
Abstract
Corrosion prevention of Built Heritage represents an important discipline within Heritage Science. Many metal materials have been widely used in antiques and contemporary art pieces, as well as Buildings and Construction. Conservation of metallic building structures is crucial because of cultural value and historical significance. This chapter is devoted for studying the use of electrochemical techniques for on-site corrosion monitoring. There is an increased interest in the area of heritage conservation due to its significance i.e., a strong commitment to maintain and preserve the conditions of the structures, its cultural value and structural integrity. From a Heritage Science conservation standpoint there is a need to establish new conservation strategies and policies, thus allowing to preserve the culture as a whole and the heritage identity. Identifying the current state of corrosion damage on contemporary built heritages is a crucial task that needs to be executed. Non-destructive and quantitative techniques for measuring corrosion are needed to detect the deterioration of structures at an early stage (to predict their residual life), and thus to decide what prevention or repair systems need to be applied. Electrochemical techniques provide virtually the only viable procedure for assessing reinforcement corrosion without removing the concrete layer. Electrochemical corrosion techniques are valuable non-destructive diagnostic tools used for evaluating reinforced concrete heritage buildings and structures, which can supply information for the restoration repairs. The expected outcome of the on-site application of electrochemical technique, is to identify the type of corrosion processes and mechanisms, which will establish a corrosion control and management protocol of existing contemporary buildings in order to ensure and guarantee its preservation.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


1 Introduction to Corrosion in Contemporary Built Heritage
Corrosion prevention of Contemporary Built Heritage is an essential part of the corrosion umbrella that not only includes construction, but also antiques and works of art. Preserving these treasures are crucial because it allows for their historical importance to remain to future generations, so the first step is to conserve and restore history by monitoring, maintaining and preventing corrosion.
Within the past decade there has been an increased interest in conserving the heritage of the 20th century due to the devastating effects and challenges corrosion poses for management and conservation practitioners. Lack of recognition (with the exception of the icons), and the absence of comprehensive research frameworks for identifying 20th-century heritage and correspondingly poor protection have resulted in the loss of many places that will be lamented in the near future.
Engineers in the corrosion field are constantly troubled with predicting the service-life of concrete structures due to climate change, but an important and useful laboratory and field technique for assessing reinforced concrete structures (RCS) corrosion is the use of electrochemical instruments. This is because it is aimed at monitoring corrosion without having to remove slabs of reinforced concrete, neither disturbing nor destroying the structure. These electrochemical techniques can assist engineers and technicians to decide on type of repairs and maintenance are required and to evaluate the corrosion state of the internal structure [1].
RCS proliferation of historical buildings has attracted concern due to the need to preserve and control its integrity, as shown in Fig. 1. Prompted by the need to rebuild historical building structures in Europe devastated in many places during World War II and taking the advantage of two decades of a flourishing economy that accelerated infrastructure development; it raises continual dilemmas about the advisability of undertaking costly repair operations or deciding the demolition and construction of new structures. The number of structures that are rehabilitated is increasing with respect to the structures already built, which is due to two main reasons: new practical uses attributed to existing built heritage structures that can be restored preserving their historical value and, because society takes into consideration the high costs that involves the demolition of existing structures [1, 2].

Corroded rebar under concrete slab
Nowadays, historical buildings are in dire need of serious overhaul to increase the value and extend the lifetime of service. Since built heritage becomes older, some conservation and preventive strategies need to be put in practice.
Usually, the rebar reinforcements are in the passive state because of concrete high pH, according to Pourbaix diagram depicted in Fig. 2 [3]. However exceptional circumstances can occur in which the passivity of the steel is destroyed, the reinforcement corroding in the active state, due to certain factors that precipitate the corrosion process, essentially, as already described in previous chapters, the carbonation of concrete and the presence of chlorides above a critical concentration threshold [1, 2]. The transition from the passive state to the active one happens then by a complex mechanism that comprises a series of successive stages.

Pourbaix diagram for iron in H2O at 25 °C
1.1 Factors Controlling Corrosion in Contemporary Built Heritage
Corrosion of heritage buildings and structures made of concrete and reinforced steel has been identified as a main reason for their deterioration. Steel reinforcements remain in a passive state when embedded in chloride-free and non-carbonated concrete, because its high alkalinity (pH higher than 12). Passive layers formed on steel reinforcement surfaces present low dissolution reaction kinetics, which lead to very low corrosion rates. Therefore negligible risk of corrosion occurs, thus providing a long-lasting service lifetime. A corrosion life-time prediction diagram is proposed by Tutti (Fig. 3).

