
Abstract
NAFLD is an emerging metabolic disease that is affecting almost 25% of the world population. NAFLD is more prevalent in obese subjects or in patients with type 2 diabetes (T2D) where the prevalence is much higher. The pathophysiology of NAFLD is still not completely understood. However, alteration in both glucose and lipid metabolism, mainly because of impairment in insulin action (i.e., insulin resistance) and insulin secretion, is among the major causes of hepatic fat accumulation (steatosis) and its progression to more severe liver disease including nonalcoholic steatohepatitis (NASH). In this chapter, I reviewed the current knowledge on insulin resistance in NAFLD and its impact on the metabolic cross talk among liver, muscle, and adipose tissue.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Insulin resistance (IR)
- Type 2 diabetes (T2D)
- Gluconeogenesis (GNG)
- Endogenous glucose production (EGP)
- Liver, muscle, adipose tissue
4.1 Introduction
NAFLD is an emerging metabolic disease that is affecting almost 25% of the world population [1]. In NAFLD there is a high prevalence of obesity (51%), metabolic syndrome (43%), and type 2 diabetes (T2D, 23%) [2]. Alteration in glucose and lipid metabolism and increased insulin resistance (IR) are highly common [3,4,5,6,7].
IR is a characteristic feature of patients with T2D and is also common in obese subjects regardless of T2D. Most of the subjects diagnosed with NAFLD are obese, so it is not surprising to find that the majority of patients with NAFLD have insulin resistance and T2D [8,9,10]. However, impaired insulin action is often detected also in nonobese NAFLD [3] that are as IR as obese and diabetic NAFLD [11].
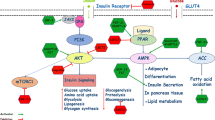
IR is the inability of a known amount of endogenous (or exogenous) insulin to stimulate glucose metabolism in several organs, in particular muscle, liver, and adipose tissue (Fig. 4.1). However, insulin exerts its effects not only on glucose but also on lipids and protein metabolism. Insulin stimulates lipogenesis and protein synthesis and inhibits lipolysis and protein catabolism. In conditions of IR, the antilipolytic effect of insulin is impaired as well as its anabolic/anticatabolic effects. Thus, IR is present not only in liver and muscle but also in adipose tissue with the consequence of overflow of fatty acids to the liver that increases the risk of NAFLD [12].

Insulin resistance (IR) is the inability of insulin to stimulate glucose metabolism in several organs, in particular muscle, liver, and adipose tissue. This results in increased glucose production (EGP) and insufficient glucose disposal. Adipose tissue IR results in increased lipolysis and overabundance of circulating fatty acids, which in turn may contribute to the worsening of insulin resistance and ectopic fat accumulation. Insulin is exerting its effects also on protein metabolism and IR results in excess muscle catabolism and increased circulating amino acids (AA)
In this chapter, I reviewed the current knowledge on IR in NAFLD and its impact on the metabolic cross talk among liver, muscle, and adipose tissue.
4.2 What Is Insulin Resistance and How We Can Measure It
Insulin is one of the most important metabolic hormones, and it is essential for the homeostasis of glucose, lipids, and protein. Insulin exerts its effects binding to its membrane receptors and is transmitted through the cell by a series of protein–protein interactions starting with the phosphorylation of insulin receptor substrates (IRS-1 and -2), which leads to the activation of PI3K and phosphorylation of Akt, which are the main signals involved in the metabolic effects of insulin [13, 14]. In the liver insulin regulates also the transcription factor Foxo1. Insulin-mediated Akt phosphorylation of Foxo1 leads to the decreased transcription of PEPCK that in turn decreases gluconeogenesis (GNG) and endogenous glucose production (EGP). Insulin also stimulates glucose uptake in muscle and liver by stimulating glycogen synthesis and glycolysis. Table 4.1 shows the most used indexes, divided in those based on samples taken during fasting state or during oral glucose tolerance test (OGTT). Hepatic and adipose tissue IR can be assessed using fasting measurements, while reliable measurements of peripheral insulin resistance are obtained using OGTT challenge.
4.2.1 Assessment of Peripheral (Muscle) Insulin Resistance
Insulin resistance in vivo is assessed in several ways. The gold standard method is the euglycemic hyperinsulinemic clamp where insulin is infused in pharmacological doses and glucose is infused along to maintain plasma glucose concentration at constant levels (around 5 mmol/L) [15]. An insulin infusion rate of 40 mU/min/m2 or higher is infused to evaluate peripheral insulin resistance since at this dose endogenous glucose production (EGP) is almost suppressed. Given the complexity of the clamp, several indexes have been developed and used to assess the degree of insulin resistance [16] and summarized in Table 4.1.
Fasting indexes are HOMA-IR and QUICKI that are based on the product of fasting glucose and insulin concentrations. They are widely used since they are measured after an overnight fasting. Recently, Isokuortti et al. have determined the HOMA-IR cutoff for NAFLD (liver fat ≥5.56%, based on the Dallas Heart Study) that was 2.0 [17]. However, this cutoff should be taken cautiously since the same authors found a large inter-laboratory variation for HOMA-IR (25%) due mainly to inter-assay variation in insulin (25%) rather than glucose (5%) measurements [17]. The most reliable indexes are based on glucose and insulin concentrations measured during the OGTT. The most used are the Matsuda index [18] and the OGIS index [19]. The last one has the advantage that is an estimate of glucose clearance based on a mathematical model. Not only OGIS correlates with both glucose disposal during the clamp but also with glucose clearance during OGTT [16]. OGIS has been used in a few studies in subjects with NAFLD and was found also associated to increased liver fibrosis [16, 20]. It should be considered that in subjects with diabetes insulin secretion is often impaired [21] and thus glucose concentrations (and consequently HOMA and OGTT indexes) are altered not because of insulin resistance but because of impaired insulin secretion [22]. For this reason, in diabetic subjects only the hyperinsulinemic euglycemic clamp or the infusion of tracers can give a reliable measurement of muscle insulin resistance.
4.2.2 Assessment of Endogenous Glucose Production and Hepatic Insulin Resistance
In fasting state glucose is produced mainly by the liver (90%) and in part by the kidney (max 10%) [23]. Endogenous glucose production (EGP) can be estimated noninvasively by the infusion of a tracer (i.e., glucose labelled with either a radioactive or a stable isotope). The euglycemic hyperinsulinemic clamp with lower doses of insulin (e.g., 10 mU/min/m2) is used together with the infusion of tracers to measure hepatic insulin resistance (given by the changes in EGP). A measure of hepatic insulin resistance (Hep-IR) is the % suppression during insulin infusion [24]. The dose response insulin-EGP is hyperbolic (Fig. 4.2) and thus the product of insulin times EGP is a surrogate measure of hepatic IR [24, 25]. For this reason, Hep-IR is often and more easily estimated in fasting state [16, 25]. Other indexes have been derived using OGTT data without tracer infusion and validated against the tracers [26, 27] (Table 4.1). However, these indexes were never tested in large cohort of subjects or after intervention (e.g., weight loss or drug).

The hyperbolic function relates insulin concentration to glucose production (EGP) (Panel a) or to FFA/Lipolysis (Panel b). As subjects become more insulin-resistant, the curve moves to the right, meaning that higher insulin concentrations are needed to maintain the same rates of lipolysis, EGP, or FFA concentrations. These relationships are true in both fasting and hyperinsulinemic state (redrawn from [24])
4.2.3 Assessment of Lipolysis and Adipose Tissue Insulin Resistance
Lipolysis, i.e., the rate of adipose tissue triglyceride (TG) hydrolysis, is measured by the infusion of labeled glycerol and calculating rate of appearance (Ra-glycerol) since the free fatty acids (FFA) can be retained and re-esterified to TG [10]. Thus FFA release reflects on in part lipolysis. On the other hand, glycerol cannot be used for TG synthesis since the adipocytes lack the enzyme glycerol kinase [10]. The hydrolysis of one mole of TG results in the release of a mole of glycerol into the systemic circulation.
