
Abstract
The TRIM protein family is emerging as a central component of mammalian antiviral innate immunity. Beginning with the identification of TRIM5α as a mammalian post-entry restriction factor against retroviruses, to the repeated observation that many TRIMs ubiquitinate and regulate signaling pathways, the past decade has witnessed an intense research effort to understand how TRIM proteins influence immunity. The list of viral families targeted directly or indirectly by TRIM proteins has grown to include adenoviruses, hepadnaviruses, picornaviruses, flaviviruses, orthomyxoviruses, paramyxoviruses, herpesviruses, rhabdoviruses and arenaviruses. We have come to appreciate how, through intense bouts of positive selection, some TRIM genes have been honed into species-specific restriction factors. Similarly, in the case of TRIMCyp, we are beginning to understand how viruses too have mutated to evade restriction, suggesting that TRIM and viruses have coevolved for millions of years of primate evolution. Recently, TRIM5α returned to the limelight when it was shown to trigger the expression of antiviral genes upon recognition of an incoming virus, a paradigm shift that demonstrated that restriction factors make excellent pathogen sensors. However, it remains unclear how many of ~100 human TRIM genes are antiviral, despite the expression of many of these genes being upregulated by interferon and upon viral infection. TRIM proteins do not conform to one type of antiviral mechanism, reflecting the diversity of viruses they target. Moreover, the cofactors of restriction remain largely enigmatic. The control of retroviral replication remains an important medical subject and provides a useful backdrop for reviewing how TRIM proteins act to repress viral replication.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Electrophoretic Mobility Shift Assay
- Murine Leukemia Virus
- Embryonal Carcinoma Cell
- Trim Protein
- TRIM22 Expression
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Tripartite motif-containing (TRIM) proteins are an ancient family of E3 ligases. They are characterized by a conserved architecture of two or three zinc-binding folds (one RING domain, one or two B-Box domains), a predicted coiled coil (collectively an RBCC), and usually a C-terminal domain that recruits specific interaction partners (Nisole et al. 2005). TRIM genes predate the emergence of metazoans (Marin 2012), appear sporadically in invertebrates (Sardiello et al. 2008), and experienced substantial species-specific expansion in vertebrates (Reddy et al. 1991; Meroni and Diez-Roux 2005; Boudinot et al. 2011). They are extremely numerous in the higher vertebrates; humans encode nearly 100 TRIM genes, several of which are so young that they are absent in other ape species (Han et al. 2011).
Several lines of evidence suggest that a large proportion of TRIM proteins have roles in innate immunity. One, TRIM genes evolve by segmental duplication events followed by selection-driven adaption. Viruses themselves are candidate sources of selective pressure, as a panel of TRIMs has been shown to inhibit the replication of particular viruses (Mallery et al. 2010; Stremlau et al. 2004; Wolf and Goff 2007; Everett et al. 2006; Taylor et al. 2011; Poole et al. 2009; Kajaste-Rudnitski et al. 2011). In turn, TRIM proteins can influence the evolution of their viral adversaries, as exemplified by TRIM5α and TRIMCyp, which are immersed in genetic conflicts with retroviruses (Ylinen et al. 2010; Price et al. 2009; Newman et al. 2006). Two, several TRIMs have been shown to influence the flux through innate signaling pathways, by ligating ubiquitin to component signal transducers (McNab et al. 2011; Kawai and Akira 2011; Tsuchida et al. 2010). Often, this activity is triggered by viral infection. Perhaps related to this, some TRIM proteins can potentiate immune signaling pathways when transfected into reporter assays, an effect comparable to known signaling transducers like MAVS/IPS-1 or TRAF6 (Uchil et al. 2013; Pertel et al. 2011; Yu et al. 2011; Versteeg et al. 2013). Three, expression of many TRIM genes is acutely sensitive to both interferon (Carthagena et al. 2009; Ozato et al. 2008; Asaoka et al. 2005) and viral infection. This is true for TRIMs from fish (van der Aa et al. 2009) to mammals (Rajsbaum et al. 2008). In one study, half of all human TRIM transcripts were sensitive to regulation by interferons (Carthagena et al. 2009). Four, TRIM genes can be polymorphic, which can have the effect of broadening the antiviral repertoire within a species (Newman et al. 2006; Wilson et al. 2008a; Kirmaier et al. 2010). Five, the most ancient of TRIM genes, represented by human TRIM37, is related to the TRAF family of ubiquitin E3 ligases (Marin 2012), which regulate innate signal transduction through their ubiquitin ligase activity. Human TRAFs bear an N-terminal RING and C-terminal MATH domain, like TRIM37, and these flank tandem zinc fingers. It is becoming apparent that TRIMs and TRAFs have some comparable roles in innate immune signaling (Xia et al. 2009; Gack et al. 2007; Pertel et al. 2011; Tsuchida et al. 2010). And six, although not evidence in itself, the TRIM family experiences rapid gene gain and loss, a phenotype indicative of adaptation that is often exhibited by immunity-related genes (Barreiro and Quintana-Murci 2010; Han et al. 2011).
In this chapter, we focus our attention on the antiretroviral properties of the TRIM family members TRIM5, TRIM22/Staf50, and TRIM28/Kap-1/TIF1β, as the best characterized TRIM proteins with antiretroviral activity. We review recent data suggesting a role for the TRIM28-related protein TRIM24 in retroviral restriction. We discuss how the biology of these proteins relates to their antiviral mechanism and comment on the diversity of retroviruses they target. Finally, we review the use of screens as a means to uncover novel antiretroviral activity in the TRIM family.
2 TRIM5α and TRIMCyp Bind Retroviral Capsids to Accelerate Their Degradation and Stimulate Innate Signaling
2.1 Combining Two Activities
TRIM5α has two known functions in the cell. The first is as a defensive retroviral capsid-binding protein that can inhibit or restrict retroviral infection (Stremlau et al. 2004; Keckesova et al. 2004; Kirmaier et al. 2010). The second is an E3 ubiquitin ligase that promotes NFκB and AP-1 transcription factor signaling (Pertel et al. 2011; Tareen and Emerman 2011). These two activities are mechanistically integrated in that E3 activity potentiates restriction of retroviral infectivity (Wu et al. 2006) while binding retroviral capsids with high avidity enhances innate signaling (Pertel et al. 2011). In this manner, TRIM5α mounts a proteasome-dependent block to viral DNA synthesis while simultaneously acting as a pattern recognition receptor (PRR) for the retroviral capsid. In this section, we discuss some of the salient and more recent findings in TRIM5α/TRIMCyp biology. For a discussion on the evolution of these proteins, we direct the reader to a recent review by Daugherty and Malik (Daugherty and Malik 2012).
2.2 The C-Terminal PRYSPRY has Surface Variable Loops that Determine Restriction Specificity
Human TRIM5 belongs to a TRIM cluster on chromosome 11p15, alongside its paralogues TRIM6, TRIM22, TRIM34, and TRIM21, which arose by gene duplication events after the divergence of placental and marsupial mammals ~180 million years ago (Sawyer et al. 2007). In humans, TRIM5α is the longest of five putative isoforms encoded by the TRIM5 gene and is unique in bearing a PRYSPRY domain, alternately called a B30.2, at its C terminus (Reymond et al. 2001; Stremlau et al. 2004). The PRYSPRY, an amalgam of PRY and SPRY subdomains, is a distorted β-sandwich, two layers of antiparallel β-sheets connected by non-structured loops. In TRIM5α, the PRYSPRY is the point of contact with the viral capsid, and it bears the telltale signs of antagonistic host–virus evolution: The variable loops are hot spots of positive selection through primate evolution and are poorly conserved in sequence and length between primates and harbor determinants of antiviral specificity. Being so mobile, these loops have eluded the efforts of crystallographers until only recently, and we now have a model of the rhesus macaque TRIM5α PRYSPRY, derived by superposition of a 1.55 Å crystal structure (solved by substituting the most dynamic variable loop (v1) with a two-amino acid linker) with NMR relaxation parameters of the variable loops (Biris et al. 2012). The TRIM5α PRYSPRY is similar to the few other PRYSPRY structures available, including that from the Drosophila SPRY-SOCS box family protein Gustavus (Woo et al. 2006a) and human TRIM21 (James et al. 2007). As previously predicted, the major differences between TRIM5 species variants cluster on one surface, comprising the longest variable loops. Intriguingly, there is striking congruence between the positions of residues in this surface that influence capsid recognition in TRIM5α, antibody binding in TRIM21, and hereditary disease when mutated in Pyrin/TRIM20 and MID1/TRIM18 (Woo et al. 2006b).
2.3 TRIM5α Recognizes Multiple Capsid Epitopes to Maintain Broad Specificity
Collectively, TRIM5α proteins restrict a surprising diversity of retroviruses, bearing capsids that are only distantly related, at least at the level of primary sequence. For example, the tamarin monkey TRIM5α restricts both spumaviruses and the betaretrovirus Mason-Pfizer monkey virus (MPMV) (Diehl et al. 2008; Yap et al. 2008); the ring-tailed lemur TRIM5α restricts feline, equine and primate lentiviruses as well as a murine gammaretrovirus (Rahm et al. 2011) human TRIM5α restricts an equine lentivirus and a murine gammaretrovirus (Stremlau et al. 2004; Keckesova et al. 2004). It transpires that different retroviral capsid proteins (CA), despite having distinct primary sequence, comprise subunits of similar helical tertiary structures, which assemble into hexamers or pentamers within symmetrical lattices (Ganser et al. 2003; Li et al. 2000; Ganser-Pornillos et al. 2004; Mortuza et al. 2008). Binding of TRIM5α to monomeric or dimeric CA is difficult to detect by conventional biochemical or yeast two-hybrid techniques, suggesting low-affinity interactions (Nisole et al. 2004; Sebastian and Luban 2005). However, mature retroviral cores, comprising complexed CA monomers, isolated from virions with gentle detergents, are bound by immobilized TRIM5α, supporting the hypothesis that TRIM5α recognizes complex multivalent epitopes in the capsid structure (Pornillos et al. 2011; Ganser-Pornillos et al. 2007; Welker et al. 2000; Sebastian and Luban 2005). Furthermore, capsid-nucleocapsid (CANC) tubes, which are built from the same hexagonal lattice as the natural HIV-1 core and are often used as structural surrogates of mature capsids, are bound by TRIM5α in vitro (Black and Aiken 2010; Li et al. 2000; Stremlau et al. 2006; Ganser et al. 1999). Within cells, TRIM5α restriction can be saturated by mature, stable retroviral cores but not by fragments of Gag (Towers et al. 2003; Cowan et al. 2002; Sebastian and Luban 2005; Forshey et al. 2005; Shi and Aiken 2006; Dodding et al. 2005), further supporting the hypothesis that TRIM5α recruitment to virus requires the intact CA complex structure. Recently, direct binding between recombinant proteins has also been observed. Recombinant rhesus macaque TRIM5α, the RING substituted for the TRIM21 RING (TRIM5-21R) to improve solubility, decorates HIV-1 CANC tubes when viewed by electron microscopy (EM). This TRIM5 chimera also sediments with capsids derived from membrane-stripped EIAV, despite HIV-1 and EIAV CAs sharing only ~25 % sequence identity (Langelier et al. 2008; Kar et al. 2008).
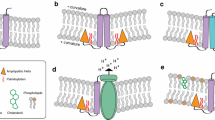
This model of multiple low-affinity interactions between TRIM5α and capsids requires multiple points of contact in the CA lattice (Ganser-Pornillos et al. 2011; Li et al. 2000). Supporting this hypothesis, two remarkably similar clefts exist in the N-terminal domains of both murine leukemia virus (MLV) and HIV-1 CA and are sampled by PRYSPRY variable loops (Fig. 1). One cleft, formed by CA α-helices 4–6 in MLV and 4–7 in HIV-1, contains determinants of viral tropism (Ohkura et al. 2011; Maillard et al. 2011; Maillard et al. 2007; Owens et al. 2004). For example, MLV CA residue 110 is a determinant for N- or B-tropism, and residues 82 and 117 toggle B-MLV sensitivity to human TRIM5α variants (Kozak and Chakraborti 1996; Maillard et al. 2007; Towers et al. 2000). HIV-1 CA mutated at residues E128 and R132 (to alanines) are less infectious in human cells but more infectious in Old World primate cells (Hatziioannou et al. 2004). Similarly, mutation of HIV-1 CA residues 83, 120, and 122—residues analogous to those in MLV that determine sensitivity to human TRIM5α—can potentiate HIV-1 restriction by human TRIM5α (Maillard et al. 2007, 2011). The second cleft, between the MLV N-terminal β-strands and α-helix 6, and HIV-1 β1 and α-helix 6, is critical for MLV restriction by rhesus TRIM5α; the L10 W mutation obscures the cleft, prevents rhesus TRIM5α binding to N-MLV CA, and allows the virus to escape restriction (Ohkura et al. 2011). Overall, the combinatorial involvement of several regions of the PRYSPRY and CA in restriction fit a model of multivalent interaction.

Residues in N-terminal domains of HIV-1 and MLV CA proteins that influence sensitivity to TRIM5α occupy two surface-exposed clefts. N-terminal CA domains (CAN) from HIV-1 (a) (PDB 1GWP) and N-MLV (b) (PDB 1U7 K). Residues that impact on TRIM5α restriction are labeled green (HIV-1) or orange (N-MLV). Both CAN are viewed from the same orientation, with the β-sheet-α-helix 6 cleft (HIV-1 and MLV) and the α-helix 4–7 (HIV-1) and α-helix 4–6 (N-MLV) clefts visible. Based on studies in (Ohkura et al. 2011) and (Maillard et al. 2011)
2.4 TRIMCyp is Also a Versatile Capsid Recognition Domain
Examination of TRIM5 variants in primate species revealed that on several occasions, the TRIM5α PRYSPRY has been replaced by a cyclophilin A (CypA) cDNA by retrotransposition. The fact that this event has occurred independently on more than one occasion is evidenced by differences in the position of the CypA cDNA, as well as differences in the splicing events that give rise to the TRIMCyp chimeric proteins in each case (Wilson et al. 2008b; Sayah et al. 2004; Nisole et al. 2004; Virgen et al. 2008; Lin and Emerman 2008; Newman et al. 2008; Brennan et al. 2008; Malfavon-Borja et al. 2013; Liao et al. 2007). It transpires that, like a PRYSPRY, the CypA domain is also a very versatile recognition domain for retroviral CA proteins and is able to switch capsid specificity through single amino acid substitutions. Whereas Aotus (Owl monkey) TRIMCyp recognizes HIV-1 and FIV (Diaz-Griffero et al. 2006b), as does the parental CypA protein (Lin and Emerman 2006), Macaca. mulatta (rhesus macaque) TRIMCyp restricts HIV-2 and HIV-1 O-groups but not HIV-1 M-group viruses (Price et al. 2009; Virgen et al. 2008; Wilson et al. 2008b). Thus, these TRIMCyps differentiate between viral lineages (HIV-1 and HIV-2) and independent HIV-1 zoonoses (HIV-1 M- and O-groups).