Tutti diagram corrosion life-time prediction for reinforced concrete structures
It was once thought that after passivity breakdown, corrosion of steel in concrete during active state was under cathodic control, i.e. limited by oxygen access and its diffusion through the concrete pores network, to the metal surface [1, 2]. This mechanism leads to icorr values around 0.1 μA/cm2 [4]. However, either carbonated or chloride-contaminated concrete, it is commonly found that corrosion current density increase up to icorr values of 10 μA/cm2. This preliminary hypothesis, is not accepted as those icorr values of 10 μA/cm2 are larger than the dissolved oxygen diffusion limit intensity within the concrete pore solution, \(i_{\text{lim,O}_{2}}\). Furthermore, those high icorr values cannot either be attributed to passive films formation.
Another mechanism of this particular case is where icorr values are found to be unusually high; thus, a controlling stage that allows a much faster dissolution process than the passive layers either the \(i_{\text{lim,O}_{2}}\), can be assured. Previous research results show that once the passive to active transition takes place the concrete resistivity, ρ, is the key factor controlling corrosion rate.
The concrete resistivity value (ρ), strongly depends on the hydration degree of concrete pores. Moisture can ranges from 103 Ω cm, for saturated conditions up to 1011 Ω cm, for dried concrete. On the other hand, the supply of oxygen and moisture plays a crucial role on the active state of reinforcements corrosion. Mainly moisture is to be responsible for the large resistivity variations since oxygen supply is continuously present. As a result, icorr shows a close dependence on concrete resistivity (ρ).
There is no consensus when establishing a threshold of ρ above which the risk of corrosion can be neglected. This is due to the lack of a clear awareness of tolerable and inadmissible corrosion rate values in reinforced concrete structures (RCS). Although it is clear and accepted with no doubt the existence of an inverse relationship between icorr and ρ.
1.2 Electrochemical Corrosion Techniques and Methods: Fundamentals and Concepts
During last decades, great advances were made on development of electrochemical techniques and devices for on-site monitoring of corrosion processes. Different corrosion characterization techniques are included in Table 1, including direct quantification methods, accelerated corrosion test and electrochemical techniques. Advantages and drawbacks are also reported for each method.
A notable advantage is that electrochemical methods provide a true value of corrosion rate. Therefore, there is no need to use a correlation factor to quantify electrochemical data measurements with real corrosion rate. Those electrochemical techniques make possible on-site monitoring of different combinations of metal and environments.
The two most common methods for obtaining quantitative information on the corrosion rate of reinforcements are the DC linear polarization resistance method and the AC electrochemical impedance spectroscopy method [1,2,3,4,5,6,7,8,9]. The ideal solution for studying RCS behavior would be to directly measure the corrosion rate of real size structures. The technique most widely used in the field, due to its simplicity, is that of potential measurements, whose most serious limitation is its exclusively qualitative nature [7]. The essential difficulty in real structures stems from the impossibility of knowing with certainty the surface area of the reinforcements affected by the electrical signals in the measuring process.
The signals are applied with small counter electrodes (CEs) and extend, while progressively decaying, to a critical length which is unknown in each case. Using a CE of the same size as the structures, with the aim of achieving a uniform distribution of the current lines, is obviously not possible.
The behavior of a RCS may be represented, in a first approximation, by the modified Randles circuit (Fig. 4), though other more complex circuits allow a better simulation of the steel-concrete system [1, 2].

