
Abstract
The renin-angiotensin-aldosterone system (RAAS) represents one of the most important contributors to vascular, cardiac, and renal pathology. Its major effector peptides, notably the octapeptide angiotensin II (AngII), induce vasoconstriction (directly and indirectly via sympathetic activation and vasopressin release) and participate in processes of vascular and cardiac hypertrophy, remodeling, inflammation, and fibrosis, directly through action on the angiotensin AT1 receptor and indirectly through aldosterone stimulation. In addition, the RAAS engenders renal salt retention and is involved in various renal pathologies. By the above mechanisms, the RAAS contributes to the development and maintenance of arterial hypertension. RAAS-induced vascular, cardiac, and renal pathologies occur already in the prehypertensive state, i.e., before overt hypertension has developed. Recently, a so-called protective arm of the renin-angiotensin system (RAS) has been described including angiotensin-converting enzyme 2 (ACE2), the angiotensin AT2 receptor, and the Ang 1–7/Mas receptor system. The “protective RAS” appears to represent an intrinsic opponent of the classical “harmful RAS,” and stimulation of this system may become an important therapeutic approach in individuals at cardiovascular risk.
Inhibitors of the “harmful” RAAS (ACE inhibitors, AT1 receptor, and aldosterone receptor antagonists) have been successfully introduced into the clinic with a broad spectrum of indications, particularly as antihypertensive agents. The TROPHY study with the AT1 receptor blocker, candesartan, and in the PHARAO study with the ACE inhibitor, ramipril, have provided evidence that the natural course from prehypertension to hypertension can be delayed, supporting the idea that in certain patient populations at risk, chronic antihypertensive treatment with an inhibitor of the RAS started already in the prehypertensive phase may exert protection by preventing the onset of high blood pressure disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
The renin-angiotensin system (RAS) plays a central role in blood pressure regulation. The main effector peptides of this system, the octapeptide angiotensin II (Ang II; Ang 1–8) and the heptapeptide angiotensin III (Ang III; Ang 2–8), act at least on four different receptor subtypes (ATR 1–4). Most of the classical angiotensin actions are mediated by the AT1 receptor (AT1R). They include generalized vasoconstriction, increased release of noradrenaline, stimulation of proximal tubular reabsorption of sodium ions, secretion of aldosterone from the adrenal cortex, and cell growth in the arterial wall and in the heart [1]. Ang II induces endothelial dysfunction, activates prooxidant and proinflammatory processes, and promotes cardiovascular remodeling, thus contributing to vascular tone regulation as well as to the development and progression of hypertension [2, 3].
In the past two decades, novel RAS peptides and receptors have been identified, including the angiotensin AT2 receptor (AT2R), angiotensin-converting enzyme 2 (ACE2), and Ang (1–7) with its G-protein-coupled receptor Mas. The AT2R and the MasR form heterodimers and are functionally closely related [4]. These components are considered as the “protective arm” of RAS because they mainly activate opposing actions compared to those mediated by the AT1R.
Prehypertension is characterized by functional and structural changes in the microcirculation. A reduction of small arterial elasticity, the earliest predictor for hypertension development [5], along with endothelial dysfunction, nitric oxide deficiency, accumulation of extracellular matrix, and inflammation, contributes to early vascular remodeling. Increased circulating and local expression of RAS components in the vasculature and subsequently enhanced Ang II production are involved in these pathological processes [6]. Experimental studies in “prehypertensive” rats provided first evidence for the unique effect of RAS interaction on vasculature and blood pressure: Inhibiting the RAS by ACE inhibitors or angiotensin AT1 receptor antagonists (ARBs) prevented the progression of hypertension and vascular remodeling in young “prehypertensive” spontaneously hypertensive rats (SHR) [7,8,9,10,11,12]. Later on, investigations in humans [13,14,15] provided evidence that interfering with the RAS not only lowered blood pressure but also improved vascular factors determining vascular tone.
The present overview deals with the role of “harmful” and “protective” arms of RAS in prehypertension particularly in the context of early vascular remodeling. Furthermore, studies on pharmacological blockade of the RAS in prehypertensive humans are discussed.
