinen L, Strange B, Lister A, Takeuchi Y, Goff SP, Towers GJ (2003) Characterization of murine leukemia virus restriction in mammals. J Virol 77(24):13403–13406
Best S, Le Tissier P, Towers G, Stoye JP (1996) Positional cloning of the mouse retrovirus restriction gene Fv1. Nature 382(6594):8269
Bishop KN, Bock M, Towers G, Stoye JP (2001) Identification of the regions of Fv1 necessary for murine leukemia virus restriction. J Virol 75(11):5182–5188
Braaten D, Franke EK, Luban J (1996) Cyclophilin A is required for an early step in the life cycle of human immunodeficiency virus type 1 before the initiation of reverse transcription. J Virol 70(6):3551–3560
Braaten D, Luban J (2001) Cyclophilin A regulates HIV-1 infectivity, as demonstrated by gene targeting in human T cells. EMBO J 20(6):1300–1309
Brennan G, Kozyrev Y, Hu SL (2008) TRIMCyp expression in Old World primates Macaca nemestrina and Macaca fascicularis. Proc Natl Acad Sci USA 105(9):3569–3574
Bukovsky AA, Weimann A, Accola MA, Gottlinger HG (1997) Transfer of the HIV-1 cyclophilin-binding site to simian immunodeficiency virus from Macaca mulatta can confer both cyclosporin sensitivity and cyclosporin dependence. Proc Natl Acad Sci USA 94(20):10943–10948
Bukrinsky MI, Sharova N, McDonald TL, Pushkarskaya T, Tarpley WG, Stevenson M (1993) Association of integrase, matrix, and reverse transcriptase antigens of human immunodeficiency virus type 1 with viral nucleic acids following acute infection. Proc Natl Acad Sci USA 90(13):6125–6129
Campbell EM, Dodding MP, Yap MW, Wu X, Gallois-Montbrun S, Malim MH, Stoye JP, Hope TJ (2007) TRIM5 alpha cytoplasmic bodies are highly dynamic structures. Mol Biol Cell 18(6):2102–2111
Campbell EM, Perez O, Anderson JL, Hope TJ (2008) Visualization of a proteasome-independent intermediate during restriction of HIV-1 by rhesus TRIM5alpha. J Cell Biol 180(3):549–561
Chelbi-Alix MK, Pelicano L, Quignon F, Koken MH, Venturini L, Stadler M, Pavlovic J, Degos L, de The H (1995) Induction of the PML protein by interferons in normal and APL cells. Leukemia 9(12):2027–2033
Cowan S, Hatziioannou T, Cunningham T, Muesing MA, Gottlinger HG, Bieniasz PD (2002) Cellular inhibitors with Fv1-like activity restrict human and simian immunodeficiency virus tropism. Proc Natl Acad Sci USA 99(18):11914–11919
DesGroseillers L, Jolicoeur P (1983) Physical mapping of the Fv-1 tropism host range determinant of BALB/c murine leukemia viruses. J Virol 48(3):685–696
Diaz-Griffero F, Kar A, Lee M, Stremlau M, Poeschla E, Sodroski J (2007a) Comparative requirements for the restriction of retrovirus infection by TRIM5alpha and TRIMCyp. Virology 369(2):400–410
Diaz-Griffero F, Kar A, Perron M, Xiang SH, Javanbakht H, Li X, Sodroski J (2007b) Modulation of retroviral restriction and proteasome inhibitor-resistant turnover by changes in the TRIM5alpha B-box 2 domain. J Virol 81(19):10362–10378
Diaz-Griffero F, Li X, Javanbakht H, Song B, Welikala S, Stremlau M, Sodroski J (2006) Rapid turnover and polyubiquitylation of the retroviral restriction factor TRIM5. Virology 349(2):300–315
Dodding MP, Bock M, Yap MW, Stoye JP (2005) Capsid processing requirements for abrogation of Fv1 and Ref1 restriction. J Virol 79(16):10571–10577
Dorfman T, Gottlinger HG (1996) The human immunodeficiency virus type 1 capsid p2 domain confers sensitivity to the cyclophilin-binding drug SDZ NIM 811. J Virol 70(9):5751–5757
Farnet CM, Haseltine WA (1991) Determination of viral proteins present in the human immunodeficiency virus type 1 preintegration complex. J Virol 65(4):1910–1915
Fassati A, Goff SP (2001) Characterization of intracellular reverse transcription complexes of human immunodeficiency virus type 1. J Virol 75(8):3626–3635