Equivalent electrical circuit (EEC), used for modeling reinforced concrete structures electrochemical response
Several researchers have explored the possibility of applying to the steel-concrete system, different corrosion rate measuring methods based on the application of short duration galvanostatic pulses [1, 2, 10], or the instantaneous discharging of a capacitor (Coulostatic method). Besides their great speed, these methods offer the advantage of causing minimal disturbance of the electrode. Following on from these valuable contributions, the objective pursued is twofold:
-
(i)
To demonstrate that it is possible to estimate the corrosion rate of steel in concrete also by means of potentiostatic pulses, and
-
(ii)
That by supposing an approximate value of the double layer capacitance, this estimation can be made with sufficient exactitude for many practical purposes without the need to know the surface area of the rebar.
2 Concrete as an Electrolyte
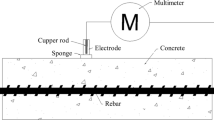
It may be surprising that concrete, a solid ceramic material with a stony appearance and consistency, behaves like an electrolyte and electrochemical methods can be applied to the study of the degradation processes of reinforcements in reinforced concrete structures (RCS). But this is indeed the case, otherwise there would be no risk of corrosion for the reinforcements. Cement mortar and concrete are porous materials, with an aqueous phase that partially fulfills its pore network and provides electrical conductivity. Water uptake through the concrete pore network increases as moisture increases. RCS immersed in water (docks, piers, onshore and offshore structures), they reach saturation conditions. Concrete conductivity varies between very wide limits depending on the degree of saturation of the pores [10, 11], that is, the amount of electrolyte present. There is the possibility that corrosion electrochemical cells form, as a result of the movement of the ions that are generated in local anodes and cathodes, that electrical signals can be applied to the reinforcements and, consequently, to measure their response and estimate their corrosion rate (Fig. 5). All electrochemical techniques for the study of corrosion are therefore applicable to metals embedded in concrete, having contributed to a considerable advance, since they began (during the 60s) to be used in the RCS, and to a better knowledge about the corrosion behavior of RCS in the most varied environments [10, 11].

a Wiring diagram for the application of the on-site electrochemical technique for reinforced concrete corrosion, including working electrode (WE), counter electrode (CE) and reference electrode (RE). b Delamination on reinforced concrete pillar caused by corrosion
2.1 On-Site Electrochemical Corrosion Methods
Corrosion processes involve the presence and transport of charged species, either ions or electrons. Reason why electrochemical methods provide a very useful tool to quantify those electrochemical processes and elucidate corrosion mechanisms. In 1957, Stern and Geary first introduced electrochemical quantitative methods to measure corrosion [5]. This methods was based on previous corrosion cells model proposed by Evans and mixed potential (Ecorr) theory established by Wagner and Traud [4].
Reviewing some electrochemical principles, it is considered electrical equivalence of both half-reactions (Eq. 1), anodic (oxidation) and cathodic (reduction) at mixed potential (Ecorr). Therefore, it is not possible to measure any corrosion current (Icorr), since the electrochemical cell is producing a zero net current (Inet), according to Eq. 2. So, half-reaction currents are equal magnitude but with opposite sign.
where, Ia and Ic, are the anodic and cathodic currents respectively. And Icorr, is the corrosion current.
An external electrical excitation signal needs to be applied to breakdown this half-reaction currents equality, thus allowing electrochemical measurements (Eq. 3). This is because of the zero net current at the mixed potential. In this way polarization signals are used.
The system (electrochemical cell) is polarized, resulting on an effective net current produced (Fig. 6). Thus, electrochemical measurement are applied [5]:

Evans diagram for an electrochemical corrosion process. The application of a small polarization breaks the existing balance in the Ecorr between Ia and Ic to enable the measure of ΔI, and to obtain the Icorr
The plotted relationship between applied voltage (potential), and the obtained electrical current, or vice versa is the polarization curve.
Potential values comprised between both anodic (Ea) and cathodic (Ec) half-reaction equilibrium potentials, have a direct relationship on the overall corrosion process, thus resulting in a current value (It), according to Eq. 3. Polarization curve and Evans diagram is depicted in Fig. 6.
A useful procedure to calculate Ecorr and icorr is the intersection method, where Tafel slopes for anodic and cathodic half-reaction are obtained using a semi-logarithmic data plot (Fig. 7), thus obtaining Tafel law, (Eq. 4):

Polarization plot, E versus log i. Tafel slopes for the anodic (βa) and cathodic (βc) half-reactions. The intersection point determines Ecorr and icorr values
where, η is polarization current, βa and βc are the anodic and cathodic Tafel slopes, respectively, and i is current density.
For those Tafel slopes to be obtained, high polarizations need to be applied, thus producing an irreversible modification on the chemistry of the electrochemical interface metal/solution, also defined as electric double-layer. As a consequence, this method becomes destructive.
Polarization curves present a linear region nearby the corrosion potential (Ecorr), i.e., for low polarization (η) values. An advantage of this fact is the use of the linear polarization resistance (LPR) method. Therefore, according to Stern and Geary, the slope at linear region is correlated to the corrosion rate. Thus, it is possible to establish a direct relationship between polarization resistance (Rp) and corrosion current density (icorr), as it is shown in Eq. 5:
where (Δi) is current density response after an excitation potential signal (ΔE) is applied or vice versa, depending upon type of control, potentiostatic or galvanostatic.
B is defined as a constant calculated from Tafel slopes βa and βc. B value usually took values comprises between the range 13–52 mV [5]. In addition, icorr is the corrosion current density (Icorr/cm2), and Rp is polarization resistance.
The advantage of the linear polarization resistance (LPR) is that is non-destructive method. It also provides a simple and fast way to evaluate corrosion rate.
This LPR method is used to obtain Rp, as it is depicted in Fig. 8. As a consequence, high polarized systems will show a low polarization resistance (Rp) slope (Δi/ΔE), so, in other words, corrosion resistance materials will present a high Rp, and a high hysteresis loop when plotting (Δi/ΔE) in a cyclic, direct and reverse scan, mode.

