
Abstract
Adenosine receptors modulate neuronal and synaptic function in a range of ways that may make them relevant to the occurrence, development and treatment of brain ischemic damage and degenerative disorders. A1 adenosine receptors tend to suppress neural activity by a predominantly presynaptic action, while A2A adenosine receptors are more likely to promote transmitter release and postsynaptic depolarization. A variety of interactions have also been described in which adenosine A1 or A2 adenosine receptors can modify cellular responses to conventional neurotransmitters or receptor agonists such as glutamate, NMDA, nitric oxide and P2 purine receptors. Part of the role of adenosine receptors seems to be in the regulation of inflammatory processes that often occur in the aftermath of a major insult or disease process. All of the adenosine receptors can modulate the release of cytokines such as interleukins and tumor necrosis factor-α from immune-competent leukocytes and glia. When examined directly as modifiers of brain damage, A1 adenosine receptor (AR) agonists, A2AAR agonists and antagonists, as well as A3AR antagonists, can protect against a range of insults, both in vitro and in vivo. Intriguingly, acute and chronic treatments with these ligands can often produce diametrically opposite effects on damage outcome, probably resulting from adaptational changes in receptor number or properties. In some cases molecular approaches have identified the involvement of ERK and GSK-3β pathways in the protection from damage. Much evidence argues for a role of adenosine receptors in neurological disease. Receptor densities are altered in patients with Alzheimer’s disease, while many studies have demonstrated effects of adenosine and its antagonists on synaptic plasticity in vitro, or on learning adequacy in vivo. The combined effects of adenosine on neuronal viability and inflammatory processes have also led to considerations of their roles in Lesch–Nyhan syndrome, Creutzfeldt–Jakob disease, Huntington’s disease and multiple sclerosis, as well as the brain damage associated with stroke. In addition to the potential pathological relevance of adenosine receptors, there are earnest attempts in progress to generate ligands that will target adenosine receptors as therapeutic agents to treat some of these disorders.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Neuroprotection
- Neurodegeneration
- Ischaemia
- Alzheimer’s disease
- β-amyloid
- Huntington’s disease
- Parkinson’s disease
- Neurotoxicity
- Aging
- Stroke
- Lesch-Nyhan syndrome
- Multiple sclerosis
- Creutzfeldt-Jacob syndrome
- Prion disease
- Acute administration
- Chronic administration
- Receptor up-regulation
- Receptor down-regulation
1 Introduction
As will be evident elsewhere in this volume, adenosine receptors are essentially ubiquitous, with almost all cell types expressing functional forms of at least one of the four known subtypes (A1, A2A, A2B, A3). Each of these subtypes has been associated with a range of actions, some of which may become over- or underexpressed, over- or underactive. Such a change in activity could lead to abnormalities of tissue function, which may be severe enough to lead to overt disease. In this chapter, the evidence for a possible contribution of adenosine receptors to the processes of neurodegeneration and neurological disorders involving neurodegeneration will be addressed, together with the potential for developing adenosine receptor ligands as therapeutic agents to modify those disorders.
2 Relevant General Features of Adenosine Receptor Actions
2.1 A1 Adenosine Receptors
A1 adenosine receptors occur throughout the central nervous system (CNS), with a high density in the hippocampus and neocortex. The widespread distribution of these receptors is seen in almost all mammalian species examined, including humans (Fastbom et al. 1986, 1987a, b). All cell types in the CNS possess these receptors, including both neurons and microglia (Goodman and Snyder 1982; Lee and Reddington 1986; Rivkees et al. 1995; Fiebich et al. 1996b; Svenningsson et al. 1997; Ochiishi et al. 1999a, b), with neuronal receptors existing on presynaptic terminals and postsynaptic membranes (Ochiishi et al. 1999a, b). Probably the most prominent consequence of activating the A1 adenosine receptor (AR) is the inhibition of neurotransmitter release from synaptic terminals, an action that has been linked to the reduction of calcium influx in response to action potential invasion of the terminals (Wu and Saggau 1997). A1ARs are able to suppress the release of a variety of neurotransmitters, including glutamate (Corradetti et al. 1984; Fastbom and Fredholm 1985; Andine et al. 1990; Butcher et al. 1990), acetylcholine (Spignoli et al. 1984; Brown et al. 1990) and dopamine (Michaelis et al. 1979; Chowdhury and Fillenz 1991). There is a significant degree of specificity in this action, however, since it seems to result primarily in a suppression of release of excitatory transmitters such as the major excitatory transmitter glutamate (Corradetti et al. 1984; Héron et al. 1993; Poli et al. 1991), rather than inhibitory transmitters such as gamma-aminobutyric acid (GABA). While a depression of GABA release can be demonstrated using A1AR agonists, the potency of these compounds and the amount of release inhibition that can be produced are far less than those that have been reported on glutamate release (Hollins and Stone 1980). This difference may be fundamentally important to understanding the relevance of adenosine receptors in neurodegeneration and neuroprotection, since the brain damage which follows strokes and traumatic (mechanical) injuries to the brain (Corsi et al. 1999a) has been attributed to a massive release of glutamate, and it is a suppression of this that may contribute to the neuroprotective efficacy of adenosine A1 (and A2A) AR. The much smaller effect on GABA release means that the risk of reinstating a degree of hyperexcitability, as a result of blocking inhibitory transmission, is greatly reduced.
Activation of A1AR reduces calcium influx, or inhibits calcium availability, as demonstrated in neuronal and cardiac tissues (Dolphin and Prestwich 1985; Fredholm and Dunwiddie 1988; Rudolphi et al. 1992; Scholz and Miller 1992). This may be related to the frequently observed ability of A1AR to modulate the potassium conductances of several types, including the ATP-sensitive potassium channels in heart and hippocampal neurons (Trussel and Jackson 1985; Regenold and Illes 1990; Hosseinzadeh and Stone 1998). There appear to be neuronal chloride conductances which are also sensitive to purines, resulting in an increased chloride influx which should contribute to neuronal inhibition in most areas of the brain (Mager et al. 1990; Schubert et al. 1991).
2.2 A2A Adenosine Receptors
A population of A2AARs is usually distinguished from A2BARs on the basis of the higher affinity of A2AARs for the agonist ligand 2-[4-(2-carboxyethyl)-phenylethylamino]-5′ N-ethyl-carboxamido-adenosine(CGS21680).CGS21680 sho ws an approximately 140-fold selectivity for A2AARs relative to A1ARs, (Bridges et al. 1988; Hutchison et al. 1989; Merkel et al. 1992). The A2AARs occur predominantly on neurons in the striatum, especially the GABAergic striatopallidal projection neurons and on cholinergic interneurons (Jarvis and Williams 1989; Schiffmann et al. 1991; Cunha et al. 1994; Kurokawa et al. 1994; Latini et al. 1996; Ongini and Fredholm 1996; Moreau and Huber 1999). They are also found in the nucleus accumbens and olfactory tubercle, and the hippocampus and cerebral cortex (Cunha et al. 1994; Dixon et al. 1996), although in the last two areas there are significant pharmacological differences between the nominally A2A sites and those classically described in striatum (Cunha et al. 1996). A broadly similar distribution exists in human brain, since, although they were initially reported to exist primarily in striatal regions (Martinez-Mir et al. 1991), subsequent work has shown their presence more widely throughout the CNS (Svenningsson et al. 1997).
There is abundant evidence from a number of biochemical and electrophysiological investigations that the activation of A2AAR promotes the release of neurotransmitters, including glutamate (Sebastiao and Ribeiro 1992; Cunha et al. 1994), an effect probably produced by increasing presynaptic calcium influx (Goncalves et al. 1997). Administration of the A2AAR agonist CGS21680 in vivo does not itself alter the extracellular levels of glutamate in the CNS, but in the rat it can increase the efflux of glutamate triggered by ischemia (Fredholm and Dunwiddie 1988; O’Regan et al. 1992). Consistent with this, the AR antagonist 5-amino-9-chloro-2-(2-furyl)-1,2,4-triazolo[1,5-c]quinazoline (CGS15943) can depress glutamate release, possibly by blocking the enhancing effect of endogenous adenosine at A2AAR (Fredholm and Dunwiddie 1988). The facilitation of release by A2AAR agonists has also been demonstrated for other transmitters such as GABA. Hence it is possible that neuroprotection by A2AAR agonists may result, at least in part, from increased extracellular levels of GABA causing generalized inhibition of cell activity, calcium influx and damage (Mayfield et al. 1993; Kurokawa et al. 1994).
2.3 A2B Adenosine Receptors
The low-affinity A2BAR was cloned in the early 1990s, and has long remained the least known adenosine receptor subtype. The A2B receptor positively couples to both adenylyl cyclase and phospholipase C (PLC), the latter occurring through Gq proteins and representing the most important pathway responsible for A2B-mediated effects (Linden et al. 1999). The A2BAR is expressed at low levels in almost all tissues including brain and spinal cord, and its low affinity for the natural ligand suggests that it could be mainly recruited under pathological conditions.
In the CNS, A2BARs have been suggested to mediate the outgrowth of dorsal spinal cord axons (Corset et al. 2000) and to interact with inflammatory cytokines in the induction of long-term brain responses to trauma and ischemia, such as reactive astrogliosis. A complex interaction between A2BAR and tumor necrosis factor alpha (TNF-α) has been reported, depending upon specific pathophysiological conditions. In particular, prolonged treatment of human astrocytes with the proinflammatory cytokine TNF-α increased the functional responsiveness of A2BAR, which, in turn, synergized with the cytokine in inducing the morphological signs of chronic reactive gliosis (Trincavelli et al. 2004). Conversely, short-term exposure of astrocytes to TNF-α caused the phosphorylation of A2BAR and impairment in their coupling to Gs proteins, with consequent decreases of cyclic adenosine monophosphate (cAMP) production. TNF-α-mediated downregulation of A2BAR was demonstrated to occur via protein kinase C (PKC) intracellular kinase. This event likely represents a defense mechanism to counteract excessive A2B receptor activation under acute damage conditions characterized by massive release of both cytokines and adenosine, such as those occurring during trauma or ischemia (Trincavelli et al. 2008).
A2BARs have been also suggested to inhibit taurine release from pituicytes, the astroglial cells of the neurohypophyses. In whole rat neurohypophyses pre- loaded with [3H]taurine, taurine efflux elicited by hypotonic shocks was about 30–50% smaller in the presence of 10 mM adenosine or 1 mM NECA (5′-N-ethylcarboxamidoadenosine). The A2BAR antagonists MRS1706 {N-(4-acetyl- phenyl)- 2-[4-(2,3,6,7 -tetrahydro-2,6-dioxo-1,3-dipropyl-1H-purin-8-yl)-phenoxy] acetamide} or alloxazine partially reversed the inhibition of release by NECA, while neither agonists of the adenosine A1,A2A or A3 ARs nor the A1AR antagonist DPCPX (8-cyclopentyl-1,3-dipropylxanthine) had any effect (Pierson et al. 2007). Based on evidence implicating taurine not only in cell osmoregulation but also in olfactory, auditory and visual development, as well as in long-term potentiation in the striatum (Warskulat et al. 2007), if confirmed by further studies, this observation may unveil entirely new pathophysiological roles for this as-yet neglected adenosine receptor subtype.
2.4 A3 Adenosine Receptors
The A3ARs (Zhou et al. 1992) have been less well studied than the A1 and A2A AR populations recognized earlier. A3 sites exist primarily in peripheral tissues, but they are believed to occur on neuronal and glial cells membranes in most species examined, including human (Jacobson 1998), although at least one group has reported failing to find either the A3 receptor protein or its mRNA in the CNS (Rivkees et al. 2000). This report was accompanied by claims that several of the purportedly selective ligands used in the functional study of A3 receptor effects actually have significant activity at A1AR that could complicate the interpretation of results, and could possible account entirely for the supposed actions attributed to A3AR.
2.5 Receptor Interactions
There is good evidence for interactions between receptors for adenosine and other neuroactive compounds. For instance, activation of N-methyl-d-aspartic acid (NMDA) receptors can inhibit the actions of A1AR agonists on presynaptic terminals (Bartrup and Stone 1990; Bartrup et al. 1991; Nikbakht and Stone 2001). In a situation in which the levels of glutamate increase significantly, therefore, there is a real danger that the protective activity of endogenous adenosine could be compromised by NMDA receptors. The direction of receptor interactions is reversed at postsynaptic sites. On hippocampal and striatal neurons, for example, adenosine can depress the activation of NMDA receptors. This action can be produced by A1 or A2AAR (de Mendonça et al. 1995; Norenberg et al. 1997, 1998; Wirkner et al. 2000, 2004; Gerevich et al. 2002). The relevance of these interactions remains unclear, as do the circumstances under which one or the other would be dominant. Thus, if an increase in the ambient levels of glutamate occurs prior to any elevation of adenosine levels, then a loss of AR-mediated protection would be expected, leading to enhanced cell damage. If, however, any increase in adenosine levels precedes a change in glutamate, the purine could limit the release of the amino acid, and block the activation of NMDA receptors by the lower amounts of glutamate present.
There may be a significant contribution to A2AAR antagonist neuroprotection by the modulation of responses to other neuroactive agents via influences directly on the receptors. Simpson et al. (1992) and later Cunha et al. (1994) reported the ability of A2AAR to antagonize the activation of A1AR, a proposal subsequently confirmed and supported by other groups (Dixon et al. 1997; O’Kane and Stone 1998; Latini et al. 1999). The A2A agonist CGS21680 inhibits neuronal responses to the A1 ligand CPA (O’Kane and Stone 1998), an action that may be related to its ability to induce a low-affinity site for the highly selective A1 receptor agonist 2-chloro-N 6-cyclopentyladenosine (CCPA) (Dixon et al. 1997). This interaction may occur between the membrane receptors themselves, or via an intermediate, diffusible messenger. Both A1 and A2AARs suppress the electrophysiological effects of glutamate or NMDA applied directly to neurons (de Mendonça et al. 1995; Norenberg et al. 1997, 1998; Gerevich et al. 2002; Wirkner et al. 2000, 2004), while CGS21680 reduces the increased postsynaptic influx of calcium induced by quinolinic acid, and the A2AAR antagonist SCH58261 increases it (Popoli et al. 2002), although another antagonist 4-(2-[7-amino-2-(2-furyl)(1,2,4)-triazolo(2,3-a)-(1,3,5)triazin-5-yl-amino]-ethyl)phenol (ZM241385) appears not to do so (Tebano et al. 2004). One implication of this interaction is that, when the extracellular level of adenosine reaches levels sufficient to activate A2AAR, as it can do after kainate administration or ischemia, it may inhibit A1 receptor function. This phenomenon may explain the curious observation that neuroprotection by ZM241385 is lessened by DPCPX (Jones et al. 1998a, b). The protection by ZM241385 could be due to its blockade of A2AAR, thus “releasing” A1AR from tonic suppression by A2AAR. If the heightened activation of A1AR were then responsible for the neuroprotection, it would be prevented by DPCPX, as observed (Jones et al. 1998a, b).
As in the case of the A2A agonists noted above, there is evidence that agonists at A3AR, administered acutely, may reduce responses to A1AR agonists, and thus decrease the protective activity of endogenous adenosine levels (von Lubitz et al. 1999a). On the contrary, a chronic activation of A3AR exerts protective effects, as detailed below (see Sect. 3.4). However, it is not known whether these effects are mediated by an opposite activity on the A1AR subtype.
Finally, there is evidence that some AR subtypes can physically interact with other neurotransmitter receptors, leading to the generation of receptor heteromers characterized by unique pharmacological properties. Yoshioka et al. (2001) co-expressed A1AR and P2Y1 receptor for ADP in HEK293 cells. These receptors co-immunoprecipitated in western blots of whole cell membrane lysates. Coexpressing the P2Y1 receptor did not alter surface expression of the A1 receptor, but it did inhibit the binding of radiolabeled A1AR agonists and antagonists in membrane preparations. This change was not seen in a mixture of membranes from cells expressing each receptor individually. Additionally, the binding of an A1AR agonist was displaced by the P2Y1 agonist ADPβS and the P2Y1 antagonist N 6-methyl-2′-deoxyadenosine-3′, 5′-bisphosphate (MRS2179) in cotransfected cells, but not in cells expressing the A1 receptor only. Globally, these data indicate formation of a functional heteromeric complex where A1ARs physically interact with P2Y1 receptors (Abbracchio et al. 2006).
A1ARs couple to Gi, mediating depression of intracellular cAMP levels, whereas P2Y1 receptors interact with Gq∕11 and have no effect on cAMP. ADPβS inhibited cAMP production in co-transfected cells only, an effect that was antagonized by the A1 antagonist DPCPX, but not by MRS2179, and was abolished by pertussis toxin. Thus, ADPβS appears to have acted via the A1AR ligand-binding site; i.e., the P2Y1∕A1 dimer has novel pharmacological properties compared with the parent receptors. Interestingly, although ADPβS induced inositol phosphate synthesis, the A1 agonist cyclopentyl adenosine (CPA) did not. Thus, dimerization did not lead to a complete change in pharmacological properties in this case.
Using confocal laser microscopy to study the subcellular distribution of the P2Y1 and A1AR, Yoshioka et al. (2001) showed that both were expressed mainly near the plasma membrane of HEK293 cells. Furthermore, there was a strong overlap in their distribution in individual cells. This was confirmed in a subsequent study using the biophysical technique of bioluminescence resonance energy transfer (Yoshioka et al. 2002b). In the absence of agonists, the receptors showed a homogeneous colocalization across the cells. Addition of ADPβS and CPA together, but not alone, induced an increase in the bioluminescence resonance energy transfer ratio over 10 min. Thus, although the receptors have a constitutive association, their coactivation increased the association. This association was also seen with native receptors in central neurons. Using confocal laser microscopy and double immunofluorescence, Yoshioka et al. (2002a) demonstrated that the P2Y1 and A1AR colocalized in neurons of the rat cortex, hippocampus, and cerebellum. A direct association was then shown by their coimmunoprecipitation in membrane extracts from these regions.
The structural requirements for the receptor–receptor interaction are not known at present. The physiological roles of the P2Y1∕A1 dimer also remain to be determined, although Nakata et al. (2003) have pointed out that its pharmacological properties resemble those of a presynaptic receptor that mediates inhibition of neurotransmitter release in some tissues. Finally, Yoshioka et al. (2001) reported that the rat P2Y2 receptor also coimmunoprecipitated with the A1 receptor when they were coexpressed in HEK293 cells. Thus, the formation of oligomers by A1AR receptors is likely to be widespread and to greatly increase the diversity of purinergic signaling.
In a similar way, A2AARs have been demonstrated to dimerize with D2 receptors, an interaction which involved peculiar peptide residues (Canals et al. 2003). The formation of A2A∕D2 receptor heteromers in the plasma membrane contributes to explain the early observation of agonist affinity loss at the D2 receptor after activation of the A2AAR (Ferré et al. 1991) and provides a molecular explanation to the functional interaction between adenosine and dopamine in basal ganglia.
2.6 Anti-inflammatory Effects
One line of argument that tissue protection by purines is more dependent on modulation of the immune system than on neurotransmitter release or activity is that protection against damage is shown in a range of tissues besides the CNS. Adenosine antagonizes the release and actions of several proinflammatory cytokines such as TNF-α and complement (Lappin and Whaley 1984; Cronstein et al. 1992; LeVraux et al. 1993; Barnes et al. 1995; Ritchie et al. 1997). A2AARs specifically inhibit the production of IL-12 by human monocytes but increase the generation of IL-10 (Link et al. 2000). This ability to modulate the relative release of several cytokines could be a significant factor in determining the overall immune profile that occurs in response to different primary activating stimuli in different inflammatory situations. Adenosine suppresses phagocytosis, free radical generation and cell adherence by white blood cells activated by immune stimulation (Cronstein et al. 1985, 1987, 1990, 1992; Burkey and Webster 1993; Cronstein 1994). There is now clear evidence that A2AAR play a major role in this form of cellular regulation (Dianzani et al. 1994; Hannon et al. 1998), probably acting via the activation of a serine/threonine protein phosphatase (Revan et al. 1996). Most strikingly, adenosine receptors protect the heart against damage occasioned by ischemia (Zhao et al. 1993; Matherne et al. 1997). Indeed, all anti-inflammatory actions of adenosine have been demonstrated in the myocardium, including suppression of TNF-α production (Meldrum et al. 1997; Wagner et al. 1998a, b; Cain et al. 1998) and regulation of neutrophil adherence to myocytes (Bullough et al. 1995). There is, however, some confusion as to the nature of the ARs involved. Human neutrophils possess A1 and A2AARs (Varani et al. 1998) and Cronstein et al. (1992) have demonstrated that both receptors are able to modulate several aspects of the immune response, including chemotaxis. Lozza et al. (1997) have suggested that A1 and A2AAR agonists are both able to protect the heart against ischemia/reperfusion injury, but there are reports that A1 agonists but not A2AAR agonists provide cardiac protection (Casati et al. 1997), whereas other groups have claimed the opposite (Cargnoni et al. 1999). The former claim is more consistent with evidence that resistance to myocardical ischemia is correlated with the level of expression of A1AR. In most cases, the two populations of receptor exhibit opposing actions, suggesting that their joint presence could be the basis of a control system in which low concentrations of adenosine, via A1AR, are normally able to enhance the sensitivity of white blood cells to immune stimuli but, at the higher concentrations likely to occur at the time of an established immune response, A2AAR can restrain the extent of cellular activity (Cronstein et al. 1992).