2 Classical Renin-Angiotensin System
2.1 AT1 Receptor
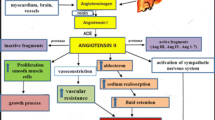
Ang II constricts precapillary arterioles by activating AT1 receptors of vascular smooth muscle cells (VSMC). Direct vasoconstriction in the kidney leads to reduced renal flow and subsequent efferent arteriole constriction resulting in increased filtration pressure. Blood pressure-driven diuresis and sodium excretion generate a feedback loop on renin release. Furthermore, Ang II facilitates peripheral noradrenergic neurotransmission by augmenting norepinephrine release from sympathetic nerve terminals and by enhancing the vascular response to norepinephrine. This facilitating effect is mediated by presynaptically localized AT1 receptors [16]. Expression of endothelin-1 in response to Ang II also contributes to vasoconstriction [17] (Fig. 22.1).

Mechanisms of RAS—mediated prehypertension. Ang angiotensin, AT1R angiotensin AT1 receptor, AT2R angiotensin AT2 receptor, ACE angiotensin converting enzyme, ALDOS aldosterone synthase, MR mineralocorticoid receptor, NADPH Nicotinamidadenindinucleotidphosphat, NO nitric oxide, NEP neprilysin, VR vasopressin receptor, V1R type 1 vasopressin receptor, Mas R Mas receptor
The pathophysiological mechanisms of vascular remodeling are attributed to an Ang II-dependent increase of NAD(P)H oxidase activity via the AT1R in endothelial and VSMCs [18, 19], thereby stimulating reactive oxygen species (ROS) and nitrogen (RNS) formation in the vessel wall [20]. ROS products such as superoxide and H2O2 may activate mitogen-activated protein kinases, tyrosine kinases, phosphatases, calcium channels, and redox-sensitive transcription factors [20]. Activation of these signaling pathways results in cell growth and expression of proinflammatory genes.
Above hypertrophic effects on the vascular wall, actions of Ang II mediated by ROS include vasoconstriction and decreased vasodilatation. The ROS, which is generated especially by NAD(P)H oxidase, causes lipid peroxidation and generation of various vasoconstricting molecules such as F2 isoprostanes. On the other hand, ROS/RNS reduce the availability of the major vasodilator NO by reacting with superoxide [21].
Furthermore, via AT1R activation, Ang II controls cellular growth, migration, and intercellular matrix deposition and hence influences chronic adaptive changes in vascular growth and remodeling. Ang II stimulates the accumulation of extracellular matrix proteins, like collagen, elastin, fibrillin, fibronectin, and proteoglycans, which induce a phenotype switch in VSMC from contractile to proliferative/synthetic [22].
2.2 Vasopressin
Acting on AT1 receptors in hypothalamus and brainstem, Ang II or Ang III influence drinking behavior, sodium intake, natriuresis, and vasopressin release [23]. Vasopressin, an antidiuretic hormone, induces volume expansion followed by elevation of blood pressure. The pressor and antidiuretic actions are mediated by different vasopressin receptor subtypes, V1a, V1b, and the V2 receptors (V1aR, V1bR, V2R). The V1aR are expressed abundantly in the vascular smooth muscle cells, and their stimulation is responsible for the vasopressor effect. Blockade of the V1aR for 4 weeks in prehypertensive SHR could attenuate the development of hypertension in adult SHR [24]. This was recently supported by an increase of plasma vasopressin and of renal V1aR gene and protein expressions parallel to hypertension development in SHR [25]. However, in well-hydrated volunteers and in patients with a mild form of essential hypertension, V1R blockade did not alter blood pressure [26, 27]. Thus, the potential contribution of vasopressin to the development of hypertension from prehypertension requires further investigations.
2.3 Aldosterone
In 1958, Franz Gross postulated a physiological link between the RAS and aldosterone secretion in the zona glomerulosa of the adrenal gland [28]. Later on, several groups of investigators confirmed that Ang II stimulates aldosterone secretion [29]. Aldosterone, the primary mineralocorticoid, acts via the mineralocorticoid receptors (MR) in the kidneys and plays a central role in the regulation of blood pressure, blood volume, and salt household. Importantly, aldosterone contributes to the pathogenesis of hypertension beyond primary aldosteronism via several pathogenetic pathways, e.g., renal sodium and water retention, increased peripheral resistance, and stimulation of the sympathetic nervous system [30]. Since aldosterone levels within the upper part of the physiological range predispose normotensive subjects to the development of hypertension [31], it can be assumed that aldosterone also contributes to prehypertension.