a Steel in concrete without chlorides metal will corrode much less than, b steel in concrete with chlorides
2.2 Potentiostatic Pulse Technique Fundamentals
When the polarization of an electrode is obtained, either by a potentiostatic pulse, a galvanostatic pulse or an amount of current is interrupted, the system is represented by the EEC in Fig. 4, where the potential decay curve is exponential, immediately after the ohmic drop takes place, according to the Eq. 6:
where
- η0:
-
maximum real polarization at the moment of current interruption, i.e., for time = 0
- ηt:
-
polarization of working electrode (WE) at time t during relaxation after current interruption.
After the current interruption, the increase in the electrochemical double layer charge density is progressively consumed by the corrosion reaction, and taking logarithms of the above equation, yields to Eq. 7:
This expression offers two ways for calculating Rp and therefore the corrosion rate through the Stern-Geary expression [5], as shown in Eq. 8:
One of the two procedures consists of determining the slope of the plot of ln ηt versus t, as has hitherto been performed [4,5,6,7,8,9,10]. The second procedure is based on the direct measurement of the time constant attributable to the corrosion process at the steel-concrete interface.
2.3 First Procedure: Estimation of Rp from the Slope of the Plot of t versus ln ηt
Given that η0 is a constant value for each test, from Eq. 9, it is deduced that the plot of t versus ln ηt is a straight line from whose slope Rp may be deduced if the capacitance value (C) is known:
Therefore, on the basis of real experience, it may be supposed without much error that the value of C is, for the system studied, between 10−4 and 10−5 F/cm2, though much higher values may be determined for rebar that corrode in active state [10], perhaps due to the effect of the existence of voluminous corrosion products at the steel-concrete interface.
A disadvantage to be considered is that the overall process mixes several partial processes with different time constants. Previous experience allows this limitation to be overcome, since it is known that Rp is of the order of 104 Ω cm2 for reinforcements that corrode in active state and around 105–106 Ω cm2 for passive rebar. By selecting an intermediate capacitance value, C, e.g. 5 × 10−5 F/cm2, it is possible to very approximately estimate the time constant of the corrosion process, Ƭ = CRp, which is the magnitude of interest. Having determined Ƭ, the time interval (around Ƭ) is defined in which, in practice, the slope of the straight line for calculating Rp by Eq. 9 must be fitted. In the plot of ln ηt versus t it would be necessary to consider tenths of a second in the case of active structures and units or tens of seconds in the case of passive structures.
2.4 Second Procedure: Direct Determination of the Time Constant
For a time t = Ƭ = CRp, Eq. 10 is reduced to:
Thus, it is possible to determine the time constant of the corrosion process, Ƭ = CRp, by measuring the time in which ηt is reduced to 37% of its initial value (Fig. 9).