The regulation of cytokines by A3AR is quite selective. Production of several cytokines, including some such as IL-1β and IL-6, which are also proinflammatory, can be modified by A3AR activation (Ramakers et al. 2006). A3AR may also suppress the oxidative burst that accompanies the response of defensive leucocytes to immune activation. They can reduce superoxide generation in human eosinophils (Ezeamuzie and Philips 1999), for example, although there is apparently no similar suppression of oxidative activity in human neutrophils (Hannon et al. 1998). The former action could be secondary to an increase in the level of antioxidant enzymes, including superoxide dismutase, which has been shown to be produced by A3AR agonists in endothelial cells (Maggirwar et al. 1994).
3 Role of Adenosine Receptors in Brain Cell Survival and in Neurodegenerative Diseases
3.1 A1 Adenosine Receptors and Neuroprotection
Although much of the interest in the therapeutic value of purine receptor ligands has centered on protection following strokes, there remains the possibility that overactivation of glutamate receptors may contribute to chronic neurodegenerative disorders such as Alzheimer’s disease and Huntington’s disease. This possibility is the rationale for studying the protective effects of agents against excitotoxins, which are frequently used as a model of stroke and neurodegenerative disease. The most commonly used excitotoxins are kainic acid and quinolinic acid, a tryptophan metabolite for which the evidence for a role in some degenerative disorders is substantial (see Stone 1993, 2001; Stone and Darlington 2002 for reviews). Not only do they produce a controllable degree and extent of injury, but the mechanisms of damage have much in common with natural causes. Thus, even the damage produced by kainic acid probably involves a presynaptic action of kainate, which induces the release of endogenous compounds such as glutamate and aspartate (Kohler et al. 1978; Ferkany et al. 1982; Lehmann et al. 1983; Jacobson and Hamberger 1985; Connick and Stone 1986; Virgili et al. 1986; Okazaki and Nadler 1988). Whether this secondary release is the primary cause of cell death or only a contributory (perhaps permissive) component is irrelevant, since the essential issue is that the inhibition of their release by an agent such as an adenosine A1AR agonist will have the same net protective activity.
Both A1AR agonists and A2AAR agonists and antagonists (see also below) can protect against kainic acid-induced damage (MacGregor and Stone 1993; Jones et al. 1998a, b). R-Phenylisopropyladenosine (R-PIA) protected against kainic acid neurotoxicity in several regions of the CNS in addition to the hippocampus (MacGregor and Stone 1993; MacGregor et al. 1993, 1996), the involvement of A1AR being further confirmed by showing that protection could be prevented by the simultaneous administration of an A1AR antagonist such as DPCPX. In addition to the use of DPCPX to confirm the involvement of A1AR, there have been several studies showing that DPCPX and other selective A1AR blockers increase the amount of neuronal damage after ischemia or the administration of excitotoxins (Rudolphi et al. 1987; von Lubitz et al. 1994a; Phillis 1995). R-PIA prevented the kainate-induced damage in areas such as the basolateral amygdala, pyriform cortex and rhinal fissure. An observation that has not been pursued, but which may be of considerable pathological and therapeutic importance, was that some areas of the brain, especially those located in more caudal regions such as the entorhinal cortex, the posteromedial cortical amygdaloid nucleus and the amygdalopyriform transition, were not protected by A1AR activation. It is still uncertain why R-PIA showed such regionally selective protection. There may be fewer A1ARs in the resistant areas, or a greater susceptibility to damage which the adenosine agonist was unable to overcome at the doses used. The protection afforded by A1ARs does not necessarily require the use of a selective exogenous agonist, since compounds which raise the concentrations of endogenous adenosine, either by inhibiting transporter function or adenosine metabolism, can also produce protection (Parkinson et al. 1994; Pazzagli et al. 1994).
There is an even greater contribution of presynaptic release in the neuronal damage caused by quinolinic acid, although its major action seems to be the activation of NMDA receptors and the generation of reactive oxygen species (Stone and Darlington 2002; Stone 2001).
A number of other studies have demonstrated protection by adenosine analogs against damage produced by toxins or excitotoxins (Arvin et al. 1989; Connick and Stone 1989; Finn et al. 1991). One especially interesting report showed that protection could be produced by the adenosine A1 receptor agonist N 6-cyclohexyladenosine (CHA) against the selective dopaminergic neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (Lau and Mouradian 1993). This protection raises the real possibility that a selective A1AR agonist could be useful in Parkinson’s disease, where a proportion of cases may be caused by the exposure of patients to exogenous toxins with molecular structures and a propensity to generate oxidative stress similar to those of MPTP. However, the mechanism of protection against MPTP remains unclear, although antagonists at NMDA receptors can also block MPTP damage, raising the possibility that glutamate receptors may play a critical role comparable to that exhibited by them in stroke-induced damage, and against which A1AR agonists are also effective.
3.2 A2A Adenosine Receptors and Neuroprotection
At variance from the clearcut neuroprotective role exerted by A1AR, contrasting data have been reported so far on the beneficial/detrimental roles mediated by A2AAR on brain cells.
As with the A1AR, very early studies indicated that agonists at A2AAR can produce protection of the CNS against several insults, including ischemia (Phillis 1995; Sheardown and Knutsen 1996), and excitotoxins such as kainate (Sperk 1994; Jones et al. 1998a, b). However, protection by CGS21680 was largely prevented by 8-(p-sulfophenyl)-theophylline (8PST), a nonselective xanthine antagonist that blocks both A1 and A2ARs. Since this antagonist does not penetrate the blood–brain barrier, it was suggested that the protective activity of CGS21680 was generated via sites on the systemic rather than central side of the barrier. The effect was believed to be primarily exerted on the vascular system, modifying blood flow to the potentially damaged regions of brain, or on white blood cells of the immune system, reducing their penetration to and activation by the early neuronal damage. This conclusion was supported by findings that administering CGS21680 directly into the hippocampus did not induce protection.
On the other hand, neuroprotection by an antagonist at A2AAR was first reported by Gao and Phillis (1994). They found that the nonselective A2AR antagonist CGS15943 protected the gerbil brain against ischemic damage, an observation later supported using the more selective compounds 8-(3-chlorostyryl)caffeine (CSC) and 4-amino-1-phenyl[1,2,4]-triazolo[4,3-a]quinoxaline (CP66,713) (Phillis 1995). Many of the earlier studies examined protection against global cerebral ischemia, but protection has also been demonstrated against focal ischemic damage (Ongini et al. 1997). More recent work has involved a range of different receptor ligands and ischemic models (von Lubitz et al. 1995b; Sheardown and Knutsen 1996; Monopoli et al. 1998). In addition, protection by A2AAR antagonists occurs against excitotoxins such as kainic acid, glutamate and quinolinic acid (Jones et al. 1998a, b). The ability of A2AAR antagonists to protect the CNS has received strong support from the generation of transgenic mice lacking these receptors. These knockout animals exhibit a significantly lower level of brain injury following excitotoxins or ischemia (Bona et al. 1997; Chen et al. 1999).
Interestingly, a possible mechanism at the basis of the neuroprotective effects of A2AAR antagonists may reside in blockade of A2AAR-mediated glutamate release by astrocytes.
Adenosine causes a two- to threefold increase in glutamate release from cultured hippocampal astrocytes (Nishizaki et al. 2002; Nishizaki 2004). Such an effect is mimicked by the A2AAR agonist CGS21680 and inhibited by the A2AAR antagonist 3,7-dimethyl-1-propargylxanthine (DMPX), but not by the A1AR antagonist 8-cyclopentyltheophylline (8-CPT) (Li et al. 2001; Nishizaki et al. 2002). These observations suggest that adenosine stimulates vesicular glutamate release from astrocytes via A2AAR. This agrees with recent findings demonstrating that the A2A receptor antagonist ZM241385 (5 nM via probe) completely prevents the increase in extracellular glutamate outflow induced by dihydrokainic acid, a blocker of glial glutamate uptake (Pintor et al. 2004).
More recently, however, the equation A2A receptor blockade = neuroprotection has appeared too simplistic (in this respect, see Popoli et al. 2007). First, it is now definitely clear that, besides mediating “bad” responses (for example, stimulation of glutamate outflow and excessive glial activation), A2AARs also promote “good” responses (such as trophic and anti-inflammatory effects). This implies that blockade of A2AAR can result in either protoxic or neuroprotective effects according to the mechanisms involved in a given experimental model.
Confirmation that A2AAR activation could be neuroprotective came with the development of more selective compounds. Thus, ZM241385 is highly selective for A2AAR, with an approximately 80-fold greater affinity at A2AAR compared with A2BAR. It has an affinity for A2AAR that is around 1,000 times greater than for A1AR (Palmer et al. 1995). When examined for its ability to protect the CNS against kainic acid, ZM241385 was as effective as the agonist ligand CGS21680. Indeed, the agonist and antagonist together produced a synergistic protection leading to the complete protection of hippocampal neurones (Jones et al. 1998a, b).
To explain these puzzling results, several hypotheses have been invoked, including different degrees of presynaptic versus postsynaptic A2A receptor blockade. The question of presynaptic versus postsynaptic sites of action of A2AAR has been explored by Blum et al. (2003a), with the conclusion that the overall response will depend on the balance of involvement of the former, at which A2AAR activation appears to be deleterious, whereas A2AAR stimulation is protective at postsynaptic sites. In line with this hypothesis, the increase in intracellular calcium levels induced by quinolinic acid in striatal neurons (an effect mediated by postsynaptic NMDA receptors) is significantly potentiated by the A2AAR antagonist 7-(2-phenylethyl)-5-amino-2-(2-furyl)-pyrazolo-[4,3-e]-1,2,4-triazolo[1,5-c]-pyrimidine (SCH58261) and prevented by the A2AAR agonist CGS21680 (Popoli et al. 2002). In agreement, CGS-21680 was reported to reduce NMDA currents in striatal neurons (Norenberg et al. 1997; Wirkner et al. 2000). Moreover, ZM241385 potentiated NMDA-induced effects in rat corticostriatal slices (Tebano et al. 2004), and the A2AAR antagonist CSC potentiated NMDA-induced toxicity in the hippocampus (Robledo et al. 1999). Thus, as far as NMDA-dependent toxicity is concerned, it seems that A2AAR activation, rather than its blockade, can exert neuroprotective effects.
However, the activity of A2AAR antagonists on the “postsynaptic side” of excitotoxicity appears to be far more problematic. At variance from protective receptors on postsynaptic neuronal cells, postsynaptic A2AARs localized on microglial inflammatory cells might play a detrimental role. In addition, A2AARs expressed by bone marrow-derived cells have been proposed as potential contributors to striatal damage induced by mitochondrial dysfunctions in Huntington’s and Parkinson’s disease (Huang et al. 2006), as was previously suggested in the ischemic context (Yu et al. 2004). In accordance with these findings, in an established in vitro model of reactive astrogliosis, blockade of A2AARs abolished growth-factor mediated astrocytic activation, an event that may potentially contribute to inflammation and neuronal damage in neurodegenerative diseases (Brambilla et al. 2003).
Finally, A2AARs can mediate neuroprotection by potentiating brain-derived neurotrophic factor (BDNF) survival signaling pathways. The first link between BDNF and adenosine was provided in 2001, with the demonstration that the activation of tropomyosin-related kinase (Trk)A receptors in PC12 cells and TrkB in hippocampal neurons could be obtained in the absence of neurotrophins by treatment with adenosine (Lee and Chao 2001). These effects were reproduced by using the adenosine agonist CGS21680 and were counteracted with the antagonist ZM241385, indicating that this transactivation by adenosine involves the A2AAR subtype. At hippocampal synapses, presynaptic activity-dependent release of adenosine, through the activation of A2AAR, facilitates BDNF modulation of synaptic transmission (for a review, see: Popoli et al. 2007). A similar positive interaction has more recently been confirmed to occur at the neuromuscular junction, which possesses both adenosine A2AAR and BDNF TrkB receptors. The following sequence of events in what concerns cooperativity between A2AAR and TrkB receptors has been suggested: A2AARs activate the PKA pathway, which promotes the action of BDNF through TrkB receptors coupled to PLCγ, leading to the enhancement of neuromuscular transmission (Pousinha et al. 2006; see also below). Preliminary data indicate that A2AARs also regulate BDNF levels in the striatum. The importance of A2AAR in regulating BDNF has recently been strengthened by the demonstration that both BDNF levels and functions are significantly reduced in the brains of A2AAR knockout (KO) mice (Popoli et al. 2007).
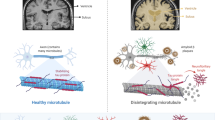
The possible detrimental/beneficial effects elicited by A2AAR activation or blockade on different brain cell populations are summarized in Fig. 1.

Schematic representation of the possible effects elicited by A2A adenosine receptor (AR) activation or blockade on different brain cell populations. In the presynaptic neurons, A2AAR blockade may exert beneficial effects through the inhibition of glutamate release. In the postsynaptic neurons, adenosine A2AARs inhibit N-methyl-d-aspartate (NMDA) receptor currents and activate tropomyosin-related kinase (Trk)B receptors, both being potentially beneficial effects. The picture is further complicated by the different effects elicited by the stimulation or blockade of A2AARs expressed on non-neuronal cells. In astrocytes, A2AAR stimulation can induce both deleterious effects by an increase in glutamate outflow (for a more detailed description of the effects elicited by A2AARs on glial-mediated modulation of glutamate outflow, see the main text), and beneficial effects through an inhibition of nitric oxide (NO) and tumor necrosis factor alpha (TNF-α) release. This latter beneficial effect has been observed also in microglial cells, although the stimulation of A2AARs can also induce potentially deleterious effects on this cell population (see also Saura et al. 2005). Finally, in bone marrow-derived cells, it seems to be the blockade of A2AARs that, through the reduction of cytokine release, can induce beneficial effects. Reproduced and modified from Popoli et al. (2007) with permission from Elsevier
3.3 A2B Adenosine Receptors and Neuroprotection
Far less is known about the role of A2BARs in neuroprotection compared to that of A1 and of A2AARs. As already mentioned, expression of A2BARs on glial cells and their lower affinity for adenosine suggests a role under emergency conditions (when adenosine levels are massively increased) in mediating long-term inflammatory changes. In line with this hypothesis, A2BARs were found to synergize with the proinflammatory cytokine TNF-α in mediating the induction of reactive astrogliosis (see Trincavelli et al. 2004).
3.4 A3 Adenosine Receptors and Neuroprotection
A dual, biphasic role of A3AR in neuroprotection has been described in several experimental models, both in vivo and in vitro. In fact, von Lubitz et al. clearly demonstrated that an acute administration of the A3AR selective agonist N 6-(3-iodobenzyl)adenosine-5′-N-methyluronamide (IB–MECA) to gerbils dramatically worsened the outcome of a subsequent ischemic episode, whereas chronic stimulation of this receptor subtype protected the animals from stroke, probably through the induction of preconditioning (von Lubitz et al. 1999b; see also below). The protective action of A3AR agonists against ischemic damage has been recently confirmed by Chen et al. (2006), who also showed that neuroprotection was completely lost in A3 knockout mice, thus demonstrating the specific involvement of this receptor subtype. Similar results have been obtained in in vitro models. In fact, in non-neuronal cells, low concentrations of the A3AR agonist 2-chloro-N 6-(3-iodobenzyl)adenosine-5′-N-methyluronamide (Cl–IB–MECA; 10 nM or 1 μM) protected against the cell death induced by selective antagonists at this receptor subtype (Yao et al. 1997). Thus, it is suggested that there is a tonic low level of A3AR activation, possibly induced by the release of endogenous adenosine, which results in cell protection. Protection by Cl–IB–MECA against cell death has been also demonstrated in primary cortical cultures subjected to oxygen–glucose deprivation (Chen et al. 2006). Opposite toxic effects can be achieved when concentrations of agonists ≥ 10 μM are used. This has been proven true in several non-neuronal cell lines, with induction of apoptosis and Bak expression (Yao et al. 1997), but also in rat cerebellar granule cells (Sei et al. 1997) and in astrocytic cultures (Abbracchio et al. 1998; Di Iorio et al. 2002), where the reduction of the Bcl-2 expression and the activation of the proapoptotic enzyme caspase 3 by Cl–IB–MECA have been demonstrated (Appel et al. 2001).
3.5 Adenosine Receptors and Therapeutic Possibilities
These various findings have aroused great interest in the search for new drugs that could be used to slow or prevent the neuronal damage that characterizes neurodegenerative conditions such as Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease. That interest is attributable not only to the efficacy of the compounds available, but also to the fact that they should be relatively free of major side effects. Whereas the use of A1AR agonists would lead to a suppression of transmitter release at many sites within the central and peripheral nervous system, whatever the physiopathological state of those sites, A2AAR antagonists should only produce effects when the receptors are being activated by endogenous adenosine. In practice, this means that A2AAR antagonists have little effect on heart rate, blood pressure, or other vital signs under normal conditions. During ischemia in the brain, however, the levels of adenosine may rise to levels at which A2AAR are activated. Stimulation of A2AAR increases the release of the excitotoxic amino acid glutamate (O’Regan et al. 1992; Popoli et al. 1995), which would tend to cause or facilitate the occurrence of damage. Under these circumstances, A2AAR antagonists should reduce the enhanced release of glutamate and thus decrease the extent of neuronal damage. Their beneficial activity would therefore be restricted to those areas of the brain experiencing ischemia, with little or no effect on other areas of the brain or peripheral tissues.
A particularly exciting aspect of A2AAR protection is that it may contribute to the long-term benefits of treating patients with Parkinson’s disease with A2A receptor antagonists. It is clear that A2AARs potently modulate cell sensitivity to dopamine receptors, accounting for the beneficial effects of adenosine antagonists in this disease (Mally and Stone 1994, 1996, 1998). This phenomenon has led to clinical trials with A2AAR antagonists in Parkinson’s disease with promising, though as-yet unpublished, results. In lower primates, A2AAR antagonists are certainly effective against toxin-induced models of the disorder (Kanda et al. 1998; Grondin et al. 1999). The occurrence of protection has been supported strongly by the demonstration that MPTP was able to produce little damage in transgenic mice engineered to be deficient in A2AAR (Ongini et al. 2001).
As noted earlier, the protective effect of A2AAR antagonists may be the result of their removal of the A2AAR suppression of A1AR (Jones et al. 1998a, b; Pedata et al. 2001). Since there is clear evidence that A1AR activation is protective, this interaction would explain both the protection by A2AAR antagonists and the blockade of that protection by A1AR antagonists (Jones et al. 1998a, b). A1AR activation suppresses excitatory transmitter amino acid release, as does blockade of A2AAR by CGS15943 (Simpson et al. 1992), whereas blockade of A1AR or activation of A2AAR enhances the release.
3.6 Molecular Basis of Neuroprotection
There has to date been little progress in identifying the molecular basis of the neuroprotective activity of adenosine receptors, quite apart from identifying the relative importance of neurons and glia in neuroprotection. Staurosporine is a well-recognized activator of apoptosis, and in many cells, including astrocytes, this activity is accompanied by caspase 3, p38 mitogen-activated protein kinase (MAPK) and glycogen synthase kinase 3β(GSK3β) activation (D’Alimonte et al. 2007). The induction of apoptosis can be prevented by CCPA at A1 receptor-selective concentrations that are blocked by DPCPX. In addition, these authors noted that CCPA induced the phosphorylative activation of Akt and thus activation of phosphatidylinositol 3-kinase (PI3K), leading to the proposal that this action caused inhibition of the staurosporine effects. The same group has reported a similarly protective action of CCPA against astrocyte apoptosis induced by the quasi-ischemic procedure of oxygen/glucose deprivation (Ciccarelli et al. 2007). Abnormalities in both the p38 and GSK3β pathways have been implicated in the neuronal damage following acute (stroke) and chronic (Alzheimer’s disease) neurodegenerative conditions, so that the modulation of adenosine A1 receptor function may have a more fundamental and direct relevance to cell protection in these cases than merely a global influence on cell excitability or transmitter release. Protection was again accompanied by activation of PI3K. Pharmacological modifiers of apoptosis led to the overall conclusion that A1 receptor activation protects by activating the PI3K and extracellular signal-regulated kinase 1 and 2 (ERK1/2) MAPK pathways.
Some of the mechanisms at the basis of A2AAR-mediated neuroprotection have already been described above (see Sect. 3.2). In addition, part of the neuroprotective effects of A2AAR may stem from the reduction of nitric oxide production. Saura et al. (2005) reported that CGS21680 potentiated the lipolysaccharide-induced increase of NOS expression and NO production in mixed neuron/glial cultures, whereas ZM241385 blocked the effect. Similarly, Fiebich et al. (1996b) have shown that the activation of A2AAR can induce the expression of COX-2, a key proinflammatory molecule giving rise to eicosanoids and, indirectly, to increased oxidative stress. The A2AAR antagonists could therefore suppress this expression as part of their neuroprotective mechanism (in this respect, see also Sect. 3.6).
The mechanisms underlying the protective effects exerted by low doses of A3AR agonists have not been clearly understood. In an in vivo model of ischemia, protection by IB–MECA appears to be associated with preservation of cytoskeletal proteins (such as microtubule-associated protein) and increased deposition of glial fibrillary acidic protein in injured areas (von Lubitz et al. 1999b). This would accord with studies in vitro, using glial cultures, in which Cl–IB–MECA induced a number of cytoskeletal changes with the formation of actin filaments (the so-called “stress fibers”) accompanied by alterations of cell morphology, such as the emission of long and thick processes in parallel with the alteration of cytoskeletal-associated RhoGTPases (Abbracchio et al. 1997). These changes resulted in a significant reduction of spontaneous apoptosis in culture (Abbracchio et al. 1998), suggesting that astrocytes exposed to nanomolar concentrations of A3AR agonists are more resistant to cell death, probably due to increased adherence to the culture substrate. Therefore, it can be envisaged that neuroprotection observed in vivo could (at least in part) be due to the beneficial effects of A3AR agonists on astrocytes, which might in turn help neurons to survive the ischemic episode.