The switch in restriction specificity from M-group HIV-1 to most isolates of O-group HIV-1 and HIV-2 in M. mulatta TRIMCyp is due to 2 amino acid changes in the Cyp domain with respect to the parental CypA sequence. Substitutions D66N and R69H, alter Cyp conformation and physical properties allowing conformational diversity not present in the parental CypA molecule (Price et al. 2009; Ylinen et al. 2010; Caines et al. 2012). Intriguingly, R69H expands antiviral specificity by allowing an open binding site conformation that allows restriction of HIV-2 but maintains the ability to hold the closed binding site conformation that can recruit and restrict HIV-1 (Fig. 2a). The second substitution in rhesus TRIMCyp D66 N focuses specificity away from HIV-1 M-group while maintaining binding and restriction of HIV-1 O-group and HIV-2 capsids, by maintaining conformational flexibility (Caines et al. 2012). The natural germline CypA is fixed in the closed binding site conformation and binds HIV-2 CA poorly. The significant difference between HIV-1 and HIV-2 capsids in regard to TRIMCyp specificity is HIV-2 bearing a cyclophilin-binding loop that is shorter by one residue (Price et al. 2009). Furthermore, the sensitivity of naturally occurring HIV-1 isolates to M. mulatta TRIMCyp can be predicted based on the residue at position CA88 of the cyclophilin-binding loop. O-group isolates bearing methionine, valine, or isoleucine at CA position 88 are sensitive to M. mulatta TRIMCyp, whereas those bearing an alanine at this position in CA are not. Alanine is found in only 7 of 66 O-group sequences but almost all M-group sequences and these viruses remain insensitive to M. mulatta TRIMCyp restriction (Price et al. 2009).

Evolution of TRIMCyp in Asian macaques. a Mutations D66 N and R69H convey the property of conformational diversity onto the Asian monkey TRIMCyp cyclophilin domain (Caines et al. 2012). Thus, the molecule exists between “open” and “closed” conformations. This diversity allows this particular TRIMCyp to restrict both HIV-2 via recruitment of HIV-2 capsid to an “open” conformation cyclophilin and HIV-1 O-group capsid via a “closed” conformation. The HIV-2 capsid derived binding peptide is shown in yellow, and the HIV-1 O-group capsid derived peptide is shown in green. The mobile cyclophilin active site loop encompassing residues 66 and 69 is shown in yellow. b Dendrogram illustrating the macaque phylogeny with the restriction specificity of the TRIMCyp species variants shown on the right. The changes to Cyp domain sequence that have occurred are shown on the branches. The specificity switches suggest waves of selective forces from pathogenic viruses during macaque evolution (Ylinen et al. 2010). c X-ray crystal structures of HIV-1 CA in complex with Cyp molecules illustrate the plasticity of Cyp recruitment to capsid (Ylinen et al. 2010). HIV-1 CA (dark green) in complex with wild-type cyclophilin A (light green) superposed on the structure of HIV-1 CA (gray) and the M. fasicularis TRIMCyp Cyp domain (yellow) that bears substitutions R69H and E143 K. Mutation to lysine attracts the peptide oxygen at position P58. This propagates changes to other Cyp active site residues, including R148 that is coordinated by the P58 peptide oxygen in CypA structure. The altered position of P58 releases the side chain of R148, allowing it to interact with capsid residues downstream of residue A88. The structure around Cyp residue 69 is not significantly changed by the R69H change in the crystal structure
Interestingly, like Aotus TRIMCyp, the M. fasicularis (cynomolgus macaque) TRIMCyp restricts HIV-1, FIV, and SIVtan but not HIV-2 (Ylinen et al. 2010). M. fasicularis TRIMCyp bears the specificity enhancing R69H substitution in the CypA domain, which is expected to broaden restriction to HIV-2. However, M. fasicularis TRIMCyp also bears a second change, in this case E143K, that focuses specificity away from HIV-2 while maintaining restriction of HIV-1 and SIVtan (Ylinen et al. 2010) (Fig. 2a). Considering the substitutions that have occurred in the TRIMCyp molecules in the context of the macaque phylogeny suggests a complex evolutionary history (Fig. 2b). Note that R69H, which expands specificity, is present in all of the macaque TRIMCyps, whereas D66N, which narrows specificity toward HIV-2, is present in M. nemestrina (pigtail macaque) and M. mulatta but not M. fasicularis. This is despite M. fascicularis being more closely related to M. mulatta than M. nemestrina and is suggestive of waves of infection that selected for alternate antiviral specificities during macaque evolution. Structural studies of the Cyp domain also illustrate the plasticity of this molecule and its ability to adapt to recruiting different lentiviral capsids. For example, although K143 is distant from the CypA active site, mutation to glutamate causes a conformational change that is propagated to the binding site, altering CA recognition (Fig. 2c) (Ylinen et al. 2010).
Despite the absence of any known extant lentiviruses in Asian macaques, these observations imply that historic lentiviruses may have provided selection pressures to drive diversification of the CypA domain of macaque TRIMCyps. Indeed, fossil records demonstrate the ancestors of Asian monkeys migrated across Europe ~5.5 million years ago, and lentiviruses are known to have been circulating in European mammals at least 12 million years ago, according to endogenized lentiviral proviruses found in rabbits and hares (Katzourakis et al. 2007; Keckesova et al. 2009; Li et al. 2009).
Most recently, a third example of a TRIMCyp fusion has been described in Old World primates (TRIMCypA3), although in this case the gene has decayed, perhaps due to a lack of recent selective advantage (Malfavon-Borja et al. 2013). Reconstruction of the ancient TRIMCyp suggested that it too had broad antiviral activity able to restrict HIV-1 and HIV-2 lineage viruses. However, the protein subsequently evolved to target unknown viruses, presumably those that circulated at this time, and restriction of the viruses we know today was lost. Why lentiviruses retain cyclophilin binding in the face of persistent restriction and TRIMCyp evolution remains the subject of some speculation. Cyclophilin A is certainly a cofactor for HIV-1 replication and probably FIV too (Franke et al. 1994; Thali et al. 1994; Lin and Emerman 2006). The interaction with cyclophilins is certainly ancient, as evidenced by the demonstration of cyclophilin A binding by the extinct endogenous lentiviruses RELIK and PSIV (Goldstone et al. 2010). It is possible that cyclophilin recruitment is preserved through necessity to recruit another cyclophilin, for example the nuclear pore–associated cyclophilin Nup358, which acts as a cofactor for HIV-1 and possibly other lentiviruses (Ocwieja et al. 2011; Schaller et al. 2011).
2.5 TRIM5α Causes Structural Damage to Capsids
An important goal in the TRIM5 field is to understand the details of the restriction mechanism and the fate of restricted virions. The best supported model suggests that TRIM5α binding to capsids causes their untimely disassembly (Zhao et al. 2011; Stremlau et al. 2006). Capsid disassembly has been measured by the so-called fate-of-capsid (FOC) assay, which correlates sensitivity to TRIM5α restriction with loss of particulate, pelletable capsid. For example, centrifugation of SIVmac-, N-MLV- or HIV-1-infected cell lysates expressing restrictive squirrel monkey, human, or rhesus TRIM5α, respectively, demonstrated a decrease in capsid sedimentation compared to non-restrictive cells (Stremlau et al. 2006; Perron et al. 2007). The loss of CA sedimentation has been interpreted as showing that TRIM5α restriction causes CA disassembly. This assay was validated with the observations that PRYSPRY deletion or HIV-1 capsid mutation H87Q that reduces restriction, restores the particulate capsid that is found in the pellet. The FOC assay has also been useful in demonstrating the importance of the TRIM5α RBCC motif in restriction. Deletion of the RING or B-Box2 prevents loss of pelletable N-MLV capsids, even though these two domains are not necessary for binding to CANC arrays in vitro (Perron et al. 2007; Zhao et al. 2011). This suggests that to mediate uncoating in cells, in addition to capsid binding, TRIM5α requires the capacity to multimerize and recruit the proteasome.
Another assay supporting a role for premature uncoating in TRIM5 restriction involves incubation of CANC tubes with cell lysates expressing TRIM5α or TRIMCyp, followed by EM. Generally, CANC tubes are shortened or damaged under such conditions (Black and Aiken 2010). Rhesus TRIM5-21R, or another recombinant TRIM5α mutant comprising simply the CC to PRYSPRY, have also been shown to alter the appearance of both CANC tubes and isolated HIV-1 cores, respectively (Zhao et al. 2011; Langelier et al. 2008). Specifically, the TRIM5 CC-PRYSPRY truncation released linear fragments of CA rather than soluble monomers (Zhao et al. 2011). Using this truncation mutant, an elegant approach to identify the regions of HIV-1 CA targeted by TRIM5α was devised, whereby hexamers in the form of CANC tubes were cross-linked by the introduction of cysteines at inter- or intra-hexamer interfaces. In this system, only the oxidation of cysteines positioned at the inter-hexamer interface prevented CC-PRYSPRY-induced CANC damage, suggesting that TRIM5α causes capsid disassembly by separating adjacent hexamers. In these experiments, a RING or B-Box2 is not required, presumably because the in vitro reactions use high protein concentrations that obviate the requirement for RING–B-Box2-mediated multimerization and any proteasome recruitment that the TRIM5N-terminus may normally orchestrate within cells.
2.6 TRIM5α Ubiquitination
If the first stage of TRIM5α restriction involves formation of a TRIM5α–capsid complex that decreases capsid integrity and prevents nuclear entry, the second stage is defined by two observations. One, it precludes viral reverse transcription (Hofmann et al. 1999; Towers et al. 2000), and two, it is overcome by proteasome inhibition (Wu et al. 2006). There is little evidence for capsid degradation during restriction (Chatterji et al. 2006; Stremlau et al. 2006; Yueh et al. 2006; Perron et al. 2007), and neither ubiquitin nor the RING domain is required for the structural damage caused to CANC tubes by TRIM5α in vitro (Zhao et al. 2011; Black and Aiken 2010). Rather, the generally accepted model of TRIM5α restriction is that TRIM5α bound to capsids recruits proteasomes that potentiate capsid disassembly (Stremlau et al. 2006; Perron et al. 2007). In agreement, proteasome inhibition in the FOC assay inhibits HIV-1 uncoating by TRIM5α/TRIMCyp and restores pelletable capsid (Diaz-Griffero et al. 2007). Indeed, TRIM5α can bind to proteasome components, for example the regulatory protein RPT1/PSMC2 (Lukic et al. 2011). The fate of the non-infectious TRIM5α–capsid complex is uncertain in the absence of the second proteasome-dependent stage of restriction. Although the virus has sufficient time and resources to undergo reverse transcription and is not rapidly uncoated, it is prevented from entering the nucleus and forming DNA circles, a surrogate marker for nuclear entry (Wu et al. 2006; Shank and Varmus 1978).
2.6.1 The Role of the RING
The molecular details of the second stage of restriction remain poorly defined. Specifically, how does TRIM5α recruit the proteasome? A role for TRIM5α ubiquitination has been established in innate immune signaling (see below) but not during restriction. TRIM5α is evidently an E3 ligase capable of autoubiquitination in vitro, and it accumulates in a ubiquitinated form under proteasome inhibition (Xu et al. 2003; Yamauchi et al. 2008; Pertel et al. 2011; Diaz-Griffero et al. 2006a). Additionally, for a protein of 57 kDa, TRIM5α has an unusually short half-life (less than an hour), indicating that it is subject to rapid turnover (Diaz-Griffero et al. 2006a; Javanbakht et al. 2005). Indeed, proteasome inhibitors simultaneously lengthen TRIM5α half-life and rescue viral reverse transcription, suggesting that rapid TRIM5α turnover might be sufficient to levy the block to reverse transcription. As an example, Aotus TRIMCyp has a short half-life and restricts before reverse transcription, whereas the artificial factor Fv1Cyp has a long half-life and blocks after DNA synthesis (Schaller et al. 2007; Yap et al. 2007). Following this argument, it is surprising that in several cases, RING deletions abrogate TRIM5α restriction completely rather than phenocopy proteasome inhibition. Rhesus TRIM5α∆RING or the C15A/C18A RING mutants have threefold longer half-lives and lose the majority of their ability to restrict HIV-1 and N-MLV, respectively (Yap et al. 2007; Stremlau et al. 2004; Diaz-Griffero et al. 2007). Furthermore, human TRIM5α∆RING loses all but a twofold restriction against N-MLV infection (Perez-Caballero et al. 2005a; Perron et al. 2007). Aotus TRIMCyp∆RING persists for ten times its usual half-life and loses 90 % of its restriction against HIV-1 (Diaz-Griffero et al. 2006a, b, 2007; Perez-Caballero et al. 2005b).
A more comprehensive mutagenesis of the rhesus TRIM5α RING, in either the putative E2-binding site or RING–RING dimerization interface, can inhibit autoubiquitination in vitro and completely inhibit restriction of HIV-1 and EIAV, suggesting that ubiquitination is required for viral restriction (Lienlaf et al. 2011). The same observation is made upon Ubc13/Uev1a depletion, the E2 heterodimer used by TRIM5α to synthesis ubiquitin chains (see below) (Pertel et al. 2011). These observations suggest that the nature of the TRIM5α–capsid complex is dependent upon ubiquitination, although the RING mutants are no less able to bind CANC tubes in vitro (Lienlaf et al. 2011). Therefore, ∆RING or cysteine mutants are inconsistent with proteasome inhibition and might behave aberrantly through misfolding, mislocalization, or dominant-negative effects and are perhaps not the best tool for dissecting TRIM5α ubiquitination. This might explain the observation that rhesus TRIM5α∆RING is unable to hetero-oligomerize wild-type TRIM5α when both proteins are over-expressed, leading to a proposed role for the RING domain in TRIM5α oligomerization (Li et al. 2011). Interestingly, other rhesus TRIM5α RING mutants have been described that retain potent restriction of HIV-1 but have lost the ability to block viral reverse transcription (Roa et al. 2012). Substitutions at residue Tyr63 in the rhesus protein, which faces into the putative E2-binding cleft, bind CANC tubes in vitro but restrict after reverse transcription and fail to dismantle HIV-1 capsids in the FOC assay, phenocopying proteasome inhibition. These mutants are likely to be informative for the dissection of the mechanism behind proteasome recruitment.
2.6.2 TRIM5 Signaling
A recent development in the field has been the suggestion that TRIM5α constitutes a critical component of an innate signaling pathway. LPS stimulation of TLR4 generates an antiviral state that inhibits the infectivity of diverse viruses including Newcastle disease virus and vesicular stomatitis virus, and TRIM5α appears to contribute to this process (Pertel et al. 2011). In this regard, TRIM5α ubiquitination has been dissected in detail and shown to involve the synthesis of lysine 63 (K63)-linked polyubiquitin using the E2 heterodimer Ubc13/Uev1a. In vitro ubiquitination reactions suggest that these ubiquitin chains are unanchored, a recently described phenomenon that is proposed to mediate TRAF6 signaling (Xia et al. 2009). In this model, unanchored K63-ubiquitin chains promote the autophosphorylation activity of transforming growth factor-β-activated kinase 1 (TAK1), resulting in IKK activation and NFκB nuclear translocation. Notably, recognition of the retroviral lattice potentiates TRIM5α ubiquitination in vitro and innate signaling in cells, broadening our appreciation of what defines a pattern recognition receptor (PRR), or indeed, a pathogen-associated molecular pattern (PAMP) which, in this case, may be the CA lattice itself. This observation parallels another intriguing discovery that TRIM5-21R dimers themselves assemble into hexameric arrays, multimerizing via the B-Box2 (Ganser-Pornillos et al. 2011). Importantly, two-dimensional hexagonal CANC crystals accelerate TRIM5-21R polymerization, further suggesting TRIM5α might dynamically respond to its target in cells and form a hexameric cage around the incoming virion (Diaz-Griffero 2011). In this regard, recognition of capsid has been shown to accelerate TRIM5α turnover (Rold and Aiken 2008), suggesting that recruitment to virions facilitates TRIM5α enzymatic activity and turnover through virus-induced multimerization.