Forshey BM, Shi J, Aiken C (2005) Structural requirements for recognition of the human immunodeficiency virus type 1 core during host restriction in owl monkey cells. J Virol 79(2):869–875
Forshey BM, von Schwedler U, Sundquist WI, Aiken C (2002) Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J Virol 76(11):5667–5677
Franke EK, Luban J (1996) Inhibition of HIV-1 replication by cyclosporine A or related compounds correlates with the ability to disrupt the Gag-cyclophilin A interaction. Virology 222(1):279–282
Franke EK, Yuan HE, Luban J (1994) Specific incorporation of cyclophilin A into HIV-1 virions. Nature 372(6504):359–362
Freemont PS (2000) RING for destruction? Curr Biol 10(2):R84–R87
Gamble TR, Vajdos FF, Yoo S, Worthylake DK, Houseweart M, Sundquist WI, Hill CP (1996) Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell 87(7):1285–1294
Goldschmidt V, Bleiber G, May M, Martinez R, Ortiz M, Telenti A (2006) Role of common human TRIM5alpha variants in HIV-1 disease progression. Retrovirology 3:54
Gongora C, Tissot C, Cerdan C, Mechti N (2000) The interferon-inducible Staf50 gene is downregulated during T cell costimulation by CD2 and CD28. J Interferon Cytokine Res 20(11):955–961
Greber UF, Singh I, Helenius A (1994) Mechanisms of virus uncoating. Trends Microbiol 2(2):52–56
Grewe C, Beck A, Gelderblom HR (1990) HIV: early virus-cell interactions. J Acquir Immune Defic Syndr 3(10):965–974
Grutter C, Briand C, Capitani G, Mittl PR, Papin S, Tschopp J, Grutter MG (2006) Structure of the PRYSPRY-domain: implications for autoinflammatory diseases. FEBS Lett 580(1):99–106
Hatziioannou T, Cowan S, Goff SP, Bieniasz PD, Towers GJ (2003) Restriction of multiple divergent retroviruses by Lv1 and Ref1. EMBO J 22(3):385–394
Hatziioannou T, Cowan S, Von Schwedler UK, Sundquist WI, Bieniasz PD (2004a) Species-specific tropism determinants in the human immunodeficiency virus type 1 capsid. J Virol 78(11):6005–6012
Hatziioannou T, Perez-Caballero D, Cowan S, Bieniasz PD (2005) Cyclophilin interactions with incoming human immunodeficiency virus type 1 capsids with opposing effects on infectivity in human cells. J Virol 79(1):176–183
Hatziioannou T, Perez-Caballero D, Yang A, Cowan S, Bieniasz PD (2004b) Retrovirus resistance factors Ref1 and Lv1 are species-specific variants of TRIM5alpha. Proc Natl Acad Sci USA 101(29):10774–10779
Himathongkham S, Luciw PA (1996) Restriction of HIV-1 (subtype B) replication at the entry step in rhesus macaque cells. Virology 219(2):485–488
Hofmann W, Schubert D, LaBonte J, Munson L, Gibson S, Scammell J, Ferrigno P, Sodroski J (1999) Species-specific, postentry barriers to primate immunodeficiency virus infection. J Virol 73(12):10020–10028
Javanbakht H, An P, Gold B, Petersen DC, O'Huigin C, Nelson GW, O'Brien SJ, Kirk GD, Detels R, Buchbinder S, Donfield S, Shulenin S, Song B, Perron MJ, Stremlau M, Sodroski J, Dean M, Winkler C (2006a) Effects of human TRIM5alpha polymorphisms on antiretroviral function and susceptibility to human immunodeficiency virus infection. Virology 354(1):15–27
Javanbakht H, Diaz-Griffero F, Stremlau M, Si Z, Sodroski J (2005) The contribution of RING and B-box 2 domains to retroviral restriction mediated by monkey TRIM5alpha. J Biol Chem 280(29):26933–26940
Javanbakht H, Yuan W, Yeung DF, Song B, Diaz-Griffero F, Li Y, Li X, Stremlau M, Sodroski J (2006b) Characterization of TRIM5alpha trimerization and its contribution to human immunodeficiency virus capsid binding. Virology 353(1):234–246
Jolicoeur P, Rassart E (1980) Effect of Fv-1 gene product on synthesis of linear and supercoiled viral DNA in cells infected with murine leukemia virus. J Virol 33(1):183–195
Kar AK, Diaz-Griffero F, Li Y, Li X, Sodroski J (2008) Biochemical and biophysical characterization of a chimeric TRIM21-TRIM5alpha protein. J Virol 82(23):11669–11681