3.7 Trophic Activity
It is possible that an important feature of adenosine receptor activation or blockade contributing to the regulation of neuronal and glial function and viability is the ability of these receptors to directly influence the growth and development of nerve and glial cells. Much of this work has been performed and reviewed by Rathbone et al. (1992, 1999). Both A1 and A2AARs can promote neuritogenesis in neuroblastoma cells, the A2AAR acting via PKA (Canals et al. 2005). Trophic effects are exerted by A2AARs via a positive synergistic interaction with BDNF prosurvival pathways (see above). This interaction occurred through activation of PI3K/Akt via a Trk-dependent mechanism, resulting in increased cell survival after nerve growth factor or brain-derived neurotrophic factor withdrawal.
The ability of adenosine to mediate trophic effects via activation of its receptors presents yet another factor to be considered in using adenosine ligands therapeutically, since antagonists might inhibit a valuable degree of structural reorganization and recovery following a brain insult or limit the degree of damage produced in a degenerative disorder.
4 Aging and Alzheimer’s Disease
4.1 Changes of Adenosine Receptors with Aging
Since ARs can modify the neural release and actions of acetylcholine, one of the neurotransmitters most intimately associated with the loss of cortical afferent neurons arising from the nucleus basalis of Meynert, and therefore the transmitter most commonly linked to the development of Alzheimer’s disease, they have attracted some attention in relation to dementias.
It is interesting to compare the range of studies that have examined adenosine receptors during the normal aging process with those that have concentrated selectively on changes found in the brains of patients with dementias. Animal studies to date have centered largely on A1AR presence and distribution in view of their ability to inhibit transmitter release. Reports of alterations with ageing have been confusing, no doubt (at least in part) due to the differing choices of species, brain region, methodology and ligands employed. Some groups have reported clear decreases in A1AR binding in limited regions of animal brain (Araki et al. 1993; Cunha et al. 1995), whereas others have found more generalized losses (Pagonopoulou and :̧def :̧def Angelatou 1992) or no change (Virus et al. 1984; Hara et al. 1992; Fredholm et al. 1998) with aging. In one of the earliest of these studies, the loss of a low-affinity subtype of A1AR was described (Corradetti et al. 1984), although binding was examined using an agonist ligand, and the pharmacological tools to explore the nature of the receptor in more detail were not yet available. A later study using gerbils classified as “middle aged” (16 months old), and which may not therefore have direct relevance for neurodegeneration in the elderly, found significant reductions of A1AR density in the hippocampus compared with young animals (1 month old), whereas increased binding was found in the neocortex (Araki et al. 1993). When changes in the presence of A1AR were studied using quantitative autoradiography in the brains of young, old, and senescent rats (3, 24, or 30 months), the density of receptors diminished with age, although the dynamics of that reduction were very different in the various brain regions examined. Thus, while a gradual decline in receptor numbers was seen in hippocampus, cortical sites were lost only after 24 months of age (Meerlo et al. 2004). Fredholm et al. (1998) noted that, while they could find no change in receptor binding, mRNA for the A1AR was decreased in aging rats, a finding which emphasizes the importance for interpretation of examining the receptor message as well as the protein and, ideally, a measure of receptor function.
Results with A2AARs have been more consistent, usually indicating a reduction in receptor binding in regions of high density such as striatum (Fredholm et al. 1998). Although these changes were statistically significant, the limited magnitude of the change (20% decrease between 6 and 99 weeks of age) leaves open the question of the functional meaning of that change in the light of the innate adaptive plasticity of the brain.
No data on the possible changes of A3AR with age are available at the moment.
4.2 Alterations of Adenosine Receptors in Alzheimer’s Patients
The examination of human brain tissue from patients who died with a confirmed diagnosis of Alzheimer’s disease seems to consistently show a loss of A1AR (Jansen et al. 1990; Kalaria et al. 1990; Ulas et al. 1993; Deckert et al. 1998), especially and most clearly in the hippocampus, a region of the brain most intimately involved in the processes of learning and memory.
Jansen et al. (1990) described a decrease in receptor densities for several neuroactive compounds in post-mortem tissue from Alzheimer’s disease patients. Losses were found in receptors for most of these, including adenosine A1ARs, which were reduced by 46% in the dentate gyrus. An autoradiographic study using DPCPX as a ligand also reported marked decreases in A1AR binding in the outer layers of the dentate gyrus, probably reflecting the loss of perforant path input (Jaarsma et al. 1991). The surprising observation was made that the CA1 and CA3 regions showed no loss of A1AR, despite clear cellular degeneration and reduced numbers of NMDA receptors. Although the difference was attributed to a dendritic location of the A1AR, the recognized association of A1AR with presynaptic terminals leaves open the question of whether the perforant path is far more profoundly affected by degeneration than intrinsic hippocampal fibers.
Ulas et al. (1993) also found a similar decrease in A1 receptor binding in the hippocampus and parahippocampal gyrus of Alzheimer individuals and age-matched controls, with a loss of binding density, though not affinity, in the dentate gyrus (molecular layer). However, decreases were also seen in the CA1 stratum oriens and outer layers of the para-hippocampal gyrus, with subnormal levels of antagonist binding in the CA3 region. Coupling to G proteins was similar in the control and patient populations, indicating a normal transduction pathway for the remaining receptors.
Striatal A1ARs are also decreased in patients with Alzheimer’s disease. Quantitative autoradiography in the post-mortem striatum indicated a reduction of A1-binding sites in Alzheimer’s disease patients compared with matched controls. No comparable change of another presynaptic site, that for kappa opiate receptors, was noted, but the loss of A1AR showed a strong correlation with the decreased activity of choline acetyltransferase measured in the same tissue samples (Ikeda et al. 1993). In contrast, the levels of A1AR and A2ARs appear to be increased in the frontal cortex, in parallel with an increased functional activity of these receptors (Albasanz et al. 2008).
In a fascinating analysis of post-mortem neocortical and hippocampal tissue from patients with Alzheimer’s disease, Angulo et al. (2003) reported a significant colocalization of A1AR with β-amyloid in senile plaques. They also showed that, in human neuroblastoma cells, activation of A1AR activated PKC, p21 Ras and ERK1/2, leading to increased formation of soluble β-amyloid fragments, raising the possibility that agonists at A1AR might be valuable drugs in the treatment of established or late-stage Alzheimer’s disease.
4.3 Adenosine Receptors and Cognition
In considering both the possible role of adenosine receptors in the symptomatology of Alzheimer’s disease, and the potential value of adenosine ligands in treatment, it is clearly important to consider not only histologically or functionally defined neuronal damage but also the reflection of that damage at the behavioral level, especially for cognition.
There is increasing epidemiological evidence for a role of adenosine receptors in cognitive decline with aging. Much of this evidence relates to the use of coffee, which, in several recent studies, has been concluded to produce a protective effect against the cognitive decline in Alzheimer’s disease (van Gelder et al. 2007; Quintana et al. 2007). It is clear, however, that the variations in methodology between studies rather confuse attempts to compare results. It is also clear that the relationship between coffee and cognition is not simple, with major questions remaining, such as the role of caffeine versus other constituents of the brew, and the existence of an optimal coffee intake, above and below which cognitive decline may be enhanced.
Despite this caveat, studies specifically focused on caffeine have reached similar conclusions. The risk of developing Alzheimer’s disease, for example, is inversely related to caffeine consumption (Maia and de Mendonça 2002). Both caffeine and ZM241385 prevent the neuronal toxicity caused by β-amyloid peptide in vitro or in vivo (Dall’lgna et al. 2003, 2007). Caffeine was also effective in the Swedish mutation transgenic mouse model of Alzheimer’s disease, in which cognitive deficits are associated with the induced overexpression of β-amyloid in the brain. Caffeine was able to reduce the β-amyloid load and behavioral indications of cognitive impairment in these mice (Arendash et al. 2006). Associated proteins such as presenilin 1 and β-secretase were also reduced. Confirmation that these effects were likely to be a direct result of actions on the neurons rather than glia or peripheral mechanisms was obtained by showing a similar reduction of β-amyloid formation in neuronal cultures with the same mutation.
Psychological studies have investigated the effects of caffeine on a range of behaviors in human subjects, including vigilance and aspects of learning, as well as in a variety of modified states, including subject age, frequency of caffeine use, level of tolerance or withdrawal, and state of sleep deprivation. However, the relevant doses and their molecular mechanism of action often remain unproven. In a representative study, Riedel et al. (1995) noted that, in healthy subjects, 250 mg of caffeine reduced the scopolamine-induced performance deficit in memory tasks. The provocative conclusion was drawn that any cognition-enhancing drug being considered for therapeutic use should be shown to be at least as active as this dose of caffeine: an amount equivalent to only three cups of coffee. Results of the many studies on caffeine are, however, often confusing. In one study of almost 1,000 people, it was reported that the consumption of (caffeinated) coffee was associated with improved cognitive performance in women, especially those aged over 80 years, but not men. A possible attribution of this finding to caffeine was made on the basis that decaffeinated coffee seemed to have no influence on cognitive function (Johnson-Kozlow et al. 2002).
It seems likely that the effects of adenosine antagonists, especially the nonselective ones such as caffeine, may have quite subtle effects on learning. Angelucci et al. (2002) suggested that this was due to an effect to improve memory retention, with less or no effect on memory acquisition, while Hauber and Bareiss (2001) showed an improvement by theophylline of spatial reference memory when acquisition was achieved under light conditions, but not in the dark.
Whereas most studies have found that agonists at A1ARs tend to impair learning and memory function (Normile and Barraco 1991; Zarrindast and Shafaghi 1994; Corodimas and Tomita 2001), there are occasional reports of learning facilitation or improvement after the acute (Hooper et al. 1996) or chronic (von Lubitz et al. 1993) administration of an agonist. Antagonists have clear ability to enhance cognition and to reverse induced cognitive deficits. One of the earliest studies on animal learning used the compound RS-(—)-8-(3-oxocyclopentyl)-1,3-dipropyl-7H-purine-2,6-dione (KFM19), an A1AR antagonist, which showed cognition-enhancing properties in a rat model (Schingnitz et al. 1991). In a more recent study of olfactory discrimination and social memory in rats, Prediger et al. (2005) demonstrated that deficits in the behaviors of both 12- and 18-month-old animals could be prevented by caffeine or ZM241385. Interestingly, A1AR blockade by DPCPX was ineffective. Similarly, DPCPX was reported not to affect the acquisition of a shock-induced avoidance task, even though caffeine, or a selective A2AAR antagonist, did so (Kopf et al. 1999). It is important to note, however, that knockout studies have not been consistent with many of the pharmacological studies using antagonists. Thus, mice lacking A1AR exhibited normal learning of spatial tasks in the water maze (Gimenez-Llort et al. 2002).
Maemoto et al. (2004) have also shown recently that a new A1AR-selective antagonist, FR194921 (2-(1-methyl-4-piperidinyl)-6-(2-phenylpyrazolo[1,5-a]pyridin-3-yl)-3(2H)-pyridazinone) was able to reverse scopolamine-induced deficits on a passive avoidance test, with little effect on behavioral paradigms related to anxiety and depression. Pitsikas and Borsini (1997) obtained similar results using the A1AR antagonist \(S\rm{-}(-)\)-8-(3-oxocyclopentyl)-1,3-dipropyl-7H-purine-2,6-dione (BIIP20). A number of detailed structure–activity studies have attempted to define the molecular requirements of A1AR antagonism that are needed for cognition enhancement (Suzuki et al. 1993).
Few studies have been performed using A2A or A3 AR ligands in human subjects. Most recently, a specific relationship between A2AAR and Alzheimer’s disease was reported by Scatena et al. (2007). The administration of SCH58261 to mice in which β-amyloid (25–35) peptide was delivered into the cerebral ventricles was found able to prevent the subsequent neuronal loss, raising the possibility of reversing the neuronal loss in Alzheimer’s disease that is attributable to β-amyloid accumulation. This is particularly interesting in relation to the report, described above, of the colocalization of A1AR with β -amyloid in senile plaques and the ability of A1AR to increase the formation of soluble β-amyloid fragments. Since there are several strands of evidence that A2AAR can inhibit the activation of A1AR (Cunha et al. 1994; Dixon et al. 1997; O’Kane and Stone 1998), it is possible that the effect of SCH58261 is the result of removing an A2AAR-mediated suppression of A1AR, unmasking the protective efficacy of A1ARs.
Part of the difficulty in accounting for the detailed mechanism of A2AAR antagonist protection lies in the fact that modulation of A2AAR results in a plethora of actions, many of which are in functional opposition to each other. Thus, while A2AAR agonists promote glutamate release (Sebastiao and Ribeiro 1996) and antagonists should therefore have a valuable action in suppressing excessive release (see also Sect. 3.2), the opposite applies to the inflammatory cytokines. Activation of A2AAR inhibits both the initial calcium influx and the subsequent release of TNF-α induced by various stimuli, including the neurotoxic HIV protein Tat (Fotheringham et al. 2004). Blockade of A2AAR should therefore increase the release of TNF-α and, presumably, related proinflammatory cytokines, thus potentially increasing cell damage. Perhaps the net neuroprotective effects of A2AAR blockade are a complex result of pro- and anti-inflammatory activities, at least some of which may be time dependent, determined by whether the antagonists are present acutely or chronically.
The A3AR agonist IB–MECA appeared to have little effect alone on measures of learning using simple tests such as spontaneous alternation and passive avoidance. However, this compound did prevent the deficits in these behaviors induced by scopolamine or dizocilpine (Rubaj et al. 2003).
4.4 The Enigma of Propentofylline
The xanthine derivative propentofylline has been the subject of research for almost 20 years, yet in many respects it remains an enigma. It is also an enigma that requires decoding, since its activity may have significance for understanding the role of adenosine receptors in health and disease. Propentofylline is a weak antagonist at adenosine receptors. Its main actions seem to be an inhibition of adenosine uptake into cells, resulting in increased extracellular concentrations, and an inhibition of cyclic AMP phosphodiesterases. But at the level of the behaving animal, its overall effect is to promote cognitive function. Propentofylline has been shown to protect against cerebral ischemia in gerbils (Dux et al. 1990).
Even in humans, this compound is an effective cognition enhancer (Noble and Wagstaff 1997), and has been found to improve cognitive function in patients with vascular dementias (Mielke et al. 1996a, b). In animal models of Alzheimer’s disease, it has been shown to prevent the cognitive impairment caused by intracerebral administration of β-amyloid (1–40) (Yamada et al. 1998). This effect was attributed to the promotion of nerve growth factor (NGF) production, which raises further questions about the relationship between this hypothesis and the activation of adenosine receptors. One possibility is that raised extracellular adenosine levels activate A2AAR, and these in turn, as shown by Heese et al. (1997), then promote the generation of NGF and other neurotrophins. The balance of activation of adenosine receptors could be tipped from A1AR to A2AAR activation by virtue of the inhibitory effect of propentofylline on phosphodiesterase (Schubert et al. 1997). This hypothesis would be consistent with the fact that propentofylline is able to suppress TNF-α production (Meiners et al. 2004), an action that could be mediated partly via the activation (direct or indirect) of A2AAR.
There may also be more direct influences of propentofylline on microglial cells which regulate their degree of inflammatory activity (McRae et al. 1994; Schubert et al. 1996; Rudolphi and Schubert 1997), though the extent to which adenosine receptors might be involved in this also remains unclear. Some of these effects are almost certainly mediated via changes in calcium dynamics within neurons and glia (McLarnon et al. 2005).
4.5 Adenosine, Homocysteinuria and Alzheimer’s Disease
Homocysteinuria has been widely linked to vascular abnormalities leading, directly or indirectly, to the compromise of neuronal function and cognitive dysfunction seen in vascular dementia and Alzheimer’s disease, and there have been suggestions that a deficiency of adenosine may contribute to the neurological manifestations of increased homocysteine levels. One of the consequences of raised extracellular homocysteine is a parallel reduction of adenosine concentrations, possibly resulting from the formation of S-adenosylhomocysteine (SAH). A strong negative correlation between plasma levels of the two compounds has been recorded in Alzheimer’s disease patients (Selley 2004). It is possible, therefore, that a raised homocysteine level could induce a fall of adenosine concentrations to the extent that activation of protective receptors, including A1 and A2AARs, is compromised.
4.6 Genetic Studies
In an attempt to assess the possible relevance to Alzheimer’s disease of mutations in the A2A receptor gene, Liu et al. (2005) have examined 174 patients and 141 controls for the presence of the 1976 T > C polymorphism. No significant differences were noted in the genotype distribution or allelic frequency of this molecule, implying that a change of A2AAR function characterized by this mutation was not likely to be a major contributor to the Alzheimer’s disease susceptibility. However, the numbers of patients are not high for this type of study, and there may be alternative polymorphisms that are more relevant.
5 Creutzfeldt–Jakob Disease
Creutzfeldt–Jakob disease (CJD) is one of the prion diseases, characterized by the presence of protease-resistant prion protein within the brain parenchyma, leading to neuronal degeneration, motor impairment and ultimately death. CJD is often considered to be the human equivalent of scrapie, a disease primarily of sheep and related animals, and bovine spongiform encephalopathy (BSE) in cattle. The involvement of transmitters and other endogenous neural molecules in the development of prion-induced brain damage has received rather little attention, other than a degree of focus on glutamate and its receptor subtypes. However, Rodriguez et al. (2006) have examined the levels of adenosine A1AR in the neocortex of 12 patients with CJD and six age-matched controls. Elevated numbers of A1AR were identified in the patient group, together with increased receptor activity in cyclic AMP assays but normal levels of mRNA, suggesting increased receptor efficacy together with a possible decrease in the rate of receptor turnover.
When similar measurements were made in mice expressing bovine BSE prion protein, a similar increase in A1AR number in the brain occurred in parallel with the appearance of prion protein and the development of motor symptoms (Rodriguez et al. 2006). A simplistic interpretation of these data would be consistent with an upregulation of A1AR function as a protective adaptation to the potentially injurious prion protein. However, it will be important to assess how the changes in A1ARs compare with changes in other purine receptors, purine transporters and purine metabolic enzymes, in addition to other ARs and other neuroactive substances, before a significant role of A1ARs can be considered in isolation.
6 Lesch–Nyhan Syndrome
Lesch–Nyhan syndrome (LNS) is the result of an X-linked deficiency of hypoxanthine–guanine phosphoribosyltransferase (HGPRT). The lack of this major purine salvage enzyme results in high levels of hypoxanthine and uric acid, the latter producing a range of consequences in peripheral tissues, such as gouty arthritis and nephrolithiasis. In some cases, especially those with a complete absence of enzyme activity, there is also involvement of the CNS, with mental retardation and self-mutilation. Since the realization that the latter behavior could be induced by the administration of high doses of caffeine, the question has arisen of whether the various behavioral symptoms are due to a lack of adenosine or its receptors. To date, in spite of the increased de novo synthesis of purines, there is little evidence for any abnormality of adenosine levels or function, but it has been found that hypoxanthine can inhibit adenosine uptake. Levels of hypoxanthine comparable with those found in LNS patients suppress the equilibrative nucleoside transporters in human leucocytes, whether they are sensitive or not to nitrobenzylthioinosine (NBTI) (Torres et al. 2004; Prior et al. 2006). An examination of ARs in a mouse HGPRT knockout model of LNS has revealed an increase in the expression of A1AR and a decrease of A2AAR in the brain (Bertelli et al. 2006). What remains unclear is whether these receptor changes are induced by the alterations of adenosine uptake, and whether either of these phenomena can account for any of the behavioral symptoms in mouse models or human patients.
7 Multiple Sclerosis
Multiple sclerosis (MS) is an autoimmune disorder that results in damage to areas of the CNS. It has been widely considered that the primary site of damage is the oligodendrocyte and myelin sheath surrounding central axons, but more recent work is beginning to indicate a significant involvement of neuronal damage, produced either directly by autoantibodies or occurring secondary to the loss of myelin.
The various adenosine receptors are effective modulators of cytokine release from immune-competent cells (Haskò and Cronstein 2004; Bours et al. 2006; Haskò et al. 2007). Adenosine levels in the blood of MS patients are lower than in controls (Mayne et al. 1999), raising the possibility that this could contribute to the induction of an autoimmune attack. The actions of adenosine on blood mononuclear cells also differ between patients and controls. Both groups of cells release similar amounts of the proinflammatory cytokines TNF-α and IL-6 in the resting state, but when activated, the increased production of TNF-α is reduced by A1AR activation in controls but not patients with MS. Conversely, A1ARs inhibit IL-6 but not TNF-α release in patients (Mayne et al. 1999). Both results are consistent with an apparently lower A1AR density in cells from MS patients (Johnston et al. 2001). These data are also reflected in transgenic mice lacking A1AR, which show a marked propensity to develop experimental allergic encephalomyelitis (EAE), a condition widely recognized as the murine equivalent of MS (Tsutsui et al. 2004). The signs and symptoms of EAE develop in parallel with increased production of proinflammatory cytokines, consistent with the inhibitory activity of A1AR activation in monocytes from control humans.