2.7 TRIM5α Cytoplasmic Bodies as Components of Restriction
Like many TRIM proteins, TRIM5α forms highly dynamic cytoplasmic bodies, the function of which is not clear (Reymond et al. 2001; Campbell et al. 2007). However, their molecular composition is slowly becoming known. For example, there is mounting evidence that TRIM5α cytoplasmic bodies are lipid-rich structures. Rhesus TRIM5α immunoprecipitates flotillin-1 and co-localizes by immunofluorescence (IF) with caveolin-1, both of which are components of lipid microdomains (Ohmine et al. 2010). Cholesterol depletion disrupts cytoplasmic bodies, and lipid starvation increases HIV-1 or N-MLV infection of rhesus macaque or human cells, respectively, suggesting that lipid-rich bodies are important for the restriction mechanism (Ohmine et al. 2010). In support of this, replenishing lipids with serum restores restriction. A recent proteomic analysis of fractionated lipid microdomains from human fibroblasts identified TRIM5α (Kim et al. 2009). Furthermore, a screen in 293T cells to identify binding partners of TRIM5α found that the factor accumulates in a detergent-insoluble fraction, potentially enriched for lipids (Hwang et al. 2010). Membranous lipid rafts are important signaling platforms (Triantafilou et al. 2011), and cytoplasmic organelles such as peroxisomes play a role in innate immune signaling (Dixit et al. 2010). Potentially, TRIM5α signaling and restriction are coordinated by lipid-rich assembly platforms, a notion which remains to be tested.
A role for TRIM5α bodies in restriction is further hinted at in the observation, demonstrated by immunofluorescence, that proteasome inhibition sequesters HIV-1 at rhesus TRIM5α bodies (Campbell et al. 2008; Danielson et al. 2012). Under these conditions, the TRIM5α bodies also stain positively for proteasomes, ubiquitin, and CA, suggesting proteasomes are recruited in order to solubilize the CA/TRIM5α complexes, which is also inferred by the FOC assay (Diaz-Griffero et al. 2006a, 2007; Campbell et al. 2008; Danielson et al. 2012; Lukic et al. 2011). Indeed, the amount of particulate capsid in the FOC assay correlates with the number of viral reverse transcripts in those cell lysates (Kim et al. 2011), demonstrating a direct relationship between the proteasome-mediated uncoating and a block to reverse transcription. Interestingly, some TRIM5α bodies are seen to form rapidly around HIV-1 in the presence of proteasome inhibitor, reaching full intensity within a minute from first appearing, suggesting TRIM5α is able to traffic to incoming virions in response to infection (Campbell et al. 2008). Indeed, TRIM5α cytoplasmic bodies are highly motile, exhibiting short saltatory or long-distance unidirectional movements at speeds of >2 μm/s (Campbell et al. 2007). They also co-localize with microtubules, as do HIV-1 cores, providing a potential mechanism for their motility and recruitment to virions (Sodeik 2002; Diaz-Griffero et al. 2006a). In the absence of proteasome inhibitors, TRIM5α, HIV-1, and proteasome co-localization is minimal, implying restriction and viral destruction ordinarily occurs rapidly via transient associations, as demonstrated by measuring the kinetics of restriction (Perron et al. 2007; Perez-Caballero et al. 2005b).
3 TRIM28 is a Transcriptional Corepressor of Endogenous and Exogenous Retroviruses
3.1 TRIM28 is a Molecular Scaffold for Heterochromatin-Inducing Enzymes
Whereas TRIM5α was investigated in the context of restriction before the discovery of its constitutive role in immune signaling, TRIM28 was first considered the requisite corepressor of Krüppel-associated box (KRAB) domain zinc finger proteins (KRAB-ZFP) (Friedman et al. 1996; Kim et al. 1996; Moosmann et al. 1996), before becoming implicated in retroviral immunity. Numbering nearly 400 members in humans and mice, KRAB-ZFPs are the largest family of transcriptional repressors in vertebrates (Emerson and Thomas 2009). Members of this superfamily bear an N-terminal KRAB domain—a conserved 75-residue repressor motif—and between 2 and 40 C-terminal C2H2 zinc finger motifs, which bind DNA and direct target specificity (Thiesen et al. 1991; Bellefroid et al. 1991). The KRAB domain recruits TRIM28 (also called KRAB-associated protein 1 (KAP1), TIF1-β, or KRIP-1), which provides a platform from which chromatin-remodeling enzymes gain access to nucleosomes (Bellefroid et al. 1991; Huntley et al. 2006; Urrutia 2003). This results in the introduction and spreading of heterochromatin at specific loci and transcriptional shutdown. Tethering TRIM28 directly to a promoter causes transcriptional silencing (Friedman et al. 1996; Sripathy et al. 2006), demonstrating the protein’s ability to deliver repression.
The various domains of TRIM28 have been attributed specific roles (Fig. 3). TRIM28 homodimerizes and binds a KRAB domain (Peng et al. 2000) via its RBCC motif, which contains two B-Boxes. Beyond the RBCC, a hydrophobic PxVxL pentapeptide binds heterochromatin protein 1 (HP1) (Ryan et al. 1999; Nielsen et al. 1999; Le Douarin et al. 1996), an interaction essential for both nuclear retention and repression (Iyengar et al. 2011; Sripathy et al. 2006). At TRIM28’s C terminus, a tandem PHD (plant homeodomain) finger and bromodomain recruit chromatin remodifiers, for example the histone 3 lysine 9 (H3K9)-specific methyltransferase SETDB1/ESET (Schultz et al. 2002) and the nucleosome-remodeling and histone deacetylation (NuRD) component CHD3/Mi2α (Schultz et al. 2001), which associates with the histone deacetylases HDAC1 and HDAC2. The mechanism by which TRIM28 recruits these enzymes is small ubiquitin-like modifier (SUMO) protein dependent. The PHD domain is a zinc-coordinating RING-like C4HC3 fold and binds Ubc9, the sole SUMO E2 conjugation enzyme. Consequently, the domain is an intramolecular E3 ligase for SUMO, autosumoylating the neighboring bromodomain (Mascle et al. 2007; Ivanov et al. 2007; Zeng et al. 2008). Mutation of target lysines abolishes TRIM28 repression activity (Mascle et al. 2007), interaction with SETDB1 and CHD3 (Ivanov et al. 2007), and prevents the introduction of H3K9me3 marks on target DNA (Ivanov et al. 2007), without impacting on KRAB–TRIM28 binding or TRIM28-DNA localization (Iyengar et al. 2011). SUMO interaction motifs (SIMs) in SETDB1 and CHD3 are necessary for both their binding to TRIM28 and optimal enzymatic activity (Ivanov et al. 2007). TRIM28 represents the first example of a PHD harboring such activity; indeed, very few E3 ligases for SUMO are currently known, with the protein inhibitor of activated STAT (PIAS) proteins being the best characterized to date (Shuai and Liu 2005). Some other TRIMs have been shown to possess SUMO E3 activity, for example TRIM19 (PML) (Boutell et al. 2003) and TRIMs 22, 27, and 32 (Chu and Yang 2011) .

Introduction of heterochromatin by TRIM28 and associated factors. The TRIM28 RBCC interacts with the KRAB domain of a KRAB-ZFP, which is targeted to a specific target sequence by DNA-binding zinc fingers. The TRIM28 plant homeodomain (PHD) is a SUMO E3 enzyme that binds the SUMO conjugation E2 enzyme Ubc9 to SUMOylate the bromodomain. Chromatin-remodifying enzymes like lysine methyltransferase SETDB1 and histone deacetylase complex NuRD bear SUMO-interacting motifs (SIM) that bind the SUMOylated bromodomain, and heterochromatin markers like H3K9me3 and deacetylated H4 are established. By binding the PxVxL pentapeptide in the TRIM28 HP1 box, HP1 proteins are recruited to H3K9me3 sites, contributing to heterochromatin propagation
The consequences of TRIM28 chromatin recruitment are still being unraveled. A high-H3K9me3, low-histone acetylation state as a result of SETDB1 and CHD3 activities reduces RNA polymerase II binding (Groner et al. 2010; Sripathy et al. 2006). Moreover, the HP1β protein binds to nearby H3K9me3 marks and recruits additional heterochromatin-inducing factors (Kwon and Workman 2011). This could enable TRIM28-mediated silencing to spread over long distances—possibly several kilobases (kb)—from the original site of repressor binding (Groner et al. 2010). Indeed, TRIM28 is shown to bind intragenic loci many kb from promoters, including the 3′ ends of genes (Iyengar et al. 2011). Another consequence of TRIM28 recruitment is de novo DNA methylation. TRIM28 and the KRAB protein Zfp57 implement the DNA methylation pattern underlying gene imprinting, the phenomenon of monoallelic gene expression in plants and mammals (Quenneville et al. 2011; Li et al. 2008; Zuo et al. 2012). DNA methylation has also been observed in a conditional gene regulation system, whereby TRIM28 is artificially targeted to a particular gene in a doxycycline-dependent manner. In this system, a KRAB domain is fused to the Escherichia coli tetracycline repressor (tTRKRAB) and co-transduced with the target gene under the control of tetracycline operator (tetO) sequences (Szulc et al. 2006; Herchenroder et al. 1999). Using this system, TRIM28 delivers irreversible transcriptional silencing if induced during the first few days of mouse development, due to the establishment of repressive promoter DNA methylation (Wiznerowicz et al. 2007). However, introduction of repressive marks using the tTRKRAB system appears to be reversible when introduced into a variety of differentiated human and mouse cells, mouse ES cells, and in adult mice (Szulc et al. 2006; Groner et al. 2012), suggesting that the nature of the repression complex might vary in a cell-type- and temporal-specific manner. Indeed, TRIM28-mediated repression and subsequent DNA methylation appears to be irreversible in mouse ES cells when the target is an endogenous retrovirus (Rowe et al. 2013a).
Surprisingly, relatively few targets of TRIM28 are known, despite a prevalence of binding sites. Genome-wide chromatin immunoprecipitation followed by sequencing (ChIP-seq profiling) suggests ~7,000 TRIM28-binding sites in the human genome (O’Geen et al. 2007), yet the expression of relatively few host genes is affected by TRIM28 depletion in cell lines (Iyengar et al. 2011; Groner et al. 2010). For example, it has been postulated that enrichment of TRIM28 and subsequent heterochromatin formation at genes encoding KRAB-ZFPs protects these loci from deleterious recombination; KRAB-ZFP genes are highly similar as a result of gene duplication (Emerson and Thomas 2009; Vogel et al. 2006). However, TRIM28 depletion only weakly affects KRAB-ZFP expression levels in cell lines. Moreover, upon TRIM28 depletion in cell lines (Iyengar et al. 2011) or deletion in ES cells (Rowe et al. 2013b), some gene transcripts are downregulated, while others are upregulated, suggestive of a role for TRIM28 in more than just transcriptional repression. This ambiguity in gene targets is in part explained by the recent finding that TRIM28 underlies an ES-specific restriction of endogenous retroviruses (ERV).
3.2 TRIM28 Mediates Repression of Endogenous Retroviruses in Embryonic Stem Cells
Nearly half of human and mouse genomes are derived from LTR (~8 %)- and non-LTR (~35 %)-containing retroelements. For example, there are around 2,500 intracisternal A-type particle (IAP) elements (a so-called class II ERV) in the mouse genome (Zhang et al. 2008). Although the majority of ERVs have accumulated mutations that render them unable to retrotranspose, many ERV LTRs represent cis-acting regulatory elements that can influence the expression of neighboring genes and require silencing (Rowe et al. 2013b). Similarly, active retroelements must be regulated in order to avoid insertional mutagenesis. Interestingly, TRIM28-knockout mice are embryonic lethal as they fail to gastrulate (Cammas et al. 2000). Furthermore, zygotic heterozygous Trim28 embryos, with just maternal Trim28 deleted, die before birth due to demethylation of various promoters (Messerschmidt et al. 2012). Wild-type embryos undergo massive genome-wide demethylation after fertilization, followed by a program of selective remethylation (Feng et al. 2010). To prevent spurious gene and ERV activation during the wave of demethylation, repression is imparted by histone methylation. It was recently shown that ERV LTRs and 5′UTRs are decorated with H3K9me3, TRIM28, and SETDB1 in mouse embryonic stem (ES) cells but not in differentiated embryonic fibroblasts (MEF) (Mikkelsen et al. 2007; Matsui et al. 2010). A conditional knockout mouse revealed the importance of TRIM28 in this process, as loss of TRIM28 in mouse ES cells simultaneously removes H3K9me3 marks and SETDB1 binding from IAP promoters (Rowe et al. 2010). Similarly, H3K9me3 is removed following deletion of SETDB1 in ES cells while having no impact on TRIM28 docking at DNA (Matsui et al. 2010), confirming SETDB1 as the downstream effector of a TRIM28-mediated histone methylation of ERVs. Accordingly, deletion of either of these factors causes large increases in both class I (MLV) and class II (IAP and MusD) ERV mRNAs in mouse ES cells but not in differentiated MEFs (Matsui et al. 2010; Rowe et al. 2010). Strikingly, IAP mRNA was upregulated by ~500-fold in TRIM28-deleted embryos (Rowe et al. 2010), perhaps underlying the embryonic lethality of TRIM28-knockout mice. Indeed, TRIM28 deletion caused a proportional increase in IAV DNA, indicating that activated ERVs are competent for reverse transcription and integration (Matsui et al. 2010), a known mechanism for genome mutagenesis (Zhang et al. 2008). Furthermore, single-stranded DNA derived from reverse transcribing endogenous retroviruses has the potential to trigger pattern recognition (Stetson et al. 2008), placing TRIM28 in a critical position in the regulation of host innate immune responses.
TRIM28-mediated histone methylation and transcriptional repression occur upstream of DNA methylation, as enrichment of SETDB1 and H3K9me3 at ERVs was unaffected by triple deletion of the three mammalian DNA methyltransferases DNMT1, DNMT3a, and DNMT3b (Matsui et al. 2010). However, TRIM28 recruitment rapidly leads to de novo DNA methylation at ERVs, which becomes critical for their silencing later in embryogenesis (Rowe et al. 2013a). TRIM28’s target within ERVs in ES cells maps to the 5′UTR, as this ~500-bp region cloned from derepressed IAP cDNAs in TRIM28-deleted ES cells, confers repression of a heterologous promoter in a position-independent manner (Rowe et al. 2010). Therefore, it would seem that a TRIM28, KRAB-ZFP repressor complex mediates an essential histone and DNA methylation of ERV 5′UTRs in mouse ES cells that sustains cell viability. It transpires that the same mechanism underlies a potent restriction of exogenous MLV transcription in the same cells.
3.3 TRIM28 is Responsible for the Stem Cell–Specific Restriction of Murine Leukemia Viruses
Of a panel of RNA and DNA viruses tested for their abilities to replicate in undifferentiated murine embryonal carcinoma (EC) cells, MLVs were unique in their inability to establish a productive infection (Teich et al. 1977). The restriction was shown to occur in the absence of viral proteins and was subsequent to integration. Production of viral RNA was limiting, implying a block to transcription or viral RNA stability (Teich et al. 1977; Loh et al. 1988). Importantly, the restriction was lost following cell differentiation (Teich et al. 1977). De novo DNA methylation (Stewart et al. 1982) and poor enhancer activity of the 5′ LTR (Feuer et al. 1989) were among several mechanisms initially demonstrated to contribute to the restriction in EC cells. However, another critical determinant of restriction was found to reside within the MLV 18 base pair proline transfer RNA (tRNAPro) primer-binding site (PBSPro) (Feuer et al. 1989; Barklis et al. 1986; Kempler et al. 1993). Substitution for a different PBS, complementary to tRNAGln (PBSGln), rescued MLV from restriction (Petersen et al. 1991).
