
Abstract
Alzheimer’s disease (AD) is the most common dementia in the elderly and its increasing prevalence presents treatment challenges. Despite a better understanding of the disease, the current mainstay of treatment cannot modify pathogenesis or effectively address the associated cognitive and memory deficits. Emerging evidence suggests adenosine G protein-coupled receptors (GPCRs) are promising therapeutic targets for Alzheimer’s disease. The adenosine A1 and A2A receptors are expressed in the human brain and have a proposed involvement in the pathogenesis of dementia. Targeting these receptors preclinically can mitigate pathogenic β-amyloid and tau neurotoxicity whilst improving cognition and memory. In this review, we provide an accessible summary of the literature on Alzheimer’s disease and the therapeutic potential of A1 and A2A receptors. Although there are no available medicines targeting these receptors approved for treating dementia, we provide insights into some novel strategies, including allosterism and the targeting of oligomers, which may increase drug discovery success and enhance the therapeutic response.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Dementias are the most prevalent neurological disorder in the aged population, affecting more than 50 million people worldwide in 2020 [1]. In the absence of effective therapies, the number of people with dementia is projected to double every 20 years, reaching 152 million in 2050 [1]. While there are hundreds of described neurodegenerative dementias, the most common is AD [2]. Sporadic AD, the most common form, seems to be multifactorial, with age, genetic, and environmental factors contributing to disease risk, manifestation, and progression [3, 4]. AD is classically defined as a dual clinicopathological entity, meaning that to fully diagnose AD requires (i) observations of a specified clinical presentation over time, related to episodic memory impairment and other cognitive, behavioural, and neuropsychiatric abnormalities and (ii) observations of specific neurological changes (e.g. neurofibrillary tangles, amyloid plaques, synaptic loss). Recent advances in amyloid positron emission tomography (PET) and structural magnetic resonance imaging (MRI) have enabled the detection of in vivo biological evidence of AD pathologies to assist the classification of presymptomatic AD stages [5,6,7].
Neuropathological alterations in Alzheimer’s disease
Compared to age-matched non-AD individuals, AD patients experience an accelerated rate of brain mass shrinkage due to neuronal death and grey matter loss; the amount of neuronal atrophy correlates with AD severity [8,9,10,11]. AD neuropathology is characterised by significant gyral shrinkage, widened sulci, and enlarged ventricles present in multiple areas within the brain, along with a significant reduction in the volume and/or cortical thickness of regions such as hippocampus and entorhinal, temporal, parietal, and frontal cortex [12,13,14,15]. There are currently three main theories regarding the mechanisms underlying neurodegeneration in AD. Although some features are shared by all three theories, each postulates a different causal event or sequence of events leading to neurodegeneration.
The amyloid cascade hypothesis posits the accumulation of toxic amyloid β (Aβ) peptides as the main cause of neurodegeneration [16]. The formation of amyloid plaques is driven by aberrant processing of amyloid precursor protein (APP) into Aβ40 and Aβ42 peptides, which normally play important physiological roles, including synaptic function, neuronal development and plasticity, and lipid homeostasis [17,18,19]. Although Aβ40 is more abundant than Aβ42 in normal conditions, in AD the ratio shifts in favour of Aβ42 generation. The formation of Aβ deposits could be due to altered Aβ42/Aβ40 ratios or the failure of Aβ clearance processes [20, 21]. Neuritic dense core Aβ plaques are deposited in the hippocampus, amygdala, and cortex, resulting in a cascade of events that include sustained inflammatory responses, imbalanced neuronal ionic homeostasis, altered kinase and phosphatase activity, tau phosphorylation, neurofibrillary tangle (NFT) formation, and neuronal and synapse loss [22,23,24]. Aβ pathology is widespread in early diseases, whereas tau pathology develops much later, suggesting changes in Aβ are the initial insult driving tau pathology. As such, the amyloid theory has become the dominant model of AD pathogenesis; guiding the development of potential AD disease-modifying treatments.
An opposing view suggests tau pathology is responsible for the initiation of AD neurodegeneration. Under normal conditions, soluble tau protein acts as a microtubule stabiliser, maintaining neuronal integrity, neurite outgrowth, and axonal transport [25,26,27]. The degree of tau phosphorylation is critical in regulating microtubule assembly and, in AD, tau protein becomes hyperphosphorylated through the actions of multiple kinases [28,29,30,31,32,33,34]. Hyperphosphorylated tau detaches from microtubules and self-aggregates to form NFTs, which disrupt axonal transport and subsequently lead to neuronal loss [35, 36] (Fig. 1). Furthermore, NFTs are often associated with Aβ plaques and chronic inflammation, as evidenced by the accumulation of activated astrocytes and microglia [37,38,39,40,41]. The tau theory is supported by the correlation between tau pathology and the degree of AD dementia, with the distribution and amount of NFTs in AD brains related to the severity and time course of the disease [42, 43]. In symptomatic subjects, NFTs are widespread and correlate well with the AD-affected functional brain circuits [44,45,46,47]. Importantly, Aβ and tau pathologies do not develop in the same brain region; thus, it is debatable whether the Aβ pathology drives tau pathology (Fig. 2). Targeting tau pathology with drugs or vaccines to inhibit tau aggregation or promote tau degradation is being investigated for both symptomatic and preventative treatment [48, 49].

Schematic diagram of brain atrophy and neuropathological alterations between normal, healthy brain versus AD brain. On the left is a healthy aged brain, while on the right, an aged Alzheimer’s brain (shaded in yellow) has marked atrophy, including widened sulci, enlarged ventricles, and gyral shrinkage. At the cellular level, dying neurons are surrounded by reactive astrocytes, activated microglia, extracellular Aβ plaques, and intracellular neurofibrillary tangles in aged AD brain. Adapted from “Pathology of Alzheimer’s Disease 2” template by BioRender.com (2022). Retrieved from https://app.biorender.com/biorender-templates

Simplified overview showing the spatiotemporal patterns of Aβ and tau pathologies in conjunction with proposed model of early and late inflammation occurrence during AD progression. Yellow and green shading in the brain indicates areas affected by tau and Aβ pathology, respectively. Extensive Aβ pathology can be detected in individuals with preclinical AD, and the extent of Aβ pathology alters minimally in symptomatic stages, whereas tau pathology develops considerably later. Further, early inflammation is likely to begin before the presence of Aβ plaques, while late inflammation should commence when the first Aβ plaques are established. The early and late inflammation overlaps at the later stage of preclinical AD. At the clinical stage, the late inflammation becomes predominant. The scheme of the evolution of the AD pathology is adapted from [304]. Created with BioRender.com
Another pathological feature shared by all AD pathogenesis theories is chronic inflammation. The inflammation theory of AD pathogenesis postulates that activated microglia and reactive astrocytes trigger AD pathogenesis and this event precedes the presence of Aβ plaques and NFTs [50]. This theory is supported by the presence of inflammatory changes very early in AD neuropathology [51, 52] (Fig. 2). Given activated microglia and astrocytes are commonly found in close proximity to Aβ and tau deposits, dysfunction of astrocytes or microglia may lead to the formation of plaques and NFTs [53,54,55,56,57]. While the precise mechanisms are yet to be defined, it is suggested intracellular accumulation of Aβ oligomers leads to preclinical AD inflammation before clinical hallmarks of AD are present [58, 59]. The imbalance between pro-inflammatory and anti-inflammatory mechanisms and a shift from neuroprotective to neurotoxic glial phenotypes further exacerbates disease pathology, leading to late clinical AD inflammation [60,61,62] (Fig. 2). While the concept of chronic inflammation has been recognised as an important AD feature, most drug discovery efforts are directed towards agents targeting Aβ peptides and tau protein. This is likely due to an incomplete understanding of mechanisms underlying microglial and astrocytic activation and the mechanistic link between amyloid, tau, and inflammatory pathologies. Despite these unresolved problems, reducing neuroinflammation has recently attracted more interest and is still under investigation [63, 64].
Glial cells in Alzheimer’s disease-associated inflammation
Microglia and astrocytes are two of the most common glial cells present in the CNS. Microglia are actively involved in regulating various aspects of neuroplasticity and mediating neuroprotection, while the principal role of astrocytes is to maintain overall brain homeostasis via the uptake and release of ions and neurotransmitters from the extracellular fluid surrounding neurons [65,66,67,68,69]. When the brain is injured, microglia and astrocytes undergo rapid activation, resulting in phagocytosis of cellular debris and production of neurotoxic and inflammatory compounds (e.g. reactive oxygen species, growth factors and cytokines) [66, 70]. In AD, chronic activation of glial cells results in the release of a variety of pro-inflammatory cytokines, chemokines, reactive oxygen species, and N-terminally truncated Aβs that impair neuronal activity and survival [71,72,73,74,75]. Analysis of neuroinflammatory gene expression in the frontal cortex of early-, mid-, and late-stage AD patients revealed activated microglia are present throughout the disease course. Additionally, there are signs of reactive astrogliosis in plaque-containing areas, with glial Aβ clearance mechanisms becoming impaired [76,77,78]. A buildup of Aβ oligomers and fibrils can activate microglial cell-surface receptors, leading to chronic activation of store-operated calcium (Ca2+) entry and upregulation of pro-inflammatory mediators [79,80,81,82,83,84]. Activated microglia and astrocytes intensify and sustain activation of each other via secretion of pro-inflammatory molecules, exacerbating chronic neuroinflammation and impairing their ability to promote neuronal survival, growth, synaptogenesis, and phagocytosis [85,86,87,88]. The best approach for anti-inflammatory use in AD is to target the activated microglia and/or reactive astrocytes to disrupt inflammation in the initial stages of AD. Indeed, there is increased interest in the expression and distribution of various GPCRs connected to microglial and astroglial activation in the AD brain and the ability to attenuate AD inflammation [89, 90].
Current status of drug development
Pharmacotherapeutic options for patients diagnosed with AD are extremely limited, most only treat symptoms rather than disease progression. As such, it is imperative to seek new AD treatments to prevent, delay, and treat AD clinical symptoms. To date, only five agents have been approved based on modest symptomatic clinical effects; the cholinesterase inhibitors tacrine, donepezil, rivastigmine, and galantamine for mild-to-moderate AD and the glutamate antagonist memantine for moderate-to-severe AD [4]. Around 20% of ongoing clinical trials are focused on new symptomatic agents aimed at enhancing cognitive function through modulation of neurotransmitter synthesis, receptor activation and reuptake [91,92,93]. For disease-modifying treatments, attention for both Aβ-targeted and tau-related therapies has increased; as of March 2022, around thirty anti-Aβ or anti-tau agents are in phase 2 or 3 trials, with many more in the preclinical stages. However, there is an increasing diversity of targets for disease-modifying therapies, including vasculature, inflammation, and metabolism, reflecting the constantly changing understanding of AD disease biology [92, 93].
Only one disease-modifying agent has been approved for AD. In 2021, the FDA controversially approved the use of the anti-Aβ monoclonal antibody aducanumab in mild cognitive impairment and early-stage Alzheimer’s disease [94,95,96,97]. While both phase III clinical trials of aducanumab were terminated early due to lack of clinical benefit, post hoc analyses revealed one trial met its outcomes of reducing cognitive and functional decline at high aducanumab doses [94,95,96,97]. Additionally, both trials revealed a marked decrease in amyloid plaques with high dose aducanumab. Newer monoclonal anti-Aβ antibodies have also shown efficacy in reducing the levels of AD biomarkers in phase II trials, but results are again mixed when considering cognitive and functional benefits [98,99,100]. Trials of other anti-Aβ or tau therapies have also had mixed results. The γ-secretase inhibitor semagacestat, anti-Aβ monoclonal antibodies bapineuzumab and solanezumab, anti-aggregation agent scyllo-inositol (ELND005), RAGE receptor inhibitor (PF-04494700), and tau aggregation inhibitor TRx0237 have displayed no clinical efficacy in phase III trials of mild-to-moderate AD patients [101,102,103,104,105,106]. Aβ peptides and tau protein remain strong candidates as therapeutic targets; however, the failure of multiple therapeutic trials highlights the need to consider other targets.
Of note, few anti-inflammatory agents have reached phase III clinical trials to date, although inflammation reflects the second most popular target for current preclinical and clinical AD drug development [92, 93]. The main reason for the delayed development of anti-inflammatory agents is due to conflicting results of epidemiological studies and clinical trial results for nonsteroidal anti-inflammatory drugs (NSAIDs). Multiple epidemiological studies indicate long-term NSAIDs usage reduces AD risk by about 50% in individuals bearing one or more ε4 alleles of apolipoprotein E (apoE), which is strongly associated with increased risk of both familial and sporadic AD [107,108,109,110,111,112]. Conversely, prospective clinical observations reported traditional NSAIDs or selective cyclooxygenase 2 (COX2) inhibitors did not slow down the cognitive decline associated with mild-to-moderate AD [113,114,115]. Interestingly, in one large prevention trial, asymptomatic participants showed reduced AD incidence 18–24 months post-NSAID use; however, NSAIDs had adverse effects in patients with cognitive impairment and/or were at a later stage of presymptomatic AD [116]. Taken together, anti-inflammatory agents seem to elicit different effects at various AD stages; anti-inflammatory therapy is beneficial in preventing AD onset but becomes completely non-beneficial in symptomatic AD patients. Counteracting inflammation at later stages by preventing the prolonged microglial and/or astroglial activation during the presymptomatic phase or earlier may be a promising protective strategy.
G protein-coupled receptors implicated in Alzheimer’s disease
Numerous studies have presented compelling results relating GPCRs to AD pathogenesis [117,118,119]. GPCRs are membrane-bound receptors that transduce external stimuli into signalling cascades within the cell and are important for numerous physiological and pathophysiological processes [120]. Gene expression profiles from AD patients’ postmortem brains via cDNA microarray analysis demonstrate transcript levels of a number of GPCRs changed dramatically, among which were inflammation-associated GPCRs, hormone receptors, and neurotransmitter receptors [121]. Given altered GPCR expression levels would influence the related biological processes, mounting evidence implicates several GPCRs in AD pathogenesis. Several GPCR families have been targeted by putative AD therapies; however, many of these discovery programs have been discontinued (Fig. 3). Of note, there are currently 35 agents targeting GPCRs in the discovery, preclinical, and clinical stages of drug discovery pipelines [122]. Among these are ligands for a range of different GPCR families including serotonergic, cannabinoid, muscarinic, opioid, glutamatergic, and purinergic receptors [122]. Drugs targeting adenosine A1 and A2A receptors are of particular interest, owing to the potential role of these receptors in inflammatory processes, as well as both tauopathy and Aβ pathologies [123, 124].

Current status of GPCR-targeted AD therapy development. Of 80 GPCR-targeting agents that have been investigated, 45 have subsequently been discontinued or development halted. Thirty-five are currently in various phases of preclinical and clinical development for AD as both disease-modifying and symptomatic agents, targeting a wide range of GPCR families. “Other” receptors include purinergic (including adenosine), adrenergic, histamine, sphingosine, calcium sensing receptor, gastric inhibitory peptide receptor, glucagon-like peptide 1 receptor, G-protein coupled bile acid receptor 1, and vasoactive intestinal polypeptide receptor. GABA, γ-amino butyric acid; mGlu, metabotropic glutamate. Data compiled using Cortellis Competitive Intelligence software from Clarivate Analytics and is current as of March 2022
Adenosine receptors
The adenosine receptors are family A GPCRs, with four structurally similar members; the adenosine A1 (A1R), the adenosine A2A (A2AR), the adenosine A2B (A2BR), and the adenosine A3 receptor (A3R) [125]. Through these receptors, adenosine exerts neuromodulatory effects to regulate essential processes (e.g. neuronal signalling, astrocytic function, learning and memory, motor function, control of sleep and arousal, and normal ageing processes) [126]. Of the four adenosine receptors, A1R and A2AR show the greatest expression in the brain and have relevance to AD [127, 128], whereas A2BR and A3R show relatively lower levels of expression [129]. To date, little is known about the role of A2BR and A3R in AD pathologies.
Adenosine A1 receptor
Mapping A1R expression in both the rat and human brain has demonstrated widespread A1R distribution, with greater abundance in the hippocampus, cerebral cortex, cerebellum, thalamus, and basal ganglia [130, 131]. In the human cerebellum, A1R density is low with strong A1R immunoreactivity observed in Purkinje cells. In contrast, in the rat cerebellum, moderate A1R expression is detected with weak labelling of Purkinje cells, suggesting these discrepancies in A1R expression may reflect species differences. In the rat brain, the highest A1R immunoreactivity was found in the large pyramidal neurons of layer 5 of the cerebral cortex and the pyramidal cells in the fields CA2-CA3 of the hippocampus [132]. Furthermore, A1Rs are most abundant in synapses, particularly in the presynaptic active zone and postsynaptic density [133, 134]. A1Rs are also present in astrocytes, microglia, and oligodendrocytes at a much lower level [135,136,137].
