
Abstract
Signaling from the epidermal growth factor receptor (EGFR) elicits multiple biological responses, including cell proliferation, migration, and survival. Receptor endocytosis and trafficking are critical physiological processes that control the strength, duration, diversification, and spatial restriction of EGFR signaling through multiple mechanisms, which we review in this chapter. These mechanisms include: (i) regulation of receptor density and activation at the cell surface; (ii) concentration of receptors into distinct nascent endocytic structures; (iii) commitment of the receptor to different endocytic routes; (iv) endosomal sorting and postendocytic trafficking of the receptor through distinct pathways, and (v) recycling to restricted regions of the cell surface. We also highlight how communication between organelles controls EGFR activity along the endocytic route. Finally, we illustrate how abnormal trafficking of EGFR oncogenic mutants, as well as alterations of the endocytic machinery, contributes to aberrant EGFR signaling in cancer.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Epidermal Growth Factor Receptor (EGFR)
- EGFR Trafficking
- EGFR Signaling
- EGFR Activation
- EGFR Ubiquitination
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
9.1 Introduction: EGFR and the ErbB Family
The epidermal growth factor receptor (EGFR) belongs to the ErbB family of receptor tyrosine kinases (RTKs), which includes three other members, ErbB2, ErbB3, and ErbB4 (Lemmon et al. 2014). At the systems level, EGFR signaling is critical for developmental processes and adult tissue regeneration, while at the cellular level it elicits a number of responses, including cell proliferation, migration, and survival (Schlessinger 2014). Gain-of-function genetic lesions in the EGFR gene, as well as alterations in the EGFR signaling cascade, are involved in several human solid tumors, such as glioblastoma, lung, head and neck, and colon cancer. Thus, the EGFR is a target of several anti-cancer therapies [Sect. 9.4.3 and Yarden and Pines (2012)].
The readout of EGFR signaling is complicated by the fact that there are seven known EGFR ligands that are active in different physiological contexts and capable of inducing specific signaling and biological outputs (Singh and Harris 2005; Wilson et al. 2009): epidermal growth factor (EGF), transforming growth factor alpha (TGFα), amphiregulin (AREG), epiregulin (EREG), heparin-binding EGF-like (HB-EGF), betacellulin (BTC), and epigen (EPG). EGF is the best-studied EGFR ligand and—together with TGFα, AREG and EPG—is specific for the EGFR, while the other ligands also bind to ErbB4. The different EGFR ligands have distinct binding kinetics (Macdonald-Obermann and Pike 2014) and differentially influence EGFR trafficking and fate (see Sect. 9.2.3.3). Importantly, EGFR overexpression in solid tumors is often associated with increased secretion of cognate ligand(s) resulting in chronic EGFR activation (see Sect. 9.4.1).
EGFR signaling is finely tuned in cells by multiple coordinated mechanisms, including regulation by phosphatases, feedback inhibitors of the kinase, and endocytosis (Lemmon and Schlessinger 2010). Besides being the major mechanism of long-term signal attenuation—via removal of receptors from the plasma membrane (PM) and their targeting to degradation—endocytosis controls the timing, type, and strength of EGFR signaling, thanks to the spatial constraints provided by intracellular compartments through which the receptor is trafficked (Sigismund et al. 2012).
In this chapter, we first describe the different mechanisms governing EGFR endocytosis and postendocytic trafficking (Sect. 9.2). We then highlight the importance of endocytosis in controlling EGFR signaling and function in physiological processes (Sect. 9.3). Finally, we discuss how cancer cells evade endocytic control of EGFR signaling, thereby, acquiring a proliferative/migratory advantage (Sect. 9.4).
9.2 Mechanisms of EGFR Endocytosis
9.2.1 EGFR Activation at the Cell Surface
The human EGFR gene is located on the short arm of chromosome 7 and encodes for a 1210-residue precursor protein, which, after cleavage of the N-terminal signal peptide, yields a mature protein of 1186 residues (Ullrich et al. 1984). Herein, we adopt the amino acid numbering of the mature EGFR form.
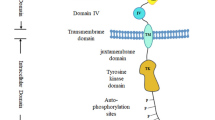
The EGFR consists of an extracellular region responsible for ligand recognition, a transmembrane (TM) domain, and an intracellular region that includes the juxtamembrane regulatory region, the kinase domain, and the intracellular C-terminal regulatory tail containing the tyrosine residues phosphorylated upon ligand binding (Lemmon and Schlessinger 2010). The intracellular region also contains lysine acceptor residues, located primarily in the kinase domain, which are critical for receptor ubiquitination (Huang et al. 2006).
Molecular details of EGFR activation at the PM have been obtained over the last decades through the combination of biological investigations and structural studies (Garrett et al. 2002; Ogiso et al. 2002) [reviewed in Lemmon et al. (2014), Kovacs et al. (2015)]. In resting cells, the EGFR continuously shifts from an open to a closed, autoinhibited, conformation. This closed state, in which intramolecular interactions prevent receptor dimerization and spurious kinase activation, is energetically favored in the absence of ligand. Thus, in resting cells, EGFR is primarily found as an autoinhibited monomer.
Ligand binding stabilizes the open EGFR conformation, which is capable of receptor dimerization, shifting the monomer–dimer equilibrium to the dimeric state (Lemmon 2009). Dimerization, in turn, determines a series of structural rearrangements that are transmitted to the cytoplasmic domain, and allow the formation of asymmetric dimers between the juxtaposed catalytic domains, finally leading to the allosteric activation of the EGFR (Zhang et al. 2006). In the active dimer, each monomer trans-autophosphorylates specific tyrosine residues in the intracytoplasmic region of the other monomer, thereby, triggering the signaling cascade (Lemmon et al. 2014).
Notably, in the absence of ligand, EGFR moieties can spontaneously form finite-lifetime dimers, whose abundance depends on cell type and EGFR expression levels (Chung et al. 2010). These preformed dimers, although primed for ligand binding and signaling, are inactive; ligand binding is still required for receptor activation and signaling (Chung et al. 2010). Importantly, EGFR overexpression, as occurs in tumors, can increase the amount of unbound homodimers (or ErbB family heterodimers) and has been proposed as a mechanism at the basis of spurious kinase activation in the absence of ligand (Chung et al. 2010). Ligand-independent kinase activation in the presence of high numbers of surface EGFRs can however be limited by phosphatases. Indeed, constitutive trafficking of unbound/inactive EGFRs to endosomes allows receptor dephosphorylation by the phosphatase PTP1B, which is resident in the endoplasmic reticulum (ER) and interacts with endosomal EGFR via so-called ER contact sites (see Sect. 9.3.1) (Baumdick et al. 2015).
Following EGFR activation, phosphorylated tyrosine residues in the intracellular tail act as docking sites for signaling molecules and endocytic adaptors, which trigger signaling and receptor endocytosis, respectively (Lemmon and Schlessinger 2010). One protein recruited to the EGFR at the PM is the E3 ligase, Cbl, which ubiquitinates lysine residues in the kinase domain (Levkowitz et al. 1998, 1999; Huang et al. 2006). EGFR ubiquitination is a critical signal in the endocytic pathway (Umebayashi et al. 2008); at the PM, it determines the endocytic route (see Sect. 9.2.2.3), while at the endosomal sorting station it targets receptors to a degradative fate (see Sect. 9.2.3.1).
9.2.2 EGFR Internalization Routes
In the absence of ligand, EGFR is internalized at a very slow rate and is mainly recycled back to the PM at a rate that is ~5–10 times higher than its constitutive endocytic rate. This results in a predominant PM location of the receptor (Dunn et al. 1986; Carpenter and Cohen 1976; Stoscheck and Carpenter 1984). The ratio of PM versus intracellular EGFR in basal conditions, however, is highly dependent on the level of EGFR expression. As expected, ligand binding and kinase activation increase the endocytic rate constant and are indeed essential for rapid EGFR endocytosis (Sorkin and Goh 2008).
Endocytosis can occur through different pathways, broadly classified as clathrin-mediated endocytosis (CME) and non-clathrin endocytosis (NCE, Fig. 9.1). The choice of these different pathways depends on the cell context, the nature of homo-/heterodimerization of the receptor, ligand concentration, and the presence of specific endocytic signals in the intracytoplasmic tail, as discussed in the following sections.

Endocytic routes and sorting of EGFR heterodimers and homodimers. At the plasma membrane (PM), EGFR dimers can be internalized by different routes. Upon ligand binding, EGFR–ErbB2 heterodimers (left) are phosphorylated and internalized via clathrin-mediated endocytosis (CME, black lines), where the internalizing pit is coated by clathrin, AP2, and endocytic adaptor proteins. EGFR–ErbB2 heterodimers are poorly ubiquitinated due to the inefficient recruitment of Cbl. Once they reach the endosomal station, ligand dissociates from the receptor due to the more acidic pH of the endosomes, and the heterodimers are almost exclusively recycled back to the PM, while being inefficiently degraded, thus sustaining signaling. At high dose of ligand, EGFR–EGFR homodimers (right) can be internalized via both CME and non-clathrin endocytosis (NCE). EGFRs entering via CME (red lines) recruit endocytic adaptors (e.g., eps15 and epsin), AP2, and signaling proteins (e.g., Grb2) and are mainly recycled back to the PM. CME is required to sustain signaling from endosomes and/or through cycles of receptor recycling. Receptor ubiquitination by Cbl is not required for CME. In parallel, a fraction of EGFR, which is extensively ubiquitinated by Cbl, in complex with Grb2, at the PM, enter the cell via NCE and is primarily targeted to the lysosome for degradation. Receptors coming from both CME and NCE reach the endosomal station, where they are subjected to further regulation by ubiquitination/deubiquitination reactions. In the endosomes, ubiquitinated EGFRs are recognized by the ESCRT-0 complex (Hrs, STAM, EPS15b), which drives the receptor to degradation
9.2.2.1 Internalization Signals
The EGFR contains several internalization motifs and signals in its intracytoplasmic region that are unmasked/activated upon EGF binding. These include two recognition motifs for the major endocytic adaptor, adaptor protein 2 (AP2), which links cargoes to the clathrin machinery: (i) the YRAL motif, responsible for recruitment of the AP2 μ subunit; (ii) the LL motif, critical for tyrosine phosphorylation of the AP2 β2 subunit, which is predicted to facilitate the interaction between this motif and AP2 (Goh and Sorkin 2013). Interestingly, mutation of these two AP2 binding motifs does not affect CME of the EGFR (Goh et al. 2010). Similarly, functional ablation of AP2 in cells only partially inhibits EGFR internalization (Hinrichsen et al. 2003; Motley et al. 2003), suggesting the existence of AP2-independent mechanisms responsible for EGFR-CME (see Sect. 9.2.2.2).
Several tyrosine residues in the EGFR cytoplasmic tail, in addition to triggering signaling events once phosphorylated, can also recruit endocytic factors (Roskoski 2014). For instance, residues pY1068/pY1086 act as a docking site for the adaptor protein, Grb2, which bridges the phosphorylated receptor to Cbl and the endocytic machinery, as well as to the RAS/MAPK signaling cascade (Goh and Sorkin 2013; Sorkin and Goh 2008). Cbl itself, besides ubiquitinating the EGFR, also acts as an adaptor molecule for several endocytic proteins involved in receptor internalization (Schmidt and Dikic 2005; Lemmon and Schlessinger 2010). As mentioned, Cbl can be recruited to the activated EGFR indirectly via Grb2 (Waterman et al. 2002; Jiang et al. 2003), In addition, Cbl can also bind directly to pY1045 (Waterman et al. 1999b). This two-pronged interaction between Cbl and the EGFR is needed for stable Cbl recruitment and efficient receptor ubiquitination (Capuani et al. 2015; Sigismund et al. 2013).
Cooperativity between the direct and indirect binding modes results in an “off–on” threshold response in receptor ubiquitination as EGF concentration increases (Sigismund et al. 2013). Indeed, while the levels of phosphorylated EGFR (EGFR-pY) increase gradually with increasing EGF concentrations, the levels of ubiquitinated receptor (EGFR-Ub) display a sigmoidal dose-response, increasing sharply between 1 and 10 ng/ml EGF before reaching a plateau (Sigismund et al. 2013). Thus, Cbl, in complex with Grb2, effectively acts as an analogical-to-digital converter that translates a linear EGF input into an “off–on” threshold response for receptor ubiquitination (Capuani et al. 2015; Sigismund et al. 2013). This ubiquitination threshold response acts as a critical signal influencing EGFR internalization in specific cellular contexts (see Sect. 9.2.2.3) and receptor degradation (see Sect. 9.2.3.1).
The impact of ubiquitination on EGFR internalization is made more complex by the fact that the EGFR is subjected to different types of ubiquitin (Ub) modifications. Mass spectrometry studies revealed that the predominant modifications are Lys63 polyUb chains and multi-monoUb, while Lys48 and Lys11 polyUb chains are less abundant (Huang et al. 2006). Lys63 and monoUb are both critical signals in trafficking (Acconcia et al. 2009), but whether they serve different functions in EGFR endocytosis remains to be determined. The relevance of Lys48 and Lys11 polyUb chains to EGFR biology is also currently unclear.
The EGFR is also modified by the Ub-like molecule, Nedd8 (Oved et al. 2006). Neddylation is catalyzed by Cbl in complex with the Nedd8-specific E2 enzyme (Ubc12), and it is thought to occur on multiple lysine residues in the kinase domain, possibly overlapping with ubiquitination sites. Nedd8 was proposed to “prime” the EGFR for further ubiquitination reactions, and to cooperate with Ub to target EGFR to degradation (Oved et al. 2006). However, the exact involvement of neddylation in EGFR biology still needs to be clarified.
9.2.2.2 Clathrin-Mediated Endocytosis
CME has been extensively studied over the last decades, the result being a high-resolution molecular picture of the process. The EGFR is internalized via CME in all cell types and at all physiological EGF concentrations (Sigismund et al. 2008, 2013; Carpentier et al. 1982; Gorden et al. 1978; Hanover et al. 1984; Sorkin and Carpenter 1993; Jiang et al. 2003). In CME, the active receptor is recognized by adaptor molecules—primarily AP2—that bridge the cargo to clathrin, driving its internalization via clathrin-coated pits [CCPs, reviewed in Kirchhausen et al. (2014), McMahon and Boucrot (2011), Fig. 9.1]. The last step of vesicle pinching from the PM is performed by the large GTPase dynamin [reviewed in Antonny et al. (2016)]. Dynamin is also part of the scission machinery in some clathrin-independent pathways (see also Sect. 9.2.2.3).
Many accessory proteins cooperate in cargo recognition, CCP formation and vesicle release, including eps15, epsin, Grb2, Cbl, and intersectins (ITSNs) (McMahon and Boucrot 2011). The involvement of so many endocytic factors in CME and the existence of distinct internalization signals in the EGFR C-terminal tail (see Sect. 9.2.2.1) have led to the notion that CME is controlled by several redundant mechanisms that together confer robustness to the system (Goh et al. 2010). Moreover, it has been hypothesized that the different endocytic proteins might be involved in the formation of distinct types of CCPs, specialized in cargo selection and targeting to specific intracellular fates (Lakadamyali et al. 2006).
In addition to internalization signals centered on the receptor, monoubiquitination of endocytic adaptors (e.g., eps15) has also been shown to be critical to EGFR-CME (Savio et al. 2016). Indeed, it has been proposed that cycles of ubiquitination (by the E3 ligase, NEDD4) and deubiquitination [by the deubiquitinating enzyme (DUB) enzyme, Usp9X] are necessary for EGFR-CME (Savio et al. 2016).
9.2.2.3 Non-clathrin Endocytosis
EGFR-NCE pathways were first observed over 30 years ago (Lund et al. 1990), but their study was hampered by their morphological heterogeneity, cell context dependency and peculiar growth condition requirements (Johannes et al. 2015). Despite their heterogeneity, the different EGFR-NCE mechanisms all share the common feature of being activated at high, nearly saturating, EGF doses (>10 ng/ml) (Boucrot et al. 2015; Lund et al. 1990; Orth et al. 2006; Sigismund et al. 2005).