The PBS makes a particularly effective target for restriction, as it is complementary to the 3′ end of a specific cellular tRNA. The tRNA is recruited to the viral RNA to act as a primer for reverse transcription. Thus, the PBS sequence is dictated by the tRNA, making escape by mutation difficult. This so-called repressor-binding site (RBS) was shown to confer restriction in an orientation- and position-independent manner, against MLV or heterologous promoters (Petersen et al. 1991; Loh et al. 1990), suggesting that the restriction was DNA rather than RNA mediated. Furthermore, DNA-binding factors were suspected based on the observation that competitive DNA bearing the M-MLV RBS could alleviate restriction (Loh et al. 1988). Similarly, exonuclease III protection assays and electrophoretic mobility shift assays (EMSA), using an M-MLV PBS for a probe, revealed that DNA binding was enriched in EC nuclear extracts (Petersen et al. 1991; Loh et al. 1990). Selection for rare escape mutants identified a single-point mutation within the RBS—referred to as the B2 RBS—that bypassed restriction in EC cells (Barklis et al. 1986), and was not bound by RBS-specific factors in EC EMSA or DNA protection experiments (Petersen et al. 1991; Loh et al. 1990). Collectively, these findings paved the way for identification of the nuclear factor/s that bound the MLV provirus to prevent transcription. In 2007, thirty years after the phenotype was first described, TRIM28 was identified by mass spectrometry (MS) as the EC-specific component of the restriction complex (Wolf and Goff 2007). Antibodies against TRIM28 were able to supershift the PBS-binding complex in EMSAs, while cell differentiation saw a decline in TRIM28 protein expression concurrent with the loss in probe binding by EMSA, explaining the differentiation state specificity of the restriction phenotype (Wolf and Goff 2007). Depletion of TRIM28 in EC or ES cells alleviated restriction of MLV, while ChIPs revealed enrichment of TRIM28 at an MLV proviral WT PBSPro, but not a B2 PBSPro, revealing the mechanism behind the escape of the B2 mutant virus.
Given this handle on the molecular composition of the MLV restriction complex, the identity of the KRAB-ZFP protein that bound the MLV PBS was revealed to be ZFP809 (Wolf and Goff 2009). EMSA was used to isolate the complex, which was cross-linked to the PBS probe, immunoprecipitated with anti-TRIM28 antibodies, resolved by SDS-PAGE and bands identified by mass spectrometry (Wolf and Goff 2009). Of three candidate proteins, only recombinant GST-tagged ZFP809 bound the MLV PBS probe in EMSA (Wolf and Goff 2009). As with TRIM28, ZFP809 depletion from EC cells attenuates restriction, while ChIP reveals enrichment of ZFP809 at the proviral PBSPro (Wolf and Goff 2009). Furthermore, the MLV restriction phenotype of the expected specificity (i.e., targeting WT PBSPro but not PBSGln) could be introduced to 293 cells by over-expression of ZFP809, and reversed by TRIM28 depletion (Wolf and Goff 2009). As expected from the model of TRIM28–KRAB-ZFP–mediated repression, TRIM28 binding to the MLV PBS is independent of its ability to bind HP1γ, although TRIM28 PxVxL mutants are unable to restrict MLV (Wolf et al. 2008a). Indeed, HP1γ was also enriched at the MLV PBSPro (Wolf and Goff 2007). Although we do not yet have a complete picture of TRIM28-mediated restriction at the PBS, the silencing of PBSPro in endogenous retroviruses (ERV), by de novo DNA methylation, was recently shown to require TRIM28 and SETDB1 (Rowe et al. 2013a), strongly suggesting TRIM28 mediated restriction mechanisms behind repression of both exogenous and endogenous retroviruses are comparable in nature.
3.4 TRIM28-Mediated Restriction of Complex Retroviruses
In a screen to identify other retroviral PBS sequences bound by the EC cell restriction complex, PBSLys-1,2, as used by spumavirus, maedi-visna virus and Mason-Pfizer monkey virus, was comparably bound to PBSPro in gel shift assays (Yamauchi et al. 1995), by a complex containing TRIM28 (Wolf et al. 2008b). Indeed, substitution of PBSPro in MLV for PBSLys-1,2 demonstrates that this PBS is also able to confer repression of viral transcription (Yamauchi et al. 1995), suggesting PBSLys-1,2-using retroviruses might also suffer silencing in EC cells. However, there is no cross-competition between PBSPro and PBSLys-1,2 probes in EMSA experiments, meaning that TRIM28 is recruited to PBSLys-1,2 by an alternative KRAB-ZFP protein (Yamauchi et al. 1995; Wolf et al. 2008b), the identity of which is not yet known. Human T cell leukemia virus 1 (HTLV-1) also uses tRNAPro for replication and is restricted in 293 cells expressing ectopic ZFP809 (Wolf et al. 2008b); interestingly, restriction was not overcome in this setting by the HTLV-1 trans-activator Tax (Wolf et al. 2008b), suggesting TRIM28 might contribute to HTLV-1 tropism.
TRIM28 has not been shown to mediate repression of the HIV-1 LTR, although there are several circumstantial observations that suggest it might. First, ectopic expression of the KRAB-ZFP ZBRK1/ZNF350 represses HIV-1 LTR-driven luciferase activity and its depletion proportionally augments HIV-1 LTR activity in 293T cells or the T cell line MT-4 (Nishitsuji et al. 2012). A target for ZBRK1 activity was mapped to the U3 region of the HIV-1 LTR; ZBRK1 bound an LTR-containing probe in EMSA and was enriched at stably integrated LTRs as shown by ChIP (Nishitsuji et al. 2012). Furthermore, the repression activity of ZBRK1 was dependent on TRIM28, as implicated by KRAB mutations that inhibit TRIM28 binding, or depletion of TRIM28. Furthermore, although the HDAC inhibitor trichostatin A (TSA) inhibited ZBRK1-mediated repression, depletion of either SETDB1 or HP1 did not (Nishitsuji et al. 2012), suggesting that TRIM28 might mediate repression of HIV-1 through histone deacetylation and not establishment of an H3K9me3-rich heterochromatin. However, it should be noted that HDAC inhibitors are effective activators of the HIV-1 LTR (Van Lint et al. 1996), and TSA treatment reduces HP1γ expression (Bartova et al. 2005), suggesting that the effects of TSA and ZBRK1 might be antagonistic and unrelated. Nonetheless, depletion of endogenous ZBRK1 also diminished a modest enrichment of TRIM28 at the HIV-1 LTR as shown by ChIP (Nishitsuji et al. 2012). Similarly, weak repression of the HIV-1 LTR has been demonstrated with the macrophage-specific KRAB-ZFP protein OTK18/ZNF175 (Carlson et al. 2004), although the effect was not abrogated by deletion of its two KRAB domains. Second, artificial targeting of KRAB domains from KOX1—a human KRAB-ZFP known to bind TRIM28 (Moosmann et al. 1996)—to the HIV-1 genome by fusing them to GAL4 or tetR DNA-binding domains inhibits HIV-1 Tat-dependent transcription in a dose-dependent manner (Herchenroder et al. 1999). In these experiments, repression was observed when KRAB binding was 6 kb from the 5′ LTR, consistent with the ability of TRIM28 to mediate long-range repression. Similarly, downregulation of HIV-1 transcription was seen with a KRAB-Tat chimera (Pengue et al. 1995), although using Tat as the DNA-targeting domain makes interpretation less straightforward. More recently, custom-designed KRAB-ZFPs that recognize endogenous HIV-1 genomic sequences were shown to levy substantial transcriptional repression in reporter assays in stimulated T cells and to restrict full-length HIV-1 replication (Reynolds et al. 2003; Segal et al. 2004), providing a proof-of-concept for the design of artificial restriction factors based on a TRIM28-recruiting KRAB domain. Third, latent HIV-1 proviruses are enriched for HP1γ and H3K9me3, and HP1γ depletion enhances both basal- and Tat-mediated transcription from an integrated LTR (du Chene et al. 2007). Fourth, HIV-1 bears a PBS complementary to cellular tRNALys-3, which mediates modest restriction when substituted for PBSPro in a recombinant MLV (Yamauchi et al. 1995). A subset of MusD retroelements, possibly targeted by mouse TRIM28, also use PBSLys-3 (Rowe et al. 2010). Together these experiments suggest that HIV-1 proviruses might be targets of KRAB-ZFP proteins and are enriched for heterochromatin signatures. It remains to be shown conclusively whether TRIM28 specifically regulates HIV-1 transcription.
3.5 TRIM28 Restricts HIV-1 Integration by Facilitating Integrase Deacetylation
A different model for TRIM28 restriction has been proposed, whereby TRIM28 induces deacetylation of HIV-1 Integrase (IN) to decrease integration efficiency. This finding is based on the observation that HIV-1 IN is acetylated at lysines in its C-terminal domain, by histone acetyl transferases (HAT) GCN5 or p300, and this modification is required for optimal integration efficiency (Cereseto et al. 2005; Terreni et al. 2010). However, the significance of HIV-1 IN acetylation is contentious, not least because substitution of purported target lysine residues in IN appears to impact on HIV-1 replication in a manner dependent on the whether IN has been epitope tagged (Terreni et al. 2010; Topper et al. 2007). Nonetheless, a yeast two-hybrid (Y2H) screen for human lymphocyte proteins that bind a constitutively acetylated IN-HAT chimera revealed TRIM28 as an IN interaction partner (Terreni et al. 2010; Allouch and Cereseto 2009; Allouch et al. 2011). Potentially, the TRIM28 bromodomain recognizes acetyl-lysine residues in IN, although structural studies of the tandem PHD–bromodomain suggest this binding affinity has been lost in TRIM28 (Zeng et al. 2008). Over-expression of TRIM28 decreases HIV-1 integrant numbers in single-cycle assays, while depletion of TRIM28 in primary lymphocytes increased the number of integrants by threefold (Allouch et al. 2011). Histone deacetylase 1 (HDAC1), able to deacetylate IN in vitro, was found to coimmunoprecipitate with the IN–TRIM28 complex (Allouch et al. 2011). Therefore, TRIM28 might recruit HDAC1—as it does the NuRD complex (see above)—to eliminate IN acetylation and integration efficiency. In support of this model, treatment of cells with HDAC inhibitors, or depletion of HDAC1, increases integration in restrictive cells. However, this observation is at odds with a described role for HDAC1 in HIV-1 reverse transcription (Sorin et al. 2009). Moreover, GCN5 depletion has little impact on HIV-1 integration (Terreni et al. 2010). Thus, this proposed role for TRIM28 in HIV-1 restriction requires further investigation.
3.6 TRIM28-Related Protein TRIM24 Represses ERV Transcription in Hepatocytes
TRIM28 is highly similar to two other TRIMs, TRIM24/TIF1α and TRIM33/TIF1γ, a TRIM subfamily sharing the RBCC–PHD–bromodomain architecture. In mouse hepatocytes, these proteins hetero-oligomerize to form complexes that antagonize transcriptional activation by the all-trans retinoic acid receptor (RAR) α. Genetic disruption of these TRIM complexes leads to large-scale transcriptional upregulation and ultimately hepatocellular carcinoma (HCC) (Khetchoumian et al. 2007; Herquel et al. 2011). Interestingly, it was recently shown that among transcripts elevated in TRIM24-knockout hepatocytes were several host ISGs, suggesting either that TRIM24 might repress ISG transcription directly or perhaps like TRIM28, it represses ERV transcription and subsequent reverse transcription—a known trigger of pattern recognition and autoimmunity (Stetson et al. 2008). Although the TRIM28-sensitive class II ERV IAPs were not expressed in TRIM24-knockout hepatocytes, a particular class of mouse retrotransposons called VL30 ERVs were strongly derepressed specifically in liver tissue (Herquel et al. 2013). Notably, these VL30 ERVs contain all-trans retinoic acid response elements in their LTRs, suggesting that TRIM24 mediates their transcriptional repression through its inhibition of RARα activity. Moreover, in a manner reminiscent of TRIM28-mediated restriction, TRIM24 binding is enriched at VL30 LTRs (Herquel et al. 2013).
Whether this example of ERV silencing by TRIM24 occurs in other species remains to be seen, and the precise mechanism of restriction requires further delineation. Nonetheless, these findings suggest that this subfamily of TRIM proteins has been employed repeatedly for retroviral restriction and the prevention of autoimmunity, which might even contribute to the oncogenic nature of TRIM24 loss. Indeed, derepressed VL30 cDNAs in TRIM24-knockout hepatocytes coincide with a modest upregulation of the ISG IRF9 (Herquel et al. 2013). In a more comprehensive screen, loss of TRIM24 function in HCC was shown to correlate with widespread upregulation of IFN signaling pathway components and ISGs (Tisserand et al. 2011)—for example Ifit genes, Gbp genes, Herc5, Isg15, Ifi204 and weak upregulation of other known retroviral restriction factors Samhd1 and Mov10—possibly exacerbated by accumulation of cytosolic retroviral cDNAs. Although TRIM24 dose not interact with KRAB domains (Abrink et al. 2001; Peng et al. 2000), its PHD and bromodomain are able to recognize histone lysines and acetyl-lysines (Tsai et al. 2010), suggesting TRIM24 likely regulates transcriptional control in a distinct manner to TRIM28. Given what we now understand about TRIM28 and TRIM24 biology, the implication is that the related protein TRIM33 might also restrict a specific set of endogenous retroviruses in a tissue-specific manner.
4 TRIM22 Represses HIV-1 LTR Transcription and is a Correlate of Viral Control in HIV-1-Infected Patients
TRIM22, also called stimulated transactivating factor of 50 kDa (Staf50), was first cloned from a library of type I IFN-upregulated cDNAs in human lymphoblastoid Daudi cells (Tissot and Mechti 1995). Like TRIM5, TRIM22 expression is strongly stimulated by type 1 interferon treatment in several cell types (Gao et al. 2009) and expression is elevated following HIV-1 infection of PBMCs (Singh et al. 2011). TRIM22 also has a PRYSPRY domain, which like TRIM5α’s PRYSPRY is highly variable between species. Indeed, phylogenetic analyses reveal that TRIM22 has evolved through periodic episodes of positive selection (Sawyer et al. 2007). Such a pattern of evolution is strongly suggestive of genetic conflict with pathogens and supports the hypothesis that TRIM22 is a restriction factor. Indeed, the positions of positively selected amino acids in TRIM22, concentrated in the PRYSPRY, are strikingly similar to the amino acid positions in TRIM5α, which determine restriction specificity in primates (see TRIM5).
TRIM22 was first recognized by its sequence homology to murine Rpt-1 (Tissot and Mechti 1995; Patarca et al. 1988), also called TRIM30α, which is one of several mouse paralogues of human TRIM5 (Tareen et al. 2009). Based on this homology and in light of a modest repression by murine TRIM30α on transcription from the HIV-1 LTR (Patarca et al. 1988), human TRIM22 was shown to mediate modest restriction against the HIV-1 LTR (Tissot and Mechti 1995). Further, type I IFN-restricted infection of HIV-1 in Jurkat cells is counteracted by TRIM22 depletion. In other studies, TRIM22 was shown to underlie a difference in HIV-1 permissivity between two clones of monocytic U937 cells (Franzoso et al. 1994; Kajaste-Rudnitski et al. 2011). While various factors had been proposed to underlie the phenotype, including lysosomal serine proteases (Franzoso et al. 1994), poor fusogenic activity in the non-permissive clone (Moriuchi et al. 1997), and possibly HIV-1 cellular coreceptor CXCR4 expression (Biswas et al. 1998), the cause of restriction in the non-permissive U937 clone remained ill defined. However, the non-permissive clone became permissive to HIV-1 replication on depletion of its constitutively expressed TRIM22, while the permissive clone became non-permissive on ectopic expression of TRIM22 (Kajaste-Rudnitski et al. 2011). TRIM22 represses basal transcription from the LTR and, unlike TRIM28-mediated repression of HTLV-1 transactivation by Tax (Wolf and Goff 2009), TRIM22 cannot overcome Tat-stimulated transcription. TRIM22 repression is also independent of the HIV-1 LTR NF-κB sites, but it does moderately interfere with the ability of TNFα, or a combination of the phorbal ester PMA and ionomycin, to stimulate HIV-1 transcription, suggesting that TRIM22 might interfere with specific signaling cascades, calcium regulation, or the binding of transcription factors like AP-1, NFAT, or Sp1 to the HIV-1 LTR. Indeed, TRIM22 activity against HIV-1 might be indirect, as, again like TRIM5α, it is able to selectively induce innate signaling pathways when over-expressed (Yu et al. 2011).