The euglycemic hyperinsulinemic clamp with lower doses of insulin (e.g., 10 mU/min/m2) is used to measure adipose tissue IR measuring the suppression of free fatty acids or lipolysis. Similar to EGP also the dose response insulin-FFA concentrations or insulin-lipolysis follow a hyperbolic curve (Fig. 4.2) [3, 24, 28, 29]. Thus, the product of insulin times FFA (Adipo-IR) or the product of insulin times Ra-glycerol (Lipo-IR) are surrogates measure of adipose tissue IR in fasting state [28]. As stated above since in subjects with diabetes insulin secretion is often impaired (especially in postprandial state) only fasting Adipo-IR is reliable while OGTT suppression of FFA does not follow a hyperbolic relationship [3, 28].
4.3 Insulin Resistance: Impact of the Liver-Pancreas Cross Talk
The pancreas has an important role in the regulation of glucose homeostasis through the secretion of vital hormones, like insulin and glucagon. The pancreas releases the hormones directly into the portal vein, and thus they first reach the liver since their primary role is the regulation of glucose production and the maintenance of glucose concentration (Fig. 4.3). In the following paragraphs, I will discuss the mechanism of insulin and glucagon secretion and the importance of the cross-talk liver-pancreas I NAFLD.

The pancreas main hormones are insulin and glucagon that are secreted into the portal vein and thus they first reach the liver and then the systemic circulation. The main actions of insulin in the liver are the inhibition of EGP and the increased lipogenesis. Glucagon increases EGP thus contributing to hepatic IR. Great part of the insulin secreted is degraded in the liver during the first pass. In NAFLD insulin clearance is decreased in proportion to hepatic fat
4.3.1 Insulin Secretion and Clearance in NAFLD
Insulin is secreted by the pancreatic β-cells in response to hyperglycemia and is important to maintain the glucose concentrations within a tight range [12]. The beta cells produce proinsulin, and the enzymatic cleavage of proinsulin results in equimolar secretion of insulin and c-peptide into the portal vein [12]. Most of secreted insulin is degraded by the liver and in part by the kidney and the muscle (around 60%); on the other hand, c-peptide is not degraded by the liver, but it is mainly excreted through the kidney [30], and for this reason it is used to estimate prehepatic insulin secretion and insulin clearance [31, 32].
The main action of insulin is to suppress EGP, to promote glycogen synthesis and store glucose in the liver and in the muscle, to increase glycolysis (Figs. 4.3 and 4.4). Moreover, the effect on the adipose tissue is to inhibit lipolysis and promote lipogenesis also by stimulating glucose uptake in the adipose tissue where it is converted to glycerol-3P and used to synthesize TG.

In response to increased blood glucose, e.g., after a meal or OGTT, insulin is increased and glucagon decreased. Ingested glucose stimulates the release of incretin hormones like GLP-1 and GIP by the intestine, which stimulate insulin secretion and inhibit glucagon release. The figure shows main action of insulin and glucagon on liver, muscle, and adipose tissue. In IR state most of insulin actions are impaired in all tissues
Increased peripheral insulin concentration is a compensatory mechanism to overcome peripheral insulin resistance since more insulin is required to have the same metabolic effects [12, 33]. The liver metabolizes most of the secreted insulin and by reducing insulin clearance acts as a modulator of peripheral insulin concentrations following the increased insulin demand due to peripheral insulin resistance. In the pathophysiology of type 2 diabetes, insulin secretion is increased following as subjects progress from NGT to IGT, but when beta cell failure causes a decrease in insulin secretion, they develop type 2 diabetes [32, 33].
In insulin resistance state insulin clearance is decreased, contributing to peripheral hyperinsulinemia [31] (Fig. 4.3). Several studies have demonstrated that subjects with NAFLD have reduced insulin clearance proportionally to the degree of liver fat [4, 25, 34, 35]. However, the mechanisms that regulate hepatic insulin clearance are still unknown. Moreover, insulin clearance is not a static process but is rather influenced by several factors, like nutrient intake and some hormones.
4.3.2 Glucagon Secretion in NAFLD
Glucagon is the other important hormone secreted by the α-cells of the pancreas, with opposite actions compared to insulin [12] (Figs. 4.3 and 4.4). The α-cells produce pro-glucagon, a 160-amino-acid polypeptide, and by enzymatic cleavage, glucagon is secreted into the portal vein [12]. Proglucagon is produced also by the intestinal L-cell [12, 36, 37] although different enzymes in a tissue-specific manner are converting proglucagon to glucagon, GLP-1, and other peptides like GLP-2 or oxyntomodulin [12].
Glucagon regulates hepatic metabolism by stimulating gluconeogenesis, glycogenolysis, and net hepatic glucose output [38,39,40] (Figs. 4.3 and 4.4). Thus, insulin and glucagon have opposite effects on glucose metabolism. In T2D fasting plasma glucagon levels are increased despite the hyperglycemia and fail to be reduced by the postprandial hyperinsulinemia observed after meal ingestion [33, 40,41,42,43]. In NAFLD, glucagon concentrations are increased [44], even in nondiabetic subjects, possibly contributing to increased EGP and hepatic insulin resistance.
4.3.3 Incretin Effect on Insulin Secretion
The incretins (glucagon-like peptide 1, GLP-1, and glucose-dependent insulinotropic polypeptide, GIP) are hormones that are secreted by the intestinal cells in response to nutrients (Fig. 4.4) and are able to potentiate the insulin secretion [45]. GLP-1 is produced from enzymatic cleavage of proglucagon produced by the intestinal epithelial L cells. However, since proglucagon is produced also by pancreatic α-cells, both GLP-1 and glucagon can be released by the pancreas [46] and the gastrointestinal tract [37].
GLP-1 and glucagon are tightly related since GLP-1 not only stimulates insulin secretion but also inhibits glucagon release (Fig. 4.3). GLP-1 modulates hepatic, but not peripheral glucose metabolism, by suppressing EGP independently of glucagon [12, 47,48,49,50,51]. A new study just showed that insulin can regulates the α-cells and promotes the release of GLP-1 in a time- and dose-dependent manner under high-glucose conditions [52]. Thus, GLP-1 is important not only because it stimulates insulin secretion but also for its independent effect on hepatic insulin resistance. This is very important since GLP-1 receptor agonists (GLP-1RA) are a new class of antidiabetic drugs that are important in reducing hepatic fat and improving hepatic insulin sensitivity by decreasing EGP and increasing hepatic glucose uptake [47, 53,54,55,56].
Only few studies have looked at GLP-1 action in NAFLD, finding that it is often impaired although not always its secretion is compromised [44, 57]. Treatment of NAFLD with GLP-1RA has been shown to be effective [53, 54, 56] although the mechanisms of action are still not completely elucidated. The effect on adipose tissue and lipolysis is controversial although the treatment with GLP-1RA is effective on weight loss [58], and improves adipo-IR and lipotoxicity [47, 54].
4.4 Muscle Insulin Resistance in NAFLD
4.4.1 Impact on Glucose Metabolism
Impaired muscle glucose uptake and disposal are the principal defects associated with peripheral insulin-resistant state (Fig. 4.1). Most of the subjects with NAFLD have reduced muscle insulin sensitivity independent of obesity or diabetes [3, 7, 16, 25, 59, 60]. Moreover, muscle IR is present long before significant TG accumulation in the liver (at 1.5%) as shown by the recent paper by Bril et al. [59].
In NAFLD, decreased insulin-stimulated glucose disposal during the hyperinsulinemic clamp is proportional to hepatic fat accumulation and mainly due to a significant reduction in non-oxidative glucose disposal comparable with the one observed in T2D without NAFLD [11, 25, 59]. Also when measured by indexes derived by OGTT, insulin resistance was higher in subjects with NAFLD, and this is evident already in children [61].
In nondiabetic subjects with NAFLD, glucose tolerance seems to be independent of the degree of hepatic steatosis, while nondiabetic NAFLD with significant fibrosis (F2–F4) has worse glucose tolerance independent of obesity [16] (Fig. 4.5). This is confirmed by the strong inverse association between insulin sensitivity measured by OGIS and degree of liver fibrosis [16, 20].