For one EGFR-NCE pathway, the dependency on high EGF concentrations has been explained at the molecular level and directly linked to the EGFR-Ub threshold response [see Sect. 9.2.2.1 and Sigismund et al. (2005, 2013)]. It was shown that activation of EGFR-NCE occurs over the same EGF concentration range (~1–10 ng/ml) as EGFR-Ub [Sigismund et al. (2013) and Sect. 9.2.2.1]. Importantly, mutations that inhibit EGFR ubiquitination also inhibit EGFR-NCE to a similar extent, showing that EGFR-Ub and NCE are mechanistically linked [Sigismund et al. (2013) and Fig. 9.1, right]. Furthermore, proteins containing ubiquitin-binding domains (UBDs), such as eps15 and epsins, are needed to recognize EGFR-Ub and to target it to internalization via NCE [Sigismund et al. (2005) and Fig. 9.1, right], further supporting the link between receptor ubiquitination and NCE.
The above-described EGFR-NCE pathway is active in different cell lines and has a relatively slow internalization rate (≤CME) and requires cholesterol-enriched PM domains, while it is caveolin-independent (Sigismund et al. 2005, 2013). Importantly, internalization through NCE versus CME has important consequences on EGFR fate and signaling (Fig. 9.1, right), as discussed in Sect. 9.3.2.
At the molecular level, EGFR-NCE requires dynamin fission activity, and the ubiquitin-binding endocytic adaptors, eps15 and epsin. However, a molecular definition of the pathway was obtained only recently. Through a proteomic approach coupled with RNAi screening, proteins previously not suspected to participate in endocytosis were identified as specific players of the pathway, among which the ER-resident protein Reticulon3, RTN3 (Caldieri et al. 2017). The pathway relies on the formation of ER–PM contact sites that depend on RTN3 function and are required for the formation/maturation of NCE tubular invaginations. Local Ca2+ release at these sites, triggered by IP3-dependent activation of ER Ca2+ channels, is needed for the completion of EGFR internalization in a positive feedback loop (Caldieri et al. 2017) (Fig. 9.2). Mechanistically, how ER–PM contacts are established, if they require a direct EGFR–RTN3 interaction and if/how EGFR ubiquitination is needed for their formation remain open issues.

Model for EGFR endocytosis. At high dose of EGF, EGFR is internalized through clathrin-mediated endocytosis (CME) and non-clathrin endocytosis (NCE) in some cell lines. NCE is mediated by tubular invaginations that, differently from clathrin-coated pits, need the establishment of RTN3-dependent contact sites with the endoplasmic reticulum (ER) in order to progress. In an initial phase, RTN3-dependent ER–PM contact sites are required for the formation/maturation of tubular invaginations; then, they act as sites of local calcium release (red circles), which is required for the fission of the tubular invagination and the completion of the internalization step. By inhibiting EGFR entry via NCE, RTN3 KD affects the subsequent EGFR targeting to the lysosomal compartment and delays receptor degradation. It is unclear whether RTN3 is also involved in ER contact sites with early/late endocytic stations, e.g., endosomes, multivesicular bodies (MVBs), lysosomes
Other EGFR-NCE pathways have been described in fibroblasts and migrating cells. For instance, a macropinocytic-like pathway involving large tubular structures originating from circular dorsal ruffles or “waves” has been observed in mouse and human fibroblasts that is thought to be critical for 3D cell migration and extracellular matrix degradation [see Sect. 9.3.2 and Orth et al. (2006)]. In addition, a fast-kinetic NCE pathway that mediates ligand-triggered uptake of different G protein-coupled receptors (GPCRs) and RTKs, including the EGFR, has been identified (Boucrot et al. 2015). This endophilin-dependent, clathrin-independent pathway, called fast endophilin-mediated endocytosis (FEME) appears to be active in a very specialized region of the migrating cell, i.e., the leading edge, and to be required for spatially restricted EGF-dependent signaling [see Sect. 9.3.2 and Boucrot et al. (2015)] (see also Sect. 9.3.2). At the molecular level, FEME requires the BAR domain-containing protein, endophilinA2, as well as dynamin for scission (Boucrot et al. 2015). This NCE pathway shows many similarities with the recently described Shiga toxin uptake pathway, which is also clathrin-independent, endophilinA2-, and dynamin-dependent (Renard et al. 2015). The EGFR modifications and/or the signaling cascade required to trigger these forms of EGFR-NCE are currently unknown.
9.2.3 EGFR Trafficking and Fate
Independent of the entry route, EGFRs internalized from the PM invariably reach the early endosomes (EEs), where they are sorted toward different fates [reviewed in Wandinger-Ness and Zerial (2014)]. Characteristic features of EEs include the presence of the small GTPase, Rab5, the Rab5 effector, EEA1, and an enrichment in phosphatidylinositol 3-phosphate [PI(3)P] (Wandinger-Ness and Zerial 2014). Receptors in EEs are either directed to recycling endosomes for recycling to the PM or targeted to the late endosomes (LEs) for degradation through the progressive conversion of Rab5-enriched EEs into Rab7-enriched LEs (Rink et al. 2005; Poteryaev et al. 2010).
EGFR can also be trafficked through endosomes positive for the Rab5 effector, APPL1 (Miaczynska et al. 2004). It is currently debated whether these APPL1-positive endosomes represent a distinct class of endosomes or an early compartment in the maturation of EEA1-positive endosomes (Kalaidzidis et al. 2015; Zoncu et al. 2009). The existence of different endosomal populations, characterized by distinct molecular markers and cargoes, raises the possibility that cargo-driven regulation of the endosomal compartment might be a mechanism for achieving signal diversification. Indeed, endosomes are dynamic structures that are tightly regulated by signaling. For instance, the EGF–EGFR complex regulates the location, number, and size of EEs (Collinet et al. 2010) and drives the formation of multivesicular bodies (MVBs) from LEs (White et al. 2006). These observations highlight the instructive role of EGFR signaling on endocytic progression and suggest that the endocytic pathway can be rearranged depending on the signaling input.
In line with this concept, EGF signaling induces the synthesis of the EGFR itself (Earp et al. 1986; Scharaw et al. 2016). Notably, continuous stimulation of cells with high EGF concentrations (but not low concentrations or pulse stimulation) increases the transport efficiency of newly synthesized EGFRs from the ER to the PM, via a mechanism involving the transcription factor, RNF11, normally localized in EEs (Scharaw et al. 2016). Upon continuous, high dose, EGF stimulation, a pool of RNF11 is found in the nucleus, where it activates transcription of the inner coat protein complex II (COPII) components, SEC23B, SEC24B, and SEC24D, which are specifically required for EGFR transport to the PM (Scharaw et al. 2016). Although the mechanism is still under investigation, it has been proposed that RNF11 might act as a “sensor” in the EEs, receiving signals from internalized EGFR to translocate to the nucleus (Scharaw et al. 2016). This scenario, if confirmed, would represent a new regulatory mechanism coupling EGFR degradation (that is significant at high EGF) with its biosynthesis and transport, to preserve EGFR levels at the PM.
Together, these findings suggest that the EGFR is not a passive passenger along the endocytic pathway, but, instead, it directly influences the nature of the pathway along its journey.
9.2.3.1 Ubiquitin-Dependent Sorting of EGFR to MVBs
The decision to target cargoes to recycling or degradation is critical for cell physiology, and the discriminating factor is cargo ubiquitination (Piper et al. 2014; Conte and Sigismund 2016). Following ubiquitination, EGFRs are actively trafficked along the degradative pathway by the ESCRT (endosomal sorting complexes required for transport) complexes [reviewed in Wollert et al. (2009), Raiborg and Stenmark (2009)]. Recycling, instead, appears to be the default pathway of internalized EGFRs, and escape from this fate is achieved through efficient receptor ubiquitination.
Once EGFR-Ub reaches the limiting membrane of the MVBs, it is recognized by the ESCRT-0 complex that is comprised of the UBD-containing proteins, Hrs (hepatocyte growth factor-regulated tyrosine kinase substrate) and STAM1/2 (signal transducing adaptor molecule 1 and 2). This complex retains EGFR-Ub in the limiting membrane, thus precluding its recycling (Wollert et al. 2009; Raiborg and Stenmark 2009). Retention of EGFR-Ub triggers a series of events leading to the sequential recruitment of ESCRT-I, ESCRT-II, and ESCRT-III complexes to the MVB membrane, which transfer the cargo to one another (Wollert et al. 2009; Raiborg and Stenmark 2009). Sorting along this pathway appears to rely on Lys63-polyubiquitination of the EGFR intracytoplasmic domain (Huang et al. 2013), which provides multiple binding sites for tandem UBDs present in ESCRT components.
Finally, ESCRT-III drives inward MVB membrane invagination leading to the formation of intraluminal vesicles (ILVs) into which EGFR-Ub is packed (Henne et al. 2013). Defective EGFR ubiquitination, or downregulation of the ESCRT components, results in inefficient incorporation of EGFR-Ub into ILVs, delayed receptor degradation and sustained signaling (Bache et al. 2003; Belleudi et al. 2009; Jekely and Rorth 2003). ILVs are then released from MVBs into the lumen of the lysosome, the main hydrolytic compartment of the cell. In addition to their hydrolytic role [reviewed in Scott et al. (2014)], lysosomes are also emerging as a signaling platform, where growth factor signaling, energy metabolism, and autophagic pathways are integrated (Settembre et al. 2013).
EGFR ubiquitination is finely regulated along the endocytic pathway by the coordinated action of E3 ligases and DUBs (Clague et al. 2012). The E3 ligase, Cbl, is recruited at the PM and remains associated with the EGFR all along the endocytic route (Umebayashi et al. 2008). This ensures maintenance of EGFR ubiquitination at later stages of trafficking when it is needed for receptor targeting to the ESCRT machinery. Besides Cbl, the E3 ligase Cullin3 (CUL3) is also implicated in postendocytic trafficking of the EGFR (Huotari et al. 2012). In particular, it was shown that CUL3, in complex with the substrate-specific adaptor, SPOPL, ubiquitinates eps15 (in complex with Hrs) in endosomes, thus, regulating its turnover (Gschweitl et al. 2016). Degradation of eps15 by CUL3 appears to be critical for MVB formation, EGFR sorting to MVBs, and receptor degradation (Gschweitl et al. 2016).
Interestingly, two eps15 isoforms appear to have different roles in EGFR recycling versus degradation. Eps15s, which lacks the ubiquitin-interacting motifs (UIMs), has been implicated in receptor recycling (Chi et al. 2011), while eps15b, which lacks the EH domains, interacts with Hrs and is involved in sorting of the EGFR to MVBs (Roxrud et al. 2008).
Several DUBs are also involved in EGFR trafficking and sorting. Some DUBs appear to act directly on the EGFR, such as AMSH (associated molecule with the SH3 domain of STAM) that removes Ub from the receptor at the endosomal level, protecting EGFR from degradation and favoring its recycling (McCullough et al. 2004; Ma et al. 2007). Similarly, OTUD7/Cezanne (Pareja et al. 2012) and USP2 (Liu et al. 2013) directly counteract Cbl-mediated EGFR ubiquitination and, consequently, receptor degradation. Other DUBs, instead, act directly on the endocytic machinery, such as Usp9x, which controls EGFR fate by deubiquitination of eps15 (Savio et al. 2016), and UBPY (also called USP8, Ub-specific Protease 8), which regulates the stability of Hrs and STAM, thereby impinging on EGFR degradation (Row et al. 2006).
9.2.3.2 Inducible Feedback Inhibitors Controlling EGFR Trafficking
Sustained treatment of cells with EGF induces a transcriptional response leading to entry into the G1 phase of the cell cycle (Avraham and Yarden 2011). In this phase, positive or negative feedback regulators of EGFR signaling are transcribed (Avraham and Yarden 2011). These include the feedback inhibitors SOCS4 and SOCS5 [members of the suppressor of cytokine signaling family (Kario et al. 2005; Nicholson et al. 2005)], and LRIG1 [leucine-rich and immunoglobulin-like domain 1 (Gur et al. 2004)], which increase ubiquitination and degradation of both active and ligand-free EGFR, restricting receptor activation. In contrast, MIG6 (mitogen-induced gene 6, also known as RALT) acts through a Ub-independent mechanism to inhibit EGFR signaling: it binds to the ligand-bound EGFR kinase domain and inhibits its allosteric activation (Anastasi et al. 2007; Zhang et al. 2007). MIG6 also drives endocytosis and degradation of inactivated EGFRs in a Ub-independent manner (Frosi et al. 2010), via an unknown mechanism.
Importantly, loss of LRIG1 and MIG6 in mice causes increased EGFR expression and aberrant cell proliferation, leading to tissue hyperplasia (Segatto et al. 2011) and, in the case of MIG6, to epithelial tumor formation (Ferby et al. 2006), highlighting the critical role of these feedback inhibitors in restricting EGFR activation.
9.2.3.3 Impact of Different EGFR Ligands and Heterodimers on Receptor Trafficking and Fate
Different EGFR ligands (see Sect. 9.3.2) can induce different signaling outputs by mechanisms that are still not fully defined. One mechanism controlling ligand-dependent signaling specificity appears to be the strength of the ligand–receptor interaction. This has been demonstrated for TGFα versus EGF, which display similar affinities for EGFR at the neutral pH of the PM, while in the mildly acidic endosomal environment (pH ~6–6.5) the affinity of TGFα drops causing ligand–receptor dissociation (Ebner and Derynck 1991; French et al. 1995). This results in EGFR inactivation, receptor dephosphorylation, Cbl detachment, and receptor deubiquitination (Longva et al. 2002). The TGFα-free EGFRs are then recycled to the cell surface. This propensity for receptor recycling following TGFα stimulation is consistent with the higher capacity of TGFα to induce mitogenic signaling compared with EGF (Waterman et al. 1998; Lenferink et al. 1998).
In contrast, the EGF–EGFR complex remains stable along the endocytic route and continues to be ubiquitinated by Cbl (Umebayashi et al. 2008) and to proceed toward the degradative compartments (Ebner and Derynck 1991; French et al. 1995). Of note, not all EGF–EGFRs are ubiquitinated and targeted for degradation; some EGF–EGFRs are recycled to the PM. The ratio between EGFR degradation versus recycling is finely regulated by EGF concentration and activation of different endocytic pathways (see Sect. 9.3.2).
Similar to TGFα, it was shown that EGFRs bound to EPI, EREG, and AREG are preferentially recycled back to the PM with little, if any, degradation, while BTC and HB-EGF efficiently induce EGFR ubiquitination and lysosomal degradation (Roepstorff et al. 2009; Stern et al. 2008; Wilson et al. 2012).
The nature of the EGFR homo-/heterodimers formed upon ligand binding can also influence receptor trafficking (Lenferink et al. 1998). For instance, compared with EGFR homodimers, heterodimers recruit inefficiently Cbl and the endocytic machinery (Baulida et al. 1996; Levkowitz et al. 1996; Waterman et al. 1999a). Moreover, ligand-binding affinity is reduced in the context of heterodimers, causing ligand dissociation in endosomes (Lenferink et al. 1998). Together, these properties cause the efficient recycling of heterodimers coupled with inefficient degradation (Fig. 9.1, left). Signaling from heterodimers is therefore more sustained and potentially more oncogenic than signaling from homodimers (see Sect. 9.4.1). Indeed, EGFR kinase active mutants in non-small-cell lung cancer (NSCLC) have been proposed to form heterodimers with ErbB2 to escape downregulation (see Sect. 9.4.1).
9.3 Control of EGFR Signaling by Endocytosis
Endocytosis is a mechanism to downregulate signaling by removing active receptors from the PM and targeting them to lysosomal degradation [reviewed in Sigismund et al. (2012)]. However, the impact of endocytosis on signaling extends beyond signal extinction. Endocytic recycling pathways are crucial for sustaining signaling and redirecting receptors to specific regions of the PM, while the distinct endocytic compartments provide temporal and spatial dimensions to the signaling cascade (Sigismund et al. 2012). These compartments serve at least two functions: (i) they sustain signaling originating at the PM by continuously recruiting the same PM signaling effectors; (ii) they facilitate the assembly of endomembrane-specific signaling platforms leading to diversification of the signaling response (see Sect. 9.3.3).