Some of this work may be translated into therapeutic application, since methylthioadenosine has now been shown to not only suppress proinflammatory cytokine production by human white blood cells but also prevent and reverse EAE in animals (Moreno et al. 2006). These effects were attributed to an interference with the activation of the nuclear transcription factor NF-kB, and the involvement of AR activation of blockade was left open. Nevertheless, the potential implications of this activity of methylthioadenosine on MS treatment will no doubt encourage much further work on its molecular basis.
8 Huntington’s Disease
Huntington’s disease (HD) is an inherited neurodegenerative disease caused by loss of neurons in the striatum—especially medium spiny neurons containing GABA and enkephalin—and cortex. These changes result in motor abnormalities such as chorea, with the development of mental and psychological deterioration. The molecular origin of the degeneration has been ascribed to the production of an abnormal form of the protein huntingtin, in which an extended polyglutamine sequence (CAG triplets at the gene level) occurs.
Among the earliest proposals for the mechanism of neurodegeneration in HD was that excessive stimulation of glutamate receptors could be responsible for neuronal damage and death (Coyle and Schwarcz 1976; Lipton and Rosenberg 1994). Indeed, a large number of studies have demonstrated that overactivation of NMDA receptors in particular can produce many of the symptoms of HD in animals. The most effective agonist in this regard is quinolinic acid (Stone 2001), an endogenous metabolite of tryptophan that, unlike glutamate itself, is a selective agonist at the NMDA receptors (Stone and Perkins 1981; Stone and Darlington 2002). Administration of quinolinic acid into the striatum produces chronic neurodegeneration, which reproduces many of the electrophysiological, histological, motor and other behavioral symptoms of human HD (Beal et al. 1986, 1991; Ferrante et al. 1993; Popoli et al. 1994, 2002). Since ARs are important regulators of glutamate-mediated neurotransmission, there have been many suggestions that adenosine may be relevant to understanding HD, either as a key to the underlying cellular actions of huntingtin, and thus the molecular basis of the disorder, or as a means to treat the development or progress of the condition.
A second major hypothesis is that mutant huntingtin induces changes in mitochondrial function, and it is this that represents the primary cellular abnormality (“gain of function” hypothesis). A number of other potentially pathogenetic events have been attributed to mutant huntingtin. For example, proteolytic cleavage of mutant huntingtin generates fragments that aggregate into the nucleus and cytoplasm, thus contributing to early neuropathology. Accumulation of proteolytic huntingtin fragments and their aggregation may also trigger a cascade of damaging processes, leading to increasing dysfunctions in neurons through oxidative injury, transcriptional dysregulation, glutamate receptor excitotoxicity and apoptotic signals (Popoli et al. 2007 and references therein). In addition to this toxicity, there may also be a “loss of function” effect due to the loss of some beneficial actions exerted by normal huntingtin, which has been shown to be antiapoptotic, essential for normal embryonic development, and stimulatory on the production of BDNF into the cortex and its delivery to the striatal targets (see Popoli et al. 2007 and references therein).
Animal models of HD have become widely used based on each of these defects, namely the intrastriatal application of quinolinic acid or the administration (intrastriatal or systemic) of the mitochondrial toxin 3-nitro-propionic acid (3-NP). In addition, there are several transgenic models involving the induced expression of mutant hungtingtin, the R6/2 model being the most commonly used (Mangiarini et al. 1996).
8.1 Adenosine Receptors in HD
As noted above, A1AR activation suppresses glutamate release from neurons. In line with the excitotoxic hypothesis, Blum et al. (2002) have reported that an A1AR agonist, referred to as an adenosine amine congener (ADAC), was able to prevent the neuronal degeneration and motor sequelae of 3-NP administration to mice. Since no protection was apparent in cell cultures, the results were interpreted to indicate an action on presynaptic sites, presumably those at which the release of glutamate could be inhibited. Conversely, the A1AR antagonist DPCPX exacerbates damage induced by a similar mitochondrial poison, malonate (Alfinito et al. 2003).
The ARs that have become of greatest interest in HD are the A2AARs. Activation of these promotes the release of glutamate, depending on the age of animals and the presence of a depolarizing stimulus (Corsi et al. 1999a, 2000), and increased numbers or functional activity of A2AARs could cause or contribute to an excitotoxic process (Domenici et al. 2007). The administration of CGS21680 itself increases extracellular glutamate levels (Popoli et al. 1995).
Consistent with this, A2AAR antagonists have been shown to reduce the toxic consequences of quinolinic acid administration, an effect correlated with a reduction of glutamate release triggered by quinolinic acid (Reggio et al. 1999; Popoli et al. 2002; Scattoni et al. 2007). A similar phenomenon has been described in R6/2 mice, in which SCH58261 reduced the motor abnormalities and loss of brain tissue (Chou et al. 2005) and glutamate release in the striatum of R6/2 mice (Gianfriddo et al. 2004).
While this provides comforting support of the concept that quinolinic acid administration provides an acceptable model of HD, it is important to establish whether glutamate release is elevated in mutant mice or HD patients and, if so, the mechanism involved. The most obvious possibility, that of raised quinolinic acid levels, has been supported directly by evidence from Guidetti et al. (2004), who measured increased amounts in patients at an early stage of HD. As to the mechanism, the presence of mutant huntingtin has been shown to reduce the uptake of glutamate by astrocytes (Behrens et al. 2002), a result that could cause increased activation of glutamate receptors, contributing to excitotoxicity.
Neuroprotection has also been demonstrated for the 3-NP model of HD. The blockade of A2AAR by CSC-protected mice treated with 3-NP against neuronal loss (Fink et al. 2004). Similarly, less cell death was seen when 3-NP was administered to A2AAR knockout mice compared with wild-type controls. Consistent with this, another inhibitor of mitochondrial complex II, malonate, produced a degeneration of striatal neurons that was also prevented by DMPX (Alfinito et al. 2003).
However, as already noted above (Fig. 6), the role of A2AARs in HD is far more complex. Activation of A2AARs has also been reported to mediate beneficial effects. The A2AAR agonist CGS21680 enhances the neurotrophic activity of growth factors such as BDNF, a key factor promoting the viability of striatal neurons, by facilitating TrkB receptor function (Lee and Chao 2001). Agonists at A2AAR are also associated with a normalization of cyclic AMP response element binding protein (CREB) in transgenic animals (Chiang et al. 2005).
Moreover, CGS21680 reduces the incidence of abnormal extracellular macromolecular deposits that are present in HD brains in a similar way to β-amyloid deposits in Alzheimer’s disease and Lewy bodies in Parkinson’s disease. In R6/2 mice, ubiquitinated deposits have indeed been demonstrated in striatal cells, both in vivo and in cell cultures, which appear to depend on the expression of mutant huntingtin protein (Chou et al. 2005). These deposits are reduced by CGS21680. In the same study, it was also noted that CGS21680 corrected the abnormally high levels of blood glucose and 5′-adenosine monophosphate (AMP)-activated protein kinase activity in the mutant mice, strongly suggesting a more fundamental role of A2AAR than had hitherto been suspected in the regulation of cellular biochemistry.
There is a significant depletion of A2AAR in the striatum of patients with HD and in transgenic mice (Blum et al. 2003b) or rats (Bauer et al. 2005) expressing mutant huntingtin. On the other hand, the density of A2AAR on blood platelets is increased in human HD patients (Varani et al. 2003), showing a significant correlation with both CAG repeat length (Maglione et al. 2006) and anticipation of symptoms between generations (Maglione et al. 2005). In a total of 126 HD gene-positive individuals, A2AARB max values were found to be robustly increased at all HD stages as well as in 32 presymptomatic subjects (Varani et al. 2007). The same abnormality is present also in other neurological diseases characterized by an extended polyglutamine sequence (polyQ), but not in non-polyQ inherited disorders (Varani et al. 2007). The same peripheral cells exhibited altered membrane fluidity, a finding that may explain the observed change in receptor density. Authors argue that the observed alteration in lymphocytes reflects the presence of the mutant protein and suggest that the measurement of the A2AAR binding activity might be of potential interest for a peripheral assessment of chemicals capable of interfering with the immediate toxic effects of the mutation.
There is clear evidence for increased activity of A2AAR associated with HD. Striatal neurons expressing mutant huntingtin were found to show increased A2AAR activation of adenylate cyclase (Varani et al. 2001), and a similar result was observed subsequently in blood cells of HD patients (Varani et al. 2003). The ability of A2AAR stimulation to raise cyclic AMP levels is also increased in R6/2 mice (Chou et al. 2005; Tarditi et al. 2006), and in animals treated with 3-NP (Blum et al. 2003a). In the Tarditi et al. study, an increase in both the number of A2AARs as well as their activation of adenylate cyclase was reported, which was apparent within a few days of birth of R6/2 mice. Both of these parameters then fell to the values seen in wild-type animals. The mRNA for A2AAR, in contrast, showed no change until 21 days postnatally, after which it decreased substantially. Two conclusions may be drawn from this work. Firstly, the mismatch between A2AAR protein and mRNA could indicate changes in factors that affect translation or transcription of the A2AAR, or which regulate receptor activity. Secondly, a loss rather than an increase of A2AAR seems to be associated with older mice in which motor symptoms of HD are beginning to occur. A further intriguing observation in this study was that in the young mice, A2AAR function was not prevented by ZM241385, whereas sensitivity to this antagonist was established in the older animals after 21 days of age. Whether this also implies differences in the regulation of receptor function, or different variant structures of the receptor protein at different ages, remains to be explored.
In conclusion, available data on the potential exploitation of A2AAR ligands in HD are controversial and reflect the complexity of A2AAR regulation in this disease (for further comments, see Popoli et al. 2007). The complex mutual relationship between AR activities mediating detrimental or beneficial effects (see also Sect. 3) makes it difficult to establish whether targeting A2AAR would really be of interest to treat HD. Further basic research is needed to solve several specific questions, in particular: (1) neuronal versus non-neuronal receptor localization, and (2), for receptors expressed in neurons, pre- versus postsynaptic sites (see Fig. 6).
9 Cerebral Ischemia and Reperfusion: Stroke
9.1 Role of A1 Adenosine Receptors
One of the earliest reports of neuroprotection against ischemia was that the nonselective agonist 2-chloroadenosine would prevent hippocampal damage in rats (Evans et al. 1987). Similar results were obtained subsequently using A1AR-selective agonists (von Lubitz et al. 1989; Phillis and O’Regan 1993; von Lubitz et al. 1995a), with suggestions that the protection could involve an inhibition of leukocyte adherence and extravasation (Grisham et al. 1989).
The finding that theophylline could increase the release of glutamate produced by ischemia certainly suggests that endogenous adenosine is exerting an inhibitory action on glutamate release (Héron et al. 1993), although this could have been due to A1 or A2A AR blockade. The simultaneous measurement of purine and glutamate release into the extracellular space of brain, together with the neuronal damage and behavioral consequences of an ischemic episode, revealed a significant relationship between these parameters, with a lower extracellular glutamate being associated with less cell damage (Melani et al. 1999). It is interesting that several nonpurine compounds that can depress the release of excitatory amino acids are also protective against ischemic damage (Ochoa et al. 1992; Graham et al. 1993). Conversely, A1AR blockade exacerbates ischemic damage (Phillis 1995).
On the other hand, it has been argued that the release of endogenous glutamate is not actually related to ischemic-induced brain damage. Systemic administration of R-PIA, CHA or an adenosine uptake inhibitor did not prevent the increase of glutamate levels in brain during ischemia (Héron et al. 1993, 1994; Cantor et al. 1992; Kano et al. 1994), although other groups have reported a decreased release using CPA (Simpson et al. 1992). The differences may depend on the pharmacokinetics of the agonists used or the model used for inducing damage.
Although little is known of the signaling pathways that underlie ischemic damage or adenosine-mediated protection in vivo, some clues may be gleaned from in vitro work. Di Capua et al. (2003), for example, found that A1AR agonism protected primary rat neurons against “chemical ischemia” (produced by iodoacetate) via the activation of protein kinase C-epsilon. The activity of A1ARs themselves may change under ischemic conditions. Adenosine A1ARs are desensitized and internalized by a period of hypoxia in brain slices (Coelho et al. 2006). A period of ischemia in vivo followed by reperfusion has been said to result in no change in the number of A1ARs or their inhibitory efficacy on presynaptic transmitter release (Shen et al. 2002), although Lai et al. (2005) have reported an increase in A1AR expression in the cerebral cortex following ischemia in Wistar rats.
9.2 Role of A2 Adenosine Receptors
The activation of A2AAR can protect neurons against ischemia-induced damage. One of the best-tested A2AAR agonists is ATL-146e, which prevents ischemic damage in the spinal cord (Cassada et al. 2001a; Reece et al. 2006) as well as damage induced by mechanical trauma (Reece et al. 2004; Okonkwo et al. 2006). This protection afforded by ATL-146e was accompanied by the normalization of several molecular markers, such as those for apoptosis (Cassada et al. 2001b), microtubule-associated protein 2 (MAP-2) and TNF-α levels (Reece et al. 2004). However, the protection is not completely prevented by ZM241385, implying that there are relevant sites of action other than A2AAR. A period of ischemia of the spinal cord does, however, induce a highly significant increase in A2AAR number, a finding that may contribute to the protective effect of A2AAR agonists. There is also a greater inhibition of TNF-α levels in postischemic spinal cord, as well as reduced platelet adhesion to endothelial cells (Cassada et al. 2002), consistent with an important role of A2AAR on blood cells
On the other hand, blockade of A2AAR is also neuroprotective against ischemic damage caused by transient or permanent arterial occlusion (Gao and Phillis 1994; Phillis 1995; Monopoli et al. 1998; Pedata et al. 2005). Confirmation of the detrimental influence of A2AAR has come from an examination of A2AAR-deficient transgenic mice (Chen et al. 1999). These animals showed substantial resistance to ischemia-induced brain damage compared with their normal littermates.
An interesting observation reported by Corsi et al. (1999a, b) is that the agonist CGS-21680 only increased the spontaneous efflux of glutamate and GABA in young (not old) rats, although it enhanced potassium-evoked release similarly in both groups of animals. This may have implications for the utility of A2AAR agonist and antagonist ligands in treating older patients after cerebral ischemia, since chronic treatment might show fewer side effects attributable to increased basal release of glutamate, while retaining neuroprotective activity against the depolarization-induced release occurring during and immediately after cerebral ischemia or trauma. The reason for the increased damage may depend, at least partly, on the increased release of glutamate and related amino acids that these compounds produced during cerebral ischemia (O’Regan et al. 1992)
It is interesting to note that, while most of the work in this area has employed adult rodents, there is some evidence that the reverse situation occurs for young animals. Thus, in neonatal rats, Aden et al. (2003) found that it was activation of A2AAR that protected against a period of hypoxia and ischemia, with A2AAR knockout mice showing greater brain damage than wild-type controls.
The release of proinflammatory cytokines such as TNF-α from macrophages is suppressed by activation of A2AAR (Kreckler et al. 2006). Work by Chen and colleagues (Yu et al. 2004), however, has revealed a fascinating insight into the sites through which protection is mediated. By generating populations of rats lacking A2AAR generally and replacing bone marrow tissue selectively with cells reconstituted to contain A2AAR, they have been able to comment directly on the roles of receptors intrinsic to the CNS relative to those in the blood. The results showed that the presence of A2AARs on blood cells alone was sufficient to reverse the protective effect of generalized A2AAR knockout, while wild-type mice given A2AAR knockout bone marrow cells were protected against ischemic damage. This illuminating study strongly suggests that the A2AARs relevant to protection against ischemic damage are those on blood cells. This may also imply that the mechanism of A2AAR antagonist protection is more strongly dependent on, for example, the release of inflammatory cytokines, than had previously been thought.
Although the A2BAR has received relatively little attention with respect to neuroprotection, its activation has a number of consequences that could well contribute significantly to the phenomenon. For example, there is evidence that its activation of p38 MAPK leads to the increased expression of IL-6 in macrophages (Fiebich et al. 1996a, 2005). Since IL-6 is a cytokine that has been reported to protect neurons against a range of insults (Bensadoun et al. 2001; Carlson et al. 1999), its production, either in central glia or peripheral cells, may result in some protective efficacy.
Brain inflammation induced in rats by a chronic intraventricular infusion of LPS was associated with a loss of neuronal A2BAR. This loss was prevented by a nitro derivative of the anti-inflammatory drug flurbiprofen, while the parent compound was inactive (Rosi et al. 2003). The authors’ conclusion was that an NO-releasing anti-inflammatory compound might be an effective inhibitor of brain inflammation in conditions such as Alzheimer’s disease, and that changes in the density of A2BAR might be involved. It is becoming increasingly clear that much more work is required to expand our knowledge of the effects of A2BAR activation or loss on the overall profile of pro- and anti-inflammatory cytokines in the brain and elsewhere, especially in relation to the net effects on neurotransmission, β-amyloid production, and neuronal or glial cell viability.
9.3 Role of A3 Adenosine Receptors
As already mentioned above (see Sect. 3.4), A3AR activation can protect isolated cells from hypoxia-induced death (Chen et al. 2006), and it reduced infarct size in rats subjected to middle cerebral artery occlusion (MCAo). Conversely, animals lacking A3AR exhibit substantially increased infarct volumes, suggesting that the activation of these receptors by endogenous adenosine normally acts as a physiological brake on those processes causing damage (Chen et al. 2006; Fedorova et al. 2003).
The chronic administration of an A3AR agonist such as IB–MECA affords protection against a subsequent period of cerebral ischemia (von Lubitz et al. 1999b, 2001).
At least part of the protective activity of A3AR agonists may involve modulation of immune-competent cells and the inflammatory reaction to cellular damage. Agonists have been shown to inhibit the generation of several proinflammatory cytokines from cells, including interleukin (IL) 10, IL-12, interferon-γ and TNF-α (Haskò et al. 1998; McWhinney et al. 1996). The latter action is sufficiently robust to have been developed as a screen for new agonist compounds (Knutsen et al. 1998). Indeed, it has been suggested that activation of A3AR may be responsible for the reported inhibition by adenosine of TNF-α secretion in the human U937 macrophage cell line (Sajjadi et al. 1996).
The opposite effects obtained on the outcome of brain ischema upon acute or chronic treatment with selective A3AR agonists are discussed below (see Sect. 9.5).
9.4 Time Course of Protection Induced by Adenosine Receptor Ligands
One of the valuable features of neuroprotection by A1AR activation is that it can be demonstrated for a period of several hours following the occurrence of a vascular or toxic insult. This is a major consideration for any drug intended for clinical use as a neuroprotectant following an acute incident such as a stroke, since the expansion of damage from a limited central region into a more extensive penumbral area occurs over a period of hours or days, and it is essential to limit the degree of that expansion if patient recovery is to be optimized. Most authorities consider that there is a window of opportunity for neuroprotection of up to several hours after the occurrence of stroke. Several A1AR-selective agonists such as R-PIA certainly exhibit protection, even when administered up to 2 h after excitotoxic insults, indicating that the neuronal network and intracellular signaling processes that contribute to damage continue to operate over this time frame (Miller et al. 1994). Against ischemia-induced damage, cyclohexyladenosine (CHA) remains protective when administered up to at least 30 min following cerebral ischemia (von Lubitz et al. 1989), and ADAC similarly has a window of efficacy of several hours after cerebral ischemia in gerbils (von Lubitz et al. 1996). This latter compound is of special interest since it seems to possess fewer of the cardiovascular side effects associated with some other A1AR agonists (Bischofberger et al. 1997), and its efficacy is still apparent when administered chronically in very low doses (von Lubitz et al. 1999a). The importance of this finding is that many other purine receptor ligands produce opposite effects when used chronically rather than in a single acute dose paradigm. Since most patients needing neuroprotection may be taking the drugs for prolonged periods of time, this could be a highly significant advantage of ADAC and related compounds.
The timing of acute administration of A3AR agonists is also important. Treatment prior to ischemia increased infarct size, while postischemic administration reduced damage, probably as a result of altered dynamics of receptor activation, on neurons, glia and blood components (von Lubitz et al. 2001).
9.5 Acute Versus Chronic Administration
Despite the evidence for a neuroprotective action of adenosine and A1AR agonists, caffeine—a nonselective antagonist at both A1 and A2 ARs—was also found to protect against ischemic damage in the CNS after its chronic administration (Rudolphi et al. 1989; Sutherland et al. 1991). Single, acute injections of more selective A1AR antagonists, including DPCPX, were also found to exacerbate ischemic damage (Phillis 1995; von Lubitz et al. 1994a), while their chronic administration reduced damage and produced neuroprotection (von Lubitz et al. 1994a). This dichotomy of response probably indicates compensatory changes of receptor density that follow the prolonged presence of any receptor ligand. However, such changes may be limited in extent, or restricted to certain cell subtypes, since no significant changes in A1AR binding were detected after chronic administration of antagonists (Traversa et al. 1994). However, others have reported that chronic administration of the AR antagonists caffeine and theophylline increase A1ARs in cerebral cortex (Murray 1982; Szot et al. 1987) and the hippocampal CA1 region (Rudolphi et al. 1989)
Chronic administration of low doses of ADAC generated the opposite result, with marked protection of the brain. The reasons for this difference from other A1AR agonists is not entirely clear, although the authors point out the substantial difference in molecular structure between ADAC and other compounds, with the implication that it may yield a different spectrum or time course of action on a range of cellular targets whose balance determines the overall production of neuronal damage or protection (von Lubitz et al. 1999a).
