
Abstract
Scleroderma (systemic sclerosis (SSc)) is to a large degree identified by the vascular features which may precede the onset of fibrosis by months or even years. Abnormalities in microvessel morphology and vascular dysfunction occur early and evolve into a distinctive vasculopathy that relentlessly advances in nearly all affected organs. This evidence suggests that endothelial cells (ECs) are the primary target in SSc and that the interactions of ECs with other cells and pathways, including cells of both the innate and adaptive immune systems, platelets and coagulation factors, vascular smooth muscle cells and fibroblasts, may take center stage in the disease pathogenesis. Therapeutically, the use of angiotensin-converting enzyme inhibitors for scleroderma renal crisis, endothelin receptor antagonists for pulmonary arterial hypertension (PAH), and phosphodiesterase inhibitors for PAH, Raynaud’s phenomenon, and digital ulcers has significantly improved the care of SSc patients. However, despite a substantial symptomatic improvement, regression of the vascular lesions has been difficult to achieve.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Angiogenesis
- Autoantibody
- Coagulation
- Endothelial cell
- Endothelin
- Epigenetics
- Microangiopathy
- Platelet
- ROS
- Vascular disease
- Vasculogenesis
Introduction
Scleroderma (systemic sclerosis (SSc)) is to a large degree identified by the vascular features which may precede the onset of fibrosis by months or even years. Abnormalities in microvessel morphology and vascular dysfunction occur early during the disease course and evolve into a distinctive vasculopathy that relentlessly advances in nearly all affected organs. This evidence suggests that endothelial cells (ECs) are the primary target in SSc and that multiple interactions of ECs with other cells and pathways, including cells of both the innate and adaptive immune systems, platelets and coagulation factors, vascular smooth muscle cells (VSMCs), and fibroblasts, position ECs in center stage of disease pathogenesis. In SSc, vascular injury includes persistent EC activation/damage and apoptosis, intimal thickening, vessel narrowing, and obliteration. These profound vascular changes lead to vascular tone dysfunction and reduced capillary blood flow, with consequent chronic tissue ischemia. The resulting tissue hypoxia induces complex cellular and molecular mechanisms in the attempt to recover EC functions including adequate tissue perfusion. Nonetheless, there is no evidence of significant compensatory angiogenesis in SSc, and the disease evolves toward irreversible structural changes in multiple vascular beds culminating in the loss of the peripheral capillary network. A critical imbalance between proangiogenic and antiangiogenic factors may be largely responsible for the lack of vascular regeneration in SSc. Besides insufficient angiogenesis, increasing evidence indicates that defective vasculogenesis with altered numbers and functional defects of bone marrow-derived endothelial progenitor cells (EPCs) may contribute to the pathogenesis of SSc vasculopathy.
Clinically, the vascular manifestations in SSc include Raynaud’s phenomenon (RP), the early edematous puffy hands, telangiectasias, digital ulcers, gastric antral vascular ectasia (GAVE), pulmonary arterial hypertension (PAH), myocardial dysfunction, and scleroderma renal crisis. Therapeutically, the use of angiotensin-converting enzyme inhibitors for scleroderma renal crisis, endothelin receptor antagonists for PAH, and phosphodiesterase inhibitors for PAH, RP, and digital ulcers have significantly improved the care of SSc patients. However, despite this symptomatic improvement, regression of the vascular lesions has been difficult to achieve.
The Vascular Problem
Vascular involvement in SSc includes a spectrum of changes that affect predominantly the microcirculation, mainly the capillaries and the small arterioles. The pathologic changes in the vascular tree range from EC activation with increased expression of adhesion molecules to EC apoptosis, capillary necrosis, intimal proliferation, and luminal occlusion that result in tissue ischemia and contribute to organ failure together with concomitant fibrosis. The current hypotheses in SSc vascular pathogenesis implicate a possible, yet unidentified, microbial trigger and an immune-mediated mechanism that results in vascular injury possibly through recognition of a putative vascular or perivascular autoantigen and the induction of autoantibodies. This chapter will review the morphologic and functional features of SSc vasculopathy with special emphasis on the mechanisms and pathologic sequences of vascular injury.
The Vascular Lesion
The prominent vascular abnormalities in SSc are typically observed in the capillaries and the small blood vessels [1]. Swelling and proliferation of the intima with mononuclear cell infiltration are seen in the small arterioles. In the capillary network, the vascular disease is characterized by distorted and irregular capillary loops in all involved organs including the skin, kidneys, lungs, heart, and muscles, reflecting the diffuse nature of the microvascular disorder in SSc, even in sites not affected by fibrosis [1, 2]. At the ultrastructural level, the earliest vascular changes in the edematous stage of the disease consist of opening of EC tight junctions, vacuolization of cytoplasm with increase in the number of basal lamina-like layers, and occasional entrapment of lymphocytes and cellular vesicles in the vessel wall [1]. Further signs of nuclear injury in association with EC membrane disruption occur in more advanced disease. Ghost vessels consisting of an intact basal lamina with remnants of ECs are occasionally noted in association with perivascular cellular infiltrates that consist of macrophages and T and B cells, with predominance of CD4+ T cells [3]. EC apoptosis was first described on ultrastructural examination of SSc skin biopsies in the early inflammatory stages of the disease, suggesting a causal association [3]. It was later noted in the University of California at Davis (UCD) lines 200/206 chickens that spontaneously develop a systemic disease that closely resembles human SSc [4]. Apoptosis of ECs is not unique to SSc vasculopathy and is frequently observed in diseases with prominent vascular involvement, such as early atherosclerotic lesions, graft rejection, thrombotic thrombocytopenic purpura, and the hemolytic uremic syndrome, suggesting that EC apoptosis may be a common prerequisite for a variety of vascular disorders [5, 6]. The fate of apoptotic ECs in SSc is not well examined. A possible defect in the orderly removal of apoptotic cells may lead to phagocytosis by dendritic cells and macrophages and subsequent presentation of cellular antigens to CD8+ T cells [7], leading to immune recognition of vascular antigens. Moreover, apoptotic ECs can activate the alternative complement and coagulation cascades leading to vascular microthrombosis and further tissue compromise [8, 9]. Typical ultrastructural SSc microvascular changes are shown in Fig. 16.1.

(a–c) Representative transmission electron microscopy (TEM) photomicrographs of SSc dermal microvessels. (a) Microvessel shows apoptosis of ECs and delaminated basal lamina (arrows). Apoptotic ECs display plasma membrane blebbing with release of apoptotic bodies. (b) Swollen ECs occluding microvessel’s lumen. (c) Microvessel with an occluded lumen shows thickened basal lamina (asterisks). EC endothelial cell. TEM; original magnification: ×6000
Increased EC thymidine labeling has been reported in SSc, particularly in the indurated phase of the disease [10]. EC thymidine labeling is seen only in the involved “fibrotic” skin, suggesting that EC changes in the uninvolved skin represent cellular activation but not true injury. Significant intimal proliferation and accumulation of proteoglycans in the arterioles and small-sized arteries are common in SSc [11]. Moreover, abnormality of the vessel wall is likely to result from increased synthesis of extracellular matrix (ECM) by intimal and adventitial fibroblasts. Transdifferentiation of ECs into profibrotic myofibroblasts via the process of endothelial–mesenchymal transition (EndoMT) may contribute further to vascular wall fibrosis [12].
The presence of vascular pathology at one site does not necessarily indicate similar pathology at other sites; for instance, the presence of vascular pulmonary pathology clinically manifesting as PAH may not be associated with vascular pathology in the kidneys nor is there any relationship between vascular changes in the gastrointestinal tract (GAVE) and vascular changes in either the lungs or the kidneys. Milder degrees of vascular changes can be seen in clinically uninvolved skin, mainly in the papillary dermal layer in association with platelets adhering to the vessel walls of the dermal microvasculature [3]. It is not known if these changes precede the development of tissue fibrosis or are independent of it. In contrast to small arterioles, large-sized vessel occlusion in SSc is unusual; however, it can occur particularly in the ulnar arteries in patients with the limited form of SSc in association with anticentromere antibodies [13]. In the arteries, intimal proliferation of a uniform and symmetrical nature leads to the formation of a neointima indistinguishable from that seen in other autoimmune diseases, chronic homograft rejection, and accelerated atherosclerosis such as restenosis after coronary bypass [14]. The medial layers are usually thinned, except in hypertensive patients where medial hypertrophy and fibrinoid deposition can be seen [15]. The capillary changes in SSc are most visible in the nail folds.
Nailfold Capillaroscopy
The first description of capillary abnormalities in SSc was published in 1925 by Brown and O’Leary who employed a simple technique to describe capillary changes typical of the microvasculature involvement in SSc [16]. In the 1970s–1980s, the seminal work by Maricq and colleagues popularized the use of wide-field capillary microscopy and described specific “SSc patterns” of capillary changes [17–22] (Fig. 16.2).

Nailfold capillaroscopy. (a) Early scleroderma pattern shows well-preserved capillary architecture and density and presence of dilated and giant capillaries. (b) Active scleroderma pattern shows frequent giant capillaries and hemorrhages, moderate loss of capillaries, and disorganization of capillary architecture. (c) Late scleroderma pattern shows severe capillary architecture disorganization with dropouts, presence of arborized capillaries, and absence of giant capillaries. (d) Normal nailfold capillaroscopic pattern (Courtesy of Cutolo et al. [22], published by Oxford University Press. With permission)
Nailfold capillaroscopy has great diagnostic value in the early stages of the disease. It is the technique of choice for the identification of patients with RP who are at risk for developing SSc [23–25].
Vascular Dysfunction in Scleroderma
The normal vascular endothelium is a single-cell thick layer that is endowed with remarkably diverse functions. ECs regulate coagulation and fibrinolysis, vascular permeability, vascular tone, and metabolism and nutrition of surrounding cells. Injury to the endothelium in SSc leads to profound vascular dysfunction that is prominent in the early stages of the disease and progressively worsens over time [3, 26]. ECs also regulate vascular tone by producing vasoconstrictive and vasodilatory molecules. Vascular dysfunction can cause an imbalance favoring the production of vasoconstrictors. Impaired endothelial-dependent vasodilation in SSc may be due both to overexpression of the potent vasoconstrictor endothelin-1 (ET-1) and reduced production of the vasodilators nitric oxide (NO) and prostacyclin.
Endothelin-1
ET-1 is a powerful dose-dependent vasoconstrictor. At low concentrations, ET-1 is vasodilator, while at higher concentrations, it mediates vasoconstriction and reduction of blood flow [27]. ET-1 is overexpressed in the skin of SSc patients [28, 29]. Moreover, the circulating levels of ET-1 and tissue concentrations of ET-1 in the lungs, kidneys, and liver are elevated in SSc [30–33].
Early studies showed that ET-1 induces fibroblast proliferation and collagen synthesis in a dose-dependent manner, potentially linking ET-1 to vascular dysfunction and fibrosis. ET-1 acts through the endothelin receptors type A (ETA) and type B (ETB) that are expressed on various cell types. An imbalanced expression of ETA and ETB receptors has been reported in SSc patients [34, 35]. The role of endogenous ET-1 in EC dysfunction was examined in tight-skin mice (TSK1), a mouse model of SSc [36]. The mesenteric arteries of TSK1 mice showed decrease responses to acetylcholine, suggesting impaired NO-independent, prostaglandin-mediated relaxation that was partially restored by the dual endothelin receptor antagonist (ERA) bosentan [37], suggesting a role for ET1 in the impaired vasodilation.
ET-1 also stimulates VSMC proliferation and contraction, possibly contributing to remodeling of the vascular wall and vasculopathic manifestations characteristic of SSc, such as PAH and digital ulcers. Indeed, clinical studies have shown that ERAs may be effective in the treatment of SSc-related PAH and in the prevention of digital ulcers [38, 39]. Treatment with the ERA bosentan may also improve peripheral microcirculation in SSc patients, as demonstrated by the shifting from the “late” to the “active” nailfold videocapillaroscopy pattern after therapy [40]. In another study, long-term treatment with bosentan in combination with iloprost reduced the progression of nailfold microvascular damage in SSc patients over a 3-year follow-up period [41].
Nitric Oxide
NO can play a dual role in the endothelium, depending on which NO synthase (NOS) catalyzes its synthesis from L-arginine. Mammals produce three NOS isoforms. Two are expressed in the endothelium; endothelial NO synthase (eNOS) is expressed in ECs, whereas inducible NO synthase (iNOS) is expressed in ECs, VSMCs, fibroblasts, macrophages, and other cell types [42]. Endothelial NOS plays an important role in maintaining vasorelaxation and in controlling vascular tone, blood pressure, and thrombosis [43]. Disruption of the endothelium reduces NO synthesis and release with significant consequences to the vasculature. Deficient endothelial-dependent relaxation in SSc is suggested by impaired maximal responses to endothelial-dependent vasodilators and normal responses to endothelial-independent dilators [44–47].
Despite the expectation that NO production would be reduced in a dysfunctional endothelium, NOx (NO and NO2) levels appear to be elevated in SSc patients, as shown by immunohistochemical studies on skin biopsies [48]. This is probably related to the upregulation of iNOS expression in association with increased levels of immunodetectable nitrotyrosine, a marker of NO-mediated free radical injury [49]. Thus, endothelial dysfunction in SSc may be in part attributable to abnormal regulation of the NO pathway.
Nature of the Endothelial Injury
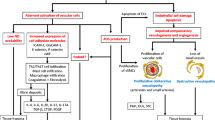
The triggers for vascular injury and dysfunction in SSc are largely unknown. However, infectious agents, EC cytotoxicity, activation and apoptosis, autoantibodies, shear stress, and ischemia/reperfusion are among the potential contributors. Current hypotheses in the pathogenesis of SSc vascular disease implicate a possible, yet unidentified, microbial trigger and an immune-mediated activation that lead to vascular injury and the generation of progressive occlusive arteriolar disease (Fig. 16.3).

Pathogenesis of SSc vasculopathy. Endothelial injury and dysfunction is initiated by the actions of free radicals and environmental, chemical, or microbial agents that injure the endothelium either directly or indirectly by the induction of immune activation and the generation of autoantibodies and activated cellular immunity. The vascular injury leads to the activation of platelet and coagulation pathways that result in vascular microthrombosis. The resulting vasculopathy is associated with intimal hyperplasia in the small arterioles, and the ensuing luminal narrowing results in tissue hypoxia and the generation of the state of chronic ischemia. Released vascular products, in association with hypoxia and ischemia, collectively contribute to the activation of resident fibroblasts that transdifferentiate into profibrotic myofibroblasts and, in turn, perpetuate the vasculopathy by triggering vascular wall fibrosis
Microbial Triggers
Infectious agents may be part of the etiology of SSc, but the evidence is indirect. In one study, patients with RP were treated with triple antibiotics to eliminate Helicobacter pylori infection, a gastric bacterium associated with other vascular diseases [50]. The treatment regimen resulted in the disappearance of RP in 17 % of the patients and reduced symptoms in 72 % [51]. Other studies evaluated the incidence of Helicobacter pylori infection in SSc patients [52]; one study showed a higher correlation, while others showed none [53, 54].
Cytomegalovirus (CMV) infection may play a role in the pathogenesis of SSc vasculopathy. CMV infects ECs and monocytes/macrophages leading to dysregulation of the immune system and upregulation of fibrogenic cytokines [14, 55], events that are also characteristic of SSc. Indirect evidence supporting a role for CMV includes the presence of CMV antibodies in serum from SSc patients that could potentially interact with autoantigens [55, 56]. Rat and mouse models show that CMV infection leads to formation of intimal lesions [57, 58]. An immune-suppressed mouse model of CMV-induced neointima development has many characteristics of SSc including EC apoptosis, myofibroblast differentiation, and increased expression of transforming growth factor-β (TGF-β) and platelet-derived growth factor (PDGF). However, a significant difference between this model and human SSc is that the vascular lesions develop in the main abdominal vessels, while in SSc, the lesions occur in the microvasculature [59].