Endocytosis is not required for all signaling outputs. Impairment of EGFR endocytosis using a dominant-negative dynamin mutant increased PLCγ and Shc activation, and, concomitantly, decreased PI3K/Akt and Erk signaling, leading to inhibition of EGF-dependent mitogenesis (Vieira et al. 1996). Similarly, inhibition of CME in HeLa cervical cancer cells by clathrin- or AP2-knockdown curtailed Erk and Akt phosphorylation, without affecting Shc phosphorylation (Sigismund et al. 2008). In contrast, in dynamin-knockout mouse fibroblasts, inhibition of EGFR internalization did not alter Erk and Akt signaling elicited by EGF stimulation (Sousa et al. 2012), suggesting that the endocytic requirement of specific signaling outputs might be cell type specific. To further complicate the picture, the EGFR can be internalized through different internalization routes with specific fates and signaling outcomes (Fig. 9.1 and Sect. 9.3.2).
9.3.1 Regulation of EGFR Activity by Phosphatases Along the Endocytic Pathway
Along the endocytic pathway, EGFR is subjected to fine-tuned regulation of its signaling by different enzymes. For instance, DUBs, by regulating EGFR ubiquitination, influence sorting to the lysosome and receptor downmodulation (see Sect. 9.2.3.1). Protein tyrosine phosphatases (PTPs) also affect signaling at different steps of the endocytic pathway [Fig. 9.3 and Lemmon et al. (2016)].

Modulation of EGFR signaling by phosphatases. At the PM, dynamic interchange of EGFR phosphorylation by the activated EGFR kinase and dephosphorylation by phosphatases allows for rapid receptor activation, while ensuring responsiveness of the system (1). At the endosomal station, phosphatases, which are activated by the EGFR in a feedback loop, serve to maintain a constant number of active EGFRs/endosome (2). Once the EGFR has been internalized and reaches the multiple vesicular bodies (MVBs), the phosphatase, PTP1B, located at the cytosolic face of the endoplasmic reticulum (ER), dephosphorylates the receptor at the ER–MVB contact sites, prior to its targeting to the intraluminal vesicles of MVBs (3). ER–MVB contact sites are tethered by annexin1—localized in the ER—through its binding to the EGFR in the MVBs. Annexin1 is regulated by calcium (Ca++) release at contact sites and is involved in intraluminal vesicle formation and MVB maturation. PTP1B is also involved in the dephosphorylation of unliganded receptors, which have been internalized via the constitutive pathway to the endosomal station where they are dephosphorylated by PTP1B at ER-endosome contact sites and are then recycled back to the PM (4). This mechanism has been proposed to limit spurious kinase activation
PTPs are active in the early phases of EGFR activation at the PM [Fig. 9.3 (1) and Kleiman et al. (2011)], although at this stage the EGFR kinase activity overwhelms their action and the receptor is rapidly phosphorylated (Capuani et al. 2015). Nonetheless, the fast phosphorylation turnover at the PM is thought to increase responsiveness, providing dynamic plasticity to the system in response to different cues (Lemmon et al. 2016). PTPs have an even more prominent role in the endosomes (Kleiman et al. 2011). Here, their action is critical for maintaining a specific amount of active receptors per endosome (Villasenor et al. 2015), which, in turn, determines the final signaling output (Fig. 9.3 (2) and Sect. 9.3.3).
How the spatial distribution of PTPs along the endocytic pathway regulates the number of active EGFRs is exemplified by the ER-localized phosphatase, PTP1B (see Sect. 9.3.4 for EGFR regulation by PTP1B at ER-endosome contact sites). This phosphatase is unevenly distributed in the cell, with lowest concentrations found at the cell periphery and highest at perinuclear area (Eden et al. 2010), where termination of signaling in LEs takes place. PTP1B dephosphorylates ligand-activated EGFRs trafficking en route toward the LEs prior to degradation in the lysosome [Fig. 9.3 (3) and Baumdick et al. (2015)], as well as EGFRs activated independently of ligand (phosphorylated at Y845) that have reached the perinuclear compartment [Fig. 9.3 (4)], prompting their recycling back to the PM. This latter mechanism is thought to suppress spurious kinase activation, while maintaining sensitivity to EGF at the PM.
These studies imply that dephosphorylation by PTPs is a way to restrict EGFR signaling and to maintain physiological levels of active receptors. This regulatory function of PTPs is in agreement with their role as tumor suppressors (Zhao et al. 2015). However, PTPs can also function as positive regulators of RTKs, as in the case of PTPD1, a FERM (four-point-one, ezrin, radixin, moesin) domain-containing PTP that has been shown to promote EGFR signaling (Cardone et al. 2004; Carlucci et al. 2010). In cell monolayers, PTPD1 is excluded from E-cadherin rich cell–cell contacts, while in isolated cells it relocalizes from the cytosol to the PM regions by binding to phosphoinositides through its FERM domain (Roda-Navarro and Bastiaens 2014). Specifically, PTPD1 is transiently recruited to EGF-induced membrane ruffles and is released just before the formation of active EGFR-containing micropinosomes. Although the exact mechanism is unclear, functional data suggest that PTPD1 has a positive role in the propagation of EGFR signaling at early stages of the pathway (Roda-Navarro and Bastiaens 2014).
9.3.2 Regulation of EGFR Signaling by the Internalization Route
The internalization route taken by the EGFR at the PM is critical in determining receptor fate. Depending on the concentration of the ligand, different endocytic pathways (CME and NCE) can be activated (see Sect. 9.2.2.3). In HeLa cells, CME and NCE counteract each other by determining opposing (recycling vs. degradation) receptor fates [Fig. 9.1, right, and Sigismund et al. (2008)]. CME, which is active at all ligand concentrations, preferentially targets the EGFR for recycling to the PM (around 70%), with a minor portion directed to lysosomes for degradation (around 30%). In contrast, NCE is sharply activated at sub-saturating EGF doses following receptor ubiquitination (Sect. 9.2.2.3) and targets the majority of EGFRs for degradation (>90%), resulting in signal attenuation in conditions of excessive stimulus (Sigismund et al. 2008). The integrated function of CME and NCE determines the final EGFR signaling response: a mechanism that also applies to other receptors, such as TGFβR (Di Guglielmo et al. 2003), Notch (Shimizu et al. 2014) and Wnt (Yamamoto et al. 2006, 2008).
The mechanisms by which CME influences EGFR signaling are multiple. By promoting recycling, CME prolongs the EGFR signaling response and protects the receptor from degradation in conditions of limited ligand availability. Additionally, receptors can be recycled to specific regions of the PM where signaling is needed. These two properties highlight CME as a mechanism providing spatial and temporal control to EGFR signaling. Consistently, CME is required for sustaining the later decay-phase of EGFR signaling and for EGFR-mediated DNA synthesis [see Sect. 9.3 (Vieira et al. 1996; Sigismund et al. 2008)].
CME also contributes to the “early phase” of EGFR signaling at the PM. A single particle tracking study investigating the correlation between EGFR mobility/aggregation at the PM and receptor signaling activity showed that immobile EGFR is clustered in CCPs that act as platforms for enhanced receptor phosphorylation and consequently signal amplification (Ibach et al. 2015). This allows the formation of local gradients of active receptors that spatially constrain EGFR signaling in response to local stimuli.
In contrast, NCE appears to be responsible for EGFR degradation and long-term signaling attenuation in conditions of high EGF in specific cellular contexts (Sigismund et al. 2008). One hypothesis is that NCE represents a mechanism to protect cells from overstimulation. Thus, loss of this route could lead to aberrant EGFR signaling and contribute to tumorigenesis.
The upstream signal triggering NCE is the sharp increase in EGFR ubiquitination at high EGF concentrations [see Sect. 9.2.2.1 and Sigismund et al. (2013)], which seals receptor fate already at the PM. A study integrating mathematical modeling and wet-laboratory experiments revealed that EGFR ubiquitination—and consequently its recruitment to NCE—is controlled by EGFR levels (Capuani et al. 2015). In physiological conditions, EGFR phosphorylation is counterbalanced by its ubiquitination, limiting receptor activation. However, at supraphysiological EGFR/EGF levels, EGFR phosphorylation and ubiquitination become uncoupled, leading to increased receptor signaling that is no longer counteracted by degradation (Capuani et al. 2015). Under these conditions, EGFR would evade NCE-mediated downmodulation, providing cancer cells with a proliferative advantage (see Sect. 9.4.1).
As in the case of CME, where CCPs were shown to function as platforms for local amplification of EGFR signaling, NCE routes have also been shown to be confined to specific PM regions where they execute polarized functions. For example, the FEME pathway (see Sect. 9.2.2.3) was shown to act locally at the leading edge of migrating cells (Boucrot et al. 2015) to ensure the rapid internalization of receptors through tubular-vesicular structures and, possibly, to promote EGF-dependent directed cell migration. Additionally, in mouse and human fibroblasts, EGFR is internalized through clathrin-independent macropinocytic-like pathways mediated by circular dorsal ruffles or “waves” in specific regions of the PM, which generate tubular-vesicular structures (Orth et al. 2006). The ability of cells to internalize large numbers of EGFRs might be relevant for signaling and polarized processes. It has been hypothesized that “waves” might contribute to three-dimensional cell migration and to extracellular matrix degradation, two critical processes in tumor cell invasion (Suetsugu et al. 2003).
9.3.3 Regulation of EGFR Signaling at the Level of the Endosomes
Endosomes, in addition to being critical sorting stations, are thought to be important platforms for signaling events, where signals elicited at the PM can be sustained and/or diversified (Villasenor et al. 2016). This notion was first proposed in the 1990s, when RTKs and connected signaling molecules were detected in endosomes (Di Guglielmo et al. 1994; Grimes et al. 1996), and was later reinforced and extended to other receptors [see, for instance, Schenck et al. (2008), Coumailleau et al. (2009), Fortian and Sorkin (2014), Calebiro (2009, #8), Ferrandon et al. (2009), Nakamura (2014, #37), Irannejad et al. (2013), Lampugnani et al. (2006)]. The concept of a “signaling endosome” originated from neurons, in which NGF binding to its receptor, TrkA, in axon terminals initiates a signaling response that is then transmitted to the neuronal cell body through a long distance, retrograde, transport of endosomes carrying activated TrkA (Grimes et al. 1996; Beattie et al. 1996; Howe and Mobley 2005; Cosker et al. 2008).
More recently, the concept of “signaling endosomes” was corroborated by studies on both RTKs and GPCRs [reviewed in Irannejad et al. (2015)]. Three mechanisms have been proposed by which endosomes control signaling: “scaffolding,” “sequestration,” and “catalysis” (Irannejad et al. 2015). In the “scaffolding” mechanism, growth factor receptors confined in endosomes engage signaling adaptors that act as a scaffold for downstream effectors, promoting their activation. For example, EGFR engages the adaptor Grb2 which recruits and activates Erk (Di Guglielmo et al. 1994; Fortian and Sorkin 2014). Similarly, GPCRs use beta-arrestin as an endosomal scaffold to continue signaling after internalization (Shenoy and Lefkowitz 2005), and phosphorylated C-Met engages the nucleotide exchange factor, Vav2, which leads to sustained Rac signaling (Menard et al. 2014).
In the “sequestration” mechanism, signal amplification is achieved by entrapping cytoplasmic, negative, signaling regulators in endosomes. For instance, in Wnt/wingless signaling, the inhibitory enzyme, GSK3, is physically sequestered into the endosomal lumen, leading to reduced cytosolic GSK3 activity and, consequently, to enhanced beta-catenin signaling (Taelman et al. 2010).
Finally, the “catalysis” mechanism involves activation of enzymes in endosomes to augment signaling. For example, heterotrimeric G proteins from GPCRs are activated not only at the PM, but also in the limiting endosomal membranes where they promote downstream signaling through production of second messengers, such as cyclic AMP (cAMP) (Irannejad et al. 2013; Tsvetanova and von Zastrow 2014). In this case, endosomal signaling acts to sustain the cAMP response observed at the PM, and to determine the final cAMP-dependent transcriptional response (Tsvetanova and von Zastrow 2014).
In the case of the EGFR, a quantitative high-resolution microscopy approach revealed that the endosomal system works as an analog–digital converter (Villasenor et al. 2015). Active phosphorylated EGFRs form clusters of ~80 molecules per EE. The endosomal fusion machinery works to keep the number of active EGFRs/endosome constant: Increasing the EGF concentration does not produce larger EGFR clusters, rather, a higher number of EGFR-positive endosomes. Notably, inhibition of endosome fusion enhances the number of EGFR clusters and determines a different signaling outcome, i.e., prolonged EGFR activation and Erk signaling response. These clusters represent the quanta of signaling that provide robustness to the cellular response in case of fluctuations in ligand or receptor levels. This mechanism applies also to other RTKs, such as the hepatocyte growth factor receptor (HGFR) and the nerve growth factor receptor (NGFR) (Villasenor et al. 2015).
Also in this case, phosphatases have a pivotal role in fine-tuning EGFR signaling: Phosphorylated EGFR in endosomes recruits and activates, through phosphorylation, the phosphatase SHP2, forming a negative feedback loop to maintain a constant number of phosphorylated EGFRs/endosome (Villasenor et al. 2015).
9.3.4 How the Endoplasmic Reticulum Modulates EGFR Signaling
Multiple cell compartments act in concert to control intracellular signals. This integrated function is mainly achieved by membrane–membrane contact sites: regions of close apposition (<30 nm) between the membranes of organelles (Phillips and Voeltz 2016). In recent years, contact sites have emerged as platforms of signaling regulation and places where materials, such as lipids and Ca2+, can be rapidly exchanged [reviewed in Levine and Patel (2016)]. ER-endosomal contact sites have been detected in mammalian cells, while ER-vacuole contact sites have been identified in yeast (Eden et al. 2010; Rocha et al. 2009; West et al. 2011). A study based on high-resolution, three-dimensional, electron microscopy showed that endosomes trafficking along microtubules are wrapped by ER tubules. These contacts are maintained, and actually increase, as endosomes traffic and mature (Friedman et al. 2013). Importantly, the ER-endosomal contact sites determine the timing and position of endosome fission events during cargo sorting (Rowland et al. 2014).
Crosstalk between the endosomal compartment and the ER also has a role in the modulation of RTK signaling. For instance, upon internalization, the EGFR—and other RTKs (e.g., insulin receptor and Met)—interacts with the phosphatase PTP1B localized at the cytosolic face of the ER (Haj et al. 2002; Sangwan et al. 2008; Romsicki et al. 2004). PTP1B regulates both constitutively internalized and ligand-activated EGFR (see Sect. 9.3.2). EGFR–PTP1B proximity was shown to occur at sites of physical contact between the ER and the limiting membrane of MVBs (Eden et al. 2010). At these sites, ER-resident PTP1B dephosphorylates MVB-localized EGFR “in trans” [Fig. 9.3 (3)]. The formation of ER–MVB contacts is mediated by annexin-1 and its Ca2+-dependent binding partner S100A11, in a Ca2+-dependent fashion (Eden et al. 2016; Kilpatrick et al. 2017). Ca2+ is released from the endolysosomal compartment by the two-pore channel (TPC), which localizes at ER-endosome contact sites and is regulated by nicotinic acid adenine dinucleotide phosphate (NAADP). Affecting these contacts delayed EGFR dephosphorylation by PTP1B and its subsequent degradation, enhancing signaling (Kilpatrick et al. 2017).
In addition to dephosphorylating internalized RTKs after endocytosis, PTP1B was also shown to act on EGFR localized at the PM, through the formation of ER–PM contact sites (Haj et al. 2012). This interaction appears to be restricted to regions of cell–cell contacts, identified as sites of PTP1B-mediated signaling regulation.