Adenosine A2A receptor
Like A1R, A2AR is also expressed throughout the brain. Highly enriched in the striatum, olfactory tubercle, and nucleus accumbens, A2AR is ubiquitously expressed in other brain regions (rat and human) at lower densities [138, 139]. In the rat basal ganglia, A2ARs are predominantly located in dendritic spines and postsynaptic densities, where A2ARs control the integration of signal responses from corticothalamic glutamatergic neurons and medium spiny GABAergic neurons [140, 141]. However, in rat cortical regions, A2ARs are predominantly located in synapses, particularly in the presynaptic active zone [142]. In contrast to A1Rs, A2ARs have a broader localisation in different types of nerve terminals. In addition to being expressed in neurons, A2ARs are also located in astrocytes [143, 144] and microglia [145].
Adenosine A1 and A2A receptor signalling
General signalling
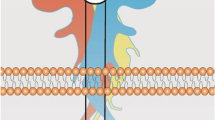
Signal transduction mediated by A1R and A2AR is largely driven by coupling to heterotrimeric G protein complexes, which are composed of a Gα subunit and Gβγ heterodimeric complex (Fig. 4) [146]. Pertussis toxin-sensitive Gi/o proteins are preferentially activated by A1R, whereas A2AR shows a preference for Gs and Golf proteins, with the latter being primarily restricted to striatal brain regions [147,148,149]. These G proteins affect the activity of adenylyl cyclase (AC), with Gαs proteins activating AC and increasing the production of the second messenger cyclic adenosine monophosphate (cAMP) from ATP [150]. Conversely, stimulation of Gαi/o proteins results in AC inhibition and thereby reduces cAMP production. Downstream targets of cAMP include cyclic nucleotide-gated ion channels (HCN), the small G protein guanine nucleotide exchange factor (EPAC) and protein kinase A (PKA), all of which play important roles in neural physiology [151, 152].

Canonical Gs or Gi/o signalling pathways upon adenosine A2A and A1 receptor activation. Activation of Gs stimulates AC, thereby leading to the production of cAMP and PKA stimulation. Gαs elicited the B-Raf-mediated activation of ERKs via Rap-1 or Ras or inhibits C-Raf-mediated activations of ERKs by phosphorylating C-Raf through PKA. Gαs can also stimulate p38 MAPK and inhibits ERK5 via PKA-dependent mechanisms. The dashed lines indicate the pathways remain unclear. In contrast, Gi/o inhibits cAMP-dependent signalling and stimulates the activities of ERK, JNK, and PI3K via βγ-subunits-dependent mechanisms. Abbreviations: AC, adenylyl cyclase; AKT, protein kinase B/Akt; ATP, adenosine triphosphate; CaMK, Ca2+/calmodulin-dependent protein kinase; cAMP, cyclic adenosine monophosphate; CREB, cAMP response element-binding protein; EPAC, exchange protein directly activated by cAMP; ERK1/2, extracellular signal-regulated kinases 1 and 2; GSK3β, glycogen synthase kinase 3β; JNK, c-Jun N-terminal kinase; MAP3K, mitogen-activated protein kinase kinase kinases; MEK1/2, mitogen-activated protein kinase kinases 1 and 2; mTOR, mammalian target of rapamycin; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; PDK1, 3-phosphoinositide dependent protein kinase-1; PI3K, phosphoinositide 3 kinase; PKA, protein kinase A; PLC, phospholipase C; Src, proto-oncogene tyrosine-protein kinase. Created with BioRender.com
In addition to cAMP signalling, both A1R and A2AR also signal via additional effectors, including mitogen-activated protein kinases (MAPK) and Akt/protein kinase B, which play a role in cell growth, survival, differentiation, and protein transcription and translation [153, 154]. A1R stimulates the extracellular signal-regulated kinases 1 and 2 (ERK1/2), c-Jun N-terminal kinases (JNK), and the p38 MAPK, and additionally phosphorylates Akt via phosphoinositide 3-kinase (PI3K) activation [155,156,157,158,159,160,161,162]. This signalling is largely driven via interactions of βγ subunits with effectors upon dissociation from Gαi/o proteins. Additionally, A1R mobilises intracellular calcium stores and activates multiple isoforms of protein kinase C, via direct βγ interactions with phospholipase C [162,163,164,165]. Similarly, A2AR also signals via ERK1/2, JNK, p38, and Akt, with these effects thought to be downstream of α subunit activity of Gs [156, 166,167,168,169,170,171,172,173]. Interestingly, some studies support a stimulatory or inhibitory effect of these second messengers by A2AR, suggesting this signalling can be conditional, depending on cell background and disease context [174, 175].
Neuronal signalling
In addition to the above-generalised signalling pathways, A1R and A2AR both stimulate additional pathways and effectors that are either brain-specific or present in only niche cell types. A2AR has been established as a modulator of neurotrophins, including brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF), which play important roles in neuronal differentiation and survival, in addition to regulating synaptic transmission and plasticity [176]. In microglia and hippocampal slices, A2AR can increase the release of both BDNF and NGF, which may be mediated by the transactivation of neurotrophic receptors [177,178,179,180,181]. A1R has also been suggested to play a role in neurotrophin signalling, although its effects are less well defined [182].
Modulation of ion channel activity represents another vital signalling mechanism for both A1R and A2AR. A1R couples to a number of ion channels mediating an overall net hyperpolarisation of neurons [183]. It is well established that A1R couples to G protein-coupled inwardly-rectifying potassium (GIRK) channels, which are a class of transmembrane proteins that facilitate potassium flux into the cytosol [184,185,186]. A1R coupling to GIRK channels is through the direct binding of βγ subunits. Additionally, A1R reduces the current of voltage-dependent calcium channels, with a proposed preference for N-type channels [187,188,189,190]. Alongside A1R, A2AR also couples to voltage-dependent calcium channels; however, A2AR has a stimulatory effect on the current [187, 191]. Both receptors can also activate ATP-sensitive potassium channels (KATP), which are present in plasma, mitochondrial, and nuclear membranes, and play a role in neuronal excitability and survival [192, 193].
Collectively, these various signalling streams result in an overall inhibitory effect of A1Rs and excitatory effects of A2ARs, allowing fine-tuning of neuronal circuitry. Activation of presynaptic A1Rs on excitatory neurons reduces neurotransmitter release and induces synaptic depression, whereas presynaptic A2ARs are involved in increasing neurotransmitter release [133, 194]. Similarly, postsynaptic A1R activation causes membrane hyperpolarisation with subsequent inhibition of neuronal firing. In contrast, activated postsynaptic A2ARs increase cellular excitability [195]. Importantly, the effects of both A1R and A2AR are not limited to the modulation of neuronal activity, these receptors also coordinate the function of additional cells, including astrocytes and microglia (see [196] for review). These effects impart additional fine-tuning of neural homeostasis and inflammatory balance.
The role of A1R and A2AR in dementia
Animal models of AD and old age, alongside human postmortem analyses, have revealed evidence of a disruption to the neural adenosine network [197, 198]. Levels of adenosine and its related metabolites are altered in the brains of patients with AD [199, 200]. Similarly, adenosine receptors (AR) change their pattern of localisation and density in affected brain regions of AD [201,202,203]. Postmortem analysis of AD patients’ brains showed reduced A1R expression at the dentate gyrus and hippocampal CA3 regions [204], which are focal points for the spread of NFTs and subsequent neuronal loss [44]. PET studies with a radiolabelled A1R antagonist have also demonstrated reduced A1R levels in the temporal cortex in patients with AD compared to elderly subjects [205]. Moreover, postmortem AD frontal cortex samples showed increased A1R and A2AR expression, compared to age-matched controls [202]. Analysis of patients with frontotemporal lobar degeneration revealed increased A2AR expression in the temporal cortex and enhanced A2AR immunoreactivity in neurons expressing tau pathology [206]. Moreover, A2AR expression is increased in the aged forebrain when compared to young subjects, with a further significant increase in AD patients [207]. Significantly increased brain A2AR levels in AD patients are coupled with peripheral platelets also reflecting an increase in A2AR [208]. Accordingly, peripheral A2AR expression could act as a biomarker for dementia and inform disease progression.
Animal and cellular models recapitulate these changes to AR expression, with increased A1R immunoreactivity in neurons with NFTs and in amyloid plaques, alongside enhanced glial A2AR expression [201]. In human neuroblastoma cells and primary rat cortical neurons, administration of Aβ25-35 increased A1R and A1R/A2AR expression, respectively [209, 210]. APP/PS1 AD mice show increased A1R and A2AR levels compared to non-transgenic mice, and rats with sporadic dementia also show elevated A2AR levels [123, 211, 212]. The 5XFAD model of AD also demonstrates increased A1R expression, highlighting changes to ARs can be found across different animal models, suggesting a central role in the pathogenesis of dementia [209]. Overall, the precise mechanisms underlying the dysregulation of A1R and A2AR in AD/dementia remain unresolved. However, it has been suggested that early in the disease process, disruption to the homeostatic levels of adenosine may impart dysfunctional regulatory feedback, which modifies the expression and locality of AR receptors [200].
Epidemiological studies have reported an inverse association between caffeine intake and AD/dementia risk [213,214,215,216,217]. Significant caffeine consumption is associated with a lowered rate of Aβ positivity as measured by PET [218]. Indeed, caffeine, acting as a nonselective A1R and A2AR receptor antagonist, reduces Aβ toxicity and enhances cognition in numerous models. The neuroprotective qualities of caffeine have been demonstrated in cultured rat and mouse neurons, where Aβ-induced neurotoxicity and tau phosphorylation was reduced, respectively [123, 219]. In APPsw transgenic mice and a rat model of sporadic dementia, caffeine promoted neuroprotection and mitigated cognitive impairment [123, 212]. Long-term administration of caffeine to AD transgenic mice also improved cognition and reduced Aβo generation and was accompanied by a modest reduction in presenilin-1 and β-secretase expression levels [220]. These neuroprotective actions have largely been ascribed to caffeine’s A2AR antagonism, a notion supported by studies using selective A2AR antagonists (e.g. KW6002, SCH58261, ZM241385, MSX-3). In animal models, including APP/PS1 mice and THY-Tau22 mice, administering selective A2AR antagonists improved memory deficits and mitigated the Aβ toxicity or tau hyperphosphorylation associated with the disease [207, 211, 221]. Moreover, the administration of human Aβ1-42 fragment reduced memory performance in rats, which was reversed by SCH58261 and KW6002 [222]. Interestingly, in this model, A2AR antagonism could not mitigate acute memory deficits induced by the muscarinic receptor antagonist scopolamine or the NMDA antagonist MK801. As such, it has been suggested the beneficial effects of A2AR antagonists on memory may not be a generalised effect but rather specific to the disease processes involved in dementia/AD.
Alongside animal studies, pharmacological A2AR blockage in neurons attenuated Aβ-induced neuronal death and further reduced synaptic loss [223]. Similarly, in cultured rat cerebellar neurons ZM241385 was neuroprotective; however, A1R antagonist CPX was ineffective, implying Aβ-induced neurotoxicity is primarily mediated through A2AR activity [224]. Additional studies have provided diverging roles for A1R. In a model using human neural cells, application of the selective A1R agonist R-PIA led to soluble APP production and increased tau phosphorylation, which was reversed by A1R-selective antagonist DPCPX [201]. In tau transgenic mice, A1R antagonist rolofylline restored memory deficits and reduced synaptic dysfunction in neural cells [225]. Given that A1R redistributes and co-localises with NFTs and Aβ plagues in AD patients, these studies suggest A1R may play a direct role in mediating some of the pathology [201, 202]. Importantly, in addition to pathological signalling, A1R activation is strongly neuroprotective in a number of settings [226] and elicits anti-inflammatory effects in chronic neuroinflammation [124, 227]. Indeed, acute administration of an A1R agonist decreased neurodegeneration in in vitro and in vivo models challenged with noxious stimuli [227]. As such, selective A1R antagonists may protect against β-amyloid and tau neurotoxicity and enhance cognition, whereas A1R agonists may impart neuroprotection.
In addition to pharmacological studies, A2AR genetic deletion or overexpression has also revealed interesting findings. In APP/PS1 mice, A2AR downregulation via shRNA restored long-term potentiation and improved memory deficits [211]. In contrast, A2AR overexpression in cortical and hippocampal neurons of rats resulted in increased glutamate release, which was associated with changes in synaptic plasticity [207]. In THY-Tau22 mice, genetic deletion of A2AR protected against spatial memory deficits, reduced neuroinflammation, and decreased tau hyperphosphorylation [221]. Conversely, selective A2AR overexpression in the forebrain of THY-Tau22 mice resulted in tau hyperphosphorylation and increased memory deficits [206]. Mice also showed increased expression of hippocampal c1q complement protein, a biomarker found in patients with frontotemporal lobar degeneration, suggesting A2AR may contribute to this process. Interestingly, upregulation of genes associated with immune responses was also found in this study, with further cell-specific enrichment analysis revealing these genes were preferentially increased in microglia. As such, microglial A2AR and its associated immune responses may play a role in the pathogenesis of dementia. Indeed, additional studies support the importance of glial cells, with A2AR being upregulated in microglia and astrocytes after treatment with Aβo [228] and conditional genetic ablation of astroglial A2AR enhancing long-term memory in young and in ageing mice [229]. Targeting the NLRP3 (nucleotide-binding oligomerisation domain-, leucine-rich repeat-, and pyrin domain-containing 3) inflammasome to modulate AD pathology has increasing interest as a therapeutic strategy [230], with emerging evidence of A2AR in microglia offers an upstream therapeutic target [231]. A2AR activation stimulates sustained NLRP3 inflammasome activity and the production of proinflammatory cytokines (e.g. IL-1β) in macrophages and primary microglia [232,233,234,235]. Notably, in preclinical models of hypoxic-ischemia and autoimmune encephalomyelitis, caffeine inhibited NLRP3 inflammasome activation and microglial activation to confer neuroprotection and attenuate disease pathology [236, 237]. Therefore, in addition to modulating neuronal signalling, targeting A2AR on microglia may present an opportunity to target neuroinflammation associated with AD.
Therapeutic paradigms for targeting adenosine receptors
A1R and A2AR: antagonism or agonism?
The growing evidence supporting a role for A1R and A2AR in dementia/AD highlights these GPCRs represent promising drug targets. Overall, the studies described predominantly indicate that A1R and A2AR antagonism may be a viable therapeutic approach to mitigate pathology and improve patient symptoms. Indeed, A1R antagonism may afford protection against β-amyloid and tau neurotoxicity and the enhancement of cognition is clearly desirable in the setting of dementia. However, the neuroprotective nature of A1R is also an important consideration. Studies showing reduced brain A1R expression in patients with AD make this particularly pertinent, as reduced A1R expression could facilitate neuronal excitotoxicity [238]. As such, it is interesting to speculate how the administration of an agonist or antagonist would fare clinically, given the divergent preclinical data. Similarly, the protection afforded by A2AR antagonism against β-amyloid neurotoxicity and memory impairment in preclinical settings makes it a desirable drug target. The A2AR antagonist istradefylline enhanced cognition in mice with amyloid pathology and is a clinically used adjunctive therapy in Parkinson’s disease [239, 240]. Istradefylline has not been tested in humans with AD/dementia; however, this may represent a worthwhile exploratory investigation given this compound is clinically available in some countries. Interestingly, A2AR agonism may be useful very early in the disease process, where its anti-inflammatory effects may dampen disease progression [241]. A2AR induction of BDNF signalling could also contribute to greater neuronal survival and maintenance of synaptic plasticity; however, these considerations remain largely unexplored.
Nonetheless, despite the promising preclinical data, the void of clinically approved drugs targeting A1R or A2AR for AD/dementias remains apparent. This may be partly owing to the need for a deeper understanding of the precise roles of A1R and A2AR in AD/dementia. Moreover, the widespread distribution of ARs and their role in fundamental biological processes is an additional hurdle for the drug discovery process. For example, A1R activation is associated with lowered heart rate and altered blood pressure [242], whilst antagonism is associated with increased seizure risk and sleep disturbance [243, 244]. A2AR antagonism, such as that mediated by istradefylline, can cause involuntary movements and hallucinations [245]. Many of these on-target side effects are centrally driven, making it difficult to dissociate the therapeutic and adverse effects. As such, prototypical AR agonists or antagonists may encounter challenges during clinical translation. Alternative pharmacological approaches that circumvent these limitations can minimise the risk of on-target adverse effects.
