
Abstract
Systemic sclerosis (SSc) is a systemic autoimmune connective tissue disease in which the combination of microvasculopathy, inflammation, autoimmunity, and fibrosis leads to severe skin- and organ-based complications. Contributing substantially to the high morbidity of SSc, peripheral microvascular abnormalities are one of the earliest hallmarks of the disease pathophysiology, clinically manifesting in severe ischemic complications including digital ulcers. In SSc, the vascular disease is characterized by a massive destructive and proliferative vasculopathy preceding the development of dermal and visceral fibrosis. So far, the molecular mechanisms leading to the observed vascular changes have not been completely elucidated. However, with the help of appropriate in vitro and in vivo models, endothelial cell injury and apoptosis, an imbalance of pro- and anti-angiogenic factors, and an insufficient angiogenesis and vasculogenesis have been identified as pathogenic key factors.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Systemic sclerosis
- Scleroderma
- Microangiopathy
- Vasculopathy
- Digital ulcers
- Angiogenesis
- Vasculogenesis
- Endothelial cells
- Apoptosis
- Endothelial progenitor cells
- Animal models
Introduction
Systemic sclerosis (SSc) is a systemic autoimmune connective tissue disease in which the combination of microvasculopathy, inflammation, and autoimmunity leads to fibrosis of the skin and the internal organs. Microvascular abnormalities are one of the earliest hallmarks of the disease, clinically manifesting in Raynaud’s phenomenon and ischemic complications including digital ulcers [1, 2], which contribute substantially to the high disease morbidity of SSc (Fig. 4.1a) [3, 4].

Clinical manifestations of the SSc microvascular disease. (a) Fingertip ulcers with signs of necrosis. (b) Nailfold capillaroscopy showing the formation of irregularly shaped dilated and bushy capillaries
In SSc, microvascular injury is characterized by a progressive reduction of capillary density, the formation of an irregular and chaotic capillary architecture (Fig. 4.1b) [5,6,7], and a proliferative vasculopathy with intimal hyperplasia, extracellular matrix (ECM) deposition, luminal narrowing, and finally vessel occlusion [8,9,10].
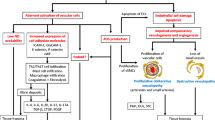
So far, the initiating events leading to the vascular dysfunction in SSc have not been elucidated. However, endothelial cell (EC) injury and apoptosis are regarded as one of the earliest and most central events in the initiation of SSc microvasculopathy [11, 12]. Furthermore, an impaired angiogenic and vasculogenic response were identified to be involved in the disease pathophysiology. An overview about the pathophysiologic mechanisms leading to the SSc vascular disease is illustrated in Fig. 4.2.

Overview of the vascular mechanisms of the SSc microvasculopathy. This figure summarizes the molecular and cellular mechanisms of the peripheral SSc microvasculopathy (see text for explanation). AECA (anti-endothelial antibodies), Ang1/2 (angiopoietin 1/2), anti-AT1R (anti-angiotensin receptor type-1), anti-ETAR (anti-endothelin receptor type A), eNOS (endothelial nitric oxide synthase), EPC (endothelial progenitor cell), ET-1 (endothelin-1), ET-R (endothelin receptor), NO (nitric oxide), PTX3 (pentraxin 3), uPA (urokinase-type plasminogen activator), uPAR (urokinase-type plasminogen activator receptor), MMP12 (matrix metalloproteinase 12), VEGF (vascular endothelial growth factor), VEGFR2 (vascular endothelial growth factor receptor 2), vWF (von Willebrand factor)
In the following, we will discuss the cellular and molecular key players as well as animal models of the SSc microvasculopathy. We will focus on the peripheral vascular mechanisms, whereas the mechanisms underlying pulmonary arterial hypertension (PAH) and scleroderma renal crisis are discussed elsewhere.
Role of the Endothelium
Apart from functioning as a barrier, the endothelium exerts major physiological effects including endocrine functions, regulation of vascular tone, platelet aggregation and leucocyte adhesion, inhibition of deposition of ECM proteins, and regulation of proliferation of vascular smooth muscle cells (VSMC) [1]. Given the pleiotropic effects, endothelial dysfunction can have severe pathological consequences.
In SSc, endothelial dysfunction is morphologically reflected by the formation of large gaps between ECs, vacuolization of EC cytoplasm, and the thickening of the capillary basement membrane [10, 13, 14]. Additionally, it manifests in increased serum levels of von Willebrand factor (vWF) and endothelin-1 (ET-1) as well as reduced levels of nitric oxide (NO) and endothelial NO synthase (eNOS) gene expression, changing the endothelial functional profile from a protective vasodilating to a vasoconstrictive cell type [1, 14]. ET-1 is a potent VSMC and fibroblast mitogen [15,16,17], whereas NO strongly inhibits VSMC proliferation [18]. Thus, in the presence of EC injury, the proliferation of VSMC is favored causing neointimal hyperplasia with luminal narrowing and ultimately vessel obliteration [10, 19].
Furthermore, the expression of cell adhesion molecules is significantly increased on the EC surface, facilitating the adhesion of inflammatory cells and subsequent tissue invasion [19], eventually leading to the characteristic histopathological feature of perivascular tissue infiltrates observed in SSc patients [20].
In addition to EC dysfunction, EC apoptosis is a central event in the pathogenesis of vasculopathy in SSc, which was firstly described in the University of California at Davis (UCD) lines 200/206 chicken model [12]. The triggering events causing EC cell death are not yet identified; however many possible pathogenic mechanisms are proposed including viral triggers, cytotoxic T cells, antibody-dependent cellular cytotoxicity, anti-endothelial antibodies (AECAs), and ischemia-reperfusion injury [11].
Depending on patient selection and detection techniques, AECAs can be detected in 22–86% of sera from SSc patients [21, 22]. These autoantibodies were shown to induce EC apoptosis in vitro and in vivo. Incubation of ECs with AECAs positive sera from SSc patients resulted in induction of apoptosis, whereas incubation with sera from healthy donors did not show an effect [23, 24]. In vivo confirmation was performed using the UCD-200/206 chicken model. AECA isolated from sera from UCD-200 animals were able to bind to ECs and to induce apoptosis in normal chicken embryos [25]. However, the molecular mechanism leading to EC apoptosis are widely unknown and need to be further elucidated. Sgonc et al. proposed that EC apoptosis is mediated via the FAS (CD95) pathway as evidenced by the inhibition of apoptosis induction using an anti-Fas ligand antibody [24]. In contrast, Bordron et al. reported that EC apoptosis occurs via FAS-independent pathways [23].