Glucose and insulin curves during OGTT in nondiabetic subjects with NAFLD according to the presence of obesity and the stage of fibrosis (a, b) or the degree of steatosis (c, d) (n = 145) (from [16] with permission)
4.4.2 Impact on Protein Metabolism
The muscle is where protein are stored. Insulin regulates also protein metabolism by stimulating protein synthesis and reducing protein catabolism. In insulin-resistant state, despite high insulin concentrations, protein catabolism is not suppressed (Fig. 4.1) and fasting amino acid concentrations, in particular the concentrations of essential amino acids, like branched chain amino acids (BCAA), are increased [62,63,64]. Several studies have reported increased fasting BCAA concentrations in NAFLD also related to the severity of this disease, in particular to presence of NASH and fibrosis [62, 65,66,67]. BCAA have been associated also to hepatic IR since they stimulate mTOR1 [64]. However, if BCAA or other amino acids are simple biomarkers of IR or active players has still to be demonstrated. What is known is that subjects with NAFLD have decreased lean body mass and are more sarcopenic compared to subjects without NAFLD [68,69,70]; this condition is worsened in subjects with fibrosis F3–F4 [70]. We have hypothesized that this might be associated to muscle IR, i.e., reduced protein net balance due to increased protein catabolism and reduced protein anabolism [62].
4.5 Hepatic Insulin Resistance in NAFLD
4.5.1 Impact on Hepatic Glucose Production
The liver is the principal organ that produces glucose (EGP) [23]. Hepatic insulin resistance is defined as a defect of insulin to suppress EGP during fasting and/or during insulin infusion (Fig. 4.1). Hepatic IR is strongly associated to hepatic fat accumulation [25, 59]. In general fasting hepatic IR is increased proportionally to the degree of hepatic steatosis and is already present even when hepatic TG are less than 5% [25, 59]. It is important to note that nondiabetic subjects with NAFLD have increased fasting hepatic insulin resistance compared to non-NAFLD and similar to T2D without NAFLD and having T2D and NAFLD further increases hepatic IR [11, 25, 59]. Ortiz-Lopez et al. reported that in NAFLD with normal glucose tolerance (NGT), insulin-mediated suppression of EGP is preserved [71]. However, it has been shown that even the % suppression is similar, the dose response insulin-EGP is shifted to the right, indicating the need of higher insulin concentrations than subjects without NAFLD to suppress EGP or in other words they are more insulin-resistant [3]. In general, having have prediabetes or T2D is in general associated to lower suppression of EGP [25, 71].
4.5.2 Impact on Gluconeogenesis
The liver produces glucose through glycogenolysis and gluconeogenesis (GNG). After overnight fasting, more than 50% of the glucose is synthesized from gluconeogenic precursors such as lactate/pyruvate, glucogenic amino acids, and glycerol [38, 72, 73]. Glycerol used for gluconeogenesis comes mainly from the hydrolysis of triglycerides in the adipose tissue, while the amino acids comes from muscular proteolysis (Fig. 4.1). Almost all amino acids are glucogenic and they are alanine, glutamic acid and glutamine, glycine, arginine, asparagine, aspartic acid, cysteine, serine, valine, phenylalanine, tyrosine, isoleucine, tryptophan, methionine, histidine, threonine, proline, while lysine and leucine are used to produce ketone bodies. Fasting endogenous glucose production is tightly regulated and in nondiabetic subjects, endogenous glucose production (EGP) by the liver is relatively similar among subjects when whole body fluxes are normalized by lean body mass [38, 74]. In diabetic subjects, EGP is increased proportionally to fasting plasma glucose [23, 33, 38].
High rates of glucose production are mainly due to increased GNG flux [38]. In NAFLD, GNG fluxes tend to be increased as a consequence of increased glycerol and amino acid concentrations [75], indicating increased peripheral lipolysis and protein catabolism. The visceral fat is often increased in these subjects, and it is related to increased insulin resistance [4]. This tissue is also highly lipolytic making VF an important contributor of glucogenic substrates since it is drained by the portal vein. We have shown that GNG flux is increased proportionally to VF, while there is no correlation between GNG and the amount of TG stored in the liver [4, 25, 76].
Hepatic IR is also due to impaired suppression of GNG since it has been shown that glycogenolytic fluxes are similarly suppressed in non-diabetic and T2D subjects during the euglycemic hyperinsulinemic clamp [77]. Insulin exerts its effects on the liver by reducing glycogenolysis and after a meal by stimulating glycogen synthesis (Fig. 4.4). The effects on the gluconeogenesis are mild and indirect since the release of most of gluconeogenic precursors (i.e., glycerol and amino acids) is insulin-dependent (Fig. 4.1) and high insulin concentrations decrease lipolysis (i.e., glycerol release from the adipose tissue) and proteolysis (i.e., amino acid release from the muscle) both in fasting and feeding state [14]. So it is likely that increased gluconeogenesis is a compensatory mechanism for the hepatic metabolism of substrates in excess [10, 75].
4.5.3 Impact on Hepatic De Novo Lipogenesis
Insulin also promotes lipogenesis and triglyceride production in the liver. Moreover, insulin promotes the de novo synthesis of fatty acid (DNL) first palmitate and then for elongation stearic acid, and for desaturation palmitoleic acid and oleic acid. The main DNL precursors are carbohydrates that, if they cannot be oxidized or stored as glycogen, are then stored as TG [10, 75, 78, 79]. DNL is increased in subjects with NAFLD, particularly after high carbohydrate and/or high fructose intake [10, 78, 80]. Donnelly et al. have estimated that in NAFLD about 26% of intrahepatic TG (IHTG) are from DNL, 59% from FFA (i.e., derived from peripheral lipolysis), and 15% from TG of the diet [80]. In subjects with NAFLD, DNL is increased up to three times the rate observed in healthy subjects [81]. However, not all subjects with NAFLD have increased DNL, particularly if they have PNPLA3 148M allele since these subjects have lower DNL and expression of the lipogenic transcription factor SREBP1c [82]. Moreover, DNL rates are highly dependent on meal composition. We have recently shown that carbohydrate overfeeding stimulated DNL by +98% and increased IHTG +33% [79]; also fat overfeeding increased IHTG, +55% if the diet was rich in saturated fat vs +15% for diet rich in unsaturated fat +15% (p < 0.05), but this was due to excess fat since the rates of hepatic DNL were unchanged compared to baseline [79].
4.6 Adipose Tissue Insulin Resistance in NAFLD
4.6.1 Impact on Lipolysis
IR is present not only in the liver and the muscle but also at the level of the adipose tissue (Fig. 4.1). The main effect of insulin in the adipose tissue is glucose uptake for triglyceride synthesis and inhibition of lipolysis. In presence of IR, there is an excess lipolysis and FFA release despite high circulating levels of insulin. This is more evident during fasting state when insulin is low [11, 28, 59, 83,84,85]. However, also diet composition seems to be implicated in the worsening of adipose tissue IR. We have shown that overfeeding with saturated fatty acids increased fasting lipolysis compared to diet with similar caloric intake but rich in unsaturated fat or carbohydrates [79]. Excess FFA from the adipose tissue determines an overflow to the liver and other organs (Fig. 4.1) which in presence of high insulin concentrations favors intracellular TG re-esterification and ectopic fat accumulation not only in the liver but also in other organs including pancreas and heart [21, 86].
4.6.2 Impact on Lipogenesis
Insulin also promotes lipogenesis and adiposity (Fig. 4.4). Excess carbohydrate promotes adipogenesis since glucose is also used as a precursor of glycerol-3P and used for TG synthesis. DNL occurs mainly in the liver although we cannot exclude that it might be active also in the adipose tissue [87]. Ectopic fat accumulates only when the subcutaneous adipose tissue is not able to store excess fat and glucose since lipogenesis is impaired [86, 88, 89]. This adipose tissue is often found inflamed, resistant to the antilipolytic effect of insulin, with increased release of pro-inflammatory adipokines and reduced secretion of adiponectin [86, 88, 89]. This not only impairs fatty acid oxidation but also promotes the synthesis of lipotoxic lipids that may act as signals that worsen IR, glucose, and lipid metabolism (see below).
4.7 Lipotoxicity, Glucotoxicity, and IR
Lipotoxicity is the accumulation of lipids that impair metabolic signaling, leading to alteration in glucose and lipid metabolism, insulin resistance, and impaired insulin secretion [90]. Impaired triglyceride synthesis or partial hydrolysis of TG can lead to the production and accumulation of lipid species like diacylglycerols (DAG) and ceramides [5, 14, 91, 92]. Production of lipotoxic metabolites like DAG can cause insulin resistance by activating PKCε [13, 93]. In humans, lipid infusion induces muscle IR by transient increase in total and cytosolic DAG content [93]. The activated PKCε binds to the insulin receptor and inhibits its tyrosine kinase activity interfering with the ability of insulin to phosphorylate IRS-2 on tyrosine residues. Hepatic cytosolic DAG were observed also in human livers of subjects with NAFLD and correlated with activation of PKCε [94]. A stepwise increase in DAG and the product/precursor ratio (TAG/DAG) was observed from normal livers to NAFL to NASH [92].