Allosterism
Allosteric ligands may overcome many of the limitations associated with traditional agonists and antagonists. Allosteric compounds bind to a topographically distinct site acting to modulate the affinity and/or efficacy of the orthosteric endogenous ligand, in this case, adenosine [246]. A larger number of pharmacological parameters drive the activity of allosteric ligands when compared with orthosteric compounds, which affords more nuanced signalling. This is evidenced by the broad categories in which they can be classed, including (i) allosteric agonists, which increase (agonist) or decrease (inverse agonist) signalling by binding to the allosteric site; (ii) positive or negative allosteric modulators (PAM or NAM), which can increase or decrease, respectively, the affinity and/or efficacy of an orthosteric ligand; and (iii) neutral allosteric ligands (NAL), which exhibit neutral cooperativity with the orthosteric ligand and have no intrinsic efficacy in their own right [247]. Moreover, the pharmacological parameters encoded by allosteric ligands can be fine-tuned by medicinal chemistry and structure-based efforts. As such, allosteric modulators belonging to the same class (PAM, NAM, or allosteric agonist) can elicit a broad spectrum of activity and thus can be rationally selected based on the underlying disease.
Allosteric ligands offer numerous advantages. Since allosteric sites typically show greater sequence divergence compared to orthosteric sites, allosteric ligands typically exhibit greater subtype selectivity [246, 248]. As such, allosteric ligands can impart efficacy and/or affinity modulation upon endogenous adenosine in a subtype-selective manner. Additionally, allosteric modulators can signal in a spatiotemporally specific manner, modifying the signalling of adenosine only where and when it is present [246, 249]. This feature is especially important in AD/dementia, as evidence suggests adenosine levels in the brain can vary in a region-specific manner [199].
Therapeutic potential of A1R and A2AR allostery
Allostery has been detected and quantified at both A1R and A2AR, although there has been significantly more traction at A1R. A large number of A1R PAMs have been discovered, with very early studies characterising the allosteric enhancer PD 81,723 [250]. The utility of A1R PAMs has been demonstrated preclinically and in a phase II trial, confirming this pharmacological approach has the translational potential [249, 251]. To date, selective A1R NAMs have not been identified. A2AR allosteric modulators are limited. A fragment-based drug discovery approach identified potential PAM and NAM scaffolds for A2AR [251,252,253]. These findings, along with recent structural advances in the AR field [254,255,256,257], are likely to accelerate the discovery of new A1R and A2AR allosteric ligands. Despite the paucity of A1R and A2AR NAMs and the lack of evaluation of these classes of compounds in AD models, the physiological advantages remain conceptually promising. For A1R and A2AR NAMs, inhibiting signalling only where there is increased adenosine tone could mitigate β-amyloid neurotoxicity and enhance cognition, whilst avoiding the risk of seizures, sleep, and motor disturbances commonly associated with antagonism. This could afford a much more nuanced fine-tuning of AR signalling, without globally reducing adenosine function across all brain areas. Furthermore, this would also likely reduce the incidence of peripheral cardiovascular side effects.
Receptor oligomerisation (homo- and heteromerization)
Another form of allostery to consider when targeting A1R and A2AR is protein–protein interactions. The traditional model of GPCR activity depicted the receptors to function exclusively as monomeric entities. Over the last two decades, increasing evidence indicates GPCRs form homomers and heteromers or higher-order oligomers as part of GPCR normal trafficking and function [258, 259]. Homomerisation describes the self-association of receptor subunits, while heteromerisation describes the association of two or more different receptor subunits, with biochemical properties demonstrably different from individual components. Recent studies support the existence of A1R homomers at the plasma membrane using bimolecular fluorescence complementation and fluorescence correlation spectroscopy [260]. Conversely, cell surface A2AR homomers were confirmed in bioluminescence resonance energy transfer (BRET) or Förster resonance energy transfer (FRET) experiments [261] and may form into oligomers with three or more A2AR protomers [262]. Heteromers and/or higher-order oligomers between different AR subtypes (for review see [263]), as well as with unrelated GPCRs receptors and signalling complexes with other membrane proteins in the brain have increased attention. For an oligomeric interaction to be considered physiologically significant, it is critical to show physical association in native tissue or primary cells and demonstrate unique ‘biochemical fingerprints’ distinct to the oligomer [264].
There are two general mechanisms by which receptor oligomerisation can influence the drug effect [265]. (1) The receptor oligomer becomes a new conduit, whereby ligand-oligomer interactions generate a unique biochemical signalling fingerprint. For example, A2AR agonists decrease the affinity and intrinsic efficacy of D2R agonists in A2AR-D2R heteromers [266]. (2) One receptor modulates ligand binding and/or signalling effects mediated by the other receptor. For example, D2R selectively confers negative cooperativity towards the A2AR antagonist SCH442416 in a A2AR-D2R heteromer, compared to when not forming heteromers or forming heteromers with A1R [267]. As such, SCH442416 would less effectively target A2AR-D2R heteromers expressed in striatopallidal neurons, compared to presynaptic A2AR-A1R heteromers localised in cortico-striatal glutamatergic neurons. Heteromers/homomers with allosteric properties provide an exciting possibility to fine-tune receptor signalling, trafficking, and pharmacological properties.
Therapeutic potential of targeting AR oligomers
Receptor oligomerisation gives rise to novel therapeutic interventions, such as co-activation or co-inhibition of both protomers and activation of one protomer while inhibiting another protomer. Targeting A2AR-D2R heteromers is a well-established therapeutic strategy for Parkinson’s disease where A2AR antagonism in conjunction with D2R agonism is highly desired. Commonly, A2AR antagonists are dosed in conjunction with L-DOPA; however, there are continuing efforts to develop bivalent ligands to co-occupy and specifically target A2AR-D2R heteromers [268,269,270]. Beyond A2AR-D2R heteromers, multiple bona fide AR heteromeric complexes are of therapeutic interest and relevant in the setting of AD (Table 1). A number of recent reviews provide in-depth coverage of the scope of AR heteromerisation [263, 271, 272].
As discussed above, A1R and A2AR are promising therapeutic targets for AD. A1R-A2AR heteromers are expressed in neurons and glia, with heteromers modulating neurotransmitter levels in the context of a tripartite synapse [273, 274]. A1R-A2AR heteromers can be considered a ‘thermostat’ for extracellular adenosine levels, with the opposing effects of Gi versus Gs coupling controlling brain cell responses depending on whether adenosine levels are high or low. Therefore, receptor oligomerisation may also raise unexpected confounds and impacts due to altering the signalling balance of heteromers. Further, oligomerisation may be cell type- or brain region-specific. For example, recent reports of A2AR-A3R heteromers show differential brain-region expression, being the highest in striatal neurons compared to the hippocampus or frontal cortex but are also found in microglia [275]. To therapeutically exploit A2AR-A3R, it would be desirable to inhibit A2AR, thereby removing A2AR functional antagonism of A3R signalling and shifting the balance towards A3R activation by endogenous adenosine. Despite being found at low levels in the brain, A3R is suggested to be neuroprotective in a number of settings, including traumatic brain injury and cerebral ischemia [276, 277]. Targeting AR oligomers linked to disease biology also offers the potential for greater selectivity. Proximity-based approaches suggest heterocomplexes between A2AR and N-methyl-D-aspartate ionotropic glutamate receptors (NMDAR) are increased in activated microglia as well as in the hippocampus of transgenic AD mice (APPsw, Ind) [278]. A2AR has a central role in modulating mGlu5, D1R, and NMDAR signalling in the hippocampus under physiological conditions [279], possibly linked to the propensity of A2AR to form heteromeric complexes.
Concluding remarks
Dementia remains a significant neurological disorder. This review has highlighted preclinical studies implicating A1R and A2AR as promising GPCR targets for Alzheimer’s disease. Overall, A1R and A2AR inhibition seems to mitigate the neurotoxicity associated with the β-amyloid accumulation and tau hyperphosphorylation and improve cognition and memory. Stimulation of A1R can also promote neuroprotection and therefore adds an additional layer of complexity in considering agonism versus antagonism when targeting this receptor. Although not explored for AD, the notion of biased agonism, which is a growing paradigm at adenosine receptors [249, 280], could conceptually be harnessed to develop an A1R compound to improve cognition but also remain neuroprotective. Indeed, although drug discovery at adenosine receptors has traditionally experienced hurdles, the novel therapeutic paradigms covered in this review, including allostery and the targeting of oligomers, present a promising future avenue of investigation. It is hoped harnessing this knowledge may increase the development and translation of clinical candidates with enhanced therapeutic responses and limited side effects.
Data availability
Not applicable.
Change history
16 November 2022
A Correction to this paper has been published: https://doi.org/10.1007/s11302-022-09906-x
References
(2020) 2020 Alzheimer’s disease facts and figures. Alzheimer's Dement 16(3):391–460. https://doi.org/10.1002/alz.12068
Association AP (2013) Neurocognitive disorders. Diagnostic and Statistical Manual of Mental Disorders, American Psychiatric Association Arlington, VA
Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, Gamst A, Holtzman DM, Jagust WJ, Petersen RC, Snyder PJ, Carrillo MC, Thies B, Phelps CH (2011) The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7(3):270–279. https://doi.org/10.1016/j.jalz.2011.03.008
Scheltens P, Blennow K, Breteler MM, de Strooper B, Frisoni GB, Salloway S, Van der Flier WM (2016) Alzheimer’s disease. Lancet 388(10043):505–517. https://doi.org/10.1016/s0140-6736(15)01124-1
Brys M, Glodzik L, Mosconi L, Switalski R, De Santi S, Pirraglia E, Rich K, Kim BC, Mehta P, Zinkowski R, Pratico D, Wallin A, Zetterberg H, Tsui WH, Rusinek H, Blennow K, de Leon MJ (2009) Magnetic resonance imaging improves cerebrospinal fluid biomarkers in the early detection of Alzheimer’s disease. J Alzheimers Dis 16(2):351–362. https://doi.org/10.3233/JAD-2009-0968
Dubois B, Feldman HH, Jacova C, Cummings JL, Dekosky ST, Barberger-Gateau P, Delacourte A, Frisoni G, Fox NC, Galasko D, Gauthier S, Hampel H, Jicha GA, Meguro K, O’Brien J, Pasquier F, Robert P, Rossor M, Salloway S, Sarazin M, de Souza LC, Stern Y, Visser PJ, Scheltens P (2010) Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol 9(11):1118–1127. https://doi.org/10.1016/s1474-4422(10)70223-4
Tapiola T, Alafuzoff I, Herukka SK, Parkkinen L, Hartikainen P, Soininen H, Pirttilä T (2009) Cerebrospinal fluid β-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain. Arch Neurol 66(3):382–389. https://doi.org/10.1001/archneurol.2008.596
Fox NC, Scahill RI, Crum WR, Rossor MN (1999) Correlation between rates of brain atrophy and cognitive decline in AD. Neurology 52(8):1687–1689. https://doi.org/10.1212/wnl.52.8.1687
Jack CR Jr, Petersen RC, Xu YC, Waring SC, O’Brien PC, Tangalos EG, Smith GE, Ivnik RJ, Kokmen E (1997) Medial temporal atrophy on MRI in normal aging and very mild Alzheimer’s disease. Neurology 49(3):786–794. https://doi.org/10.1212/wnl.49.3.786
Piguet O, Double KL, Kril JJ, Harasty J, Macdonald V, McRitchie DA, Halliday GM (2009) White matter loss in healthy ageing: a postmortem analysis. Neurobiol Aging 30(8):1288–1295. https://doi.org/10.1016/j.neurobiolaging.2007.10.015
Stout JC, Jernigan TL, Archibald SL, Salmon DP (1996) Association of dementia severity with cortical gray matter and abnormal white matter volumes in dementia of the Alzheimer type. Arch Neurol 53(8):742–749. https://doi.org/10.1001/archneur.1996.00550080056013
deToledo-Morrell L, Stoub TR, Bulgakova M, Wilson RS, Bennett DA, Leurgans S, Wuu J, Turner DA (2004) MRI-derived entorhinal volume is a good predictor of conversion from MCI to AD. Neurobiol Aging 25(9):1197–1203. https://doi.org/10.1016/j.neurobiolaging.2003.12.007
Dickerson BC, Stoub TR, Shah RC, Sperling RA, Killiany RJ, Albert MS, Hyman BT, Blacker D, Detoledo-Morrell L (2011) Alzheimer-signature MRI biomarker predicts AD dementia in cognitively normal adults. Neurol 76(16):1395–1402. https://doi.org/10.1212/WNL.0b013e3182166e96
Du AT, Schuff N, Amend D, Laakso MP, Hsu YY, Jagust WJ, Yaffe K, Kramer JH, Reed B, Norman D, Chui HC, Weiner MW (2001) Magnetic resonance imaging of the entorhinal cortex and hippocampus in mild cognitive impairment and Alzheimer’s disease. J Neurol Neurosurg Psychiatry 71(4):441–447. https://doi.org/10.1136/jnnp.71.4.441
Jack CR Jr, Petersen RC, Xu YC, O’Brien PC, Smith GE, Ivnik RJ, Boeve BF, Waring SC, Tangalos EG, Kokmen E (1999) Prediction of AD with MRI-based hippocampal volume in mild cognitive impairment. Neurol 52(7):1397–1403. https://doi.org/10.1212/wnl.52.7.1397
Hardy JA, Higgins GA (1992) Alzheimer’s disease: the amyloid cascade hypothesis. Sci 256(5054):184–185. https://doi.org/10.1126/science.1566067
Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, Lieberburg I, Koo EH, Schenk D, Teplow DB et al (1992) Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nat 359(6393):322–325. https://doi.org/10.1038/359322a0