Underscoring the role of AECA in digital ulcers, the presence of AECA antibodies in SSc patients correlates significantly with an increase in the incidence of digital ischemia [26, 27]. However, the epitopes targeted by AECA are still unknown, and the presence of these autoantibodies is not restricted exclusively to SSc. AECA are additionally detected in other autoimmune diseases including systemic vasculitis and systemic lupus erythematosus (SLE) [28]. Currently, it is under debate whether the occurrence of AECA is an epiphenomenon associated with vascular injury or whether AECA have a pathogenic role in SSc [21, 29].
Apart from AECA , stimulating autoantibodies targeting the endothelin receptor type A (ETAR) and the angiotensin receptor type-1 (AT1R) expressed on ECs were implicated in the disease pathophysiology, especially in the development of pulmonary vasculopathy and digital ulcers, and also predicted survival [30, 31]. Becker et al. found evaluated levels of these autoantibodies in patients with SSc-PAH and connective tissue disease (CTD)-PAH as compared to other PAH types. Pathophysiological effects favoring the pathogenesis of SSc were analyzed using microvascular endothelial cells, fibroblasts, peripheral blood mononuclear cells, and IgG transfer into mice. As a smaller study could partially not reproduce some of the findings in SSc-PAH, further confirmation in other cohorts will be important [32, 33].
Angiogenesis and Vasculogenesis
By limiting the vascular supply, EC injury and apoptosis with resulting loss of capillaries are known to trigger tissue hypoxia [19]. Hypoxia is a potent inducer of vascular repair mechanisms including angiogenesis and vasculogenesis.
Angiogenesis
Angiogenesis, the formation of new blood vessels from pre-existing ones, is known to be impaired in SSc. Paradoxically to the severe reduction in the capillary density in SSc, there is an insufficient angiogenic response in SSc [7, 19, 34]. To ensure an effective angiogenesis, the expression of pro- and anti-angiogenic factors needs to be regulated strictly in a temporal and spatial manner. In SSc, this control is disturbed [19]. Vascular endothelial growth factor A (VEGF-A, hereafter referred to as VEGF), one of the key physiological pro-angiogenic factors, is drastically upregulated in different cell types of the skin from SSc patients including keratinocytes, endothelial cells, and dermal fibroblasts [35]. Additionally, the expression of its receptors, especially VEGFR2, is significantly increased on ECs in skin biopsies derived from SSc patients [2, 35]. In line with the results obtained from tissue biopsies, increased VEGF levels were also detected in sera from SSc patients compared to healthy controls [36, 37]. In addition, patients with fingertip ulcers showed increased VEGF serum levels than healthy controls. However, VEGF levels were lower as compared to patients without digital ulcerations, revealing that VEGF has a protective role against ischemic complications once a threshold serum level of VEGF is exceeded [37]. Under normal physiologic conditions, VEGF is involved in many steps of the angiogenic process by increasing vascular permeability, stimulation of EC proliferation and migration, and promotion of tube formation [7, 38, 39]. Therefore, a dysregulation of VEGF expression could have severe effects on the process of angiogenesis. Prolonged overexpression of VEGF causes an exaggerated angiogenic response with uncontrolled vessel fusion resulting in chaotic vessel architectures with giant capillaries. In addition to the overall upregulation of VEGF expression, the time kinetic of the VEGF overexpression is critical [7, 39] as a too short VEGF response results in an unstable vessel formation as well [40].
Recently, two different splice variants of VEGF were identified, VEGF165 and VEGF165b possessing pro- or anti-angiogenic properties, respectively. Manetti et al. [41] showed that the elevated levels of VEGF in serum and skin of SSc patients are based on the overexpression of the anti-angiogenic VEGF165b isoform revealing that a switch from pro- to anti-angiogenic splice variants may be the main cause for the insufficient angiogenic response in SSc [2]. Manetti and coworkers could link this switch to the increased expression levels of transforming growth factor-beta 1 (TGF-β1) and the serine/arginine protein 55 (SrP55) VEGF splice factor [41, 42].
Recently, the angiopoietin (Ang)/Tie2 system was found to be involved in the disturbed angiogenesis in SSc. Tie2 is tyrosine kinase receptor expressed on the cell surface of ECs with Ang1 and Ang2 as the main ligands [43, 44]. Whereas Ang1 is expressed by many cell types, including pericytes, VSMCs, and fibroblasts, the expression of Ang2 seems to be restricted to ECs [45].
While Ang1 inherits vasoprotective and anti-inflammatory properties and is crucial for vessel maturation and vessel integrity, Ang2 occupies vessel-destabilizing properties and functions at a natural antagonist of Tie2 and Ang1 [46]. However, the functions of Ang2 are contextual and are regulated by VEGF [44, 47,48,49]. Furthermore, it was shown that VEGF induces shedding of membrane-bound Tie2. Soluble Tie2 (sTie2) can bind to Ang1 and 2 and inhibit Ang/Tie2 signaling [47].
In SSc, it was shown that Ang1 and Ang2 are differentially expressed in the sera from SSc patients with significantly decreased levels of Ang1 and increased levels of Ang2 [43]. The increased serum levels of Ang2 correlated significantly with disease severity and activity leading to the hypothesis that an altered expression of Ang1 and Ang2 might contribute to the vascular disease of SSc. Furthermore, also increased serum levels of sTie2 were detected in a subgroup of SSc patients and the elevated levels correlated with the occurrence of PAH and the frequency of microhemorrhages in nailfold capillaroscopy [50].
In line with the results obtained for serum levels of Ang2 and sTie2, Dunne et al. [45] measured increased plasma levels of these molecules. However, in contrast to Michalska-Jakubus et al. [43], they detected increased levels of Ang1. Nevertheless, taken all three studies together, there is strong evidence that an imbalance of angiopoietins and Tie2 might play a key role in the pathogenesis of the SSc microvasculopathy.