Parvovirus B19 has also been implicated in the pathogenesis of SSc. Parvovirus B19 is much more commonly found as an active infection in SSc patients (4 %) than the general population (0.6 %) [60]. Parvovirus B19 was detected in bone marrow biopsies from a high percentage of SSc patients (57 %), who did not have an active B19 infection [61]. Further studies found a correlation between B19 expression levels and the severity of endothelial dysfunction in SSc [62]. This suggests a potential role for this virus in SSc pathogenesis.
Endothelial Cell Cytotoxicity, Apoptosis, and Activation
The evidence of EC injury in SSc patients prompted studies to investigate the presence of potential cytotoxic factors in their serum. Some studies suggest that the cytotoxic effect of SSc plasma on ECs is antibody dependent [63–65]. Sgonc et al. showed that activated natural killer (NK) cells are required for antiendothelial cell antibody (AECA)-dependent apoptosis in human dermal microvascular ECs (MVECs) [66]. Two different cytotoxic mechanisms can be stimulated by NK cells: a granzyme/perforin synergistic mechanism and Fas/FasL interaction [67].
Kahaleh et al. reported a circulating proteolytic EC cytotoxic factor in plasma of SSc patients [68]. In a later study, they identified a granular enzyme, thought to be granzyme A that significantly inhibited EC growth [69].
Granzyme B (GrB), a serine protease found in cytoplasmic granules of NK cells and T lymphocytes, is protective against various pathogens [70, 71]. Some studies demonstrate that GrB cleaves vitronectin, fibronectin, and laminin, three proteins that have a primary role in ECM function, and consequently inhibits cell migration, spreading, and invasion [72]. Other studies have shown that the proteolytic activity of GrB and other proteases contained in T-cell granules cleaves plasminogen and plasmin into angiostatin fragments [73], which have multiple consequences in the ECM; plasminogen cleavage in the kringle domains produces antiangiogenic angiostatin, the pool of plasminogen that can be converted to plasmin is reduced, and subsequently, plasmin activity and its effects on regulating ECM composition become limited. Evidence indicates that this mechanism may be enhanced in SSc plasma contributing to increased levels of angiostatin [73].
A number of ECM cleavage products are inhibitors of angiogenesis; they all induce EC apoptosis. These antiangiogenic fragments include the collagen XV and XVIII breakdown product endostatin [74], collagen IV breakdown product tumstatin [75] and canstatin [76], plasminogen breakdown product angiostatin [73, 77], and 18-kDa prolactin fragment [78], among others [79]. Under normal conditions, antiangiogenic factors are tightly regulated to maintain a balance with proangiogenic factors. In addition to elevated plasma angiostatin levels [73], in SSc, there is evidence of elevated levels of endostatin [80, 81] which suggests an imbalance that favors angiogenesis inhibitors with proapoptotic activity.
Antibody-Dependent Cellular Cytotoxicity
Autoimmunity is a major component of SSc. Interestingly, all autoantigens are redistributed and clustered in blebs or apoptotic bodies on the cell surface of apoptotic cells despite their varied functions, structures, and cellular localization [82]. This led Casciola-Rosen et al. to propose that reactive oxygen species (ROS) in the blebs and apoptotic bodies may produce fragments of autoantigens which expose cryptic epitopes recognized by T cells [82]. This hypothesis has been supported by others showing that posttranslational protein alterations expose cryptic cleavage sites with significance in selection of autoantigens [83, 84]. The role of GrB cleavage of autoantigens is suggested by the presence of autoantibodies that recognize GrB-generated autoantigen fragments particularly in limited SSc (lSSc) patients with ischemic digital loss [85–87].
Antiendothelial Cell Antibodies (AECAs)
Circulating autoantibodies to ECs (AECAs) are found in 44–84 % of SSc patients in clinical trials [88, 89]. A greater incidence of AECAs is found in patients with digital ischemia or PAH [90, 91]. Interestingly, AECAs bind distinctly different micro- and macrovascular EC antigens in SSc patients with associated PAH and in patients with idiopathic PAH as compared to SSc patients without PAH [92]. The variant affinities could potentially be used as a predictor of more advanced vascular disease in SSc patients.
The role of AECAs in the induction of EC apoptosis is suggested by findings in the avian model of SSc (UCD 200/2006 chickens) [4]. EC apoptosis was noted following the transfer of AECAs into normal chick embryos [93]; however, apoptosis was also noted in other cell types making it difficult to determine if apoptosis results directly from AECAs binding or if it is part of normal embryogenesis.
SSc sera containing autoantibodies were shown to induce EC apoptosis [94]. A caspase inhibitor could only partially inhibit the apoptosis, suggesting that additional factors are involved in the induction of apoptosis. One such factor may be the ECM microfibrillar protein fibrillin-1. Autoantibodies to fibrillin-1 are found in a large proportion of SSc patients [95]. Fibrillin-1 expression was detected in apoptotic ECs (identified in the blebs and apoptotic bodies) exposed to SSc serum containing AECAs. These data suggest that AECAs found in SSc serum may induce fibrillin-1 expression and EC apoptosis, but the evidence is indirect.
Besides apoptosis, more recently, it has been reported that AECAs may play an important role in activating ECs [96]. Arends et al. demonstrated that immunoglobulins G (IgG) from AECA-positive SSc patients, patients with idiopathic PAH, and patients with systemic lupus erythematosus activate MVECs with increased expression of intercellular adhesion molecule-1 (ICAM-1) [96]. However, disease specificity for this observation is not established. Moreover, Wolf et al. reported significant elevation of anti-ICAM-1 IgM antibodies in the sera of both lSSc and diffuse SSc (dSSc) patients, while elevated anti-ICAM-1 IgG antibodies were found only in lSSc patients [97]. Moreover, anti-ICAM-1 IgG antibodies bind to ECs leading to a significant increase in ROS production [97].
Shear Stress and SSc Vascular Disease
Intimal hypertrophy and vascular remodeling are major contributors to vascular complications and mortality in SSc [98–100]. Key factors that contribute to vascular wall remodeling are shear stress, chronic hypoxia, and inflammation.
Increased shear stress impacts many aspects of the vascular wall. It can enhance EC and VSMC proliferation, increase levels of inflammatory molecules such as interleukin-8 (IL-8), promote ROS and collagen production, and inhibit eNOS levels and EC repair.
Increased cyclic stretch, which accompanies shear stress, plays an important role in regulating EC phenotype and gene expression by inducing ROS production. This leads to increased permeability, production of vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF), apoptosis and cytoskeletal remodeling and increased production of ECM, matrix-degrading enzymes, and inflammation [101, 102]. In this manner, increased cyclic stretch could partially explain the increased formation of ECM in disorders such as SSc. Cyclic stretch causes a much greater loss of endothelial barrier function in response to VEGF stimulation compared to no stretch controls; this effect is partially ameliorated by adding a ROS scavenger to the cells [101].
Reperfusion and Oxidative Injury
The concept that ischemia/reperfusion generates ROS was reported by McCord in 1985 [103]. Ischemia/reperfusion associated with RP leads to ROS generation that may contribute to vascular endothelial damage. Reactive oxygen and nitrogen species can contribute to vascular damage through peroxidation of lipids, oxidation of proteins, or a flawed antioxidant defense system such as that described for NO. Thus, significant decrease in total plasma antioxidant capacity was noted in SSc patients [104]. Moreover, antioxidant levels inversely correlated with the severity of RP and with capillary loss, suggesting that oxidative stress worsens in advanced microvasculature disease. In addition, increased levels of oxidative stress markers were reported and correlated with the overall severity of the disease [105]. A role for autoantibodies in deficient oxidative stress defense was suggested by the presence of autoantibodies that inhibit the oxidative stress repair enzyme methionine sulfoxide reductase A and B (MSRA and MSRB [106–108]) in SSc patients [109]. The antibody titer correlated with renal vascular damage and inversely correlated with pulmonary function [109].
Agonistic Antibodies to the Platelet-Derived Growth Factor Receptor and Generation of ROS
Baroni et al. identified autoantibodies in the sera of SSc patients that interact with and activate the receptor for platelet-derived growth factor (PDGFR), resulting in ROS accumulation in target cells that can potentially account for tissue injury [110]. Furthermore, these antibodies are capable of upregulating the expression of α-smooth muscle actin (α-SMA) and type I collagen in fibroblasts. Of note, these genes are characteristically overexpressed in SSc fibroblasts. It is interesting to note that PDGFR plays an important role in angiogenesis related to ischemic tissue and tumor growth [111]; nevertheless, the possible vascular consequences of the receptor activation in SSc are not known. Moreover, subsequent reports were unable to confirm the presence of stimulatory anti-PDGFR antibodies in SSc sera. Differences in the methodologies of these studies might account for the discrepant results, and further investigations are needed to further dissect the role of stimulatory anti-PDGFR antibodies in SSc [112].
Epigenetics and SSc Vasculopathy
The basis for EC dysfunction and altered cellular phenotype in SSc has not been determined, but primary metabolic abnormalities, responses to abnormal environmental signals, and clonal selection have all been hypothesized to play a role [113]. Nonetheless, SSc cellular abnormalities persist in multiple generations in vitro. The persistence of dysfunctional phenotype outside the disease environment suggests the possibility of an in vivo imprinting of disease phenotype that is inherited and is transmitted from one generation to the next. The term epigenetics describes all inherited changes in gene expression that are not coded in the DNA sequence itself. The two major mechanisms that are known to mediate epigenetic changes are DNA methylation and histone modification. More recently, microRNAs (miRNAs) have been identified as a major contributor to gene expression through posttranscriptional mechanisms [114] and thus were added to the epigenetic machinery.
Methylation of the CpG dinucleotides has long been recognized as a major epigenetic modification of the mammalian genome and is implicated in imprinting, X chromosome inactivation, embryonic development, defense against retroviral sequences, transcriptional repression of certain genes, and carcinogenesis [115, 116]. CpG islands are stretches of DNA located within the promoter regions of about 40 % of mammalian genes. The methylation of CpG islands leads to a stable, heritable repression of transcription of the affected gene. DNA methylation is the most widely studied epigenetic mechanism in autoimmune diseases and is considered the core epigenetic control mechanism. DNA methylation is accomplished by the de novo DNA methyltransferases (DNMTs) DNMT3a and DNMT3b, while an existing methylation pattern is maintained during cell division and thus inherited in proliferating cells by DNMT1 [117].
There is emerging evidence suggesting that alteration of DNA methylation profiles at global or gene-specific levels may contribute to SSc pathogenesis. Endothelial nitric oxide synthase gene (NOS3) expression is reduced in SSc skin and cultured ECs [118]. Data suggest that heavy methylation of the CpG sites in the promoter region of NOS3 gene leads to gene repression and that the addition of DNMT1 inhibitor 5-azacytidine leads to normalization of NOS3 expression. The significance of underexpression of NOS3 is illustrated by the fact that NOS3-null mice are characterized by systemic and pulmonary hypertension, impaired wound healing and angiogenesis, and impaired mobilization of stem and endothelial precursors, leading to failure of neovascularization [119, 120]. Another endothelial gene that appears to be regulated by epigenetic control is bone morphogenic protein receptor II (BMPRII). BMPs are members of the TGF-β superfamily of proteins that coordinate cell proliferation, differentiation, and survival. The latter is particularly true for ECs, since BMP signaling through BMPRII favors EC survival and apoptosis resistance. An inactivating mutation in the BMPRII gene has been linked to primary pulmonary hypertension suggesting a crucial role in the development of PAH-related vasculopathy [121]. In SSc, significant decrease in the expression levels of BMPRII in ECs and skin was described [122]. This was shown to be associated with enhanced responses of SSc ECs to apoptotic signals, including serum starvation and oxidation injury. Sequencing the BMPRII promoter region after bisulfite conversion demonstrated heavy methylation of CpG sites in BMPRII promoter in SSc ECs. BMPRII expression levels and the enhanced EC apoptotic responses were normalized by the addition of 5-azacytidine. These data suggest that epigenetic repression of BMPRII gene may play a central role in EC vulnerability to apoptosis and that impaired BMPRII signaling in SSc ECs may contribute to the pathogenesis of SSc vasculopathy [122].
The exact nature of epigenetic triggers in SSc is not known, but both hypoxia and oxidation injury are suggested triggers, as well as nutrients and other environmental factors [123].
Defective Vascular Regeneration in SSc
Vasculopathy in SSc is systemic, progressive, and often irreversible. This suggests that structural changes culminating in the obliteration and loss of the microvasculature are not normally repaired by either a compensatory growth of new vessels from existing vessels (angiogenesis) or de novo formation of new vessels (vasculogenesis). Both defective angiogenic pathways, as well as abnormalities in the release of bone marrow-derived progenitor cells, which have the potential to initiate vascular repair, have been identified in SSc patients.
Defective Angiogenesis: The Proangiogenic to Antiangiogenic Switch
Tissue ischemia and hypoxia are usually the main triggers for angiogenesis through the upregulation of proangiogenic factors, which then initiate angiogenic sprouting from preexisting microvessels by inducing vasodilation, proliferation, and migration of ECs, invasion of the surrounding extracellular matrix, and formation and stabilization of the vascular lumen of newly formed capillary vessels [124]. However, despite chronic tissue ischemia and progressive loss of microvessels in multiple vascular beds, compensatory angiogenesis is dysregulated and insufficient, and it does not allow vascular recovery in the course of SSc [125, 126].
Increasing evidence indicates that a dysregulated expression of a large array of proangiogenic and antiangiogenic factors may be largely responsible for the impaired angiogenic response found in patients with SSc [125, 126]. The nailfold capillaroscopic changes observed in the course of SSc may be explained by the action of different factors on angiogenesis. In the early stages of the disease, a proinflammatory state and an increased production of proangiogenic factors may stimulate angiogenesis. As a result, capillaroscopic analysis of the nailfold bed demonstrates the presence of microhemorrhages and tortuous, giant capillary loops, which are immature and instable microvessels presumably formed during an uncontrolled angiogenic response. This short proangiogenic response is followed by a dramatic impairment of the angiogenic process which might in part be explained by the action of several antiangiogenic factors, ultimately resulting in reduced capillary density and extensive avascular areas. Thus, the dramatic switch from proangiogenic to antiangiogenic characteristics suggests that the angiogenic process becomes impaired.
However, the functional role of angiogenic mediators in the disease pathogenesis is still poorly understood. Interestingly, while both several proangiogenic and antiangiogenic factors are overexpressed in SSc [125, 126], there appears to be an imbalance in the ratio of these mediators, favoring inhibition of angiogenesis and progressive vascular disease (Table 16.1).
Among the proangiogenic factors, VEGF is considered one of the most potent regulators of physiologic and pathologic angiogenesis and is overexpressed in most angiogenic conditions. Surprisingly, several studies have demonstrated enhanced expression of VEGF in both the skin and the circulation of SSc patients throughout different disease stages [125, 127–131]. The paradox is that VEGF levels, rather than being associated with evidence of angiogenesis, actually correlate with progressive microvascular loss and disease progression. In fact, a positive correlation was found between VEGF levels and the severity of nailfold capillary loss as well as with the extent of skin fibrosis measured by Rodnan skin thickness score [130]. Moreover, VEGFR-1 and VEGFR-2 are also upregulated on dermal ECs in SSc-affected skin [129, 131]. Nevertheless, there is also evidence suggesting that VEGF receptor signaling may be impaired in SSc based on the observation that dermal MVECs isolated from SSc patients show impaired response to VEGF in vitro [132].