The effects of acute and chronic treatment with A2AAR ligands show less disparity than in the case of the A1AR ligands described above. Overall, the qualitative effects of agonists and antagonists are similar whether they are administered acutely or chronically. This assertion would be consistent with evidence that receptor numbers and affinities change little in vivo (von Lubitz et al. 1995b) or in vitro (Abbracchio et al. 1992) in the continued presence of A2AAR ligands.
As mentioned above (see Sects. 3.4 and 3.5), the acute administration of agonists at A3ARs, and their application to neurons in cell culture, does appear to induce neuronal death (Sei et al. 1997). In addition, an A3AR agonist can potentiate the degree of CNS damage following cerebral ischemia (von Lubitz et al. 1994b). On the other hand, the maintained presence or chronic intermittent administration of A3AR agonists produces protection, probably as a result of compensatory adaptations in the number or sensitivity of receptors. Thus, acute administration of the selective agonist ligand IB–MECA significantly enhanced the extent of brain damage following ischemia in gerbils. Chronic administration of the same compound, however, resulted in a highly significant reduction in the ischemic damage (von Lubitz et al. 1994b; 1999b; Chen et al. 2006).
9.6 Therapeutic Implications of Preconditioning
Relatively short periods of hypoxia, hypoglycemia or ischemia can result in protection of tissues against a subsequent and more severe insult. This is the phenomenon of preconditioning. Neuronal preconditioning has been demonstrated using both in vivo and in vitro preparations (Schurr et al. 1986; Khaspekov et al. 1998). One factor contributing to this is a change in the number of A1ARs, which increases after the preconditioning period, probably as an adaptive protective development against further ischemia (Zhou et al. 2004). Adenosine is known to be involved in preconditioning, mainly through its opening of KATP channels (Yao and Gross 1994). In many models, even those in which it is induced by an anesthetic agent such as isoflurane (Liu et al. 2006), preconditioning can be prevented almost completely by A1AR blockers (Hiraide et al. 2001; Nakamura et al. 2002; Yoshida et al. 2004; Pugliese et al. 2003), although A2AARs seem to contribute little to the phenomenon. The lack of involvement of A2AARs is also surprising given the foregoing discussion on the clear neuroprotective activity of A2AAR antagonists (Phillis 1995; Jones et al. 1998a, b), although it is likely that the differences between in vitro slice preparations and in vivo studies are largely responsible for this difference. Interestingly, A3AR antagonists can enhance neuronal recovery after simulated ischemia in vitro, consistent with the work quoted above that acute activation of A3AR worsens ischemic damage in vivo (Pugliese et al. 2007).
10 Prospects for Adenosine Receptor-Based Therapeutics
In summary, there is an increasingly acceptable rationale, at the cellular, biochemical and behavioral levels, for believing that changes in AR function might contribute to the symptoms and possibly progression of neurodegenerative disorders (Ribeiro et al. 2002), and that ligands acting at the various ARs may have a potential role in the therapeutic treatment of some of those disorders (Muller 1997, 2000; Mally and Stone 1998; Broadley 2000; Press et al. 2007; Baraldi et al. 2008). Of course, no receptor population is likely to function in isolation in the CNS. The activation or blockade of other neurotransmitter receptors may have significant effects on the number or efficacy of ARs.
For example, von Lubitz et al. (1995a) tested combinations of ligands acting at NMDA and A1AR, using either acute or chronic treatments. The results revealed changes of animal responses with combined treatments that suggested important interactions between NMDA and A1AR contributing to the changes of seizure generation and motor impairment. There were parallel changes of A1AR density which indicated that the interactions were occurring at a deeper level of cellular function than merely a degree of nonspecificity in the ligand efficacy at the different receptors.
If ARs are indeed significant contributors to neuroprotection by responding to altered endogenous levels of the purine, or if they are used as targets for therapeutic agents that act directly upon them, it will be necessary to obtain information on the manner in which those receptors behave throughout the period of insult and subsequently. A range of factors, such as acidity, oxygen levels, cytokines, peptides, growth factors, and undoubtedly many more, could act to modify receptor responsiveness in a fashion that reduces or enhances the expected efficacy of agonists or antagonists. Examples of this include the report that tissue oxidation reduces the affinity of A1AR antagonists, but not agonists, although the density of binding sites was decreased for both (Oliveira et al. 1995). Changes in the balance of agonist to antagonist activity could be produced in this way, which could significantly alter the anticipated response to ligands.
The ability of AR antagonists to reverse cognitive dysfunction has been taken to indicate that they may have a standalone place in the treatment of dementias. However, given the undoubted existence of this and many other receptor interaction phenomena, the generalized loss of neurons that can occur with aging or disease, and the complexity of neuronal interactions that underlie cognitive performance, it is likely that future attention will shift to compounds that retain specificity of action but act at a defined number of different sites (Van der Schyf et al. 2006). In this context, however, it seems likely that blockade of ARs would be one of the more valuable sites to include in the profile of optimum targets.
For the treatment of ischemic damage, however, there is clearly a potential use for A1AR agonists and A2AAR antagonists. An alternative approach to using conventional agonists for stroke-induced brain damage could be to inhibit adenosine kinase. This enzyme is a major route for the removal of adenosine, converting it to AMP. Consequently, the overexpression of the kinase results in an exacerbation of ischemic damage (Pignataro et al. 2007), whereas inhibition has been found to raise extracellular adenosine levels and produce protection against damage (Jiang et al. 1997).
Whichever strategic approach is used, and whichever receptor subtype is selected, it seems likely that ARs will in the future represent a valuable series of targets for protection of the brain against a range of insults. However, we will have to solve some pending issues concerning the opposite (beneficial versus detrimental) effects exerted by some AR subtypes depending on their cellular and/or pre- versus postsynaptic localization. This especially applies to the A2AAR subtype (see Fig. 1). Blockade of A2AAR can result either in protoxic or neuroprotective effects according to the mechanisms involved in a given experimental model and, in some cases, to the disease stage. In this respect, it is envisaged that notable advances will be achieved by the availability of transgenic mice bearing selective defects of A2AAR on specific cell populations (Yu et al. 2004). The use of these mice will help in addressing the therapeutical use of A2AAR ligands in not only HD but also all other neurodegenerative diseases characterized by a dysfunction of the adenosinergic system.
Abbreviations
- ADAC:
-
Adenosine amine congener
- AMP:
-
Adenosine monophosphate
- AR:
-
Adenosine receptor
- BDNF:
-
Brain-derived neurotrophic factor
- BIIP20:
-
S-( − )-8-(3-Oxocyclopentyl)-1,3-dipropyl-7H-purine-2,6-dione
- cAMP:
-
Cyclic adenosine monophosphate
- CCPA:
-
2-Chloro-N 6-cyclopentyladenosine
- CGS15943:
-
5-Amino-9-chloro-2-(2-furyl)-1,2,4-triazolo[1,5-c]quinazoline
- CGS21680:
-
2-[4-(2-Carboxyethyl)-phenylethylamino]-5′ N-ethyl-carbox amido-adenosine
- CHA:
-
N 6-Cyclohexyladenosine
- CJD:
-
Creutzfeldt–Jakob disease
- Cl-IB-MECA:
-
2-Chloro-N 6-(3-iodobenzyl)adenosine-5′-N-methyluronamide
- CNS:
-
Central nervous system
- CP66,713:
-
4-Amino-1-phenyl[1,2,4]-triazolo[4,3-a]quinoxaline
- CPA:
-
Cyclopentyl adenosine
- 8-CPT:
-
8-Cyclopentyltheophylline
- CREB:
-
Cyclic AMP responsive element binding protein
- CSC:
-
8-(3-Chloro styryl)caffeine
- DMPX:
-
3,7-Dimethyl-1-propargylxanthine
- DPCPX:
-
8-Cyclopentyl-1,3-dipropylxanthine
- EAE:
-
Allergic encephalomyelitis
- ERK1/2:
-
Extracellular signal-regulated kinases 1 and 2
- GABA:
-
Gamma-aminobutyric acid
- HD:
-
Huntington’s disease
- HGPRT:
-
Hypoxanthine-guanine phosphoribosyltransferase
- IB–MECA:
-
N 6-(3-Iodobenzyl)adenosine-5′-N-methyluronamide
- IL:
-
Interleukin
- KFM19:
-
RS-( − )-8-(3-oxocyclopentyl)-1,3-dipropyl-7H-purine-2,6-dione
- LNS:
-
Lesch–Nyhan syndrome
- MAP-2:
-
Microtubule-associated protein 2
- MAPK:
-
Mitogen-activated protein kinases
- MCAo:
-
Middle cerebral artery occlusion
- MPTP:
-
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MRS2179:
-
N 6-Methyl-2′-deoxyadenosine-3′, 5′-bisphosphate
- MRS1706:
-
N-(4-Acetylphenyl)-2-[4-(2,3,6,7-tetrahydro-2,6-dioxo-1,3- dipropyl-1H-purin-8-yl)-phenoxy]acetamide
- MS:
-
Multiple sclerosis
- NBTI:
-
Nitrobenzylthioinosine
- NECA:
-
5′-N-Ethylcarboxamidoadenosine
- NGF:
-
Nerve growth factor
- NMDA:
-
N-Methyl-d-aspartate
- 3-NP:
-
3-Nitro-propionic acid
- PKC:
-
Protein kinase C
- PLC:
-
Phospholipase C
- R-PIA:
-
R-Phenylisopropyladenosine
- SAH:
-
S-Adenosylhomocysteine
- SCH58261:
-
7-(2-Phenylethyl)-5-amino-2-(2-furyl)-pyrazolo-[4,3-e]-1,2,4-triazolo[1,5-c]-pyrimidine
- TNF-α:
-
Tumor necrosis factor alpha
- Trk:
-
Tropomyosin-related kinase
- ZM241385:
-
4-(2-[7-Amino-2-(2-furyl)(1,2,4)-triazolo(2,3-a)-(1,3,5)triazin-5-yl-amino]ethyl)phenol
References
Abbracchio MP, Fogliatto G, Paoletti AM, Rovati GE, Cattabeni F (1992) Prolonged invitro exposure of rat-brain slices to adenosine-analogs: selective desensitization of adenosine-A(1) but not adenosine-A(2) receptors. Eur J Pharmacol 227:317–324
Abbracchio MP, Rainaldi G, Giammarioli AM, Ceruti S, Brambilla R, Cattabeni F, Barbieri D, Franceschi C, Jacobson KA, Malorni W (1997) The A3 adenosine receptor mediates cell spreading, reorganisation of actin cytoskeleton and distribution of Bcl-x(L): studies in human astroglioma cells. Biochem Biophys Res Commun 241:297–304
Abbracchio MP, Ceruti S, Brambilla R, Barbieri D, Camurri A, Franceschi C, Giammarioli AM, Jacobson KA, Cattabeni F, Malorni W (1998) Adenosine A3 receptors and viability of astrocytes. Drug Dev Res 45:379–386
Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Knight GE, Fumagalli M, Gachet C, Jacobson KA, Weisman GA (2006) International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol Rev 58:281–341
Aden U, Halldner L, Lagercrantz H, Dalmau I, Ledent C, Fredholm BB (2003) Aggravated brain damage after hypoxic ischemia in immature adenosine A(2A) knockout mice. Stroke 34: 739–744
Albasanz JL, Perez S, Barrachina M, Ferrer I, Martin M (2008) Up-regulation of adenosine receptors in the frontal cortex in Alzheimer’s disease. Brain Pathol 18:211–219
Alfinito PD, Wang SP, Manzino L, Rijhsinghani S, Zeevalk GD, Sonsalla PK (2003) Adenosinergic protection of dopaminergic and GABAergic neurons against mitochondrial inhibition through receptors located in the substantia nigra and striatum, respectively. J Neurosci 23:10982–10987
Andiné P, Rudolphi KA, Fredholm BB, Hagberg H (1990) Effect of propentofylline (HWA285) in extracellular purines and excitatory amino acids in CA1 of rat hippocampus during transient ischaemia. Br J Pharmacol 100:814–818
Angelucci ME, Cesário C, Hiroi RH, Rosalen PL, Da Cunha C (2002) Effects of caffeine on learning and memory in rats tested in the Morris water maze. Braz J Med Biol Res 35: 1201–1208
Angulo E, Casado V, Mallol J, Canela EI, Vinals F, Ferrer I, Lluis C, Franco R (2003) A(1) adenosine receptors accumulate in neurodegenerative structures in Alzheimer disease and mediate both amyloid precursor protein processing and tau phosphorylation and translocation. Brain Pathol 13:440–451
Appel E, Kazimirsky G, Ashkenazi E, Kim SG, Jacobson KA, Brodie C (2001) Roles of BCL-2 an caspase 3 in the adenosine A3 receptor-induced apoptosis. J Mol Neurosci 17:285–292
Araki T, Kato H, Kanai Y, Kogure K (1993) Selective changes of neurotransmitter receptors in middle-aged gerbil brain. Neurochem Intern 23:541–548
Arendash GW, Schleif W, Rezai-Zadeh K, Jackson EK, Zacharia LC, Cracchiolo JR, Shippy D, Tan J (2006) Caffeine protects Alzheimer’s mice against cognitive impairment and reduces brain β-amyloid production. Neuroscience 142:941–952
Arvin B, Neville LF, Pan J, Roberts PJ (1989) 2-Chloroadenosine attenuates kainic acid-induced toxicity within the rat striatum: relationship to release of glutamate and Ca2 + influx. Br J Pharmacol 98:225–235
Baraldi PG, Tabrizi MA, Gessi S, Borea PA (2008) Adenosine receptor antagonists: translating medicinal chemistry and pharmacology into clinical utility. Chem Rev 108:238–263
Barnes CR, Mandell GL, Carper HT, Luong S, Sullivan GW (1995) Adenosine modulation of TNFα-induced neutrophil activation. Biochem Pharmacol 50:1851–1857
Bartrup JT, Stone TW (1990) Activation of NMDA receptor-coupled channels suppresses the inhibitory action of adenosine on hippocampal slices. Brain Res 530:330–334
Bartrup JT, Addae JI, Stone TW (1991) Interaction between adenosine and excitatory agonists in rat hippocampal slices. Brain Res 564:323–327
Bauer A, Zilles K, Matusch A, Holzmann C, Riess O, von Horsten S (2005) Regional and subtype selective changes of neurotransmitter receptor density in a rat transgenic for the Huntington’s disease mutation. J Neurochem 94:639–650
Beal MF, Kowall NW, Ellison DW, Mazurek MF, Swartz KJ, Martin JB (1986) Replication of the neurochemical characteristics of Huntington’s disease by quinolinic acid. Nature 321:168–171
Beal MF, Ferrante RJ, Swartz KJ, Kowall NW (1991) Chronic quinolinic acid lesions in rats closely resemble Huntington’s disease. J Neurosci 11:1649–1659
Behrens PF, Franz P, Woodman B, Lindenberg KS, Landwehrmeyer GB (2002) Impaired glutamate transport and glutamate–glutamine cycling: downstream effects of the Huntington mutation. Brain 125:1908–1922
Bensadoun JC, de Almeida LP, Dréano M, Aebischer P, Déglon N (2001) Neuroprotective effect of interleukin-6 and IL6/IL6R chimera in the quinolinic acid rat model of Huntington’s syndrome. Eur J Neurosci 14:1753–1761
Bertelli M, Cecchin S, Lapucci C, Jacomelli G, Jinnah HA, Pandolfo M, Micheli V (2006) Study of the adenosinergic system in the brain of HPRT knockout mouse (Lesch–Nyhan disease). Clin Chim Acta 373:104–107
Bischofberger N, Jacobson KA, von Lubitz DKJE (1997) Adenosine A(1) receptor agonists as clinically viable agents for treatment of ischemic brain disorders. Ann NY Acad Sci 825:23–29
Blum D, Gall D, Galas MC, d’Alcantara P, Bantubungi K, Schiffmann, SN (2002) The adenosine A(1) receptor agonist adenosine amine congener exerts a neuroprotective effect against the development of striatal lesions and motor impairments in the 3-nitropropionic acid model of neurotoxicity. J Neurosci 22:9122–9133
Blum D, Galas MC, Pintor A, Brouillet E, Ledent C, Muller CE, Bantubungi K, Galluzzo M, Gall D, Cuvelier L, Rolland AS, Popoli P, Schiffmann SN (2003a) A dual role of adenosine A(2A) receptors in 3-nitropropionic acid-induced striatal lesions: implications for the neuroprotective potential of A(2)A antagonists. J Neurosci 23:5361–5369
Blum D, Hourez R, Galas MC, Popoli P, Schiffmann SN (2003b) Adenosine receptors and Huntington’s disease: implications for pathogenesis and therapeutics. Lancet Neurol 2:366–374
Bona E, Adén U, Gilland E, Fredholm BB, Hagberg H (1997) Neonatal cerebral hypoxia-ischaemia: the effect of adenosine receptor antagonists. Neuropharmacology 9:1327–1338
Bours MJ, Swennen EL, Di Virgilio F, Cronstein BN, Dagnelie PC (2006) Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther 112:358–404
Brambilla R, Cottini L, Fumagalli M, Ceruti S, Abbracchio MP (2003) Blockade of A2A adenosine receptors prevents basic fibroblast growth factor-induced reactive astrogliosis in rat striatal primary astrocytes. Glia 43:190–194
Bridges AJ, Bruns RF, Ortwine DF, Priebe SR, Szotek DL, Trivedi BK (1988) N 6-[2-(3,5-Dimethoxyphenyl)-2-(2-methylphenyl)-ethyl]adenosine and its uronamide derivatives. Novel adenosine agonists and antagonists with both high affinity and high selectivity for the adenosine A2 receptor. J Med Chem 31:1282–1285
Broadley KJ (2000) Drugs modulating adenosine receptors as potential therapeutic agents for cardiovascular diseases. Exp Opin Ther Pat 10:1669–1692
Brown SJ, James S, Reddington M, Richardson PJ (1990) Both A1 and A2A purine receptors regulate striatal acetylcholine release. J Neurochem 55:31–38
Bullough DA, Magill MJ, Firestein GS, Mullane KM (1995) Adenosine activates A2 receptors to inhibit neutrophil adhesion and injury to isolated cardiac myocytes. J Immunol 155:2579–2586
Burkey TH, Webster RD (1993) Adenosine inhibits fMLP-stimulated adherence and superoxide anion generation by human neutrophils at an early step in signal transduction. Biochem Biophys Acta 1175:312–318
Butcher SP, Bullock R, Graham DI, McCulloch J (1990) Correlation between amino acid release and neuropathologic outcome in rat brain following middle cerebral artery occlusion. Stroke 21:1727–1733
Cain BS, Meldrum DR, Dinarello CA, Meng X, Banerjee A, Harken AH (1998) Adenosine reduces cardiac TNF-α production and human myocardial injury following ischaemia-reperfusion. J Surg Res 76:117–123
Canals M, Marcellino D, Fanelli F, Ciruela F, de Benedetti P, Goldberg SR, Neve K, Fuxe K, Agnati LF, Woods AS, Ferré S, Lluis C, Bouvier M, Franco R (2003) Adenosine A2A-dopamine D2 receptor–receptor heteromerization: qualitative and quantitative assessment by fluorescence and bioluminescence energy transfer. J Biol Chem 278:46741–46749
Canals M, Angulo E, Casado V, Canela EI, Mallol J, Vinals F, Staines W, Tinner B, Hillion J, Agnati L, Fuxe K, Ferre S, Lluis C, Franco R (2005) Molecular mechanisms involved in the adenosine A(1) and A(2A) receptor-induced neuronal differentiation in neuroblastoma cells and striatal primary cultures. J Neurochem 92:337–348
Cantor SL, Zornow MH, Miller LP, Yaksh TL (1992) The effect of cyclohexyladenosine on the periischemic increases of hippocampal glutamate and glycine in the rabbit. J Neurochem 59:1884–1892
Cargnoni A, Ceconi C, Boraso A, Bernocchi P, Monopoli A, Curello S, Ferrari R (1999) Role of A2A receptors in the modulation of myocardial reperfusion damage. J Cardiovasc Pharmacol 33:883–893
Carlson NG, Wieggel WA, Chen J, Bacchi A, Rogers SW, Gahring LC (1999) Inflammatory cytokines IL-1α, IL-1β, IL-6, and TNF-α impart neuroprotection to an excitotoxin through distinct pathways. J Immunol 163:3963–3968