The effects of aldosterone on blood pressure regulation extend beyond increased intravascular fluid retention and volume overload. Aldosterone modulates vascular tone by upregulation of the AT1R, by limiting bioavailability of endothelial NO, by increasing pressor responses to catecholamines, and by impairing the vasodilatory response to acetylcholine [32]. In addition, aldosterone excess activates inflammation and oxidative stress alters fibrinolysis by increasing plasminogen activator inhibitor-1 expression [33] and promotes vascular hypertrophy followed by increased arterial stiffness [34]. All these cellular pathways, regulated by aldosterone via the MR and by Ang II via its AT1R, can reinforce each other [35].
In an experimental model of prehypertension in young SHR, treatment with the MR antagonist, spironolactone resulted in prolonged blood pressure reduction and decreased collagen deposition [36]. Nevertheless, compared to the AT1R antagonist, losartan, the transient effect of spironolactone treatment was less impressive.
3 “Protective” Arm of the RAS
Recently, attention has been paid to the “protective” arm of RAS [37] that consists of several angiotensin peptides and their fragments and receptors with actions at least partly opposing the classical RAS concept. Some of these angiotensin peptides, related enzymes, and receptors are of particular interest because they play a protective role in the cardiovascular system.
Angiotensin-converting enzyme 2 (ACE2) has been described to be a potent negative regulator of the RAS, counterbalancing the multiple functions of ACE [38]. ACE2 converts the decapeptide, angiotensin I, to angiotensin Ang (1–9), which can be further converted by ACE to a shorter peptide, Ang (1–7). Alternatively, Ang (1–7) can also be formed directly from Ang I via neutral endopeptidase (NEP, neprilysin). Interestingly, in prehypertensive SHR, the ACE2 levels are reduced [39].
Ang (1–7) evokes a range of acute central and peripheral effects such as vasodilatation, inhibition of VSMC proliferation, and inhibition of vasopressin release [40]. Although some of these effects depend on the acute activation of eNOS or inhibition of NADPH oxidase [41, 42], others may point to a potential role of Ang (1–7) in endothelial regeneration [43].
Furthermore, Ang (1–7) is known to be the endogenous ligand for the Mas receptor, a seven-transmembrane domain G-protein-coupled receptor sharing a 31% sequence identity with the AT2R [38, 44]. Other studies have suggested that the Mas receptor can heterodimerize with AT1R to inhibit the effects of Ang II [45]. A recent study shows heterodimerization and close functional relationship of the Mas R and the AT2R [4]. Mas receptor activation promotes often opposing effects to those of the AT1R such as anti-inflammation, antiproliferation [46], and blood pressure reduction as shown in DOCA-salt-induced hypertension in rats [47].
Ang IV (3–8) is formed via the cleavage of Ang III (Ang 2–8) by aminopeptidase B or N. Ang IV was reported to activate anti-inflammation and antiproliferation through a poorly defined AT4R and to induce vasodilatation and vascular protection via eNOS activation and subsequent NO release [48]. In addition, chronic treatment with Ang IV improved endothelial dysfunction in ApoE-deficient mice. This vasoprotective effect most likely resulted from increased NO bioavailability [49].
The angiotensin AT2 receptor (AT2R) is much less expressed under basal conditions compared to the AT1R. However, in cardiovascular diseases, such as hypertension or left ventricular hypertrophy, the AT2R expression is upregulated [3, 50]. The AT2R is a seven-transmembrane domain G-coupled receptor [51] that acts via several intracellular signaling pathways such as NO/cGMP activation [52], inhibition of mitogen-activated protein kinases (MAPKs) by protein phosphatases [53], phospholipase A2 stimulation [54], or disruption of AT1R signaling by AT1R-AT2R heterodimerization [55]. Similar to the MasR, AT2R activation promotes often opposing effects to those of the AT1R such as anti-inflammation, vasodilatation, and cell proliferation [1]. Activated AT2R also inhibits sympathetic activity [56] and through the phosphorylation of MAP kinase counteracts AT1R-mediated actions [57]. Notably, the AT2R mediates activation of bradykinin/NO/cGMP system in endothelial cells [58], in the heart [59] and in the aorta of prehypertensive stroke-prone spontaneously hypertensive rats (SHR-SP) [52]. In SHR-SP, the AT2-mediated increase in aortic cGMP is mediated by bradykinin B2 receptors, which activate NO synthase, followed by NO production and formation of the cGMP. cGMP, in turn, exerts antihypertensive and tissue protective effects such as vasodilatation, natriuresis, and antigrowth [60]. In addition, AT2 knockout mice have slightly elevated blood pressure, low basal levels of renal bradykinin and cGMP, as well as low NO production [61]. Conversely, AT2 receptor overexpression activated the vascular kinin system and caused vasodilatation [62]. In humans, the AT2-mediated vasorelaxation has been directly demonstrated in isolated coronary artery [63] and gluteal vasculatures [64]. Whereas acute vasodilator role of AT2R is well described, chronic decrease of blood pressure seems to be minimal after AT2R stimulation [65, 66].