Karageorgos L, Li P, Burrell C (1993) Characterization of HIV replication complexes early after cell-to-cell infection. AIDS Res Hum Retroviruses 9(9):817–823
Keckesova Z, Ylinen LM, Towers GJ (2004) The human and African green monkey TRIM5alpha genes encode Ref1 and Lv1 retroviral restriction factor activities. Proc Natl Acad Sci USA 101(29):10780–10785
Keckesova Z, Ylinen LM, Towers GJ (2006) Cyclophilin A renders human immunodeficiency virus type 1 sensitive to Old World monkey but not human TRIM5 alpha antiviral activity. J Virol 80(10):4683–4690
Kootstra NA, Munk C, Tonnu N, Landau NR, Verma IM (2003) Abrogation of postentry restriction of HIV-1-based lentiviral vector transduction in simian cells. Proc Natl Acad Sci USA 100(3):1298–1303
Kozak CA, Chakraborti A (1996) Single amino acid changes in the murine leukemia virus capsid protein gene define the target of Fv1 resistance. Virology 225(2):300–305
Langelier CR, Sandrin V, Eckert DM, Christensen DE, Chandrasekaran V, Alam SL, Aiken C, Olsen JC, Kar AK, Sodroski JG, Sundquist WI (2008) Biochemical characterization of a recombinant TRIM5alpha protein that restricts human immunodeficiency virus type 1 replication. J Virol 82(23):11682–11694
Li X, Li Y, Stremlau M, Yuan W, Song B, Perron M, Sodroski J (2006a) Functional replacement of the RING, B-box 2, and coiled-coil domains of tripartite motif 5alpha (TRIM5alpha) by heterologous TRIM domains. J Virol 80(13):6198–6206
Li X, Sodroski J (2008) The TRIM5alpha B-box 2 domain promotes cooperative binding to the retroviral capsid by mediating higher-order self-association. J Virol 82(23):11495–11502
Li Y, Li X, Stremlau M, Lee M, Sodroski J (2006b) Removal of arginine 332 allows human TRIM5alpha to bind human immunodeficiency virus capsids and to restrict infection. J Virol 80(14):6738–6744
Liao CH, Kuang YQ, Liu HL, Zheng YT, Su B (2007) A novel fusion gene, TRIM5-Cyclophilin A in the pig-tailed macaque determines its susceptibility to HIV-1 infection. AIDS 21(Suppl 8):S19–S26
Lilly F (1970) Fv-2: identification and location of a second gene governing the spleen focus response to Friend leukemia virus in mice. J Natl Cancer Inst 45(1):163–169
Lin TY, Emerman M (2006) Cyclophilin A interacts with diverse lentiviral capsids. Retrovirology 3:70
Lin TY, Emerman M (2008) Determinants of cyclophilin A-dependent TRIM5 alpha restriction against HIV-1. Virology 379(2):335–341
Luban J, Bossolt KL, Franke EK, Kalpana GV, Goff SP (1993) Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell 73(6):1067–1078
Masters SL, Yao S, Willson TA, Zhang JG, Palmer KR, Smith BJ, Babon JJ, Nicola NA, Norton RS, Nicholson SE (2006) The SPRY domain of SSB-2 adopts a novel fold that presents conserved Par-4-binding residues. Nat Struct Mol Biol 13(1):77–84
Meroni G, Diez-Roux G (2005) TRIM/RBCC, a novel class of ‘single protein RING finger’ E3 ubiquitin ligases. Bioessays 27(11):1147–1157
Miller MD, Farnet CM, Bushman FD (1997) Human immunodeficiency virus type 1 preintegration complexes: studies of organization and composition. J Virol 71(7):5382–5390
Mische CC, Javanbakht H, Song B, Diaz-Griffero F, Stremlau M, Strack B, Si Z, Sodroski J (2005) Retroviral restriction factor TRIM5alpha is a trimer. J Virol 79(22):14446–14450
Munk C, Brandt SM, Lucero G, Landau NR (2002) A dominant block to HIV-1 replication at reverse transcription in simian cells. Proc Natl Acad Sci USA 99(21):13843–13848
Nakayama EE, Miyoshi H, Nagai Y, Shioda T (2005) A specific region of 37 amino acid residues in the SPRY (B30.2) domain of African green monkey TRIM5alpha determines species-specific restriction of simian immunodeficiency virus SIVmac infection. J Virol 79(14):8870–8877