Interestingly, in dermal microvessels of SSc patients, an imbalance favoring the expression of Ang2 was observed. Additionally, the expression of membrane-bound Tie2 (mTie) was substantially reduced, since compared with healthy controls, only 5.5% vs. 90% of dermal microvessels expressed mTie2 [51]. Furthermore, in line with the results from Michalska-Jakubus et al. [43], a reduced Ang1/Ang2 ratio in patients’ sera was detected, which was associated with an increase of sTie2, thus indicating that the loss of mTie2 occurred due to increased shedding. Notably, the results obtained in SSc patients were mirrored in VEGF transgenic (tg) mice [51]. In VEGF tg mice, chronically elevated levels of VEGF lead to the development of microvasculopathy and dermal fibrosis, reminiscent of SSc [52]. In contrast, nonvascular models of SSc such as the model of bleomycin-induced skin fibrosis or the TSK1 (tight skin 1) model did not show the same alterations of the Ang/Tie2 system underlining its importance for the vascular pathology. Thus, these studies suggest a complex interplay between VEGF and the Ang/Tie2 system in the pathogenesis of the SSc microvasculopathy.
In addition to VEGF and the Ang/Tie2 system, the urokinase-type plasminogen activator (uPA)-uPA receptor (uPAR) system plays an important role in the impaired angiogenesis in SSc. Activated by VEGF, uPAR is centrally involved in the angiogenic process by catalyzing the cleavage of plasminogen to plasmin, which in turn activates matrix metalloproteinases (MMPs) resulting in ECM proteolysis [53, 54]. This leads to the degradation of the basement membrane enabling EC migration and invasion during the process of angiogenesis [53]. Microvascular ECs (MVECs) derived from SSc patients show higher expression levels of uPAR on their cell surface as compared to healthy MVECs. However, in SSc uPAR functions are impaired due to MMP-12-dependent truncation of uPAR in between the binding domain of uPA [55]. Thus, the receptor loses its ability to bind uPA compromising MVECs proliferation and invasion and therefore the angiogenic process in SSc [55]. MMP-12 is constitutively overexpressed in SSc MVECS [55, 56]. Transfecting normal MVECs with MMP-12 led to similar results with loss of EC proliferation, and invasion, and disturbed capillary formation [57]. In addition to overexpression of MMP-12, MVECs express higher amounts of pentraxin 3 (PTX3), a multifunctional pattern recognition receptor suppressing fibroblast growth factor-2 (FGF-2)-dependent EC proliferation and neovascularization [58, 59]. Silencing of MMP-12 and PTX3 in SSc MVECs using small interfering RNAs (siRNAs) restored the angiogenic potential of these cells [57]. Recently, PTX3 was identified to be a useful biomarker predicting the presence and future occurrences of digital ulcerations [59]. Furthermore, downregulation of several pro-angiogenic factors, including tissue kallikreins 9, 11, and 12, is accompanied with the dysregulated angiogenic response in SSc [60].
Moreover, recently, we could show that micro-RNAs (miRNAs) might play a pivotal role in the microvasculopathy in SSc. Specifically, we could identify miR-139b as a key molecule involved in the pathogenesis of the microvascular disease in SSc [61]. MiR-139b was downregulated in SSc cultured fibroblasts and SSc skin biopsies, and this downregulation was associated with an overexpression of uPA. By inducing VSMC proliferation and inhibiting VSMC apoptosis, the increased levels of uPA contributed to the proliferative microvasculopathy characteristic for SSc in an uPAR-independent manner. These findings suggest a dual role of the uPA-uPAR system in the SSc vasculopathy, with (1) an impaired angiogenesis with decreased proliferation and migration capacity of MVECs due to the lack of functional uPAR on the cell surface of MVECs and (2) an increased VSMC proliferation and decreased apoptosis of VSMCs leading to intimal hyperplasia occurring independently of uPAR [61].
Vasculogenesis
Apart from an insufficient angiogenesis, a dysregulated vasculogenesis is an additional pathophysiologic mechanism of microvasculopathy in SSc [7]. Vasculogenesis describes the formation of new vessels from circulating precursor cells, including endothelial progenitor cells (EPCs) independent of pre-existing vessels mostly occurring during development [2]. EPCs, also termed late EPCs, are bone marrow-derived cells, which can also be detected to a lesser extent in the adult peripheral blood circulation. They can be characterized as CD14-, CD133+, CD34+, and VEGF2R+ cells [62,63,64]. Using this marker combination, conflicting results concerning the number of EPCs in peripheral blood from SSc patients were received. Whereas Kuwana et al. [63] detected reduced numbers of EPCs in SSc patients, Del Papa et al. [62] measured significantly increased numbers of EPCs. Subgroup analysis of the SSc patient cohort revealed a negative correlation between the EPCs counts and the disease duration [34, 62]. As the patient cohort of Kuwana et al. was mainly composed of late-stage SSc patients, the discrepancy observed between both studies may be referred to the diseases duration of the selected SSc patients [62]. In line with the results from Del Papa and coworkers, Avouac et al. [65] also measured increased circulating EPCs counts in SSc, proposing an increased mobilization of these cells from the bone marrow. Additionally, they found a positive correlation between low EPC numbers and diseases severity as well as the past occurrence and the presence of digital ulcers, suggesting an increased homing of EPCs in later diseases stages [65]. However, recently further conflicting results were reported by two additional studies [66, 67]. They detected significantly decreased circulating EPCs counts also in SSc patients with early disease stages.
These contradictory results might be based on the different methodologies used to analyze EPCs, the use of different combinations of surface markers for EPC characterization, and differences in the subsets of patients, medications, and disease durations between the study cohorts [34, 66,67,68].
In contrast to the conflicting results obtained for the cell numbers of EPCs, consistent results were reported for the functional defects of EPCs in the peripheral blood as well as bone marrow from SSc patients. It was found that the potential of SSc EPCs to differentiate to mature ECs is impaired [63]. Recently, Kuwana and Okazaki reported that the neovascularization capacity of SSc EPCs is affected in vivo as well [69]. Tumors from mice that received transplants of SSc-derived CD133+ cells showed fewer vessel formations with incorporation of human EPC-derived mature endothelial cells as compared to tumors from mice receiving transplants of CD133+ cells from healthy controls [69].