Potentiostatic pulse technique, a non-destructive method to determine polarization resistance, Rp
As C is directly proportional to the surface area of the capacitor (electrochemical double layer) and Rp is inversely proportional, Ƭ is independent of this magnitude. Consequently, if the C value per unit of surface area is inserted in the preceding equations, the Rp of the rebar per unit of surface area will be calculated, without the total number or diameter of rebars being of importance.
As a result, the possible interference of other partial processes besides corrosion must be of little importance, or must occur in very different times to those used in the measurements, in order not to mask the potential decay due to the corrosion process.
The direct measurement of Ƭ to determine Rp applying galvanostatic pulses [1, 2]; though previously determining the interfacial capacitance from the charge injected, the potential shift achieved, and the surface area of the steel. The authors of the present work have shown that it is possible to estimate, with galvanostatic pulses, reliable values of Rp in RCSs without the need to know the surface area of the rebar, considering an approximate value of the capacitance.
2.5 EIS: An Alternating Current (AC) Technique
Electrochemical Impedance Spectroscopy (EIS), is an advantageous technique for on-site measurement of corrosion rate in Contemporary Built Heritage. The use of EIS, represents a non-destructive technique that allows to elucidate corrosion process mechanisms.
The EIS measurements determine the impedance magnitude (Z) of an electrochemical system, which allows to obtain polarization resistance values [12,13,14,15]. Therefore corrosion rate is determined for steel reinforcements in concrete in a non-destructive way.
The application of small potential (E) excitation signals, using alternating current over a wide frequency range (ω), allows to obtain the impedance value. The Z value is composed of a real (Zreal) and an imaginary (Zimag) part, (Eq. 11). Graphical chart includes real part plotted on x-axis and negative imaginary part on y-axis, resulting on the so-called Nyquist plot. Each point on the plot will represent the impedance Z value for a given single frequency (Fig. 10).

a Nyquist diagram obtained using EIS, a non-destructive AC-electrochemical technique. b Ranldes electrical equivalent circuit (EEC), use to model electrochemical impedance response of the interface
On the Nyquist plot the impedance can be represented as a vector of length |Z|. The angle between this vector and the x-axis is the phase angle ∅.
Low frequency data are on the right side of the plot and higher frequencies are on the left. This is true for EIS data where impedance usually falls as frequency rises (this is not true of all circuits).
The Nyquist plot represents the results from the RC circuit [16, 17]. The obtained semicircle is characteristic of a single time constant. Electrochemical Impedance plots often contain several time constants. Often only a portion of one or more of their semicircles is seen.
The electrochemical double layer is formed because the presence of an interface. For steel in concrete, interfaces are the result of different nature composition system, in this case, contact area of steel reinforcements and concrete contitutes an interface, but also corrosion products will enable extra interfaces, between those and the steel and also in between corrosion products and the concrete itself [18]. Double-layers at interfaces behave as an electrical circuits, thus when an external potential is applied they get charged with time-delay between its input and output.
Electrochemical interfaces they work as capacitors, therefore when potential (V) signals are applied to a discharged capacitor, the capacitor will charge while if voltage is release, the capacitor discharges in the opposite direction. The accumulated charge (Q) on the capacitor is obtained by Eq. 12:
The time delay between charging either discharging process is known as the time constant (Ƭ) of the electric circuit for each single electrochemical system. The resultant time constant depends upon the reactive components either capacitive or inductive connected to it and is a measurement of the response time. This transient response time constant is obtained by Eq. 13:
Considering transient response of the system, it appears to be crucial achieving and steady-state situation to study the steel-concrete system, in order to get robust and reproducible results. A common problem in EIS measurements and their analysis is drift in the system being measured. In practice, a steady state can be difficult to achieve (Fig. 11). The electrochemical cell can change through adsorption of solution impurities, growth of an oxide layer, build-up of reaction products in solution, coating degradation, temperature changes, diffusion processes, among others. EIS analysis tools may provide inaccurate results on a system, if measurements are performed on non-steady-state conditions.

Nyquist plot obtained using EIS technique for steel reinforced concrete
In polarization processes on electrochemical systems, applied excitation potential (Vc), has a direct relationship with capacitor charge, Vc = Q/C. If transient response is considered, the potential across the electrochemical double layer, considered as a capacitor (Vc), presents a narrow dependence on the applied external potential (Vext) at any instant in time during the charging period is given as (Eq. 14):
where
- V c :
-
is the voltage across the capacitor
- V s :
-
is the supply voltage
- t:
-
is the elapsed time since the application of the supply voltage
- CR p :
-
is the time constant (Ƭ) of the RC charging circuit.
After a period equivalent to 4 time constants, (4 Ƭ) the capacitor in this RC charging circuit is virtually fully charged and the voltage across the capacitor is now approx. 98% of its maximum value, 0.98 × Vs. The time period taken for the capacitor to reach this 4 Ƭ point is known as the transient period.
After a time of 5T the capacitor is now fully charged and the voltage across the capacitor, (Vc) is equal to the supply voltage, (Vs). As the capacitor is fully charged no more current flows in the circuit. The time period after this 5 Ƭ point is known as the steady state.
This time constant represents the value depending on type of electrochemical process involved in the corrosion reaction. Therefore, determination of the time constant will allow elucidation of polarization resistance (Rp). According to Stern and Geary equation [5], that Rp will provide a corrosion rate value.
3 Corrosion Control in Built Heritage
By retarding either the anodic or cathodic reactions the rate of corrosion can be reduced, thus avoiding corrosion products to form on the metal surface (Fig. 12). This can be achieved in several ways.