As an explanation for the “VEGF paradox,” it has been shown that VEGF-A primary transcript can be alternatively spliced in its terminal exon, producing two distinct mRNA splice variants that are translated to the proangiogenic VEGF165 and antiangiogenic VEGF165b isoforms. Of interest, Manetti et al. reported that the increase in VEGF in SSc skin was the result of a significant increase in the antiangiogenic VEGF165b isoform instead of VEGF165 [133]. Moreover, they provided evidence that circulating levels of VEGF165b were raised in SSc and that these increased levels were both early and persistent features of the disease. In addition, MVECs isolated from SSc skin constitutively expressed and released higher levels of VEGF165b than did MVECs from healthy individuals [133]. Interestingly, VEGF165 and VEGF165b isoforms bind to the tyrosine kinase receptor VEGFR-2 with the same affinity, but binding of VEGF165b results in an insufficient tyrosine phosphorylation/activation of VEGFR-2 and incomplete or transient downstream signaling, which leads to an impaired angiogenic response [127]. Accordingly, SSc MVECs expressed higher levels of VEGFR-2 but also showed impaired phosphorylation/activation of this receptor, reduced activation of extracellular signal-regulated kinase (ERK)1/2, and impaired capillary morphogenesis in vitro [133]. Of note, treatment with recombinant VEGF165b, as well as with conditioned media from SSc MVECs, inhibited VEGF165-mediated VEGFR-2 phosphorylation, ERK1/2 signaling, and capillary morphogenesis in healthy MVECs, and these antiangiogenic effects could be abrogated by the administration of anti-VEGF165b neutralizing antibodies. The angiogenic potential of SSc MVECs is significantly impaired, but can be restored by the addition of combination of high-dose recombinant VEGF165 and anti-VEGF165b-blocking antibodies [133]. Finally, it is interesting to note that the switch from proangiogenic to antiangiogenic VEGF isoforms may be driven by TGF-β and the SRp55 splicing factor [133]. In another recent study, Manetti et al. [134] have reported that plasma levels of the antiangiogenic VEGF165b isoform correlated with the severity of capillary architectural loss and derangement, further suggesting that VEGF165b may participate in the loss of microvessels in SSc (Fig. 16.4).

In most angiogenic conditions, such as cancers, proangiogenic VEGF165 predominates, whereas in deficient angiogenic conditions, such as SSc, the balance shifts to favor the expression of the VEGF165b antiangiogenic isoform. The two isoforms bind to VEGFR-2 with the same affinity, but binding of VEGF165b results in an insufficient tyrosine phosphorylation/activation of VEGFR-2 and incomplete or transient downstream signaling, which lead to impaired angiogenic response
A dysregulated expression of different chemokines and their receptors has also been linked to disturbed angiogenesis in SSc. In particular, antiangiogenic chemokines lacking the enzyme-linked receptor (ELR) motif, such as monokine induced by interferon-γ (IFN-γ) (MIG/CXCL9) and IFN-inducible protein 10 (IP-10/CXCL10), are elevated in SSc serum, whereas the expression of their receptor CXCR3 is downregulated on SSc dermal ECs [135]. In contrast, proangiogenic CXCL16 and its receptor CXCR6 are elevated in SSc serum and on SSc dermal ECs, respectively [135]. Thus, these findings may suggest that angiogenic chemokine receptor expression is likely regulated in an effort to promote angiogenesis.
Elevated levels of antiangiogenic pentraxin 3 (PTX3), a multifunctional pattern recognition protein that can suppress fibroblast growth factor 2 (FGF-2) proangiogenic function, have been associated with vascular manifestations of SSc [136]. Circulating PTX3 and FGF-2 levels were significantly higher in SSc patients than in healthy subjects. Moreover, circulating levels of PTX3 are elevated in SSc patients with digital ulcers or PAH, while FGF-2 is reduced in SSc patients with PAH. Multivariate analysis identified elevated PTX3 as an independent parameter associated with the presence of digital ulcers and PAH, and PTX3 levels were a useful predictor of future occurrences of digital ulcers. Reduced FGF-2 was independently associated with the presence of PAH [136].
In another recent study, Manetti et al. investigated the possible involvement of epidermal growth factor-like domain 7 (EGFL7), a unique proangiogenic molecule which is predominantly expressed and secreted by ECs and their progenitors and controls vascular development and integrity [137]. Circulating levels and dermal expression of EGFL7 were significantly decreased in SSc patients. Remarkably, the decrease of serum EGFL7 levels in SSc patients correlated with the severity of nailfold capillary abnormalities and presence of digital ulcers. In contrast to constitutive endothelial expression of EGFL7 in healthy skin, EGFL7 was found to be strongly reduced or even undetectable in SSc dermal microvessels. Furthermore, EGFL7 protein was found to be significantly downregulated in vitro in dermal MVECs obtained from SSc patients compared with control cells. Thus, it has been suggested that the loss of EGFL7 expression might contribute to the development and progression of peripheral microvascular damage and defective vascular repair process in SSc patients [137].
Defective Vasculogenesis
In the past, it had been thought that impaired angiogenesis and increased apoptosis of mature ECs were exclusively responsible for the microvascular abnormalities in SSc. However, several studies published over the last few years suggest that impairment of vasculogenesis (i.e., the generation of new blood vessels by stem or progenitor cells) and vascular repair may be also involved in SSc capillary loss, as shown by altered numbers and functional defects of circulating EPCs [125]. In fact, new blood vessels can be formed in the adult not only by the sprouting of fully differentiated ECs but also by recruitment of circulating progenitor cells, independently of the preexisting vasculature [138]. In particular, a subset of bone marrow-derived CD34+ progenitor cells can acquire the characteristics of mature ECs, express EC markers, and incorporate into new capillary vessels at sites of ischemia [138]. Postnatal vasculogenesis contributes to vascular healing in response to vascular injury through the processes of rapid re-endothelialization of denuded vessels and collateral vessel formation in ischemic tissues. In this process, EPCs (identified as CD34+/CD133+/VEGFR-2+ cells) are mobilized from their bone marrow niches into the circulation in response to stress- and/or damage-related signals and migrate through the bloodstream and home to the sites of vascular injury, where they extravasate through the endothelium and contribute to the formation of neovessels and the repair of damaged vasculature working in concert with preexisting mature ECs.
Several studies have analyzed the levels of circulating EPCs in the peripheral blood of SSc patients in comparison with healthy controls and/or other rheumatic conditions [125, 139, 140]. Although conflicting results have been reported, a possible correlation between reduction in the number of EPCs and severity of peripheral vascular manifestations has been demonstrated [141]. Moreover, it has been shown that SSc serum may induce EPC apoptosis and that this might account, at least in part, for the decreased numbers of circulating EPCs in SSc patients [142]. AECAs may also be directly responsible for apoptosis of EPCs in SSc [143]. In another study, EPC (defined as CD34+/CD133+/CD309+) counts were significantly lower in SSc patients with digital ulcers or PAH and correlated negatively with circulating levels of antiangiogenic PTX3 [136]. Interestingly, PTX3 inhibited differentiation of EPCs in vitro [136]. Moreover, EPCs from SSc patients exhibited an impaired in vivo neovascularization capacity in a model of cotransplantation of immunomagnetically sorted circulating EPCs and murine colon carcinoma CT-26 cells beneath the skin of severe combined immunodeficiency (SCID) mice [144].
In addition to CD34+ EPCs, it has been proposed that bone marrow-derived CD14+ monocytes can serve as a subset of EPCs because of their expression of endothelial markers and ability to promote neovascularization in vitro and in vivo [145]. However, the current consensus is that monocytic cells do not give rise to ECs in vivo, but rather function as support cells, by promoting vascular formation and repair through their recruitment to the site of vascular injury, secretion of proangiogenic factors, and differentiation into mural cells (VSMCs and pericytes). These CD14+ monocytes that function in a supporting role in vascular repair are now termed monocytic proangiogenic hematopoietic cells (PHCs) [145]. Yamaguchi et al. recently showed that in patients with SSc, circulating monocytic PHCs increase dramatically in association with enhanced angiogenic potency, suggesting that these compensatory effects may be induced in response to defective vascular repair machinery [145].
Cipriani et al. examined the in vitro capacity of bone marrow-derived mesenchymal stem cells (MSCs) to differentiate toward the EC lineage [146]. In SSc patients, the percentage of VEGFR-2+ and CXCR4+ MSCs and endothelial-like MSCs was significantly lower than in controls. Accordingly, both SSc MSCs and endothelial-like MSCs displayed impaired responses to VEGF- and stromal cell-derived factor-1 (SDF-1)-induced migration, invasion and capillary-like structure formation on Matrigel, as well as an early senescence [146]. These data collectively suggest that endothelial repair may be affected in SSc starting from bone marrow stem niches. However, despite an impaired commitment toward the endothelial phenotype, Guiducci et al. [147] demonstrated that SSc bone marrow-derived MSCs exhibit an intact, or even potentiated, proangiogenic paracrine machinery that is suitable for autologous therapeutic approaches. Indeed, SSc MSCs overexpress different proangiogenic growth factors and are able to promote dermal EC sprouting angiogenesis in vitro [147]. In agreement with these findings, the same authors reported that intravenous infusion of culture-expanded autologous bone marrow-derived MSCs could regenerate the peripheral vascular network in a case of SSc complicated by acute gangrene of the extremities [148]. Other observations have provided support for the efficacy of MSCs in treating intractable digital ulcers, where autologous injection of MSCs led to a reduction in the size of the ulcers, an increase in blood flow, and the formation of new capillaries [149].
The Plasminogen Activator Pathway
The plasminogen activator (PA) pathway plays key roles in angiogenesis. Plasminogen is a precursor of proangiogenic plasmin [150] and a precursor of antiangiogenic angiostatin [77]; therefore, it plays a complicated role in the regulation of vascular homeostasis. Plasminogen can be cleaved at the carboxy terminus by PA to produce plasmin, a proteolytic and fibrinolytic protease. Its proteolytic activity activates many key proangiogenic factors including VEGF [151], TGF-β [152] and matrix metalloproteinases (MMPs) [153]. Plasminogen can also be cleaved within its amino terminus kringle domains by a number of proteases [154, 155]. The cleaved kringle domain fragments are angiostatin of various sizes with different antiangiogenic activity. Thus, alterations in plasminogen processing can have a profound effect on angiogenic homeostasis.
Plasmin activity is reduced in SSc while the amount of circulating angiostatin is increased [73]. This proangiogenic/antiangiogenic imbalance was manifested by reduced migration and proliferation of normal human dermal MVECs when exposed to SSc patients’ plasma. Interestingly, exposure of normal dermal MVECs to angiostatin in amounts similar to those detected in SSc plasma resulted in a significant impairment of cell migration and ability to form vascular structures in collagen gels [73].
The PA pathway is further implicated in SSc by studies that examined the importance of urokinase plasminogen activator receptor (uPAR) in ECs isolated from SSc patients’ skin biopsies. uPAR plays an important role in cell motility, adhesion, and matrix invasion through its binding interactions with urokinase plasminogen activator (uPA) [156], vitronectin [157], and intracellular signaling mediators, such as the integrin receptors [158]. D’Alessio et al. have shown that in dermal MVECs isolated from SSc skin, uPAR undergoes truncation between domains 1 and 2, a cleavage that is known to impair uPAR functions [132]. The uPAR cleavage in SSc MVECs was associated with the overexpression of MMP-12 [132]. Indeed, overproduction of MMP-12 by SSc dermal MVECs and fibroblasts accounts for endothelial uPAR cleavage leading to impaired uPA-induced MVECs migration, invasion, proliferation, and capillary morphogenesis on Matrigel [132]. Strikingly, treatment with an anti-MMP-12 monoclonal antibody was able to restore the angiogenic activity of normal MVECs treated with conditioned media from SSc MVECs [132, 159]. Furthermore, uPAR cleavage in SSc MVECs results in loss of integrin-mediated uPAR connection with the actin cytoskeleton [159]. The uncoupling of cleaved uPAR from β2 integrins impairs the activation of the small Rho GTPases Rac and Cdc42 and thus inhibits uPAR-dependent cytoskeletal rearrangement and cell motility, and ultimately resulting in impaired angiogenesis [159]. In a subsequent study, the same authors reported that MMP-12 gene silencing could in part restore the ability of SSc MVECs to produce capillary structures in vitro [160]. Moreover, elevated levels of MMP-12 in the circulation of SSc patients were associated with the severity of nailfold capillary abnormalities and the presence of digital ulcers [161]. Interestingly, elevated MMP-12 may suppress angiogenesis not only through the cleavage and subsequent inactivation of uPAR [132] but also through the proteolysis of plasminogen and generation of angiostatin [162].
Microarray Studies Revealing Altered Balance of Proangiogenic and Antiangiogenic Factors in SSc
Gene expression levels of proangiogenic and antiangiogenic factors have been analyzed in microarray studies which compared the transcriptome profiling of dermal MVECs isolated from skin biopsies from normal individuals and SSc patients [163, 164]. One microarray gene expression study detected important differences between normal and SSc MVECs in the kallikrein gene family [163]. Proangiogenic kallikreins 9, 11, and 12 were downregulated in SSc MVECs, whereas antiangiogenic kallikrein 3 was upregulated. The microarray data were further validated in experiments using normal MVECs treated with antibodies against kallikreins 9, 11, and 12 and subsequently analyzed in migration, proliferation, and capillary morphogenesis functional assays. All three antibodies were able to block angiogenesis in healthy MVECs [163]. Among the multiple kallikrein family members, tissue kallikrein (also known as kallikrein 1 or “true” tissue kallikrein) is a serine protease that cleaves kininogen and thereby regulates the kininogen–kinin pathway. Tissue kallikrein synthetized at the blood vessels acts through kinins which modulate a broad spectrum of vascular functions, playing an important role in the regulation of vascular homeostasis and angiogenesis [165–167]. Del Rosso et al. found that tissue kallikrein circulating levels are increased in SSc, particularly in patients with signs of early and active vascular disease, suggesting a role in the development of SSc microvascular abnormalities [168].
A second microarray gene expression study examined the expression of 14,000 genes in MVECs from skin biopsies obtained from normal subjects and dSSc patients [164]. Genes were categorized into functional groups based on gene ontology. Of those 14,000 genes, SSc MVECs overexpressed a number of proangiogenic transcripts but also a variety of genes that have a negative effect on angiogenesis [164]. Conversely, several genes that promote cell migration and adhesion to the ECM were downregulated in SSc MVECs, suggesting an anti-invasive phenotype of these cells [164]. In particular, the angiogenesis inhibitor PTX3, which is known to inhibit the proangiogenic effects of FGF-2, was strongly upregulated in SSc MVECs compared with normal MVECs [164]. Interestingly, in a subsequent study, Margheri et al. reported that silencing of PTX3 in SSc MVECs significantly increased their invasion in Matrigel and could in part restore their ability to produce capillary tubes in vitro [160]. Moreover, the 14,000 gene microarray study reported a reduced expression of desmoglein-2 (DSG2) in SSc MVECs [164]. Thus, to identify the role of DSG2 downregulation in the defective angiogenesis characteristic of SSc, Giusti et al. [169] studied the effect of silencing DSG2 in normal MVECs using DSG2-siRNA. The authors reported impaired actin stress fiber formation and diminished capillary morphogenesis in vitro. Of note, transfection of DSG2 into SSc MVECs restored their angiogenic properties in vitro to normal levels [169].
Avouac et al. investigated the gene expression profile of circulating EPC-derived ECs in normal and hypoxic conditions [170]. Their data revealed important gene expression changes in EPC-derived ECs from SSc patients, characterized by a proadhesive, proinflammatory, and activated phenotype [170].
Finally, the gene array study of Tinazzi et al. showed that circulating ECs from SSc patients display an altered expression of genes involved in the control of apoptosis and angiogenesis which may be modified by iloprost infusion [171].