As we discussed (Sect. 9.2.2.3), ER–PM contact sites are also critical at early step of EGFR endocytosis. Indeed, the ER-resident protein RTN3 mediates the formation of contacts between the ER and sites of EGFR internalization at the PM at high ligand concentration (Fig. 9.2), a mechanism that leads to EGFR-NCE receptor degradation and signal extinction (Caldieri et al. 2017). Thus, ER contact sites control EGFR fate at multiple levels; e.g., at the PM and the endosomal stations. Ca2+ signaling appears to be involved in both cases, although through different mechanisms (Kilpatrick et al. 2017; Caldieri et al. 2017). Whether RTN3 is acting only at PM–ER interface or it has a role also at later step is unclear. Furthermore, a possible interplay between PTP1B and RTN3-dependent ER–PM contact sites in EGFR regulation remains to be established.
Finally, ER-based ubiquitination has been proposed to regulate levels of newly synthesized ErbB3 receptors by promoting their ER-associated degradation (ERAD) (Fry et al. 2011). Indeed, the ER-localized E3 ubiquitin ligase, Nrdp1, interacts with and ubiquitinates the nascent form of ErbB3, thereby regulating the steady-state levels of the receptor (Fry et al. 2011). Whether this mechanism also applies to the EGFR or other RTKs remains to be established.
9.4 EGFR Trafficking and Cancer
In addition to being a critical regulator of physiological cellular processes, EGFR signaling has a crucial role in the development and progression of many types of cancer; a condition where normal cellular homeostasis is subverted (Zwick et al. 2001). The first evidence linking altered EGFR signaling to cancer came in the early eighties when the viral-erbB (v-erbB) oncogene product was found to be homologous to the amplified EGFR gene in the human A431 epidermal carcinoma cell line (Ullrich et al. 1984). Since then, numerous studies characterizing the role of the EGFR in cancer have been conducted. The emerging concept is that there is a tight relationship between the oncogenic forms of the receptor and the trafficking routes the protein takes inside the cell.
9.4.1 How Different Oncogenic Forms of EGFR Are Influenced by Trafficking
Neoplastic transformation induced by the EGFR can be triggered by gene amplification and/or protein overexpression, mutations, or in-frame deletions (Roskoski 2014). These genetic lesions frequently occur concomitantly with increased EGFR ligand production triggered by autocrine or paracrine loops (Wilson et al. 2009). Autocrine secretion is often the result of positive feedback loops downstream of excessive EGFR activation that ultimately lead to the induction of the promoter of EGF family ligands (Avraham and Yarden 2011). Additionally, some solid tumors upregulate metalloproteases leading to enhanced cleavage of EGF ligand precursors (Wilson et al. 2009).
EGFR genetic alterations have been reported to cause altered trafficking of the receptor, which contributes to aberrant signaling and oncogenesis. For instance, gene amplification or receptor overexpression leads to increased EGFR density on the PM, which favors receptor dimerization and spurious kinase activation (Wiley 1988; Sawano et al. 2002; Chung et al. 2010). It has been suggested that the excessive number of activated EGFRs causes saturation of the endocytic machinery, increasing the residence time of surface EGFRs, delaying downregulation and, ultimately, leading to sustained signal (French et al. 1994; Wiley 1988).
Additionally, saturation of the endocytic/ubiquitination machinery has been proposed as a mechanism underlying sustained signaling in EGFR-overexpressing cancer cells (Capuani et al. 2015). As receptor levels increase, there is a progressive uncoupling between EGFR phosphorylation and ubiquitination (see Sect. 9.3.2). This uncoupling is due to the limiting amount of Cbl, which becomes saturated in conditions of high numbers of activated EGFR (i.e., EGFR overexpression coupled with ligand overproduction). This situation is predicted to cause sustained EGFR signaling and impaired receptor downregulation, which, however, can be partially restored by overexpressing Cbl in the cell (Capuani et al. 2015). Thus, Cbl is the weak and critical element in the system and, consistently, escape from Cbl-dependent degradation is one of the most common mechanisms enacted by oncogenic EGFR mutants (see below).
Finally, EGFR overexpression favors heterodimerization with the other ErbB family members, which influences trafficking (Arteaga and Engelman 2014). In particular, heterodimerization of EGFR with the ligand–orphan receptor ErbB2 enhances recycling [Fig. 9.1, left, and Ebner and Derynck (1991), French et al. (1995)]. ErbB2, besides being constitutively active when engaged in a heterodimer, evades ubiquitination, thereby, favoring recycling and sustained signaling over degradation and signal attenuation (see Sect. 9.2.3.3). Therefore, the formation of EGFR–ErbB2 heterodimers shifts the signaling output toward proliferation (Lenferink et al. 1998; Worthylake et al. 1999).
One of the best-described mechanisms of oncogenic activation of the EGFR is mutation. Large genetic rearrangements, as well as single base mutations, have been described, which produce oncogenic forms of the EGFR, whose expression often correlates with poor prognosis (Yarden and Pines 2012). In some cases, aberrant endocytosis and trafficking of these mutated receptors have been shown to contribute to their deregulated signaling (Yarden and Pines 2012).
A well-characterized truncated form of the EGFR is EGFRvIII, which has been detected in brain, most glioblastomas, and lung, breast and ovarian cancers (Ekstrand et al. 1992; Moscatello et al. 1995; Wong et al. 1992). EGFRvIII is a deletion mutant that lacks exons 2–7, resulting in a receptor with a truncated extracellular domain. This truncation mutant dimerizes and undergoes autophosphorylation in absence of ligands, while being poorly internalized and efficiently recycled back to the PM rather than being degraded. The end result is the excessive and sustained activation of the EGFR signaling cascade (Grandal et al. 2007). Although the truncation affects the extracellular portion of the receptor, the sustained EGFR activation is thought to be caused by impairment of receptor ubiquitination due to hypo-phosphorylation of pY1045, the direct Cbl binding site. This leads to slow receptor turnover and increased signaling (Schmidt et al. 2003; Han et al. 2006; Grandal et al. 2007). Despite possessing the same intracytoplasmic tail as the wild-type receptor, EGFRvIII activates different signaling networks compared to the wild-type receptor possibly attributable to altered internalization and trafficking (Johnson et al. 2012).
Reduced downmodulation of EGFR has also been described for other mutant forms of the receptor, EGFRvIV and EGFRvV, which lack the portion of the cytoplasmic tail encompassing the Cbl binding site, Y1045 (Roskoski 2014). These mutants, whose activation is still ligand-dependent, retain the potential to modulate oncogenic signaling pathways, e.g., Ras/MAPK signaling, commonly elicited by the wild-type receptor (Grovdal et al. 2004).
Other somatic activating mutations in the EGFR have been identified in NSCLC and patients carrying these mutations are treated with EGFR kinase inhibitors (e.g., Gefinitib and Erlotinib) as the first-line therapy (Lynch et al. 2004; Paez et al. 2004; Pao et al. 2004; Roskoski 2014). These activating mutations appear to lock the receptor in an active conformational state, causing ligand-independent firing and signaling up to 50-fold above the basal unliganded receptor activity (Yun et al. 2007). The EGFR-L834R mutant exemplifies the connection between EGFR ubiquitination-dependent trafficking and human cancers. EGFR–L834R possesses an intact Cbl binding site that is more highly phosphorylated compared with the wild-type receptor. Nevertheless, Cbl recruitment and receptor ubiquitination are impaired, causing reduced degradation and sustained activation of downstream signaling molecules, including Ras, MEK, and Erk (Kon et al. 2014; Shtiegman et al. 2007). One hypothesis to explain these observations is that EGFR–L834R forms heterodimers with ErbB2, even in the absence of ligand (Kon et al. 2014). NCSLC EGFR mutants in exons 18–21 also show a higher propensity to heterodimerize with ErbB3 (Rothenberg et al. 2008), which, as in the case of ErbB2 heterodimers, might divert EGFR mutants from a degradative toward a recycling fate, thereby enhancing signaling.
9.4.2 Mutations in Trafficking Genes Influencing EGFR Oncogenic Potential
Besides EGFR mutations that affect Cbl recruitment and activity toward the receptor, Cbl itself is mutated in human cancers [reviewed in Sigismund et al. (2012)]. Missense homozygous mutations of Cbl targeting its E3 ligase activity have been described in ~5% of myeloid neoplasms (Caligiuri et al. 2007; Dunbar et al. 2008; Sargin et al. 2007). In these cases, however, Cbl activity is primarily directed toward the RTK, FLT3 (Grand et al. 2009; Sargin et al. 2007; Sanada et al. 2009), with no connection to EGFR ubiquitination and trafficking. Similarly, heterozygous germline mutations of Cbl are found in patients affected by Noonan Syndrome (NS), a clinically variable disease [reviewed in Allanson (2007), Tartaglia et al. (2011)]. As in myeloid malignancies, Cbl mutations are missense mutations that alter the region responsible for ligase activity, but in this case they are heterozygous and thus predicted to act in a dominant-negative fashion (Martinelli et al. 2010). When overexpressed in COS-1 cells, these mutants affect EGFR ubiquitination and cause prolonged Ras–MAPK signaling (Martinelli et al. 2010). However, the relevance of these mutations to EGFR ubiquitination and trafficking in vivo has not yet been established.
In addition to Cbl, several oncogenes have been proposed to influence EGFR signaling by altering its trafficking, thereby contributing to EGFR oncogenic potential. For instance, in NSCLC cell lines, Src, a non-receptor tyrosine kinase (nRTK) that is directly activated by the EGFR (and other RTKs), cooperates with mutated EGFR to generate aberrant signaling and to induce cell transformation (Chung et al. 2009; Leung et al. 2009). Furthermore, aberrant Src activation, as observed in many cancer cells or cells transformed by the viral oncogene, v-Src, interferes with Cbl-mediated EGFR ubiquitination and receptor downmodulation (Bao et al. 2003; Wu et al. 2003; Feng et al. 2006).
Another oncogene that influences EGFR endocytosis is ACK1 (activated Cdc42-associated Kinase), a nRTK that interacts with EGFR-Ub through its UBD (Shen et al. 2007), facilitating receptor degradation (Kelley and Weed 2012). When mutated, ACK1 retains EGFR at the PM, sustaining its signaling (Chua et al. 2010; Kelley and Weed 2012). Similarly, the oncogenic form of Vav, a Rho GTPase guanine nucleotide exchange factor (GEF), causes increased Erk and Akt phosphorylation upon EGFR activation by delaying receptor endocytosis (Thalappilly et al. 2010).
In addition to PM signaling, aberrant EGFR signaling from intracellular compartments can also be oncogenic. This was shown in cancer cells with loss of function mutations of the Von Hippel-Lindau (VHL) protein or in cells subjected to hypoxic conditions (Wang et al. 2009). In both cases, reduced expression of the Rab5 effector, rabaptin-5, was observed, which determines inefficient Rab5-mediated endosome fusion and persistent retention of active EGFR in EEs, leading to prolonged pro-survival signaling from intracellular compartments (Wang et al. 2009).
Finally, alterations of proteins not directly involved in EGFR regulation, but active in other cellular pathways subverted in cancer, can interfere with EGFR (and MET) signaling (Muller et al. 2009, 2013). This is the case of p53 gain-of-function mutants that have lost tumor-suppressor activity, but have acquired endocytosis-related phenotypes, which interfere with EGFR trafficking and signaling. Expression of these mutants enhances co-trafficking and recycling of the β1-integrin/EGFR complex, via a mechanism dependent on the Rab11-effector, Rab-coupling protein (RCP), resulting in constitutive activation of EGFR/integrin signaling. Consequently, mutant p53 expression promotes tumor cell invasion, random cell migration and metastatic dissemination (Muller et al. 2009).
In conclusion, there are several, although scattered, evidences linking the oncogenic potential of known endocytic/signaling molecules to an altered EGFR trafficking. Yet, a direct conclusive link is missing. Given the potential relevance of this issue to cancer, future investigations are warranted.
9.4.3 Pharmacological Targeting of EGFR: Harnessing EGFR Endocytosis
Given the crucial role of the EGFR in different cancers, much effort has been placed on the discovery of target-specific drugs that modulate its activity (Arteaga and Engelman 2014). These include monoclonal humanized antibodies (mAbs) directed against the extracellular domain of the EGFR and selective small molecule inhibitors that target the intracellular tyrosine kinase domain.
By targeting the ATP-binding domain of the EGFR, small molecule inhibitors impair phosphorylation of the receptor C-terminal tail causing repression of ligand-induced signals (Arteaga and Engelman 2014). Interestingly, these inhibitors show a higher affinity for mutated forms of EGFR, meaning inhibition is achieved at lower drug concentrations compared to those needed for inhibition of the wild-type receptor (Carey et al. 2006). Examples of small molecule EGFR inhibitors include Gefinitib, Erlotinib, and Afatinib, which are approved for lung cancer treatment (Hirsch et al. 2013; Cohen et al. 2005; Thatcher et al. 2005). Interestingly, EGFR kinase inhibitors, like Gefitinib, increase the formation of inactive dimers through an inside-out signaling transmitted from the kinase domain to the extracellular dimerization domain (Arteaga et al. 1997; Gan et al. 2007). Gefitinib-induced EGFR dimers display increased ligand-binding affinity and peculiar binding kinetics (Bjorkelund et al. 2011). Whether the increase in dimer formation might stimulate EGFR endocytosis and, thus, contribute to signal extinction in parallel to kinase inhibition, remains to be clarified.
Cetuximab and Panitumumab are the most widely used EGFR-neutralizing mAbs. Their effectiveness has been proven in the treatment of head and neck cancer, and metastatic colon cancer (Peeters et al. 2015; Licitra et al. 2013; Pierotti et al. 2010). Mechanistically, these mAbs act by preventing ligand binding, thereby, inhibiting receptor activation and downstream signaling (Bou-Assaly and Mukherji 2010; Dubois and Cohen 2009; Vincenzi et al. 2008). The mAbs also induce EGFR dimerization and, thus, it has been proposed that they stimulate EGFR endocytosis and downmodulation (Fan et al. 1993). However, experiments with radiolabeled Cetuximab showed that antibody-bound EGFRs are internalized at a lower rate compared with ligand-induced endocytosis and are more efficiently recycled compared with EGF-bound dimers (Jaramillo et al. 2006). Interestingly, the combination of anti-EGFR antibodies directed against non-overlapping antigens was more efficient in interfering with ligand binding, and in accelerating EGFR endocytosis and degradation (Friedman et al. 2005; Pedersen et al. 2010) or increasing receptor recycling (Spangler et al. 2010). Although the mechanism is still unclear, combinatorial EGFR antibody treatment might improve anti-tumor efficacy through the regulation of EGFR trafficking.
9.5 Concluding Remarks
A wealth of evidence points to the relevance of endocytosis and trafficking in determining EGFR signaling outcome and in governing cell behavior, as also supported by the frequent alterations of EGFR endocytic and trafficking routes in human cancers. Yet, there are many aspects of the EGFR pathway that still need to be decoded, both in physiological contexts as well as in cancer. A major challenge is to clarify how EGFR signaling is interpreted in space and time, and how it is integrated with other cellular processes and signaling pathways to determine a specific cellular outcome. This should be clarified not only at the population level, but also at single-cell level. Indeed, single-cell heterogeneity in a population context was shown to be critical for the final cellular response (Elowitz et al. 2002; Frechin et al. 2015; Snijder et al. 2009). Notably, EGFR endocytosis and its downstream signaling are strongly population context dependent (Cohen-Saidon et al. 2009; Snijder et al. 2009; Liberali et al. 2014).