Bishop GM, Robinson SR (2004) Physiological roles of amyloid-beta and implications for its removal in Alzheimer’s disease. Drugs Aging 21(10):621–630. https://doi.org/10.2165/00002512-200421100-00001
Grimm MO, Grimm HS, Hartmann T (2007) Amyloid beta as a regulator of lipid homeostasis. Trends Mol Med 13(8):337–344. https://doi.org/10.1016/j.molmed.2007.06.004
Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y (1994) Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43). Neuron 13(1):45–53. https://doi.org/10.1016/0896-6273(94)90458-8
Miller DL, Papayannopoulos IA, Styles J, Bobin SA, Lin YY, Biemann K, Iqbal K (1993) Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer’s disease. Arch Biochem Biophys 301(1):41–52. https://doi.org/10.1006/abbi.1993.1112
Beyreuther K, Masters CL (1991) Amyloid precursor protein (APP) and beta A4 amyloid in the etiology of Alzheimer’s disease: precursor-product relationships in the derangement of neuronal function. Brain Pathol 1(4):241–251. https://doi.org/10.1111/j.1750-3639.1991.tb00667.x
Hardy J, Allsop D (1991) Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 12(10):383–388. https://doi.org/10.1016/0165-6147(91)90609-v
Selkoe DJ (1991) The molecular pathology of Alzheimer’s disease. Neuron 6(4):487–498. https://doi.org/10.1016/0896-6273(91)90052-2
Caceres A, Kosik KS (1990) Inhibition of neurite polarity by tau antisense oligonucleotides in primary cerebellar neurons. Nature 343(6257):461–463. https://doi.org/10.1038/343461a0
Drubin DG, Kirschner MW (1986) Tau protein function in living cells. J Cell Biol 103(6 Pt 2):2739–2746. https://doi.org/10.1083/jcb.103.6.2739
Lace GL, Wharton SB, Ince PG (2007) A brief history of tau: the evolving view of the microtubule-associated protein tau in neurodegenerative diseases. Clin Neuropathol 26(2):43–58. https://doi.org/10.5414/npp26043
Alvarez A, Toro R, Cáceres A, Maccioni RB (1999) Inhibition of tau phosphorylating protein kinase cdk5 prevents beta-amyloid-induced neuronal death. FEBS Lett 459(3):421–426. https://doi.org/10.1016/s0014-5793(99)01279-x
Lee G, Thangavel R, Sharma VM, Litersky JM, Bhaskar K, Fang SM, Do LH, Andreadis A, Van Hoesen G, Ksiezak-Reding H (2004) Phosphorylation of tau by fyn: implications for Alzheimer’s disease. J Neurosci 24(9):2304–2312. https://doi.org/10.1523/jneurosci.4162-03.2004
Lindwall G, Cole RD (1984) Phosphorylation affects the ability of tau protein to promote microtubule assembly. J Biol Chem 259(8):5301–5305
Lucas JJ, Hernández F, Gómez-Ramos P, Morán MA, Hen R, Avila J (2001) Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. Embo j 20(1–2):27–39. https://doi.org/10.1093/emboj/20.1.27
Rickle A, Bogdanovic N, Volkman I, Winblad B, Ravid R, Cowburn RF (2004) Akt activity in Alzheimer’s disease and other neurodegenerative disorders. NeuroReport 15(6):955–959. https://doi.org/10.1097/00001756-200404290-00005
Hensley K, Floyd RA, Zheng NY, Nael R, Robinson KA, Nguyen X, Pye QN, Stewart CA, Geddes J, Markesbery WR, Patel E, Johnson GV, Bing G (1999) p38 kinase is activated in the Alzheimer’s disease brain. J Neurochem 72(5):2053–2058. https://doi.org/10.1046/j.1471-4159.1999.0722053.x
Zhu X, Castellani RJ, Takeda A, Nunomura A, Atwood CS, Perry G, Smith MA (2001) Differential activation of neuronal ERK, JNK/SAPK and p38 in Alzheimer disease: the ‘two hit’ hypothesis. Mech Ageing Dev 123(1):39–46. https://doi.org/10.1016/s0047-6374(01)00342-6
Alonso AC, Zaidi T, Grundke-Iqbal I, Iqbal K (1994) Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci USA 91(12):5562–5566. https://doi.org/10.1073/pnas.91.12.5562
Ruben GC, Iqbal K, Wisniewski HM, Johnson JE Jr, Grundke-Iqbal I (1993) Alzheimer neurofibrillary tangles contain 2.1 nm filaments structurally identical to the microtubule-associated protein tau: a high-resolution transmission electron microscope study of tangles and senile plaque core amyloid. Brain Res 602(2):164–179. https://doi.org/10.1016/0006-8993(92)91092-s
Braak H, Braak E, Grundke-Iqbal I, Iqbal K (1986) Occurrence of neuropil threads in the senile human brain and in Alzheimer’s disease: a third location of paired helical filaments outside of neurofibrillary tangles and neuritic plaques. Neurosci Lett 65(3):351–355. https://doi.org/10.1016/0304-3940(86)90288-0
Cras P, Kawai M, Siedlak S, Perry G (1991) Microglia are associated with the extracellular neurofibrillary tangles of Alzheimer disease. Brain Res 558(2):312–314. https://doi.org/10.1016/0006-8993(91)90783-r
Goedert M, Spillantini MG, Jakes R, Crowther RA, Vanmechelen E, Probst A, Götz J, Bürki K, Cohen P (1995) Molecular dissection of the paired helical filament. Neurobiol Aging 16(3):325–334. https://doi.org/10.1016/0197-4580(95)00017-9
Probst A, Ulrich J, Heitz PU (1982) Senile dementia of Alzheimer type: astroglial reaction to extracellular neurofibrillary tangles in the hippocampus. An immunocytochemical and electron-microscopic study. Acta Neuropathol 57(1):75–79. https://doi.org/10.1007/bf00688880
Goedert M, Spillantini MG (2006) A century of Alzheimer’s disease. Sci 314(5800):777–781. https://doi.org/10.1126/science.1132814
Bennett DA, Schneider JA, Wilson RS, Bienias JL, Arnold SE (2004) Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch Neurol 61(3):378–384. https://doi.org/10.1001/archneur.61.3.378
Gómez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT (1997) Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol 41(1):17–24. https://doi.org/10.1002/ana.410410106
Braak H, Braak E (1997) Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging 18(4):351–357. https://doi.org/10.1016/s0197-4580(97)00056-0
Knopman DS, Parisi JE, Salviati A, Floriach-Robert M, Boeve BF, Ivnik RJ, Smith GE, Dickson DW, Johnson KA, Petersen LE, McDonald WC, Braak H, Petersen RC (2003) Neuropathology of cognitively normal elderly. J Neuropathol Exp Neurol 62(11):1087–1095. https://doi.org/10.1093/jnen/62.11.1087
Price JL, Morris JC (1999) Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann Neurol 45(3):358–368. https://doi.org/10.1002/1531-8249(199903)45:3%3c358::aid-ana12%3e3.0.co;2-x
Savva GM, Wharton SB, Ince PG, Forster G, Matthews FE, Brayne C (2009) Age, neuropathology, and dementia. N Engl J Med 360(22):2302–2309. https://doi.org/10.1056/NEJMoa0806142
Boutajangout A, Ingadottir J, Davies P, Sigurdsson EM (2011) Passive immunization targeting pathological phospho-tau protein in a mouse model reduces functional decline and clears tau aggregates from the brain. J Neurochem 118(4):658–667. https://doi.org/10.1111/j.1471-4159.2011.07337.x
Boutajangout A, Wisniewski T (2014) Tau-based therapeutic approaches for Alzheimer’s disease - a mini-review. Gerontology 60(5):381–385. https://doi.org/10.1159/000358875
McGeer PL, McGeer EG (2013) The amyloid cascade-inflammatory hypothesis of Alzheimer disease: implications for therapy. Acta Neuropathol 126(4):479–497. https://doi.org/10.1007/s00401-013-1177-7
Brosseron F, Krauthausen M, Kummer M, Heneka MT (2014) Body fluid cytokine levels in mild cognitive impairment and Alzheimer’s disease: a comparative overview. Mol Neurobiol 50(2):534–544. https://doi.org/10.1007/s12035-014-8657-1
Tarkowski E, Andreasen N, Tarkowski A, Blennow K (2003) Intrathecal inflammation precedes development of Alzheimer’s disease. J Neurol Neurosurg Psychiatry 74(9):1200–1205. https://doi.org/10.1136/jnnp.74.9.1200
Benzing WC, Wujek JR, Ward EK, Shaffer D, Ashe KH, Younkin SG, Brunden KR (1999) Evidence for glial-mediated inflammation in aged APP(SW) transgenic mice. Neurobiol Aging 20(6):581–589. https://doi.org/10.1016/s0197-4580(99)00065-2
Castellani RJ, Lee HG, Zhu X, Perry G, Smith MA (2008) Alzheimer disease pathology as a host response. J Neuropathol Exp Neurol 67(6):523–531. https://doi.org/10.1097/NEN.0b013e318177eaf4
Graeber MB, Streit WJ (2010) Microglia: biology and pathology. Acta Neuropathol 119(1):89–105. https://doi.org/10.1007/s00401-009-0622-0
Heppner FL, Ransohoff RM, Becher B (2015) Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci 16(6):358–372. https://doi.org/10.1038/nrn3880
Wyss-Coray T, Mucke L (2002) Inflammation in neurodegenerative disease–a double-edged sword. Neuron 35(3):419–432. https://doi.org/10.1016/s0896-6273(02)00794-8
Koistinaho M, Lin S, Wu X, Esterman M, Koger D, Hanson J, Higgs R, Liu F, Malkani S, Bales KR, Paul SM (2004) Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat Med 10(7):719–726. https://doi.org/10.1038/nm1058
Simard AR, Soulet D, Gowing G, Julien JP, Rivest S (2006) Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 49(4):489–502. https://doi.org/10.1016/j.neuron.2006.01.022
Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T (2000) Inflammation and Alzheimer’s disease. Neurobiol Aging 21(3):383–421. https://doi.org/10.1016/s0197-4580(00)00124-x
Hickman SE, Allison EK, El Khoury J (2008) Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J Neurosci 28(33):8354–8360. https://doi.org/10.1523/jneurosci.0616-08.2008
Jimenez S, Baglietto-Vargas D, Caballero C, Moreno-Gonzalez I, Torres M, Sanchez-Varo R, Ruano D, Vizuete M, Gutierrez A, Vitorica J (2008) Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer’s disease: age-dependent switch in the microglial phenotype from alternative to classic. J Neurosci 28(45):11650–11661. https://doi.org/10.1523/jneurosci.3024-08.2008
Chiang K, Koo EH (2014) Emerging therapeutics for Alzheimer’s disease. Annu Rev Pharmacol Toxicol 54:381–405. https://doi.org/10.1146/annurev-pharmtox-011613-135932
Wyss-Coray T, Rogers J (2012) Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med 2(1):a006346–a006346. https://doi.org/10.1101/cshperspect.a006346
Graeber MB, Christie MJ (2012) Multiple mechanisms of microglia: a gatekeeper’s contribution to pain states. Exp Neurol 234(2):255–261. https://doi.org/10.1016/j.expneurol.2012.01.007
Henkel JS, Beers DR, Zhao W, Appel SH (2009) Microglia in ALS: the good, the bad, and the resting. J Neuroimmune Pharmacol 4(4):389–398. https://doi.org/10.1007/s11481-009-9171-5
Ji K, Akgul G, Wollmuth LP, Tsirka SE (2013) Microglia actively regulate the number of functional synapses. PLoS ONE 8(2):e56293. https://doi.org/10.1371/journal.pone.0056293
Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Sci 308(5726):1314–1318. https://doi.org/10.1126/science.1110647
Sofroniew MV, Vinters HV (2010) Astrocytes: biology and pathology. Acta Neuropathol 119(1):7–35. https://doi.org/10.1007/s00401-009-0619-8
Eng LF, Ghirnikar RS (1994) GFAP and astrogliosis. Brain Pathol 4(3):229–237. https://doi.org/10.1111/j.1750-3639.1994.tb00838.x
London A, Cohen M, Schwartz M (2013) Microglia and monocyte-derived macrophages: functionally distinct populations that act in concert in CNS plasticity and repair. Front Cell Neurosci 7:34. https://doi.org/10.3389/fncel.2013.00034
Abramov AY, Canevari L, Duchen MR (2003) Changes in intracellular calcium and glutathione in astrocytes as the primary mechanism of amyloid neurotoxicity. J Neurosci 23(12):5088–5095. https://doi.org/10.1523/jneurosci.23-12-05088.2003
Blasko I, Veerhuis R, Stampfer-Kountchev M, Saurwein-Teissl M, Eikelenboom P, Grubeck-Loebenstein B (2000) Costimulatory effects of interferon-gamma and interleukin-1beta or tumor necrosis factor alpha on the synthesis of Abeta1–40 and Abeta1–42 by human astrocytes. Neurobiol Dis 7(6 Pt B):682–689. https://doi.org/10.1006/nbdi.2000.0321
Fuller S, Steele M, Münch G (2010) Activated astroglia during chronic inflammation in Alzheimer’s disease–do they neglect their neurosupportive roles? Mutat Res 690(1–2):40–49. https://doi.org/10.1016/j.mrfmmm.2009.08.016
Oberstein TJ, Spitzer P, Klafki HW, Linning P, Neff F, Knölker HJ, Lewczuk P, Wiltfang J, Kornhuber J, Maler JM (2015) Astrocytes and microglia but not neurons preferentially generate N-terminally truncated Aβ peptides. Neurobiol Dis 73:24–35. https://doi.org/10.1016/j.nbd.2014.08.031
Sudduth TL, Schmitt FA, Nelson PT, Wilcock DM (2013) Neuroinflammatory phenotype in early Alzheimer’s disease. Neurobiol Aging 34(4):1051–1059. https://doi.org/10.1016/j.neurobiolaging.2012.09.012
Kuruppu S, Rajapakse NW, Parkington HC, Smith AI (2017) The characteristics of astrocyte on Aβ clearance altered in Alzheimer’s disease were reversed by anti-inflammatory agent (+)-2-(1-hydroxyl-4-oxocyclohexyl) ethyl caffeate. Am J Transl Res 9(7):3514–3516
Söllvander S, Nikitidou E, Brolin R, Söderberg L, Sehlin D, Lannfelt L, Erlandsson A (2016) Accumulation of amyloid-β by astrocytes result in enlarged endosomes and microvesicle-induced apoptosis of neurons. Mol Neurodegener 11(1):38. https://doi.org/10.1186/s13024-016-0098-z
Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, White CL 3rd, Araoz C (1989) Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci USA 86(19):7611–7615. https://doi.org/10.1073/pnas.86.19.7611
Fillit H, Ding WH, Buee L, Kalman J, Altstiel L, Lawlor B, Wolf-Klein G (1991) Elevated circulating tumor necrosis factor levels in Alzheimer’s disease. Neurosci Lett 129(2):318–320. https://doi.org/10.1016/0304-3940(91)90490-k
Hoffmann A, Kann O, Ohlemeyer C, Hanisch UK, Kettenmann H (2003) Elevation of basal intracellular calcium as a central element in the activation of brain macrophages (microglia): suppression of receptor-evoked calcium signaling and control of release function. J Neurosci 23(11):4410–4419
Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE (2009) CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J Neurosci 29(38):11982–11992. https://doi.org/10.1523/jneurosci.3158-09.2009
Toescu EC, Möller T, Kettenmann H, Verkhratsky A (1998) Long-term activation of capacitative Ca2+ entry in mouse microglial cells. Neuroscience 86(3):925–935. https://doi.org/10.1016/S0306-4522(98)00123-7
Vom Berg J, Prokop S, Miller KR, Obst J, Kälin RE, Lopategui-Cabezas I, Wegner A, Mair F, Schipke CG, Peters O, Winter Y, Becher B, Heppner FL (2012) Inhibition of IL-12/IL-23 signaling reduces Alzheimer’s disease-like pathology and cognitive decline. Nat Med 18(12):1812–1819. https://doi.org/10.1038/nm.2965
Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ (2010) Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 330(6012):1774. https://doi.org/10.1126/science.1197623
Stalder M, Deller T, Staufenbiel M, Jucker M (2001) 3D-Reconstruction of microglia and amyloid in APP23 transgenic mice: no evidence of intracellular amyloid. Neurobiol Aging 22(3):427–434. https://doi.org/10.1016/s0197-4580(01)00209-3