Other lipotoxic compounds are ceramides and in general saturated fat. However, total hepatic ceramides are often similar among NAFLD/NASH and controls [92, 94]. This is likely because increased hepatic ceramide accumulation and/or de novo synthesis is more associated to presence of insulin resistance rather than NAFLD due to genetic predisposition [91, 95].
Lipotoxicity has been almost exclusively attributed to saturated fat that either comes from the diet or is synthesized from de novo lipogenesis (DNL). Studies in cells have shown that the incubation with oleic acid (18:1) results in immediate incorporation into triglyceride (TG) and increases TG accumulation. On the other hand, the incubation with palmitic acid (C16:0) results in poor incorporation into triglyceride and causes apoptosis [96]. The co-incubation of C18:1 and C16:0 reduces apoptosis and stimulates palmitate incorporation into TG [96]. However, when triglyceride synthesis is impaired, e.g., in cells from Dgat1 null mice, both incubation with oleate and palmitate leads to lipotoxicity [96], indicating that accumulation of excess FA in cellular triglyceride stores may be protective against lipotoxicity.
Glucotoxicity, i.e., the toxic effects of hyperglycemia and excess carbohydrate intake on cells and tissues, is as harmful as lipotoxicity [5]. As previously discussed, hyperglycemia and excess carbohydrate intake can favor DNL, i.e., synthesis of palmitate (a saturated fat and a precursor of ceramides and other lipotoxic lipids) [5, 79]. Glucotoxicity and lipotoxicity are closely interrelated, and both contribute to the deterioration of insulin resistance and impaired insulin secretion [5, 90]. In particular, glucotoxicity alters IRS-1 signal, promotes JNK activation, and determines IR not only in liver but also in muscle, initiating a vicious cycle [5].
4.8 Genetic Vs Metabolic NAFLD
Although NAFLD is not a genetic disease, several polymorphisms have been associated to increased risk of development and progression of NAFLD showing that subjects carrying the gene variant for PNPLA3, hypo-betalipoproteinemia, DGAT, or TM6SF2 are more likely to have NAFLD [97,98,99]. An interesting observation was that although these subjects have NAFLD, their insulin-resistant state is no different from subjects without the gene variant and no NAFLD [4, 98]. Moreover, when subjects with NAFLD homozygous either for the rs738409 PNPLA3 G allele (PNPLA3-148MM) or the C allele (PNPLA3-148II) were placed on a hypocaloric low-carbohydrate diet for 6 days, those at high risk of NAFLD (with the G allele) had a better metabolic outcome with higher decrease in steatosis and better improvement in peripheral IR despite similar weight loss [100]. The PNPLA3 protein has lipase activity towards TG in hepatocytes and retinyl esters in hepatic stellate cells; the I148M substitution leads to a loss of function promoting intrahepatic TG accumulation [101]. PNPLA3 variant was not associated to alteration in peripheral lipolysis or hepatic fatty acid oxidation when subjects with NAFLD were matched for hepatic triglyceride accumulation [100]. On the other hand in TM6SF2 E167K variant carriers hepatic lipid synthesis from unsaturated fatty acids is impaired [79] and together with reduced VLDL secretion could contribute to increased intrahepatic TG [102].
However, it should be noted that different mechanisms explain the pathophysiology of metabolic NAFLD vs genetic NAFLD. In metabolic NAFLD, the subcutaneous adipose tissue is not able to store excess caloric intake, then fat accumulates as ectopic fat in other tissues like liver, muscle, pancreas, and heart [86, 88, 89]. This has also been supported by genetic studies [103, 104]; using integrative genomic approaches these authors have found that a cluster of genes associated with insulin resistance (of which the most important is PPARG) was also associated to a reduced capacity of subcutaneous tissue to expand, resulting in ectopic fat accumulation, NAFLD, and higher visceral-to-subcutaneous adipose tissue ratio [103, 104].
4.9 Insulin Resistance and NAFLD: Chicken or Egg?
Although it is recognized that IR is strongly associated to NAFLD, if IR precedes/causes NAFLD [105] or the other way around [106] has been long debated. Recent cross-sectional studies have shown that impairment in peripheral insulin sensitivity is present already in subjects with minimal hepatic TG accumulation (i.e., less than 5%) [59]. Currently there are no longitudinal studies that have properly addressed this point. This is due to many reasons, mainly because (1) data are lacking, as NAFLD has been recognized as a metabolic disease only in recent years and (2) the assessment of presence of IR is no trivial (see previous paragraphs). Metabolic studies in subjects carrying genetic risk factors for NAFLD and overfeeding studies involving non-IR subjects helped answering, at list in part, this question. It is now recognized that NAFLD has two main phenotypes: genetic (type 1) vs metabolic (type 2) NAFLD but only metabolic NAFLD is associated to IR [4, 7, 107].
Overfeeding/inactivity studies of non-IR subjects helped understanding the mechanism of development of NAFLD/IR. Several overfeeding studies have shown that the decrease in insulin sensitivity precedes the development of NAFLD. Knudesen et al. showed that 14 days of inactivity and overfeeding (+50%) induced IR as early as day 3, while body fat and visceral fat were increased significantly only after 14 days [108]. The recent paper by Peterson et al. has shown that overfeeding by 40% for 8 weeks (56 days) decreased peripheral glucose disposal, in particular nonoxidative disposal rate, at low (10 mU/min·m2) but not at high (50 mU/min·m2) insulin infusion rates and although it increased body weight by 7.6 kg (of which +4.2 kg of body fat), there was no clinically significant change in hepatic fat that was 1.5% at baseline and 2.2% at the end of study [109]. However, also visceral fat was low at baseline (0.58 kg) and 0.94 kg at the end of study.
Although large prospective studies on this topic are still lacking, it seems that only subjects with the “metabolic” NAFLD are more insulin-resistant and at increased risk of T2D [9,10,11], while subjects with the “genetic” NAFLD are more at risk of HCC and chronic liver disease [7, 99].
4.10 Conclusions
In subjects with NAFLD, IR is more pronounced at the level of the muscle, where glucose uptake is reduced, but it is present also in the liver, where insulin does not properly suppress hepatic glucose production, and in the adipose tissue, where peripheral lipolysis is high despite high insulin concentrations [8, 25, 59]. Genetic NAFLD dissociates from metabolic NAFLD since very often these subjects do not have IR or increased DNL.
Abbreviations
- AA:
-
Amino acids
- AKT:
-
Protein kinase B
- BCAA:
-
Branched chain amino acids
- DAG:
-
Diacylglycerols
- DNL:
-
De novo lipogenesis
- EGP:
-
Endogenous glucose production
- FFA:
-
Free fatty acids
- FOXO1:
-
Forkhead box protein O1
- GNG:
-
Gluconeogenesis
- Hep-IR:
-
Hepatic insulin resistance
- IHTG:
-
Intrahepatic TG
- IR:
-
Insulin resistance
- IRS-1 and IRS-2:
-
Insulin receptor substrates
- OGTT:
-
Oral glucose tolerance test
- PEPCK:
-
Phosphoenol-pyruvate carboxykinase
- Ra:
-
Rate of appearance
- T2D:
-
Type 2 diabetes
- TG:
-
Triglyceride
References
Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, George J, Bugianesi E. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2018;15(1):11–20. https://doi.org/10.1038/nrgastro.2017.109.
Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64(1):73–84. https://doi.org/10.1002/hep.28431.
Bugianesi E, Gastaldelli A, Vanni E, Gambino R, Cassader M, Baldi S, Ponti V, Pagano G, Ferrannini E, Rizzetto M. Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: sites and mechanisms. Diabetologia. 2005;48(4):634–42. https://doi.org/10.1007/s00125-005-1682-x.
Gaggini M, Morelli M, Buzzigoli E, DeFronzo RA, Bugianesi E, Gastaldelli A. Non-alcoholic fatty liver disease (NAFLD) and its connection with insulin resistance, dyslipidemia, atherosclerosis and coronary heart disease. Nutrients. 2013;5(5):1544–60. https://doi.org/10.3390/nu5051544.
Mota M, Banini BA, Cazanave SC, Sanyal AJ. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism. 2016;65(8):1049–61. https://doi.org/10.1016/j.metabol.2016.02.014.