There is also pressing need to follow the trafficking and fate of individual receptors in unperturbed conditions (i.e., without ablation of critical factors or treatment with chemical inhibitors), in order to illuminate the contribution of the different factors to EGFR endocytosis in physiological settings. This area of investigation, which is being greatly advanced by technologies for single-molecule tracking, is particularly relevant for endocytosis. Endocytosis is a highly modular process with many alternative (and redundant) signals, adaptors, and fission machineries. As a consequence, it is highly plastic and can be efficiently and rapidly rewired through adaptive modifications of the availability of endocytic factors, PM lipid/cholesterol level, and changes in membrane tension. Consequently, compensation among different endocytic pathways is likely and it has actually has been reported (Kalia et al. 2006; Nevins and Thurmond 2006; Damke et al. 1995; Guha et al. 2003; Chaudhary et al. 2014), rendering the analysis in unperturbed conditions highly needed.
Finally, increasingly advanced models of EGFR signaling and endocytosis are needed to achieve system-level understanding. Mathematical models of both the EGFR signaling cascade (Kholodenko et al. 1999) and EGFR trafficking (Sorkin et al. 1991; Wiley et al. 1991) have been generated in the past; however, they were treated initially as separated processes. Attempts to integrate EGFR activation, ubiquitination, and trafficking were undertaken only more recently and have unveiled peculiar, unexpected, characteristics of the system [see for instance Wiley et al. (2003), Resat et al. (2003), Capuani et al. (2015), Kleiman et al. (2011)]. Such an approach is critical, as it will also help to identify the weak elements of the network that are hijacked by cancer cells and that could represent critical points of therapeutic intervention.
References
Acconcia F, Sigismund S, Polo S (2009) Ubiquitin in trafficking: the network at work. Exp Cell Res 315(9):1610–1618. https://doi.org/10.1016/j.yexcr.2008.10.014
Allanson JE (2007) Noonan syndrome. Am J Med Genet C Semin Med Genet 145C(3):274–279. https://doi.org/10.1002/ajmg.c.30138
Anastasi S, Baietti MF, Frosi Y, Alema S, Segatto O (2007) The evolutionarily conserved EBR module of RALT/MIG6 mediates suppression of the EGFR catalytic activity. Oncogene 26(57):7833–7846. https://doi.org/10.1038/sj.onc.1210590
Antonny B, Burd C, De Camilli P, Chen E, Daumke O, Faelber K, Ford M, Frolov VA, Frost A, Hinshaw JE, Kirchhausen T, Kozlov MM, Lenz M, Low HH, McMahon H, Merrifield C, Pollard TD, Robinson PJ, Roux A, Schmid S (2016) Membrane fission by dynamin: what we know and what we need to know. EMBO J 35(21):2270–2284. https://doi.org/10.15252/embj.201694613
Arteaga CL, Engelman JA (2014) ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 25(3):282–303. https://doi.org/10.1016/j.ccr.2014.02.025
Arteaga CL, Ramsey TT, Shawver LK, Guyer CA (1997) Unliganded epidermal growth factor receptor dimerization induced by direct interaction of quinazolines with the ATP binding site. J Biol Chem 272(37):23247–23254
Avraham R, Yarden Y (2011) Feedback regulation of EGFR signalling: decision making by early and delayed loops. Nat Rev Mol Cell Biol 12(2):104–117. https://doi.org/10.1038/nrm3048
Bache KG, Brech A, Mehlum A, Stenmark H (2003) Hrs regulates multivesicular body formation via ESCRT recruitment to endosomes. J Cell Biol 162(3):435–442. https://doi.org/10.1083/jcb.200302131
Bao J, Gur G, Yarden Y (2003) Src promotes destruction of c-Cbl: implications for oncogenic synergy between Src and growth factor receptors. Proc Natl Acad Sci U S A 100(5):2438–2443. https://doi.org/10.1073/pnas.0437945100
Baulida J, Kraus MH, Alimandi M, Di Fiore PP, Carpenter G (1996) All ErbB receptors other than the epidermal growth factor receptor are endocytosis impaired. J Biol Chem 271(9):5251–5257
Baumdick M, Bruggemann Y, Schmick M, Xouri G, Sabet O, Davis L, Chin JW, Bastiaens PI (2015) EGF-dependent re-routing of vesicular recycling switches spontaneous phosphorylation suppression to EGFR signaling. Elife 4. https://doi.org/10.7554/elife.12223
Beattie EC, Zhou J, Grimes ML, Bunnett NW, Howe CL, Mobley WC (1996) A signaling endosome hypothesis to explain NGF actions: potential implications for neurodegeneration. Cold Spring Harb Symp Quant Biol 61:389–406
Belleudi F, Leone L, Maggio M, Torrisi MR (2009) Hrs regulates the endocytic sorting of the fibroblast growth factor receptor 2b. Exp Cell Res 315(13):2181–2191. https://doi.org/10.1016/j.yexcr.2009.03.022
Bjorkelund H, Gedda L, Barta P, Malmqvist M, Andersson K (2011) Gefitinib induces epidermal growth factor receptor dimers which alters the interaction characteristics with (1)(2)(5)I-EGF. PLoS ONE 6(9):e24739. https://doi.org/10.1371/journal.pone.0024739
Bou-Assaly W, Mukherji S (2010) Cetuximab (erbitux). AJNR Am J Neuroradiol 31(4):626–627. https://doi.org/10.3174/ajnr.A2054
Boucrot E, Ferreira AP, Almeida-Souza L, Debard S, Vallis Y, Howard G, Bertot L, Sauvonnet N, McMahon HT (2015) Endophilin marks and controls a clathrin-independent endocytic pathway. Nature 517(7535):460–465. https://doi.org/10.1038/nature14067
Caldieri G, Barbieri E, Nappo G, Raimondi A, Bonora M, Conte A, Verhoef LGGC, Confalonieri S, Malabarba MG, Bianchi F, Cuomo A, Bonaldi T, Martini E, Mazza D, Pinton P, Tacchetti C, Polo S, Di Fiore PP, Sigismund S (2017) Reticulon 3-dependent ER-PM contact sites control EGFR nonclathrin endocytosis. Science 356(6338):617–624. https://doi.org/10.1126/science.aah6152
Caligiuri MA, Briesewitz R, Yu J, Wang L, Wei M, Arnoczky KJ, Marburger TB, Wen J, Perrotti D, Bloomfield CD, Whitman SP (2007) Novel c-CBL and CBL-b ubiquitin ligase mutations in human acute myeloid leukemia. Blood 110(3):1022–1024. https://doi.org/10.1182/blood-2006-12-061176
Calebiro D, Nikolaev VO, Gagliani MC, de Filippis T, Dees C, Tacchetti C, Persani L, Lohse MJ (2009) Persistent cAMP-signals triggered by internalized G-protein-coupled receptors. PLoS Biol 7(8):e1000172. https://doi.org/10.1371/journal.pbio.1000172
Capuani F, Conte A, Argenzio E, Marchetti L, Priami C, Polo S, Di Fiore PP, Sigismund S, Ciliberto A (2015) Quantitative analysis reveals how EGFR activation and downregulation are coupled in normal but not in cancer cells. Nat Commun 6:7999. https://doi.org/10.1038/ncomms8999
Cardone L, Carlucci A, Affaitati A, Livigni A, DeCristofaro T, Garbi C, Varrone S, Ullrich A, Gottesman ME, Avvedimento EV, Feliciello A (2004) Mitochondrial AKAP121 binds and targets protein tyrosine phosphatase D1, a novel positive regulator of src signaling. Mol Cell Biol 24(11):4613–4626. https://doi.org/10.1128/MCB.24.11.4613-4626.2004
Carey KD, Garton AJ, Romero MS, Kahler J, Thomson S, Ross S, Park F, Haley JD, Gibson N, Sliwkowski MX (2006) Kinetic analysis of epidermal growth factor receptor somatic mutant proteins shows increased sensitivity to the epidermal growth factor receptor tyrosine kinase inhibitor, erlotinib. Cancer Res 66(16):8163–8171. https://doi.org/10.1158/0008-5472.CAN-06-0453
Carlucci A, Porpora M, Garbi C, Galgani M, Santoriello M, Mascolo M, di Lorenzo D, Altieri V, Quarto M, Terracciano L, Gottesman ME, Insabato L, Feliciello A (2010) PTPD1 supports receptor stability and mitogenic signaling in bladder cancer cells. J Biol Chem 285(50):39260–39270. https://doi.org/10.1074/jbc.M110.174706
Carpenter G, Cohen S (1976) 125I-labeled human epidermal growth factor. Binding, internalization, and degradation in human fibroblasts. J Cell Biol 71(1):159–171
Carpentier JL, Gorden P, Anderson RG, Goldstein JL, Brown MS, Cohen S, Orci L (1982) Co-localization of 125I-epidermal growth factor and ferritin-low density lipoprotein in coated pits: a quantitative electron microscopic study in normal and mutant human fibroblasts. J Cell Biol 95(1):73–77
Chaudhary N, Gomez GA, Howes MT, Lo HP, McMahon KA, Rae JA, Schieber NL, Hill MM, Gaus K, Yap AS, Parton RG (2014) Endocytic crosstalk: cavins, caveolins, and caveolae regulate clathrin-independent endocytosis. PLoS Biol 12(4):e1001832. https://doi.org/10.1371/journal.pbio.1001832
Chi S, Cao H, Wang Y, McNiven MA (2011) Recycling of the epidermal growth factor receptor is mediated by a novel form of the clathrin adaptor protein Eps15. J Biol Chem 286(40):35196–35208. https://doi.org/10.1074/jbc.M111.247577
Chua BT, Lim SJ, Tham SC, Poh WJ, Ullrich A (2010) Somatic mutation in the ACK1 ubiquitin association domain enhances oncogenic signaling through EGFR regulation in renal cancer derived cells. Mol Oncol 4(4):323–334. https://doi.org/10.1016/j.molonc.2010.03.001
Chung BM, Dimri M, George M, Reddi AL, Chen G, Band V, Band H (2009) The role of cooperativity with Src in oncogenic transformation mediated by non-small cell lung cancer-associated EGF receptor mutants. Oncogene 28(16):1821–1832. https://doi.org/10.1038/onc.2009.31
Chung I, Akita R, Vandlen R, Toomre D, Schlessinger J, Mellman I (2010) Spatial control of EGF receptor activation by reversible dimerization on living cells. Nature 464(7289):783–787. https://doi.org/10.1038/nature08827
Clague MJ, Liu H, Urbe S (2012) Governance of endocytic trafficking and signaling by reversible ubiquitylation. Dev Cell 23(3):457–467. https://doi.org/10.1016/j.devcel.2012.08.011
Cohen MH, Johnson JR, Chen YF, Sridhara R, Pazdur R (2005) FDA drug approval summary: erlotinib (Tarceva) tablets. Oncologist 10(7):461–466. https://doi.org/10.1634/theoncologist.10-7-461
Cohen-Saidon C, Cohen AA, Sigal A, Liron Y, Alon U (2009) Dynamics and variability of ERK2 response to EGF in individual living cells. Mol Cell 36(5):885–893. https://doi.org/10.1016/j.molcel.2009.11.025
Collinet C, Stoter M, Bradshaw CR, Samusik N, Rink JC, Kenski D, Habermann B, Buchholz F, Henschel R, Mueller MS, Nagel WE, Fava E, Kalaidzidis Y, Zerial M (2010) Systems survey of endocytosis by multiparametric image analysis. Nature 464(7286):243–249. https://doi.org/10.1038/nature08779
Conte A, Sigismund S (2016) Chapter Six—The ubiquitin network in the control of EGFR Endocytosis and Signaling. Prog Mol Biol Transl Sci 141:225–276. https://doi.org/10.1016/bs.pmbts.2016.03.002
Cosker KE, Courchesne SL, Segal RA (2008) Action in the axon: generation and transport of signaling endosomes. Curr Opin Neurobiol 18(3):270–275. https://doi.org/10.1016/j.conb.2008.08.005
Coumailleau F, Furthauer M, Knoblich JA, Gonzalez-Gaitan M (2009) Directional Delta and Notch trafficking in Sara endosomes during asymmetric cell division. Nature 458(7241):1051–1055. https://doi.org/10.1038/nature07854
Damke H, Baba T, van der Bliek AM, Schmid SL (1995) Clathrin-independent pinocytosis is induced in cells overexpressing a temperature-sensitive mutant of dynamin. J Cell Biol 131(1):69–80
Di Guglielmo GM, Baass PC, Ou WJ, Posner BI, Bergeron JJ (1994) Compartmentalization of SHC, GRB2 and mSOS, and hyperphosphorylation of Raf-1 by EGF but not insulin in liver parenchyma. EMBO J 13(18):4269–4277
Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL (2003) Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nat Cell Biol 5(5):410–421. https://doi.org/10.1038/ncb975
Dubois EA, Cohen AF (2009) Panitumumab. Br J Clin Pharmacol 68(4):482–483. https://doi.org/10.1111/j.1365-2125.2009.03492.x
Dunbar AJ, Gondek LP, O’Keefe CL, Makishima H, Rataul MS, Szpurka H, Sekeres MA, Wang XF, McDevitt MA, Maciejewski JP (2008) 250K single nucleotide polymorphism array karyotyping identifies acquired uniparental disomy and homozygous mutations, including novel missense substitutions of c-Cbl, in myeloid malignancies. Cancer Res 68(24):10349–10357. https://doi.org/10.1158/0008-5472.CAN-08-2754
Dunn WA, Connolly TP, Hubbard AL (1986) Receptor-mediated endocytosis of epidermal growth factor by rat hepatocytes: receptor pathway. J Cell Biol 102(1):24–36
Earp HS, Austin KS, Blaisdell J, Rubin RA, Nelson KG, Lee LW, Grisham JW (1986) Epidermal growth factor (EGF) stimulates EGF receptor synthesis. J Biol Chem 261(11):4777–4780
Ebner R, Derynck R (1991) Epidermal growth factor and transforming growth factor-alpha: differential intracellular routing and processing of ligand-receptor complexes. Cell Regul 2(8):599–612
Eden ER, White IJ, Tsapara A, Futter CE (2010) Membrane contacts between endosomes and ER provide sites for PTP1B-epidermal growth factor receptor interaction. Nat Cell Biol 12(3):267–272. https://doi.org/10.1038/ncb2026
Eden ER, Sanchez-Heras E, Tsapara A, Sobota A, Levine TP, Futter CE (2016) Annexin A1 tethers membrane contact sites that mediate ER to endosome cholesterol transport. Dev Cell 37(5):473–483. https://doi.org/10.1016/j.devcel.2016.05.005
Ekstrand AJ, Sugawa N, James CD, Collins VP (1992) Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc Natl Acad Sci U S A 89(10):4309–4313
Elowitz MB, Levine AJ, Siggia ED, Swain PS (2002) Stochastic gene expression in a single cell. Science 297(5584):1183–1186. https://doi.org/10.1126/science.1070919
Fan Z, Masui H, Altas I, Mendelsohn J (1993) Blockade of epidermal growth factor receptor function by bivalent and monovalent fragments of 225 anti-epidermal growth factor receptor monoclonal antibodies. Cancer Res 53(18):4322–4328
Feng Q, Baird D, Peng X, Wang J, Ly T, Guan JL, Cerione RA (2006) Cool-1 functions as an essential regulatory node for EGF receptor- and Src-mediated cell growth. Nat Cell Biol 8(9):945–956. https://doi.org/10.1038/ncb1453
Ferby I, Reschke M, Kudlacek O, Knyazev P, Pante G, Amann K, Sommergruber W, Kraut N, Ullrich A, Fassler R, Klein R (2006) Mig6 is a negative regulator of EGF receptor-mediated skin morphogenesis and tumor formation. Nat Med 12(5):568–573. https://doi.org/10.1038/nm1401