Casati C, Forlani A, Lozza G, Monopoli A (1997) Hemodynamic changes do not mediate the cardioprotection induced by the A1 adenosine receptor agonist CCPA in the rabbit. Pharmacol Res 35:51–55
Cassada DC, Tribble CG, Kaza AK, Fiser SM, Long SM, Linden J, Rieger JM, Kron IL, Kern JA (2001a) Adenosine analogue reduces spinal cord reperfusion injury in a time-dependent fashion. Surgery 130:230–235
Cassada DC, Tribble CG, Laubach VE, Nguyen BN, Rieger JM, Linden J, Kaza AK, Long SM, Kron IL, Kern JA (2001b) An adenosine A(2A) agonist, ATL-146e, reduces paralysis and apoptosis during rabbit spinal cord reperfusion. J Vasc Surg 34:482–488
Cassada DC, Tribble CG, Long SM, Laubach VE, Kaza AK, Linden J, Nguyen BN, Rieger JM, Fiser SM, Kron IL, Kern JA (2002) Adenosine A2(A) analogue ATL-146e reduces systemic tumor necrosing factor-α and spinal cord capillary platelet-endothelial cell adhesion molecule-1 expression after spinal cord ischemia. J Vasc Surg 35, 994–998
Chen JF, Huang Z, Ma J, Zhu J, Moratalla R, Standaert D, Moskowitz MA, Fink JS, Schwarzschild MA (1999) A2A adenosine receptor deficiency attenuates brain injury induced by transient focal ischaemia in mice. J Neurosci 19:9192–9200
Chen G-J, Harvey BK, Shen H, Chou J, Victor A, Wang Y (2006) Activation of adenosine A3 receptors reduces ischemic brain injury in rodents. J Neurosci Res 84:1848–1855
Chiang MC, Lee YC, Huang CL, Chern YJ (2005) cAMP-response element-binding protein contributes to suppression of the A(2A) adenosine receptor promoter by mutant huntingtin with expanded polyglutamine residues. J Biol Chem 280:14331–14340
Chou SY, Lee YC, Chen HM, Chiang MC, Lai HL, Chang HH, Wu YC, Sun CN, Chien CL, Lin YS, Wang SC, Tung YY, Chang C, Chern YJ (2005) CGS21680 attenuates symptoms of Huntington’s disease in a transgenic mouse model. J Neurochem 93:310–320
Chowdhury M, Fillenz M (1991) Presynaptic adenosine A2 and NMDA receptors regulate dopamine synthesis in rat striatal synaptosomes. J Neurochem 56:1783–1788
Ciccarelli R, D’Alimonte I, Ballerini P, D’Auro M, Nargi E, Buccella S, Di Iorio P, Bruno V, Nicoletti F, Caciagli F (2007) Molecular signalling mediating the protective effect of A(1) adenosine and mGlu3 metabotropic glutamate receptor activation against apoptosis by oxygen/glucose deprivation in cultured astrocytes. Mol Pharmacol 71:1369–1380
Coelho JE, Rebola N, Fragata I, Ribeiro JA, De Mendonca A, Cunha RA (2006) Hypoxia-induced desensitization and internalization of adenosine A(1) receptors in the rat hippocampus. Neuroscience 138:1195–1203
Connick JH, Stone TW (1986) The effects of kainate, β-kainate and quinolinic acids on the release of endogenous amino acids from rat brain slices. Biochem Pharmacol 35:3631–3635
Connick JH, Stone TW (1989) Quinolinic acid neurotoxicity: protection by intracerebral phenylisopropyl adenosine (PIA) and potentiation by hypotension. Neurosci Lett 101:191–196
Corodimas KP, Tomita H (2001) Adenosine A1 receptor activation selectively impairs the acquisition of contextual fear conditioning in rats. Behav Neurosci 115(6):1283–1290
Corradetti R, Lo Conte G, Moroni F, Passani MB, Pepeu G (1984) Adenosine decreases aspartate and glutamate release from rat hippocampal slices. Eur J Pharmacol 140:19–26
Corset V, Nguyen-Ba-Charvet KT, Forcet C, Moyse E, Chetodal, A, Mehlen P (2000) Netrin-1-mediated axon outgrowth and cAMP production requires interaction with adenosine A2B receptor. Nature 407:747–750
Corsi C, Melani A, Bianchi L, Pepeu G, Pedata F (1999a) Striatal A2A adenosine receptors differentially regulate spontaneous and K + -evoked glutamate release in vivo in young and aged rats. Neuroreport 10:687–691
Corsi C, Melani A, Bianchi L, Pepeu G, Pedata F (1999b) Effect of adenosine A2A stimulation on GABA release from the striatum of young and aged rats in vivo. Neuroreport 10:3933–3937
Corsi C, Melani A, Bianchi L, Pedata F (2000) Striatal A2A adenosine receptor antagonism differentially modifies striatal glutamate outflow in vivo in young and aged rats. Neuroreport 11:2591–2595
Coyle JT, Schwarcz R (1976) Lesion of striatal neurones with kainic acid provides a model for Huntington’s chorea. Nature 263:244–246
Cronstein BN (1994) Adenosine, an endogenous anti-inflammatory agent. J Appl Physiol 76:5–13
Cronstein BN, Rosenstein ED, Kramer SB, Weissmann G, Hirschhorn R (1985) Adenosine: a physiologic modulator of superoxide anion generation by human neutrophils. Adenosine acts via an A2 receptor on human neutrophils. J Immunol 135:1366–1371
Cronstein BN, Kubersky SM, Weissmann G, Hirschhorn R (1987) Engagement of adenosine receptors inhibits hydrogen peroxide-release by activated human neutrophils. Clin Immunol Immunopathol 42:76–85
Cronstein BN, Daguma L, Nichols D, Hutchison AJ, Williams M (1990) The adenosine/neutrophil paradox resolved: human neutrophils possess both A1 and A2 receptors that both promote chemotaxis and inhibit O2 generation respectively. J Clin Invest 85:1150–1157
Cronstein BN, Levin RI, Philips M, Hirschhorn R, Abramson SB, Weissmann G (1992) Neutrophil adherence to endothelium is enhanced via adenosine A1 receptors and inhibited via adenosine A2 receptors. J Immunol 148:2201–2206
Cunha RA, Johansson B, van der Ploeg I, Sebastião AM, Ribeiro JA, Fredholm BB (1994) Evidence for functionally important adenosine A2a receptors in the rat hippocampus. Brain Res 649:208–216
Cunha RA, Constantino MC, Sebastião AM, Ribeiro JA (1995) Modification of A1 and A2a adenosine receptor binding in aged striatum, hippocampus and cortex of the rat. Neuroreport 6:1583–1588
Cunha RA, Johansson B, Constantino MD, Sebastião AM, Fredholm BB (1996) Evidence for high affinity binding sites for the adenosine A2A receptor agonist [3H]CGS21680 in the rat hippocampus and cerebral cortex that are different from striatal A2A receptors. Naunyn–Schmiedeberg’s Arch Pharmacol 353:261–271
D’Alimonte I, Ballerini P, Nargi E, Buccella S, Giuliani P, Di Iorio P, Caciagli, F (2007) Staurosporine-induced apoptosis in astrocytes is prevented by A(1) adenosine receptor activation. Neurosci Lett 418:66–71
Dall’lgna OP, Porciuncula LO, Souza DO, Cunha RA, Lara DR (2003) Neuroprotection by caffeine and adenosine A(2A) receptor blockade of β-amyloid neurotoxicity. Br J Pharmacol 138: 1207–1209
Dall’Igna OP, Fett P, Gomes MW, Souza DO, Cunha RA, Lara DR (2007) Caffeine and adenosine A(2a) receptor antagonists prevent β-amyloid (25–35)-induced cognitive deficits in mice. Exp Neurol 203:241–245
Deckert J, Abel F, Kunig G, Hartmann J, Senitz D, Maier H, Ransmayr G, Riederer P (1998) Loss of human hippocampal adenosine A(1) receptors in dementia: evidence for lack of specificity. Neurosci Lett 244:1–4
De Mendonca A, Sebastiao AM, Ribeiro JA (1995) Inhibition of NMDA receptor-mediated currents in isolated rat hippocampal-neurons by adenosine A(1) receptor activation. Neuroreport 6:1097–1100
Dianzani C, Brunelleschi S, Viano I, Fantozzi R (1994) Adenosine modulation of primed human neutrophils. Eur J Pharmacol 263:223–226
Di-Capua N, Sperling O, Zoref-Shani E (2003) Protein kinase C-epsilon is involved in the adenosine-activated signal transduction pathway conferring protection against ischemia-reperffision injury in primary rat neuronal cultures. J Neurochem 84:409–412
Di Iorio P, Kleywegt S, Ciccarelli R, Traversa U, Andrew CM, Crocker CE, Werstiuk ES, Rathbone MP (2002) Mechanisms of apoptosis induced by purine nucleosides in astrocytes. Glia 38: 179–190
Dixon AK, Gubitz AK, Sirinathsinghji DJ, Richardson PJ, Freeman TC (1996) Tissue distribution of adenosine receptor mRNAs in the rat. Br J Pharmacol 118:1461–1468
Dixon AK, Widdowson L, Richardson PJ (1997) Desensitisation of the adenosine A1 receptor by the A2A receptor in the rat striatum. J Neurochem 69:315–321
Dolphin AC, Prestwich SA (1985) Pertussis toxin reverses adenosine inhibition of neuronal glutamate release. Nature 316:148–150
Domenici MR, Scattoni ML, Martire A, Lastoria G, Potenza RL, Borioni A, Venerosi A, Calamandrei G, Popoli P (2007) Behavioral and electrophysiological effects of the adenosine A2A receptor antagonist SCH58261 in R6/2 Huntington’s disease mice. Neurobiol Disease 28:197–205
Dux E, Fastbom J, Ungerstedt U, Rudolphi K, Fredholm BB (1990) Protective effect of adenosine and a novel xanthine derivative propentofylline on the cell damage after bilateral carotid occlusion in the gerbil hippocampus. Brain Res 516:248–256
Evans MC, Swan JH, Meldrum BS (1987) An adenosine analog, 2-chloroadenosine, protects against long-term development of ischemic cell loss in the rat hippocampus. Neurosci Lett 83:287–292
Ezeamuzie CI, Philips E (1999) Adenosine A3 receptors on human eosinophils mediate inhibition of degranulation and superoxide anion release. Br J Pharmacol 127:188–194
Fastbom J, Fredholm BB (1985) Inhibition of (3H)glutamate release from rat hippocampal slices by L-PIA. Acta Physiol Scand 125:121–123
Fastbom J, Pazos A, Probst A, Palacios JM (1986) Adenosine A1-receptors in human brain: characterisation and autoradiographic visualisation. Neurosci Lett 65:127–132
Fastbom J, Pazos A, Palacios JM (1987a) The distribution of adenosine A1 receptors and 5′-nucleotidase in the brain of some commonly used experimental animals. Neuroscience 22:813–826
Fastbom J, Pazos A, Probst A, Palacios JM (1987b) Adenosine A1 receptors in the human brain: a quantitative autoradiographic study. Neuroscience 22:827–839
Fedorova IM, Jacobson MA, Basile A, Jacobson KA (2003) Behavioral characterization of mice lacking the A(3) adenosine receptor: sensitivity to hypoxic neurodegeneration. Cell Mol Neurobiol 23:431–447
Ferkany JW, Zaczek R, Coyle JT (1982) Kainic acid stimulates excitatory amino acid neurotransmitter release at presynaptic receptors. Nature 298:757–759
Ferrante RJ, Kowall NW, Cipolloni PB, Storey E, Beal MF (1993) Excitotoxin lesions in primates as a model for Huntington’s disease: histopathologic and neurochemical characterization. Exp Neurol 119:46–71
Ferré S, von Euler G, Johansson B, Fredholm BB, Fuxe K (1991) Stimulation of high-affinity adenosine A2 receptors decreases the affinity of dopamine D2 receptors in rat striatal membranes. Proc Natl Acad Sci U S A 88:7238–7241
Fiebich BL, Biber K, Gyufko K, Berger R, Bauer J, vanCalker D (1996a) Adenosine A(2b) receptors mediate an increase in interleukin (IL)-6 mRNA and IL-6 protein synthesis in human astroglioma cells. J Neurochem 66:1426–1431
Fiebich BL, Biber K, Lieb K, vanCalker D, Berger M, Bauer J, GebickeHaerter PJ (1996b) Cyclooxygenase-2 expression in rat microglia is induced by adenosine A(2a)-receptors. Glia 18:152–160
Fiebich BL, Akundi RS, Biber K, Hamke M, Schmidt C, Butcher RD, van Calker D, Willmroth F (2005) IL-6 expression induced by adenosine A(2b) receptor stimulation in U373MG cells depends on p38 mitogen activated kinase and protein kinase C. Neurochem Intern 46:501–512
Fink JS, Kalda A, Ryu H, Stack EC, Schwarzschild MA, Chen JF, Ferrante RJ (2004) Genetic and pharmacological inactivation of the adenosine A(2A) receptor attenuates 3-nitropropionic acid-induced striatal damage. J Neurochem 88:538–544
Finn SF, Swartz KJ, Beal MF (1991) 2-Chloroadenosine attenuates NMDA, kainate and quisqualate toxicity. Neurosci Lett 126:191–194
Fotheringham J, Mayne M, Holden C, Nath A, Geiger JD (2004) Adenosine receptors control HIV-1 Tat-induced inflammatory responses through protein phosphatase. Virology 327:186–195
Fredholm BB, Dunwiddie TV (1988) How does adenosine inhibit transmitter release? Trends Pharmacol Sci 9:130–134
Fredholm BB, Johansson B, Lindstrom K, Wahlstrom G (1998) Age-dependent changes in adenosine receptors are not modified by life-long intermittent alcohol administration. Brain Res 791:177–185
Gao Y, Phillis JW (1994) CGS 15943, an adenosine A2 receptor antagonist, reduces cerebral ischemic injury in the Mongolian gerbil. Life Sci 55:PL61–PL65
Gerevich Z, Wirkner K, Illes P (2002) Adenosine A(2A) receptors inhibit the N-methyl-D-aspartate component of excitatory synaptic currents in rat striatal neurons. Eur J Pharmacol 451:161–164
Gianfriddo M, Melani A, Turchi D, Giovannini MG, Pedata F (2004) Adenosine and glutamate extracellular concentrations and mitogen-activated protein kinases in the striatum of Huntington transgenic mice. Selective antagonism of adenosine A(2A) receptors reduces transmitter outflow. Neurobiol Dis 17:77–88
Giménez-Llort L, Fernández-Teruel A, Escorihuela RM, Fredholm BB, Tobeña A, Pekny M, Johansson B (2002) Mice lacking the adenosine A1 receptor are anxious and aggressive, but are normal learners with reduced muscle strength and survival rate. Eur J Neurosci 16:547–550
Goncalves ML, Cunha RA, Ribeiro JA (1997) Adenosine A2A receptors facilitate 45Ca2 + uptake through class A calcium channels in rat hippocampal CA3 but not CA1 synaptosomes. Neurosci Lett 238:73–77
Goodman RR, Snyder SH (1982) Autoradiographic localisation of adenosine receptors in rat brain using [3H]cyclohexyladenosine. J Neurosci 2:1230–1241
Graham SH, Chen J, Sharp FR, Simon RP (1993) Limiting ischaemic injury by inhibition of excitatory amino acid release. J Cereb Blood Flow Metab 13:88–97
Grisham MB, Hernandez LA, Granger DN (1989) Adenosine inhibits ischemia-reperfusion-induced leukocyte adherence and extravasation. Am J Physiol 257:H1334–H1339
Grondin R, Bédard PJ, Hadj Tahar A, Grégoire L, Mori A, Kase H (1999) Antiparkinsonian effect of a new selective adenosine A2A receptor antagonist in MPTP-treated monkeys. Neurology 52:1673–1677
Guidetti P, Luthi-Carter RE, Augood SJ, Schwarcz R (2004) Neostriatal and cortical quinolinate levels are increased in early grade Huntington’s disease. Neurobiol Dis 17:455–461
Hannon JP, Bray-French KM, Phillips RM, Fozard JR (1998) Further pharmacological characterization of the adenosine receptor subtype mediating inhibition of oxidative burst in human isolated neutrophils. Drug Dev Res 43:214–224
Hara H, Onodera H, Kato H, Kogure K (1992) Effects of aging on signal transmission and transduction systems in the gerbil brain: morphological and autoradiographic study. Neuroscience 46:475–488
Haskó G, Cronstein BN (2004) Adenosine: an endogenous regulator of innate immunity. Trends Immunol 25(1):33–39
Haskó G, Németh ZH, Vizi ES, Salzman AL, Szabó C (1998) An agonist of adenosine A3 receptors decreases interleukin-12 and interferon-gamma production and prevents lethality in endotoxemic mice. Eur J Pharmacol 358:261–268
Haskó G, Pacher P, Deitch EA, Vizi ES (2007) Shaping of monocyte and macrophage function by adenosine receptors. Pharmacol Ther 113:264–275
Hauber W, Bareiss A (2001) Facilitative effects of an adenosine A1∕A2 receptor blockade on spatial memory performance of rats: selective enhancement of reference memory retention during the light period. Behav Brain Res 118(1):43–52
Heese K, Fiebich BL, Bauer J, Otten U (1997) Nerve growth factor (NGF) expression in rat microglia is induced by adenosine A(2a)-receptors. Neurosci Lett 231:83–86
Héron A, Lasbennes F, Seylez J (1993) Adenosine modulation of amino acid release in rat hippocampus during ischaemia and veratridine depolarisation. Brain Res 608:27–32
Héron A, Lekieffre D, Le Peillet E, Lasbennes F, Seylaz J, Plotkine M, Boulu RG (1994) Effects of an A1 adenosine receptor agonist on the neurochemical, behavioural and histological consequences of ischaemia. Brain Res 641:217–224
Hiraide T, Katsura K, Muramatsu H, Asano G, Katayama Y (2001) Adenosine receptor antagonists cancelled the ischemic tolerance phenomenon in gerbil. Brain Res 910:94–98
Hollins C, Stone TW (1980) Adenosine inhibition of GABA release from slices of rat cerebral cortex. Br J Pharmacol 69:107–112
Hooper N, Fraser C, Stone TW (1996) Effects of purine analogues on spontaneous alternation in mice. Psychopharmacology 123(3):250–257
Hosseinzadeh H, Stone TW (1998) Tolbutamide blocks postsynaptic but not presynaptic effects of adenosine on hippocampal CA1 neurons. J Neural Transm 105:161–172
Huang QY, Wei C, Yu LQ, Coelho JE, Shen HY, Kalda A, Linden J, Chen JF (2006) Adenosine A2A receptors in bone marrow-derived cells but not in forebrain neurons are important contributors to 3-nitropropionic acid-induced striatal damage as revealed by cell-type-selective inactivation. J Neurosci 26:11371–11378
Hutchison AJ, Webb RL, Oei HH, Ghai GR, Zimmerman MB, Williams M (1989) CGS21680, an A2 selective adenosine receptor agonist with preferential hypotensive activity. J Pharmacol Exp Ther 251:47–55
Ikeda M, Mackay KB, Dewar D, Mcculloch J (1993) Differential alterations in adenosine-A(1) and kappa-1-opioid receptors in the striatum in Alzheimer’s-disease. Brain Res 616:211–217
Jaarsma D, Sebens JB, Korf J (1991) Reduction of adenosine-A1-receptors in the perforant pathway terminal zone in Alzheimer hippocampus. Neurosci Lett 121:111–114
Jacobson KA (1998) Adenosine A3 receptors: novel ligands and paradoxical effects. Trends Pharmacol Sci 19:184–191
Jacobson I, Hamberger A (1985) Kainic acid-induced changes of extracellular amino acid levels, evoked potentials and EEG activity in the rabbit olfactory bulb. Brain Res 348:289–296
Jansen KLR, Faull RLM, Dragunow M, Synek BL (1990) Alzheimer’s disease: changes in hippocampal N-methyl-D-aspartate, quisqualate, neurotensin, adenosine, benzodiazepine, serotonin and opioid receptors—an autoradiographic study. Neuroscience 39:613–627
Jarvis MF, Williams M (1989) Direct autoradiographic localisation of adenosine A2 receptors in the brain using the A2-selective agonist, [3H]CGS21680. Eur J Pharmacol 168:243–246
Jiang N, Kowaluk EA, Lee CH, Mazdiyasni H, Chopp M (1997) Adenosine kinase inhibition protects brain against transient focal ischemia in rats. Eur J Pharmacol 320:131–137
Johnson-Kozlow M, Kritz-Silverstein D, Barrett-Connor E, Morton D (2002) Coffee consumption and cognitive function among older adults. Am J Epidemiol 156:842–850
Johnston JB, Silva C, Gonzalez G, Holden J, Warren KG, Metz LM, Power C (2001) Diminished adenosine A1 receptor expression on macrophages in brain and blood of patients with multiple sclerosis. Ann Neurol 49:650–658
Jones PA, Smith RA, Stone TW (1998a) Protection against intrahippocampal kainate excitotoxicity by intracerebral administration of an adenosine A2A receptor antagonist. Brain Res 800: 328–335
Jones PA, Smith RA, Stone TW (1998b) Protection against kainate-induced excitotoxicity by adenosine A2A receptor agonists and antagonists. Neuroscience 85:229–237
Kalaria RN, Sromek S, Wilcox BJ, Unnerstall JR (1990) Hippocampal adenosine-A1-receptors are decreased in Alzheimer’s-disease. Neurosci Lett 118:257–260
Kanda T, Jackson MJ, Smith LA, Pearce RK, Nakamura J, Kase H, Kuwana Y, Jenner P (1998) Adenosine A2A antagonist: a novel antiparkinsonian agent that does not provoke dyskinesia in parkinsonian monkeys. Ann Neurol 43:507–513
Kano T, Katayama Y, Kawamata T, Hirota H, Tsubokawa T (1994) Propentofylline administered by microdialysis attenuates ischaemia-induced hippocampal damage but not excitatory amino acid release in gerbils. Brain Res 641:149–154