Corroded rebars showing the oxide scale
3.1 Conditioning the Metal
This can be sub-divided into two main groups: (a) Surface treatment and coating the metal, in order to interpose a corrosion resistant coating between metal and environment. The coating may consist of: another metal, e.g. zinc or tin coatings on steel, a protective coating derived from the metal itself, anodic coating or protective native oxide, organic coatings, such as resins, polymers, paints, enamel, oils and greases. The action of protective coatings is often more complex than simply providing a barrier between metal and environment [19]. Paints may contain a corrosion inhibitor. By galvanizing process, zinc coating in iron or steel confers cathodic protection. (b) Alloying the metal to produce a more corrosion resistant alloy, e.g. stainless steel, in which ordinary steel is alloyed with chromium and nickel. Stainless steel is protected by an invisibly thin, naturally formed film of chromium oxide Cr2O3.
3.2 Conditioning the Corrosive Environment
Influencing the cathodic reaction (oxygen reduction) from water systems in the pH range 6.5–8.5 provides a solution for corrosion to be absent. The removal of oxygen could be achieved by the use of strong reducing agents e.g. sulfite. However, for open evaporative cooling systems this approach to corrosion prevention is not practical since fresh oxygen from the atmosphere will have continual access.
The use of corrosion inhibitors compounds is an effective and cost viable solution for corrosion control. Corrosion inhibitors are chemical additives, whose, when added to a corrosive environment, reduce the corrosion rate [20].
Corrosion inhibitors work in one of the following ways:
Anodic inhibitors, as the name implies an anodic inhibitor interferes with the anodic process, dissolution of metal.
If an anodic inhibitor is not present at a concentration level sufficient to block off all the anodic sites, localized attack such as pitting corrosion can become a serious problem due to the oxidizing nature of the inhibitor which raises the metal potential and encourages the anodic reaction (Eq. 15). Other examples of anodic inhibitors include orthophosphate, nitrite, ferricyanide and silicates.
Cathodic inhibitors, the major cathodic reaction in corrosion processes is the reduction of oxygen, (Eq. 16).
There are other cathodic reactions and additives that suppress these reactions called cathodic inhibitors. They function by reducing the available area for the cathodic reaction. This is often achieved by precipitating an insoluble species onto the cathodic sites. Zinc ions are used as cathodic inhibitors because of the precipitation of Zn(OH)2 at cathodic sites as a consequence of the localized high pH.
Mixed inhibitors, because of the danger of pitting when using anodic inhibitors alone, it became common practice to incorporate a cathodic inhibitor into formulated performance was obtained by a combination of inhibitors interfering both anodic and cathodic reactions [21]. This corrosion inhibition process is generally referred to a ‘synergism’ and demonstrates the synergistic action which exists between zinc and chromate ions.
Other classification refers to adsorption type corrosion inhibitors. Many organic corrosion inhibitors work by an adsorption mechanism. The resultant film of chemisorbed inhibitor is then responsible for protection either by physically blocking the surface from the corrosion environment or by retarding the electrochemical processes. The main functional groups capable of forming chemisorbed bonds with metal surfaces are amino (–NH2), carboxyl (–COOH), and phosphonate (–PO3H2) although other functional groups or atoms can form co-ordinate bonds with metal surfaces.
3.3 Electrochemical Control
Since corrosion is an electrochemical process its progress may be studied by measuring the changes which occur in metal potential with time or with applied electrical currents. Conversely, the rate of corrosion reactions may be controlled by passing anodic or cathodic currents into the metal. If, for example, electrons are passed into the metal and reach the metal/electrolyte interface (a cathodic current) the anodic reaction will be stifled while the cathodic reaction rate increases. This process is called cathodic protection and can only be applied if there is a suitable conducting medium such as earth or water through which a current can flow to the metal to be protected in most soils or natural waters corrosion of steel is prevented if the potential of the metal surface is lowered by 300 or 400 mV. Cathodic protection may be achieved by using a DC power supply (impressed current) or