Wegiel J, Wang KC, Imaki H, Rubenstein R, Wronska A, Osuchowski M, Lipinski WJ, Walker LC, LeVine H (2001) The role of microglial cells and astrocytes in fibrillar plaque evolution in transgenic APP(SW) mice. Neurobiol Aging 22(1):49–61. https://doi.org/10.1016/s0197-4580(00)00181-0
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541(7638):481–487. https://doi.org/10.1038/nature21029
Hamby ME, Coppola G, Ao Y, Geschwind DH, Khakh BS, Sofroniew MV (2012) Inflammatory mediators alter the astrocyte transcriptome and calcium signaling elicited by multiple G-protein-coupled receptors. J Neurosci 32(42):14489. https://doi.org/10.1523/JNEUROSCI.1256-12.2012
Haque ME, Kim I-S, Jakaria M, Akther M, Choi D-K (2018) Importance of GPCR-mediated microglial activation in Alzheimer’s disease. Front Cell Neurosci 12:258–258. https://doi.org/10.3389/fncel.2018.00258
Zhang F, Zhong R-j, Cheng C, Li S, Le W-d (2020) New therapeutics beyond amyloid-β and tau for the treatment of Alzheimer’s disease. Acta Pharmacologica Sinica. https://doi.org/10.1038/s41401-020-00565-5
Cummings J, Lee G, Zhong K, Fonseca J, Taghva K (2021) Alzheimer’s disease drug development pipeline: 2021. Alzheimers Dement (N Y) 7(1):e12179. https://doi.org/10.1002/trc2.12179
van Bokhoven P, de Wilde A, Vermunt L, Leferink PS, Heetveld S, Cummings J, Scheltens P, Vijverberg EGB (2021) The Alzheimer’s disease drug development landscape. Alzheimers Res Ther 13(1):186. https://doi.org/10.1186/s13195-021-00927-z
Haeberlein SB, Salloway S, Aisen P, Frederik B, Castrillo-Viguera C, Chen T, Cohen S, Hansson O, He P, Iwatsubo T, Mallinkrodt C, Mummery CJ, Muralidharan KK, Nisenbaum L, Rajagovindan R, Vellas B, Wu S, Yang L, Tian Y (2021) Evaluation of aducanumab efficacy in early Alzheimer’s disease. Paper presented at: 15th International Conference on Alzheimer's & Parkinson's Diseases virtual conference; March 9–14, 2021
Cummings J, Aisen P, Lemere C, Atri A, Sabbagh M, Salloway S (2021) Aducanumab produced a clinically meaningful benefit in association with amyloid lowering. Alzheimers Res Ther 13(1):98. https://doi.org/10.1186/s13195-021-00838-z
Knopman DS, Jones DT (2019) Greicius MD (2021) Failure to demonstrate efficacy of aducanumab: an analysis of the EMERGE and ENGAGE trials as reported by Biogen. Alzheimers Dement 17(4):696–701. https://doi.org/10.1002/alz.12213
Tampi RR, Forester BP, Agronin M (2021) Aducanumab: evidence from clinical trial data and controversies. Drugs Context 10.https://doi.org/10.7573/dic.2021-7-3
Klein G, Delmar P, Voyle N, Rehal S, Hofmann C, Abi-Saab D, Andjelkovic M, Ristic S, Wang G, Bateman R, Kerchner GA, Baudler M, Fontoura P, Doody R (2019) Gantenerumab reduces amyloid-beta plaques in patients with prodromal to moderate Alzheimer’s disease: a PET substudy interim analysis. Alzheimer’s res ther 11(1):101. https://doi.org/10.1186/s13195-019-0559-z
Mintun MA, Lo AC, Duggan Evans C, Wessels AM, Ardayfio PA, Andersen SW, Shcherbinin S, Sparks J, Sims JR, Brys M, Apostolova LG, Salloway SP, Skovronsky DM (2021) Donanemab in early Alzheimer’s disease. N Engl J Med 384(18):1691–1704. https://doi.org/10.1056/NEJMoa2100708
Swanson CJ, Zhang Y, Dhadda S, Wang J, Kaplow J, Lai RYK, Lannfelt L, Bradley H, Rabe M, Koyama A, Reyderman L, Berry DA, Berry S, Gordon R, Kramer LD, Cummings JL (2021) A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Abeta protofibril antibody. Alzheimers Res Ther 13(1):80. https://doi.org/10.1186/s13195-021-00813-8
Doody RS, Farlow M, Aisen PS (2014) Phase 3 trials of solanezumab and bapineuzumab for Alzheimer’s disease. N Engl J Med 370(15):1460. https://doi.org/10.1056/NEJMc1402193
Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, He F, Sun X, Thomas RG, Aisen PS, Siemers E, Sethuraman G, Mohs R (2013) A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med 369(4):341–350. https://doi.org/10.1056/NEJMoa1210951
Galasko D, Bell J, Mancuso JY, Kupiec JW, Sabbagh MN, van Dyck C, Thomas RG, Aisen PS (2014) Clinical trial of an inhibitor of RAGE-Aβ interactions in Alzheimer disease. Neurol 82(17):1536–1542. https://doi.org/10.1212/wnl.0000000000000364
Gauthier S, Feldman HH, Schneider LS, Wilcock GK, Frisoni GB, Hardlund JH, Moebius HJ, Bentham P, Kook KA, Wischik DJ, Schelter BO, Davis CS, Staff RT, Bracoud L, Shamsi K, Storey JM, Harrington CR, Wischik CM (2016) Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: a randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet 388(10062):2873–2884. https://doi.org/10.1016/s0140-6736(16)31275-2
Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, Sabbagh M, Honig LS, Porsteinsson AP, Ferris S, Reichert M, Ketter N, Nejadnik B, Guenzler V, Miloslavsky M, Wang D, Lu Y, Lull J, Tudor IC, Liu E, Grundman M, Yuen E, Black R, Brashear HR (2014) Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med 370(4):322–333. https://doi.org/10.1056/NEJMoa1304839
Vandenberghe R, Rinne JO, Boada M, Katayama S, Scheltens P, Vellas B, Tuchman M, Gass A, Fiebach JB, Hill D, Lobello K, Li D, McRae T, Lucas P, Evans I, Booth K, Luscan G, Wyman BT, Hua L, Yang L, Brashear HR, Black RS (2016) Bapineuzumab for mild to moderate Alzheimer’s disease in two global, randomized, phase 3 trials. Alzheimers Res Ther 8(1):18. https://doi.org/10.1186/s13195-016-0189-7
Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261(5123):921–923
McGeer PL, Schulzer M, McGeer EG (1996) Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: a review of 17 epidemiologic studies. Neurology 47(2):425–432. https://doi.org/10.1212/wnl.47.2.425
Stewart WF, Kawas C, Corrada M, Metter EJ (1997) Risk of Alzheimer’s disease and duration of NSAID use. Neurology 48(3):626–632. https://doi.org/10.1212/wnl.48.3.626
Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD (1993) Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA 90(5):1977–1981
Vlad SC, Miller DR, Kowall NW, Felson DT (2008) Protective effects of NSAIDs on the development of Alzheimer disease. Neurology 70(19):1672–1677. https://doi.org/10.1212/01.wnl.0000311269.57716.63
Zannis VI, Breslow JL, Utermann G, Mahley RW, Weisgraber KH, Havel RJ, Goldstein JL, Brown MS, Schonfeld G, Hazzard WR, Blum C (1982) Proposed nomenclature of apoE isoproteins, apoE genotypes, and phenotypes. J Lipid Res 23(6):911–914
Aisen PS (2002) Evaluation of selective COX-2 inhibitors for the treatment of Alzheimer’s disease. J Pain Symptom Manage 23(4 Suppl):S35-40. https://doi.org/10.1016/s0885-3924(02)00374-3
Aisen PS, Schafer KA, Grundman M, Pfeiffer E, Sano M, Davis KL, Farlow MR, Jin S, Thomas RG, Thal LJ (2003) Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA 289(21):2819–2826. https://doi.org/10.1001/jama.289.21.2819
Martin BK, Szekely C, Brandt J, Piantadosi S, Breitner JC, Craft S, Evans D, Green R, Mullan M (2008) Cognitive function over time in the Alzheimer’s Disease Anti-inflammatory Prevention Trial (ADAPT): results of a randomized, controlled trial of naproxen and celecoxib. Arch Neurol 65(7):896–905. https://doi.org/10.1001/archneur.2008.65.7.nct70006
Breitner JC, Baker LD, Montine TJ, Meinert CL, Lyketsos CG, Ashe KH, Brandt J, Craft S, Evans DE, Green RC, Ismail MS, Martin BK, Mullan MJ, Sabbagh M, Tariot PN (2011) Extended results of the Alzheimer’s disease anti-inflammatory prevention trial. Alzheimers Dement 7(4):402–411. https://doi.org/10.1016/j.jalz.2010.12.014
Dal Pra I, Armato U, Chiarini A (2019) Family C G-protein-coupled receptors in Alzheimer’s disease and therapeutic implications. Front Pharmacol 10:1282. https://doi.org/10.3389/fphar.2019.01282
Thathiah A, De Strooper B (2011) The role of G protein-coupled receptors in the pathology of Alzheimer’s disease. Nat Rev Neurosci 12(2):73–87. https://doi.org/10.1038/nrn2977
Zhao J, Deng Y, Jiang Z, Qing H (2016) G Protein-Coupled Receptors (GPCRs) in Alzheimer’s disease: a focus on BACE1 related GPCRs. Front Aging Neurosci 8:58. https://doi.org/10.3389/fnagi.2016.00058
Rosenbaum DM, Rasmussen SG, Kobilka BK (2009) The structure and function of G-protein-coupled receptors. Nature 459(7245):356–363. https://doi.org/10.1038/nature08144
Blalock EM, Geddes JW, Chen KC, Porter NM, Markesbery WR, Landfield PW (2004) Incipient Alzheimer’s disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc Natl Acad Sci USA 101(7):2173
Cortellis (2022) Cortellis Drug Discovery Intelligence. https://www.cortelliscom/intelligence/homedo Accessed 17 March 2022
Arendash GW, Schleif W, Rezai-Zadeh K, Jackson EK, Zacharia LC, Cracchiolo JR, Shippy D, Tan J (2006) Caffeine protects Alzheimer’s mice against cognitive impairment and reduces brain beta-amyloid production. Neuroscience 142(4):941–952. https://doi.org/10.1016/j.neuroscience.2006.07.021
Giunta S, Andriolo V, Castorina A (2014) Dual blockade of the A1 and A2A adenosine receptor prevents amyloid beta toxicity in neuroblastoma cells exposed to aluminum chloride. Int J Biochem Cell Biol 54:122–136. https://doi.org/10.1016/j.biocel.2014.07.009
Fredholm BB, Arslan G, Halldner L, Kull B, Schulte G, Wasserman W (2000) Structure and function of adenosine receptors and their genes. Naunyn Schmiedebergs Arch Pharmacol 362(4–5):364–374
Dunwiddie TV, Masino SA (2001) The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci 24:31–55. https://doi.org/10.1146/annurev.neuro.24.1.31
Costenla AR, Cunha RA, de Mendonça A (2010) Caffeine, adenosine receptors, and synaptic plasticity. J Alzheimers Dis 20(Suppl 1):S25-34. https://doi.org/10.3233/jad-2010-091384
Costenla AR, Diógenes MJ, Canas PM, Rodrigues RJ, Nogueira C, Maroco J, Agostinho PM, Ribeiro JA, Cunha RA, de Mendonça A (2011) Enhanced role of adenosine A(2A) receptors in the modulation of LTP in the rat hippocampus upon ageing. Eur J Neurosci 34(1):12–21. https://doi.org/10.1111/j.1460-9568.2011.07719.x
Fredholm BB, Ijzerman AP, Jacobson KA, Klotz K-N, Linden J (2001) International Union of Pharmacology. XXV. Nomenclature and Classification of Adenosine Receptors. Pharmacol Rev 53(4):527
Rivkees SA, Price SL, Zhou FC (1995) Immunohistochemical detection of A1 adenosine receptors in rat brain with emphasis on localization in the hippocampal formation, cerebral cortex, cerebellum, and basal ganglia. Brain Res 677(2):193–203. https://doi.org/10.1016/0006-8993(95)00062-u
Schindler M, Harris CA, Hayes B, Papotti M, Humphrey PP (2001) Immunohistochemical localization of adenosine A1 receptors in human brain regions. Neurosci Lett 297(3):211–215. https://doi.org/10.1016/s0304-3940(00)01643-8
Ochiishi T, Saitoh Y, Yukawa A, Saji M, Ren Y, Shirao T, Miyamoto H, Nakata H, Sekino Y (1999) High level of adenosine A1 receptor-like immunoreactivity in the CA2/CA3a region of the adult rat hippocampus. Neurosci 93(3):955–967. https://doi.org/10.1016/s0306-4522(99)00179-7
Rebola N, Pinheiro PC, Oliveira CR, Malva JO, Cunha RA (2003) Subcellular localization of adenosine A(1) receptors in nerve terminals and synapses of the rat hippocampus. Brain Res 987(1):49–58. https://doi.org/10.1016/s0006-8993(03)03247-5
Tetzlaff W, Schubert P, Kreutzberg GW (1987) Synaptic and extrasynaptic localization of adenosine binding sites in the rat hippocampus. Neurosci 21(3):869–875. https://doi.org/10.1016/0306-4522(87)90043-1
Biber K, Klotz KN, Berger M, Gebicke-Härter PJ, van Calker D (1997) Adenosine A1 receptor-mediated activation of phospholipase C in cultured astrocytes depends on the level of receptor expression. J Neurosci 17(13):4956–4964. https://doi.org/10.1523/jneurosci.17-13-04956.1997
Gebicke-Haerter PJ, Christoffel F, Timmer J, Northoff H, Berger M, Van Calker D (1996) Both adenosine A1- and A2-receptors are required to stimulate microglial proliferation. Neurochem Int 29(1):37–42
Othman T, Yan H, Rivkees SA (2003) Oligodendrocytes express functional A1 adenosine receptors that stimulate cellular migration. Glia 44(2):166–172. https://doi.org/10.1002/glia.10281
Dixon AK, Gubitz AK, Sirinathsinghji DJ, Richardson PJ, Freeman TC (1996) Tissue distribution of adenosine receptor mRNAs in the rat. Br J Pharmacol 118(6):1461–1468. https://doi.org/10.1111/j.1476-5381.1996.tb15561.x
Mishina M, Ishiwata K, Kimura Y, Naganawa M, Oda K, Kobayashi S, Katayama Y, Ishii K (2007) Evaluation of distribution of adenosine A2A receptors in normal human brain measured with [11C]TMSX PET. Synapse 61(9):778–784. https://doi.org/10.1002/syn.20423
Hettinger BD, Lee A, Linden J, Rosin DL (2001) Ultrastructural localization of adenosine A2A receptors suggests multiple cellular sites for modulation of GABAergic neurons in rat striatum. J Comp Neurol 431(3):331–346. https://doi.org/10.1002/1096-9861(20010312)431:3%3c331::aid-cne1074%3e3.0.co;2-w
Rodrigues RJ, Alfaro TM, Rebola N, Oliveira CR, Cunha RA (2005) Co-localization and functional interaction between adenosine A(2A) and metabotropic group 5 receptors in glutamatergic nerve terminals of the rat striatum. J Neurochem 92(3):433–441. https://doi.org/10.1111/j.1471-4159.2004.02887.x
Rebola N, Canas PM, Oliveira CR, Cunha RA (2005) Different synaptic and subsynaptic localization of adenosine A2A receptors in the hippocampus and striatum of the rat. Neuroscience 132(4):893–903. https://doi.org/10.1016/j.neuroscience.2005.01.014
Li XX, Nomura T, Aihara H, Nishizaki T (2001) Adenosine enhances glial glutamate efflux via A2a adenosine receptors. Life Sci 68(12):1343–1350. https://doi.org/10.1016/s0024-3205(00)01036-5
Nishizaki T, Nagai K, Nomura T, Tada H, Kanno T, Tozaki H, Li XX, Kondoh T, Kodama N, Takahashi E, Sakai N, Tanaka K, Saito N (2002) A new neuromodulatory pathway with a glial contribution mediated via A(2a) adenosine receptors. Glia 39(2):133–147. https://doi.org/10.1002/glia.10100
Fiebich BL, Biber K, Lieb K, van Calker D, Berger M, Bauer J, Gebicke-Haerter PJ (1996) Cyclooxygenase-2 expression in rat microglia is induced by adenosine A2a-receptors. Glia 18(2):152–160. https://doi.org/10.1002/(sici)1098-1136(199610)18:2%3c152::aid-glia7%3e3.0.co;2-2
Oldham WM, Hamm HE (2008) Heterotrimeric G protein activation by G-protein-coupled receptors. Nat Rev Mol Cell Biol 9(1):60–71. https://doi.org/10.1038/nrm2299
Fredholm BB, IJ AP, Jacobson KA, Klotz KN, Linden J (2001) International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev 53(4):527–552