So far, the pathophysiologic mechanisms leading to the impaired EPC functions and numbers have not been fully elucidated. Recently, Zhu et al. [67] proposed that autoantibody-induced EPC apoptosis might be a potential cause. They could demonstrate that incubation of EPCs with SSc sera induced EPC apoptosis. Since the depletion of the IgG fractions in the sera prevented the apoptosis of EPC, these results indicate that autoantibodies might be involved in this process.
In addition, we found a link between EC apoptosis and reduced numbers of circulating angiogenic cells (CACs) [2, 70]. CACs, also termed as early EPCs , are a subpopulation of EPCs and are regarded as transdifferentiated CD14+ monocytes acquiring EC characteristics under defined culture conditions [34, 71]. CACs are strong promotors of the angiogenic response by inducing migration and sprouting of ECs without the capacity of forming blood vessels [72]. We could show that CACs phagocytize microparticles derived from apoptotic ECs leading to the initiation of the apoptotic cascade in this EPC subtype via sphingomyelinase-/ceramide-dependent pathways [70].
In conclusion, functional impairment as well as disturbed EPC counts might play a critical role in the SSc microvascular disease. However, EPC counts in SSc patients are still unresolved, and the underlying mechanisms leading to the impaired EPC numbers and functions remain enigmatic.
Animal Models of Microvasculopathy
To study the molecular basis of the SSc microvasculopathy and to serve as platforms for therapeutic proof of concept-studies, animal models reflecting the characteristic microvascular features of SSc are of outmost importance. An overview of the existing vascular SSc models is depicted in Table 4.1.
UCD-200/206 Chickens
UCD-200/206 chickens spontaneously develop inherited scleroderma covering the full spectrum of the SSc disease symptoms, including fibrosis of the skin and visceral organs, the presence of autoantibodies, and perivascular mononuclear infiltrates as well as obliterative vasculopathy [73,74,75]. These chickens develop the first symptoms 1 to 2 weeks post-hatch starting with the induction of EC apoptosis by AECA followed by the occurrence of perivascular infiltrates and the development of skin and organ fibrosis [12, 76]. Sgonc and coworkers showed that the primary event of the vascular disease in these chickens is AECA-dependent EC apoptosis resulting in severe capillary loss and chronic ischemia manifesting in necrosis of the combs and digits [12, 77].
Despite the fact that the UCD chickens display the entire hallmarks of the SSc pathophysiology, this model finds only relatively limited application in SSc research due to the clinical heterogeneity, the high costs of maintenance, the prolonged generation times, and the different genetic background as compared to humans [2]. Very recently, this model was applied to test the therapeutic efficacy of local administration of VEGF121 bound to a fibrin matrix enabling the cell-demanded release of VEGF from the matrix, which is stopped as soon as it is no longer required. Upon VEGF121-fibrin treatment, 79.3% of all lesions showed clinical improvement with prevention of digital ulcerations when applied early in the disease course. Simultaneously, the angiogenic response was increased reflecting in an increase microvascular density as well as increased numbers of ECs in the combs and neck lesions treated with VEGF121-fibrin [78].
Friend Leukemia Integration-1 (Fli-1)-Deficient Mice
Fli-1 is a member of the Ets (E26 transformation-specific) transcription factor family. It is highly expressed in the endothelial cell and hematopoietic cell lineage and to a lesser extent in dermal fibroblasts [79]. Fli-1 is a negative regulator of type 1 collagen expression [80, 81]. In SSc skin biopsies, the expression of Fli-1 is significantly reduced in ECs and even undetectable in fibroblasts, and the observed downregulation correlates with the induction of collagen synthesis [81]. In order to assess the role of Fli-1 downregulation in the SSc vascular disease, Asano et al. generated conditional Fli-1 knockout mice with a deficiency of Fli-1 restricted to ECs [82]. These Fli-1 ECKO mice show severe abnormalities of the skin vasculature with arteriolar stenosis, formation of micro-aneurysms, and capillary dilation [79, 82]. In addition, vessel stability is compromised and vascular permeability is significantly increased [82] emphasizing the role of Fli-1 in the microvasculopathy observed in SSc.
Fli-1 deficiency in these mice is associated with a reduced expression of vascular endothelial cadherin (VE-cadherin), platelet/endothelial cell adhesion molecule (PECAM-1), platelet-derived growth factor (PDGF-B), and sphingosine 1 phosphate (S1P1) receptors, as well as an increased expression of MMP-9 in ECs [79, 82]. In accordance with the results in this animal model, reduced expression of VE-cadherins and PECAM-1 and increased levels of MMP-9 in ECs are also found in SSc patients revealing that this mouse model recapitulates important features of microvasculopathy in SSc and supports the hypothesis that Fli-1 is involved in the pathogenesis of the SSc vasculopathy [79].
Sclerodermatous Graft-Versus-Host Disease
The murine model of sclerodermatous graft-versus-host disease (Scl GVHD) displays important features of the SSc pathophysiology including dermal and pulmonary fibrosis, severe dermal mononuclear infiltration, and a significant upregulation of TGF-β1 and procollagen 1 mRNA levels in the dermis [83,84,85]. In the Scl GVHD model , lethally irradiated Balb/c mice are transplanted with B10.D2 bone marrow and spleen cells differing in minor histocompatibility antigens [84, 85]. The manifestations of the first SSc symptoms become apparent at day 21 after bone marrow transplantation [84]. However, signs of pulmonary and peripheral vasculopathy were not detectable in this model. In 2004, Ruzek and coworkers [86] introduced a modified Scl GVDH model , manifesting microvascular changes as well. In contrast to the original model, RAG-2 knockout mice lacking B and T cells are used as recipients for transfer B10.D2 spleen cells to induce the GVH SSc disease. Besides dermal and visceral fibrosis, these mice show a significant reduction in the ratio of total vessel to lumen area in skin and kidney indicating vasoconstriction. Intimal hyperplasia due to proliferation of VSMCs was identified as a major contributor to the observed vessel occlusion. Apart from luminal narrowing, these mice also show evaluated levels of ET-1 and an altered SMC morphology.
uPAR Deficient Mice
While originally constructed by Dewerchin and coworkers [87] to study the in vivo role of soluble versus receptor-bound uPA, Manetti and coworkers recently proposed uPAR-deficient mice as a novel model for SSc peripheral vasculopathy [88]. Apart from the development of dermal and lung fibrosis, these mice show a significant reduction in the dermal capillary density which was accompanied by a significant increase in EC apoptosis. However, signs of proliferative vasculopathy characterized by neointimal hyperplasia of dermal and pulmonary vessels were not detected in these knockout mice. Interestingly, uPAR expression was also significantly downregulated in skin biopsies from SSc patients revealing that inactivation of uPAR is recapitulating the SSc disease pathophysiology.