Ferrandon S, Feinstein TN, Castro M, Wang B, Bouley R, Potts JT, Gardella TJ, Vilardaga JP (2009) Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat Chem Biol 5(10):734–742. https://doi.org/10.1038/nchembio.206
Fortian A, Sorkin A (2014) Live-cell fluorescence imaging reveals high stoichiometry of Grb2 binding to the EGF receptor sustained during endocytosis. J Cell Sci 127(Pt 2):432–444. https://doi.org/10.1242/jcs.137786
Frechin M, Stoeger T, Daetwyler S, Gehin C, Battich N, Damm EM, Stergiou L, Riezman H, Pelkmans L (2015) Cell-intrinsic adaptation of lipid composition to local crowding drives social behaviour. Nature 523(7558):88–91. https://doi.org/10.1038/nature14429
French AR, Sudlow GP, Wiley HS, Lauffenburger DA (1994) Postendocytic trafficking of epidermal growth factor-receptor complexes is mediated through saturable and specific endosomal interactions. J Biol Chem 269(22):15749–15755
French AR, Tadaki DK, Niyogi SK, Lauffenburger DA (1995) Intracellular trafficking of epidermal growth factor family ligands is directly influenced by the pH sensitivity of the receptor/ligand interaction. J Biol Chem 270(9):4334–4340
Friedman LM, Rinon A, Schechter B, Lyass L, Lavi S, Bacus SS, Sela M, Yarden Y (2005) Synergistic down-regulation of receptor tyrosine kinases by combinations of mAbs: implications for cancer immunotherapy. Proc Natl Acad Sci U S A 102(6):1915–1920. https://doi.org/10.1073/pnas.0409610102
Friedman JR, Dibenedetto JR, West M, Rowland AA, Voeltz GK (2013) Endoplasmic reticulum-endosome contact increases as endosomes traffic and mature. Mol Biol Cell 24(7):1030–1040. https://doi.org/10.1091/mbc.E12-10-0733
Frosi Y, Anastasi S, Ballaro C, Varsano G, Castellani L, Maspero E, Polo S, Alema S, Segatto O (2010) A two-tiered mechanism of EGFR inhibition by RALT/MIG6 via kinase suppression and receptor degradation. J Cell Biol 189(3):557–571. https://doi.org/10.1083/jcb.201002032
Fry WH, Simion C, Sweeney C, Carraway KL 3rd (2011) Quantity control of the ErbB3 receptor tyrosine kinase at the endoplasmic reticulum. Mol Cell Biol 31(14):3009–3018. https://doi.org/10.1128/MCB.05105-11
Gan HK, Walker F, Burgess AW, Rigopoulos A, Scott AM, Johns TG (2007) The epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor AG1478 increases the formation of inactive untethered EGFR dimers. Implications for combination therapy with monoclonal antibody 806. J Biol Chem 282(5):2840–2850. https://doi.org/10.1074/jbc.M605136200
Garrett TP, McKern NM, Lou M, Elleman TC, Adams TE, Lovrecz GO, Zhu HJ, Walker F, Frenkel MJ, Hoyne PA, Jorissen RN, Nice EC, Burgess AW, Ward CW (2002) Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor alpha. Cell 110(6):763–773
Goh LK, Sorkin A (2013) Endocytosis of receptor tyrosine kinases. Cold Spring Harb Perspect Biol 5(5):a017459. https://doi.org/10.1101/cshperspect.a017459
Goh LK, Huang F, Kim W, Gygi S, Sorkin A (2010) Multiple mechanisms collectively regulate clathrin-mediated endocytosis of the epidermal growth factor receptor. J Cell Biol 189(5):871–883. https://doi.org/10.1083/jcb.201001008
Gorden P, Carpentier JL, Cohen S, Orci L (1978) Epidermal growth factor: morphological demonstration of binding, internalization, and lysosomal association in human fibroblasts. Proc Natl Acad Sci U S A 75(10):5025–5029
Grand FH, Hidalgo-Curtis CE, Ernst T, Zoi K, Zoi C, McGuire C, Kreil S, Jones A, Score J, Metzgeroth G, Oscier D, Hall A, Brandts C, Serve H, Reiter A, Chase AJ, Cross NC (2009) Frequent CBL mutations associated with 11q acquired uniparental disomy in myeloproliferative neoplasms. Blood 113(24):6182–6192. https://doi.org/10.1182/blood-2008-12-194548
Grandal MV, Zandi R, Pedersen MW, Willumsen BM, van Deurs B, Poulsen HS (2007) EGFRvIII escapes down-regulation due to impaired internalization and sorting to lysosomes. Carcinogenesis 28(7):1408–1417. https://doi.org/10.1093/carcin/bgm058
Grimes ML, Zhou J, Beattie EC, Yuen EC, Hall DE, Valletta JS, Topp KS, LaVail JH, Bunnett NW, Mobley WC (1996) Endocytosis of activated TrkA: evidence that nerve growth factor induces formation of signaling endosomes. J Neurosci 16(24):7950–7964
Grovdal LM, Stang E, Sorkin A, Madshus IH (2004) Direct interaction of Cbl with pTyr 1045 of the EGF receptor (EGFR) is required to sort the EGFR to lysosomes for degradation. Exp Cell Res 300(2):388–395. https://doi.org/10.1016/j.yexcr.2004.07.003
Gschweitl M, Ulbricht A, Barnes CA, Enchev RI, Stoffel-Studer I, Meyer-Schaller N, Huotari J, Yamauchi Y, Greber UF, Helenius A, Peter M (2016) A SPOPL/Cullin-3 ubiquitin ligase complex regulates endocytic trafficking by targeting EPS15 at endosomes. Elife 5:e13841. https://doi.org/10.7554/eLife.13841
Guha A, Sriram V, Krishnan KS, Mayor S (2003) Shibire mutations reveal distinct dynamin-independent and -dependent endocytic pathways in primary cultures of Drosophila hemocytes. J Cell Sci 116(Pt 16):3373–3386. https://doi.org/10.1242/jcs.00637
Gur G, Rubin C, Katz M, Amit I, Citri A, Nilsson J, Amariglio N, Henriksson R, Rechavi G, Hedman H, Wides R, Yarden Y (2004) LRIG1 restricts growth factor signaling by enhancing receptor ubiquitylation and degradation. EMBO J 23(16):3270–3281. https://doi.org/10.1038/sj.emboj.7600342
Haj FG, Verveer PJ, Squire A, Neel BG, Bastiaens PI (2002) Imaging sites of receptor dephosphorylation by PTP1B on the surface of the endoplasmic reticulum. Science 295(5560):1708–1711. https://doi.org/10.1126/science.1067566
Haj FG, Sabet O, Kinkhabwala A, Wimmer-Kleikamp S, Roukos V, Han HM, Grabenbauer M, Bierbaum M, Antony C, Neel BG, Bastiaens PI (2012) Regulation of signaling at regions of cell-cell contact by endoplasmic reticulum-bound protein-tyrosine phosphatase 1B. PLoS ONE 7(5):e36633. https://doi.org/10.1371/journal.pone.0036633
Han W, Zhang T, Yu H, Foulke JG, Tang CK (2006) Hypophosphorylation of residue Y1045 leads to defective downregulation of EGFRvIII. Cancer Biol Ther 5(10):1361–1368
Hanover JA, Willingham MC, Pastan I (1984) Kinetics of transit of transferrin and epidermal growth factor through clathrin-coated membranes. Cell 39(2 Pt 1):283–293
Henne WM, Stenmark H, Emr SD (2013) Molecular mechanisms of the membrane sculpting ESCRT pathway. Cold Spring Harb Perspect Biol 5(9). https://doi.org/10.1101/cshperspect.a016766
Hinrichsen L, Harborth J, Andrees L, Weber K, Ungewickell EJ (2003) Effect of clathrin heavy chain- and alpha-adaptin-specific small inhibitory RNAs on endocytic accessory proteins and receptor trafficking in HeLa cells. J Biol Chem 278(46):45160–45170. https://doi.org/10.1074/jbc.M307290200
Hirsch FR, Janne PA, Eberhardt WE, Cappuzzo F, Thatcher N, Pirker R, Choy H, Kim ES, Paz-Ares L, Gandara DR, Wu YL, Ahn MJ, Mitsudomi T, Shepherd FA, Mok TS (2013) Epidermal growth factor receptor inhibition in lung cancer: status 2012. J Thorac Oncol 8(3):373–384. https://doi.org/10.1097/JTO.0b013e31827ed0ff
Howe CL, Mobley WC (2005) Long-distance retrograde neurotrophic signaling. Curr Opin Neurobiol 15(1):40–48. https://doi.org/10.1016/j.conb.2005.01.010
Huang F, Kirkpatrick D, Jiang X, Gygi S, Sorkin A (2006) Differential regulation of EGF receptor internalization and degradation by multiubiquitination within the kinase domain. Mol Cell 21(6):737–748. https://doi.org/10.1016/j.molcel.2006.02.018
Huang F, Zeng X, Kim W, Balasubramani M, Fortian A, Gygi SP, Yates NA, Sorkin A (2013) Lysine 63-linked polyubiquitination is required for EGF receptor degradation. Proc Natl Acad Sci U S A 110(39):15722–15727. https://doi.org/10.1073/pnas.1308014110
Huotari J, Meyer-Schaller N, Hubner M, Stauffer S, Katheder N, Horvath P, Mancini R, Helenius A, Peter M (2012) Cullin-3 regulates late endosome maturation. Proc Natl Acad Sci U S A 109(3):823–828. https://doi.org/10.1073/pnas.1118744109
Ibach J, Radon Y, Gelleri M, Sonntag MH, Brunsveld L, Bastiaens PI, Verveer PJ (2015) Single particle tracking reveals that EGFR signaling activity is amplified in clathrin-coated pits. PLoS ONE 10(11):e0143162. https://doi.org/10.1371/journal.pone.0143162
Irannejad R, Tomshine JC, Tomshine JR, Chevalier M, Mahoney JP, Steyaert J, Rasmussen SG, Sunahara RK, El-Samad H, Huang B, von Zastrow M (2013) Conformational biosensors reveal GPCR signalling from endosomes. Nature 495(7442):534–538. https://doi.org/10.1038/nature12000
Irannejad R, Tsvetanova NG, Lobingier BT, von Zastrow M (2015) Effects of endocytosis on receptor-mediated signaling. Curr Opin Cell Biol 35:137–143. https://doi.org/10.1016/j.ceb.2015.05.005
Jaramillo ML, Leon Z, Grothe S, Paul-Roc B, Abulrob A, O’Connor McCourt M (2006) Effect of the anti-receptor ligand-blocking 225 monoclonal antibody on EGF receptor endocytosis and sorting. Exp Cell Res 312(15):2778–2790. https://doi.org/10.1016/j.yexcr.2006.05.008
Jekely G, Rorth P (2003) Hrs mediates downregulation of multiple signalling receptors in Drosophila. EMBO Rep 4(12):1163–1168. https://doi.org/10.1038/sj.embor.7400019
Jiang X, Huang F, Marusyk A, Sorkin A (2003) Grb2 regulates internalization of EGF receptors through clathrin-coated pits. Mol Biol Cell 14(3):858–870. https://doi.org/10.1091/mbc.E02-08-0532
Johannes L, Parton RG, Bassereau P, Mayor S (2015) Building endocytic pits without clathrin. Nat Rev Mol Cell Biol 16(5):311–321. https://doi.org/10.1038/nrm3968
Johnson H, Del Rosario AM, Bryson BD, Schroeder MA, Sarkaria JN, White FM (2012) Molecular characterization of EGFR and EGFRvIII signaling networks in human glioblastoma tumor xenografts. Mol Cell Proteomics 11(12):1724–1740. https://doi.org/10.1074/mcp.M112.019984
Kalaidzidis I, Miaczynska M, Brewinska-Olchowik M, Hupalowska A, Ferguson C, Parton RG, Kalaidzidis Y, Zerial M (2015) APPL endosomes are not obligatory endocytic intermediates but act as stable cargo-sorting compartments. J Cell Biol 211(1):123–144. https://doi.org/10.1083/jcb.201311117
Kalia M, Kumari S, Chadda R, Hill MM, Parton RG, Mayor S (2006) Arf6-independent GPI-anchored protein-enriched early endosomal compartments fuse with sorting endosomes via a Rab5/phosphatidylinositol-3’-kinase-dependent machinery. Mol Biol Cell 17(8):3689–3704. https://doi.org/10.1091/mbc.E05-10-0980
Kario E, Marmor MD, Adamsky K, Citri A, Amit I, Amariglio N, Rechavi G, Yarden Y (2005) Suppressors of cytokine signaling 4 and 5 regulate epidermal growth factor receptor signaling. J Biol Chem 280(8):7038–7048. https://doi.org/10.1074/jbc.M408575200
Kelley LC, Weed SA (2012) Cortactin is a substrate of activated Cdc42-associated kinase 1 (ACK1) during ligand-induced epidermal growth factor receptor downregulation. PLoS ONE 7(8):e44363. https://doi.org/10.1371/journal.pone.0044363
Kholodenko BN, Demin OV, Moehren G, Hoek JB (1999) Quantification of short term signaling by the epidermal growth factor receptor. J Biol Chem 274(42):30169–30181
Kilpatrick BS, Eden ER, Hockey LN, Yates E, Futter CE, Patel S (2017) An endosomal NAADP-sensitive two-pore Ca2+ channel regulates ER-endosome membrane contact sites to control growth factor signaling. Cell Rep 18(7):1636–1645. https://doi.org/10.1016/j.celrep.2017.01.052
Kirchhausen T, Owen D, Harrison SC (2014) Molecular structure, function, and dynamics of clathrin-mediated membrane traffic. Cold Spring Harb Perspect Biol 6(5):a016725. https://doi.org/10.1101/cshperspect.a016725
Kleiman LB, Maiwald T, Conzelmann H, Lauffenburger DA, Sorger PK (2011) Rapid phospho-turnover by receptor tyrosine kinases impacts downstream signaling and drug binding. Mol Cell 43(5):723–737. https://doi.org/10.1016/j.molcel.2011.07.014
Kon S, Kobayashi N, Satake M (2014) Altered trafficking of mutated growth factor receptors and their associated molecules: implication for human cancers. Cell Logist 4:e28461. https://doi.org/10.4161/cl.28461
Kovacs E, Zorn JA, Huang Y, Barros T, Kuriyan J (2015) A structural perspective on the regulation of the epidermal growth factor receptor. Annu Rev Biochem 84:739–764. https://doi.org/10.1146/annurev-biochem-060614-034402
Lakadamyali M, Rust MJ, Zhuang X (2006) Ligands for clathrin-mediated endocytosis are differentially sorted into distinct populations of early endosomes. Cell 124(5):997–1009. https://doi.org/10.1016/j.cell.2005.12.038
Lampugnani MG, Orsenigo F, Gagliani MC, Tacchetti C, Dejana E (2006) Vascular endothelial cadherin controls VEGFR-2 internalization and signaling from intracellular compartments. J Cell Biol 174(4):593–604. https://doi.org/10.1083/jcb.200602080
Lemmon MA (2009) Ligand-induced ErbB receptor dimerization. Exp Cell Res 315(4):638–648. https://doi.org/10.1016/j.yexcr.2008.10.024
Lemmon MA, Schlessinger J (2010) Cell signaling by receptor tyrosine kinases. Cell 141(7):1117–1134. https://doi.org/10.1016/j.cell.2010.06.011
Lemmon MA, Schlessinger J, Ferguson KM (2014) The EGFR family: not so prototypical receptor tyrosine kinases. Cold Spring Harb Perspect Biol 6(4):a020768. https://doi.org/10.1101/cshperspect.a020768
Lemmon MA, Freed DM, Schlessinger J, Kiyatkin A (2016) The dark side of cell signaling: positive roles for negative regulators. Cell 164(6):1172–1184. https://doi.org/10.1016/j.cell.2016.02.047
Lenferink AE, Pinkas-Kramarski R, van de Poll ML, van Vugt MJ, Klapper LN, Tzahar E, Waterman H, Sela M, van Zoelen EJ, Yarden Y (1998) Differential endocytic routing of homo- and hetero-dimeric ErbB tyrosine kinases confers signaling superiority to receptor heterodimers. EMBO J 17(12):3385–3397. https://doi.org/10.1093/emboj/17.12.3385