Corvol JC, Studler JM, Schonn JS, Girault JA, Hervé D (2001) Galpha(olf) is necessary for coupling D1 and A2a receptors to adenylyl cyclase in the striatum. J Neurochem 76(5):1585–1588. https://doi.org/10.1046/j.1471-4159.2001.00201.x
Kull B, Svenningsson P, Fredholm BB (2000) Adenosine A(2A) receptors are colocalized with and activate g(olf) in rat striatum. Mol Pharmacol 58(4):771–777. https://doi.org/10.1124/mol.58.4.771
Hurley JH (1999) Structure, mechanism, and regulation of mammalian adenylyl cyclase. J Biol Chem 274(12):7599–7602. https://doi.org/10.1074/jbc.274.12.7599
Kopperud R, Krakstad C, Selheim F, Døskeland SO (2003) cAMP effector mechanisms. Novel twists for an ‘old’ signaling system. FEBS Lett 546(1):121–126. https://doi.org/10.1016/s0014-5793(03)00563-5
Wainger BJ, DeGennaro M, Santoro B, Siegelbaum SA, Tibbs GR (2001) Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature 411(6839):805–810. https://doi.org/10.1038/35081088
Cargnello M, Roux PP (2011) Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev 75(1):50–83. https://doi.org/10.1128/MMBR.00031-10
Manning BD, Cantley LC (2007) AKT/PKB signaling: navigating downstream. Cell 129(7):1261–1274. https://doi.org/10.1016/j.cell.2007.06.009
Schulte G, Fredholm BB (2003) Signalling from adenosine receptors to mitogen-activated protein kinases. Cell Signal 15(9):813–827. https://doi.org/10.1016/s0898-6568(03)00058-5
Schulte G, Fredholm BB (2000) Human adenosine A(1), A(2A), A(2B), and A(3) receptors expressed in Chinese hamster ovary cells all mediate the phosphorylation of extracellular-regulated kinase 1/2. Mol Pharmacol 58(3):477–482
Migita H, Kominami K, Higashida M, Maruyama R, Tuchida N, McDonald F, Shimada F, Sakurada K (2008) Activation of adenosine A1 receptor-induced neural stem cell proliferation via MEK/ERK and Akt signaling pathways. J Neurosci Res 86(13):2820–2828. https://doi.org/10.1002/jnr.21742
Brust TB, Cayabyab FS, Zhou N, MacVicar BA (2006) p38 mitogen-activated protein kinase contributes to adenosine A1 receptor-mediated synaptic depression in area CA1 of the rat hippocampus. J Neurosci 26(48):12427–12438. https://doi.org/10.1523/JNEUROSCI.4052-06.2006
Brust TB, Cayabyab FS, MacVicar BA (2007) C-Jun N-terminal kinase regulates adenosine A1 receptor-mediated synaptic depression in the rat hippocampus. Neuropharmacol 53(8):906–917. https://doi.org/10.1016/j.neuropharm.2007.09.001
Xie KQ, Zhang LM, Cao Y, Zhu J, Feng LY (2009) Adenosine A(1) receptor-mediated transactivation of the EGF receptor produces a neuroprotective effect on cortical neurons in vitro. Acta Pharmacol Sin 30(7):889–898. https://doi.org/10.1038/aps.2009.80
Gervitz LM, Nalbant D, Williams SC, Fowler JC (2002) Adenosine-mediated activation of Akt/protein kinase B in the rat hippocampus in vitro and in vivo. Neurosci Lett 328(2):175–179. https://doi.org/10.1016/s0304-3940(02)00495-0
Baltos JA, Gregory KJ, White PJ, Sexton PM, Christopoulos A, May LT (2016) Quantification of adenosine A(1) receptor biased agonism: implications for drug discovery. Biochem Pharmacol 99:101–112. https://doi.org/10.1016/j.bcp.2015.11.013
Dickenson JM, Hill SJ (1998) Involvement of G-protein betagamma subunits in coupling the adenosine A1 receptor to phospholipase C in transfected CHO cells. Eur J Pharmacol 355(1):85–93. https://doi.org/10.1016/s0014-2999(98)00468-3
Freund S, Ungerer M, Lohse MJ (1994) A1 adenosine receptors expressed in CHO-cells couple to adenylyl cyclase and to phospholipase C. Naunyn Schmiedebergs Arch Pharmacol 350(1):49–56. https://doi.org/10.1007/BF00180010
Akbar M, Okajima F, Tomura H, Shimegi S, Kondo Y (1994) A single species of A1 adenosine receptor expressed in Chinese hamster ovary cells not only inhibits cAMP accumulation but also stimulates phospholipase C and arachidonate release. Mol Pharmacol 45(5):1036–1042
Che J, Chan ES, Cronstein BN (2007) Adenosine A2A receptor occupancy stimulates collagen expression by hepatic stellate cells via pathways involving protein kinase A, Src, and extracellular signal-regulated kinases 1/2 signaling cascade or p38 mitogen-activated protein kinase signaling pathway. Mol Pharmacol 72(6):1626–1636. https://doi.org/10.1124/mol.107.038760
Chen YC, Huang SH, Wang SM (2008) Adenosine-stimulated adrenal steroidogenesis involves the adenosine A2A and A2B receptors and the Janus kinase 2-mitogen-activated protein kinase kinase-extracellular signal-regulated kinase signaling pathway. Int J Biochem Cell Biol 40(12):2815–2825. https://doi.org/10.1016/j.biocel.2008.05.016
Seidel MG, Klinger M, Freissmuth M, Holler C (1999) Activation of mitogen-activated protein kinase by the A(2A)-adenosine receptor via a rap1-dependent and via a p21(ras)-dependent pathway. J Biol Chem 274(36):25833–25841. https://doi.org/10.1074/jbc.274.36.25833
Sexl V, Mancusi G, Holler C, Gloria-Maercker E, Schutz W, Freissmuth M (1997) Stimulation of the mitogen-activated protein kinase via the A2A-adenosine receptor in primary human endothelial cells. J Biol Chem 272(9):5792–5799. https://doi.org/10.1074/jbc.272.9.5792
Ahmad A, Schaack JB, White CW, Ahmad S (2013) Adenosine A2A receptor-dependent proliferation of pulmonary endothelial cells is mediated through calcium mobilization, PI3-kinase and ERK1/2 pathways. Biochem Biophys Res Commun 434(3):566–571. https://doi.org/10.1016/j.bbrc.2013.03.115
Mori Y, Higuchi M, Masuyama N, Gotoh Y (2004) Adenosine A2A receptor facilitates calcium-dependent protein secretion through the activation of protein kinase A and phosphatidylinositol-3 kinase in PC12 cells. Cell Struct Funct 29(4):101–110. https://doi.org/10.1247/csf.29.101
Perez-Aso M, Fernandez P, Mediero A, Chan ES, Cronstein BN (2014) Adenosine 2A receptor promotes collagen production by human fibroblasts via pathways involving cyclic AMP and AKT but independent of Smad2/3. FASEB J 28(2):802–812. https://doi.org/10.1096/fj.13-241646
Boucher M, Pesant S, Falcao S, de Montigny C, Schampaert E, Cardinal R, Rousseau G (2004) Post-ischemic cardioprotection by A2A adenosine receptors: dependent of phosphatidylinositol 3-kinase pathway. J Cardiovasc Pharmacol 43(3):416–422. https://doi.org/10.1097/00005344-200403000-00013
Giambelluca MS, Pouliot M (2017) Early tyrosine phosphorylation events following adenosine A2A receptor in human neutrophils: identification of regulated pathways. J Leukoc Biol 102(3):829–836. https://doi.org/10.1189/jlb.2VMA1216-517R
Hirano D, Aoki Y, Ogasawara H, Kodama H, Waga I, Sakanaka C, Shimizu T, Nakamura M (1996) Functional coupling of adenosine A2a receptor to inhibition of the mitogen-activated protein kinase cascade in Chinese hamster ovary cells. Biochem J 316(Pt 1):81–86. https://doi.org/10.1042/bj3160081
Chao MV (2003) Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci 4(4):299–309. https://doi.org/10.1038/nrn1078
Wiese S, Jablonka S, Holtmann B, Orel N, Rajagopal R, Chao MV, Sendtner M (2007) Adenosine receptor A2A-R contributes to motoneuron survival by transactivating the tyrosine kinase receptor TrkB. Proc Natl Acad Sci USA 104(43):17210–17215. https://doi.org/10.1073/pnas.0705267104
Fontinha BM, Diogenes MJ, Ribeiro JA, Sebastiao AM (2008) Enhancement of long-term potentiation by brain-derived neurotrophic factor requires adenosine A2A receptor activation by endogenous adenosine. Neuropharmacol 54(6):924–933. https://doi.org/10.1016/j.neuropharm.2008.01.011
Diogenes MJ, Fernandes CC, Sebastiao AM, Ribeiro JA (2004) Activation of adenosine A2A receptor facilitates brain-derived neurotrophic factor modulation of synaptic transmission in hippocampal slices. J Neurosci 24(12):2905–2913. https://doi.org/10.1523/JNEUROSCI.4454-03.2004
Tebano MT, Martire A, Potenza RL, Gro C, Pepponi R, Armida M, Domenici MR, Schwarzschild MA, Chen JF, Popoli P (2008) Adenosine A(2A) receptors are required for normal BDNF levels and BDNF-induced potentiation of synaptic transmission in the mouse hippocampus. J Neurochem 104(1):279–286. https://doi.org/10.1111/j.1471-4159.2007.05046.x
Heese K, Fiebich BL, Bauer J, Otten U (1997) Nerve growth factor (NGF) expression in rat microglia is induced by adenosine A2a-receptors. Neurosci Lett 231(2):83–86. https://doi.org/10.1016/s0304-3940(97)00545-4
Ciccarelli R, Di Iorio P, Bruno V, Battaglia G, D’Alimonte I, D’Onofrio M, Nicoletti F, Caciagli F (1999) Activation of A(1) adenosine or mGlu3 metabotropic glutamate receptors enhances the release of nerve growth factor and S-100beta protein from cultured astrocytes. Glia 27(3):275–281
Haas HL, Selbach O (2000) Functions of neuronal adenosine receptors. Naunyn Schmiedebergs Arch Pharmacol 362(4–5):375–381. https://doi.org/10.1007/s002100000314
Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y (2010) Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 90(1):291–366. https://doi.org/10.1152/physrev.00021.2009
Clark BD, Kurth-Nelson ZL, Newman EA (2009) Adenosine-evoked hyperpolarization of retinal ganglion cells is mediated by G-protein-coupled inwardly rectifying K+ and small conductance Ca2+-activated K+ channel activation. J Neurosci 29(36):11237–11245. https://doi.org/10.1523/JNEUROSCI.2836-09.2009
Kim CS, Johnston D (2015) A1 adenosine receptor-mediated GIRK channels contribute to the resting conductance of CA1 neurons in the dorsal hippocampus. J Neurophysiol 113(7):2511–2523. https://doi.org/10.1152/jn.00951.2014
Umemiya M, Berger AJ (1994) Activation of adenosine A1 and A2 receptors differentially modulates calcium channels and glycinergic synaptic transmission in rat brainstem. Neuron 13(6):1439–1446. https://doi.org/10.1016/0896-6273(94)90429-4
Yawo H, Chuhma N (1993) Preferential inhibition of omega-conotoxin-sensitive presynaptic Ca2+ channels by adenosine autoreceptors. Nature 365(6443):256–258. https://doi.org/10.1038/365256a0
Wu LG, Saggau P (1994) Adenosine inhibits evoked synaptic transmission primarily by reducing presynaptic calcium influx in area CA1 of hippocampus. Neuron 12(5):1139–1148. https://doi.org/10.1016/0896-6273(94)90321-2
Mynlieff M, Beam KG (1994) Adenosine acting at an A1 receptor decreases N-type calcium current in mouse motoneurons. J Neurosci 14(6):3628–3634
Gubitz AK, Widdowson L, Kurokawa M, Kirkpatrick KA, Richardson PJ (1996) Dual signalling by the adenosine A2a receptor involves activation of both N- and P-type calcium channels by different G proteins and protein kinases in the same striatal nerve terminals. J Neurochem 67(1):374–381. https://doi.org/10.1046/j.1471-4159.1996.67010374.x
Li Q, Puro DG (2001) Adenosine activates ATP-sensitive K(+) currents in pericytes of rat retinal microvessels: role of A1 and A2a receptors. Brain Res 907(1–2):93–99. https://doi.org/10.1016/s0006-8993(01)02607-5
Heurteaux C, Lauritzen I, Widmann C, Lazdunski M (1995) Essential role of adenosine, adenosine A1 receptors, and ATP-sensitive K+ channels in cerebral ischemic preconditioning. Proc Natl Acad Sci USA 92(10):4666–4670. https://doi.org/10.1073/pnas.92.10.4666
Popoli P, Betto P, Reggio R, Ricciarello G (1995) Adenosine A2A receptor stimulation enhances striatal extracellular glutamate levels in rats. Eur J Pharmacol 287(2):215–217. https://doi.org/10.1016/0014-2999(95)00679-6
Thompson SM, Haas HL, Gähwiler BH (1992) Comparison of the actions of adenosine at pre- and postsynaptic receptors in the rat hippocampus in vitro. J Physiol 451:347–363. https://doi.org/10.1113/jphysiol.1992.sp019168
Boison D, Chen JF, Fredholm BB (2010) Adenosine signaling and function in glial cells. Cell Death Differ 17(7):1071–1082. https://doi.org/10.1038/cdd.2009.131
Burnstock G (2016) An introduction to the roles of purinergic signalling in neurodegeneration, neuroprotection and neuroregeneration. Neuropharmacol 104:4–17. https://doi.org/10.1016/j.neuropharm.2015.05.031
Woods LT, Ajit D, Camden JM, Erb L, Weisman GA (2016) Purinergic receptors as potential therapeutic targets in Alzheimer’s disease. Neuropharmacol 104:169–179. https://doi.org/10.1016/j.neuropharm.2015.10.031
Alonso-Andres P, Albasanz JL, Ferrer I, Martin M (2018) Purine-related metabolites and their converting enzymes are altered in frontal, parietal and temporal cortex at early stages of Alzheimer’s disease pathology. Brain Pathol 28(6):933–946. https://doi.org/10.1111/bpa.12592
Chang CP, Wu KC, Lin CY, Chern Y (2021) Emerging roles of dysregulated adenosine homeostasis in brain disorders with a specific focus on neurodegenerative diseases. J Biomed Sci 28(1):70. https://doi.org/10.1186/s12929-021-00766-y
Angulo E, Casadó V, Mallol J, Canela EI, Viñals F, Ferrer I, Lluis C, Franco R (2003) A1 adenosine receptors accumulate in neurodegenerative structures in Alzheimer disease and mediate both amyloid precursor protein processing and tau phosphorylation and translocation. Brain Pathol 13(4):440–451. https://doi.org/10.1111/j.1750-3639.2003.tb00475.x
Albasanz JL, Perez S, Barrachina M, Ferrer I, Martín M (2008) Up-regulation of adenosine receptors in the frontal cortex in Alzheimer’s disease. Brain Pathol 18(2):211–219. https://doi.org/10.1111/j.1750-3639.2007.00112.x
Ikeda M, Mackay KB, Dewar D, McCulloch J (1993) Differential alterations in adenosine A1 and kappa 1 opioid receptors in the striatum in Alzheimer’s disease. Brain Res 616(1–2):211–217. https://doi.org/10.1016/0006-8993(93)90211-5
Kalaria RN, Sromek S, Wilcox BJ, Unnerstall JR (1990) Hippocampal adenosine A1 receptors are decreased in Alzheimer’s disease. Neurosci Lett 118(2):257–260. https://doi.org/10.1016/0304-3940(90)90641-l
Fukumitsu N, Ishii K, Kimura Y, Oda K, Hashimoto M, Suzuki M, Ishiwata K (2008) Adenosine A(1) receptors using 8-dicyclopropylmethyl-1-[(11)C]methyl-3-propylxanthine PET in Alzheimer’s disease. Ann Nucl Med 22(10):841–847. https://doi.org/10.1007/s12149-008-0185-5
Carvalho K, Faivre E, Pietrowski MJ, Marques X, Gomez-Murcia V, Deleau A, Huin V, Hansen JN, Kozlov S, Danis C, Temido-Ferreira M, Coelho JE, Meriaux C, Eddarkaoui S, Gras SL, Dumoulin M, Cellai L, Neuro CEBBB, Landrieu I, Chern Y, Hamdane M, Buee L, Boutillier AL, Levi S, Halle A, Lopes LV, Blum D (2019) Exacerbation of C1q dysregulation, synaptic loss and memory deficits in tau pathology linked to neuronal adenosine A2A receptor. Brain 142(11):3636–3654. https://doi.org/10.1093/brain/awz288