Role of Perivascular Cells
Pericytes are located on the abluminal side of ECs in capillaries, precapillary arterioles, and postcapillary venules [172–174]. They are in close spatial proximity to and provide partial coverage for ECs [172]. Pericyte cell bodies and ECs are contained within the same basement membrane [175]. Vascular stabilization or maturation requires maintenance of established vessels and EC coverage by pericytes or VSMCs around the endothelial layer during angiogenesis [176].
Pericytes are contractile cells [177] which enable them to participate in hemodynamic regulation of microvascular blood flow and permeability by modifying (increase or decrease) inflammatory leakage at EC junctions. Additionally, pericytes contract in response to vasoactive mediators; ET-1, norepinephrine, angiotensin II, and bradykinin stimulate pericyte contraction, while NO, adenosine, adrenergic antagonists, and lipopolysaccharide induce pericyte relaxation [178–181].
Vascular permeability can be regulated by the extent of EC coverage by pericytes or by the extent of pericyte contraction that modifies EC intercellular junctions [182–185]. Pericyte coverage of EC junctions prevents leakage of proteins and cells from the vessel wall [174].
Pericytes are considered to be progenitor cells with the potential to differentiate into different cell types such as fibroblasts and myofibroblasts [186–189]; thus, they may link vascular injury to tissue fibrosis. Microvascular pericytes express PDGF receptors in wound healing and dermal scarring tissue [190, 191], as well as in early stages of SSc [192–194]. In another study [195], myofibroblasts were found in SSc skin biopsies, but not in normal skin. Two markers of myofibroblast differentiation were expressed in both pericytes and myofibroblasts in SSc but not in control biopsies [195], suggesting that pericytes may represent a source of myofibroblasts during dermal fibrosis. Asano et al. reported a decreased pericyte coverage of dermal capillaries in SSc [196].
Besides fibroblasts, myofibroblasts, and pericytes, recent evidence indicates that other stromal cell types may be implicated in the pathophysiology of SSc. Telocytes, formerly called interstitial Cajal-like cells, are a distinct population of stromal cells that are characterized by very long cytoplasmic processes [197]. Telocytes act as supporting cells by forming a scaffold that defines the correct three-dimensional organization of tissues/organs during prenatal life and during tissue repair/renewal in postnatal life. It has been suggested that telocytes are involved in intercellular signaling that could influence the transcriptional activity of neighboring cells, either directly, by cell-to-cell contacts, or indirectly, by shedding microvesicles and exosomes or by secreting paracrine signaling molecules, including miRNAs [197]. Recently, Manetti et al. reported that telocytes display severe ultrastructural damage suggestive of ischemia-induced cell degeneration and are progressively lost from clinically affected SSc skin [198]. The authors proposed that, in SSc skin, the progressive loss of telocytes contributes to the altered three-dimensional organization of the ECM and may reduce the control of fibroblast and myofibroblast activity, thus favoring the fibrotic process [198]. Since telocytes are abundant in perivascular location, where they intimately surround the vessel basement membrane, and produce proangiogenic factors and participate to vascular stability [197], presumably their loss may even be linked to microvascular abnormalities in SSc. Immunofluorescence and transmission electron microscopy photomicrographs of telocytes and microvessels in healthy and SSc skin are shown in Fig. 16.5.

(a, b) Representative photomicrographs of skin sections from healthy controls and SSc patients double immunostained for CD34 (green) and CD31 (red) and counterstained with DAPI (blue) to identify the nuclei. Telocytes are identified as CD34-positive/CD31-negative stromal cells, while endothelial cells are CD34/CD31 double positive. (a) Telocytes are numerous in healthy dermis, where they surround the microvessels (arrows). (b) No telocytes can be observed in SSc affected dermis. Original magnification: ×40. (c, d) Representative transmission electron microscopy (TEM) photomicrographs of healthy and SSc skin. (c) Healthy skin. The thin and long varicose processes of a perivascular telocyte (TC) encircle the basal lamina of a blood microvessel (arrows). (d) SSc skin. Telocytes are not identifiable around an occluded microvessel. Only a few cell debris are observed. The vessel basal lamina is thickened (asterisk). EC endothelial cell. TEM; Original magnification: ×6000
Vascular Wall Remodeling in SSc
Vascular remodeling follows microvascular injury and damage. Intimal and medial thickening and adventitial fibrosis are the common forms of remodeling found in SSc [149]. Intimal lesions change the vessel function and alter the composition and organization of the ECM. VSMCs are likely responsible for generating the fibrotic intimal lesions [149]. Under normal conditions, they assume a contractile or differentiated phenotype and regulate vessel diameter and blood flow. In pathological conditions, VSMCs become synthetic or dedifferentiated and generate intimal lesions through their secretion of ECM proteins in the absence of their contractile function [199–201]. Dysfunctional ECs, infiltrating leukocytes, and altered ECM in SSc provide the cues for intimal VSMCs to proliferate and form a fibrotic vascular lesion. In a recent study, Arts et al. demonstrated that a unique SSc IgG may induce growth and profibrotic state in VSMCs through the activation of the epidermal growth factor receptor signaling cascade [202].
Another theory is that transdifferentiation of ECs via the process of TGF-β-induced EndoMT may preferentially lead to subendothelial accumulation of myofibroblasts and fibrotic tissue with subsequent neointimal thickening, leading to a fibroproliferative vasculopathy [12, 203].
The ECM and basement membrane provide a support scaffold that is essential for blood vessel stability. Adhesion of ECs to the ECM enables them to undergo migration, proliferation, and morphogenesis, which are all necessary for neovascularization [204]. Degradation of the ECM/basement membrane leads to vessel collapse/regression [205–208]. Proteases that degrade the ECM/basement membrane play a key role in matrix remodeling during normal wound healing as well as in vascular diseases such as SSc.
The proteolytic activity of plasmin modifies many aspects of the ECM. It is the primary protease that degrades fibrin [207, 208], an ECM protein that forms a supportive scaffold for angiogenic vessels [209–211]. Fibrin, the major constituent of provisional matrix, enables ECs to adhere, spread, and proliferate [212, 213]. Fibrin or accumulated fibrinogen can be broken down by plasmin to negatively regulate angiogenesis [214, 215]. Reduced plasmin in SSc patients has been reported [73].
In addition to its own proteolytic activity, plasmin contributes to matrix remodeling by activating numerous MMPs [216, 217]. Of those, MMP-1, -3, -9, -10, and -13 promote capillary network regression followed by EC apoptosis [218, 219]. Several laboratories have examined plasma from SSc patients for the presence of various MMPs. One study reported that MMP-9 was downregulated in SSc patients, particularly those with PAH [220]. In another study, MMP-9 concentrations were greater in SSc plasma than controls [221].
Vascular Hypoxia in the Pathogenesis of SSc
Capillary rarefaction and disturbed blood flow, as well as excessive ECM accumulation, cause chronic tissue hypoxia in SSc. It was postulated that the avascular antiangiogenic areas visible in nailfold beds may result from tissue hypoxia and that the enlargement of capillaries may be a localized compensatory proangiogenic response to tissue hypoxia [125, 129]. In fact, intradermal skin oxygenation measurements verified hypoxic conditions in SSc patients with more advanced disease as compared to those with early disease and controls. Interestingly, expression of HIF-1α, a hypoxia-inducible transcriptional factor that upregulates VEGF, in the skin of SSc patients was lower than in controls. Additionally, HIF-1α expression did not correlate with VEGF expression pattern. Thus, it is concluded that increased VEGF expression in SSc patients is not stimulated by hypoxia, but could be the basis of megacapillaries [129]. However, subsequent studies demonstrated that the increase in VEGF expression in SSc skin was mainly the result of a significant increase in the antiangiogenic VEGF165b isoform instead of proangiogenic VEGF165 [133].
The expression levels of VEGF, VEGFR-1, and VEGFR-2 have been examined also in late-outgrowth EPCs isolated from SSc patients and normal controls [222]. This study showed reduced mRNA and protein expression of VEGFR-1 in SSc EPCs under hypoxic conditions. Serum levels of soluble VEGFR-1 were significantly lower in SSc patients compared to controls, while soluble VEGF levels were elevated in SSc patients. Thickened skin in SSc may lead to hypoxia, as suggested by oxygen measurement in SSc skin [129, 223]. In both studies, the lowest oxygen levels were found in the thickened fibrotic skin. DNA microarray studies in human pulmonary arterial ECs exposed to normoxic or hypoxic conditions and in ECs infected with a constitutively active HIF-1α or β-galactosidase and exposed to nonhypoxic conditions [224] showed that genes upregulated by HIF-1α regulate collagen biosynthesis. The list includes COL1A2, COL4A1, COL4A2, COL5A1, COL9A1, COL18A1, procollagen prolyl hydroxylases, lysyl oxidase, and lysyl hydroxylases [224]. Thus, hypoxia could actively contribute to the fibroproliferative vasculopathy with intimal thickening characteristic of SSc.
In a recent study, Makarenko et al. [225] demonstrated that exposing human MVECs to intermittent hypoxia induces cell dysfunction associated with an increase in ROS formation and ERK phosphorylation. Of note, inhibition of the ERK signaling pathway and the use of antioxidants prevented the effect of intermittent hypoxia on MVECs [225]. This observation argues for a possible role for antioxidants in prevention of MVEC dysfunction mediated by intermittent hypoxia in vascular disorders such as SSc.
Relationship of Vasculopathy to Fibrosis
SSc is a connective tissue disease with markedly increased levels of collagen deposition that causes tissue fibrosis. Fibroblasts acquire abnormal characteristics that include increased proliferation, a switch to the activated state (myofibroblasts), and synthesis of abnormally high levels of collagen and other ECM components [226]. Excessive ECM deposition and accumulation in the heart, lung, vessels, skin, and kidney result in uncontrolled organ fibrosis and organ dysfunction, which are a major part of the SSc pathology. Despite the abnormal fibroblast activities that lead to the fibrotic state in SSc patients, the defective vasculature response to injury is considered to occur before fibrosis [149]. There are various hypotheses as to the mechanism by which the vascular system may contribute to fibrosis. One theory proposes that cytokines secreted by activated ECs form a gradient that attracts fibroblasts to the blood vessels which, in turn, stimulate fibroblast proliferation, activation, as well as synthesis and secretion of collagen. In support to this hypothesis, the deposition of ECM appears mostly clustered in perivascular regions in early fibrotic lesions. Another proposed mechanism is that ECs become permeable, which results in interstitial secretion of factors that stimulate fibroblast proliferation, activation, and production of collagen [226].
Two factors that have been implicated in the link between SSc vasculopathy and fibrosis are TGF-β and connective tissue growth factor (CTGF/CCN2). The TGF-β-dependent mechanism is linked to EC apoptosis and myofibroblast differentiation. Apoptotic ECs recruit phagocytes, particularly macrophages that engulf the apoptotic cells. This event stimulates upregulation of TGF-β, which promotes profibrotic myofibroblast differentiation and apoptosis resistance [227].
Another proposed mechanism is centered on CTGF and is independent of TGF-β pathway [228]. CTGF is a fibrinogenic protein that is constitutively expressed in dermal fibroblasts isolated from dSSc patients [229, 230]. Furthermore, CTGF injected subcutaneously into neonatal NIH Swiss mice stimulates tissue fibrosis without TGF-β upregulation. Further delineation of this mechanism was performed in C3H mice injected subcutaneously with serum-free medium conditioned by apoptotic murine aortic ECs. The mice that received the apoptotic ECs developed thickening of the skin with elevated expression of α-SMA, vimentin, and collagen and myofibroblast differentiation. Myofibroblast differentiation was reversed when caspase-3 expression was inhibited in ECs. CTGF was identified as the sole fibrinogenic protein whose expression was upregulated in mice treated with apoptotic ECs [228].
The notion that MVECs may play a central role in the pathogenesis of SSc, including activation of the fibrotic program, was reemphasized recently by a report from Serratì et al. [231], who demonstrated that conditioned media obtained from cultured SSc MVECs, but not control MVECs, evoked fibroblast activation as suggested by the overexpression of α-SMA, vimentin, and type I collagen. Moreover, the activated fibroblasts exhibited an aggressive mesenchymal cell behavior by upregulating MMP-2 and MMP-9 expression and acquiring the ability to invade Matrigel. The effect of SSc MVEC conditioned media on fibroblasts was mediated by elevated CTGF/CCN2, which in turn activates TGF-β. Interestingly, the use of CTGF/CCN2 inhibitor and TGF-β inhibitor peptides reversed fibroblast activation induced by SSc MVEC conditioned media [231].
Vascular injury can activate platelets to release serotonin and other profibrotic mediators, which ensures efficient and self-limited wound healing under physiologic conditions. By contrast, chronic vasculopathy may cause persistent increase in serotonin levels and perpetuate profibrotic tissue repair processes, thus linking vascular disease and fibrosis in SSc [232]. Indeed, serotonin stimulates fibroblasts to release excessive amounts of ECM proteins. The profibrotic effects of serotonin are exclusively mediated via 5HT2B receptors which are overexpressed in SSc, suggesting an increased sensitivity of SSc fibroblasts to the profibrotic effects of serotonin [232].
Finally, an important link between vasculopathy and fibrosis in SSc pathogenesis may be represented by chronic overexpression of VEGF. In fact, Maurer et al. reported that double (+/+) VEGF transgenic mice spontaneously develop significant skin fibrosis, indicating profibrotic effect of VEGF in a gene-dosing manner [233]. Moreover, in vitro analysis revealed that VEGF is able to directly induce collagen synthesis in dermal fibroblasts [233].
Animal Models of SSc Vasculopathy
Animal models provided invaluable tools for unraveling the cellular and molecular events that drive the development of tissue fibrosis. All SSc models were discovered because of their fibrotic phenotype [234, 235]. However, only few animal models of SSc replicate the full spectrum of the human disease, including the fibrotic, vascular, inflammatory, and autoimmune features. Endothelial dysfunction occurs in various models [236]; however, it is not clear if EC dysfunction is a primary or secondary event in animals with chronic hypoxia and intense tissue fibrosis.
UCD 200/206 Chickens
This chicken model manifests the entire spectrum of the clinical, histopathological, and serological features of SSc. EC apoptosis, induced by AECA-dependent cellular cytotoxicity, is considered to be a primary event in this model. Other immunologic features include the presence of antinuclear antibodies, anticardiolipin antibodies, and rheumatoid factors [237]. However, it has been difficult to use this model because of the high variability of disease course, the long generation time of chicken, the challenges in housing chicken, and the lack of easy availability of chicken reagents.
Chronic Graft-Versus-Host Disease (GVHD)
This model is produced by the transplantation of immunologically incompatible spleen and bone marrow cells in a susceptible host. Tissue fibrosis is noted after host tissue infiltration by donor T cells and monocytes/macrophages. A very interesting chronic GVHD model, avoiding the need for irradiation, has been produced using the Rag-2 mouse, an immune-deficient mouse secondary to the loss of ability to rearrange immunoglobulin and T-cell receptor genes. In this model, not only fibrosis is noted but also vasoconstriction and intimal hyperplasia in both skin and kidney. In addition, there is an increase in ET-1 and adhesion molecule expression with circulating antinuclear antibodies and anti-Scl-70 antibodies in more than 90 % of these mice [238].
Endothelial Fli1-Deficient Mouse
Friend leukemia integration-1 (Fli1) is a member of the Ets family of transcription factors that represses the transcription of collagen genes via Sp1-dependent pathway. Embryonic fibroblasts from Fli1-deficient mice exhibit significantly increased type I collagen expression levels in association with increased CTGF and decreased MMP-1 expression. Interestingly, a mouse model with a conditional deletion of Fli1 in ECs (Fli1 ECKO) develops disorganized dermal vascular network with greatly compromised vessel integrity and markedly increased vessel permeability. Downregulation of VE-cadherin and platelet-endothelial cell adhesion molecule 1, impaired development of basement membrane, and decreased pericyte coverage in dermal microvessels were also noted [196]. This phenotype is consistent with a role of Fli1 as a regulator of vessel maturation and stabilization. Importantly, a reduced expression level of Fli1 in SSc ECs was noted suggesting that Fli1 may play a critical role in the development of SSc vasculopathy [196].