Nevertheless, the AT2R has consistently been shown to be important in the prevention of vascular remodeling. In experimental studies performed in prehypertensive rats, AT2R stimulation with a selective AT2R agonist, compound 21 [67], reduced vascular fibrosis [68] and improved endothelial function and vascular composition by reducing oxidative stress, collagen content, fibronectin, and inflammatory cell infiltration [69]. AT2R stimulation also protected against nephropathy in doxorubicin-treated rats [70] and in 2K1C hypertension [71]. Furthermore, in a mouse model of type 1 diabetes, AT2R showed microvascular vasodilator properties [72].
In addition, AT2R exerts an anti-remodeling effect with regard to atherosclerotic lesions [73] and neointimal formation [74]. Iwai and colleague [75] demonstrated that AT2R/ApoE-double knockout mice fed a high-cholesterol diet display exaggerated atherosclerotic lesion development parallel with increased NADPH oxidase activity and superoxide production when compared to ApoE knockout mice. In humans, AT2Rs are expressed in the atherosclerotic and aneurysmatic lesions being mainly localized in the endothelium of vasa vasorum [76].
Taken collectively, an AT2 receptor-mediated increase in production of vasodilators (nitric oxide, cGMP), as well as the antigrowth and antifibrotic and anti-inflammatory features of this receptor, might contribute to blood pressure lowering and prevent remodeling in prehypertension.
4 Pharmacological Blockade of the RAS in Prehypertension
In view of the above-described contribution of the RAS to pathological changes in the vasculature and other target organs, given the availability of pharmacological inhibitors of this system and stimulated by experimental data in spontaneous hypertensive rats [7, 11, 12], the idea was borne to delay or even prevent the development of hypertension in prehypertensive individuals via pharmacological blockade of the RAS.
These considerations led to the conception of the so-called TROPHY (Trial of Preventing Hypertension) study “Feasibility of treating prehypertension with an angiotensin-receptor blocker” by Stevo Julius and colleagues [14]. The aim of this clinical trial was to investigate “… whether pharmacological treatment of prehypertension prevents or postpones stage 1 hypertension.”
Participants with systolic blood pressure, between 130 and 139 mmHg and diastolic blood pressures of 89 mmHg or lower, were treated for 2 years with the angiotensin AT1 receptor blocker, candesartan, or with placebo followed by placebo for 2 years for both groups. When a participant became hypertensive (stage 1), he or she was continued on candesartan. Advice for “healthy living” to reduce blood pressure was given to both groups throughout the study.
Data from 772 participants could be analyzed, roughly half and with respect to groups. During the first 2 years, 154 participants reached the endpoint in the placebo group, compared to only 53 participants in the candesartan group, corresponding to a risk reduction of more than 66%. After 4 years, 240 individuals had developed hypertension in the placebo group, compared to 208 in the candesartan group. Thus, there was still a significant risk reduction of 16% in the group that had been started on candesartan.
The results of this trial demonstrate, first, that prehypertension can indeed be considered a precursor of hypertension in a substantial number of individuals (nearly two thirds) and, second, that a period of early intervention with an inhibitor of the RAS can delay the appearance of hypertension.
While the design of this study appeared relatively straightforward and the results on first glance quite clear, TROPHY fueled a lot of discussion and received positive as well as negative critiques.
On the negative side, the authors were criticized for using an “odd clinical endpoint” [77]. Without going into too much detail here, this point was answered by the authors in a reappraisal of their outcome data using the criteria of the “Joint National Committee on Hypertension (JNC)” [78]. There were only very minor differences between this analysis and that of the original report.
Even more serious was the criticism that TROPHY did, according to scenarios developed by its authors in an interim report [79], not prevent or delay the development of hypertension but instead caused a “slow unmasking” of hypertension [77]. Indeed, although the endpoints in both groups were still significantly different with less incidence of hypertension in the candesartan group, the slope of the cumulative incidence curve rose promptly after 2 years when candesartan treatment was replaced by placebo. Continuing on their respective slopes, the curves of both groups would have probably met after another 2 years or so. Thus, the study did indeed not show that hypertension can be prevented by a transient pharmacological intervention but that it can be delayed. The authors, although playing with the thought of prevention on several occasions, did not make this claim in the abstract of their original paper but just mention that “treatment with candesartan reduced the risk of incident hypertension during the study period” which is certainly not over-interpreting the data.