by obtaining electrons from the anodic dissolution of a metal low in the galvanic series such as aluminum, zinc or magnesium (sacrificial anodes). Similar protection is obtained when steel is coated with a layer of zinc. Even at scratches or cut edges where some bare metal is exposed the zinc is able to pass protective current through the thin layer of surface moisture. In certain chemical environments it is sometimes possible to achieve anodic protection, passing a current which takes electrons out of the metal and raises its potential. Initially this stimulates anodic corrosion, but in favorable circumstances this will be followed by the formation of a protective oxidized passive surface film.
References
Bastidas DM et al (2007) A quantitative study of concrete-embedded steel corrosion using potentiostatic pulses. Corrosion 63:1094–1100
González JA (2007) F.N. Speller Award lecture: prediction of reinforced concrete structure durability by electrochemical techniques. Corrosion 63:811–818
Pourbaix M (1974) Atlas of electrochemical equilibria in aqueous solutions, 2nd English edn. National Association of Corrosion Engineers, Houston, TX, USA
ASTM G59–97 (2014) Standard test method for conducting potentiodynamic polarization resistance measurements. ASTM International, West Conshohocken, PA, USA
Stern M, Geary AL (1957) Electrochemical polarization I. A theoretical analysis of the shape of polarization curves. J Electrochem Soc 104:56–63
Wagner C, Traud W (1938) The analysis of corrosion procedures through the interaction of electrochemical partial procedures and on the potential difference of mixed electrodes. Z Elektroch 44:391–402
Evans UR (1946) Metallic corrosion, passivation and protection. In: Arnold E (ed). London, UK
Birbilis N, Cherry BW, Forsyth M, Nairn KM (2001) A consideration of the limitation of polarization resistance method to determine corrosion status of concrete reinforcement. Proc Conf Eng Mater, 277–282
Scully JR (2000) Polarization resistance method for determination of instantaneous corrosion rates. Corrosion 56:199–218
Bastidas DM, Medina E (2013) Armaduras de acero inoxidable “Stainless steel reinforcements”. Cedinox (ed). Madrid, Spain
Bastidas DM et al (2008) Electrochemical rehabilitation methods for reinforced concrete structures: advantages and pitfalls. Corros Eng Sci Technol 43:248–255
Cano E, Lafuente D, Bastidas DM (2010) Use of EIS for the evaluation of the protective properties of coatings for metallic cultural heritage: a review. J Solid State Electrochem 14:381–391
Feliu V, González JA, Feliu S (2007) Corrosion estimates from the transient response to a potential step. Corros Sci 49:3242–3255
Feliu V, González JA, Feliu S (2004) Algorithm for extracting corrosion parameters from the response of the steel-concrete system to a current pulse. J Electrochem Soc 151:B134–B140
González JA, Miranda JM, Feliu S (2004) Considerations on reproducibility of potential and corrosion rate measurements in reinforced concrete. Corros Sci 46:2467–2485
Feliu V, González JA, Andrade C, Feliu S (1998) Equivalent circuit for modelling the steel-concrete interface. I. Experimental evidence and theoretical predictions. Corros Sci 40:975–993
Feliu V, González JA, Andrade C, Feliu S (1998) Equivalent circuit for modelling the steel-concrete interface. II. Complications in applying the Stern-Geary equation to corrosion rate determinations. Corros Sci 40:995–1006
González JA, Feliu S, Rodríguez P (1997) Threshold steel corrosion rates for durability problems in reinforced structures. Corrosion 53:65–71
Bastidas DM et al (2015) Corrosion inhibition mechanism of phosphates for early-age reinforced mortar in the presence of chlorides. Cem Concr Comp 61:1–6
Bastidas DM et al (2010) A prediction study of hydroxyapatite entrapment ability in concrete. Constr Build Mater 24:2646–2649
Bastidas DM et al (2013) Comparative study of three sodium phosphates as corrosion inhibitors for steel reinforcements. Cem Concr Comp 43:31–38
Acknowledgements
Authors gratefully acknowledge The University of Akron for funding support.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Bastidas, D.M., Coleman, S. (2018). On-Site Corrosion Diagnosis and Its Control by Electrochemical Techniques in Contemporary Built Heritage. In: Bastidas, D., Cano, E. (eds) Advanced Characterization Techniques, Diagnostic Tools and Evaluation Methods in Heritage Science. Springer, Cham. https://doi.org/10.1007/978-3-319-75316-4_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-75316-4_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-75315-7
Online ISBN: 978-3-319-75316-4
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)