Leung EL, Tam IY, Tin VP, Chua DT, Sihoe AD, Cheng LC, Ho JC, Chung LP, Wong MP (2009) SRC promotes survival and invasion of lung cancers with epidermal growth factor receptor abnormalities and is a potential candidate for molecular-targeted therapy. Mol Cancer Res 7(6):923–932. https://doi.org/10.1158/1541-7786.MCR-09-0003
Levine TP, Patel S (2016) Signalling at membrane contact sites: two membranes come together to handle second messengers. Curr Opin Cell Biol 39:77–83. https://doi.org/10.1016/j.ceb.2016.02.011
Levkowitz G, Klapper LN, Tzahar E, Freywald A, Sela M, Yarden Y (1996) Coupling of the c-Cbl protooncogene product to ErbB-1/EGF-receptor but not to other ErbB proteins. Oncogene 12(5):1117–1125
Levkowitz G, Waterman H, Zamir E, Kam Z, Oved S, Langdon WY, Beguinot L, Geiger B, Yarden Y (1998) c-Cbl/Sli-1 regulates endocytic sorting and ubiquitination of the epidermal growth factor receptor. Genes Dev 12(23):3663–3674
Levkowitz G, Waterman H, Ettenberg SA, Katz M, Tsygankov AY, Alroy I, Lavi S, Iwai K, Reiss Y, Ciechanover A, Lipkowitz S, Yarden Y (1999) Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c-Cbl/Sli-1. Mol Cell 4(6):1029–1040
Liberali P, Snijder B, Pelkmans L (2014) A hierarchical map of regulatory genetic interactions in membrane trafficking. Cell 157(6):1473–1487. https://doi.org/10.1016/j.cell.2014.04.029
Licitra L, Storkel S, Kerr KM, Van Cutsem E, Pirker R, Hirsch FR, Vermorken JB, von Heydebreck A, Esser R, Celik I, Ciardiello F (2013) Predictive value of epidermal growth factor receptor expression for first-line chemotherapy plus cetuximab in patients with head and neck and colorectal cancer: analysis of data from the EXTREME and CRYSTAL studies. Eur J Cancer 49(6):1161–1168. https://doi.org/10.1016/j.ejca.2012.11.018
Liu Z, Zanata SM, Kim J, Peterson MA, Di Vizio D, Chirieac LR, Pyne S, Agostini M, Freeman MR, Loda M (2013) The ubiquitin-specific protease USP2a prevents endocytosis-mediated EGFR degradation. Oncogene 32(13):1660–1669. https://doi.org/10.1038/onc.2012.188
Longva KE, Blystad FD, Stang E, Larsen AM, Johannessen LE, Madshus IH (2002) Ubiquitination and proteasomal activity is required for transport of the EGF receptor to inner membranes of multivesicular bodies. J Cell Biol 156(5):843–854. https://doi.org/10.1083/jcb.200106056
Lund KA, Opresko LK, Starbuck C, Walsh BJ, Wiley HS (1990) Quantitative analysis of the endocytic system involved in hormone-induced receptor internalization. J Biol Chem 265(26):15713–15723
Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA (2004) Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 350(21):2129–2139. https://doi.org/10.1056/NEJMoa040938
Ma YM, Boucrot E, Villen J, el Affar B, Gygi SP, Gottlinger HG, Kirchhausen T (2007) Targeting of AMSH to endosomes is required for epidermal growth factor receptor degradation. J Biol Chem 282(13):9805–9812. https://doi.org/10.1074/jbc.M611635200
Macdonald-Obermann JL, Pike LJ (2014) Different epidermal growth factor (EGF) receptor ligands show distinct kinetics and biased or partial agonism for homodimer and heterodimer formation. J Biol Chem 289(38):26178–26188. https://doi.org/10.1074/jbc.M114.586826
Martinelli S, De Luca A, Stellacci E, Rossi C, Checquolo S, Lepri F, Caputo V, Silvano M, Buscherini F, Consoli F, Ferrara G, Digilio MC, Cavaliere ML, van Hagen JM, Zampino G, van der Burgt I, Ferrero GB, Mazzanti L, Screpanti I, Yntema HG, Nillesen WM, Savarirayan R, Zenker M, Dallapiccola B, Gelb BD, Tartaglia M (2010) Heterozygous germline mutations in the CBL tumor-suppressor gene cause a Noonan syndrome-like phenotype. Am J Hum Genet 87(2):250–257. https://doi.org/10.1016/j.ajhg.2010.06.015
McCullough J, Clague MJ, Urbe S (2004) AMSH is an endosome-associated ubiquitin isopeptidase. J Cell Biol 166(4):487–492. https://doi.org/10.1083/jcb.200401141
McMahon HT, Boucrot E (2011) Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol 12(8):517–533. https://doi.org/10.1038/nrm3151
Menard L, Parker PJ, Kermorgant S (2014) Receptor tyrosine kinase c-Met controls the cytoskeleton from different endosomes via different pathways. Nat Commun 5:3907. https://doi.org/10.1038/ncomms4907
Miaczynska M, Christoforidis S, Giner A, Shevchenko A, Uttenweiler-Joseph S, Habermann B, Wilm M, Parton RG, Zerial M (2004) APPL proteins link Rab5 to nuclear signal transduction via an endosomal compartment. Cell 116(3):445–456
Moscatello DK, Holgado-Madruga M, Godwin AK, Ramirez G, Gunn G, Zoltick PW, Biegel JA, Hayes RL, Wong AJ (1995) Frequent expression of a mutant epidermal growth factor receptor in multiple human tumors. Cancer Res 55(23):5536–5539
Motley A, Bright NA, Seaman MN, Robinson MS (2003) Clathrin-mediated endocytosis in AP-2-depleted cells. J Cell Biol 162(5):909–918. https://doi.org/10.1083/jcb.200305145
Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, Lukashchuk N, Gillespie DA, Ludwig RL, Gosselin P, Cromer A, Brugge JS, Sansom OJ, Norman JC, Vousden KH (2009) Mutant p53 drives invasion by promoting integrin recycling. Cell 139(7):1327–1341. https://doi.org/10.1016/j.cell.2009.11.026
Muller PA, Trinidad AG, Timpson P, Morton JP, Zanivan S, van den Berghe PV, Nixon C, Karim SA, Caswell PT, Noll JE, Coffill CR, Lane DP, Sansom OJ, Neilsen PM, Norman JC, Vousden KH (2013) Mutant p53 enhances MET trafficking and signalling to drive cell scattering and invasion. Oncogene 32(10):1252–1265. https://doi.org/10.1038/onc.2012.148
Nakamura N, Lill JR, Phung Q, Jiang Z, Bakalarski C, de Mazière A, Klumperman J, Schlatter M, Delamarre L, Mellman I (2014) Endosomes are specialized platforms for bacterial sensing and NOD2 signalling. Nature 509(7499):240–244. https://doi.org/10.1038/nature13133
Nevins AK, Thurmond DC (2006) Caveolin-1 functions as a novel Cdc42 guanine nucleotide dissociation inhibitor in pancreatic beta-cells. J Biol Chem 281(28):18961–18972. https://doi.org/10.1074/jbc.M603604200
Nicholson SE, Metcalf D, Sprigg NS, Columbus R, Walker F, Silva A, Cary D, Willson TA, Zhang JG, Hilton DJ, Alexander WS, Nicola NA (2005) Suppressor of cytokine signaling (SOCS)-5 is a potential negative regulator of epidermal growth factor signaling. Proc Natl Acad Sci U S A 102(7):2328–2333. https://doi.org/10.1073/pnas.0409675102
Ogiso H, Ishitani R, Nureki O, Fukai S, Yamanaka M, Kim JH, Saito K, Sakamoto A, Inoue M, Shirouzu M, Yokoyama S (2002) Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell 110(6):775–787
Orth JD, Krueger EW, Weller SG, McNiven MA (2006) A novel endocytic mechanism of epidermal growth factor receptor sequestration and internalization. Cancer Res 66(7):3603–3610. https://doi.org/10.1158/0008-5472.CAN-05-2916
Oved S, Mosesson Y, Zwang Y, Santonico E, Shtiegman K, Marmor MD, Kochupurakkal BS, Katz M, Lavi S, Cesareni G, Yarden Y (2006) Conjugation to Nedd8 instigates ubiquitylation and down-regulation of activated receptor tyrosine kinases. J Biol Chem 281(31):21640–21651. https://doi.org/10.1074/jbc.M513034200
Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M (2004) EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304(5676):1497–1500. https://doi.org/10.1126/science.1099314
Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, Mardis E, Kupfer D, Wilson R, Kris M, Varmus H (2004) EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A 101(36):13306–13311. https://doi.org/10.1073/pnas.0405220101
Pareja F, Ferraro DA, Rubin C, Cohen-Dvashi H, Zhang F, Aulmann S, Ben-Chetrit N, Pines G, Navon R, Crosetto N, Kostler W, Carvalho S, Lavi S, Schmitt F, Dikic I, Yakhini Z, Sinn P, Mills GB, Yarden Y (2012) Deubiquitination of EGFR by Cezanne-1 contributes to cancer progression. Oncogene 31(43):4599–4608. https://doi.org/10.1038/onc.2011.587
Pedersen MW, Jacobsen HJ, Koefoed K, Hey A, Pyke C, Haurum JS, Kragh M (2010) Sym004: a novel synergistic anti-epidermal growth factor receptor antibody mixture with superior anticancer efficacy. Cancer Res 70(2):588–597. https://doi.org/10.1158/0008-5472.CAN-09-1417
Peeters M, Karthaus M, Rivera F, Terwey JH, Douillard JY (2015) Panitumumab in metastatic colorectal cancer: the importance of tumour RAS status. Drugs 75(7):731–748. https://doi.org/10.1007/s40265-015-0386-x
Phillips MJ, Voeltz GK (2016) Structure and function of ER membrane contact sites with other organelles. Nat Rev Mol Cell Biol 17(2):69–82. https://doi.org/10.1038/nrm.2015.8
Pierotti MA, Negri T, Tamborini E, Perrone F, Pricl S, Pilotti S (2010) Targeted therapies: the rare cancer paradigm. Mol Oncol 4(1):19–37. https://doi.org/10.1016/j.molonc.2009.10.003
Piper RC, Dikic I, Lukacs GL (2014) Ubiquitin-dependent sorting in endocytosis. Cold Spring Harb Perspect Biol 6(1). https://doi.org/10.1101/cshperspect.a016808
Poteryaev D, Datta S, Ackema K, Zerial M, Spang A (2010) Identification of the switch in early-to-late endosome transition. Cell 141(3):497–508. https://doi.org/10.1016/j.cell.2010.03.011
Raiborg C, Stenmark H (2009) The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 458(7237):445–452. https://doi.org/10.1038/nature07961
Renard HF, Simunovic M, Lemiere J, Boucrot E, Garcia-Castillo MD, Arumugam S, Chambon V, Lamaze C, Wunder C, Kenworthy AK, Schmidt AA, McMahon HT, Sykes C, Bassereau P, Johannes L (2015) Endophilin-A2 functions in membrane scission in clathrin-independent endocytosis. Nature 517(7535):493–496. https://doi.org/10.1038/nature14064
Resat H, Ewald JA, Dixon DA, Wiley HS (2003) An integrated model of epidermal growth factor receptor trafficking and signal transduction. Biophys J 85(2):730–743
Rink J, Ghigo E, Kalaidzidis Y, Zerial M (2005) Rab conversion as a mechanism of progression from early to late endosomes. Cell 122(5):735–749. https://doi.org/10.1016/j.cell.2005.06.043
Rocha N, Kuijl C, van der Kant R, Janssen L, Houben D, Janssen H, Zwart W, Neefjes J (2009) Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7-RILP-p150 Glued and late endosome positioning. J Cell Biol 185(7):1209–1225. https://doi.org/10.1083/jcb.200811005
Roda-Navarro P, Bastiaens PI (2014) Dynamic recruitment of protein tyrosine phosphatase PTPD1 to EGF stimulation sites potentiates EGFR activation. PLoS ONE 9(7):e103203. https://doi.org/10.1371/journal.pone.0103203
Roepstorff K, Grandal MV, Henriksen L, Knudsen SL, Lerdrup M, Grovdal L, Willumsen BM, van Deurs B (2009) Differential effects of EGFR ligands on endocytic sorting of the receptor. Traffic 10(8):1115–1127. https://doi.org/10.1111/j.1600-0854.2009.00943.x
Romsicki Y, Reece M, Gauthier JY, Asante-Appiah E, Kennedy BP (2004) Protein tyrosine phosphatase-1B dephosphorylation of the insulin receptor occurs in a perinuclear endosome compartment in human embryonic kidney 293 cells. J Biol Chem 279(13):12868–12875. https://doi.org/10.1074/jbc.M309600200
Roskoski R Jr (2014) The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res 79:34–74. https://doi.org/10.1016/j.phrs.2013.11.002
Rothenberg SM, Engelman JA, Le S, Riese DJ 2nd, Haber DA, Settleman J (2008) Modeling oncogene addiction using RNA interference. Proc Natl Acad Sci U S A 105(34):12480–12484. https://doi.org/10.1073/pnas.0803217105
Row PE, Prior IA, McCullough J, Clague MJ, Urbe S (2006) The ubiquitin isopeptidase UBPY regulates endosomal ubiquitin dynamics and is essential for receptor down-regulation. J Biol Chem 281(18):12618–12624. https://doi.org/10.1074/jbc.M512615200
Rowland AA, Chitwood PJ, Phillips MJ, Voeltz GK (2014) ER contact sites define the position and timing of endosome fission. Cell 159(5):1027–1041. https://doi.org/10.1016/j.cell.2014.10.023
Roxrud I, Raiborg C, Pedersen NM, Stang E, Stenmark H (2008) An endosomally localized isoform of Eps15 interacts with Hrs to mediate degradation of epidermal growth factor receptor. J Cell Biol 180(6):1205–1218. https://doi.org/10.1083/jcb.200708115
Sanada M, Suzuki T, Shih LY, Otsu M, Kato M, Yamazaki S, Tamura A, Honda H, Sakata-Yanagimoto M, Kumano K, Oda H, Yamagata T, Takita J, Gotoh N, Nakazaki K, Kawamata N, Onodera M, Nobuyoshi M, Hayashi Y, Harada H, Kurokawa M, Chiba S, Mori H, Ozawa K, Omine M, Hirai H, Nakauchi H, Koeffler HP, Ogawa S (2009) Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms. Nature 460(7257):904–908. https://doi.org/10.1038/nature08240
Sangwan V, Paliouras GN, Abella JV, Dube N, Monast A, Tremblay ML, Park M (2008) Regulation of the Met receptor-tyrosine kinase by the protein-tyrosine phosphatase 1B and T-cell phosphatase. J Biol Chem 283(49):34374–34383. https://doi.org/10.1074/jbc.M805916200
Sargin B, Choudhary C, Crosetto N, Schmidt MH, Grundler R, Rensinghoff M, Thiessen C, Tickenbrock L, Schwable J, Brandts C, August B, Koschmieder S, Bandi SR, Duyster J, Berdel WE, Muller-Tidow C, Dikic I, Serve H (2007) Flt3-dependent transformation by inactivating c-Cbl mutations in AML. Blood 110(3):1004–1012. https://doi.org/10.1182/blood-2007-01-066076
Savio MG, Wollscheid N, Cavallaro E, Algisi V, Di Fiore PP, Sigismund S, Maspero E, Polo S (2016) USP9X controls EGFR fate by deubiquitinating the endocytic adaptor Eps15. Curr Biol 26(2):173–183. https://doi.org/10.1016/j.cub.2015.11.050
Sawano A, Takayama S, Matsuda M, Miyawaki A (2002) Lateral propagation of EGF signaling after local stimulation is dependent on receptor density. Dev Cell 3(2):245–257
Scharaw S, Iskar M, Ori A, Boncompain G, Laketa V, Poser I, Lundberg E, Perez F, Beck M, Bork P, Pepperkok R (2016) The endosomal transcriptional regulator RNF11 integrates degradation and transport of EGFR. J Cell Biol 215(4):543–558. https://doi.org/10.1083/jcb.201601090
Schenck A, Goto-Silva L, Collinet C, Rhinn M, Giner A, Habermann B, Brand M, Zerial M (2008) The endosomal protein Appl1 mediates Akt substrate specificity and cell survival in vertebrate development. Cell 133(3):486–497. https://doi.org/10.1016/j.cell.2008.02.044