The authors further conclude cautiously that “treatment of prehypertension is feasible.” This statement, too, is justified by the data of their study, but does it make sense, clinically? Would it imply that, if taken seriously, 25 million prehypertensive US Americans would have to be treated pharmacologically with an inhibitor of the RAS notwithstanding the “rest of the world”? Would the usual lifestyle adaptations (weight loss, salt restriction, exercise and dietary modifications) as more or less authoritatively advocated around the world not have the same effect without “chemistry”?
Kjeldsen et al. [80] argue against this by alluding to the fact that the prevalence of prehypertension has increased despite intensive efforts to promote such healthy lifestyles [81]. They argue further that, just taking the US American population, of the 25 million US Americans with TROPHY-like blood pressures, almost 16 million will become hypertensive over the next 4 years according to the experience from the TROPHY placebo group. Should one not intervene as early as possible in these individuals given the fact that prehypertension already carries pathological abnormalities in cardiovascular structure and function? If one follows this argument, the question is not any more whether or not it is possible to delay the onset of hypertension by transient pharmacological intervention, but to prevent hypertension altogether by early-onset, continuous treatment in prehypertensive individuals. Kjeldsen et al. [80] deliver a strong argument in favor of such early intervention: If one uses the absolute difference in risk reduction between groups in TROPHY, one can calculate that four individuals with prehypertension need to be treated to prevent one case of hypertension in 2 years.
Two years after TROPHY, another clinical study, named PHARAO (Prevention of Hypertension with the Angiotensin-converting enzyme inhibitor RAmipril in patients with high-nOrmal blood pressure), was published [15].
The objective was quite similar to the one in TROPHY, namely, to address “whether the progression to manifest hypertension in patients with high-normal blood pressure can be prevented with treatment.” The study included 505 individuals in the ramipril and 503 individuals in the placebo group, lasted 3 years and, in addition, used ambulatory blood pressure monitoring to confirm the diagnosis of hypertension. After 3 years of treatment, 153 individuals in the ramipril group (30.7%) and 216 (42.9%) in the placebo group reached the primary endpoint (relative risk reduction 34.4%; p < 0.0001). Ramipril also reduced the incidence of office hypertension in participants with high-normal blood pressure established by ambulatory blood pressure monitoring (ABPM). The authors concluded that “treatment of patients with high-normal office blood pressure with the angiotensin-converting enzyme inhibitor was well tolerated, and significantly reduced the risk of progression to manifest hypertension.” Analysis of the data further revealed that ramipril not only shifted the incidence of manifest hypertension downward in a parallel manner to the placebo group but, in addition, diminished the slope of the graph during the treatment period. This was interpreted to mean that the ACE inhibitor not only lowered blood pressure per se but also interfered with the vascular or neurohumoral factors determining vascular tone.
As with the angiotensin AT1 receptor blocker, candesartan, in TROPHY, it is now a member of another class of RAS inhibitors, the ACE inhibitor ramipril, which yielded such a preventive antihypertensive effect suggesting that a specific interference with the harmful arm of the RAS would reduce the risk toward manifest hypertension. However, in the absence of comparable studies with other classes of antihypertensive drugs, this idea, despite its theoretical plausibility, remains speculative.
References
Unger T. The angiotensin type 2 receptor: variations on an enigmatic theme. J Hypertens. 1999;17(12 Pt 2):1775–86.
Kaschina E, Unger T. Angiotensin AT1/AT2 receptors: regulation, signalling and function. Blood Press. 2003;12:70–88.
Namsolleck P, Recarti C, Foulquier S, Steckelings UM, Unger T. AT(2) receptor and tissue injury: therapeutic implications. Curr Hypertens Rep. 2014;16:416.
Leonhardt J, Villela DC, Teichmann A, Münter LM, Mayer MC, Mardahl M, et al. Evidence for heterodimerization and functional interaction of the angiotensin type 2 receptor and the receptor MAS. Hypertension. 2017;69(6):1128–35.