Fra-2 Transgenic Mouse
The fos-related antigen (Fra-2) belongs to the activator protein 1 (AP1) family of transcription factors. Fra-2 is overexpressed in SSc tissues and is shown to act as a novel downstream mediator of the profibrotic effects of TGF-β and PDGF. Mice with ectopic expression of Fra-2 in various organs develop generalized fibrosis and inflammation that is most pronounced in the lungs. Moreover, Fra-2 transgenic mice develop severe loss of small blood vessels in the skin that is paralleled by progressive skin fibrosis. Fra-2 transgenic mice display various features of SSc vasculopathy with decreased dermal capillary density and early EC apoptosis [239]. Interestingly, dermal EC apoptosis appears to occur before the onset of tissue fibrosis. The suppression of Fra-2 by small interfering RNA prevents EC apoptosis and improves parameters of EC angiogenic potential. However, no proliferative vasculopathy was detectable in the skin [239]. Conversely, Fra-2 transgenic mice have been reported to develop proliferative vasculopathy in the lungs closely resembling SSc-related PAH [240]. Thus, Fra-2 transgenic mice may be a potentially useful model for the investigation of the link between vasculopathy and fibrosis and for the exploration of the mechanisms of vasculopathy in different organs.
uPAR-Deficient Mouse
The cleavage/inactivation of uPAR is a crucial step in fibroblast-to-myofibroblast transition and has been implicated in SSc peripheral microvasculopathy [132, 159, 241]. Therefore, Manetti et al. investigated whether uPAR gene knockout in mice could result in fibrosis and peripheral microvasculopathy resembling human SSc [242]. The skin of uPAR-deficient mice displays most of the typical pathological features of SSc. Dermal thickness, collagen content, and myofibroblast counts were significantly greater in uPAR-deficient mice than in wild-type mice. Similar to SSc, the skin of uPAR-deficient mice was characterized by the presence of thickened, closely packed and irregularly distributed collagen bundles, abundant perivascular fibrosis, and partial replacement of subcutaneous fat with connective tissue. Moreover, in the dermis of uPAR-deficient mice, there was a marked overexpression of the SSc-related profibrotic factors TGF-β, CTGF/CCN2, and ET-1. Of note, in uPAR-deficient mice, dermal fibrosis was paralleled by EC apoptosis and severe loss of microvessels similar to what is seen in SSc peripheral microvasculopathy [242]. In addition, uPAR-deficient mice also develop progressive lung fibrosis resembling the nonspecific interstitial pneumonia pattern of SSc. However, no evidence of vessel intima-media proliferation was reported either in the skin or the lungs [242]. Further studies are required to ascertain whether uPAR-deficient mice may develop SSc-like features in other internal organs. In this regard, it was reported that uPAR deficiency accelerates renal fibrosis in obstructive nephropathy [243].
Conclusions
The pathogenesis of the vascular disease in SSc appears to involve many cell types and signaling pathways. A complex interaction between ECs, immune cells, VSMCs, fibroblasts, pericytes, and the ECM is likely to contribute to the vascular reactivity, remodeling, and occlusive disease of SSc. EC activation/injury is an early and probably initiating event in disease pathogenesis. The exact mechanism for the widespread vascular disease in SSc is still unknown, but EC injury induced by an infectious agent, immune-mediated cytotoxicity, AECAs, and/or ischemia–reperfusion have all been suggested. Moreover, substantial evidence indicates that angiogenic and vasculogenic repair machineries are impaired and do not allow vascular recovery. The downstream effects of vascular dysfunction adversely affect organ function and determine clinical outcomes. Understanding the mechanisms underlying these processes provides the rationale for novel therapeutic strategies and specific targeted therapy. The definition of prefibrotic vascular lesions may have future therapeutic and preventive implications for SSc. Development of means to quantify endothelial injury and the activity of the vascular lesions are essential for the monitoring of therapies designed to block further vascular injury.
References
Fleischmajer R, Perlish JS, Shaw KV, Pirozzi DJ. Skin capillary changes in early systemic scleroderma. Electron microscopy and “in vitro” autoradiography with tritiated thymidine. Arch Dermatol. 1976;112(11):1553–7.
Grassi W, Core P, Carlino G, Blasetti P, Cervini M. Labial capillary microscopy in systemic sclerosis. Ann Rheum Dis. 1993;52(8):564–9.
Fleischmajer R, Perlish JS. Capillary alterations in scleroderma. J Am Acad Dermatol. 1980;2(2):161–70.
Sgonc R, Gruschwitz MS, Dietrich H, Recheis H, Gershwin ME, Wick G. Endothelial cell apoptosis is a primary pathogenetic event underlying skin lesions in avian and human scleroderma. J Clin Invest. 1996;98(3):785–92.
Chen F, Eriksson P, Kimura T, Herzfeld I, Valen G. Apoptosis and angiogenesis are induced in the unstable coronary atherosclerotic plaque. Coron Artery Dis. 2005;16(3):191–7.
Laurence J, Mitra D, Steiner M, Staiano-Coico L, Jaffe E. Plasma from patients with idiopathic and human immunodeficiency virus-associated thrombotic thrombocytopenic purpura induces apoptosis in microvascular endothelial cells. Blood. 1996;87(8):3245–54.
Albert ML, Pearce SF, Francisco LM, Sauter B, Roy P, Silverstein RL, Bhardwaj N. Immature dendritic cells phagocytose apoptotic cells via alphavbeta5 and CD36, and cross-present antigens to cytotoxic T lymphocytes. J Exp Med. 1998;188(7):1359–68.
Greeno EW, Bach RR, Moldow CF. Apoptosis is associated with increased cell surface tissue factor procoagulant activity. Lab Invest. 1996;75(2):281–9.
Tsuji S, Kaji K, Nagasawa S. Activation of the alternative pathway of human complement by apoptotic human umbilical vein endothelial cells. J Biochem. 1994;116(4):794–800.
Fleischmajer R, Perlish JS. [3H]Thymidine labeling of dermal endothelial cells in scleroderma. J Invest Dermatol. 1977;69(4):379–82.
Rodnan GP, Myerowitz RL, Justh GO. Morphologic changes in the digital arteries of patients with progressive systemic sclerosis (scleroderma) and Raynaud phenomenon. Medicine (Baltimore). 1980;59(6):393–408.
Manetti M, Guiducci S, Matucci-Cerinic M. The origin of the myofibroblast in fibroproliferative vasculopathy: does the endothelial cell steer the pathophysiology of systemic sclerosis? Arthritis Rheum. 2011;63(8):2164–7.
Youssef P, Englert H, Bertouch J. Large vessel occlusive disease associated with CREST syndrome and scleroderma. Ann Rheum Dis. 1993;52(6):464–6.
Pandey JP, LeRoy EC. Human cytomegalovirus and the vasculopathies of autoimmune diseases (especially scleroderma), allograft rejection, and coronary restenosis. Arthritis Rheum. 1998;41(1):10–5.
Cannon PJ, Hassar M, Case DB, Casarella WJ, Sommers SC, LeRoy EC. The relationship of hypertension and renal failure in scleroderma (progressive systemic sclerosis) to structural and functional abnormalities of the renal cortical circulation. Medicine (Baltimore). 1974;53(1):1–46.
Brown GE, O’Leary PA. Skin capillaries in scleroderma. Arch Intern Med. 1925;36:73–88.
Anderson ME, Allen PD, Moore T, Hillier V, Taylor CJ, Herrick AL. Computerized nailfold video capillaroscopy – a new tool for assessment of Raynaud’s phenomenon. J Rheumatol. 2005;32(5):841–8.
Maricq HR, LeRoy EC. Patterns of finger capillary abnormalities in connective tissue disease by “wide-field” microscopy. Arthritis Rheum. 1973;16(5):619–28.
Blann AD, Illingworth K, Jayson MI. Mechanisms of endothelial cell damage in systemic sclerosis and Raynaud’s phenomenon. J Rheumatol. 1993;20(8):1325–30.
Carpentier PH, Maricq HR. Microvasculature in systemic sclerosis. Rheum Dis Clin North Am. 1990;16(1):75–91.
Bukhari M, Herrick AL, Moore T, Manning J, Jayson MI. Increased nailfold capillary dimensions in primary Raynaud’s phenomenon and systemic sclerosis. Br J Rheumatol. 1996;35(11):1127–31.
Cutolo M, Sulli A, Secchi ME, Paolino S, Pizzorni C. Nailfold capillaroscopy is useful for the diagnosis and follow-up of autoimmune rheumatic diseases. A future tool for the analysis of microvascular heart involvement? Rheumatology (Oxford). 2006;45 Suppl 4:iv43–6.
Meli M, Gitzelmann G, Koppensteiner R, Amann-Vesti BR. Predictive value of nailfold capillaroscopy in patients with Raynaud’s phenomenon. Clin Rheumatol. 2006;25(2):153–8.
Miniati I, Guiducci S, Conforti ML, Rogai V, Fiori G, Cinelli M, Saccardi R, Guidi S, Bosi A, Tyndall A, Matucci-Cerinic M. Autologous stem cell transplantation improves microcirculation in systemic sclerosis. Ann Rheum Dis. 2009;68(1):94–8.
Albrecht HP, Hiller D, Hornstein OP, Bühler-Singer S, Mück M, Gruschwitz M. Microcirculatory functions in systemic sclerosis: additional parameters for therapeutic concepts? J Invest Dermatol. 1993;101(2):211–5.
Prescott RJ, Freemont AJ, Jones CJ, Hoyland J, Fielding P. Sequential dermal microvascular and perivascular changes in the development of scleroderma. J Pathol. 1992;166(3):255–63.
Lippton HL, Hauth TA, Summer WR, Hyman AL. Endothelin produces pulmonary vasoconstriction and systemic vasodilation. J Appl Physiol (1985). 1989;66(2):1008–12.
Vancheeswaran R, Azam A, Black C, Dashwood MR. Localization of endothelin-1 and its binding sites in scleroderma skin. J Rheumatol. 1994;21(7):1268–76.
Vancheeswaran R, Magoulas T, Efrat G, Wheeler-Jones C, Olsen I, Penny R, Black CM. Circulating endothelin-1 levels in systemic sclerosis subsets; a marker of fibrosis or vascular dysfunction? J Rheumatol. 1994;21(10):1838–44.
Cambrey AD, Harrison NK, Dawes KE, Southcott AM, Black CM, du Bois RM, Laurent GJ, McAnulty RJ. Increased levels of endothelin-1 in bronchoalveolar lavage fluid from patients with systemic sclerosis contribute to fibroblast mitogenic activity in vitro. Am J Respir Cell Mol Biol. 1994;11(4):439–45.
Kawaguchi Y, Suzuki K, Hara M, Hidaka T, Ishizuka T, Kawagoe M, Nakamura H. Increased endothelin-1 production in fibroblasts derived from patients with systemic sclerosis. Ann Rheum Dis. 1994;53(8):506–10.
Morelli S, Ferri C, Di Francesco L, Baldoncini R, Carlesimo M, Bottoni U, Properzi G, Santucci A. Plasma endothelin-1 levels in patients with systemic sclerosis: influence of pulmonary or systemic arterial hypertension. Ann Rheum Dis. 1995;54(9):730–4.
Morelli S, Ferri C, Polettini E, Bellini C, Gualdi GF, Pittoni V, Valesini G, Santucci A. Plasma endothelin-1 levels, pulmonary hypertension, and lung fibrosis in patients with systemic sclerosis. Am J Med. 1995;99(3):255–60.
Abraham D, Distler O. How does endothelial cell injury start? The role of endothelin in systemic sclerosis. Arthritis Res Ther. 2007;9 Suppl 2:S2.
Frommer KW, Müller-Ladner U. Expression and function of ETA and ETB receptors in SSc. Rheumatology (Oxford). 2008;47 Suppl 5:v27–8.
Kasturi KN, Shibata S, Muryoi T, Bona CA. Tight-skin mouse an experimental model for scleroderma. Int Rev Immunol. 1994;11(3):253–71.
Richard V, Solans V, Favre J, Henry JP, Lallemand F, Thuillez C, Marie I. Role of endogenous endothelin in endothelial dysfunction in murine model of systemic sclerosis: tight skin mice 1. Fundam Clin Pharmacol. 2008;22(6):649–55.
Cozzi F, Pigatto E, Rizzo M, Favaro M, Zanatta E, Cardarelli S, Riato L, Punzi L. Low occurrence of digital ulcers in scleroderma patients treated with bosentan for pulmonary arterial hypertension: a retrospective case-control study. Clin Rheumatol. 2013;32(5):679–83.
Matucci-Cerinic M, Denton CP, Furst DE, Mayes MD, Hsu VM, Carpentier P, Wigley FM, Black CM, Fessler BJ, Merkel PA, Pope JE, Sweiss NJ, Doyle MK, Hellmich B, Medsger Jr TA, Morganti A, Kramer F, Korn JH, Seibold JR. Bosentan treatment of digital ulcers related to systemic sclerosis: results from the RAPIDS-2 randomised, double-blind, placebo-controlled trial. Ann Rheum Dis. 2011;70(1):32–8.
Guiducci S, Bellando Randone S, Bruni C, Carnesecchi G, Maresta A, Iannone F, Lapadula G, Matucci Cerinic M. Bosentan fosters microvascular de-remodelling in systemic sclerosis. Clin Rheumatol. 2012;31(12):1723–5.
Cutolo M, Zampogna G, Vremis L, Smith V, Pizzorni C, Sulli A. Long-term effects of endothelin receptor antagonism on microvascular damage evaluated by nailfold capillaroscopic analysis in systemic sclerosis. J Rheumatol. 2013;40(1):40–5.
Ignarro LJ. Biosynthesis and metabolism of endothelium-derived nitric oxide. Annu Rev Pharmacol Toxicol. 1990;30:535–60.
Ignarro LJ. Endothelium-derived nitric oxide: actions and properties. FASEB J. 1989;3(1):31–6.
Agostoni A, Marasini B, Biondi ML, Bassani C, Cazzaniga A, Bottasso B, Cugno M. L-arginine therapy in Raynaud’s phenomenon? Int J Clin Lab Res. 1991;21(2):202–3.
Cailes J, Winter S, du Bois RM, Evans TW. Defective endothelially mediated pulmonary vasodilation in systemic sclerosis. Chest. 1998;114(1):178–84.
Livi R, Teghini L, Generini S, Matucci-Cerinic M. The loss of endothelium-dependent vascular tone control in systemic sclerosis. Chest. 2001;119(2):672–3.
Matucci-Cerinic M, Pietrini U, Marabini S. Local venomotor response to intravenous infusion of substance P and glyceryl trinitrate in systemic sclerosis. Clin Exp Rheumatol. 1990;8(6):561–5.
Cotton SA, Herrick AL, Jayson MI, Freemont AJ. Endothelial expression of nitric oxide synthases and nitrotyrosine in systemic sclerosis skin. J Pathol. 1999;189(2):273–8.
Dooley A, Gao B, Bradley N, Abraham DJ, Black CM, Jacobs M, Bruckdorfer KR. Abnormal nitric oxide metabolism in systemic sclerosis: increased levels of nitrated proteins and asymmetric dimethylarginine. Rheumatology (Oxford). 2006;45(6):676–84.
Mendall MA, Goggin PM, Molineaux N, Levy J, Toosy T, Strachan D, Camm AJ, Northfield TC. Relation of Helicobacter pylori infection and coronary heart disease. Br Heart J. 1994;71(5):437–9.