Narayan S, Young JA (2004) Reconstitution of retroviral fusion and uncoating in a cell-free system. Proc Natl Acad Sci USA 101(20):7721–7726
Newman RM, Hall L, Connole M, Chen GL, Sato S, Yuste E, Diehl W, Hunter E, Kaur A, Miller GM, Johnson WE (2006) Balancing selection and the evolution of functional polymorphism in Old World monkey TRIM5alpha. Proc Natl Acad Sci USA 103(50):19134–19139
Newman RM, Hall L, Kirmaier A, Pozzi LA, Pery E, Farzan M, O'Neil SP, Johnson W (2008) Evolution of a TRIM5-CypA splice isoform in old world monkeys. PLoS Pathog 4(2):e1000003
Nisole S, Lynch C, Stoye JP, Yap MW (2004) A Trim5-cyclophilin A fusion protein found in owl monkey kidney cells can restrict HIV-1. Proc Natl Acad Sci USA 101(36):13324–13328
Nisole S, Stoye JP, Saib A (2005) TRIM family proteins: retroviral restriction and antiviral defence. Nat Rev Microbiol 3(10):799–808
Ohkura S, Yap MW, Sheldon T, Stoye JP (2006). All three variable regions of the TRIM5alpha B30.2 domain can contribute to the specificity of retrovirus restriction. J Virol 80(17):8554–8565
Orimo A, Tominaga N, Yoshimura K, Yamauchi Y, Nomura M, Sato M, Nogi Y, Suzuki M, Suzuki H, Ikeda K, Inoue S, Muramatsu M (2000) Molecular cloning of ring finger protein 21 (RNF21)/interferon-responsive finger protein (ifp1), which possesses two RING-B box-coiled coil domains in tandem. Genomics 69(1):143–149
Ou CY, Boone LR, Koh CK, Tennant RW, Yang WK (1983) Nucleotide sequences of gag-pol regions that determine the Fv-1 host range property of BALB/c N-tropic and B-tropic murine leukemia viruses. J Virol 48(3):779–784
Owens CM, Song B, Perron MJ, Yang PC, Stremlau M, Sodroski J (2004) Binding and susceptibility to postentry restriction factors in monkey cells are specified by distinct regions of the human immunodeficiency virus type 1 capsid. J Virol 78(10):5423–5437
Owens CM, Yang PC, Gottlinger H, Sodroski J (2003) Human and simian immunodeficiency virus capsid proteins are major viral determinants of early, postentry replication blocks in simian cells. J Virol 77(1):726–731
Perez-Caballero D, Hatziioannou T, Yang A, Cowan S, Bieniasz PD (2005a) Human tripartite motif 5alpha domains responsible for retrovirus restriction activity and specificity. J Virol 79(14):8969–8978
Perez-Caballero D, Hatziioannou T, Zhang F, Cowan S, Bieniasz PD (2005b) Restriction of human immunodeficiency virus type 1 by TRIM-CypA occurs with rapid kinetics and independently of cytoplasmic bodies, ubiquitin, and proteasome activity. J Virol 79(24):15567–15572
Perron MJ, Stremlau M, Lee M, Javanbakht H, Song B, Sodroski J (2007) The human TRIM5alpha restriction factor mediates accelerated uncoating of the N-tropic murine leukemia virus capsid. J Virol 81(5):2138–2148
Perron M J, Stremlau M, Sodroski J (2006) Two surface-exposed elements of the B30.2/SPRY domain as potency determinants of N-tropic murine leukemia virus restriction by human TRIM5alpha. J Virol 80(11):5631–5636
Perron MJ, Stremlau M, Song B, Ulm W, Mulligan RC, Sodroski J (2004) TRIM5alpha mediates the postentry block to N-tropic murine leukemia viruses in human cells. Proc Natl Acad Sci USA 101(32):11827–11832
Pickart CM (2001) Mechanisms underlying ubiquitination. Annu Rev Biochem 70:503–533
Pincus T, Hartley JW, Rowe WP (1971) A major genetic locus affecting resistance to infection with murine leukemia viruses. I. Tissue culture studies of naturally occurring viruses. J Exp Med 133(6):1219–1233
Pryciak PM, Varmus HE (1992) Fv-1 restriction and its effects on murine leukemia virus integration in vivo and in vitro. J Virol 66(10):5959–5966
Reymond A, Meroni G, Fantozzi A, Merla G, Cairo S, Luzi L, Riganelli D, Zanaria E, Messali S, Cainarca S, Guffanti A, Minucci S, Pelicci PG, Ballabio A (2001) The tripartite motif family identifies cell compartments. EMBO J 20(9):2140–2151