VEGF tg Mice
VEGF tg mice selectively overexpress VEGF under the control of the keratin 14 promotor in basal epidermal keratinocytes [89] leading to increased dermal levels of VEGF. As VEGF overexpression represents a key feature in the pathophysiology of the SSc vascular disease, these mice became interesting as a model of microvasculopathy in SSc. Recently, we reported that VEGF tg mice show an increase in the number of dermal microvessels as well as in the vessel wall thickness characteristic for a proliferative vasculopathy [52]. However, these changes were more pronounced in heterozygous than homozygous transgenic mice, suggesting that the pro-angiogenic effects of VEGF are dose-dependent and too high levels of VEGF, as in SSc patients, could have adverse effects. Notably, apart from vascular changes, homozygous VEGF overexpression led to the spontaneous development of skin fibrosis with dermal thickening and collagen accumulation [52].
TβRIIΔk-fib Mice
TβRIIΔk-fib mice selectively overexpress the kinase-deficient type II transforming growth factor-beta (TGF-β) receptor on fibroblasts under a pro-α2(I) collagen promoter [90, 91]. This overexpression leads to a ligand-dependent upregulation of TGF-β signaling inducing the spontaneous development of dermal and pulmonary fibrosis. These mice additionally show macrovascular changes with aortic adventitial fibrosis, VSMC attenuation, and altered vasoreactivity of isolated aortic rings [92]. Besides the macrovascular changes, these mice develop a constitutive pulmonary vasculopathy with neointimal hyperplasia, perivascular infiltration and collagen deposition, and a mild elevation in right ventricular systolic pressure (RVSP), resembling the chronic hypoxia model of PAH [93]. However, they do not show an endothelial phenotype characteristic for human PAH. Administrating the VEGF receptor tyrosine kinase inhibitor SU5416 aggravates the observed pulmonary vasculopathy with PAH, reflected in the obliteration of small and medium-sized pulmonary vessel and the upregulation of luminal cell proliferation. Furthermore, treatment with SU5416 induces EC apoptosis, which was followed by an apoptosis-resistant EC proliferation and a marked raise of the RVSP, and the development of a right ventricular hypertrophy, closely resembling human PAH [93, 94]. The manifestation of peripheral microvascular changes has not been reported yet. However, this mouse model represents a promising animal model for the SSc pulmonary vasculopathy and PAH.
Fos-Related Antigen 2 (Fra-2) tg Mice
Fra-2 tg mice, generated by Eferl and coworkers [95], ectopically express the transcription factor Fra-2 of the activator protein-1 (AP-1) family under the control of the ubiquitous major histocompatibility complex class I antigen H2Kb promotor. Fra-2 mice simultaneously develop microvascular alterations as well as dermal and visceral fibrosis closely resembling the pathophysiologic characteristics of human SSc [96].
Fra-2 tg mice display prominent features of the SSc vascular disease, including increased EC apoptosis, progressive vessel loss, and a severe proliferative pulmonary vasculopathy resembling PAH [96,97,98].
Starting at 12 weeks of age, Fra-2 tg mice show a significant reduction in the capillary density which is paralleled by the development of skin fibrosis characterized by a time-dependent increase in the dermal thickness reaching its peak at week 16 [97]. Of note, the manifestation of the peripheral vasculopathy in these mice is preceded by the appearance of EC apoptosis [97]. This is in accordance with the results obtained in SSc patients and in the UCD-200/206 chicken model, underscoring the hypothesis that EC apoptosis is one of the initiating events in the SSc vasculopathy [96].
Further emphasizing the role of the Fra-2 in the pathophysiology of microvasculopathy, Fra-2 is overexpressed in ECs and VSMCS in the skin from Fra-2 tg mice and SSc patients [97].
In addition to the pronounced peripheral vasculopathy, Fra-2 tg mice display a severe proliferative pulmonary vasculopathy with obliteration of pulmonary arteries characterized by a marked formation of neointima due to proliferation of VSMCS and especially myofibroblasts [95, 98]. Interestingly, these vascular alterations start to manifest at week 12 and precede the development of pulmonary fibrosis by 2 to 3 weeks [95, 98]. In contrast to the peripheral vasculopathy, elevated levels of EC apoptosis were not detected in the lungs [96]. Typical for PAH, this severe pulmonary vasculopathy was accompanied with an increased RVSP in Fra-2 tg mice as compared to wildtype littermates [99]. Manifesting at week 8, this raise in the RVSP steadily persisted in these mice and was not associated with an increase in the systemic arterial pressure [99].
So far, Fra-2 tg mice are the only murine SSc model displaying in a time-dependent manner the development of the destructive and proliferative vasculopathy characteristic for the human SSc microvascular disease.
Conclusion
In SSc, microvascular abnormalities become apparent at earliest diseases stages, clinically manifesting in ischemic complications including digital ulcers. Due to the high morbidity associated with these peripheral manifestations, the elucidation of the underlying molecular mechanism is of outmost importance in order to allow for the development of effective therapeutic interventions.
To date, the pathophysiologic mechanisms of the SSc vascular disease have not been fully clarified. Using in vitro and in vivo models, EC cell injury and apoptosis, an imbalance of pro- and anti-angiogenic factors, and an insufficient angiogenic and vasculogenic response have been identified as potential disease mechanisms. However, a comprehensive picture of the disease pathophysiology has not been obtained yet, and future research is needed to unravel the underlying connections.
Key Messages
-
Microvascular changes are one of the earliest hallmarks of the SSc pathophysiology clinically manifesting in severe ischemic complications such as digital ulcers.
-
In SSc, the destructive and proliferative vasculopathy precede the development of dermal and visceral fibrosis.
-
Endothelial cell injury and apoptosis are regarded as one of the initiating events in the SSc microvasculopathy.