Stefan N, Fritsche A, Schick F, Haring HU. Phenotypes of prediabetes and stratification of cardiometabolic risk. Lancet Diabetes Endocrinol. 2016;4(9):789–98. https://doi.org/10.1016/S2213-8587(16)00082-6.
Yki-Jarvinen H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinology. 2014;2(11):901–10. https://doi.org/10.1016/S2213-8587(14)70032-4.
Gastaldelli A, Kozakova M, Hojlund K, Flyvbjerg A, Favuzzi A, Mitrakou A, Balkau B, RISC Investigators. Fatty liver is associated with insulin resistance, risk of coronary heart disease, and early atherosclerosis in a large European population. Hepatology. 2009;49(5):1537–44. https://doi.org/10.1002/hep.22845.
Lallukka S, Yki-Jarvinen H. Non-alcoholic fatty liver disease and risk of type 2 diabetes. Best Pract Res Clin Endocrinol Metab. 2016;30(3):385–95. https://doi.org/10.1016/j.beem.2016.06.006.
Saponaro C, Gaggini M, Carli F, Gastaldelli A. The subtle balance between lipolysis and Lipogenesis: a critical point in metabolic homeostasis. Nutrients. 2015;7(11):9453–74. https://doi.org/10.3390/nu7115475.
Brouwers B, Schrauwen-Hinderling VB, Jelenik T, Gemmink A, Havekes B, Bruls Y, Dahlmans D, Roden M, Hesselink MKC, Schrauwen P. Metabolic disturbances of non-alcoholic fatty liver resemble the alterations typical for type 2 diabetes. Clin Sci (Lond). 2017;131(15):1905–17. https://doi.org/10.1042/CS20170261.
Gastaldelli A, Gaggini M, DeFronzo R. Glucose kinetics: an update and novel insights into its regulation by glucagon and GLP-1. Curr Opin Clin Nutr Metab Care. 2017;20(4):300–9. https://doi.org/10.1097/MCO.0000000000000384.
Postic C, Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J Clin Invest. 2008;118(3):829–38. https://doi.org/10.1172/JCI34275.
Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148(5):852–71. https://doi.org/10.1016/j.cell.2012.02.017.
DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol. 1979;237(3):E214–23. https://doi.org/10.1152/ajpendo.1979.237.3.E214.
Rosso C, Mezzabotta L, Gaggini M, Salomone F, Gambino R, Marengo A, Saba F, Vanni E, Younes R, Saponaro C, Buzzigoli E, Caviglia GP, Abate ML, Smedile A, Rizzetto M, Cassader M, Gastaldelli A, Bugianesi E. Peripheral insulin resistance predicts liver damage in nondiabetic subjects with nonalcoholic fatty liver disease. Hepatology. 2016;63(1):107–16. https://doi.org/10.1002/hep.28287.
Isokuortti E, Zhou Y, Peltonen M, Bugianesi E, Clement K, Bonnefont-Rousselot D, Lacorte JM, Gastaldelli A, Schuppan D, Schattenberg JM, Hakkarainen A, Lundbom N, Jousilahti P, Mannisto S, Keinanen-Kiukaanniemi S, Saltevo J, Anstee QM, Yki-Jarvinen H. Use of HOMA-IR to diagnose non-alcoholic fatty liver disease: a population-based and inter-laboratory study. Diabetologia. 2017;60:1873. https://doi.org/10.1007/s00125-017-4340-1.
Matsuda M, DeFronzo RA. Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care. 1999;22(9):1462–70.
Mari A, Pacini G, Murphy E, Ludvik B, Nolan JJ. A model-based method for assessing insulin sensitivity from the oral glucose tolerance test. Diabetes Care. 2001;24(3):539–48.
Svegliati-Baroni G, Bugianesi E, Bouserhal T, Marini F, Ridolfi F, Tarsetti F, Ancarani F, Petrelli E, Peruzzi E, Lo Cascio M, Rizzetto M, Marchesini G, Benedetti A. Post-load insulin resistance is an independent predictor of hepatic fibrosis in virus C chronic hepatitis and in non-alcoholic fatty liver disease. Gut. 2007;56(9):1296–301. https://doi.org/10.1136/gut.2006.107946.
Gastaldelli A. Role of beta-cell dysfunction, ectopic fat accumulation and insulin resistance in the pathogenesis of type 2 diabetes mellitus. Diabetes Res Clin Pract. 2011;93(Suppl 1):S60–5. https://doi.org/10.1016/S0168-8227(11)70015-8.
Mari A, Pacini G, Brazzale AR, Ahren B. Comparative evaluation of simple insulin sensitivity methods based on the oral glucose tolerance test. Diabetologia. 2005;48(4):748–51.
Cherrington AD. Banting lecture 1997. Control of glucose uptake and release by the liver in vivo. Diabetes. 1999;48(5):1198–214.
Groop LC, Bonadonna RC, DelPrato S, Ratheiser K, Zyck K, Ferrannini E, DeFronzo RA. Glucose and free fatty acid metabolism in non-insulin-dependent diabetes mellitus. Evidence for multiple sites of insulin resistance. J Clin Invest. 1989;84(1):205–13. https://doi.org/10.1172/JCI114142.
Gastaldelli A, Cusi K, Pettiti M, Hardies J, Miyazaki Y, Berria R, Buzzigoli E, Sironi AM, Cersosimo E, Ferrannini E, Defronzo RA. Relationship between hepatic/visceral fat and hepatic insulin resistance in nondiabetic and type 2 diabetic subjects. Gastroenterology. 2007;133(2):496–506.
Abdul-Ghani MA, Matsuda M, Balas B, DeFronzo RA. Muscle and liver insulin resistance indexes derived from the oral glucose tolerance test. Diabetes Care. 2007;30(1):89–94. https://doi.org/10.2337/dc06-1519.
Vangipurapu J, Stancakova A, Pihlajamaki J, Kuulasmaa TM, Kuulasmaa T, Paananen J, Kuusisto J, Ferrannini E, Laakso M. Association of indices of liver and adipocyte insulin resistance with 19 confirmed susceptibility loci for type 2 diabetes in 6,733 non-diabetic Finnish men. Diabetologia. 2011;54(3):563–71. https://doi.org/10.1007/s00125-010-1977-4.
Gastaldelli A, Gaggini M, DeFronzo RA. Role of adipose tissue insulin resistance in the natural history of type 2 diabetes: results from the San Antonio metabolism study. Diabetes. 2017;66(4):815–22. https://doi.org/10.2337/db16-1167.
Sondergaard E, Jensen MD. Quantification of adipose tissue insulin sensitivity. J Investig Med. 2016;64(5):989–91. https://doi.org/10.1136/jim-2016-000098.
Duckworth WC, Bennett RG, Hamel FG. Insulin degradation: progress and potential. Endocr Rev. 1998;19(5):608–24. https://doi.org/10.1210/edrv.19.5.0349.
Byrne MM, Sturis J, Polonsky KS. Insulin secretion and clearance during low-dose graded glucose infusion. Am J Physiol. 1995;268(1 Pt 1):E21–7.
Gastaldelli A, Ferrannini E, Miyazaki Y, Matsuda M, DeFronzo RA, San Antonio Metabolism Study. Beta-cell dysfunction and glucose intolerance: results from the San Antonio metabolism (SAM) study. Diabetologia. 2004;47(1):31–9. https://doi.org/10.1007/s00125-003-1263-9.
DeFronzo RA. Banting lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes. 2009;58(4):773–95. https://doi.org/10.2337/db09-9028.
Bril F, Lomonaco R, Orsak B, Ortiz-Lopez C, Webb A, Tio F, Hecht J, Cusi K. Relationship between disease severity, hyperinsulinemia, and impaired insulin clearance in patients with nonalcoholic steatohepatitis. Hepatology. 2014;59(6):2178–87. https://doi.org/10.1002/hep.26988.
Kotronen A, Juurinen L, Tiikkainen M, Vehkavaara S, Yki-Jarvinen H. Increased liver fat, impaired insulin clearance, and hepatic and adipose tissue insulin resistance in type 2 diabetes. Gastroenterology. 2008;135(1):122–30. https://doi.org/10.1053/j.gastro.2008.03.021.