In vivo support for an association between HIV-1 replication and TRIM22 expression comes from the observation that TRIM22 transcripts are elevated in HIV-1-positive patients versus controls, and their levels positively correlate with type I IFN levels (Singh et al. 2011). In the same cohort, TRIM22 levels inversely correlated with HIV-1 plasma viral load and positively correlated with CD4 + T cell count, findings that suggest the protein may be involved in control of viral replication but may simply illustrate that TRIM22 expression levels act as a marker of type I IFN levels. It is interesting that there is also evidence for TRIM22 being a transcriptional repressor at the hepatitis B virus core promoter (Gao et al. 2009), ubiquitinating the 3C protease of encephalomyocarditis virus (Eldin et al. 2009) and ubiquitinating influenza A virus nucleoprotein (Di Pietro et al. 2013).
The mechanism of TRIM22-mediated restriction of HIV-1 remains undefined. It is apparently RING independent (Kajaste-Rudnitski et al. 2011), suggesting that repression does not require ubiquitin conjugation E2 enzyme–binding activity. Unlike TRIM28, TRIM22 is not a well-characterized transcriptional corepressor, and it lacks the SUMO-regulated PHD and bromodomains that in TRIM28 recruit chromatin-remodifying enzymes (Ivanov et al. 2007) (see TRIM28). Presumably, the TRIM22 PRYSPRY forms a protein–protein interface, as it does in TRIM5α. However, TRIM22 is predominantly nuclear (Kajaste-Rudnitski et al. 2011) and localizes to nuclear bodies (Sivaramakrishnan et al. 2009), as does another nuclear antiviral TRIM protein, TRIM19/PML (Reichelt et al. 2011), although these may be different nuclear bodies. It is interesting that TRIM22′s PRYSPRY appears to regulate nuclear localization (Sivaramakrishnan et al. 2009; Herr et al. 2009; Gao et al. 2009; Yu et al. 2011), in conjunction with a bipartite NLS located between the coiled coil and PRYSPRY (Tissot and Mechti 1995). Like TRIM19, TRIM22 nuclear localization is required for its restriction activity (Reichelt et al. 2011; Gao et al. 2009). In particular, residues Val493 and Cys494 at the C terminus of human TRIM22 regulate nuclear body formation, potentially by mediating interaction with other nuclear body proteins. Whether the PRYSPRY is necessary for the observed repression of the HIV-1 LTR is an important question that remains to be answered.
Finally, TRIM22 has been reported to inhibit HIV-1 particle release in a variety of type I IFN stimulated cell lines, although only weak effects on HIV-1 replication were described (Barr et al. 2008; Ohmine et al. 2011). In fact, HIV-1 Gag downregulation from plasma membranes has been described following over-expression of a variety of TRIMs (Ohmine et al. 2011; Uchil et al. 2008), but the phenotypes are comparably weak, suggesting artifacts of transient over-expression.
5 Do Other TRIM Family Proteins Inhibit Retroviral Replication?
Of the ~100 human TRIM genes, only the four discussed have been described as having any significant impact on the life cycles of retroviruses. Do any other TRIM proteins counteract retroviruses? TRIM proteins cluster into discrete monophyletic clades (Han et al. 2011; Marin 2012), the members of which share domain organization (Ozato et al. 2008). Where one clade member is antiviral, it is tempting to speculate that other members might also have evolved immune activity. The assumption is not entirely unfounded, as TRIM5 and TRIM22, paralogues on human chromosome 11p15, have both evolved by positive selection in their PRYSPRY domains and both possess antiretroviral activities, albeit with apparently unrelated mechanisms. Moreover, TRIM21, the most distantly related TRIM in this cluster, is a restriction factor in a similar vein to TRIM5α. TRIM21 targets an early stage of adenoviral replication, although this is through recruitment of intracellular neutralizing antibodies attached to incoming viral particles rather than through recruitment of the viral capsid itself (Mallery et al. 2010).
An obvious obstacle in the search for novel antiretroviral function is having a sensitive virus and a replication assay in hand. Nonetheless, expression screens have been used to uncover retroviral restriction activities in other TRIM proteins. In one screen, 36 human TRIMs were co-transfected with MLV or HIV-1 vectors and 13 of these decreased viral titre in the supernatants of TRIM-expressing cells (Uchil et al. 2008). Human TRIM8, 15, 19, 25, 26, 28, and 35 reduced MLV titre; human TRIM15, 26, and 32 reduced HIV-1 titre. Although the restriction mechanisms have not yet been fully examined, evidence suggests restriction by these TRIM proteins might proceed via novel mechanisms. For example, TRIM15 restriction of MLV was shown to require only its B-Box2, which can immunoprecipitate the MLV Gag polyprotein (Uchil et al. 2008).
TRIM19/PML is a component of nuclear bodies and is thought to be a restriction factor against various RNA and DNA viruses. PML has been suggested to exit the nucleus upon HIV-1 infection and interact with the HIV-1 preintegration complex (PIC) (Turelli et al. 2001), but this work has not yet been replicated. TRIM32 was identified by yeast two-hybrid as a binding partner of HIV-1 Tat (Fridell et al. 1995) and has since been shown to target STING/MITA, the signal adaptor, for K63-linked ubiquitination, enhancing type I IFN production (Zhang et al. 2012). TRIM25 stimulates another signaling protein, the triphosphorylated viral RNA receptor retinoic acid-inducible gene 1 (RIG-I), also polyubiquitinating it with K63-linked chains (Gack et al. 2007). A growing number of TRIM proteins are thought to influence signaling pathways through their E3 ligase activity (Kawai and Akira 2011; McNab et al. 2011), and a large number are shown to interact with ubiquitination machinery (Napolitano et al. 2011). Moreover, stimulation of innate signaling pathways by TRIM over-expression has been demonstrated for a large number of TRIM members (Versteeg et al. 2013), which in some cases might contribute to restriction activities, as suggested for MLV restriction by TRIM1 and TRIM62 (Uchil et al. 2013). TRIM1 has been shown to be both a weak (Yap et al. 2004) and strong (Uchil et al. 2008) CA-specific MLV restriction factor in similar assays, although TRIM1 depletion in human cells had no effect on the infectivity of a wide panel of retroviruses (Zhang et al. 2006), arguing against an important antiviral role. Significantly more TRIMs affect MLV replication over HIV-1 in these screens, which has been ascribed to cis-acting sequence differences between the two viral LTRs (Uchil et al. 2013). Specifically, NFκB sites in the HIV-1 LTR might confer sensitivity to the signaling induced by various TRIMs (Uchil et al. 2013). Whether this elaborate mechanism turns out to explain other retroviral tropism remains to be seen. However, the generation of an antiviral state by many TRIM family proteins might mean that a far larger number of these have antiretroviral activity than we are currently aware.
6 Concluding Remarks
The recurrent evolutionary adoption of TRIM proteins for defense against diverse retroviruses is a testament to the versatility and utility of this protein superfamily. Yet even within the small number of TRIM proteins that have been well characterized to target retroviral replication, there is significant variation in antiviral mechanism. For example, there appears to be a differential requirement for enzymatic activity. TRIM5α requires RING-dependent E3 ligase activity for complete restriction, while TRIM22 does not. However, a role for ubiquitination in restriction by either of these proteins has not been formally demonstrated. TRIM28 requires SUMO ligase activity, but the E3 is its C-terminal PHD rather than N-terminal RING domain (Zeng et al. 2008). The TRIM28 RBCC is necessary for interaction with a KRAB-ZFP, but the role of the RING in this relationship is not yet defined. TRIM24 binds ERV LTRs and cellular genes bearing all-trans RARα response elements, interfering with RAR activity, and in this manner indirectly inhibits VL30 ERV transcription (Herquel et al. 2013). Whether TRIM24 catalytic activity contributes to RAR antagonism is not yet known. Clearly, there is much scope for further research into the role of TRIM enzymatic activity in restriction.
TRIM5 and TRIM22 demonstrate signs of positive selection that, in the case of TRIM5, is a result of direct contact with an evolving viral protein (Sawyer et al. 2007). The TRIM22 PRYSPRY has not yet been implicated in restriction per se, although it does seem to influence the protein’s cellular localization (Gao et al. 2009). TRIM28 instead interacts exclusively with host factors and is a constitutive mediator of host transcriptional regulation, suggesting purifying selection has dominated its evolution. Indeed, it is the KRAB-ZFPs that evolve through positive selection in their DNA-binding domains (Rowe et al. 2010), indicative of an interaction with viral targets. Potentially, the regulatory KRAB-ZFP network evolved primarily as an antiviral defense before its adoption by the host for constitutive gene control.
The involvement of TRIMs in signaling cascades also appears to be a feature of the family that remains understudied. TRIM5 and perhaps TRIM22 are able to regulate specific signaling cascades and thereby contribute to the antiviral state in an indirect manner. An increasing number of TRIMs have been shown to possess similar activities (Kawai and Akira 2011). Whether TRIM22, like TRIM5, triggers innate signaling upon restriction has not yet been shown, but will require further understanding of the TRIM22 restriction mechanism. TRIM24 directly binds and represses the Stat1 promoter, which contains RAR response elements, and thus the expression of many STAT1-regulated ISGs is upregulated upon TRIM24 depletion (Tisserand et al. 2011), although depression of VL30 ERVs might also contribute to this. Whether TRIM28 suppresses the stimulation of innate immunity due to prevention of cytoplasmic retroviral cDNA accumulation remains to be seen. Or perhaps, TRIM28 might trigger and potentiate an antiviral state upon recognition of recently endogenized retroviruses. That TRIM22, TRIM24, and TRIM28 are nuclear proteins reinforces the notion that any effect they have on innate signaling will be quite distinct from that of cytoplasmic TRIM5α.
TRIM5 and TRIM22 appear to be largely autonomous restriction factors, able to confer a restriction phenotype upon permissive cells (Kajaste-Rudnitski et al. 2011; Stremlau et al. 2004). Presumably, this is because their cofactors for restriction, for example the ubiquitination machinery for TRIM5, are ubiquitous in eukaryotic cells. Exceptions have been suggested, however—TRIM5 might be a less effective restriction factor in 293T cells owing to suboptimal expression of SUMO-1 (Arriagada et al. 2011). Conversely, TRIM28 requires expression of the correct KRAB-ZFP in order to target a retrovirus (Wolf and Goff 2009), suggesting the specificity of its repressive activity can be redirected by altering expression of various KRAB-ZFPs. Custom-designed KRAB-ZFPs might represent a future avenue of therapy for the repression of exogenous retroviruses like HIV-1.
References
Abrink M, Ortiz JA, Mark C, Sanchez C, Looman C, Hellman L, Chambon P, Losson R (2001) Conserved interaction between distinct Kruppel-associated box domains and the transcriptional intermediary factor 1 beta. Proc Natl Acad Sci U S A 98(4):1422–1426
Allouch A, Cereseto A (2009) Identification of cellular factors binding to acetylated HIV-1 integrase. Amino Acids 41:1137–1145
Allouch A, Di Primio C, Alpi E, Lusic M, Arosio D, Giacca M, Cereseto A (2011) The TRIM family protein KAP1 inhibits HIV-1 integration. Cell Host Microbe 9(6):484–495
Arriagada G, Muntean LN, Goff SP (2011) SUMO-interacting motifs of human TRIM5α are important for antiviral activity. PLoS Pathog 7(4):e1002019
Asaoka K, Ikeda K, Hishinuma T, Horie-Inoue K, Takeda S, Inoue S (2005) A retrovirus restriction factor TRIM5α is transcriptionally regulated by interferons. Biochem Biophys Res Commun 338(4):1950–1956
Barklis E, Mulligan RC, Jaenisch R (1986) Chromosomal position or virus mutation permits retrovirus expression in embryonal carcinoma cells. Cell 47(3):391–399
Barr SD, Smiley JR, Bushman FD (2008) The interferon response inhibits HIV particle production by induction of TRIM22. PLoS Pathog 4(2):e1000007
Barreiro LB, Quintana-Murci L (2010) From evolutionary genetics to human immunology: how selection shapes host defence genes. Nat Rev Genet 11(1):17–30
Bartova E, Pachernik J, Harnicarova A, Kovarik A, Kovarikova M, Hofmanova J, Skalnikova M, Kozubek M, Kozubek S (2005) Nuclear levels and patterns of histone H3 modification and HP1 proteins after inhibition of histone deacetylases. J Cell Sci 118(Pt 21):5035–5046
Bellefroid EJ, Poncelet DA, Lecocq PJ, Revelant O, Martial JA (1991) The evolutionarily conserved Kruppel-associated box domain defines a subfamily of eukaryotic multifingered proteins. Proc Natl Acad Sci USA 88(9):3608–3612
Biris N, Yang Y, Taylor AB, Tomashevski A, Guo M, Hart PJ, Diaz-Griffero F, Ivanov DN (2012) Structure of the rhesus monkey TRIM5alpha PRYSPRY domain, the HIV capsid recognition module. Proc Natl Acad Sci USA 109(33):13278–13283
Biswas P, Mengozzi M, Mantelli B, Delfanti F, Brambilla A, Vicenzi E, Poli G (1998) 1,25-Dihydroxyvitamin D3 upregulates functional CXCR4 human immunodeficiency virus type 1 coreceptors in U937 minus clones: NF-kappaB-independent enhancement of viral replication. J Virol 72(10):8380–8383
Black LR, Aiken C (2010) TRIM5alpha disrupts the structure of assembled HIV-1 capsid complexes in vitro. J Virol 84(13):6564–6569
Boudinot P, van der Aa LM, Jouneau L, Du Pasquier L, Pontarotti P, Briolat V, Benmansour A, Levraud JP (2011) Origin and evolution of TRIM proteins: new insights from the complete TRIM repertoire of zebrafish and pufferfish. PLoS One 6(7):e22022