-
Despite the severe loss of microvessels, there is an insufficient angiogenic response characterized by an imbalance between pro- and anti-angiogenic factors, as well as an insufficient vasculogenesis due to impaired EPCs functions and potentially numbers.
-
To study the molecular basis of microvasculopathy in SSc, animal models, which cover distinct pathophysiologic aspects, are of outmost importance.
References
Abraham D, Distler O. How does endothelial cell injury start? The role of endothelin in systemic sclerosis. Arthritis Res Ther. 2007;9(Suppl 2):S2.
Suliman Y, Distler O. Novel aspects in the pathophysiology of peripheral vasculopathy in systemic sclerosis. Curr Rheumatol Rev. 2014;9:237–44.
Guiducci S, Giacomelli R, Cerinic MM. Vascular complications of scleroderma. Autoimmun Rev. 2007;6:520–3.
Steen V, et al. Digital ulcers: overt vascular disease in systemic sclerosis. Rheumatology (Oxford, England). 2009;48(Suppl 3):iii19–24.
Cutolo M, Matucci Cerinic M. Nailfold capillaroscopy and classification criteria for systemic sclerosis. Clin Exp Rheumatol. 2007;25:663–5.
Cutolo M, Sulli A, Smith V. Assessing microvascular changes in systemic sclerosis diagnosis and management. Nat Rev Rheumatol. 2010;6:578–87.
Distler JHW, Gay S, Distler O. Angiogenesis and vasculogenesis in systemic sclerosis. Rheumatology (Oxford, England). 2006;45(Suppl 3):iii26–7.
D’Angelo WA, et al. Pathologic observations in systemic sclerosis (scleroderma). Am J Med. 1969;46:428–40.
Fleming JN, et al. Is scleroderma a vasculopathy? Curr Rheumatol Rep. 2009;11:103–10.
Pattanaik D, Brown M, Postlethwaite AE. Vascular involvement in systemic sclerosis (scleroderma). J Inflamm Res. 2011;4:105–25.
Kahaleh B. The microvascular endothelium in scleroderma. Rheumatology (Oxford, England). 2008;47(Suppl 5):v14–5.
Sgonc R, et al. Endothelial cell apoptosis is a primary pathogenetic event underlying skin lesions in avian and human scleroderma. J Clin Invest. 1996;98:785–92.
Fleischmajer R, et al. Skin capillary changes in early systemic sclerodermaelectron microscopy and “in vitro” autoradiography with tritiated thymidine. Arch Dermatol. 1976;112:1553–7.
Kahaleh MB. Vascular involvement in systemic sclerosis (SSc). Clin Exp Rheumatol. 2004;22:S19–23.
Alberts GF, et al. Constitutive endothelin-1 overexpression promotes smooth muscle cell proliferation via an external autocrine loop. J Biol Chem. 1994;269:10112–8.
Gallelli L, et al. Endothelin-1 induces proliferation of human lung fibroblasts and IL-11 secretion through an ETA receptor-dependent activation of map kinases. J Cell Biochem. 2005;96:858–68.
Piacentini L, et al. Endothelin-1 stimulates cardiac fibroblast proliferation through activation of protein kinase C. J Mol Cell Cardiol. 2000;32:565–76.
Ahanchi SS, Tsihlis ND, Kibbe MR. The role of nitric oxide in the pathophysiology of intimal hyperplasia. J Vasc Surg. 2007;45(Suppl A):A64–73.
Guiducci S, et al. Mechanisms of vascular damage in SSc–implications for vascular treatment strategies. Rheumatology (Oxford, England). 2008;47(Suppl 5):v18–20.
Varga J, Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest. 2007;117:557–67.
Mihai C, Tervaert JWC. Anti-endothelial cell antibodies in systemic sclerosis. Ann Rheum Dis. 2010;69:319–24.
Renaudineau Y, et al. Anti-endothelial cell antibodies in systemic sclerosis. Clin Diagn Lab Immunol. 1999;6(2):156–60.
Bordron A, et al. The binding of some human antiendothelial cell antibodies induces endothelial cell apoptosis. J Clin Invest. 1998;101:2029–35.
Sgonc R, et al. Endothelial cell apoptosis in systemic sclerosis is induced by antibody-dependent cell-mediated cytotoxicity via CD95. Arthritis Rheum. 2000;43(11):2550–62.
Worda M, et al. In vivo analysis of the apoptosis-inducing effect of anti-endothelial cell antibodies in systemic sclerosis by the chorionallantoic membrane assay. Arthritis Rheum. 2003;48:2605–14.
Negi VS, et al. Antiendothelial cell antibodies in scleroderma correlate with severe digital ischemia and pulmonary arterial hypertension. J Rheumatol. 1998;25:462–6.
Pignone A, et al. Anti-endothelial cell antibodies in systemic sclerosis: significant association with vascular involvement and alveolo-capillary impairment. Clin Exp Rheumatol. 1998;16:527–32.
Belizna C, Tervaert JW. Specificity, pathogenecity, and clinical value of antiendothelial cell antibodies. Semin Arthritis Rheum. 1997;27:98–109.
Belizna C, et al. Antiendothelial cell antibodies in vasculitis and connective tissue disease. Ann Rheum Dis. 2006;65:1545–50.
Becker MO, et al. Vascular receptor autoantibodies in pulmonary arterial hypertension associated with systemic sclerosis. Am J Respir Crit Care Med. 2014;190(7):808–17.
Riemekasten G, et al. Involvement of functional autoantibodies against vascular receptors in systemic sclerosis. Ann Rheum Dis. 2011;70(3):530–6.
Bourji KI, et al. Vascular receptor autoantibodies in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2015;191(5):602.
Distler O. Vascular receptor autoantibodies in pulmonary arterial hypertension associated with systemic sclerosis. J Club. [cited 2017 07.03.2017]; Available from: https://www.thoracic.org/members/assemblies/assemblies/pc/journal-club/vascular-receptor-autoantibodies-in-pulmonary-arterial-hypertension-associated-with-systemic-sclerosis.php.
Distler JHW, et al. Endothelial progenitor cells: novel players in the pathogenesis of rheumatic diseases. Arthritis Rheum. 2009;60:3168–79.