Campbell JE, Drucker DJ. Islet alpha cells and glucagon—critical regulators of energy homeostasis. Nat Rev Endocrinol. 2015;11(6):329–38. https://doi.org/10.1038/nrendo.2015.51.
Wewer Albrechtsen NJ, Kuhre RE, Pedersen J, Knop FK, Holst JJ. The biology of glucagon and the consequences of hyperglucagonemia. Biomark Med. 2016;10(11):1141–51. https://doi.org/10.2217/bmm-2016-0090.
Gastaldelli A, Baldi S, Pettiti M, Toschi E, Camastra S, Natali A, Landau BR, Ferrannini E. Influence of obesity and type 2 diabetes on gluconeogenesis and glucose output in humans: a quantitative study. Diabetes. 2000;49(8):1367–73.
Magnusson I, Rothman DL, Gerard DP, Katz LD, Shulman GI. Contribution of hepatic glycogenolysis to glucose production in humans in response to a physiological increase in plasma glucagon concentration. Diabetes. 1995;44(2):185–9.
Roden M, Perseghin G, Petersen KF, Hwang JH, Cline GW, Gerow K, Rothman DL, Shulman GI. The roles of insulin and glucagon in the regulation of hepatic glycogen synthesis and turnover in humans. J Clin Invest. 1996;97(3):642–8. https://doi.org/10.1172/JCI118460.
Cherrington AD, Chiasson JL, Liljenquist JE, Jennings AS, Keller U, Lacy WW. The role of insulin and glucagon in the regulation of basal glucose production in the postabsorptive dog. J Clin Invest. 1976;58(6):1407–18. https://doi.org/10.1172/JCI108596.
Reaven GM, Chen YD, Golay A, Swislocki AL, Jaspan JB. Documentation of hyperglucagonemia throughout the day in nonobese and obese patients with noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1987;64(1):106–10. https://doi.org/10.1210/jcem-64-1-106.
Shah P, Vella A, Basu A, Basu R, Schwenk WF, Rizza RA. Lack of suppression of glucagon contributes to postprandial hyperglycemia in subjects with type 2 diabetes mellitus. J Clin Endocrinol Metab. 2000;85(11):4053–9. https://doi.org/10.1210/jcem.85.11.6993.
Junker AE, Gluud L, Holst JJ, Knop FK, Vilsboll T. Diabetic and nondiabetic patients with nonalcoholic fatty liver disease have an impaired incretin effect and fasting hyperglucagonaemia. J Intern Med. 2016;279(5):485–93. https://doi.org/10.1111/joim.12462.
Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132(6):2131–57. https://doi.org/10.1053/j.gastro.2007.03.054.
Marchetti P, Lupi R, Bugliani M, Kirkpatrick CL, Sebastiani G, Grieco FA, Del Guerra S, D'Aleo V, Piro S, Marselli L, Boggi U, Filipponi F, Tinti L, Salvini L, Wollheim CB, Purrello F, Dotta F. A local glucagon-like peptide 1 (GLP-1) system in human pancreatic islets. Diabetologia. 2012;55(12):3262–72. https://doi.org/10.1007/s00125-012-2716-9.
Gastaldelli A, Gaggini M, Daniele G, Ciociaro D, Cersosimo E, Tripathy D, Triplitt C, Fox P, Musi N, DeFronzo R, Iozzo P. Exenatide improves both hepatic and adipose tissue insulin resistance: a dynamic positron emission tomography study. Hepatology. 2016;64(6):2028–37. https://doi.org/10.1002/hep.28827.
Gupta NA, Mells J, Dunham RM, Grakoui A, Handy J, Saxena NK, Anania FA. Glucagon-like peptide-1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatology. 2010;51(5):1584–92. https://doi.org/10.1002/hep.23569.
Prigeon RL, Quddusi S, Paty B, D’Alessio DA. Suppression of glucose production by GLP-1 independent of islet hormones: a novel extrapancreatic effect. Am J Physiol Endocrinol Metab. 2003;285(4):E701–7. https://doi.org/10.1152/ajpendo.00024.2003.
Seghieri M, Rebelos E, Gastaldelli A, Astiarraga BD, Casolaro A, Barsotti E, Pocai A, Nauck M, Muscelli E, Ferrannini E. Direct effect of GLP-1 infusion on endogenous glucose production in humans. Diabetologia. 2013;56(1):156–61. https://doi.org/10.1007/s00125-012-2738-3.
Svegliati-Baroni G, Saccomanno S, Rychlicki C, Agostinelli L, De Minicis S, Candelaresi C, Faraci G, Pacetti D, Vivarelli M, Nicolini D, Garelli P, Casini A, Manco M, Mingrone G, Risaliti A, Frega GN, Benedetti A, Gastaldelli A. Glucagon-like peptide-1 receptor activation stimulates hepatic lipid oxidation and restores hepatic signalling alteration induced by a high-fat diet in nonalcoholic steatohepatitis. Liver Int. 2011;31(9):1285–97. https://doi.org/10.1111/j.1478-3231.2011.02462.x.
Liu P, Song J, Liu H, Yan F, He T, Wang L, Shen H, Hou X, Chen L. Insulin regulates glucagon-like peptide-1 secretion by pancreatic alpha cells. Endocrine. 2018;62:394. https://doi.org/10.1007/s12020-018-1684-3.
Armstrong MJ, Gaunt P, Aithal GP, Barton D, Hull D, Parker R, Hazlehurst JM, Guo K, team Lt, Abouda G, Aldersley MA, Stocken D, Gough SC, Tomlinson JW, Brown RM, Hubscher SG, Newsome PN. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet. 2016;387(10019):679–90. https://doi.org/10.1016/S0140-6736(15)00803-X.
Armstrong MJ, Hull D, Guo K, Barton D, Hazlehurst JM, Gathercole LL, Nasiri M, Yu J, Gough SC, Newsome PN, Tomlinson JW. Glucagon-like peptide 1 decreases lipotoxicity in non-alcoholic steatohepatitis. J Hepatol. 2016;64(2):399–408. https://doi.org/10.1016/j.jhep.2015.08.038.
Cersosimo E, Gastaldelli A, Cervera A, Wajcberg E, Sriwijilkamol A, Fernandez M, Zuo P, Petz R, Triplitt C, Musi N, DeFronzo RA. Effect of exenatide on splanchnic and peripheral glucose metabolism in type 2 diabetic subjects. J Clin Endocrinol Metab. 2011;96(6):1763–70. https://doi.org/10.1210/jc.2010-2146.
Matikainen N, Soderlund S, Bjornson E, Pietilainen K, Hakkarainen A, Lundbom N, Taskinen MR, Boren J. Liraglutide treatment improves postprandial lipid metabolism and cardiometabolic risk factors in humans with adequately controlled type 2 diabetes: a single-Centre randomised controlled study. Diabetes Obes Metab. 2018;21:84. https://doi.org/10.1111/dom.13487.
Bernsmeier C, Meyer-Gerspach AC, Blaser LS, Jeker L, Steinert RE, Heim MH, Beglinger C. Glucose-induced glucagon-like peptide 1 secretion is deficient in patients with non-alcoholic fatty liver disease. PLoS One. 2014;9(1):e87488. https://doi.org/10.1371/journal.pone.0087488.
Vilsboll T, Christensen M, Junker AE, Knop FK, Gluud LL. Effects of glucagon-like peptide-1 receptor agonists on weight loss: systematic review and meta-analyses of randomised controlled trials. BMJ. 2012;344:d7771. https://doi.org/10.1136/bmj.d7771.
Bril F, Barb D, Portillo-Sanchez P, Biernacki D, Lomonaco R, Suman A, Weber MH, Budd JT, Lupi ME, Cusi K. Metabolic and histological implications of intrahepatic triglyceride content in nonalcoholic fatty liver disease. Hepatology. 2017;65(4):1132–44. https://doi.org/10.1002/hep.28985.
Fabbrini E, Magkos F, Mohammed BS, Pietka T, Abumrad NA, Patterson BW, Okunade A, Klein S. Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proc Natl Acad Sci U S A. 2009;106(36):15430–5. https://doi.org/10.1073/pnas.0904944106.
Bedogni G, Gastaldelli A, Manco M, De Col A, Agosti F, Tiribelli C, Sartorio A. Relationship between fatty liver and glucose metabolism: a cross-sectional study in 571 obese children. Nutr Metab Cardiovasc Dis. 2012;22(2):120–6. https://doi.org/10.1016/j.numecd.2010.05.003.