Gasbarrini A, Massari I, Serricchio M, Tondi P, De Luca A, Franceschi F, Ojetti V, Dal Lago A, Flore R, Santoliquido A, Gasbarrini G, Pola P. Helicobacter pylori eradication ameliorates primary Raynaud’s phenomenon. Dig Dis Sci. 1998;43(8):1641–5.
Aragona P, Magazzù G, Macchia G, Bartolone S, Di Pasquale G, Vitali C, Ferreri G. Presence of antibodies against Helicobacter pylori and its heat-shock protein 60 in the serum of patients with Sjögren’s syndrome. J Rheumatol. 1999;26(6):1306–11.
Savarino V, Sulli A, Zentilin P, Raffaella Mele M, Cutolo M. No evidence of an association between Helicobacter pylori infection and Raynaud phenomenon. Scand J Gastroenterol. 2000;35(12):1251–4.
Sulli A, Seriolo B, Savarino V, Cutolo M. Lack of correlation between gastric Helicobacter pylori infection and primary or secondary Raynaud’s phenomenon in patients with systemic sclerosis. J Rheumatol. 2000;27(7):1820–1.
Vaughan JH, Shaw PX, Nguyen MD, Medsger Jr TA, Wright TM, Metcalf JS, Leroy EC. Evidence of activation of 2 herpesviruses, Epstein-Barr virus and cytomegalovirus, in systemic sclerosis and normal skins. J Rheumatol. 2000;27(3):821–3.
Neidhart M, Kuchen S, Distler O, Brühlmann P, Michel BA, Gay RE, Gay S. Increased serum levels of antibodies against human cytomegalovirus and prevalence of autoantibodies in systemic sclerosis. Arthritis Rheum. 1999;42(2):389–92.
Presti RM, Pollock JL, Dal Canto AJ, O’Guin AK, Virgin 4th HW. Interferon gamma regulates acute and latent murine cytomegalovirus infection and chronic disease of the great vessels. J Exp Med. 1998;188(3):577–88.
Zhou YF, Shou M, Harrell RF, Yu ZX, Unger EF, Epstein SE. Chronic non-vascular cytomegalovirus infection: effects on the neointimal response to experimental vascular injury. Cardiovasc Res. 2000;45(4):1019–25.
Hamamdzic D, Harley RA, Hazen-Martin D, LeRoy EC. MCMV induces neointima in IFN-gammaR/mice: intimal cell apoptosis and persistent proliferation of myofibroblasts. BMC Musculoskelet Disord. 2001;2:3.
Ferri C, Longombardo G, Azzi A, Zakrzewska K. Parvovirus B19 and systemic sclerosis. Clin Exp Rheumatol. 1999;17(2):267–8.
Magro CM, Nuovo G, Ferri C, Crowson AN, Giuggioli D, Sebastiani M. Parvoviral infection of endothelial cells and stromal fibroblasts: a possible pathogenetic role in scleroderma. J Cutan Pathol. 2004;31(1):43–50.
Zakrzewska K, Corcioli F, Carlsen KM, Giuggioli D, Fanci R, Rinieri A, Ferri C, Azzi A. Human parvovirus B19 (B19V) infection in systemic sclerosis patients. Intervirology. 2009;52(5):279–82.
Drenk F, Deicher HR. Pathophysiological effects of endothelial cytotoxic activity derived from sera of patients with progressive systemic sclerosis. J Rheumatol. 1988;15(3):468–74.
Holt CM, Lindsey N, Moult J, Malia RG, Greaves M, Hume A, Rowell NR, Hughes P. Antibody-dependent cellular cytotoxicity of vascular endothelium: characterization and pathogenic associations in systemic sclerosis. Clin Exp Immunol. 1989;78(3):359–65.
Penning CA, Cunningham J, French MA, Harrison G, Rowell NR, Hughes P. Antibody-dependent cellular cytotoxicity of human vascular endothelium in systemic sclerosis. Clin Exp Immunol. 1984;57(3):548–56.
Sgonc R, Gruschwitz MS, Boeck G, Sepp N, Gruber J, Wick G. Endothelial cell apoptosis in systemic sclerosis is induced by antibody-dependent cell-mediated cytotoxicity via CD95. Arthritis Rheum. 2000;43(11):2550–62.
Trapani JA. Dual mechanisms of apoptosis induction by cytotoxic lymphocytes. Int Rev Cytol. 1998;182:111–92.
Kahaleh MB, Leroy EC. Endothelial injury in scleroderma. A protease mechanism. J Lab Clin Med. 1983;101(4):553–60.
Kahaleh MB, Fan PS. Mechanism of serum-mediated endothelial injury in scleroderma: identification of a granular enzyme in scleroderma skin and sera. Clin Immunol Immunopathol. 1997;83(1):32–40.
Müllbacher A, Waring P, Tha Hla R, Tran T, Chin S, Stehle T, Museteanu C, Simon MM. Granzymes are the essential downstream effector molecules for the control of primary virus infections by cytolytic leukocytes. Proc Natl Acad Sci U S A. 1999;96(24):13950–5.
Müller U, Sobek V, Balkow S, Hölscher C, Müllbacher A, Museteanu C, Mossmann H, Simon MM. Concerted action of perforin and granzymes is critical for the elimination of Trypanosoma cruzi from mouse tissues, but prevention of early host death is in addition dependent on the FasL/Fas pathway. Eur J Immunol. 2003;33(1):70–8.
Buzza MS, Zamurs L, Sun J, Bird CH, Smith AI, Trapani JA, Froelich CJ, Nice EC, Bird PI. Extracellular matrix remodeling by human granzyme B via cleavage of vitronectin, fibronectin, and laminin. J Biol Chem. 2005;280(25):23549–58.
Mulligan-Kehoe MJ, Drinane MC, Mollmark J, Casciola-Rosen L, Hummers LK, Hall A, Rosen A, Wigley FM, Simons M. Antiangiogenic plasma activity in patients with systemic sclerosis. Arthritis Rheum. 2007;56(10):3448–58.
Sasaki T, Larsson H, Tisi D, Claesson-Welsh L, Hohenester E, Timpl R. Endostatins derived from collagens XV and XVIII differ in structural and binding properties, tissue distribution and anti-angiogenic activity. J Mol Biol. 2000;301(5):1179–90.
Maeshima Y, Sudhakar A, Lively JC, Ueki K, Kharbanda S, Kahn CR, Sonenberg N, Hynes RO, Kalluri R. Tumstatin, an endothelial cell-specific inhibitor of protein synthesis. Science. 2002;295(5552):140–3.
Kamphaus GD, Colorado PC, Panka DJ, Hopfer H, Ramchandran R, Torre A, Maeshima Y, Mier JW, Sukhatme VP, Kalluri R. Canstatin, a novel matrix-derived inhibitor of angiogenesis and tumor growth. J Biol Chem. 2000;275(2):1209–15.
Cao Y, Ji RW, Davidson D, Schaller J, Marti D, Söhndel S, McCance SG, O’Reilly MS, Llinás M, Folkman J. Kringle domains of human angiostatin. Characterization of the anti-proliferative activity on endothelial cells. J Biol Chem. 1996;271(46):29461–7.
Ferrara N, Clapp C, Weiner R. The 16K fragment of prolactin specifically inhibits basal or fibroblast growth factor stimulated growth of capillary endothelial cells. Endocrinology. 1991;129(2):896–900.
Staton CA, Lewis CE. Angiogenesis inhibitors found within the haemostasis pathway. J Cell Mol Med. 2005;9(2):286–302.
Hebbar M, Peyrat JP, Hornez L, Hatron PY, Hachulla E, Devulder B. Increased concentrations of the circulating angiogenesis inhibitor endostatin in patients with systemic sclerosis. Arthritis Rheum. 2000;43(4):889–93.
Hummers LK, Hall A, Wigley FM, Simons M. Abnormalities in the regulators of angiogenesis in patients with scleroderma. J Rheumatol. 2009;36(3):576–82.
Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. 1994;179(4):1317–30.
Sercarz EE, Lehmann PV, Ametani A, Benichou G, Miller A, Moudgil K. Dominance and crypticity of T cell antigenic determinants. Annu Rev Immunol. 1993;11:729–66.
Zhou Z, Ménard HA. Autoantigenic posttranslational modifications of proteins: does it apply to rheumatoid arthritis? Curr Opin Rheumatol. 2002;14(3):250–3.
Andrade F, Roy S, Nicholson D, Thornberry N, Rosen A, Casciola-Rosen L. Granzyme B directly and efficiently cleaves several downstream caspase substrates: implications for CTL-induced apoptosis. Immunity. 1998;8(4):451–60.
Casciola-Rosen L, Andrade F, Ulanet D, Wong WB, Rosen A. Cleavage by granzyme B is strongly predictive of autoantigen status: implications for initiation of autoimmunity. J Exp Med. 1999;190(6):815–26.
Schachna L, Wigley FM, Morris S, Gelber AC, Rosen A, Casciola-Rosen L. Recognition of Granzyme B-generated autoantigen fragments in scleroderma patients with ischemic digital loss. Arthritis Rheum. 2002;46(7):1873–84.
Youinou P, Revelen R, Bordron A. Is antiendothelial cell antibody the murder weapon in systemic sclerosis? Clin Exp Rheumatol. 1999;17(1):35–6.
Hill MB, Phipps JL, Cartwright RJ, Milford Ward A, Greaves M, Hughes P. Antibodies to membranes of endothelial cells and fibroblasts in scleroderma. Clin Exp Immunol. 1996;106(3):491–7.
Negi VS, Tripathy NK, Misra R, Nityanand S. Antiendothelial cell antibodies in scleroderma correlate with severe digital ischemia and pulmonary arterial hypertension. J Rheumatol. 1998;25(3):462–6.
García de la Peña-Lefebvre P, Chanseaud Y, Tamby MC, Reinbolt J, Batteux F, Allanore Y, Kahan A, Meyer O, Benveniste O, Boyer O, Guillevin L, Boissier MC, Mouthon L. IgG reactivity with a 100-kDa tissue and endothelial cell antigen identified as topoisomerase 1 distinguishes between limited and diffuse systemic sclerosis patients. Clin Immunol. 2004;111(3):241–51.
Tamby MC, Chanseaud Y, Humbert M, Fermanian J, Guilpain P, Garcia-de-la-Peña-Lefebvre P, Brunet S, Servettaz A, Weill B, Simonneau G, Guillevin L, Boissier MC, Mouthon L. Anti-endothelial cell antibodies in idiopathic and systemic sclerosis associated pulmonary arterial hypertension. Thorax. 2005;60(9):765–72.
Worda M, Sgonc R, Dietrich H, Niederegger H, Sundick RS, Gershwin ME, Wick G. In vivo analysis of the apoptosis-inducing effect of anti-endothelial cell antibodies in systemic sclerosis by the chorioallantoic membrane assay. Arthritis Rheum. 2003;48(9):2605–14.
Bordron A, Dueymes M, Levy Y, Jamin C, Leroy JP, Piette JC, Shoenfeld Y, Youinou PY. The binding of some human antiendothelial cell antibodies induces endothelial cell apoptosis. J Clin Invest. 1998;101(10):2029–35.
Tan FK, Arnett FC, Reveille JD, Ahn C, Antohi S, Sasaki T, Nishioka K, Bona CA. Autoantibodies to fibrillin 1 in systemic sclerosis: ethnic differences in antigen recognition and lack of correlation with specific clinical features or HLA alleles. Arthritis Rheum. 2000;43(11):2464–71.
Arends SJ, Damoiseaux JG, Duijvestijn AM, Debrus-Palmans L, Boomars KA, Brunner-La Rocca HP, Cohen Tervaert JW, van Paassen P. Functional implications of IgG anti-endothelial cell antibodies in pulmonary arterial hypertension. Autoimmunity. 2013;46(7):463–70.
Wolf SI, Howat S, Abraham DJ, Pearson JD, Lawson C. Agonistic anti-ICAM-1 antibodies in scleroderma: activation of endothelial pro-inflammatory cascades. Vascul Pharmacol. 2013;59(1–2):19–26.
Al-Dhaher FF, Pope JE, Ouimet JM. Determinants of morbidity and mortality of systemic sclerosis in Canada. Semin Arthritis Rheum. 2010;39(4):269–77.
Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972–2002. Ann Rheum Dis. 2007;66(7):940–4.
Mathai SC, Hummers LK, Champion HC, Wigley FM, Zaiman A, Hassoun PM, Girgis RE. Survival in pulmonary hypertension associated with the scleroderma spectrum of diseases: impact of interstitial lung disease. Arthritis Rheum. 2009;60(2):569–77.
Birukov KG. Cyclic stretch, reactive oxygen species, and vascular remodeling. Antioxid Redox Signal. 2009;11(7):1651–67.
Grote K, Flach I, Luchtefeld M, Akin E, Holland SM, Drexler H, Schieffer B. Mechanical stretch enhances mRNA expression and proenzyme release of matrix metalloproteinase-2 (MMP-2) via NAD(P)H oxidase-derived reactive oxygen species. Circ Res. 2003;92(11):e80–6.
McCord JM. Oxygen-derived free radicals in postischemic tissue injury. N Engl J Med. 1985;312(3):159–63.
Riccieri V, Spadaro A, Fuksa L, Firuzi O, Saso L, Valesini G. Specific oxidative stress parameters differently correlate with nailfold capillaroscopy changes and organ involvement in systemic sclerosis. Clin Rheumatol. 2008;27(2):225–30.
Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39(1):44–84.
Kuschel L, Hansel A, Schönherr R, Weissbach H, Brot N, Hoshi T, Heinemann SH. Molecular cloning and functional expression of a human peptide methionine sulfoxide reductase (hMsrA). FEBS Lett. 1999;456(1):17–21.
Ogawa F, Sander CS, Hansel A, Oehrl W, Kasperczyk H, Elsner P, Shimizu K, Heinemann SH, Thiele JJ. The repair enzyme peptide methionine-S-sulfoxide reductase is expressed in human epidermis and upregulated by UVA radiation. J Invest Dermatol. 2006;126(5):1128–34.
Prentice HM, Moench IA, Rickaway ZT, Dougherty CJ, Webster KA, Weissbach H. MsrA protects cardiomyocytes against hypoxia/reoxygenation induced cell death. Biochem Biophys Res Commun. 2008;366(3):775–8.
Ogawa F, Shimizu K, Hara T, Muroi E, Komura K, Takenaka M, Hasegawa M, Fujimoto M, Takehara K, Sato S. Autoantibody against one of the antioxidant repair enzymes, methionine sulfoxide reductase A, in systemic sclerosis: association with pulmonary fibrosis and vascular damage. Arch Dermatol Res. 2010;302(1):27–35.
Baroni SS, Santillo M, Bevilacqua F, Luchetti M, Spadoni T, Mancini M, Fraticelli P, Sambo P, Funaro A, Kazlauskas A, Avvedimento EV, Gabrielli A. Stimulatory autoantibodies to the PDGF receptor in systemic sclerosis. N Engl J Med. 2006;354(25):2667–76.
Edelberg JM, Lee SH, Kaur M, Tang L, Feirt NM, McCabe S, Bramwell O, Wong SC, Hong MK. Platelet-derived growth factor-AB limits the extent of myocardial infarction in a rat model: feasibility of restoring impaired angiogenic capacity in the aging heart. Circulation. 2002;105(5):608–13.
Classen JF, Henrohn D, Rorsman F, Lennartsson J, Lauwerys BR, Wikström G, Rorsman C, Lenglez S, Franck-Larsson K, Tomasi JP, Kämpe O, Vanthuyne M, Houssiau FA, Demoulin JB. Lack of evidence of stimulatory autoantibodies to platelet-derived growth factor receptor in patients with systemic sclerosis. Arthritis Rheum. 2009;60(4):1137–44.
Derk CT, Jimenez SA. Systemic sclerosis: current views of its pathogenesis. Autoimmun Rev. 2003;2(4):181–91.
Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, Ruvkun G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403(6772):901–6.