Distler O, et al. Uncontrolled expression of vascular endothelial growth factor and its receptors leads to insufficient skin angiogenesis in patients with systemic sclerosis. Circ Res. 2004;95:109–16.
Choi J-J, et al. Elevated vascular endothelial growth factor in systemic sclerosis. J Rheumatol. 2003;30:1529–33.
Distler O, et al. Angiogenic and angiostatic factors in systemic sclerosis: increased levels of vascular endothelial growth factor are a feature of the earliest disease stages and are associated with the absence of fingertip ulcers. Arthritis Res. 2002;4:R11.
Byrne AM, Bouchier-Hayes DJ, Harmey JH. Angiogenic and cell survival functions of Vascular Endothelial Growth Factor (VEGF). J Cell Mol Med. 2005;9:777–94.
Müller-Ladner U, et al. Mechanisms of vascular damage in systemic sclerosis. Autoimmunity. 2009;42:587–95.
Dor Y, et al. Conditional switching of VEGF provides new insights into adult neovascularization and pro-angiogenic therapy. EMBO J. 2002;21:1939–47.
Manetti M, et al. Overexpression of VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, leads to insufficient angiogenesis in patients with systemic sclerosis. Circ Res. 2011;109:e14–26.
Manetti M, et al. Impaired angiogenesis in systemic sclerosis: the emerging role of the antiangiogenic VEGF(165)b splice variant. Trends Cardiovasc Med. 2011;21:204–10.
Michalska-Jakubus M, et al. Angiopoietins-1 and -2 are differentially expressed in the sera of patients with systemic sclerosis: high angiopoietin-2 levels are associated with greater severity and higher activity of the disease. Rheumatology (Oxford). 2011;50(4):746–55.
Yancopoulos GD, et al. Vascular-specific growth factors and blood vessel formation. Nature. 2000;407(6801):242–8.
Dunne J, Keen K, Van Eeden S. Circulating angiopoietin and Tie-2 levels in systemic sclerosis. Rheumatol Int. 2013;33(2):475–84.
Maisonpierre PC, et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 1997;277(5322):55–60.
Findley CM, et al. VEGF induces Tie2 shedding via a phosphoinositide 3-kinase/Akt dependent pathway to modulate Tie2 signaling. Arterioscler Thromb Vasc Biol. 2007;27(12):2619–26.
Lobov IB, Brooks PC, Lang RA. Angiopoietin-2 displays VEGF-dependent modulation of capillary structure and endothelial cell survival in vivo. Proc Natl Acad Sci U S A. 2002;99(17):11205–10.
Augustin HG, et al. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat Rev Mol Cell Biol. 2009;10(3):165–77.
Noda S, et al. Serum Tie2 levels: clinical association with microangiopathies in patients with systemic sclerosis. J Eur Acad Dermatol Venereol. 2011;25(12):1476–9.
Moritz F, et al. Tie2 as a novel key factor of microangiopathy in systemic sclerosis. Arthritis Res Ther. 2017;19(1):105.
Maurer B, et al. Vascular endothelial growth factor aggravates fibrosis and vasculopathy in experimental models of systemic sclerosis. Ann Rheum Dis. 2014;73:1880–7.
Breuss JM, Uhrin P. VEGF-initiated angiogenesis and the uPA/uPAR system. Cell Adhes Migr. 2012;6:535–615.
Smith HW, Marshall CJ. Regulation of cell signalling by uPAR. Nat Rev Mol Cell Biol. 2010;11:23–36.
D’Alessio S, et al. Matrix metalloproteinase 12-dependent cleavage of urokinase receptor in systemic sclerosis microvascular endothelial cells results in impaired angiogenesis. Arthritis Rheum. 2004;50:3275–85.
Margheri F, et al. Domain 1 of the urokinase-type plasminogen activator receptor is required for its morphologic and functional, beta2 integrin-mediated connection with actin cytoskeleton in human microvascular endothelial cells: failure of association in systemic sclerosis. Arthritis Rheum. 2006;54:3926–38.
Margheri F, et al. Modulation of the angiogenic phenotype of normal and systemic sclerosis endothelial cells by gain-loss of function of pentraxin 3 and matrix metalloproteinase 12. Arthritis Rheum. 2010;62:2488–98.
Rusnati M, et al. Selective recognition of fibroblast growth factor-2 by the long pentraxin PTX3 inhibits angiogenesis. Blood. 2004;104:92–9.
Shirai Y, et al. Elevated levels of pentraxin 3 in systemic sclerosis: associations with vascular manifestations and defective vasculogenesis. Arthritis Rheumatol (Hoboken, NJ). 2015;67:498–507.
Giusti B, et al. The antiangiogenic tissue kallikrein pattern of endothelial cells in systemic sclerosis. Arthritis Rheum. 2005;52:3618–28.
Iwamoto N, et al. Downregulation of miR-193b in systemic sclerosis regulates the proliferative vasculopathy by urokinase-type plasminogen activator expression. Ann Rheum Dis. 2016;75(1):303–10.
Del Papa N, et al. Bone marrow endothelial progenitors are defective in systemic sclerosis. Arthritis Rheum. 2006;54:2605–15.
Kuwana M, et al. Defective vasculogenesis in systemic sclerosis. Lancet. 2004;364:603–10.
Brunasso AMG, Massone C. Update on the pathogenesis of Scleroderma: focus on circulating progenitor cells. F1000Research. 2016;5:F1000 Faculty Rev-723.
Avouac J, et al. Circulating endothelial progenitor cells in systemic sclerosis: association with disease severity. Ann Rheum Dis. 2008;67:1455–60.
Andrigueti FV, et al. Decreased numbers of endothelial progenitor cells in patients in the early stages of systemic sclerosis. Microvasc Res. 2015;98:82–7.
Zhu S, et al. Transcriptional regulation of Bim by FOXO3a and Akt mediates scleroderma serum-induced apoptosis in endothelial progenitor cells. Circulation. 2008;118:2156–65.
Distler JH, et al. EULAR Scleroderma Trials and Research group statement and recommendations on endothelial precursor cells. Ann Rheum Dis. 2009;68(2):163–8.
Kuwana M, Okazaki Y. Brief report: impaired in vivo neovascularization capacity of endothelial progenitor cells in patients with systemic sclerosis. Arthritis Rheumatol (Hoboken, NJ). 2014;66:1300–5.