Rold CJ, Aiken C (2008) Proteasomal degradation of TRIM5alpha during retrovirus restriction. PLoS Pathog 4(5):e1000074
Sawyer SL, Wu LI, Akey JM, Emerman M, Malik HS (2006) High-frequency persistence of an impaired allele of the retroviral defense gene TRIM5alpha in humans. Curr Biol 16(1):95–100
Sawyer SL, Wu LI, Emerman M, Malik HS (2005) Positive selection of primate TRIM5alpha identifies a critical species-specific retroviral restriction domain. Proc Natl Acad Sci USA 102(8):2832–2837
Sayah DM, Sokolskaja E, Berthoux L, Luban J (2004) Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature 430(6999):569–573
Schaller T, Hue S, Towers GJ (2007) An active TRIM5 protein in rabbits indicates a common antiviral ancestor for mammalian TRIM5 proteins. J Virol 81(21):11713–11721
Schnell JD, Hicke L (2003) Non-traditional functions of ubiquitin and ubiquitin-binding proteins. J Biol Chem 278(38):35857–35860
Sebastian S, Luban J (2005) TRIM5alpha selectively binds a restriction-sensitive retroviral capsid. Retrovirology 2:40
Shibata R, Sakai H, Kawamura M, Tokunaga K, Adachi A (1995) Early replication block of human immunodeficiency virus type 1 in monkey cells. J Gen Virol 76 ( Pt 11):2723–2730
Si Z, Vandegraaff N, O'Huigin C, Song B, Yuan W, Xu C, Perron M, Li X, Marasco WA, Engelman A, Dean M, Sodroski J (2006) Evolution of a cytoplasmic tripartite motif (TRIM) protein in cows that restricts retroviral infection. Proc Natl Acad Sci USA 103(19):7454–7459
Sokolskaja E, Berthoux L, Luban J (2006) Cyclophilin A and TRIM5alpha independently regulate human immunodeficiency virus type 1 infectivity in human cells. J Virol 80(6):2855–2862
Sokolskaja E, Sayah DM, Luban J (2004) Target cell cyclophilin A modulates human immunodeficiency virus type 1 infectivity. J Virol 78(23):12800–12808
Song B, Diaz-Griffero F, Park DH, Rogers T, Stremlau M, Sodroski J (2005a) TRIM5alpha association with cytoplasmic bodies is not required for antiretroviral activity. Virology 343(2):201–2011
Song B, Gold B, O'Huigin C, Javanbakht H, Li X, Stremlau M, Winkler C, Dean M, Sodroski J (2005b) The B30.2(SPRY) domain of the retroviral restriction factor TRIM5alpha exhibits lineage-specific length and sequence variation in primates. J Virol 79(10):6111–6121
Song B, Javanbakht H, Perron M, Park DH, Stremlau M, Sodroski J (2005c) Retrovirus restriction by TRIM5alpha variants from Old World and New World primates. J Virol 79(7):3930–3937
Speelmon EC, Livingston-Rosanoff D, Li SS, Vu Q, Bui J, Geraghty DE, Zhao LP, McElrath MJ (2006) Genetic association of the antiviral restriction factor TRIM5alpha with human immunodeficiency virus type 1 infection. J Virol 80(5):2463–2471
Stoye JP (2002) An intracellular block to primate lentivirus replication. Proc Natl Acad Sci USA 99(18):11549–11551
Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J (2004) The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 427(6977):848–853
Stremlau M, Perron M, Lee M, Li Y, Song B, Javanbakht H, Diaz-Griffero F, Anderson DJ, Sundquist WI, Sodroski J (2006a) Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5alpha restriction factor. Proc Natl Acad Sci USA 103(14):5514–5519
Stremlau M, Perron M, Welikala S, Sodroski J (2005) Species-specific variation in the B30.2(SPRY) domain of TRIM5alpha determines the potency of human immunodeficiency virus restriction. J Virol 79(5):3139–3145
Stremlau M, Song B, Javanbakht H, Perron M, Sodroski J (2006b) Cyclophilin A: an auxiliary but not necessary cofactor for TRIM5alpha restriction of HIV-1. Virology 351(1):112–120
Sveda MM, Soeiro R (1976) Host restriction of Friend leukemia virus: synthesis and integration of the provirus. Proc Natl Acad Sci USA 73(7):2356–2360