Razin A, Cedar H. DNA methylation and gene expression. Microbiol Rev. 1991;55(3):451–8.
Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349(21):2042–54.
Richardson B. Primer: epigenetics of autoimmunity. Nat Clin Pract Rheumatol. 2007;3(9):521–7.
Romero LI, Zhang DN, Cooke JP, Ho HK, Avalos E, Herrera R, Herron GS. Differential expression of nitric oxide by dermal microvascular endothelial cells from patients with scleroderma. Vasc Med. 2000;5(3):147–58.
Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature. 1995;377(6546):239–42.
Lee PC, Salyapongse AN, Bragdon GA, Shears 2nd LL, Watkins SC, Edington HD, Billiar TR. Impaired wound healing and angiogenesis in eNOS-deficient mice. Am J Physiol. 1999;277(4 Pt 2):H1600–8.
Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. 2012;122(12):4306–13.
Wang Y, Kahaleh B. Epigenetic repression of bone morphogenetic protein receptor II expression in scleroderma. J Cell Mol Med. 2013;17(10):1291–9.
Altorok N, Almeshal N, Wang Y, Kahaleh B. Epigenetics, the holy grail in the pathogenesis of systemic sclerosis. Rheumatology (Oxford). 2015;54(10):1759–70.
Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6(4):389–95.
Manetti M, Guiducci S, Ibba-Manneschi L, Matucci-Cerinic M. Mechanisms in the loss of capillaries in systemic sclerosis: angiogenesis versus vasculogenesis. J Cell Mol Med. 2010;14(6A):1241–54.
Rabquer BJ, Koch AE. Angiogenesis and vasculopathy in systemic sclerosis: evolving concepts. Curr Rheumatol Rep. 2012;14(1):56–63.
Manetti M, Guiducci S, Ibba-Manneschi L, Matucci-Cerinic M. Impaired angiogenesis in systemic sclerosis: the emerging role of the antiangiogenic VEGF(165)b splice variant. Trends Cardiovasc Med. 2011;21(7):204–10.
Distler O, Del Rosso A, Giacomelli R, Cipriani P, Conforti ML, Guiducci S, Gay RE, Michel BA, Brühlmann P, Müller-Ladner U, Gay S, Matucci-Cerinic M. Angiogenic and angiostatic factors in systemic sclerosis: increased levels of vascular endothelial growth factor are a feature of the earliest disease stages and are associated with the absence of fingertip ulcers. Arthritis Res. 2002;4(6):R11.
Distler O, Distler JH, Scheid A, Acker T, Hirth A, Rethage J, Michel BA, Gay RE, Müller-Ladner U, Matucci-Cerinic M, Plate KH, Gassmann M, Gay S. Uncontrolled expression of vascular endothelial growth factor and its receptors leads to insufficient skin angiogenesis in patients with systemic sclerosis. Circ Res. 2004;95(1):109–16.
Choi JJ, Min DJ, Cho ML, Min SY, Kim SJ, Lee SS, Park KS, Seo YI, Kim WU, Park SH, Cho CS. Elevated vascular endothelial growth factor in systemic sclerosis. J Rheumatol. 2003;30(7):1529–33.
Mackiewicz Z, Sukura A, Povilenaité D, Ceponis A, Virtanen I, Hukkanen M, Konttinen YT. Increased but imbalanced expression of VEGF and its receptors has no positive effect on angiogenesis in systemic sclerosis skin. Clin Exp Rheumatol. 2002;20(5):641–6.
D’Alessio S, Fibbi G, Cinelli M, Guiducci S, Del Rosso A, Margheri F, Serratì S, Pucci M, Kahaleh B, Fan P, Annunziato F, Cosmi L, Liotta F, Matucci-Cerinic M, Del Rosso M. Matrix metalloproteinase 12-dependent cleavage of urokinase receptor in systemic sclerosis microvascular endothelial cells results in impaired angiogenesis. Arthritis Rheum. 2004;50(10):3275–85.
Manetti M, Guiducci S, Romano E, Ceccarelli C, Bellando-Randone S, Conforti ML, Ibba-Manneschi L, Matucci-Cerinic M. Overexpression of VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, leads to insufficient angiogenesis in patients with systemic sclerosis. Circ Res. 2011;109(3):e14–26.
Manetti M, Guiducci S, Romano E, Bellando-Randone S, Lepri G, Bruni C, Conforti ML, Ibba-Manneschi L, Matucci-Cerinic M. Increased plasma levels of the VEGF165b splice variant are associated with the severity of nailfold capillary loss in systemic sclerosis. Ann Rheum Dis. 2013;72(8):1425–7.
Rabquer BJ, Tsou PS, Hou Y, Thirunavukkarasu E, Haines 3rd GK, Impens AJ, Phillips K, Kahaleh B, Seibold JR, Koch AE. Dysregulated expression of MIG/CXCL9, IP-10/CXCL10 and CXCL16 and their receptors in systemic sclerosis. Arthritis Res Ther. 2011;13(1):R18.
Shirai Y, Okazaki Y, Inoue Y, Tamura Y, Yasuoka H, Takeuchi T, Kuwana M. Elevated levels of pentraxin 3 in systemic sclerosis: associations with vascular manifestations and defective vasculogenesis. Arthritis Rheumatol. 2015;67(2):498–507.
Manetti M, Guiducci S, Romano E, Avouac J, Rosa I, Ruiz B, Lepri G, Bellando-Randone S, Ibba-Manneschi L, Allanore Y, Matucci-Cerinic M. Decreased expression of the endothelial cell-derived factor EGFL7 in systemic sclerosis: potential contribution to impaired angiogenesis and vasculogenesis. Arthritis Res Ther. 2013;15(5):R165.
Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, Witzenbichler B, Schatteman G, Isner JM. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275(5302):964–7.
Kuwana M, Okazaki Y, Yasuoka H, Kawakami Y, Ikeda Y. Defective vasculogenesis in systemic sclerosis. Lancet. 2004;364(9434):603–10.
Allanore Y, Batteux F, Avouac J, Assous N, Weill B, Kahan A. Levels of circulating endothelial progenitor cells in systemic sclerosis. Clin Exp Rheumatol. 2007;25(1):60–6.
Avouac J, Juin F, Wipff J, Couraud PO, Chiocchia G, Kahan A, Boileau C, Uzan G, Allanore Y. Circulating endothelial progenitor cells in systemic sclerosis: association with disease severity. Ann Rheum Dis. 2008;67(10):1455–60.
Zhu S, Evans S, Yan B, Povsic TJ, Tapson V, Goldschmidt-Clermont PJ, Dong C. Transcriptional regulation of Bim by FOXO3a and Akt mediates scleroderma serum-induced apoptosis in endothelial progenitor cells. Circulation. 2008;118(21):2156–65.
Del Papa N, Quirici N, Scavullo C, Gianelli U, Corti L, Vitali C, Ferri C, Giuggioli D, Manfredi A, Maglione W, Onida F, Colaci M, Bosari S, Lambertenghi Deliliers G. Antiendothelial cell antibodies induce apoptosis of bone marrow endothelial progenitors in systemic sclerosis. J Rheumatol. 2010;37(10):2053–63.
Kuwana M, Okazaki Y. Brief report: impaired in vivo neovascularization capacity of endothelial progenitor cells in patients with systemic sclerosis. Arthritis Rheumatol. 2014;66(5):1300–5.
Yamaguchi Y, Kuwana M. Proangiogenic hematopoietic cells of monocytic origin: roles in vascular regeneration and pathogenic processes of systemic sclerosis. Histol Histopathol. 2013;28(2):175–83.
Cipriani P, Guiducci S, Miniati I, Cinelli M, Urbani S, Marrelli A, Dolo V, Pavan A, Saccardi R, Tyndall A, Giacomelli R, Cerinic MM. Impairment of endothelial cell differentiation from bone marrow-derived mesenchymal stem cells: new insight into the pathogenesis of systemic sclerosis. Arthritis Rheum. 2007;56(6):1994–2004.
Guiducci S, Manetti M, Romano E, Mazzanti B, Ceccarelli C, Dal Pozzo S, Milia AF, Bellando-Randone S, Fiori G, Conforti ML, Saccardi R, Ibba-Manneschi L, Matucci-Cerinic M. Bone marrow-derived mesenchymal stem cells from early diffuse systemic sclerosis exhibit a paracrine machinery and stimulate angiogenesis in vitro. Ann Rheum Dis. 2011;70(11):2011–21.
Guiducci S, Porta F, Saccardi R, Guidi S, Ibba-Manneschi L, Manetti M, Mazzanti B, Dal Pozzo S, Milia AF, Bellando-Randone S, Miniati I, Fiori G, Fontana R, Amanzi L, Braschi F, Bosi A, Matucci-Cerinic M. Autologous mesenchymal stem cells foster revascularization of ischemic limbs in systemic sclerosis: a case report. Ann Intern Med. 2010;153(10):650–4.
Matucci-Cerinic M, Kahaleh B, Wigley FM. Review: evidence that systemic sclerosis is a vascular disease. Arthritis Rheum. 2013;65(8):1953–62.
Pepper MS. Role of the matrix metalloproteinase and plasminogen activator-plasmin systems in angiogenesis. Arterioscler Thromb Vasc Biol. 2001;21(7):1104–17.
Keck RG, Berleau L, Harris R, Keyt BA. Disulfide structure of the heparin binding domain in vascular endothelial growth factor: characterization of posttranslational modifications in VEGF. Arch Biochem Biophys. 1997;344(1):103–13.
Ihn H. The role of TGF-beta signaling in the pathogenesis of fibrosis in scleroderma. Arch Immunol Ther Exp (Warsz). 2002;50(5):325–31.
Jinnin M, Ihn H, Mimura Y, Asano Y, Yamane K, Tamaki K. Effects of hepatocyte growth factor on the expression of type I collagen and matrix metalloproteinase-1 in normal and scleroderma dermal fibroblasts. J Invest Dermatol. 2005;124(2):324–30.
Lijnen HR, Ugwu F, Bini A, Collen D. Generation of an angiostatin-like fragment from plasminogen by stromelysin-1 (MMP-3). Biochemistry. 1998;37(14):4699–702.
O’Reilly MS, Wiederschain D, Stetler-Stevenson WG, Folkman J, Moses MA. Regulation of angiostatin production by matrix metalloproteinase-2 in a model of concomitant resistance. J Biol Chem. 1999;274(41):29568–71.
Vassalli JD, Baccino D, Belin D. A cellular binding site for the Mr 55,000 form of the human plasminogen activator, urokinase. J Cell Biol. 1985;100(1):86–92.
Wei Y, Waltz DA, Rao N, Drummond RJ, Rosenberg S, Chapman HA. Identification of the urokinase receptor as an adhesion receptor for vitronectin. J Biol Chem. 1994;269(51):32380–8.
Ragno P. The urokinase receptor: a ligand or a receptor? Story of a sociable molecule. Cell Mol Life Sci. 2006;63(9):1028–37.
Margheri F, Manetti M, Serratì S, Nosi D, Pucci M, Matucci-Cerinic M, Kahaleh B, Bazzichi L, Fibbi G, Ibba-Manneschi L, Del Rosso M. Domain 1 of the urokinase-type plasminogen activator receptor is required for its morphologic and functional, beta2 integrin-mediated connection with actin cytoskeleton in human microvascular endothelial cells: failure of association in systemic sclerosis endothelial cells. Arthritis Rheum. 2006;54(12):3926–38.
Margheri F, Serratì S, Lapucci A, Chillà A, Bazzichi L, Bombardieri S, Kahaleh B, Calorini L, Bianchini F, Fibbi G, Del Rosso M. Modulation of the angiogenic phenotype of normal and systemic sclerosis endothelial cells by gain-loss of function of pentraxin 3 and matrix metalloproteinase 12. Arthritis Rheum. 2010;62(8):2488–98.
Manetti M, Guiducci S, Romano E, Bellando-Randone S, Conforti ML, Ibba-Manneschi L, Matucci-Cerinic M. Increased serum levels and tissue expression of matrix metalloproteinase-12 in patients with systemic sclerosis: correlation with severity of skin and pulmonary fibrosis and vascular damage. Ann Rheum Dis. 2012;71(6):1064–72.
Xu Z, Shi H, Li Q, Mei Q, Bao J, Shen Y, Xu J. Mouse macrophage metalloelastase generates angiostatin from plasminogen and suppresses tumor angiogenesis in murine colon cancer. Oncol Rep. 2008;20(1):81–8.
Giusti B, Serratì S, Margheri F, Papucci L, Rossi L, Poggi F, Magi A, Del Rosso A, Cinelli M, Guiducci S, Kahaleh B, Matucci-Cerinic M, Abbate R, Fibbi G, Del Rosso M. The antiangiogenic tissue kallikrein pattern of endothelial cells in systemic sclerosis. Arthritis Rheum. 2005;52(11):3618–28.
Giusti B, Fibbi G, Margheri F, Serratì S, Rossi L, Poggi F, Lapini I, Magi A, Del Rosso A, Cinelli M, Guiducci S, Kahaleh B, Bazzichi L, Bombardieri S, Matucci-Cerinic M, Gensini GF, Del Rosso M, Abbate R. A model of anti-angiogenesis: differential transcriptosome profiling of microvascular endothelial cells from diffuse systemic sclerosis patients. Arthritis Res Ther. 2006;8(4):R115.
Bhoola KD, Figueroa CD, Worthy K. Bioregulation of kinins: kallikreins, kininogens, and kininases. Pharmacol Rev. 1992;44(1):1–80.
Plendl J, Snyman C, Naidoo S, Sawant S, Mahabeer R, Bhoola KD. Expression of tissue kallikrein and kinin receptors in angiogenic microvascular endothelial cells. Biol Chem. 2000;381(11):1103–15.
Emanueli C, Madeddu P. Targeting kinin receptors for the treatment of tissue ischaemia. Trends Pharmacol Sci. 2001;22(9):478–84.
Del Rosso A, Distler O, Milia AF, Emanueli C, Ibba-Manneschi L, Guiducci S, Conforti ML, Generini S, Pignone A, Gay S, Madeddu P, Matucci-Cerinic M. Increased circulating levels of tissue kallikrein in systemic sclerosis correlate with microvascular involvement. Ann Rheum Dis. 2005;64(3):382–7.
Giusti B, Margheri F, Rossi L, Lapini I, Magi A, Serratì S, Chillà A, Laurenzana A, Magnelli L, Calorini L, Bianchini F, Fibbi G, Abbate R, Del Rosso M. Desmoglein-2-integrin Beta-8 interaction regulates actin assembly in endothelial cells: deregulation in systemic sclerosis. PLoS One. 2013;8(7):e68117.
Avouac J, Cagnard N, Distler JH, Schoindre Y, Ruiz B, Couraud PO, Uzan G, Boileau C, Chiocchia G, Allanore Y. Insights into the pathogenesis of systemic sclerosis based on the gene expression profile of progenitor-derived endothelial cells. Arthritis Rheum. 2011;63(11):3552–62.
Tinazzi E, Dolcino M, Puccetti A, Rigo A, Beri R, Valenti MT, Corrocher R, Lunardi C. Gene expression profiling in circulating endothelial cells from systemic sclerosis patients shows an altered control of apoptosis and angiogenesis that is modified by iloprost infusion. Arthritis Res Ther. 2010;12(4):R131.
Allt G, Lawrenson JG. Pericytes: cell biology and pathology. Cells Tissues Organs. 2001;169(1):1–11.
Sima AA, Chakrabarti S, Garcia-Salinas R, Basu PK. The BB-rat; an authentic model of human diabetic retinopathy. Curr Eye Res. 1985;4(10):1087–92.
Sims DE. Recent advances in pericyte biology – implications for health and disease. Can J Cardiol. 1991;7(10):431–43.