Distler JHW, et al. Induction of apoptosis in circulating angiogenic cells by microparticles. Arthritis Rheum. 2011;63:2067–77.
Rehman J, et al. Peripheral blood “endothelial progenitor cells” are derived from monocyte/macrophages and secrete angiogenic growth factors. Circulation. 2003;107:1164–9.
Mukai N, et al. A comparison of the tube forming potentials of early and late endothelial progenitor cells. Exp Cell Res. 2008;314:430–40.
Gershwin ME, et al. Characterization of a spontaneous disease of white leghorn chickens resembling progressive systemic sclerosis (scleroderma). J Exp Med. 1981;153:1640–59.
van de Water J, Gershwin ME. Animal model of human disease. Avian scleroderma. An inherited fibrotic disease of white Leghorn chickens resembling progressive systemic sclerosis. Am J Pathol. 1985;120:478–82.
van de Water J, et al. Identification of T cells in early dermal lymphocytic infiltrates in avian scleroderma. Arthritis Rheum. 1989;32:1031–40.
Nguyen VA, et al. Endothelial injury in internal organs of University of California at Davis line 200 (UCD 200) chickens, an animal model for systemic sclerosis (Scleroderma). J Autoimmun. 2000;14:143–9.
Beyer C, et al. Animal models of systemic sclerosis: prospects and limitations. Arthritis Rheum. 2010;62:2831–44.
Allipour Birgani S, et al. Efficient therapy of ischaemic lesions with VEGF121-fibrin in an animal model of systemic sclerosis. Ann Rheum Dis. 2016;75(7):1399–406.
Asano Y, Bujor AM, Trojanowska M. The impact of Fli1 deficiency on the pathogenesis of systemic sclerosis. J Dermatol Sci. 2010;59:153–62.
Czuwara-Ladykowska J, et al. Fli-1 inhibits collagen type I production in dermal fibroblasts via an Sp1-dependent pathway. J Biol Chem. 2001;276:20839–48.
Kubo M, et al. Persistent down-regulation of Fli1, a suppressor of collagen transcription, in fibrotic scleroderma skin. Am J Pathol. 2003;163:571–81.
Asano Y, et al. Endothelial Fli1 deficiency impairs vascular homeostasis: a role in scleroderma vasculopathy. Am J Pathol. 2010;176:1983–98.
Jaffee BD, Claman HN. Chronic graft-versus-host disease (GVHD) as a model for scleroderma. I. Description of model systems. Cell Immunol. 1983;77(1):1–12.
McCormick LL, et al. Anti-TGF-beta treatment prevents skin and lung fibrosis in murine sclerodermatous graft-versus-host disease: a model for human scleroderma. J Immunol. 1999;163(10):5693–9.
Zhang Y, et al. Murine sclerodermatous graft-versus-host disease, a model for human scleroderma: cutaneous cytokines, chemokines, and immune cell activation. J Immunol. 2002;168(6):3088–98.
Ruzek MC, et al. A modified model of graft-versus-host–induced systemic sclerosis (scleroderma) exhibits all major aspects of the human disease. Arthritis Rheum. 2004;50(4):1319–31.
Dewerchin M, et al. Generation and characterization of urokinase receptor-deficient mice. J Clin Invest. 1996;97(3):870–8.
Manetti M, et al. Inactivation of urokinase-type plasminogen activator receptor (uPAR) gene induces dermal and pulmonary fibrosis and peripheral microvasculopathy in mice: a new model of experimental scleroderma? Ann Rheum Dis. 2014;73(9):1700–9.
Detmar M, et al. Increased microvascular density and enhanced leukocyte rolling and adhesion in the skin of VEGF transgenic mice. J Investig Dermatol. 1998;111(1):1–6.
Denton CP, et al. Activation of key profibrotic mechanisms in transgenic fibroblasts expressing kinase-deficient type II Transforming growth factor-{beta} receptor (T{beta}RII{delta}k). J Biol Chem. 2005;280(16):16053–65.
Denton CP, et al. Fibroblast-specific expression of a kinase-deficient type II transforming growth factor beta (TGFbeta) receptor leads to paradoxical activation of TGFbeta signaling pathways with fibrosis in transgenic mice. J Biol Chem. 2003;278(27):25109–19.
Derrett-Smith EC, et al. Systemic vasculopathy with altered vasoreactivity in a transgenic mouse model of scleroderma. Arthritis Res Ther. 2010;12(2):R69.
Derrett-Smith EC, et al. Endothelial injury in a transforming growth factor β–dependent mouse model of scleroderma induces pulmonary arterial hypertension. Arthritis Rheum. 2013;65(11):2928–39.
Gilbane AJ, et al. Impaired bone morphogenetic protein receptor II signaling in a transforming growth factor-β–dependent mouse model of pulmonary hypertension and in systemic sclerosis. Am J Respir Crit Care Med. 2015;191(6):665–77.
Eferl R, et al. Development of pulmonary fibrosis through a pathway involving the transcription factor Fra-2/AP-1. Proc Natl Acad Sci. 2008;105:10525–30.
Maurer B, Distler JHW, Distler O. The Fra-2 transgenic mouse model of systemic sclerosis. Vasc Pharmacol. 2013;58:194–201.
Maurer B, et al. Transcription factor fos-related antigen-2 induces progressive peripheral vasculopathy in mice closely resembling human systemic sclerosis. Circulation. 2009;120:2367–76.
Maurer B, et al. Fra-2 transgenic mice as a novel model of pulmonary hypertension associated with systemic sclerosis. Ann Rheum Dis. 2012;71:1382–7.
Biasin V, et al. Meprin β, a novel mediator of vascular remodelling underlying pulmonary hypertension. J Pathol. 2014;233(1):7–17.
Acknowledgments
This work was financially supported by the Swiss National Science Foundation (Grant: CRSII3_154490).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Schniering, J., Maurer, B., Distler, O. (2019). Vascular Mechanisms of Systemic Sclerosis. In: Matucci-Cerinic, M., Denton, C. (eds) Atlas of Ulcers in Systemic Sclerosis. Springer, Cham. https://doi.org/10.1007/978-3-319-98477-3_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-98477-3_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-98475-9
Online ISBN: 978-3-319-98477-3
eBook Packages: MedicineMedicine (R0)