Tareen SU, Sawyer SL, Malik HS, Emerman M (2009) An expanded clade of rodent Trim5 genes. Virology 385(2):473–483
Thali M, Bukovsky A, Kondo E, Rosenwirth B, Walsh CT, Sodroski J, Gottlinger HG (1994) Functional association of cyclophilin A with HIV-1 virions. Nature 372(6504):363–365
Tissot C, Mechti N (1995) Molecular cloning of a new interferon-induced factor that represses human immunodeficiency virus type 1 long terminal repeat expression. J Biol Chem 270(25):14891–14898
Torok M, Etkin LD (2001) Two B or not two B? Overview of the rapidly expanding B-box family of proteins. Differentiation 67(3):63–71
Towers G, Bock M, Martin S, Takeuchi Y, Stoye JP, Danos O (2000) A conserved mechanism of retrovirus restriction in mammals. Proc Natl Acad Sci USA 97(22):12295–12299
Towers GJ (2005) Control of viral infectivity by tripartite motif proteins. Hum Gene Ther 16(10):1125–1132
Towers GJ, Hatziioannou T, Cowan S, Goff SP, Luban J, Bieniasz PD (2003) Cyclophilin A modulates the sensitivity of HIV-1 to host restriction factors. Nat Med 9(9):1138–1143
van Manen D, Rits MA, Beugeling C, van Dort K, Schuitemaker H, Kootstra NA (2008) The effect of Trim5 polymorphisms on the clinical course of HIV-1 infection. PLoS Pathog 4(2):e18
Virgen CA, Kratovac Z, Bieniasz PD, Hatziioannou T (2008) Independent genesis of chimeric TRIM5-cyclophilin proteins in two primate species. Proc Natl Acad Sci USA 105(9):3563–3568
Whitcomb JM, Hughes SH (1992) Retroviral reverse transcription and integration: progress and problems. Annu Rev Cell Biol 8:275–306
Wilson SJ, Webb BL, Ylinen LM, Verschoor E, Heeney JL, Towers GJ (2008) Independent evolution of an antiviral TRIMCyp in rhesus macaques. Proc Natl Acad Sci USA 105(9):3557–3562
Woo JS, Imm JH, Min CK, Kim KJ, Cha SS, Oh BH (2006) Structural and functional insights into the B30.2/SPRY domain. EMBO J 25(6):1353–1363
Wu X, Anderson JL, Campbell EM, Joseph AM, Hope TJ (2006) Proteasome inhibitors uncouple rhesus TRIM5alpha restriction of HIV-1 reverse transcription and infection. Proc Natl Acad Sci USA 103(19):7465–7470
Xu L, Yang L, Moitra PK, Hashimoto K, Rallabhandi P, Kaul S, Meroni G, Jensen JP, Weissman AM, D'Arpa P (2003) BTBD1 and BTBD2 colocalize to cytoplasmic bodies with the RBCC/tripartite motif protein, TRIM5delta. Exp Cell Res 288(1):84–93
Yamauchi K, Wada K, Tanji K, Tanaka M, Kamitani T (2008) Ubiquitination of E3 ubiquitin ligase TRIM5 alpha and its potential role. FEBS J 275(7):1540–1555
Yap MW, Nisole S, Lynch C, Stoye JP (2004) Trim5alpha protein restricts both HIV-1 and murine leukemia virus. Proc Natl Acad Sci USA 101(29):10786–10791
Yap MW, Nisole S, Stoye JP (2005) A single amino acid change in the SPRY domain of human Trim5alpha leads to HIV-1 restriction. Curr Biol 15(1):73–78
Ylinen LM, Keckesova Z, Webb BL, Gifford RJ, Smith TP, Towers GJ (2006) Isolation of an active Lv1 gene from cattle indicates that tripartite motif protein-mediated innate immunity to retroviral infection is widespread among mammals. J Virol 80(15):7332–7338
Young JC, Agashe VR, Siegers K, Hartl FU (2004) Pathways of chaperone-mediated protein folding in the cytosol. Nat Rev Mol Cell Biol 5(10):781–791
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2009 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Song, B. (2009). TRIM5alpha. In: Spearman, P., Freed, E. (eds) HIV Interactions with Host Cell Proteins. Current Topics in Microbiology and Immunology, vol 339. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-02175-6_3
Download citation
DOI: https://doi.org/10.1007/978-3-642-02175-6_3
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-02174-9
Online ISBN: 978-3-642-02175-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)