Temido-Ferreira M, Ferreira DG, Batalha VL, Marques-Morgado I, Coelho JE, Pereira P, Gomes R, Pinto A, Carvalho S, Canas PM, Cuvelier L, Buee-Scherrer V, Faivre E, Baqi Y, Muller CE, Pimentel J, Schiffmann SN, Buee L, Bader M, Outeiro TF, Blum D, Cunha RA, Marie H, Pousinha PA, Lopes LV (2020) Age-related shift in LTD is dependent on neuronal adenosine A2A receptors interplay with mGluR5 and NMDA receptors. Mol Psychiatry 25(8):1876–1900. https://doi.org/10.1038/s41380-018-0110-9
Merighi S, Battistello E, Casetta I, Gragnaniello D, Poloni TE, Medici V, Cirrincione A, Varani K, Vincenzi F, Borea PA, Gessi S (2021) Upregulation of cortical A2A adenosine receptors is reflected in platelets of patients with Alzheimer’s disease. J Alzheimers Dis 80(3):1105–1117. https://doi.org/10.3233/JAD-201437
Liu Z, Wang F, Tang M, Zhao Y, Wang X (2019) Amyloid beta and tau are involved in sleep disorder in Alzheimer’s disease by orexin A and adenosine A(1) receptor. Int J Mol Med 43(1):435–442. https://doi.org/10.3892/ijmm.2018.3935
Castillo CA, Ballesteros-Yanez I, Leon-Navarro DA, Albasanz JL, Martin M (2021) Early effects of the soluble amyloid beta25–35 peptide in rat cortical neurons: modulation of signal transduction mediated by adenosine and group I metabotropic glutamate receptors. Int J Mol Sci 22(12). https://doi.org/10.3390/ijms22126577
Viana da Silva S, Haberl MG, Zhang P, Bethge P, Lemos C, Goncalves N, Gorlewicz A, Malezieux M, Goncalves FQ, Grosjean N, Blanchet C, Frick A, Nagerl UV, Cunha RA, Mulle C (2016) Early synaptic deficits in the APP/PS1 mouse model of Alzheimer’s disease involve neuronal adenosine A2A receptors. Nat Commun 7:11915. https://doi.org/10.1038/ncomms11915
Espinosa J, Rocha A, Nunes F, Costa MS, Schein V, Kazlauckas V, Kalinine E, Souza DO, Cunha RA, Porciúncula LO (2013) Caffeine consumption prevents memory impairment, neuronal damage, and adenosine A2A receptors upregulation in the hippocampus of a rat model of sporadic dementia. J Alzheimer’s dis : JAD 34(2):509–518. https://doi.org/10.3233/jad-111982
Cao C, Loewenstein DA, Lin X, Zhang C, Wang L, Duara R, Wu Y, Giannini A, Bai G, Cai J, Greig M, Schofield E, Ashok R, Small B, Potter H, Arendash GW (2012) High blood caffeine levels in MCI linked to lack of progression to dementia. J Alzheimers Dis 30(3):559–572. https://doi.org/10.3233/JAD-2012-111781
Lindsay J, Laurin D, Verreault R, Hébert R, Helliwell B, Hill GB, McDowell I (2002) Risk factors for Alzheimer’s disease: a prospective analysis from the Canadian Study of Health and Aging. Am J Epidemiol 156(5):445–453. https://doi.org/10.1093/aje/kwf074
Maia L, de Mendonça A (2002) Does caffeine intake protect from Alzheimer’s disease? Eur J Neurol 9(4):377–382. https://doi.org/10.1046/j.1468-1331.2002.00421.x
Ritchie K, Carrière I, de Mendonça A, Portet F, Dartigues JF, Rouaud O, Barberger-Gateau P, Ancelin ML (2007) The neuroprotective effects of caffeine. Neurol 69(6):536. https://doi.org/10.1212/01.wnl.0000266670.35219.0c
Santos C, Lunet N, Azevedo A, de Mendonça A, Ritchie K, Barros H (2010) Caffeine intake is associated with a lower risk of cognitive decline: a cohort study from Portugal. J Alzheimers Dis 20(Suppl 1):S175-185. https://doi.org/10.3233/jad-2010-091303
Kim JW, Byun MS, Yi D, Lee JH, Jeon SY, Jung G, Lee HN, Sohn BK, Lee JY, Kim YK, Shin SA, Sohn CH, Lee DY, Group KR (2019) Coffee intake and decreased amyloid pathology in human brain. Transl Psychiatry 9(1):270. https://doi.org/10.1038/s41398-019-0604-5
Currais A, Kato K, Canuet L, Ishii R, Tanaka T, Takeda M, Soriano S (2011) Caffeine modulates tau phosphorylation and affects Akt signaling in postmitotic neurons. J Mol Neurosci 43(3):326–332. https://doi.org/10.1007/s12031-010-9444-8
Arendash GW, Mori T, Cao C, Mamcarz M, Runfeldt M, Dickson A, Rezai-Zadeh K, Tane J, Citron BA, Lin X, Echeverria V, Potter H (2009) Caffeine reverses cognitive impairment and decreases brain amyloid-beta levels in aged Alzheimer’s disease mice. J Alzheimer’s dis : JAD 17(3):661–680. https://doi.org/10.3233/jad-2009-1087
Laurent C, Burnouf S, Ferry B, Batalha VL, Coelho JE, Baqi Y, Malik E, Mariciniak E, Parrot S, Van der Jeugd A, Faivre E, Flaten V, Ledent C, D’Hooge R, Sergeant N, Hamdane M, Humez S, Muller CE, Lopes LV, Buee L, Blum D (2016) A2A adenosine receptor deletion is protective in a mouse model of tauopathy. Mol Psychiatry 21(1):97–107. https://doi.org/10.1038/mp.2014.151
Cunha GM, Canas PM, Melo CS, Hockemeyer J, Muller CE, Oliveira CR, Cunha RA (2008) Adenosine A2A receptor blockade prevents memory dysfunction caused by beta-amyloid peptides but not by scopolamine or MK-801. Exp Neurol 210(2):776–781. https://doi.org/10.1016/j.expneurol.2007.11.013
Canas PM, Porciúncula LO, Cunha GM, Silva CG, Machado NJ, Oliveira JM, Oliveira CR, Cunha RA (2009) Adenosine A2A receptor blockade prevents synaptotoxicity and memory dysfunction caused by beta-amyloid peptides via p38 mitogen-activated protein kinase pathway. J Neurosci 29(47):14741–14751. https://doi.org/10.1523/jneurosci.3728-09.2009
Dall’Igna OP, Porciúncula LO, Souza DO, Cunha RA, Lara DR (2003) Neuroprotection by caffeine and adenosine A2A receptor blockade of beta-amyloid neurotoxicity. Br J Pharmacol 138(7):1207–1209. https://doi.org/10.1038/sj.bjp.0705185
Dennissen FJ, Anglada-Huguet M, Sydow A, Mandelkow E, Mandelkow EM (2016) Adenosine A1 receptor antagonist rolofylline alleviates axonopathy caused by human Tau DeltaK280. Proc Natl Acad Sci USA 113(41):11597–11602. https://doi.org/10.1073/pnas.1603119113
Cunha RA (2005) Neuroprotection by adenosine in the brain: from A(1) receptor activation to A (2A) receptor blockade. Purinergic Signal 1(2):111–134. https://doi.org/10.1007/s11302-005-0649-1
Paul S, Elsinga PH, Ishiwata K, Dierckx RA, van Waarde A (2011) Adenosine A(1) receptors in the central nervous system: their functions in health and disease, and possible elucidation by PET imaging. Curr Med Chem 18(31):4820–4835. https://doi.org/10.2174/092986711797535335
Matos M, Augusto E, Machado NJ, dos Santos-Rodrigues A, Cunha RA, Agostinho P (2012) Astrocytic adenosine A2A receptors control the amyloid-beta peptide-induced decrease of glutamate uptake. J Alzheimers Dis 31(3):555–567. https://doi.org/10.3233/JAD-2012-120469
Orr AG, Hsiao EC, Wang MM, Ho K, Kim DH, Wang X, Guo W, Kang J, Yu GQ, Adame A, Devidze N, Dubal DB, Masliah E, Conklin BR, Mucke L (2015) Astrocytic adenosine receptor A2A and Gs-coupled signaling regulate memory. Nat Neurosci 18(3):423–434. https://doi.org/10.1038/nn.3930
Van Zeller M, Dias D, Sebastiao AM, Valente CA (2021) NLRP3 inflammasome: a starring role in amyloid-beta- and tau-driven pathological events in Alzheimer’s disease. J Alzheimers Dis 83(3):939–961. https://doi.org/10.3233/JAD-210268
Merighi S, Nigro M, Travagli A, Pasquini S, Borea PA, Varani K, Vincenzi F, Gessi S (2022) A2A adenosine receptor: a possible therapeutic target for Alzheimer’s disease by regulating NLRP3 inflammasome activity? Int J Mol Sci 23(9). https://doi.org/10.3390/ijms23095056
Ouyang X, Ghani A, Malik A, Wilder T, Colegio OR, Flavell RA, Cronstein BN, Mehal WZ (2013) Adenosine is required for sustained inflammasome activation via the A(2)A receptor and the HIF-1alpha pathway. Nat Commun 4:2909. https://doi.org/10.1038/ncomms3909
Zhao W, Ma L, Cai C, Gong X (2019) Caffeine inhibits NLRP3 inflammasome activation by suppressing MAPK/NF-kappaB and A2aR signaling in LPS-induced THP-1 macrophages. Int J Biol Sci 15(8):1571–1581. https://doi.org/10.7150/ijbs.34211
Kovacs EG, Alatshan A, Budai MM, Czimmerer Z, Biro E, Benko S (2021) Caffeine has different immunomodulatory effect on the cytokine expression and NLRP3 inflammasome function in various human macrophage subpopulations. Nutr 13(7). https://doi.org/10.3390/nu13072409
Du H, Tan Y, Li CH, Zhao Y, Li P, Ning YL, Gao RB, Wang B, Peng Y, Tan SW, Huang ZZ, Chen X, Yang N, Shan FB, Xiong RP, Zhou YG (2022) High glutamate concentration reverses the inhibitory effect of microglial adenosine 2A receptor on NLRP3 inflammasome assembly and activation. Neurosci Lett 769:136431. https://doi.org/10.1016/j.neulet.2021.136431
Yang L, Yu X, Zhang Y, Liu N, Xue X, Fu J (2022) Caffeine treatment started before injury reduces hypoxic-ischemic white-matter damage in neonatal rats by regulating phenotypic microglia polarization. Pediatr Res. https://doi.org/10.1038/s41390-021-01924-6
Wang HQ, Song KY, Feng JZ, Huang SY, Guo XM, Zhang L, Zhang G, Huo YC, Zhang RR, Ma Y, Hu QZ, Qin XY (2022) Caffeine inhibits activation of the NLRP3 inflammasome via autophagy to attenuate microglia-mediated neuroinflammation in experimental autoimmune encephalomyelitis. J Mol Neurosci 72(1):97–112. https://doi.org/10.1007/s12031-021-01894-8
Cunha RA (2016) How does adenosine control neuronal dysfunction and neurodegeneration? J Neurochem 139(6):1019–1055. https://doi.org/10.1111/jnc.13724
Orr AG, Lo I, Schumacher H, Ho K, Gill M, Guo W, Kim DH, Knox A, Saito T, Saido TC, Simms J, Toddes C, Wang X, Yu GQ, Mucke L (2018) Istradefylline reduces memory deficits in aging mice with amyloid pathology. Neurobiol Dis 110:29–36. https://doi.org/10.1016/j.nbd.2017.10.014
Navarro G, Borroto-Escuela DO, Fuxe K, Franco R (2016) Purinergic signaling in Parkinson’s disease. Relevance for treat Neuropharmacol 104:161–168. https://doi.org/10.1016/j.neuropharm.2015.07.024
Marzagalli R, Castorina A (2015) The seeming paradox of adenosine receptors as targets for the treatment of Alzheimer’s disease: agonists or antagonists? Neural Regen Res 10(2):205–207. https://doi.org/10.4103/1673-5374.152370
Headrick JP, Ashton KJ, Rose’meyer RB, Peart JN (2013) Cardiovascular adenosine receptors: expression, actions and interactions. Pharmacol Ther 140(1):92–111. https://doi.org/10.1016/j.pharmthera.2013.06.002
Teerlink JR, Iragui VJ, Mohr JP, Carson PE, Hauptman PJ, Lovett DH, Miller AB, Pina IL, Thomson S, Varosy PD, Zile MR, Cleland JG, Givertz MM, Metra M, Ponikowski P, Voors AA, Davison BA, Cotter G, Wolko D, Delucca P, Salerno CM, Mansoor GA, Dittrich H, O’Connor CM, Massie BM (2012) The safety of an adenosine A(1)-receptor antagonist, rolofylline, in patients with acute heart failure and renal impairment: findings from PROTECT. Drug Saf 35(3):233–244. https://doi.org/10.2165/11594680-000000000-00000
Bjorness TE, Greene RW (2009) Adenosine and sleep. Curr Neuropharmacol 7(3):238–245. https://doi.org/10.2174/157015909789152182
Chen JF, Cunha RA (2020) The belated US FDA approval of the adenosine A2A receptor antagonist istradefylline for treatment of Parkinson’s disease. Purinergic Signal 16(2):167–174. https://doi.org/10.1007/s11302-020-09694-2
May LT, Leach K, Sexton PM, Christopoulos A (2007) Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol 47:1–51. https://doi.org/10.1146/annurev.pharmtox.47.120505.105159
Christopoulos A, Changeux JP, Catterall WA, Fabbro D, Burris TP, Cidlowski JA, Olsen RW, Peters JA, Neubig RR, Pin JP, Sexton PM, Kenakin TP, Ehlert FJ, Spedding M, Langmead CJ (2014) International Union of Basic and Clinical Pharmacology. XC. multisite pharmacology: recommendations for the nomenclature of receptor allosterism and allosteric ligands. Pharmacol Rev 66(4):918–947. https://doi.org/10.1124/pr.114.008862
Wootten D, Christopoulos A, Sexton PM (2013) Emerging paradigms in GPCR allostery: implications for drug discovery. Nat Rev Drug Discov 12(8):630–644. https://doi.org/10.1038/nrd4052
Vecchio EA, Baltos JA, Nguyen ATN, Christopoulos A, White PJ, May LT (2018) New paradigms in adenosine receptor pharmacology: allostery, oligomerization and biased agonism. Br J Pharmacol 175(21):4036–4046. https://doi.org/10.1111/bph.14337
Bruns RF, Fergus JH (1990) Allosteric enhancement of adenosine A1 receptor binding and function by 2-amino-3-benzoylthiophenes. Mol Pharmacol 38(6):939–949
Göblyös A (1808) Ijzerman AP (2011) Allosteric modulation of adenosine receptors. Biochim Biophys Acta 5:1309–1318. https://doi.org/10.1016/j.bbamem.2010.06.013
Giorgi I, Biagi G, Bianucci AM, Borghini A, Livi O, Leonardi M, Pietra D, Calderone V, Martelli A (2008) N6–1,3-diphenylurea derivatives of 2-phenyl-9-benzyladenines and 8-azaadenines: synthesis and biological evaluation as allosteric modulators of A2A adenosine receptors. Eur J Med Chem 43(8):1639–1647. https://doi.org/10.1016/j.ejmech.2007.10.021
Chen D, Errey JC, Heitman LH, Marshall FH, Ijzerman AP, Siegal G (2012) Fragment screening of GPCRs using biophysical methods: identification of ligands of the adenosine A(2A) receptor with novel biological activity. ACS Chem Biol 7(12):2064–2073. https://doi.org/10.1021/cb300436c
Draper-Joyce CJ, Bhola R, Wang J, Bhattarai A, Nguyen ATN, Cowie-Kent I, O’Sullivan K, Chia LY, Venugopal H, Valant C, Thal DM, Wootten D, Panel N, Carlsson J, Christie MJ, White PJ, Scammells P, May LT, Sexton PM, Danev R, Miao Y, Glukhova A, Imlach WL, Christopoulos A (2021) Positive allosteric mechanisms of adenosine A1 receptor-mediated analgesia. Nat 597(7877):571–576. https://doi.org/10.1038/s41586-021-03897-2
Draper-Joyce CJ, Khoshouei M, Thal DM, Liang YL, Nguyen ATN, Furness SGB, Venugopal H, Baltos JA, Plitzko JM, Danev R, Baumeister W, May LT, Wootten D, Sexton PM, Glukhova A, Christopoulos A (2018) Structure of the adenosine-bound human adenosine A1 receptor-Gi complex. Nature 558(7711):559–563. https://doi.org/10.1038/s41586-018-0236-6
Garcia-Nafria J, Lee Y, Bai X, Carpenter B, Tate CG (2018) Cryo-EM structure of the adenosine A2A receptor coupled to an engineered heterotrimeric G protein. Elife 7.https://doi.org/10.7554/eLife.35946
Garcia-Nafria J, Tate CG (2019) Cryo-EM structures of GPCRs coupled to Gs, Gi and Go. Mol Cell Endocrinol 488:1–13. https://doi.org/10.1016/j.mce.2019.02.006
Ferré S, Baler R, Bouvier M, Caron MG, Devi LA, Durroux T, Fuxe K, George SR, Javitch JA, Lohse MJ, Mackie K, Milligan G, Pfleger KD, Pin JP, Volkow ND, Waldhoer M, Woods AS, Franco R (2009) Building a new conceptual framework for receptor heteromers. Nat Chem Biol 5(3):131–134. https://doi.org/10.1038/nchembio0309-131
Ferré S, Casadó V, Devi LA, Filizola M, Jockers R, Lohse MJ, Milligan G, Pin JP, Guitart X (2014) G protein-coupled receptor oligomerization revisited: functional and pharmacological perspectives. Pharmacol Rev 66(2):413–434. https://doi.org/10.1124/pr.113.008052