Gaggini M, Carli F, Rosso C, Buzzigoli E, Marietti M, Della Latta V, Ciociaro D, Abate ML, Gambino R, Cassader M, Bugianesi E, Gastaldelli A. Altered amino acid concentrations in NAFLD: impact of obesity and insulin resistance. Hepatology. 2018;67(1):145–58. https://doi.org/10.1002/hep.29465.
Lynch CJ, Adams SH. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat Rev Endocrinol. 2014;10(12):723–36. https://doi.org/10.1038/nrendo.2014.171.
Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, Haqq AM, Shah SH, Arlotto M, Slentz CA, Rochon J, Gallup D, Ilkayeva O, Wenner BR, Yancy WS Jr, Eisenson H, Musante G, Surwit RS, Millington DS, Butler MD, Svetkey LP. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009;9(4):311–26. https://doi.org/10.1016/j.cmet.2009.02.002.
Kalhan SC, Guo L, Edmison J, Dasarathy S, McCullough AJ, Hanson RW, Milburn M. Plasma metabolomic profile in nonalcoholic fatty liver disease. Metabolism. 2011;60(3):404–13. https://doi.org/10.1016/j.metabol.2010.03.006.
Lake AD, Novak P, Shipkova P, Aranibar N, Robertson DG, Reily MD, Lehman-McKeeman LD, Vaillancourt RR, Cherrington NJ. Branched chain amino acid metabolism profiles in progressive human nonalcoholic fatty liver disease. Amino Acids. 2015;47(3):603–15. https://doi.org/10.1007/s00726-014-1894-9.
Sunny NE, Kalavalapalli S, Bril F, Garrett TJ, Nautiyal M, Mathew JT, Williams CM, Cusi K. Cross-talk between branched-chain amino acids and hepatic mitochondria is compromised in nonalcoholic fatty liver disease. Am J Physiol Endocrinol Metab. 2015;309(4):E311–9. https://doi.org/10.1152/ajpendo.00161.2015.
Koo BK, Kim D, Joo SK, Kim JH, Chang MS, Kim BG, Lee KL, Kim W. Sarcopenia is an independent risk factor for non-alcoholic steatohepatitis and significant fibrosis. J Hepatol. 2017;66(1):123–31. https://doi.org/10.1016/j.jhep.2016.08.019.
Lee MJ, Kim EH, Bae SJ, Kim GA, Park SW, Choe J, Jung CH, Lee WJ, Kim HK, Crohn’s, Colitis Association in D-G. Age-related decrease in skeletal muscle mass is an independent risk factor for incident NAFLD: a 10-year retrospective cohort study. Gut Liver. 2018;13:67. https://doi.org/10.5009/gnl18070.
Petta S, Ciminnisi S, Di Marco V, Cabibi D, Camma C, Licata A, Marchesini G, Craxi A. Sarcopenia is associated with severe liver fibrosis in patients with non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2017;45(4):510–8. https://doi.org/10.1111/apt.13889.
Ortiz-Lopez C, Lomonaco R, Orsak B, Finch J, Chang Z, Kochunov VG, Hardies J, Cusi K. Prevalence of prediabetes and diabetes and metabolic profile of patients with nonalcoholic fatty liver disease (NAFLD). Diabetes Care. 2012;35(4):873–8. https://doi.org/10.2337/dc11-1849.
Landau BR, Wahren J, Chandramouli V, Schumann WC, Ekberg K, Kalhan SC. Contributions of gluconeogenesis to glucose production in the fasted state. J Clin Invest. 1996;98(2):378–85. https://doi.org/10.1172/JCI118803.
Roden M, Stingl H, Chandramouli V, Schumann WC, Hofer A, Landau BR, Nowotny P, Waldhausl W, Shulman GI. Effects of free fatty acid elevation on postabsorptive endogenous glucose production and gluconeogenesis in humans. Diabetes. 2000;49(5):701–7.
Natali A, Toschi E, Camastra S, Gastaldelli A, Groop L, Ferrannini E. Determinants of postabsorptive endogenous glucose output in non-diabetic subjects. European Group for the Study of Insulin Resistance (EGIR). Diabetologia. 2000;43(10):1266–72.
Hyotylainen T, Jerby L, Petaja EM, Mattila I, Jantti S, Auvinen P, Gastaldelli A, Yki-Jarvinen H, Ruppin E, Oresic M. Genome-scale study reveals reduced metabolic adaptability in patients with non-alcoholic fatty liver disease. Nat Commun. 2016;7:8994. https://doi.org/10.1038/ncomms9994.
Gastaldelli A, Miyazaki Y, Pettiti M, Buzzigoli E, Mahankali S, Ferrannini E, DeFronzo RA. Separate contribution of diabetes, total fat mass, and fat topography to glucose production, gluconeogenesis, and glycogenolysis. J Clin Endocrinol Metab. 2004;89(8):3914–21. https://doi.org/10.1210/jc.2003-031941.
Gastaldelli A, Toschi E, Pettiti M, Frascerra S, Quinones-Galvan A, Sironi AM, Natali A, Ferrannini E. Effect of physiological hyperinsulinemia on gluconeogenesis in nondiabetic subjects and in type 2 diabetic patients. Diabetes. 2001;50(8):1807–12. https://doi.org/10.2337/diabetes.50.8.1807.
Jacome-Sosa MM, Parks EJ. Fatty acid sources and their fluxes as they contribute to plasma triglyceride concentrations and fatty liver in humans. Curr Opin Lipidol. 2014;25(3):213–20. https://doi.org/10.1097/MOL.0000000000000080.
Luukkonen PK, Sadevirta S, Zhou Y, Kayser B, Ali A, Ahonen L, Lallukka S, Pelloux V, Gaggini M, Jian C, Hakkarainen A, Lundbom N, Gylling H, Salonen A, Oresic M, Hyotylainen T, Orho-Melander M, Rissanen A, Gastaldelli A, Clement K, Hodson L, Yki-Jarvinen H. Saturated fat is more metabolically harmful for the human liver than unsaturated fat or simple sugars. Diabetes Care. 2018;41(8):1732–9. https://doi.org/10.2337/dc18-0071.
Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115(5):1343–51. https://doi.org/10.1172/JCI23621.
Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014;146(3):726–35. https://doi.org/10.1053/j.gastro.2013.11.049. S0016-5085(13)01730-7 [pii].
Mancina RM, Matikainen N, Maglio C, Soderlund S, Lundbom N, Hakkarainen A, Rametta R, Mozzi E, Fargion S, Valenti L, Romeo S, Taskinen MR, Boren J. Paradoxical dissociation between hepatic fat content and de novo lipogenesis due to PNPLA3 sequence variant. J Clin Endocrinol Metab. 2015;100(5):E821–5. https://doi.org/10.1210/jc.2014-4464.
Bell LN, Wang J, Muralidharan S, Chalasani S, Fullenkamp AM, Wilson LA, Sanyal AJ, Kowdley KV, Neuschwander-Tetri BA, Brunt EM, McCullough AJ, Bass NM, Diehl AM, Unalp-Arida A, Chalasani N, Nonalcoholic Steatohepatitis Clinical Research Network. Relationship between adipose tissue insulin resistance and liver histology in nonalcoholic steatohepatitis: a pioglitazone versus vitamin E versus placebo for the treatment of nondiabetic patients with nonalcoholic steatohepatitis trial follow-up study. Hepatology. 2012;56(4):1311–8. https://doi.org/10.1002/hep.25805.
Gastaldelli A, Harrison SA, Belfort-Aguilar R, Hardies LJ, Balas B, Schenker S, Cusi K. Importance of changes in adipose tissue insulin resistance to histological response during thiazolidinedione treatment of patients with nonalcoholic steatohepatitis. Hepatology. 2009;50(4):1087–93. https://doi.org/10.1002/hep.23116.
Lomonaco R, Ortiz-Lopez C, Orsak B, Webb A, Hardies J, Darland C, Finch J, Gastaldelli A, Harrison S, Tio F, Cusi K. Effect of adipose tissue insulin resistance on metabolic parameters and liver histology in obese patients with nonalcoholic fatty liver disease. Hepatology. 2012;55(5):1389–97. https://doi.org/10.1002/hep.25539.
Gaggini M, Carli F, Gastaldelli A. The color of fat and its central role in the development and progression of metabolic diseases. Horm Mol Biol Clin Investig. 2017;31(1). https://doi.org/10.1515/hmbci-2017-0060.