Mandarino LJ, Sundarraj N, Finlayson J, Hassell HR. Regulation of fibronectin and laminin synthesis by retinal capillary endothelial cells and pericytes in vitro. Exp Eye Res. 1993;57(5):609–21.
Hirschi KK, D’Amore PA. Control of angiogenesis by the pericyte: molecular mechanisms and significance. EXS. 1997;79:419–28.
Herman IM, D’Amore PA. Microvascular pericytes contain muscle and nonmuscle actins. J Cell Biol. 1985;101(1):43–52.
Edelman DA, Jiang Y, Tyburski J, Wilson RF, Steffes C. Pericytes and their role in microvasculature homeostasis. J Surg Res. 2006;135(2):305–11.
Hirschi KK, Rohovsky SA, D’Amore PA. PDGF, TGF-beta, and heterotypic cell-cell interactions mediate endothelial cell-induced recruitment of 10T1/2 cells and their differentiation to a smooth muscle fate. J Cell Biol. 1998;141(3):805–14.
Kelley C, D’Amore P, Hechtman HB, Shepro D. Microvascular pericyte contractility in vitro: comparison with other cells of the vascular wall. J Cell Biol. 1987;104(3):483–90.
Tilton RG, Kilo C, Williamson JR. Pericyte-endothelial relationships in cardiac and skeletal muscle capillaries. Microvasc Res. 1979;18(3):325–35.
Cuevas P, Gutierrez-Diaz JA, Reimers D, Dujovny M, Diaz FG, Ausman JI. Pericyte endothelial gap junctions in human cerebral capillaries. Anat Embryol (Berl). 1984;170(2):155–9.
Miller FN, Sims DE. Contractile elements in the regulation of macromolecular permeability. Fed Proc. 1986;45(2):84–8.
Murphy DD, Wagner RC. Differential contractile response of cultured microvascular pericytes to vasoactive agents. Microcirculation. 1994;1(2):121–8.
Shepro D, Morel NM. Pericyte physiology. FASEB J. 1993;7(11):1031–8.
Carmeliet P. Blood vessels and nerves: common signals, pathways and diseases. Nat Rev Genet. 2003;4(9):710–20.
Louissaint Jr A, Rao S, Leventhal C, Goldman SA. Coordinated interaction of neurogenesis and angiogenesis in the adult songbird brain. Neuron. 2002;34(6):945–60.
Palmer TD, Willhoite AR, Gage FH. Vascular niche for adult hippocampal neurogenesis. J Comp Neurol. 2000;425(4):479–94.
Rønnov-Jessen L, Petersen OW, Koteliansky VE, Bissell MJ. The origin of the myofibroblasts in breast cancer. Recapitulation of tumor environment in culture unravels diversity and implicates converted fibroblasts and recruited smooth muscle cells. J Clin Invest. 1995;95(2):859–73.
Sundberg C, Ivarsson M, Gerdin B, Rubin K. Pericytes as collagen-producing cells in excessive dermal scarring. Lab Invest. 1996;74(2):452–66.
Sundberg C, Ljungström M, Lindmark G, Gerdin B, Rubin K. Microvascular pericytes express platelet-derived growth factor-beta receptors in human healing wounds and colorectal adenocarcinoma. Am J Pathol. 1993;143(5):1377–88.
Rajkumar VS, Sundberg C, Abraham DJ, Rubin K, Black CM. Activation of microvascular pericytes in autoimmune Raynaud’s phenomenon and systemic sclerosis. Arthritis Rheum. 1999;42(5):930–41.
Jelaska A, Korn JH. Role of apoptosis and transforming growth factor beta1 in fibroblast selection and activation in systemic sclerosis. Arthritis Rheum. 2000;43(10):2230–9.
Sappino AP, Masouyé I, Saurat JH, Gabbiani G. Smooth muscle differentiation in scleroderma fibroblastic cells. Am J Pathol. 1990;137(3):585–91.
Rajkumar VS, Howell K, Csiszar K, Denton CP, Black CM, Abraham DJ. Shared expression of phenotypic markers in systemic sclerosis indicates a convergence of pericytes and fibroblasts to a myofibroblast lineage in fibrosis. Arthritis Res Ther. 2005;7(5):R1113–23.
Asano Y, Stawski L, Hant F, Highland K, Silver R, Szalai G, Watson DK, Trojanowska M. Endothelial Fli1 deficiency impairs vascular homeostasis: a role in scleroderma vasculopathy. Am J Pathol. 2010;176(4):1983–98.
Cretoiu SM, Popescu LM. Telocytes revisited. Biomol Concepts. 2014;5(5):353–69.
Manetti M, Guiducci S, Ruffo M, Rosa I, Faussone-Pellegrini MS, Matucci-Cerinic M, Ibba-Manneschi L. Evidence for progressive reduction and loss of telocytes in the dermal cellular network of systemic sclerosis. J Cell Mol Med. 2013;17(4):482–96.
Frid MG, Dempsey EC, Durmowicz AG, Stenmark KR. Smooth muscle cell heterogeneity in pulmonary and systemic vessels. Importance in vascular disease. Arterioscler Thromb Vasc Biol. 1997;17(7):1203–9.
Gittenberger-de Groot AC, DeRuiter MC, Bergwerff M, Poelmann RE. Smooth muscle cell origin and its relation to heterogeneity in development and disease. Arterioscler Thromb Vasc Biol. 1999;19(7):1589–94.
Owens GK. Molecular control of vascular smooth muscle cell differentiation. Acta Physiol Scand. 1998;164(4):623–35.
Arts MR, Baron M, Chokr N, Fritzler MJ, Canadian Scleroderma Research Group (CSRG), Servant MJ. Systemic sclerosis immunoglobulin induces growth and a pro-fibrotic state in vascular smooth muscle cells through the epidermal growth factor receptor. PLoS One. 2014;9(6):e100035.
Jimenez SA. Role of endothelial to mesenchymal transition in the pathogenesis of the vascular alterations in systemic sclerosis. ISRN Rheumatol. 2013;2013:835948.
Rhodes JM, Simons M. The extracellular matrix and blood vessel formation: not just a scaffold. J Cell Mol Med. 2007;11(2):176–205.
Davis GE, Saunders WB. Molecular balance of capillary tube formation versus regression in wound repair: role of matrix metalloproteinases and their inhibitors. J Investig Dermatol Symp Proc. 2006;11(1):44–56.
Davis GE, Senger DR. Endothelial extracellular matrix: biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ Res. 2005;97(11):1093–107.
Senger DR. Molecular framework for angiogenesis: a complex web of interactions between extravasated plasma proteins and endothelial cell proteins induced by angiogenic cytokines. Am J Pathol. 1996;149(1):1–7.
Vernon RB, Sage EH. Between molecules and morphology. Extracellular matrix and creation of vascular form. Am J Pathol. 1995;147(4):873–83.
Dvorak HF, Nagy JA, Berse B, Brown LF, Yeo KT, Yeo TK, Dvorak AM, van de Water L, Sioussat TM, Senger DR. Vascular permeability factor, fibrin, and the pathogenesis of tumor stroma formation. Ann N Y Acad Sci. 1992;667:101–11.
Dvorak HF, Senger DR, Dvorak AM. Fibrin as a component of the tumor stroma: origins and biological significance. Cancer Metastasis Rev. 1983;2(1):41–73.
Nagy JA, Brown LF, Senger DR, Lanir N, Van de Water L, Dvorak AM, Dvorak HF. Pathogenesis of tumor stroma generation: a critical role for leaky blood vessels and fibrin deposition. Biochim Biophys Acta. 1989;948(3):305–26.
Cheresh DA, Berliner SA, Vicente V, Ruggeri ZM. Recognition of distinct adhesive sites on fibrinogen by related integrins on platelets and endothelial cells. Cell. 1989;58(5):945–53.
Suehiro K, Gailit J, Plow EF. Fibrinogen is a ligand for integrin alpha5beta1 on endothelial cells. J Biol Chem. 1997;272(8):5360–6.
Sahni A, Altland OD, Francis CW. FGF-2 but not FGF-1 binds fibrin and supports prolonged endothelial cell growth. J Thromb Haemost. 2003;1(6):1304–10.
Sahni A, Francis CW. Plasmic degradation modulates activity of fibrinogen-bound fibroblast growth factor-2. J Thromb Haemost. 2003;1(6):1271–7.
Bobik A, Tkachuk V. Metalloproteinases and plasminogen activators in vessel remodeling. Curr Hypertens Rep. 2003;5(6):466–72.
Garcia-Touchard A, Henry TD, Sangiorgi G, Spagnoli LG, Mauriello A, Conover C, Schwartz RS. Extracellular proteases in atherosclerosis and restenosis. Arterioscler Thromb Vasc Biol. 2005;25(6):1119–27.
Davis GE, Pintar Allen KA, Salazar R, Maxwell SA. Matrix metalloproteinase-1 and -9 activation by plasmin regulates a novel endothelial cell-mediated mechanism of collagen gel contraction and capillary tube regression in three-dimensional collagen matrices. J Cell Sci. 2001;114(Pt 5):917–30.
Saunders WB, Bayless KJ, Davis GE. MMP-1 activation by serine proteases and MMP-10 induces human capillary tubular network collapse and regression in 3D collagen matrices. J Cell Sci. 2005;118(Pt 10):2325–40.
Giannelli G, Iannone F, Marinosci F, Lapadula G, Antonaci S. The effect of bosentan on matrix metalloproteinase-9 levels in patients with systemic sclerosis-induced pulmonary hypertension. Curr Med Res Opin. 2005;21(3):327–32.
Kim WU, Min SY, Cho ML, Hong KH, Shin YJ, Park SH, Cho CS. Elevated matrix metalloproteinase-9 in patients with systemic sclerosis. Arthritis Res Ther. 2005;7(1):R71–9.
Avouac J, Wipff J, Goldman O, Ruiz B, Couraud PO, Chiocchia G, Kahan A, Boileau C, Uzan G, Allanore Y. Angiogenesis in systemic sclerosis: impaired expression of vascular endothelial growth factor receptor 1 in endothelial progenitor-derived cells under hypoxic conditions. Arthritis Rheum. 2008;58(11):3550–61.
Silverstein JL, Steen VD, Medsger Jr TA, Falanga V. Cutaneous hypoxia in patients with systemic sclerosis (scleroderma). Arch Dermatol. 1988;124(9):1379–82.
Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, Garcia JG, Semenza GL. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood. 2005;105(2):659–69.
Makarenko VV, Usatyuk PV, Yuan G, Lee MM, Nanduri J, Natarajan V, Kumar GK, Prabhakar NR. Intermittent hypoxia-induced endothelial barrier dysfunction requires ROS-dependent MAP kinase activation. Am J Physiol Cell Physiol. 2014;306(8):C745–52.
Kahaleh MB. The role of vascular endothelium in fibroblast activation and tissue fibrosis, particularly in scleroderma (systemic sclerosis) and pachydermoperiostosis (primary hypertrophic osteoarthropathy). Clin Exp Rheumatol. 1992;10 Suppl 7:51–6.
Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3(5):349–63.
Lauber K, Bohn E, Kröber SM, Xiao YJ, Blumenthal SG, Lindemann RK, Marini P, Wiedig C, Zobywalski A, Baksh S, Xu Y, Autenrieth IB, Schulze-Osthoff K, Belka C, Stuhler G, Wesselborg S. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell. 2003;113(6):717–30.
Holmes A, Abraham DJ, Chen Y, Denton C, Shi-wen X, Black CM, Leask A. Constitutive connective tissue growth factor expression in scleroderma fibroblasts is dependent on Sp1. J Biol Chem. 2003;278(43):41728–33.
Shi-Wen X, Leask A, Abraham D. Regulation and function of connective tissue growth factor/CCN2 in tissue repair, scarring and fibrosis. Cytokine Growth Factor Rev. 2008;19(2):133–44.
Serratì S, Chillà A, Laurenzana A, Margheri F, Giannoni E, Magnelli L, Chiarugi P, Dotor J, Feijoo E, Bazzichi L, Bombardieri S, Kahaleh B, Fibbi G, Del Rosso M. Systemic sclerosis endothelial cells recruit and activate dermal fibroblasts by induction of a connective tissue growth factor (CCN2)/transforming growth factor β-dependent mesenchymal-to-mesenchymal transition. Arthritis Rheum. 2013;65(1):258–69.
Dees C, Akhmetshina A, Zerr P, Reich N, Palumbo K, Horn A, Jüngel A, Beyer C, Krönke G, Zwerina J, Reiter R, Alenina N, Maroteaux L, Gay S, Schett G, Distler O, Distler JH. Platelet-derived serotonin links vascular disease and tissue fibrosis. J Exp Med. 2011;208(5):961–72.
Maurer B, Distler A, Suliman YA, Gay RE, Michel BA, Gay S, Distler JH, Distler O. Vascular endothelial growth factor aggravates fibrosis and vasculopathy in experimental models of systemic sclerosis. Ann Rheum Dis. 2014;73(10):1880–7.
Christner PJ, Jimenez SA. Animal models of systemic sclerosis: insights into systemic sclerosis pathogenesis and potential therapeutic approaches. Curr Opin Rheumatol. 2004;16(6):746–52.
Smith GP, Chan ES. Molecular pathogenesis of skin fibrosis: insight from animal models. Curr Rheumatol Rep. 2010;12(1):26–33.
Marie I, Bény JL. Endothelial dysfunction in murine model of systemic sclerosis: tight-skin mice 1. J Invest Dermatol. 2002;119(6):1379–87.
Sgonc R. The vascular perspective of systemic sclerosis: of chickens, mice and men. Int Arch Allergy Immunol. 1999;120(3):169–76.
Ruzek MC, Jha S, Ledbetter S, Richards SM, Garman RD. A modified model of graft-versus-host-induced systemic sclerosis (scleroderma) exhibits all major aspects of the human disease. Arthritis Rheum. 2004;50(4):1319–31.
Maurer B, Busch N, Jüngel A, Pileckyte M, Gay RE, Michel BA, Schett G, Gay S, Distler J, Distler O. Transcription factor fos-related antigen-2 induces progressive peripheral vasculopathy in mice closely resembling human systemic sclerosis. Circulation. 2009;120(23):2367–76.
Maurer B, Reich N, Juengel A, Kriegsmann J, Gay RE, Schett G, Michel BA, Gay S, Distler JH, Distler O. Fra-2 transgenic mice as a novel model of pulmonary hypertension associated with systemic sclerosis. Ann Rheum Dis. 2012;71(8):1382–7.
Bernstein AM, Twining SS, Warejcka DJ, Tall E, Masur SK. Urokinase receptor cleavage: a crucial step in fibroblast-to-myofibroblast differentiation. Mol Biol Cell. 2007;18(7):2716–27.
Manetti M, Rosa I, Milia AF, Guiducci S, Carmeliet P, Ibba-Manneschi L, Matucci-Cerinic M. Inactivation of urokinase-type plasminogen activator receptor (uPAR) gene induces dermal and pulmonary fibrosis and peripheral microvasculopathy in mice: a new model of experimental scleroderma? Ann Rheum Dis. 2014;73(9):1700–9.
Zhang G, Kim H, Cai X, López-Guisa JM, Alpers CE, Liu Y, Carmeliet P, Eddy AA. Urokinase receptor deficiency accelerates renal fibrosis in obstructive nephropathy. J Am Soc Nephrol. 2003;14(5):1254–71.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Science+Business Media New York
About this chapter
Cite this chapter
Manetti, M., Kahaleh, B. (2017). Mechanisms of Vascular Disease. In: Varga, J., Denton, C., Wigley, F., Allanore, Y., Kuwana, M. (eds) Scleroderma. Springer, Cham. https://doi.org/10.1007/978-3-319-31407-5_16
Download citation
DOI: https://doi.org/10.1007/978-3-319-31407-5_16
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-31405-1
Online ISBN: 978-3-319-31407-5
eBook Packages: MedicineMedicine (R0)