Khaspekov L, Shamloo M, Victorov I, Wieloch T (1998) Sublethal in vitro glucose-oxygen deprivation protects cultured hippocampal neurons against a subsequent severe insult. Neuroreport 9:1273–1276
Knutsen LJS, Sheardown MJ, Roberts SM, Mogensen JP, Olsen UB, Thomsen C, Bowler AN (1998) Adenosine A1 and A3 selective N-alkoxypurines as novel cytokine modulators and neuroprotectants. Drug Dev Res 45:214–221
Kohler C, Schwarcz R, Fuxe K (1978) Perforant path transections protect hippocampal granule cells from kainate lesion. Neurosci Lett 10:241–246
Kopf SR, Melani A, Pedata F, Pepeu G (1999) Adenosine and memory storage: effect of A(1) and A(2) receptor antagonists. Psychopharmacology 146:214–219
Kreckler LM, Wan TC, Ge ZD, Auchampach JA (2006) Adenosine inhibits tumor necrosis factor-alpha release from mouse peritoneal macrophages via A(2A) and A(2B) but not the A(3) adenosine receptor. J Pharmacol Exp Ther 317:172–180
Kurokawa M, Kirk IP, Kirkpatrick KA, Kase H, Richardson PJ (1994) Inhibition by KF17837 of adenosine A2A receptor-mediated modulation of striatal GABA and acetylcholine release. Br J Pharmacol 113:43–48
Lai DM, Tu YK, Liu IM, Cheng JT (2005) Increase of adenosine A1 receptor gene expression in cerebral ischemia of Wistar rats. Neurosci Lett 387:59–61
Lappin D, Whaley K (1984) Adenosine A2 receptors on human monocytes modulate C2 production. Clin Exp Immunol 57:454–460
Latini S, Pazzagli M, Pepeu G, Pedata F (1996) A2 adenosine receptors: their presence and neuromodulatory role in the CNS. Gen Pharmacol 27:925–933
Latini S, Bordoni F, Corradetti R, Pepeu G, Pedata F (1999) Effect of A2A adenosine receptor stimulation and antagonism on synaptic depression induced by in vitro ischaemia in rat hippocampal slices. Br J Pharmacol 128:1035–1044
Lau Y-S, Mouradian MM (1993) Protection against acute MPT-induced dopamine depletion in mice by adenosine. J Neurochem 60:768–771
Lee FS, Chao MV (2001) Activation of Trk neurotrophin receptors in the absence of neurotrophins. Proc Natl Acad Sci USA 98:3555–3560
Lee KS, Reddington M (1986) Autoradiographic evidence for multiple CNS binding sites for adenosine derivative. Neuroscience 19:535–549
Lehmann A, Isacsson H, Hamberger A (1983) Effects of in vivo administration of kainic acid on the extracellular amino acid pool in the rabbit hippocampus. J Neurochem 40:1314–1320
LeVraux V, Chen YL, Masson I, De Sousa M, Giroud JP, Florentin I, Chauvelot-Moachon L (1993) Inhibition of human monocyte TNF production by adenosine receptor agonists. Life Sci 52:1917–1924
Li XX, Nomura T, Aihara H, Nishizaki T (2001) Adenosine enhances glial glutamate efflux via A2A adenosine receptors. Life Sci 68:1343–1350
Linden J, Thai T, Figler H, Jin X, Robeva AS (1999) Characterization of human A2B adenosine receptors: radioligand binding, western blotting, and coupling to Gq in human embryonic kidney 293 cells and HMC-1 mast cells. Mol Pharmacol 56:705–713
Link AA, Kino T, Worth JA, McGuire JL, Crane ML, Chrousos GP, Wilder RL, Elenkov IJ (2000) Ligand activation of the adenosine A2A receptors inhibits IL-12 production by human monocytes. J Immunol 164:436–442
Lipton SA, Rosenberg PA (1994) Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med 330:613–622
Liu HC, Hon CJ, Liu TY, Tsai, SJ (2005) Association analysis of adenosine A2a receptor 1976T > C polymorphisms and Alzheimer’s disease. Eur Neurol 53:99–100
Liu YH, Xiong L, Chen SY, Wang Q (2006) Isoflurane tolerance against focal cerebral ischemia is attenuated by adenosine A(1) receptor antagonists. Can J Anaes 53:194–201
Lozza G, Conti A, Ongini E, Monopoli A (1997) Cardioprotective effects of adenosine A1 and A2A receptor agonists in the isolated heart. Pharmacol Res 35:57–64
MacGregor DG, Stone TW (1993) Inhibition by the adenosine analogue, (R)-N 6-phenylisopropyladenosine, of kainic acid neurotoxicity in rat hippocampus after systemic administration. Br J Pharmacol 109:316–321
MacGregor DG, Miller WJ, Stone TW (1993) Mediation of the neuroprotective action of R-phenylisopropyladenosine through a centrally located adenosine A1 receptor. Br J Pharmacol 110:470–476
MacGregor DG, Jones PA, Maxwell WL, Graham DI, Stone TW (1996) Prevention by a purine analogue of kainate-induced neuropathology in rat hippocampus. Brain Res 725:115–120
Maemoto T, Tada M, Mihara T, Ueyama N, Matsuoka H, Harada K, Yamaji T, Shirakawa K, Kuroda S, Akahane A, Iwashita A, Matsuoka N, Mutoh S (2004) Pharmacological characterization of FR194921, a new potent, selective, and orally active antagonist for central adenosine A(1) receptors. J Pharmacol Sci 96:42–52
Mager R, Ferroni S, Schubert P (1990) Adenosine modulates a voltage-dependent chloride conductance in cultured hippocampal neurons. Brain Res 532:58–62
Maggirwar SB, Dhanraj DN, Somani SM, Ramkumar V (1994) Adenosine acts as an endogenous activator of the cellular antioxidant defense system. Biochem Biophys Res Commun 201: 508–515
Maglione V, Giallonardo P, Cannella M, Martino T, Frati L, Squitieri F (2005) Adenosine A(2A) receptor dysfunction correlates with age at onset anticipation in blood platelets of subjects with Huntington’s disease. Am J Med Genet B Neuropsychiatr Genet 139B:101–105
Maglione V, Cannella M, Martino T, De Blasi A, Frati L, Squitieri F (2006) The platelet maximum number of A2A-receptor binding sites (Bmax) linearly correlates with age at onset and CAG repeat expansion in Huntington’s disease patients with predominant chorea. Neurosci Lett 393:27–30
Maia L, de Mendonça A (2002) Does caffeine intake protect from Alzheimer’s disease? Eur J Neurol 9:377–382
Mally J, Stone TW (1994) The effect of theophylline on parkinsonian symptoms. J Pharm Pharmacol 46:515–517
Mally J, Stone TW (1996) Potential role of adenosine antagonist therapy in pathological tremor disorders. Pharmacol Ther 72:243–250
Mally J, Stone TW (1998) Potential of adenosine A(2A) receptor antagonists in the treatment of movement disorders. CNS Drugs 10:311–320
Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier T, Lehrach H, Davies SW, Bates GP (1996) Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87:493–506
Martinez-Mir MI, Probst A, Palacios JM (1991) Adenosine A2 receptors: selective localisation in the human basal ganglia and alterations with disease. Neuroscience 42:697–706
Matherne GP, Linden J, Byford AM, Gauthier NS, Headrick JP (1997) Transgenic A1 adenosine receptor overexpression increases myocardial resistance to ischaemia. Proc Nat Acad Sci USA 94:6541–6546
Mayfield RD, Suzuki F, Zahniser NR (1993) Adenosine A2A receptor modulation of electrically evoked endogenous GABA release from rat globus pallidus. J Neurochem 60:2334–2337
Mayne M, Shepel PN, Jiang Y, Geiger JD, Power C (1999) Dysregulation of adenosine A(1) receptor-mediated cytokine expression in peripheral blood mononuclear cells from multiple sclerosis patients. Ann Neurol 45:633–639
McLarnon JG, Choi HB, Lue LF, Walker DG, Kim SU (2005) Perturbations in calcium-mediated signal transduction in microglia from Alzheimer’s disease patients. J Neurosci Res 81:426–435
McRae A, Rudolphi KA, Schubert P (1994) Propentofylline depresses amyloid and Alzheimer’s CSF microglial antigens after ischaemia. Neuroreport 5:1193–1196
McWhinney CD, Dudley MW, Bowlin TL, Peet NP, Schook L, Bradshaw M, De M, Borcherding DR, Edwards CK 3rd (1996) Activation of adenosine A3 receptors on macrophages inhibits TNFα. Eur J Pharmacol 310:209–216
Meerlo P, Roman V, Farkas E, Keijser JN, Nyakas C, Luiten PGM (2004) Ageing-related decline in adenosine A1 receptor binding in the rat brain: an autoradiographic study. J Neurosci Res 78:742–748
Meiners I, Hauschildt S, Nieber K, Munch G (2004) Pentoxyphylline and propentophylline are inhibitors of TNF-ot release in monocytes activated by advanced glycation endproducts. J Neural Transm 111:441–447
Melani A, Pantoni L, Corsi C, Bianchi L, Monopoli A, Bertorelli R, Pepeu G, Pedata F (1999) Striatal outflow of adenosine, excitatory amino acids, GABA, and taurine in awake, freely moving rats after middle cerebral artery occlusion: correlation with neurological deficit and histopathological damage. Stroke 30:2448–2454
Meldrum DR, Cain BS, Cleveland JC Jr, Meng X, Ayala A, Banerjee A, Harken AH (1997) Adenosine decreases post-ischemic cardiac TNF-α production: anti-inflammatory implications for preconditioning and transplantation. Immunology 92:472–477
Merkel LA, Lappe RW, Rivera LM, Cox BF, Perrone MH (1992) Demonstration of vasorelaxant activity with an A1-selective adenosine agonist in porcine coronary artery: involvement of potassium channels. J Pharmacol Exp Ther 260:437–443
Michaelis ML, Michaelis EK, Myers SL (1979) Adenosine modulation of synaptosomal dopamine release. Life Sci 24:2083–2092
Mielke R, Kessler J, Szelies B, Herholz K, Wienhard K, Heiss WD (1996a) Vascular dementia: perfusional and metabolic disturbances and effects of therapy. J Neural Transm 47:183–191
Mielke R, Kittner B, Ghaemi M, Kessler J, Szelies B, Herholz K, Heiss WD (1996b) Propentofylline improves regional cerebral glucose metabolism and neuropsychologic performance in vascular dementia. J Neurol Sci 141:59–64
Miller WJ, Macgregor DG, Stone TW (1994) Time-course of purine protection against kainate-induced increase in hippocampal [H-3] PK11195 binding. Brain Res Bull 34:133–136
Monopoli A, Lozza G, Forlani A, Mattavelli A, Ongini E (1998) Blockade of adenosine A2A receptors by SCH58261 results in neuroprotective effects in cerebral ischaemia in rats. Neuroreport 9:3955–3959
Moreau JL, Huber G (1999) Central adenosine A2A receptors: an overview. Brain Res Rev 31: 65–82
Moreno MB, Hevia H, Santamaria M, Sepulcre J, Munoz J, Garcia-Trevijano ER, Berasain C, Corrales FJ, Avila MA, Villoslada P (2006) Methylthioadenosine reverses brain autoimmune disease. Ann Neurol 60:323–334
Muller CE (1997) A(1)-adenosine receptor antagonists. Exp Opin Ther Pat 7:419–440
Muller CE (2000) A(2A) adenosine receptor antagonists—future drugs for Parkinson’s disease? Drugs Future 25:1043–1052
Murray TF (1982) Up-regulation of rat cortical adenosine receptors following chronic administration of theophylline. Eur J Pharmacol 82:113–114
Nakamura M, Nakakimura K, Matsumoto M, Sakabe T (2002) Rapid tolerance to focal cerebral ischemia in rats is attenuated by adenosine A(1) receptor antagonist. J Cereb Blood Flow Metab 22:161–170
Nakata H, Yoshioka K, Saitoh O (2003) Hetero-oligomerization between adenosine A1 and P2Y1 receptors in living cells: formation of ATP-sensitive adenosine receptors. Drug Dev Res 58:340–349
Nikbakht M-R, Stone TW (2001) Activation of NMDA receptors suppresses the presynaptic effects of adenosine. Br J Pharmacol 133:155P
Nishizaki T (2004) ATP- and adenosine-mediated signaling in the central nervous system: adenosine stimulates glutamate release from astrocytes via A2A adenosine receptors J Pharmacol Sci 94:100–102
Nishizaki T, Nagai K, Nomura T, Tada H, Kanno T, Tozaki H, Li XX, Kondoh T, Kodama N, Takahashi E, Sakai N, Tanaka K, Saito N (2002) A new neuromodulatory pathway with a glial contribution mediated via A(2a) adenosine receptors. Glia 39:133–47
Noble S, Wagstaff AJ (1997) Propentofylline. CNS Drugs 8:257–264
Norenberg W, Wirkner K, Illes P (1997) Effect of adenosine and some of its structural analogues on the conductance of NMDA receptor channels in a subset of rat neostriatal neurones. Br J Pharmacol 122:71–80
Norenberg W, Wirkner K, Assmann H, Richter M, Illes P (1998) Adenosine A(2A) receptors inhibit the conductance of NMDA receptor channels in rat neostriatal neurons. Aminoacids 14:33–39
Normile HJ, Barraco RA (1991) N 6-Cyclopentyladenosine impairs passive avoidance retention by selective action at A1 receptors. Brain Res Bull 27:101–104
O’Kane EM, Stone TW (1998) Interactions between A1 and A2 adenosine receptor-mediated responses in the rat hippocampus in vitro. Eur J Pharmacol 362:17–25
O’Regan MH, Simpson RE, Perkins LM, Phillis JW (1992) The selective A2 adenosine receptor agonist CGS21680 enhances excitatory amino acid release from the ischaemic rat cerebral cortex. Neurosci Lett 138:169–172
Ochiishi T, Chen L, Yukawa A, Saitoh Y, Sekino Y, Arai T, Nakata H, Miyamoto H (1999a) Cellular localisation of adenosine A1 receptors in rat forebrain: immunohistochemical analysis using adenosine A1 receptor-specific monoclonal antibody. J Comp Neurol 411:301–316
Ochiishi T, Saitoh Y, Yukawa A, Saji M, Ren Y, Shirao T, Miyamoto H, Nakata H, Sekino Y (1999b) High levels of adenosine A1 receptor-like immunoreactivity in the CA2/CA3a region of the adult rat hippocampus. Neuroscience 93:955–967
Ochoa CM, Jackson TA, Aaron CS, Lahti RA, Strain GM, Von Voigtlander PF (1992) Antagonism of kainic acid lesions in the mouse hippocampus by U-54494A and U-50488H. Life Sci 51:1135–1143
Okazaki MM, Nadler JV (1988) Protective effects of mossy fibre lesions against kainic acid induced seizures and neuronal degeneration. Neuroscience 26:763–781
Okonkwo DO, Reece TB, Laurent JJ, Hawkins AS, Ellman PI, Linden J, Kron IL, Tribble CG, Stone JR, Kern JA (2006) A comparison of adenosine A(2A) agonism and methylprednisolone in attenuating neuronal damage and improving functional outcome after experimental traumatic spinal cord injury in rabbits. J Neurosurg 4:64–70
Oliveira JC, Constantino MD, Sebastiao AM, Ribeiro JA (1995) Ascorbate/Fe3 + -induced peroxidation and inhibition of the binding of A1 adenosine receptor ligands in rat-brain membranes. Neurochem Int 26:263–268
Ongini E, Fredholm BB (1996) Pharmacology of adenosine A2A receptors. Trends Pharmacol Sci 17:364–372
Ongini E, Adami M, Ferri C, Bertorelli R (1997) Adenosine A2A receptors and neuroprotection. Ann NY Acad Sci 825:30–48
Ongini E, Monopoli A, Impagnatiello F, Fredduzzi S, Schwarzschild M, Chen JF (2001) Dual actions of A2A adenosine receptor antagonists on motor dysfunction and neurodegenerative processes. Drug Dev Res 52:379–386
Pagonopoulou O, Angelatou F (1992) Reduction of A1 adenosine receptors in cortex, hippocampus and cerebellum in ageing mouse brain. Neuroreport 3:735–737
Palmer TM, Poucher SM, Jacobson KA, Stiles GL (1995) 125I-4-[7-amino-2-{2-furyl} {1,2,4}triazolo{2,3-a} {1,3,5}triazin-5-yl-amino]ethyl)phenol, a high affinity antagonist radioligand selective for the A2A adenosine receptor. Mol Pharmacol 48:970–974
Parkinson FE, Rudolphi KA, Fredholm BB (1994) Propentofylline: a nucleoside transport inhibitor with neuroprotective effects in cerebral ischaemia. Gen Pharmacol 25:1053–1058
Pazzagli M, Corsi C, Latini S, Pedata F, Pepeu G (1994) In vivo regulation of extracellular adenosine levels in the cerebral cortex by NMDA and muscarinic receptors. Eur J Pharmacol 254:277–282
Pedata F, Corsi C, Melani A, Bordoni F, Latini S (2001) Adenosine extracellular brain concentrations and role of A2A receptors in ischemia. Ann N Y Acad Sci 939:74–84
Pedata F, Gianfriddo M, Turchi D, Melani A (2005) The protective effect of adenosine A(2A) receptor antagonism in cerebral ischemia. Neurol Res 27:169–174
Phillis JW (1995) The effects of selective A1 and A2A adenosine receptor antagonists on cerebral ischemic injury in the gerbil. Brain Res 705:79–84
Phillis JW, O’Regan MH (1993) Prevention of ischemic brain injury by adenosine receptor activation. Drug Dev Res 28:390–394
Pierson PM, Peteri-Brunbäck B, Pisani DF, Abbracchio MP, Mienville JM, Rosso L (2007) A(2b) receptor mediates adenosine inhibition of taurine efflux from pituicytes. Biol Cell 99:445–454
Pignataro G, Simon RP, Boison D (2007) Transgenic overexpression of adenosine kinase aggravates cell death in ischemia. J Cereb Blood Flow Metab 27:1–5
Pintor A, Galluzzo M, Grieco R, Pèzzola A, Reggio R, Popoli P (2004) Adenosine A2A receptor antagonists prevent the increase in striatal glutamate levels induced by glutamate uptake inhibitors. J Neurochem 89:152–156
Pitsikas N, Borsini F (1997) The adenosine A1 receptor antagonist BIIP 20 counteracts scopolamine-induced behavioral deficits in the passive avoidance task in the rat. Eur J Pharmacol 328:19–22
Poli A, Lucchi R, Vibio M, Barnabei O (1991) Adenosine and glutamate modulate each other’s release from rat hippocampal synaptosomes. J Neurochem 57:298–306
Popoli P, Pèzzola A, Domenici MR, Sagratella S, Diana G, Caporali MG, Bronzetti E, Vega J, Scotti de Carolis A (1994) Behavioral and electrophysiological correlates of the quinolinic acid rat model of Huntington’s disease in rats. Brain Res Bull 35:329–335
Popoli P, Betto P, Reggio R, Ricciarello G (1995) Adenosine A(2A) receptor stimulation enhances striatal extracellular glutamate levels in rats. Eur J Pharmacol 287:215–217
Popoli P, Pintor A, Domenici MR, Frank C, Tebano MT, Pèzzola A, Scarchilli L, Quarta D, Reggio R, Malchiodi-Albedi F, Falchi M, Massotti M (2002) Blockade of striatal adenosine A2A receptor reduces, through a presynaptic mechanism, quinolinic acid-induced excitotoxicity: possible relevance to neuroprotective interventions in neurodegenerative diseases of the striatum. J Neurosci 22:1967–1975
Popoli P, Blum D, Martire A, Ledent C, Ceruti S, Abbracchio MP (2007) Functions, dysfunctions and possible therapeutic relevance of adenosine A2A receptors in Huntington’s disease. Prog Neurobiol 81:331–348
Pousinha PA, Diogenes JM, Ribeiro AJ, Sebastiao AM (2006) Triggering of BDNF facilitatory action on neuromuscular transmission by adenosine A2A receptors. Neurosci Lett 404:143–147
Prediger RDS, Batista LC, Takahashi RN (2005) Caffeine reverses age-related deficits in olfactory discrimination and social recognition memory in rats: involvement of adenosine A(1) and A(2A) receptors. Neurobiol Ageing 26:957–964
Press NJ, Gessi S, Borea PA, Polosa R (2007) Therapeutic potential of adenosine receptor antagonists and agonists. Exp Opin Ther Pat 17:979–991
Prior C, Torres RJ, Puig JG (2006) Hypoxanthine effect on equilibrative and concentrative adenosine transport in human lymphocytes. Implications in the pathogenesis of Lesch–Nyhan syndrome. Nucleosides Nucleotides Nucl Acids 25:1065–1069
Pugliese AM, Latini S, Corradetti R, Pedata F (2003) Brief, repeated, oxygen-glucose deprivation episodes protect neurotransmission from a longer ischemic episode in the in vitro hippocampus: role of adenosine receptors. Br J Pharmacol 140:305–314
Pugliese AM, Coppi E, Volpini R, Cristalli G, Corradetti R, Jeong LS, Jacobson KA, Pedata F (2007) Role of adenosine A(3) receptors on CA1 hippocampal transmission during oxygen-glucose deprivation episodes of different duration. Biochem Pharmacol 74:768–779
Quintana JLB, Allam MF, Del Castillo AS, Navajas RFC (2007) Alzheimer’s disease and coffee: a review. Neurol Res 29:91–95
Ramakers BP, Riksen NP, Rongen GA, van der Hoeven JG, Smits P, Pickkers P (2006) The effect of adenosine receptor agonists on cytokine release by human mononuclear cells depends on the specific Toll-like receptor subtype used for stimulation. Cytokine 35:95–99