Boutell C, Orr A, Everett RD (2003) PML residue lysine 160 is required for the degradation of PML induced by herpes simplex virus type 1 regulatory protein ICP0. J Virol 77(16):8686–8694
Brennan G, Kozyrev Y, Hu SL (2008) TRIMCyp expression in old world primates Macaca nemestrina and Macaca fascicularis. Proc Natl Acad Sci USA 105(9):3569–3574
Caines ME, Bichel K, Price AJ, McEwan WA, Towers GJ, Willett BJ, Freund SM, James LC (2012) Diverse HIV viruses are targeted by a conformationally dynamic antiviral. Nat Struct Mol Biol 19(4):411–416
Cammas F, Mark M, Dolle P, Dierich A, Chambon P, Losson R (2000) Mice lacking the transcriptional corepressor TIF1beta are defective in early postimplantation development. Development 127(13):2955–2963
Campbell EM, Dodding MP, Yap MW, Wu X, Gallois-Montbrun S, Malim MH, Stoye JP, Hope TJ (2007) TRIM5 alpha cytoplasmic bodies are highly dynamic structures. Mol Biol Cell 18(6):2102–2111
Campbell EM, Perez O, Anderson JL, Hope TJ (2008) Visualization of a proteasome-independent intermediate during restriction of HIV-1 by rhesus TRIM5alpha. J Cell Biol 180(3):549–561
Carlson KA, Leisman G, Limoges J, Pohlman GD, Horiba M, Buescher J, Gendelman HE, Ikezu T (2004) Molecular characterization of a putative antiretroviral transcriptional factor, OTK18. J Immunol 172(1):381–391
Carthagena L, Bergamaschi A, Luna JM, David A, Uchil PD, Margottin-Goguet F, Mothes W, Hazan U, Transy C, Pancino G, Nisole S (2009) Human TRIM gene expression in response to interferons. PLoS One 4(3):e4894
Cereseto A, Manganaro L, Gutierrez MI, Terreni M, Fittipaldi A, Lusic M, Marcello A, Giacca M (2005) Acetylation of HIV-1 integrase by p300 regulates viral integration. EMBO J 24(17):3070–3081
Chatterji U, Bobardt MD, Gaskill P, Sheeter D, Fox H, Gallay PA (2006) Trim5alpha accelerates degradation of cytosolic capsid associated with productive HIV-1 entry. J Biol Chem 281(48):37025–37033
Chu Y, Yang X (2011) SUMO E3 ligase activity of TRIM proteins. Oncogene 30(9):1108–1116
Cowan S, Hatziioannou T, Cunningham T, Muesing MA, Gottlinger HG, Bieniasz PD (2002) Cellular inhibitors with Fv1-like activity restrict human and simian immunodeficiency virus tropism. Proc Natl Acad Sci USA 99(18):11914–11919
Danielson CM, Cianci GC, Hope TJ (2012) Recruitment and dynamics of proteasome association with rhTRIM5α cytoplasmic complexes during HIV-1 infection. Traffic 13(9):1206–1217
Daugherty MD, Malik HS (2012) Rules of engagement: molecular insights from host-virus arms races. Annu Rev Genet 46:677–700
Di Pietro A, Kajaste-Rudnitski A, Oteiza A, Nicora L, Towers GJ, Mechti N, Vicenzi E (2013) TRIM22 inhibits influenza A virus infection by targeting the viral nucleoprotein for degradation. J Virol 87(8):4523–4533
Diaz-Griffero F (2011) Caging the beast: TRIM5alpha binding to the HIV-1 core. Viruses 3(5):423–428
Diaz-Griffero F, Kar A, Lee M, Stremlau M, Poeschla E, Sodroski J (2007) Comparative requirements for the restriction of retrovirus infection by TRIM5α and TRIMCyp. Virology 369(2):400–410
Diaz-Griffero F, Li X, Javanbakht H, Song B, Welikala S, Stremlau M, Sodroski J (2006a) Rapid turnover and polyubiquitylation of the retroviral restriction factor TRIM5. Virology 349(2):300–315
Diaz-Griffero F, Vandegraaff N, Li Y, McGee-Estrada K, Stremlau M, Welikala S, Si Z, Engelman A, Sodroski J (2006b) Requirements for capsid-binding and an effector function in TRIMCyp-mediated restriction of HIV-1. Virology 351(2):404–419
Diehl WE, Stansell E, Kaiser SM, Emerman M, Hunter E (2008) Identification of postentry restrictions to Mason-Pfizer monkey virus infection in New World monkey cells. J Virol 82(22):11140–11151
Dixit E, Boulant S, Zhang Y, Lee AS, Odendall C, Shum B, Hacohen N, Chen ZJ, Whelan SP, Fransen M, Nibert ML, Superti-Furga G, Kagan JC (2010) Peroxisomes are signaling platforms for antiviral innate immunity. Cell 141(4):668–681
Dodding MP, Bock M, Yap MW, Stoye JP (2005) Capsid processing requirements for abrogation of Fv1 and Ref1 restriction. J Virol 79(16):10571–10577
du Chene I, Basyuk E, Lin YL, Triboulet R, Knezevich A, Chable-Bessia C, Mettling C, Baillat V, Reynes J, Corbeau P, Bertrand E, Marcello A, Emiliani S, Kiernan R, Benkirane M (2007) Suv39H1 and HP1gamma are responsible for chromatin-mediated HIV-1 transcriptional silencing and post-integration latency. EMBO J 26(2):424–435
Eldin P, Papon L, Oteiza A, Brocchi E, Lawson TG, Mechti N (2009) TRIM22 E3 ubiquitin ligase activity is required to mediate antiviral activity against encephalomyocarditis virus. J Gen Virol 90(Pt 3):536–545
Emerson RO, Thomas JH (2009) Adaptive evolution in zinc finger transcription factors. PLoS Genet 5(1):e1000325
Everett RD, Rechter S, Papior P, Tavalai N, Stamminger T, Orr A (2006) PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J Virol 80(16):7995–8005
Feng S, Jacobsen SE, Reik W (2010) Epigenetic reprogramming in plant and animal development. Science 330(6004):622–627
Feuer G, Taketo M, Hanecak RC, Fan H (1989) Two blocks in Moloney murine leukemia virus expression in undifferentiated F9 embryonal carcinoma cells as determined by transient expression assays. J Virol 63(5):2317–2324
Forshey BM, Shi J, Aiken C (2005) Structural requirements for recognition of the human immunodeficiency virus type 1 core during host restriction in owl monkey cells. J Virol 79(2):869–875
Franke EK, Yuan HE, Luban J (1994) Specific incorporation of cyclophilin A into HIV-1 virions. Nature 372(6504):359–362
Franzoso G, Biswas P, Poli G, Carlson LM, Brown KD, Tomita-Yamaguchi M, Fauci AS, Siebenlist UK (1994) A family of serine proteases expressed exclusively in myelo-monocytic cells specifically processes the nuclear factor-kappa B subunit p65 in vitro and may impair human immunodeficiency virus replication in these cells. J Exp Med 180(4):1445–1456
Fridell RA, Harding LS, Bogerd HP, Cullen BR (1995) Identification of a novel human zinc finger protein that specifically interacts with the activation domain of lentiviral Tat proteins. Virology 209(2):347–357
Friedman JR, Fredericks WJ, Jensen DE, Speicher DW, Huang XP, Neilson EG, Rauscher FJ 3rd (1996) KAP-1, a novel corepressor for the highly conserved KRAB repression domain. Genes Dev 10(16):2067–2078
Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, Jung JU (2007) TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446(7138):916–920
Ganser BK, Cheng A, Sundquist WI, Yeager M (2003) Three-dimensional structure of the M-MuLV CA protein on a lipid monolayer: a general model for retroviral capsid assembly. EMBO J 22(12):2886–2892
Ganser BK, Li S, Klishko VY, Finch JT, Sundquist WI (1999) Assembly and analysis of conical models for the HIV-1 core. Science 283(5398):80–83
Ganser-Pornillos BK, Chandrasekaran V, Pornillos O, Sodroski JG, Sundquist WI, Yeager M (2011) Hexagonal assembly of a restricting TRIM5alpha protein. Proc Natl Acad Sci USA 108(2):534–539
Ganser-Pornillos BK, Cheng A, Yeager M (2007) Structure of full-length HIV-1 CA: a model for the mature capsid lattice. Cell 131(1):70–79
Ganser-Pornillos BK, von Schwedler UK, Stray KM, Aiken C, Sundquist WI (2004) Assembly properties of the human immunodeficiency virus type 1 CA protein. J Virol 78(5):2545–2552
Gao B, Duan Z, Xu W, Xiong S (2009) Tripartite motif-containing 22 inhibits the activity of hepatitis B virus core promoter, which is dependent on nuclear-located RING domain. Hepatology 50(2):424–433
Goldstone DC, Yap MW, Robertson LE, Haire LF, Taylor WR, Katzourakis A, Stoye JP, Taylor IA (2010) Structural and functional analysis of prehistoric lentiviruses uncovers an ancient molecular interface. Cell Host Microbe 8(3):248–259
Groner AC, Meylan S, Ciuffi A, Zangger N, Ambrosini G, Denervaud N, Bucher P, Trono D (2010) KRAB-zinc finger proteins and KAP1 can mediate long-range transcriptional repression through heterochromatin spreading. PLoS Genet 6(3):e1000869
Groner AC, Tschopp P, Challet L, Dietrich JE, Verp S, Offner S, Barde I, Rodriguez I, Hiiragi T, Trono D (2012) The Kruppel-associated box repressor domain can induce reversible heterochromatization of a mouse locus in vivo. J Biol Chem 287(30):25361–25369
Han K, Lou DI, Sawyer SL (2011) Identification of a genomic reservoir for new TRIM genes in primate genomes. PLoS Genet 7(12):e1002388
Hatziioannou T, Cowan S, Von Schwedler UK, Sundquist WI, Bieniasz PD (2004) Species-specific tropism determinants in the human immunodeficiency virus type 1 capsid. J Virol 78(11):6005–6012
Herchenroder O, Hahne JC, Meyer WK, Thiesen HJ, Schneider J (1999) Repression of the human immunodeficiency virus type 1 promoter by the human KRAB domain results in inhibition of virus production. Biochim Biophys Acta 1445(2):216–223
Herquel B, Ouararhni K, Khetchoumian K, Ignat M, Teletin M, Mark M, Bechade G, Van Dorsselaer A, Sanglier-Cianferani S, Hamiche A, Cammas F, Davidson I, Losson R (2011) Transcription cofactors TRIM24, TRIM28, and TRIM33 associate to form regulatory complexes that suppress murine hepatocellular carcinoma. Proc Natl Acad Sci USA 108(20):8212–8217
Herquel B, Ouararhni K, Martianov I, Le Gras S, Ye T, Keime C, Lerouge T, Jost B, Cammas F, Losson R, Davidson I (2013) Trim24-repressed VL30 retrotransposons regulate gene expression by producing noncoding RNA. Nat Struct Mol Biol
Herr AM, Dressel R, Walter L (2009) Different subcellular localisations of TRIM22 suggest species-specific function. Immunogenetics 61(4):271–280
Hofmann W, Schubert D, LaBonte J, Munson L, Gibson S, Scammell J, Ferrigno P, Sodroski J (1999) Species-specific, postentry barriers to primate immunodeficiency virus infection. J Virol 73(12):10020–10028
Huntley S, Baggott DM, Hamilton AT, Tran-Gyamfi M, Yang S, Kim J, Gordon L, Branscomb E, Stubbs L (2006) A comprehensive catalog of human KRAB-associated zinc finger genes: insights into the evolutionary history of a large family of transcriptional repressors. Genome Res 16(5):669–677
Hwang CY, Holl J, Rajan D, Lee Y, Kim S, Um M, Kwon KS, Song B (2010) Hsp70 interacts with the retroviral restriction factor TRIM5alpha and assists the folding of TRIM5alpha. J Biol Chem 285(10):7827–7837
Ivanov AV, Peng H, Yurchenko V, Yap KL, Negorev DG, Schultz DC, Psulkowski E, Fredericks WJ, White DE, Maul GG, Sadofsky MJ, Zhou MM, Rauscher FJ 3rd (2007) PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol Cell 28(5):823–837
Iyengar S, Ivanov AV, Jin VX, Rauscher FJ 3rd, Farnham PJ (2011) Functional analysis of KAP1 genomic recruitment. Mol Cell Biol 31(9):1833–1847
James LC, Keeble AH, Khan Z, Rhodes DA, Trowsdale J (2007) Structural basis for PRYSPRY-mediated tripartite motif (TRIM) protein function. Proc Natl Acad Sci USA 104(15):6200–6205
Javanbakht H, Diaz-Griffero F, Stremlau M, Si Z, Sodroski J (2005) The contribution of RING and B-box 2 domains to retroviral restriction mediated by monkey TRIM5α. J Biol Chem 280(29):26933–26940
Kajaste-Rudnitski A, Marelli SS, Pultrone C, Pertel T, Uchil PD, Mechti N, Mothes W, Poli G, Luban J, Vicenzi E (2011) TRIM22 inhibits HIV-1 transcription independently of its E3 ubiquitin ligase activity, Tat, and NF-kappaB-responsive long terminal repeat elements. J Virol 85(10):5183–5196
Kar AK, Diaz-Griffero F, Li Y, Li X, Sodroski J (2008) Biochemical and biophysical characterization of a chimeric TRIM21–TRIM5α protein. J Virol 82(23):11669–11681
Katzourakis A, Tristem M, Pybus OG, Gifford RJ (2007) Discovery and analysis of the first endogenous lentivirus. Proc Natl Acad Sci USA 104(15):6261–6265
Kawai T, Akira S (2011) Regulation of innate immune signalling pathways by the tripartite motif (TRIM) family proteins. EMBO Mol Med 3(9):513–527
Keckesova Z, Ylinen LM, Towers GJ (2004) The human and African green monkey TRIM5α genes encode Ref1 and Lv1 retroviral restriction factor activities. Proc Natl Acad Sci USA 101(29):10780–10785
Keckesova Z, Ylinen LM, Towers GJ, Gifford RJ, Katzourakis A (2009) Identification of a RELIK orthologue in the European hare (Lepus europaeus) reveals a minimum age of 12 million years for the lagomorph lentiviruses. Virology 384(1):7–11
Kempler G, Freitag B, Berwin B, Nanassy O, Barklis E (1993) Characterization of the moloney murine leukemia virus stem cell-specific repressor binding site. Virology 193(2):690–699
Khetchoumian K, Teletin M, Tisserand J, Mark M, Herquel B, Ignat M, Zucman-Rossi J, Cammas F, Lerouge T, Thibault C, Metzger D, Chambon P, Losson R (2007) Loss of Trim24 (Tif1α) gene function confers oncogenic activity to retinoic acid receptor alpha. Nat Genet 39(12):1500–1506
Kim J, Tipper C, Sodroski J (2011) Role of TRIM5α RING domain E3 ubiquitin ligase activity in capsid disassembly, reverse transcription blockade, and restriction of simian immunodeficiency virus. J Virol 85(16):8116–8132
Kim SS, Chen YM, O’Leary E, Witzgall R, Vidal M, Bonventre JV (1996) A novel member of the RING finger family, KRIP-1, associates with the KRAB-A transcriptional repressor domain of zinc finger proteins. Proc Natl Acad Sci USA 93(26):15299–15304
Kim SY, Wang TK, Singh RD, Wheatley CL, Marks DL, Pagano RE (2009) Proteomic identification of proteins translocated to membrane microdomains upon treatment of fibroblasts with the glycosphingolipid, C8-beta-D-lactosylceramide. Proteomics 9(18):4321–4328
Kirmaier A, Wu F, Newman RM, Hall LR, Morgan JS, O’Connor S, Marx PA, Meythaler M, Goldstein S, Buckler-White A, Kaur A, Hirsch VM, Johnson WE (2010) TRIM5 suppresses cross-species transmission of a primate immunodeficiency virus and selects for emergence of resistant variants in the new species. PLoS Biol 8(8):e1000462
Kozak CA, Chakraborti A (1996) Single amino acid changes in the murine leukemia virus capsid protein gene define the target of Fv1 resistance. Virology 225(2):300–305
Kwon SH, Workman JL (2011) The changing faces of HP1: From heterochromatin formation and gene silencing to euchromatic gene expression: HP1 acts as a positive regulator of transcription. BioEssays 33(4):280–289