Briddon SJ, Gandía J, Amaral OB, Ferré S, Lluís C, Franco R, Hill SJ, Ciruela F (2008) Plasma membrane diffusion of G protein-coupled receptor oligomers. Biochim Biophys Acta 1783(12):2262–2268. https://doi.org/10.1016/j.bbamcr.2008.07.006
Canals M, Burgueño J, Marcellino D, Cabello N, Canela EI, Mallol J, Agnati L, Ferré S, Bouvier M, Fuxe K, Ciruela F, Lluis C, Franco R (2004) Homodimerization of adenosine A2A receptors: qualitative and quantitative assessment by fluorescence and bioluminescence energy transfer. J Neurochem 88(3):726–734. https://doi.org/10.1046/j.1471-4159.2003.02200.x
Gandia J, Galino J, Amaral OB, Soriano A, Lluís C, Franco R, Ciruela F (2008) Detection of higher-order G protein-coupled receptor oligomers by a combined BRET-BiFC technique. FEBS Lett 582(20):2979–2984. https://doi.org/10.1016/j.febslet.2008.07.045
Franco R, Cordomi A, Llinas Del Torrent C, Lillo A, Serrano-Marin J, Navarro G, Pardo L (2021) Structure and function of adenosine receptor heteromers. Cell Mol Life Sci 78(8):3957–3968. https://doi.org/10.1007/s00018-021-03761-6
Pin JP, Neubig R, Bouvier M, Devi L, Filizola M, Javitch JA, Lohse MJ, Milligan G, Palczewski K, Parmentier M, Spedding M (2007) International Union of Basic and Clinical Pharmacology. LXVII. Recommendations for the recognition and nomenclature of G protein-coupled receptor heteromultimers. Pharmacol Rev 59(1):5–13. https://doi.org/10.1124/pr.59.1.5
Kenakin T, Miller LJ (2010) Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev 62(2):265–304. https://doi.org/10.1124/pr.108.000992
Ferre S, Quiroz C, Woods AS, Cunha R, Popoli P, Ciruela F, Lluis C, Franco R, Azdad K, Schiffmann SN (2008) An update on adenosine A2A-dopamine D2 receptor interactions: implications for the function of G protein-coupled receptors. Curr Pharm Des 14(15):1468–1474. https://doi.org/10.2174/138161208784480108
Orru M, Bakešová J, Brugarolas M, Quiroz C, Beaumont V, Goldberg SR, Lluís C, Cortés A, Franco R, Casadó V, Canela EI, Ferré S (2011) Striatal pre- and postsynaptic profile of adenosine A(2A) receptor antagonists. PLoS ONE 6(1):e16088. https://doi.org/10.1371/journal.pone.0016088
Jorg M, May LT, Mak FS, Lee KC, Miller ND, Scammells PJ, Capuano B (2015) Synthesis and pharmacological evaluation of dual acting ligands targeting the adenosine A2A and dopamine D2 receptors for the potential treatment of Parkinson’s disease. J Med Chem 58(2):718–738. https://doi.org/10.1021/jm501254d
Pulido D, Casado-Anguera V, Gomez-Autet M, Llopart N, Moreno E, Casajuana-Martin N, Ferre S, Pardo L, Casado V, Royo M (2022) Heterobivalent ligand for the adenosine A2A-dopamine D2 receptor heteromer. J Med Chem 65(1):616–632. https://doi.org/10.1021/acs.jmedchem.1c01763
Soriano A, Ventura R, Molero A, Hoen R, Casadó V, Cortés A, Fanelli F, Albericio F, Lluís C, Franco R, Royo M (2009) Adenosine A2A receptor-antagonist/dopamine D2 receptor-agonist bivalent ligands as pharmacological tools to detect A2A–D2 receptor heteromers. J Med Chem 52(18):5590–5602. https://doi.org/10.1021/jm900298c
Borroto-Escuela DO, Carlsson J, Ambrogini P, Narváez M, Wydra K, Tarakanov AO, Li X, Millón C, Ferraro L, Cuppini R, Tanganelli S, Liu F, Filip M, Diaz-Cabiale Z, Fuxe K (2017) Understanding the role of GPCR heteroreceptor complexes in modulating the brain networks in health and disease. Front Cell Neurosci 11(37). https://doi.org/10.3389/fncel.2017.00037
Ferré S, Franco R (2010) Oligomerization of G-protein-coupled receptors: a reality. Curr Op Pharmacol 10(1):1–5. https://doi.org/10.1016/j.coph.2009.11.002
Cristovao-Ferreira S, Navarro G, Brugarolas M, Perez-Capote K, Vaz SH, Fattorini G, Conti F, Lluis C, Ribeiro JA, McCormick PJ, Casado V, Franco R, Sebastiao AM (2013) A1R–A2AR heteromers coupled to Gs and G i/0 proteins modulate GABA transport into astrocytes. Purinergic Signal 9(3):433–449. https://doi.org/10.1007/s11302-013-9364-5
Ciruela F, Casadó V, Rodrigues RJ, Luján R, Burgueño J, Canals M, Borycz J, Rebola N, Goldberg SR, Mallol J, Cortés A, Canela EI, López-Giménez JF, Milligan G, Lluis C, Cunha RA, Ferré S, Franco R (2006) Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1–A2A receptor heteromers. J Neurosci 26(7):2080–2087. https://doi.org/10.1523/jneurosci.3574-05.2006
Lillo A, Raich I, Lillo J, Perez-Olives C, Navarro G, Franco R (2022) Expression of the adenosine A2A-A3 receptor heteromer in different brain regions and marked upregulation in the microglia of the transgenic APPSw,Ind Alzheimer’s disease model. Biomed 10(2). https://doi.org/10.3390/biomedicines10020214
Farr SA, Cuzzocrea S, Esposito E, Campolo M, Niehoff ML, Doyle TM, Salvemini D (2020) Adenosine A3 receptor as a novel therapeutic target to reduce secondary events and improve neurocognitive functions following traumatic brain injury. J Neuroinflammation 17(1):339. https://doi.org/10.1186/s12974-020-02009-7
Chen GJ, Harvey BK, Shen H, Chou J, Victor A, Wang Y (2006) Activation of adenosine A3 receptors reduces ischemic brain injury in rodents. J Neurosci Res 84(8):1848–1855. https://doi.org/10.1002/jnr.21071
Franco R, Rivas-Santisteban R, Casanovas M, Lillo A, Saura CA, Navarro G (2020) Adenosine A2A receptor antagonists affects NMDA glutamate receptor function. Potential to Address Neurodegeneration in Alzheimer's Disease. Cells 9(5). https://doi.org/10.3390/cells9051075
Krania P, Dimou E, Bantouna M, Kouvaros S, Tsiamaki E, Papatheodoropoulos C, Sarantis K, Angelatou F (2018) Adenosine A2A receptors are required for glutamate mGluR5- and dopamine D1 receptor-evoked ERK1/2 phosphorylation in rat hippocampus: involvement of NMDA receptor. J Neurochem 145(3):217–231. https://doi.org/10.1111/jnc.14268
McNeill SM, Baltos JA, White PJ, May LT (2021) Biased agonism at adenosine receptors. Cell Signal 82:109954. https://doi.org/10.1016/j.cellsig.2021.109954
Ciruela F, Escriche M, Burgueno J, Angulo E, Casado V, Soloviev MM, Canela EI, Mallol J, Chan WY, Lluis C, McIlhinney RA, Franco R (2001) Metabotropic glutamate 1alpha and adenosine A1 receptors assemble into functionally interacting complexes. J Biol Chem 276(21):18345–18351. https://doi.org/10.1074/jbc.M006960200
Toms NJ, Roberts PJ (1999) Group 1 mGlu receptors elevate [Ca2+]i in rat cultured cortical type 2 astrocytes: [Ca2+]i synergy with adenosine A1 receptors. Neuropharmacology 38(10):1511–1517. https://doi.org/10.1016/s0028-3908(99)00090-8
Yoshioka K, Hosoda R, Kuroda Y, Nakata H (2002) Hetero-oligomerization of adenosine A1 receptors with P2Y1 receptors in rat brains. FEBS Lett 531(2):299–303. https://doi.org/10.1016/S0014-5793(02)03540-8
Fredholm BB, Assender JW, Irenius E, Kodama N, Saito N (2003) Synergistic effects of adenosine A1 and P2Y receptor stimulation on calcium mobilization and PKC translocation in DDT1 MF-2 cells. Cell Mol Neurobiol 23(3):379–400. https://doi.org/10.1023/A:1023644822539
Tonazzini I, Trincavelli ML, Montali M, Martini C (2008) Regulation of A1 adenosine receptor functioning induced by P2Y1 purinergic receptor activation in human astroglial cells. J Neurosci Res 86(13):2857–2866. https://doi.org/10.1002/jnr.21727
Tonazzini I, Trincavelli ML, Storm-Mathisen J, Martini C, Bergersen LH (2007) Co-localization and functional cross-talk between A1 and P2Y1 purine receptors in rat hippocampus. Eur J Neurosci 26(4):890–902. https://doi.org/10.1111/j.1460-9568.2007.05697.x
Lillo A, Martinez-Pinilla E, Reyes-Resina I, Navarro G, Franco R (2020) Adenosine A2A and A3 receptors are able to interact with each other. A further piece in the puzzle of adenosine receptor-mediated signaling. Int J Mol Sci 21(14). https://doi.org/10.3390/ijms21145070
Ferré S, Karcz-Kubicha M, Hope BT, Popoli P, Burgueño J, Gutiérrez MA, Casadó V, Fuxe K, Goldberg SR, Lluis C, Franco R, Ciruela F (2002) Synergistic interaction between adenosine A2A and glutamate mGlu5 receptors: implications for striatal neuronal function. Proc Natl Acad Sci USA 99(18):11940–11945. https://doi.org/10.1073/pnas.172393799
Adams CL, Cowen MS, Short JL, Lawrence AJ (2008) Combined antagonism of glutamate mGlu5 and adenosine A2A receptors interact to regulate alcohol-seeking in rats. Int J Neuropsychopharmacol 11(2):229–241. https://doi.org/10.1017/S1461145707007845
Bogenpohl JW, Ritter SL, Hall RA, Smith Y (2012) Adenosine A2A receptor in the monkey basal ganglia: ultrastructural localization and colocalization with the metabotropic glutamate receptor 5 in the striatum. J Comp Neurol 520(3):570–589. https://doi.org/10.1002/cne.22751
Nishi A, Liu F, Matsuyama S, Hamada M, Higashi H, Nairn AC, Greengard P (2003) Metabotropic mGlu5 receptors regulate adenosine A2A receptor signaling. Proc Natl Acad Sci USA 100(3):1322–1327. https://doi.org/10.1073/pnas.0237126100
Kachroo A, Orlando LR, Grandy DK, Chen JF, Young AB, Schwarzschild MA (2005) Interactions between metabotropic glutamate 5 and adenosine A2A receptors in normal and parkinsonian mice. J Neurosci 25(45):10414–10419. https://doi.org/10.1523/jneurosci.3660-05.2005
Hillion J, Canals M, Torvinen M, Casado V, Scott R, Terasmaa A, Hansson A, Watson S, Olah ME, Mallol J, Canela EI, Zoli M, Agnati LF, Ibanez CF, Lluis C, Franco R, Ferre S, Fuxe K (2002) Coaggregation, cointernalization, and codesensitization of adenosine A2A receptors and dopamine D2 receptors. J Biol Chem 277(20):18091–18097. https://doi.org/10.1074/jbc.M107731200
Trincavelli ML, Daniele S, Orlandini E, Navarro G, Casadó V, Giacomelli C, Nencetti S, Nuti E, Macchia M, Huebner H, Gmeiner P, Rossello A, Lluís C, Martini C (2012) A new D2 dopamine receptor agonist allosterically modulates A(2A) adenosine receptor signalling by interacting with the A(2A)/D2 receptor heteromer. Cell Signal 24(4):951–960. https://doi.org/10.1016/j.cellsig.2011.12.018
Canals M, Marcellino D, Fanelli F, Ciruela F, de Benedetti P, Goldberg SR, Neve K, Fuxe K, Agnati LF, Woods AS, Ferré S, Lluis C, Bouvier M, Franco R (2003) Adenosine A2A-dopamine D2 receptor-receptor heteromerization: qualitative and quantitative assessment by fluorescence and bioluminescence energy transfer. J Biol Chem 278(47):46741–46749. https://doi.org/10.1074/jbc.M306451200
Kamiya T, Saitoh O, Yoshioka K, Nakata H (2003) Oligomerization of adenosine A2A and dopamine D2 receptors in living cells. Biochem Biophys Res Commun 306(2):544–549. https://doi.org/10.1016/s0006-291x(03)00991-4
Trifilieff P, Rives ML, Urizar E, Piskorowski RA, Vishwasrao HD, Castrillon J, Schmauss C, Slättman M, Gullberg M, Javitch JA (2011) Detection of antigen interactions ex vivo by proximity ligation assay: endogenous dopamine D2-adenosine A2A receptor complexes in the striatum. Biotech 51(2):111–118. https://doi.org/10.2144/000113719
Tanganelli S, Sandager Nielsen K, Ferraro L, Antonelli T, Kehr J, Franco R, Ferré S, Agnati LF, Fuxe K, Scheel-Krüger J (2004) Striatal plasticity at the network level. Focus on adenosine A2A and D2 interactions in models of Parkinson’s Disease. Parkinsonism Relat Disord 10(5):273–280. https://doi.org/10.1016/j.parkreldis.2004.02.015
Carriba P, Ortiz O, Patkar K, Justinova Z, Stroik J, Themann A, Müller C, Woods AS, Hope BT, Ciruela F, Casadó V, Canela EI, Lluis C, Goldberg SR, Moratalla R, Franco R, Ferré S (2007) Striatal adenosine A2A and cannabinoid CB1 receptors form functional heteromeric complexes that mediate the motor effects of cannabinoids. Neuropsychopharmacol 32(11):2249–2259. https://doi.org/10.1038/sj.npp.1301375
Soria G, Castañé A, Berrendero F, Ledent C, Parmentier M, Maldonado R, Valverde O (2004) Adenosine A2A receptors are involved in physical dependence and place conditioning induced by THC. Eur J Neurosci 20(8):2203–2213. https://doi.org/10.1111/j.1460-9568.2004.03682.x
Moreno E, Chiarlone A, Medrano M, Puigdellívol M, Bibic L, Howell LA, Resel E, Puente N, Casarejos MJ, Perucho J, Botta J, Suelves N, Ciruela F, Ginés S, Galve-Roperh I, Casadó V, Grandes P, Lutz B, Monory K, Canela EI, Lluís C, McCormick PJ, Guzmán M (2017) Singular location and signaling profile of adenosine A2A-cannabinoid CB1 receptor heteromers in the dorsal striatum. Neuropsychopharmacol 43(5):964–977. https://doi.org/10.1038/npp.2017.12
Lerner TN, Horne EA, Stella N, Kreitzer AC (2010) Endocannabinoid signaling mediates psychomotor activation by adenosine A2A antagonists. J Neurosci 30(6):2160–2164. https://doi.org/10.1523/jneurosci.5844-09.2010
Justinová Z, Ferré S, Redhi GH, Mascia P, Stroik J, Quarta D, Yasar S, Müller CE, Franco R, Goldberg SR (2011) Reinforcing and neurochemical effects of cannabinoid CB1 receptor agonists, but not cocaine, are altered by an adenosine A2A receptor antagonist. Addict Biol 16(3):405–415. https://doi.org/10.1111/j.1369-1600.2010.00258.x
Jack CR Jr, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, Shaw LM, Vemuri P, Wiste HJ, Weigand SD, Lesnick TG, Pankratz VS, Donohue MC, Trojanowski JQ (2013) Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 12(2):207–216. https://doi.org/10.1016/S1474-4422(12)70291-0
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. Research on AR signalling in Dr Gregory’s lab was supported by an Australian Research Council Future Fellowship (FT170100392) and a National Health and Medical Research Council (Australia) project grant: APP1123722. Research on AR signalling in Dr May’s lab was supported by a National Heart Foundation Future Leader Fellowship (101857) and National Health and Medical Research Council (Australia) project grant: APP1147291. Ongoing research into GPCR signalling in AD and AR signalling is supported by NHMRC Ideas grants APP2002947 and APP2013629, respectively. P.NH.T was supported by an Australian Government Research Training Program (RTP) scholarship.
Author information
Authors and Affiliations
Contributions
P.N.H.T., J.B. and S.D.H. wrote the manuscript. P.N.H.T. prepared Figs. 1, 2, and 4. S.D.H. prepared Fig. 3. P.N.H.T. and K.J.G. compiled Table 1. All authors contributed to the design of the manuscript. All authors critically edited and reviewed the manuscript.
Corresponding authors
Ethics declarations
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Trinh, P.N.H., Baltos, JA., Hellyer, S.D. et al. Adenosine receptor signalling in Alzheimer’s disease. Purinergic Signalling 18, 359–381 (2022). https://doi.org/10.1007/s11302-022-09883-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-022-09883-1