Rathbone MP, Middlemiss PJ, Kim JK, Gysbers JW, Deforge SP, Smith RW, Hughes DW (1992) Adenosine and its nucleotides stimulate proliferation of chick astrocytes and human astrocytoma cells. Neurosci Res 13:1–17
Rathbone MP, Middlemiss PJ, Gysbers JW, Andrew C, Herman MAR, Reed JK, Ciccarelli R, Di Iorio P, Caciagli F (1999) Trophic effects of purines in neurons and glial cells. Prog Neurobiol 59:663–690
Reece TB, Okonkwo DO, Ellman PI, Warren PS, Smith RL, Hawkins AS, Linden J, Kron IL, Tribble CG, Kern JA (2004) The evolution of ischemic spinal cord injury in function, cytoarchitecture, and inflammation and the effects of adenosine A(2A) receptor activation. J Thorac Cardiovasc Surg 128:925–932
Reece TB, Kron IL, Okonkwo DO, Laurent JJ, Tache-Leon C, Maxey TS, Ellman PI, Linden J, Tribble CG, Kern JA (2006) Functional and cytoarchitectural spinal cord protection by ATL-146e after ischemia/reperfusion is mediated by adenosine receptor agonism. J Vasc Surg 44:392–397
Regenold JT, Illes P (1990) Inhibitory adenosine A1-receptors on rat locus coeruleus neurones. An intracellular electrophysiological study. Naunyn–Schmied Arch Pharmacol 341:225–231
Reggio R, Pezzola A, Popoli P (1999) The intrastratial injection of an adenosine A(2) receptor antagonist prevents frontal cortex EEG abnormalities in a rat model of Huntington’s disease. Brain Res 831:315–318
Revan S, Montesinos MC, Naime D, Landau S, Cronstein BN (1996) Adenosine A2 receptor occupancy regulates stimulated neutrophil function via activation of a serine/threonine protein phosphatase. J Biol Chem 271:17114–17118
Ribeiro JA, Sebastiao AM, de Mendonça A (2002) Adenosine receptors in the nervous system: pathophysiological implications. Prog Neurobiol 68:377–392
Riedel W, Hogervorst E, Leboux R, Verhey F, Vanpraag H, Jolles J (1995) Caffeine attenuates scopolamine-induced memory impairment in humans. Psychopharmacology 122:158–168
Ritchie PK, Spangelo BL, Krzymowski DK, Rossiter TB, Kurth E, Judd AM (1997) Adenosine increases interleukin-6 release and decreases TNF release from rat adrenal zona glomerulosa cells, ovarian cells, anterior pituitary cells and peritoneal macrophages. Cytokine 9:187–198
Rivkees SA, Price SL, Zhou FC (1995) Immunohistochemical detection of A1 adenosine receptors in rat brain with emphasis on localisation in the hippocampal formation, cerebral cortex, cerebellum and basal ganglia. Brain Res 677:193–203
Rivkees SA, Thevananther S, Hao H (2000) Are A3 adenosine receptors expressed in the brain? Neuroreport 11:1025–1030
Robledo P, Ursu G, Mahy N (1999) Effects of adenosine and gamma-aminobutyric acid A receptor antagonists on N-methyl-D-aspartate induced neurotoxicity in the rat hippocampus. Hippocampus 9:527–533
Rodriguez A, Martin M, Albasanz JL, Barrachina M, Espinosa JC, Torres JM, Ferrer I (2006) Adenosine A(1) receptor protein levels and activity is increased in the cerebral cortex in Creutzfeldt–Jakob disease and in bovine spongiform encephalopathy-infected bovine-PrP mice. J Neuropathol Exp Neurol 65:964–975
Rosi S, McGann K, Hauss-Wegrzyniak B, Wenk GL (2003) The influence of brain inflammation upon neuronal adenosine A(2B) receptors. J Neurochem 86:220–227
Rubaj A, Zgodzinski W, Sieklucka-Dziuba M (2003) The influence of adenosine A3 receptor agonist: IB-MECA, on scopolamine- and MK-801-induced memory impairment. Behav Brain Res 141:11–17
Rudolphi KA, Schubert P (1997) Modulation of neuronal and glial cell function by adenosine and neuroprotection in vascular dementia. Behav Brain Res 83:123–128
Rudolphi KA, Keil M, Hinze HJ (1987) Effect of theophylline on ischemically induced hippocampal damage in Mongolian gerbils: a behavioural and histopathological study. J Cereb Blood Flow Metab 7:74–81
Rudolphi KA, Keil M, Fastbom J, Fredholm BB (1989) Ischemic damage in gerbil hippocampus is reduced following upregulation of adenosine (A1) receptors by caffeine treatment. Neurosci Lett 103:275–280
Rudolphi KA, Schubert P, Parkinson FE, Fredholm BB (1992) Adenosine and brain ischaemia. Cerebrovasc Brain Metab Rev 4:346–369
Sajjadi FG, Takabayashi K, Foster AC, Domingo RC, Firestein GS (1996) Inhibition of TNF-α expression by adenosine: role of A3 adenosine receptors. J Immunol 156:3435–3442
Saura J, Angulo E, Ejarque A, Casado V, Tusell JM, Moratalla R, Chen JF, Schwarzschild MA, Lluis C, Franco R, Serratosa J (2005) Adenosine A(2A) receptor stimulation potentiates nitric oxide release by activated microglia. J Neurochem 95:919–929
Scatena R, Martorana GE, Bottoni P, Botta G, Pastore P, Giardina B (2007) An update on pharmacological approaches to neurodegenerative diseases. Exp Opin Invest Drugs 16:59–72
Scattoni ML, Valanzano A, Pezzola A, De March Z, Fusco FR, Popoli P, Calamandrei G (2007) Adenosine A(2A) receptor blockade before striatal excitotoxic lesions prevents long term behavioural disturbances in the quinolinic rat model of Huntington’s disease. Behav Brain Res 176:216–221
Schiffmann SN, Jacobs O, Vanderhaegen JJ (1991) Striatal restricted adenosine A2 receptor (RDC8) is expressed by enkephalin but not by substance P neurons: an in situ hybridisation histochemistry study. J Neurochem 57:1062–1067
Schingnitz G, Kufnermuhl U, Ensinger H, Lehr E, Kuhn FJ (1991) Selective A1-antagonists for treatment of cognitive deficits. Nucleosides Nucleotides 10:1067–1076
Scholz KP, Miller RJ (1992) Inhibition of quantal transmitter release in the absence of calcium influx by a G protein-linked adenosine receptor at hippocampal synapses. Neuron 8:1139–1150
Schubert P, Ferroni S, Mager R (1991) Pharmacological blockade of chloride pumps on chloride channels reduces the adenosine-mediated depression of stimulus train-evoked calcium fluxes in rat hippocampal slices. Neurosci Lett 124:174–177
Schubert P, Ogata T, Ferroni S, McRae A, Nakamura Y, Rudolphi K (1996) Modulation of glial cell signaling by adenosine and pharmacological reinforcement: a neuroprotective strategy? Mol Chem Neuropathol 28:185–190
Schubert P, Ogata T, Rudolphi K, Marchini C, McRae A, Ferroni S (1997) Support of homeostatic glial cell signaling: a novel therapeutic approach by propentofylline. Ann NY Acad Sci 826:337–347
Schurr A, Reid KH, Tseng MT, West C, Rigor BM (1986) Adaptation of adult brain-tissue to anoxia and hypoxia invitro. Brain Res 374:244–248
Sebastiao AM, Ribeiro JA (1992) Evidence for the presence of excitatory A2 adenosine receptors in the rat hippocampus. Neurosci Lett 138:41–44
Sebastiao AM, Ribeiro JA (1996) Adenosine A(2) receptor-mediated excitatory actions on the nervous system. Progr Neurobiol 48:167–189
Sei Y, von Lubitz DKJE, Abbracchio MP, Ji X-D, Jacobson KA (1997) Adenosine A3 receptor agonist-induced neurotoxicity in rat cerebellar granule neurons. Drug Dev Res 40:267–273
Selley ML (2004) Increased homocysteine and decreased adenosine formation in Alzheimer’s disease. Neurol Res 26:554–557
Sheardown MJ, Knutsen LJS (1996) Unexpected neuroprotection observed with the adenosine A2A receptor agonist CGS21680. Drug Dev Res 39:108–114
Shen H, Zhang L, Yuen D, Logan R, Jung BP, Zhang G, Eubanks JH (2002) Expression and function of A1 adenosine receptors in the rat hippocampus following transient forebrain ischemia. Neuroscience 114:547–556
Simpson RE, O’Regan MH, Perkins LM, Phillis JW (1992) Excitatory transmitter amino acid release from the ischaemic rat cerebral cortex: effects of adenosine recptor agonists and antagonists. J Neurochem 58:1683–1690
Sperk G (1994) Kainic acid seizures in the rat. Prog Neurobiol 42:1–32
Spignoli G, Pedata F, Pepeu G (1984) A1 and A2 adenosine receptors modulate acetylcholine release from brain slices. Eur J Pharmacol 97:341–342
Stone TW (1993) The neuropharmacology of quinolinic acid and kynurenic acid. Pharmacol Rev 45:309–379
Stone TW (2001) Kynurenines in the CNS: from endogenous obscurity to clinical relevance. Prog Neurobiol 64:185–218
Stone TW, Darlington LG (2002) Endogenous kynurenines as targets for drug discovery and development. Nat Rev Drug Discov 1:609–620
Stone TW, Perkins MN (1981) Quinolinic acid: a potent endogenous excitant at amino acid receptors in the CNS. Eur J Pharmacol 72:411–412
Sutherland GR, Peeling J, Lesiuk HJ, Brownstone RM, Rydzy M, Saunders JK, Geiger JD (1991) The effects of caffeine on ischemic neuronal injury as determined by magnetic-resonance-imaging and histopathology. Neuroscience 42:171–182
Suzuki F, Shimada J, Shiozaki S, Ichikawa S, Ishii A, Nakamura J, Nonaka H, Kobayashi H, Fuse E (1993) Adenosine A1 antagonists. 3. Structure–activity relationships on amelioration against scopolamine- or N 6-((R)-phenylisopropyl)adenosine-induced cognitive disturbance. J Med Chem 36:2508–2518
Svenningsson P, Hall H, Sedvall G, Fredholm BB (1997) Distribution of adenosine receptors in the postmortem human brain: an extended autoradiographic study. Synapse 27:322–335
Szot P, Sanders RC, Murray TF (1987) Theophylline-induced up-regulation of adenosine-A1-receptors associated with reduced sensitivity to convulsants. Neuropharmacology 26: 1173–1180
Tarditi A, Camurri A, Varani K, Borea PA, Woodman B, Bates G, Cattaneo E, Abbracchio MP (2006) Early and transient alteration of adenosine A(2A) receptor signaling in a mouse model of Huntington disease. Neurobiol Dis 23:44–53
Tebano MT, Pintor A, Frank C, Domenici MR, Martire A, Pepponi R, Potenza RL, Grieco R, Popoli P (2004) Adenosine A(2A) receptor blockade differentially influences excitotoxic mechanisms at pre- and postsynaptic sites in the rat striatum. J Neurosci Res 77:100–107
Torres RJ, DeAntonio I, Prior C, Puig JG (2004) Effects of hypoxanthine on adenosine transport in human lymphocytes. Implications in the pathogenesis of Lesch–Nyhan syndrome. Nucleosides Nucleotides Nucl Acids 23:1177–1179
Traversa U, Rosati AM, Chiara F, Rodolfo V (1994) Effects of chronic administration of adenosine antagonists on adenosine A1 and A2A receptors in mouse brain. In vivo 8:1073–1078
Trincavelli ML, Marroni M, Tuscano D, Ceruti S, Mazzola A, Mitro N, Abbracchio MP, Martini C (2004) Regulation of A2B adenosine receptor functioning by tumour necrosis factor a in human astroglial cells. J Neurochem 91:1180–1190
Trincavelli ML, Tonazzini I, Montali M, Abbracchio MP, Martini C (2008) Short-term TNF-α treatment induced A2B adenosine receptor desensitization in human astroglial cells. J Cell Biochem 104:150–161
Trussel LD, Jackson MB (1985) Adenosine activated potassium conductance in cultured striatal neurones. Proc Nat Acad Sci USA 82:4857–4861
Tsutsui S, Schnermann J, Noorbakhsh F, Henry S, Yong VW, Winston BW, Warren K, Power C (2004) A1 adenosine receptor upregulation and activation attenuates neuroinflammation and demyelination in a model of multiple sclerosis. J Neurosci 24:1521–1529
Ulas J, Brunner LC, Nguyen L, Cotman CW (1993) Reduced density of adenosine-A1-receptors and preserved coupling of adenosine-A1-receptors to G-proteins in Alzheimer hippocampus: a quantitative autoradiographic study. Neuroscience 52:843–854
Van der Schyf CJ, Gal S, Geldenhuys WJ, Youdim MBH (2006) Multifunctional neuroprotective drugs targeting monoamine oxidase inhibition, iron chelation, adenosine receptors, and cholinergic and glutamatergic action for neurodegenerative diseases. Expert Opin Invest Drugs 15:873- 886
van Gelder BM, Buijsse B, Tijhuis M, Kalmijn S, Giampaoli S, Nissinen A, Kromhout D (2007) Coffee consumption is inversely associated with cognitive decline in elderly European men: the FINE Study. Eur J Clin Nutr 61:226–232
Varani K, Gessi S, Dionisotti S, Ongini E, Borea PA (1998) [3H]-SCH58261 labelling of functional A2A receptors in human neutrophil membranes. Br J Pharmacol 123:1723–1731
Varani K, Rigamonti D, Sipione S, Camurri A, Borea PA, Cattabeni F, Abbracchio MP, Cattaneo E (2001) Aberrant amplification of A(2A) receptor signaling in striatal cells expressing mutant huntingtin. FASEB J 15:1245–1247
Varani K, Abbracchio MP, Cannella M, Cislaghi G, Giallonardo P, Mariotti C, Cattabriga E, Cattabeni F, Borea PA, Squitieri F, Cattaneo E (2003) Aberrant A2A receptor function in peripheral blood cells in Huntington’s disease. FASEB J 17:2148–2150
Varani K, Bachoud-Lévi AC, Mariotti C, Tarditi A, Abbracchio MP, Gasperi V, Borea PA, Dolbeau G, Gellera C, Solari A, Rosser A, Naji J, Handley O, Maccarrone M, Peschanski M, DiDonato S, Cattaneo E (2007) Biological abnormalities of peripheral A(2A) receptors in a large representation of polyglutamine disorders and Huntington’s disease stages. Neurobiol Dis 27:36–43
Virgili M, Poli A, Contestabile A, Migani P, Barnabei O (1986) Synaptosomal release of newly synthesised or recently accumulated amino acids; differential effects of kainic acid on naturally occurring excitatory amino acids and on [D-3H]-aspartate. Neurochem Int 9:29–33
Virus RM, Baglajewski T, Radulovacki M (1984) [3H]N 6-(L-Phenylisopropyl) adenosine binding in brains from young and old rats. Neurobiol Aging 5:61–62
von Lubitz DKEJ, Dambrosia JM, Redmond DJ (1989) Protective effect of cyclohexyladenosine in treatment of cerebral ischaemia in gerbils. Neuroscience 30:451–462
von Lubitz DK, Paul IA, Bartus RT, Jacobson KA (1993) Effects of chronic administration of adenosine A1 receptor agonist and antagonist on spatial learning and memory. Eur J Pharmacol 249:271–280
von Lubitz DK, Lin RC, Melman N, Ji XD, Carter MF, Jacobson KA (1994a) Chronic administration of selective adenosine A1 receptor agonist or antagonist in cerebral ischaemia. Eur J Pharmacol 256:161–167
von Lubitz DK, Lin RC, Popik P, Carter MF, Jacobson KA (1994b) Adenosine A3 receptor stimulation and cerebral ischaemia. Eur J Pharmacol 263:59–67
von Lubitz DKJE, Kim J, Beenhakker M, Carter MF, Lin RCS, Meshulam Y, Daly JW, Shi D, Zhou LM, Jacobson KA (1995a) Chronic NMDA receptor stimulation: therapeutic implications of its effect on adenosine A(1) receptors. Eur J Pharmacol 283:185–192
von Lubitz DKJE, Lin RC-S, Jacobson KA (1995b) Cerebral ischaemia in gerbils: effects of acute and chronic treatment with adenosine A2A receptor agonist and antagonist. Eur J Pharmacol 287:295–302
von Lubitz DK, Lin RC, Paul IA, Beenhakker M, Boyd M, Bischofberger N, Jacobson KA (1996) Postischemic administration of adenosine amine congener (ADAC): analysis of recovery in gerbils. Eur J Pharmacol 316:171–179
von Lubitz DJKE, Lin RCS, Bischofberger N, Beenhakker M, Boyd M, Lipartowska R, Jacobson KA (1999a) Protection against ischemic damage by adenosine amine congener, a potent and selective adenosine A1 receptor agonist. Eur J Pharmacol 369:313–317
von Lubitz DKJE, Lin RC-S, Boyd M, Bischofberger N, Jacobson KA (1999b) Chronic administration of adenosine A3 receptor agonist and cerebral ischaemia: neuronal and glial effects. Eur J Pharmacol 367:157–163
Von Lubitz DKJE, Simpson KL, Lin RCS (2001) Right thing at a wrong time? Adenosine A(3) receptors and cerebroprotection in stroke. Neuroprotective Agents 939:85–96
Wagner DR, Combes A, McTiernan C, Sanders VJ, Lemster B, Feldman AM (1998a) Adenosine inhibits lipopolysaccharide-induced cardiac expression of TNFα. Circ Res 82:47–56
Wagner DR, McTiernan C, Sanders VJ, Feldman AM (1998b) Adenosine inhibits lipopolysaccharide-induced secretion of TNF-α in the failing human heart. Circulation 97:521–524
Warskulat U, Heller-Stilb B, Oermann E, Zilles K, Haas H, Lang F, Häussinger D (2007) Phenotype of the taurine transporter knockout mouse. Methods Enzymol 428:439–58
Wirkner K, Assmann H, Koles l, Gerevich Z, Franke H, Norenberg, Boehm R, Illes P (2000) Inhibition by adenosine A(2A) receptors of NMDA but not AMPA currents in rat neostriatal neurons. Br J Pharmacol 130:259–269
Wirkner K, Gerevich Z, Krause T, Gunther A, Koles L, Schneider D, Norenberg W, Illes P (2004) Adenosine A(2A) receptor-induced inhibition of NMDA and GABA(A) receptor-mediated synaptic currents in a subpopulation of rat striatal neurons. Neuropharmacology 46:994–1007
Wu LG, Saggau P (1997) Presynaptic inhibition of elicited neurotransmitter release. Trends Neurosci 20:204–212
Yamada K, Tanaka T, Senzaki K, Kameyama T, Nabeshima T (1998) Propentofylline improves learning and memory deficits in rats induced by β-amyloid protein-(1–40). Eur J Pharmacol 349:15–22
Yao ZH, Gross GJ (1994) The ATP-dependent potassium channel - an endogenous cardioprotective mechanism. J Cardiovasc Pharmacol 24: S28–S34
Yao Y, Sei Y, Abbracchio MP, Jiang JL, Kim YC, Jacobson KA (1997) Adenosine A3 receptor agonists protect HL-60 and U-937 cells from apoptosis induced by A3 antagonists. Biochem Biophys Res Commun 232:317–22
Yoshida M, Nakakimura K, Cui YJ, Matsumoto M, Sakabe T (2004) Adenosine a, receptor antagonist and mitochondrial ATP-sensitive potassium channel blocker attenuate the tolerance to focal cerebral ischemia in rats. J Cereb Blood Flow Metab 24:771–779
Yoshioka K, Saitoh O, and Nakata H (2001) Heteromeric association creates a P2Y-like adenosine receptor. Proc Natl Acad Sci USA 98:7617–7622
Yoshioka K, Hosada R, Kuroda Y, Nakata H (2002a) Hetero-oligomerization of adenosine A1 receptors with P2Y1 receptors in rat brains. FEBS Lett 531:299–303
Yoshioka K, Saitoh O, Nakata H (2002b) Agonist-promoted heteromeric oligomerization between adenosine A1 and P2Y1 receptors in living cells. FEBS Lett 523:147–151
Yu LQ, Huang Z, Mariani J, Wang Y, Moskowitz M, Chen JF (2004) Selective inactivation or reconstitution of adenosine A2A receptors in bone marrow cells reveals their significant contribution to the development of ischemic brain injury. Nat Med 10:1081–1087
Zarrindast MR, Shafaghi B (1994) Effects of adenosine receptor agonists and antagonists on acquisition of passive avoidance learning. Eur J Pharmacol 256:233–239
Zhao ZQ, McGee S, Nakanishi K, Toombs CF, Johnston WE, Ashar MS, Vinten-Johansen J (1993) Receptor-mediated cardioprotective effects of endogenous adenosine are exerted primarily during reperfusion after coronary occlusion in the rabbit. Circulation 88:709–719
Zhou AM, Li WB, Li QJ, Liu HQ, Feng RF, Zhao HG (2004) A short cerebral ischemic preconditioning up-regulates adenosine receptors in the hippocampal CA1 region of rats. Neurosci Res 48:397–404
Zhou QY, Li C, Olah ME, Johnson RA, Stiles GL, Civelli O (1992) Molecular cloning and characterisation of an adenosine receptor: the A3 receptor. Proc Nat Acad Sci USA 89:7432–7436
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2009 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Stone, T.W., Ceruti, S., Abbracchio, M.P. (2009). Adenosine Receptors and Neurological Disease: Neuroprotection and Neurodegeneration. In: Wilson, C., Mustafa, S. (eds) Adenosine Receptors in Health and Disease. Handbook of Experimental Pharmacology, vol 193. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-540-89615-9_17
Download citation
DOI: https://doi.org/10.1007/978-3-540-89615-9_17
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-540-89614-2
Online ISBN: 978-3-540-89615-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)