Schlessinger J (2014) Receptor tyrosine kinases: legacy of the first two decades. Cold Spring Harb Perspect Biol 6(3). https://doi.org/10.1101/cshperspect.a008912
Schmidt MH, Dikic I (2005) The Cbl interactome and its functions. Nat Rev Mol Cell Biol 6(12):907–918. https://doi.org/10.1038/nrm1762
Schmidt MH, Furnari FB, Cavenee WK, Bogler O (2003) Epidermal growth factor receptor signaling intensity determines intracellular protein interactions, ubiquitination, and internalization. Proc Natl Acad Sci U S A 100(11):6505–6510. https://doi.org/10.1073/pnas.1031790100
Scott CC, Vacca F, Gruenberg J (2014) Endosome maturation, transport and functions. Semin Cell Dev Biol 31:2–10. https://doi.org/10.1016/j.semcdb.2014.03.034
Segatto O, Anastasi S, Alema S (2011) Regulation of epidermal growth factor receptor signalling by inducible feedback inhibitors. J Cell Sci 124(Pt 11):1785–1793. https://doi.org/10.1242/jcs.083303
Settembre C, Fraldi A, Medina DL, Ballabio A (2013) Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol 14(5):283–296. https://doi.org/10.1038/nrm3565
Shen F, Lin Q, Gu Y, Childress C, Yang W (2007) Activated Cdc42-associated kinase 1 is a component of EGF receptor signaling complex and regulates EGF receptor degradation. Mol Biol Cell 18(3):732–742. https://doi.org/10.1091/mbc.E06-02-0142
Shenoy SK, Lefkowitz RJ (2005) Seven-transmembrane receptor signaling through beta-arrestin. Sci STKE 2005(308):cm10. https://doi.org/10.1126/stke.2005/308/cm10
Shimizu H, Woodcock SA, Wilkin MB, Trubenova B, Monk NA, Baron M (2014) Compensatory flux changes within an endocytic trafficking network maintain thermal robustness of Notch signaling. Cell 157(5):1160–1174. https://doi.org/10.1016/j.cell.2014.03.050
Shtiegman K, Kochupurakkal BS, Zwang Y, Pines G, Starr A, Vexler A, Citri A, Katz M, Lavi S, Ben-Basat Y, Benjamin S, Corso S, Gan J, Yosef RB, Giordano S, Yarden Y (2007) Defective ubiquitinylation of EGFR mutants of lung cancer confers prolonged signaling. Oncogene 26(49):6968–6978. https://doi.org/10.1038/sj.onc.1210503
Sigismund S, Woelk T, Puri C, Maspero E, Tacchetti C, Transidico P, Di Fiore PP, Polo S (2005) Clathrin-independent endocytosis of ubiquitinated cargos. Proc Natl Acad Sci U S A 102(8):2760–2765. https://doi.org/10.1073/pnas.0409817102
Sigismund S, Argenzio E, Tosoni D, Cavallaro E, Polo S, Di Fiore PP (2008) Clathrin-mediated internalization is essential for sustained EGFR signaling but dispensable for degradation. Dev Cell 15(2):209–219. https://doi.org/10.1016/j.devcel.2008.06.012
Sigismund S, Confalonieri S, Ciliberto A, Polo S, Scita G, Di Fiore PP (2012) Endocytosis and signaling: cell logistics shape the eukaryotic cell plan. Physiol Rev 92(1):273–366. https://doi.org/10.1152/physrev.00005.2011
Sigismund S, Algisi V, Nappo G, Conte A, Pascolutti R, Cuomo A, Bonaldi T, Argenzio E, Verhoef LG, Maspero E, Bianchi F, Capuani F, Ciliberto A, Polo S, Di Fiore PP (2013) Threshold-controlled ubiquitination of the EGFR directs receptor fate. EMBO J 32(15):2140–2157. https://doi.org/10.1038/emboj.2013.149
Singh AB, Harris RC (2005) Autocrine, paracrine and juxtacrine signaling by EGFR ligands. Cell Signal 17(10):1183–1193. https://doi.org/10.1016/j.cellsig.2005.03.026
Snijder B, Sacher R, Ramo P, Damm EM, Liberali P, Pelkmans L (2009) Population context determines cell-to-cell variability in endocytosis and virus infection. Nature 461(7263):520–523. https://doi.org/10.1038/nature08282
Sorkin A, Carpenter G (1993) Interaction of activated EGF receptors with coated pit adaptins. Science 261(5121):612–615
Sorkin A, Goh LK (2008) Endocytosis and intracellular trafficking of ErbBs. Exp Cell Res 314(17):3093–3106. https://doi.org/10.1016/j.yexcr.2008.08.013
Sorkin A, Krolenko S, Kudrjavtceva N, Lazebnik J, Teslenko L, Soderquist AM, Nikolsky N (1991) Recycling of epidermal growth factor-receptor complexes in A431 cells: identification of dual pathways. J Cell Biol 112(1):55–63
Sousa LP, Lax I, Shen H, Ferguson SM, De Camilli P, Schlessinger J (2012) Suppression of EGFR endocytosis by dynamin depletion reveals that EGFR signaling occurs primarily at the plasma membrane. Proc Natl Acad Sci U S A 109(12):4419–4424. https://doi.org/10.1073/pnas.1200164109
Spangler JB, Neil JR, Abramovitch S, Yarden Y, White FM, Lauffenburger DA, Wittrup KD (2010) Combination antibody treatment down-regulates epidermal growth factor receptor by inhibiting endosomal recycling. Proc Natl Acad Sci U S A 107(30):13252–13257. https://doi.org/10.1073/pnas.0913476107
Stern KA, Place TL, Lill NL (2008) EGF and amphiregulin differentially regulate Cbl recruitment to endosomes and EGF receptor fate. Biochem J 410(3):585–594. https://doi.org/10.1042/BJ20071505
Stoscheck CM, Carpenter G (1984) Characterization of the metabolic turnover of epidermal growth factor receptor protein in A-431 cells. J Cell Physiol 120(3):296–302. https://doi.org/10.1002/jcp.1041200306
Suetsugu S, Yamazaki D, Kurisu S, Takenawa T (2003) Differential roles of WAVE1 and WAVE2 in dorsal and peripheral ruffle formation for fibroblast cell migration. Dev Cell 5(4):595–609
Taelman VF, Dobrowolski R, Plouhinec JL, Fuentealba LC, Vorwald PP, Gumper I, Sabatini DD, De Robertis EM (2010) Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell 143(7):1136–1148. https://doi.org/10.1016/j.cell.2010.11.034
Tartaglia M, Gelb BD, Zenker M (2011) Noonan syndrome and clinically related disorders. Best Pract Res Clin Endocrinol Metab 25(1):161–179. https://doi.org/10.1016/j.beem.2010.09.002
Thalappilly S, Soubeyran P, Iovanna JL, Dusetti NJ (2010) VAV2 regulates epidermal growth factor receptor endocytosis and degradation. Oncogene 29(17):2528–2539. https://doi.org/10.1038/onc.2010.1
Thatcher N, Chang A, Parikh P, Rodrigues Pereira J, Ciuleanu T, von Pawel J, Thongprasert S, Tan EH, Pemberton K, Archer V, Carroll K (2005) Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer). Lancet 366(9496):1527–1537. https://doi.org/10.1016/S0140-6736(05)67625-8
Tsvetanova NG, von Zastrow M (2014) Spatial encoding of cyclic AMP signaling specificity by GPCR endocytosis. Nat Chem Biol 10(12):1061–1065. https://doi.org/10.1038/nchembio.1665
Ullrich A, Coussens L, Hayflick JS, Dull TJ, Gray A, Tam AW, Lee J, Yarden Y, Libermann TA, Schlessinger J et al (1984) Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Nature 309(5967):418–425
Umebayashi K, Stenmark H, Yoshimori T (2008) Ubc4/5 and c-Cbl continue to ubiquitinate EGF receptor after internalization to facilitate polyubiquitination and degradation. Mol Biol Cell 19(8):3454–3462. https://doi.org/10.1091/mbc.E07-10-0988
Vieira AV, Lamaze C, Schmid SL (1996) Control of EGF receptor signaling by clathrin-mediated endocytosis. Science 274(5295):2086–2089
Villasenor R, Nonaka H, Del Conte-Zerial P, Kalaidzidis Y, Zerial M (2015) Regulation of EGFR signal transduction by analogue-to-digital conversion in endosomes. Elife 4. https://doi.org/10.7554/elife.06156
Villasenor R, Kalaidzidis Y, Zerial M (2016) Signal processing by the endosomal system. Curr Opin Cell Biol 39:53–60. https://doi.org/10.1016/j.ceb.2016.02.002
Vincenzi B, Schiavon G, Silletta M, Santini D, Tonini G (2008) The biological properties of cetuximab. Crit Rev Oncol Hematol 68(2):93–106. https://doi.org/10.1016/j.critrevonc.2008.07.006
Wandinger-Ness A, Zerial M (2014) Rab proteins and the compartmentalization of the endosomal system. Cold Spring Harb Perspect Biol 6(11):a022616. https://doi.org/10.1101/cshperspect.a022616
Wang Y, Roche O, Yan MS, Finak G, Evans AJ, Metcalf JL, Hast BE, Hanna SC, Wondergem B, Furge KA, Irwin MS, Kim WY, Teh BT, Grinstein S, Park M, Marsden PA, Ohh M (2009) Regulation of endocytosis via the oxygen-sensing pathway. Nat Med 15(3):319–324. https://doi.org/10.1038/nm.1922
Waterman H, Sabanai I, Geiger B, Yarden Y (1998) Alternative intracellular routing of ErbB receptors may determine signaling potency. J Biol Chem 273(22):13819–13827
Waterman H, Alroy I, Strano S, Seger R, Yarden Y (1999a) The C-terminus of the kinase-defective neuregulin receptor ErbB-3 confers mitogenic superiority and dictates endocytic routing. EMBO J 18(12):3348–3358. https://doi.org/10.1093/emboj/18.12.3348
Waterman H, Levkowitz G, Alroy I, Yarden Y (1999b) The RING finger of c-Cbl mediates desensitization of the epidermal growth factor receptor. J Biol Chem 274(32):22151–22154
Waterman H, Katz M, Rubin C, Shtiegman K, Lavi S, Elson A, Jovin T, Yarden Y (2002) A mutant EGF-receptor defective in ubiquitylation and endocytosis unveils a role for Grb2 in negative signaling. EMBO J 21(3):303–313. https://doi.org/10.1093/emboj/21.3.303
West M, Zurek N, Hoenger A, Voeltz GK (2011) A 3D analysis of yeast ER structure reveals how ER domains are organized by membrane curvature. J Cell Biol 193(2):333–346. https://doi.org/10.1083/jcb.201011039
White IJ, Bailey LM, Aghakhani MR, Moss SE, Futter CE (2006) EGF stimulates annexin 1-dependent inward vesiculation in a multivesicular endosome subpopulation. EMBO J 25(1):1–12. https://doi.org/10.1038/sj.emboj.7600759
Wiley HS (1988) Anomalous binding of epidermal growth factor to A431 cells is due to the effect of high receptor densities and a saturable endocytic system. J Cell Biol 107(2):801–810
Wiley HS, Herbst JJ, Walsh BJ, Lauffenburger DA, Rosenfeld MG, Gill GN (1991) The role of tyrosine kinase activity in endocytosis, compartmentation, and down-regulation of the epidermal growth factor receptor. J Biol Chem 266(17):11083–11094
Wiley HS, Shvartsman SY, Lauffenburger DA (2003) Computational modeling of the EGF-receptor system: a paradigm for systems biology. Trends Cell Biol 13(1):43–50
Wilson KJ, Gilmore JL, Foley J, Lemmon MA, Riese DJ 2nd (2009) Functional selectivity of EGF family peptide growth factors: implications for cancer. Pharmacol Ther 122(1):1–8. https://doi.org/10.1016/j.pharmthera.2008.11.008
Wilson KJ, Mill C, Lambert S, Buchman J, Wilson TR, Hernandez-Gordillo V, Gallo RM, Ades LM, Settleman J, Riese DJ 2nd (2012) EGFR ligands exhibit functional differences in models of paracrine and autocrine signaling. Growth Factors 30(2):107–116. https://doi.org/10.3109/08977194.2011.649918
Wollert T, Yang D, Ren X, Lee HH, Im YJ, Hurley JH (2009) The ESCRT machinery at a glance. J Cell Sci 122(Pt 13):2163–2166. https://doi.org/10.1242/jcs.029884
Wong AJ, Ruppert JM, Bigner SH, Grzeschik CH, Humphrey PA, Bigner DS, Vogelstein B (1992) Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc Natl Acad Sci U S A 89(7):2965–2969
Worthylake R, Opresko LK, Wiley HS (1999) ErbB-2 amplification inhibits down-regulation and induces constitutive activation of both ErbB-2 and epidermal growth factor receptors. J Biol Chem 274(13):8865–8874
Wu WJ, Tu S, Cerione RA (2003) Activated Cdc42 sequesters c-Cbl and prevents EGF receptor degradation. Cell 114(6):715–725
Yamamoto H, Komekado H, Kikuchi A (2006) Caveolin is necessary for Wnt-3a-dependent internalization of LRP6 and accumulation of beta-catenin. Dev Cell 11(2):213–223. https://doi.org/10.1016/j.devcel.2006.07.003
Yamamoto H, Sakane H, Michiue T, Kikuchi A (2008) Wnt3a and Dkk1 regulate distinct internalization pathways of LRP6 to tune the activation of beta-catenin signaling. Dev Cell 15(1):37–48. https://doi.org/10.1016/j.devcel.2008.04.015
Yarden Y, Pines G (2012) The ERBB network: at last, cancer therapy meets systems biology. Nat Rev Cancer 12(8):553–563. https://doi.org/10.1038/nrc3309
Yun CH, Boggon TJ, Li Y, Woo MS, Greulich H, Meyerson M, Eck MJ (2007) Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 11(3):217–227. https://doi.org/10.1016/j.ccr.2006.12.017
Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J (2006) An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 125(6):1137–1149. https://doi.org/10.1016/j.cell.2006.05.013
Zhang X, Pickin KA, Bose R, Jura N, Cole PA, Kuriyan J (2007) Inhibition of the EGF receptor by binding of MIG6 to an activating kinase domain interface. Nature 450(7170):741–744. https://doi.org/10.1038/nature05998
Zhao S, Sedwick D, Wang Z (2015) Genetic alterations of protein tyrosine phosphatases in human cancers. Oncogene 34(30):3885–3894. https://doi.org/10.1038/onc.2014.326
Zoncu R, Perera RM, Balkin DM, Pirruccello M, Toomre D, De Camilli P (2009) A phosphoinositide switch controls the maturation and signaling properties of APPL endosomes. Cell 136(6):1110–1121. https://doi.org/10.1016/j.cell.2009.01.032
Zwick E, Bange J, Ullrich A (2001) Receptor tyrosine kinase signalling as a target for cancer intervention strategies. Endocr Relat Cancer 8(3):161–173
Acknowledgements
We thank Rosalind Gunby for critically reading the manuscript. This work was supported by grants from the Associazione Italiana per la Ricerca sul Cancro to PPDF (IG 14404 and MCO 10.000); MIUR (the Italian Ministry of University and Scientific Research), the Italian Ministry of Health, and The Monzino Foundation to PPDF; and the WWCR (Worldwide Cancer Research) to SS (16-1245).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Caldieri, G., Malabarba, M.G., Di Fiore, P.P., Sigismund, S. (2018). EGFR Trafficking in Physiology and Cancer. In: Lamaze, C., Prior, I. (eds) Endocytosis and Signaling. Progress in Molecular and Subcellular Biology, vol 57. Springer, Cham. https://doi.org/10.1007/978-3-319-96704-2_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-96704-2_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-96703-5
Online ISBN: 978-3-319-96704-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)