Guo ZK, Cella LK, Baum C, Ravussin E, Schoeller DA. De novo lipogenesis in adipose tissue of lean and obese women: application of deuterated water and isotope ratio mass spectrometry. Int J Obes Relat Metab Disord. 2000;24(7):932–7.
Fabbrini E, Yoshino J, Yoshino M, Magkos F, Tiemann Luecking C, Samovski D, Fraterrigo G, Okunade AL, Patterson BW, Klein S. Metabolically normal obese people are protected from adverse effects following weight gain. J Clin Invest. 2015;125(2):787–95. https://doi.org/10.1172/JCI78425.
Stefan N, Schick F, Haring HU. Causes, characteristics, and consequences of metabolically unhealthy normal weight in humans. Cell Metab. 2017;26(2):292–300. https://doi.org/10.1016/j.cmet.2017.07.008.
Gastaldelli A, Ferrannini E. Chapter 3: pathophysiology of prediabetes: role of lipotoxicity? In: Bergman M, editor. Global health perspectives in prediabetes and diabetes prevention, vol. 1. New Jersey: World Scientific; 2014. p. 31–48. https://doi.org/10.1142/9789814603324_0003.
Luukkonen PK, Zhou Y, Sadevirta S, Leivonen M, Arola J, Oresic M, Hyotylainen T, Yki-Jarvinen H. Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. J Hepatol. 2016;64(5):1167–75. https://doi.org/10.1016/j.jhep.2016.01.002.
Puri P, Baillie RA, Wiest MM, Mirshahi F, Choudhury J, Cheung O, Sargeant C, Contos MJ, Sanyal AJ. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology. 2007;46(4):1081–90. https://doi.org/10.1002/hep.21763.
Szendroedi J, Yoshimura T, Phielix E, Koliaki C, Marcucci M, Zhang D, Jelenik T, Muller J, Herder C, Nowotny P, Shulman GI, Roden M. Role of diacylglycerol activation of PKCtheta in lipid-induced muscle insulin resistance in humans. Proc Natl Acad Sci U S A. 2014;111(26):9597–602. https://doi.org/10.1073/pnas.1409229111.
Ter Horst KW, Gilijamse PW, Versteeg RI, Ackermans MT, Nederveen AJ, la Fleur SE, Romijn JA, Nieuwdorp M, Zhang D, Samuel VT, Vatner DF, Petersen KF, Shulman GI, Serlie MJ. Hepatic diacylglycerol-associated protein kinase Cepsilon translocation links hepatic steatosis to hepatic insulin resistance in humans. Cell Rep. 2017;19(10):1997–2004. https://doi.org/10.1016/j.celrep.2017.05.035.
Apostolopoulou M, Gordillo R, Koliaki C, Gancheva S, Jelenik T, De Filippo E, Herder C, Markgraf D, Jankowiak F, Esposito I, Schlensak M, Scherer PE, Roden M. Specific hepatic sphingolipids relate to insulin resistance, oxidative stress, and inflammation in nonalcoholic steatohepatitis. Diabetes Care. 2018;41(6):1235–43. https://doi.org/10.2337/dc17-1318.
Listenberger LL, Han X, Lewis SE, Cases S, Farese RV Jr, Ory DS, Schaffer JE. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A. 2003;100(6):3077–82. https://doi.org/10.1073/pnas.0630588100.
Anstee QM, Day CP. The genetics of NAFLD. Nat Rev Gastroenterol Hepatol. 2013;10(11):645–55. https://doi.org/10.1038/nrgastro.2013.182.
Petaja EM, Yki-Jarvinen H. Definitions of normal liver fat and the association of insulin sensitivity with acquired and genetic NAFLD—A systematic review. Int J Mol Sci. 2016;17(5). https://doi.org/10.3390/ijms17050633.
Sookoian S, Pirola CJ. Genetic predisposition in nonalcoholic fatty liver disease. Clin Mol Hepatol. 2017;23(1):1–12. https://doi.org/10.3350/cmh.2016.0109.
Sevastianova K, Kotronen A, Gastaldelli A, Perttila J, Hakkarainen A, Lundbom J, Suojanen L, Orho-Melander M, Lundbom N, Ferrannini E, Rissanen A, Olkkonen VM, Yki-Jarvinen H. Genetic variation in PNPLA3 (adiponutrin) confers sensitivity to weight loss-induced decrease in liver fat in humans. Am J Clin Nutr. 2011;94(1):104–11. https://doi.org/10.3945/ajcn.111.012369.
Trepo E, Romeo S, Zucman-Rossi J, Nahon P. PNPLA3 gene in liver diseases. J Hepatol. 2016;65(2):399–412. https://doi.org/10.1016/j.jhep.2016.03.011.
Dongiovanni P, Petta S, Maglio C, Fracanzani AL, Pipitone R, Mozzi E, Motta BM, Kaminska D, Rametta R, Grimaudo S, Pelusi S, Montalcini T, Alisi A, Maggioni M, Karja V, Boren J, Kakela P, Di Marco V, Xing C, Nobili V, Dallapiccola B, Craxi A, Pihlajamaki J, Fargion S, Sjostrom L, Carlsson LM, Romeo S, Valenti L. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology. 2015;61(2):506–14. https://doi.org/10.1002/hep.27490.
Lotta LA, Gulati P, Day FR, Payne F, Ongen H, van de Bunt M, Gaulton KJ, Eicher JD, Sharp SJ, Luan J, De Lucia RE, Stewart ID, Wheeler E, Willems SM, Adams C, Yaghootkar H, Consortium EP-I, Cambridge FC, Forouhi NG, Khaw KT, Johnson AD, Semple RK, Frayling T, Perry JR, Dermitzakis E, McCarthy MI, Barroso I, Wareham NJ, Savage DB, Langenberg C, O'Rahilly S, Scott RA. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat Genet. 2017;49(1):17–26. https://doi.org/10.1038/ng.3714.
Yaghootkar H, Scott RA, White CC, Zhang W, Speliotes E, Munroe PB, Ehret GB, Bis JC, Fox CS, Walker M, Borecki IB, Knowles JW, Yerges-Armstrong L, Ohlsson C, Perry JR, Chambers JC, Kooner JS, Franceschini N, Langenberg C, Hivert MF, Dastani Z, Richards JB, Semple RK, Frayling TM. Genetic evidence for a normal-weight “metabolically obese” phenotype linking insulin resistance, hypertension, coronary artery disease, and type 2 diabetes. Diabetes. 2014;63(12):4369–77. https://doi.org/10.2337/db14-0318.
Marra F, Gastaldelli A, Svegliati Baroni G, Tell G, Tiribelli C. Molecular basis and mechanisms of progression of non-alcoholic steatohepatitis. Trends Mol Med. 2008;14(2):72–81. https://doi.org/10.1016/j.molmed.2007.12.003.
Yki-Jarvinen H. Liver fat in the pathogenesis of insulin resistance and type 2 diabetes. Dig Dis. 2010;28(1):203–9. https://doi.org/10.1159/000282087.
Gastaldelli A. Insulin resistance and reduced metabolic flexibility: cause or consequence of NAFLD? Clin Sci. 2017;131(22):2701–4. https://doi.org/10.1042/CS20170987.
Knudsen SH, Hansen LS, Pedersen M, Dejgaard T, Hansen J, Hall GV, Thomsen C, Solomon TP, Pedersen BK, Krogh-Madsen R. Changes in insulin sensitivity precede changes in body composition during 14 days of step reduction combined with overfeeding in healthy young men. J Appl Physiol (1985). 2012;113(1):7–15. https://doi.org/10.1152/japplphysiol.00189.2011.
Peterson CM, Zhang B, Johannsen DL, Ravussin E. Eight weeks of overfeeding alters substrate partitioning without affecting metabolic flexibility in men. Int J Obes. 2017;41(6):887–93. https://doi.org/10.1038/ijo.2017.58.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Gastaldelli, A. (2020). NAFLD and Insulin Resistance: A Multisystemic Disease. In: Bugianesi, E. (eds) Non-Alcoholic Fatty Liver Disease. Springer, Cham. https://doi.org/10.1007/978-3-319-95828-6_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-95828-6_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-95827-9
Online ISBN: 978-3-319-95828-6
eBook Packages: MedicineMedicine (R0)