Langelier CR, Sandrin V, Eckert DM, Christensen DE, Chandrasekaran V, Alam SL, Aiken C, Olsen JC, Kar AK, Sodroski JG, Sundquist WI (2008) Biochemical characterization of a recombinant TRIM5alpha protein that restricts human immunodeficiency virus type 1 replication. J Virol 82(23):11682–11694
Le Douarin B, Nielsen AL, Garnier JM, Ichinose H, Jeanmougin F, Losson R, Chambon P (1996) A possible involvement of TIF1 alpha and TIF1 beta in the epigenetic control of transcription by nuclear receptors. EMBO J 15(23):6701–6715
Li J, Han K, Xing J, Kim HS, Rogers J, Ryder OA, Disotell T, Yue B, Batzer MA (2009) Phylogeny of the macaques (Cercopithecidae: Macaca) based on Alu elements. Gene 448(2):242–249
Li S, Hill CP, Sundquist WI, Finch JT (2000) Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature 407(6802):409–413
Li X, Ito M, Zhou F, Youngson N, Zuo X, Leder P, Ferguson-Smith AC (2008) A maternal-zygotic effect gene, Zfp57, maintains both maternal and paternal imprints. Dev Cell 15(4):547–557
Li X, Yeung DF, Fiegen AM, Sodroski J (2011) Determinants of the higher order association of the restriction factor TRIM5α and other tripartite motif (TRIM) proteins. J Biol Chem 286(32):27959–27970
Liao CH, Kuang YQ, Liu HL, Zheng YT, Su B (2007) A novel fusion gene, TRIM5-cyclophilin A in the pig-tailed macaque determines its susceptibility to HIV-1 infection. AIDS 21(Suppl 8):S19–S26
Lienlaf M, Hayashi F, Di Nunzio F, Tochio N, Kigawa T, Yokoyama S, Diaz-Griffero F (2011) Contribution of E3-ubiquitin ligase activity to HIV-1 restriction by TRIM5α(rh): structure of the RING domain of TRIM5alpha. J Virol 85(17):8725–8737
Lin TY, Emerman M (2006) Cyclophilin A interacts with diverse lentiviral capsids. Retrovirology 3:70
Lin TY, Emerman M (2008) Determinants of cyclophilin A-dependent TRIM5αrestriction against HIV-1. Virology 379(2):335–341
Loh TP, Sievert LL, Scott RW (1988) Negative regulation of retrovirus expression in embryonal carcinoma cells mediated by an intragenic domain. J Virol 62(11):4086–4095
Loh TP, Sievert LL, Scott RW (1990) Evidence for a stem cell-specific repressor of moloney murine leukemia virus expression in embryonal carcinoma cells. Mol Cell Biol 10(8):4045–4057
Lukic Z, Hausmann S, Sebastian S, Rucci J, Sastri J, Robia SL, Luban J, Campbell EM (2011) TRIM5alpha associates with proteasomal subunits in cells while in complex with HIV-1 virions. Retrovirology 8:93
Maillard PV, Reynard S, Serhan F, Turelli P, Trono D (2007) Interfering residues narrow the spectrum of MLV restriction by human TRIM5α. PLoS Pathog 3(12):e200
Maillard PV, Zoete V, Michielin O, Trono D (2011) Homology-based identification of capsid determinants that protect HIV1 from human TRIM5α restriction. J Biol Chem 286(10):8128–8140
Malfavon-Borja R, Wu LI, Emerman M, Malik HS (2013) Birth, decay, and reconstruction of an ancient TRIMCyp gene fusion in primate genomes. Proc Natl Acad Sci USA 110:E583–592
Mallery DL, McEwan WA, Bidgood SR, Towers GJ, Johnson CM, James LC (2010) Antibodies mediate intracellular immunity through tripartite motif-containing 21 (TRIM21). Proc Natl Acad Sci USA 107(46):19985–19990
Marin I (2012) Origin and diversification of TRIM ubiquitin ligases. PLoS One 7(11):e50030
Mascle XH, Germain-Desprez D, Huynh P, Estephan P, Aubry M (2007) Sumoylation of the transcriptional intermediary factor 1beta (TIF1beta), the Co-repressor of the KRAB Multifinger proteins, is required for its transcriptional activity and is modulated by the KRAB domain. J Biol Chem 282(14):10190–10202
Matsui T, Leung D, Miyashita H, Maksakova IA, Miyachi H, Kimura H, Tachibana M, Lorincz MC, Shinkai Y (2010) Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature 464(7290):927–931
McNab FW, Rajsbaum R, Stoye JP, O’Garra A (2011) Tripartite-motif proteins and innate immune regulation. Curr Opin Immunol 23(1):46–56
Meroni G, Diez-Roux G (2005) TRIM/RBCC, a novel class of ‘single protein RING finger’ E3 ubiquitin ligases. BioEssays 27(11):1147–1157
Messerschmidt DM, de Vries W, Ito M, Solter D, Ferguson-Smith A, Knowles BB (2012) Trim28 is required for epigenetic stability during mouse oocyte to embryo transition. Science 335(6075):1499–1502
Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, Lee W, Mendenhall E, O’Donovan A, Presser A, Russ C, Xie X, Meissner A, Wernig M, Jaenisch R, Nusbaum C, Lander ES, Bernstein BE (2007) Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448(7153):553–560
Moosmann P, Georgiev O, Le Douarin B, Bourquin JP, Schaffner W (1996) Transcriptional repression by RING finger protein TIF1 beta that interacts with the KRAB repressor domain of KOX1. Nucleic Acids Res 24(24):4859–4867
Moriuchi H, Moriuchi M, Arthos J, Hoxie J, Fauci AS (1997) Promonocytic U937 subclones expressing CD4 and CXCR4 are resistant to infection with and cell-to-cell fusion by T-cell-tropic human immunodeficiency virus type 1. J Virol 71(12):9664–9671
Mortuza GB, Dodding MP, Goldstone DC, Haire LF, Stoye JP, Taylor IA (2008) Structure of B-MLV capsid amino-terminal domain reveals key features of viral tropism, gag assembly and core formation. J Mol Biol 376(5):1493–1508
Napolitano LM, Jaffray EG, Hay RT, Meroni G (2011) Functional interactions between ubiquitin E2 enzymes and TRIM proteins. Biochem J 434(2):309–319
Newman RM, Hall L, Connole M, Chen GL, Sato S, Yuste E, Diehl W, Hunter E, Kaur A, Miller GM, Johnson WE (2006) Balancing selection and the evolution of functional polymorphism in old world monkey TRIM5alpha. Proc Natl Acad Sci USA 103(50):19134–19139
Newman RM, Hall L, Kirmaier A, Pozzi LA, Pery E, Farzan M, O’Neil SP, Johnson W (2008) Evolution of a TRIM5-CypA splice isoform in old world monkeys. PLoS Pathog 4(2):e1000003
Nielsen AL, Ortiz JA, You J, Oulad-Abdelghani M, Khechumian R, Gansmuller A, Chambon P, Losson R (1999) Interaction with members of the heterochromatin protein 1 (HP1) family and histone deacetylation are differentially involved in transcriptional silencing by members of the TIF1 family. EMBO J 18(22):6385–6395
Nishitsuji H, Abe M, Sawada R, Takaku H (2012) ZBRK1 represses HIV-1 LTR-mediated transcription. FEBS Lett 586(20):3562–3568
Nisole S, Lynch C, Stoye JP, Yap MW (2004) A Trim5-cyclophilin A fusion protein found in owl monkey kidney cells can restrict HIV-1. Proc Natl Acad Sci U S A 101(36):13324–13328
Nisole S, Stoye JP, Saib A (2005) TRIM family proteins: retroviral restriction and antiviral defence. Nat Rev Microbiol 3(10):799–808
O’Geen H, Squazzo SL, Iyengar S, Blahnik K, Rinn JL, Chang HY, Green R, Farnham PJ (2007) Genome-wide analysis of KAP1 binding suggests autoregulation of KRAB-ZNFs. PLoS Genet 3(6):e89
Ocwieja KE, Brady TL, Ronen K, Huegel A, Roth SL, Schaller T, James LC, Towers GJ, Young JA, Chanda SK, Konig R, Malani N, Berry CC, Bushman FD (2011) HIV integration targeting: a pathway involving Transportin-3 and the nuclear pore protein RanBP2. PLoS Pathog 7(3):e1001313
Ohkura S, Goldstone DC, Yap MW, Holden-Dye K, Taylor IA, Stoye JP (2011) Novel escape mutants suggest an extensive TRIM5α binding site spanning the entire outer surface of the murine leukemia virus capsid protein. PLoS Pathog 7(3):e1002011
Ohmine S, Sakuma R, Sakuma T, Thatava T, Solis GP, Ikeda Y (2010) Cytoplasmic body component TRIM5α requires lipid-enriched microdomains for efficient HIV-1 restriction. J Biol Chem 285(45):34508–34517
Ohmine S, Sakuma R, Sakuma T, Thatava T, Takeuchi H, Ikeda Y (2011) The antiviral spectra of TRIM5alpha orthologues and human TRIM family proteins against lentiviral production. PLoS One 6(1):e16121
Owens CM, Song B, Perron MJ, Yang PC, Stremlau M, Sodroski J (2004) Binding and susceptibility to postentry restriction factors in monkey cells are specified by distinct regions of the human immunodeficiency virus type 1 capsid. J Virol 78(10):5423–5437
Ozato K, Shin DM, Chang TH, Morse HC 3rd (2008) TRIM family proteins and their emerging roles in innate immunity. Nat Rev Immunol 8(11):849–860
Patarca R, Freeman GJ, Schwartz J, Singh RP, Kong QT, Murphy E, Anderson Y, Sheng FY, Singh P, Johnson KA et al (1988) rpt-1, an intracellular protein from helper/inducer T cells that regulates gene expression of interleukin 2 receptor and human immunodeficiency virus type 1. Proc Natl Acad Sci USA 85(8):2733–2737
Peng H, Begg GE, Schultz DC, Friedman JR, Jensen DE, Speicher DW, Rauscher FJ 3rd (2000) Reconstitution of the KRAB-KAP-1 repressor complex: a model system for defining the molecular anatomy of RING-B box-coiled-coil domain-mediated protein–protein interactions. J Mol Biol 295(5):1139–1162
Pengue G, Caputo A, Rossi C, Barbanti-Brodano G, Lania L (1995) Transcriptional silencing of human immunodeficiency virus type 1 long terminal repeat-driven gene expression by the Kruppel-associated box repressor domain targeted to the transactivating response element. J Virol 69(10):6577–6580
Perez-Caballero D, Hatziioannou T, Yang A, Cowan S, Bieniasz PD (2005a) Human tripartite motif 5α domains responsible for retrovirus restriction activity and specificity. J Virol 79(14):8969–8978
Perez-Caballero D, Hatziioannou T, Zhang F, Cowan S, Bieniasz PD (2005b) Restriction of human immunodeficiency virus type 1 by TRIM-CypA occurs with rapid kinetics and independently of cytoplasmic bodies, ubiquitin, and proteasome activity. J Virol 79(24):15567–15572
Perron MJ, Stremlau M, Lee M, Javanbakht H, Song B, Sodroski J (2007) The human TRIM5α restriction factor mediates accelerated uncoating of the N-tropic murine leukemia virus capsid. J Virol 81(5):2138–2148
Pertel T, Hausmann S, Morger D, Zuger S, Guerra J, Lascano J, Reinhard C, Santoni FA, Uchil PD, Chatel L, Bisiaux A, Albert ML, Strambio-De-Castillia C, Mothes W, Pizzato M, Grutter MG, Luban J (2011) TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 472(7343):361–365
Petersen R, Kempler G, Barklis E (1991) A stem cell-specific silencer in the primer-binding site of a retrovirus. Mol Cell Biol 11(3):1214–1221
Poole E, Groves I, MacDonald A, Pang Y, Alcami A, Sinclair J (2009) Identification of TRIM23 as a cofactor involved in the regulation of NF-kappaB by human cytomegalovirus. J Virol 83(8):3581–3590
Pornillos O, Ganser-Pornillos BK, Yeager M (2011) Atomic-level modelling of the HIV capsid. Nature 469(7330):424–427
Price AJ, Marzetta F, Lammers M, Ylinen LM, Schaller T, Wilson SJ, Towers GJ, James LC (2009) Active site remodeling switches HIV specificity of antiretroviral TRIMCyp. Nat Struct Mol Biol 16(10):1036–1042
Quenneville S, Verde G, Corsinotti A, Kapopoulou A, Jakobsson J, Offner S, Baglivo I, Pedone PV, Grimaldi G, Riccio A, Trono D (2011) In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions. Mol Cell 44(3):361–372
Rahm N, Yap M, Snoeck J, Zoete V, Munoz M, Radespiel U, Zimmermann E, Michielin O, Stoye JP, Ciuffi A, Telenti A (2011) Unique spectrum of activity of prosimian TRIM5α against exogenous and endogenous retroviruses. J Virol 85(9):4173–4183
Rajsbaum R, Stoye JP, O’Garra A (2008) Type I interferon-dependent and -independent expression of tripartite motif proteins in immune cells. Eur J Immunol 38(3):619–630
Reddy BA, Kloc M, Etkin L (1991) The cloning and characterization of a maternally expressed novel zinc finger nuclear phosphoprotein (xnf7) in Xenopus laevis. Dev Biol 148(1):107–116
Reichelt M, Wang L, Sommer M, Perrino J, Nour AM, Sen N, Baiker A, Zerboni L, Arvin AM (2011) Entrapment of viral capsids in nuclear PML cages is an intrinsic antiviral host defense against varicella-zoster virus. PLoS Pathog 7(2):e1001266
Reymond A, Meroni G, Fantozzi A, Merla G, Cairo S, Luzi L, Riganelli D, Zanaria E, Messali S, Cainarca S, Guffanti A, Minucci S, Pelicci PG, Ballabio A (2001) The tripartite motif family identifies cell compartments. EMBO J 20(9):2140–2151
Reynolds L, Ullman C, Moore M, Isalan M, West MJ, Clapham P, Klug A, Choo Y (2003) Repression of the HIV-1 5′ LTR promoter and inhibition of HIV-1 replication by using engineered zinc-finger transcription factors. Proc Natl Acad Sci USA 100(4):1615–1620
Roa A, Hayashi F, Yang Y, Lienlaf M, Zhou J, Shi J, Watanabe S, Kigawa T, Yokoyama S, Aiken C, Diaz-Griffero F (2012) RING domain mutations uncouple TRIM5alpha restriction of HIV-1 from inhibition of reverse transcription and acceleration of uncoating. J Virol 86(3):1717–1727
Rold CJ, Aiken C (2008) Proteasomal degradation of TRIM5α during retrovirus restriction. PLoS Pathog 4(5):e1000074
Rowe HM, Friedli M, Offner S, Verp S, Mesnard D, Marquis J, Aktas T, Trono D (2013a) De novo DNA methylation of endogenous retroviruses is shaped by KRAB-ZFPs/KAP1 and ESET. Development 140(3):519–529
Rowe HM, Jakobsson J, Mesnard D, Rougemont J, Reynard S, Aktas T, Maillard PV, Layard-Liesching H, Verp S, Marquis J, Spitz F, Constam DB, Trono D (2010) KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 463(7278):237–240
Rowe HM, Kapopoulou A, Corsinotti A, Fasching L, Macfarlan TS, Tarabay Y, Viville S, Jakobsson J, Pfaff SL, Trono D (2013b) TRIM28 repression of retrotransposon-based enhancers is necessary to preserve transcriptional dynamics in embryonic stem cells. Genome Res 23:452–461
Ryan RF, Schultz DC, Ayyanathan K, Singh PB, Friedman JR, Fredericks WJ, Rauscher FJ 3rd (1999) KAP-1 corepressor protein interacts and colocalizes with heterochromatic and euchromatic HP1 proteins: a potential role for Kruppel-associated box-zinc finger proteins in heterochromatin-mediated gene silencing. Mol Cell Biol 19(6):4366–4378
Sardiello M, Cairo S, Fontanella B, Ballabio A, Meroni G (2008) Genomic analysis of the TRIM family reveals two groups of genes with distinct evolutionary properties. BMC Evol Biol 8:225
Sawyer SL, Emerman M, Malik HS (2007) Discordant evolution of the adjacent antiretroviral genes TRIM22 and TRIM5 in mammals. PLoS Pathog 3(12):e197
Sayah DM, Sokolskaja E, Berthoux L, Luban J (2004) Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature 430(6999):569–573
Schaller T, Ocwieja KE, Rasaiyaah J, Price AJ, Brady TL, Roth SL, Hue S, Fletcher AJ, Lee K, KewalRamani VN, Noursadeghi M, Jenner RG, James LC, Bushman FD, Towers GJ (2011) HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog 7(12):e1002439
Schaller T, Ylinen LM, Webb BL, Singh S, Towers GJ (2007) Fusion of cyclophilin A to Fv1 enables cyclosporine-sensitive restriction of human and feline immunodeficiency viruses. J Virol 81(18):10055–10063
Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ 3rd (2002) SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev 16(8):919–932
Schultz DC, Friedman JR, Rauscher FJ 3rd (2001) Targeting histone deacetylase complexes via KRAB-zinc finger proteins: the PHD and bromodomains of KAP-1 form a cooperative unit that recruits a novel isoform of the Mi-2α subunit of NuRD. Genes Dev 15(4):428–443
Sebastian S, Luban J (2005) TRIM5α selectively binds a restriction-sensitive retroviral capsid. Retrovirology 2:40
Segal DJ, Goncalves J, Eberhardy S, Swan CH, Torbett BE, Li X, Barbas CF 3rd (2004) Attenuation of HIV-1 replication in primary human cells with a designed zinc finger transcription factor. J Biol Chem 279(15):14509–14519
Shank PR, Varmus HE (1978) Virus-specific DNA in the cytoplasm of avian sarcoma virus-infected cells is a precursor to covalently closed circular viral DNA in the nucleus. J Virol 25(1):104
Shi J, Aiken C (2006) Saturation of TRIM5α-mediated restriction of HIV-1 infection depends on the stability of the incoming viral capsid. Virology 350(2):493–500
Shuai K, Liu B (2005) Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat Rev Immunol 5(8):593–605
Singh R, Gaiha G, Werner L, McKim K, Mlisana K, Luban J, Walker BD, Karim SS, Brass AL, Ndung’u T (2011) Association of TRIM22 with the type 1 interferon response and viral control during primary HIV-1 infection. J Virol 85(1):208–216
Sivaramakrishnan G, Sun Y, Rajmohan R, Lin VC (2009) B30.2/SPRY domain in tripartite motif-containing 22 is essential for the formation of distinct nuclear bodies. FEBS Lett 583(12):2093–2099
Sodeik B (2002) Unchain my heart, baby let me go–the entry and intracellular transport of HIV. J Cell Biol 159(3):393–395
Sorin M, Cano J, Das S, Mathew S, Wu X, Davies KP, Shi X, Cheng SW, Ott D, Kalpana GV (2009) Recruitment of a SAP18-HDAC1 complex into HIV-1 virions and its requirement for viral replication. PLoS Pathog 5(6):e1000463
Sripathy SP, Stevens J, Schultz DC (2006) The KAP1 corepressor functions to coordinate the assembly of de novo HP1-demarcated microenvironments of heterochromatin required for KRAB zinc finger protein-mediated transcriptional repression. Mol Cell Biol 26(22):8623–8638
Stetson DB, Ko JS, Heidmann T, Medzhitov R (2008) Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134(4):587–598
Stewart CL, Stuhlmann H, Jahner D, Jaenisch R (1982) De novo methylation, expression, and infectivity of retroviral genomes introduced into embryonal carcinoma cells. Proc Natl Acad Sci USA 79(13):4098–4102
Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J (2004) The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in old world monkeys. Nature 427(6977):848–853
Stremlau M, Perron M, Lee M, Li Y, Song B, Javanbakht H, Diaz-Griffero F, Anderson DJ, Sundquist WI, Sodroski J (2006) Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5α restriction factor. Proc Natl Acad Sci USA 103(14):5514–5519
Szulc J, Wiznerowicz M, Sauvain MO, Trono D, Aebischer P (2006) A versatile tool for conditional gene expression and knockdown. Nat Methods 3(2):109–116
Tareen SU, Emerman M (2011) Human Trim5α has additional activities that are uncoupled from retroviral capsid recognition. Virology 409(1):113–120
Tareen SU, Sawyer SL, Malik HS, Emerman M (2009) An expanded clade of rodent Trim5 genes. Virology 385(2):473–483
Taylor RT, Lubick KJ, Robertson SJ, Broughton JP, Bloom ME, Bresnahan WA, Best SM (2011) TRIM79alpha, an interferon-stimulated gene product, restricts tick-borne encephalitis virus replication by degrading the viral RNA polymerase. Cell Host Microbe 10(3):185–196
Teich NM, Weiss RA, Martin GR, Lowy DR (1977) Virus infection of murine teratocarcinoma stem cell lines. Cell 12(4):973–982
Terreni M, Valentini P, Liverani V, Gutierrez MI, Di Primio C, Di Fenza A, Tozzini V, Allouch A, Albanese A, Giacca M, Cereseto A (2010) GCN5-dependent acetylation of HIV-1 integrase enhances viral integration. Retrovirology 7:18
Thali M, Bukovsky A, Kondo E, Rosenwirth B, Walsh CT, Sodroski J, Gottlinger HG (1994) Functional association of cyclophilin A with HIV-1 virions. Nature 372(6504):363–365
Thiesen HJ, Bellefroid E, Revelant O, Martial JA (1991) Conserved KRAB protein domain identified upstream from the zinc finger region of Kox 8. Nucleic Acids Res 19(14):3996
Tisserand J, Khetchoumian K, Thibault C, Dembele D, Chambon P, Losson R (2011) Tripartite motif 24 (Trim24/Tif1α) tumor suppressor protein is a novel negative regulator of interferon (IFN)/signal transducers and activators of transcription (STAT) signaling pathway acting through retinoic acid receptor alpha (Raralpha) inhibition. J Biol Chem 286(38):33369–33379
Tissot C, Mechti N (1995) Molecular cloning of a new interferon-induced factor that represses human immunodeficiency virus type 1 long terminal repeat expression. J Biol Chem 270(25):14891–14898
Topper M, Luo Y, Zhadina M, Mohammed K, Smith L, Muesing MA (2007) Posttranslational acetylation of the human immunodeficiency virus type 1 integrase carboxyl-terminal domain is dispensable for viral replication. J Virol 81(6):3012–3017
Towers G, Bock M, Martin S, Takeuchi Y, Stoye JP, Danos O (2000) A conserved mechanism of retrovirus restriction in mammals. Proc Natl Acad Sci USA 97(22):12295–12299
Towers GJ, Hatziioannou T, Cowan S, Goff SP, Luban J, Bieniasz PD (2003) Cyclophilin A modulates the sensitivity of HIV-1 to host restriction factors. Nat Med 9(9):1138–1143
Triantafilou M, Lepper PM, Olden R, Dias Ide S, Triantafilou K (2011) Location, location, location: is membrane partitioning everything when it comes to innate immune activation? Mediators Inflamm 2011:186093
Tsai WW, Wang Z, Yiu TT, Akdemir KC, Xia W, Winter S, Tsai CY, Shi X, Schwarzer D, Plunkett W, Aronow B, Gozani O, Fischle W, Hung MC, Patel DJ, Barton MC (2010) TRIM24 links a non-canonical histone signature to breast cancer. Nature 468(7326):927–932
Tsuchida T, Zou J, Saitoh T, Kumar H, Abe T, Matsuura Y, Kawai T, Akira S (2010) The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity 33(5):765–776
Turelli P, Doucas V, Craig E, Mangeat B, Klages N, Evans R, Kalpana G, Trono D (2001) Cytoplasmic recruitment of INI1 and PML on incoming HIV preintegration complexes: interference with early steps of viral replication. Mol Cell 7(6):1245–1254
Uchil PD, Hinz A, Siegel S, Coenen-Stass A, Pertel T, Luban J, Mothes W (2013) TRIM protein-mediated regulation of inflammatory and innate immune signaling and its association with antiretroviral activity. J Virol 87(1):257–272
Uchil PD, Quinlan BD, Chan WT, Luna JM, Mothes W (2008) TRIM E3 ligases interfere with early and late stages of the retroviral life cycle. PLoS Pathog 4(2):e16
Urrutia R (2003) KRAB-containing zinc-finger repressor proteins. Genome Biol 4(10):231
van der Aa LM, Levraud JP, Yahmi M, Lauret E, Briolat V, Herbomel P, Benmansour A, Boudinot P (2009) A large new subset of TRIM genes highly diversified by duplication and positive selection in teleost fish. BMC Biol 7:7
Van Lint C, Emiliani S, Ott M, Verdin E (1996) Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J 15(5):1112–1120
Versteeg GA, Rajsbaum R, Sanchez-Aparicio MT, Maestre AM, Valdiviezo J, Shi M, Inn KS, Fernandez-Sesma A, Jung J, Garcia-Sastre A (2013) The E3-ligase TRIM family of proteins regulates signaling pathways triggered by innate immune pattern-recognition receptors. Immunity 38(2):384–398
Virgen CA, Kratovac Z, Bieniasz PD, Hatziioannou T (2008) Independent genesis of chimeric TRIM5-cyclophilin proteins in two primate species. Proc Natl Acad Sci USA 105(9):3563–3568
Vogel MJ, Guelen L, de Wit E, Peric-Hupkes D, Loden M, Talhout W, Feenstra M, Abbas B, Classen AK, van Steensel B (2006) Human heterochromatin proteins form large domains containing KRAB-ZNF genes. Genome Res 16(12):1493–1504
Welker R, Hohenberg H, Tessmer U, Huckhagel C, Krausslich HG (2000) Biochemical and structural analysis of isolated mature cores of human immunodeficiency virus type 1. J Virol 74(3):1168–1177
Wilson SJ, Webb BL, Maplanka C, Newman RM, Verschoor EJ, Heeney JL, Towers GJ (2008a) Rhesus macaque TRIM5 alleles have divergent antiretroviral specificities. J Virol 82(14):7243–7247
Wilson SJ, Webb BL, Ylinen LM, Verschoor E, Heeney JL, Towers GJ (2008b) Independent evolution of an antiviral TRIMCyp in rhesus macaques. Proc Natl Acad Sci USA 105(9):3557–3562
Wiznerowicz M, Jakobsson J, Szulc J, Liao S, Quazzola A, Beermann F, Aebischer P, Trono D (2007) The Kruppel-associated box repressor domain can trigger de novo promoter methylation during mouse early embryogenesis. J Biol Chem 282(47):34535–34541
Wolf D, Cammas F, Losson R, Goff SP (2008a) Primer binding site-dependent restriction of murine leukemia virus requires HP1 binding by TRIM28. J Virol 82(9):4675–4679
Wolf D, Goff SP (2007) TRIM28 mediates primer binding site-targeted silencing of murine leukemia virus in embryonic cells. Cell 131(1):46–57
Wolf D, Goff SP (2009) Embryonic stem cells use ZFP809 to silence retroviral DNAs. Nature 458(7242):1201–1204
Wolf D, Hug K, Goff SP (2008b) TRIM28 mediates primer binding site-targeted silencing of Lys1,2 tRNA-utilizing retroviruses in embryonic cells. Proc Natl Acad Sci USA 105(34):12521–12526
Woo JS, Imm JH, Min CK, Kim KJ, Cha SS, Oh BH (2006a) Structural and functional insights into the B30.2/SPRY domain. EMBO J 25(6):1353–1363
Woo JS, Suh HY, Park SY, Oh BH (2006b) Structural basis for protein recognition by B30.2/SPRY domains. Mol Cell 24(6):967–976
Wu X, Anderson JL, Campbell EM, Joseph AM, Hope TJ (2006) Proteasome inhibitors uncouple rhesus TRIM5α restriction of HIV-1 reverse transcription and infection. Proc Natl Acad Sci USA 103(19):7465–7470
Xia ZP, Sun L, Chen X, Pineda G, Jiang X, Adhikari A, Zeng W, Chen ZJ (2009) Direct activation of protein kinases by unanchored polyubiquitin chains. Nature 461(7260):114–119
Xu L, Yang L, Moitra PK, Hashimoto K, Rallabhandi P, Kaul S, Meroni G, Jensen JP, Weissman AM, D’Arpa P (2003) BTBD1 and BTBD2 colocalize to cytoplasmic bodies with the RBCC/tripartite motif protein, TRIM5delta. Exp Cell Res 288(1):84–93
Yamauchi K, Wada K, Tanji K, Tanaka M, Kamitani T (2008) Ubiquitination of E3 ubiquitin ligase TRIM5α and its potential role. FEBS J 275(7):1540–1555
Yamauchi M, Freitag B, Khan C, Berwin B, Barklis E (1995) Stem cell factor binding to retrovirus primer binding site silencers. J Virol 69(2):1142–1149
Yap MW, Lindemann D, Stanke N, Reh J, Westphal D, Hanenberg H, Ohkura S, Stoye JP (2008) Restriction of foamy viruses by primate Trim5α. J Virol 82(11):5429–5439
Yap MW, Mortuza GB, Taylor IA, Stoye JP (2007) The design of artificial retroviral restriction factors. Virology 365(2):302–314
Yap MW, Nisole S, Lynch C, Stoye JP (2004) Trim5α protein restricts both HIV-1 and murine leukemia virus. Proc Natl Acad Sci USA 101(29):10786–10791
Ylinen LM, Price AJ, Rasaiyaah J, Hue S, Rose NJ, Marzetta F, James LC, Towers GJ (2010) Conformational adaptation of Asian macaque TRIMCyp directs lineage specific antiviral activity. PLoS Pathog 6(8):e1001062
Yu S, Gao B, Duan Z, Xu W, Xiong S (2011) Identification of tripartite motif-containing 22 (TRIM22) as a novel NF-kappaB activator. Biochem Biophys Res Commun 410(2):247–251
Yueh A, Leung J, Bhattacharyya S, Perrone LA, de los Santos K, Pu SY, Goff SP (2006) Interaction of moloney murine leukemia virus capsid with Ubc9 and PIASy mediates SUMO-1 addition required early in infection. J Virol 80(1):342–352
Zeng L, Yap KL, Ivanov AV, Wang X, Mujtaba S, Plotnikova O, Rauscher FJ 3rd, Zhou MM (2008) Structural insights into human KAP1 PHD finger-bromodomain and its role in gene silencing. Nat Struct Mol Biol 15(6):626–633
Zhang F, Hatziioannou T, Perez-Caballero D, Derse D, Bieniasz PD (2006) Antiretroviral potential of human tripartite motif-5 and related proteins. Virology 353(2):396–409
Zhang J, Hu MM, Wang YY, Shu HB (2012) TRIM32 protein modulates type I interferon induction and cellular antiviral response by targeting MITA/STING protein for K63-linked ubiquitination. J Biol Chem 287(34):28646–28655
Zhang Y, Maksakova IA, Gagnier L, van de Lagemaat LN, Mager DL (2008) Genome-wide assessments reveal extremely high levels of polymorphism of two active families of mouse endogenous retroviral elements. PLoS Genet 4(2):e1000007
Zhao G, Ke D, Vu T, Ahn J, Shah VB, Yang R, Aiken C, Charlton LM, Gronenborn AM, Zhang P (2011) Rhesus TRIM5α disrupts the HIV-1 capsid at the inter-hexamer interfaces. PLoS Pathog 7(3):e1002009
Zuo X, Sheng J, Lau HT, McDonald CM, Andrade M, Cullen DE, Bell FT, Iacovino M, Kyba M, Xu G, Li X (2012) Zinc finger protein ZFP57 requires its co-factor to recruit DNA methyltransferases and maintains DNA methylation imprint in embryonic stem cells via its transcriptional repression domain. J Biol Chem 287(3):2107–2118
Acknowledgments
We are very grateful to Helen Rowe, Leo James, and Stéphane Hué for critically reading the manuscript. Work in the Towers lab is funded by the Wellcome Trust, the Medical Research Council, and the UCL/UCLH National Institute of Health Research Biomedical Research Centre.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Fletcher, A.J., Towers, G.J. (2013). Inhibition of Retroviral Replication by Members of the TRIM Protein Family. In: Cullen, B. (eds) Intrinsic Immunity. Current Topics in Microbiology and Immunology, vol 371. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-37765-5_2
Download citation
DOI: https://doi.org/10.1007/978-3-642-37765-5_2
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-37764-8
Online ISBN: 978-3-642-37765-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)