Peralta CA, Adeney KL, Shlipak MG, Jacobs D Jr, Duprez D, Bluemke D, et al. Structural and functional vascular alterations and incident hypertension in normotensive adults: the Multi-Ethnic Study of Atherosclerosis. Am J Epidemiol. 2010;171:63–71.
Paul M, Poyan Mehr A, Kreutz R. Physiology of local renin-angiotensin systems. Physiol Rev. 2006;86(3):747–803.
Harrap SB, Van der Merwe WM, Griffin SA, Macpherson F, Lever AF. Brief angiotensin converting enzyme inhibitor treatment in young spontaneously hypertensive rats reduces blood pressure long-term. Hypertension. 1990;16:603–14.
Lundie MJ, Friberg P, Kline RL, Adams MA. Long-term inhibition of the renin-angiotensin system in genetic hypertension: analysis of the impact on blood pressure and cardiovascular structural changes. J Hypertens. 1997;15:339–48.
Lee RM, Berecek KH, Tsoporis J, McKenzie R, Triggle CR. Prevention of hypertension and vascular changes by captopril treatment. Hypertension. 1991;17:141–50.
Unger T, Mattfeldt T, Lamberty V, Bock P, Mall G, Linz W, Schölkens BA, Gohlke P. Effect of early onset angiotensin converting enzyme inhibition on myocardial capillaries. Hypertension. 1992;20:478–82.
Wu JN, Berecek KH. Prevention of genetic hypertension by early treatment of spontaneously hypertensive rats with the angiotensin converting enzyme inhibitor captopril. Hypertension. 1993;22:139–46.
Unger T, Rettig R. Development of genetic hypertension. Is there a “critical phase”? Hypertension. 1990;16(6):615.
Schiffrin EL, Deng LY, Larochelle P. Effects of a beta-blocker or a converting enzyme inhibitor on resistance arteries in essential hypertension. Hypertension. 1994;23:83–91.
Julius S, Nesbitt SD, Egan BM, Weber MA, Michelson EL, Kaciroti N, et al. Trial of Preventing Hypertension (TROPHY) Study Investigators. Feasibility of treating prehypertension with an angiotensin-receptor blocker. N Engl J Med. 2006;20(354):1685–97.
Lüders S, Schrader J, Berger J, Unger T, Zidek W, Böhm M, et al. The PHARAO study: prevention of hypertension with the angiotensin-converting enzyme inhibitor ramipril in patients with high-normal blood pressure: a prospective, randomized, controlled prevention trial of the German Hypertension League. J Hypertens. 2008;26:1487–96.
Balt JC, Mathy MJ, Pfaffendorf M, van Zwieten PA. Sympatho-inhibitory properties of various AT1 receptor antagonists. J Hypertens Suppl. 2002;20:S3–11.
Hahn AW, Regenass S, Kern F, Bühler FR, Resink TJ. Expression of soluble and insoluble fibronectin in rat aorta: effects of angiotensin II and endothelin-1. Biochem Biophys Res Commun. 1993;192:189–97.
Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994;74:1141–8.
Zafari AM, Ushio-Fukai M, Akers M, Yin Q, Shah A, Harrison DG, Taylor WR, Griendling KK. Novel role of NADH/NADPH oxidase-derived hydrogen peroxide in angiotensin 11-induced hypertrophy of rat smooth muscle cells. Hypertension. 1998;32:488–95.
Montezano AC, Touyz RM. Reactive oxygen species, vascular Noxs, and hypertension: focus on translational and clinical research. Antioxid Redox Signal. 2014;20:164–82.
Robert Li Y. Free radical biomedicine: principles, clinical correlations, and methodologies: Bentham eBooks; 2012. https://doi.org/10.2174/97816080532231120101.
Lacolley P, Regnault V, Nicoletti A, Li Z, Michel JB. The vascular smooth muscle cell in arterial pathology: a cell that can take on multiple roles. Cardiovasc Res. 2012;15(95):194–204.
Culman J, Baulmann J, Blume A, Unger T. The renin-angiotensin system in the brain: an update. J Renin Angiotensin Aldosterone Syst. 2001;2:96–102.
Burrell LM, Phillips PA, Risvanis J, Aldred KL, Hutchins AM, Johnston CI. Attenuation of genetic hypertension after short-term vasopressin V1A receptor antagonism. Hypertension. 1995;26:828–34.
Burrell LM, Risvanis J, Dean RG, Patel SK, Velkoska E, Johnston CI. Age-dependent regulation of renal vasopressin V(1A) and V2 receptors in rats with genetic hypertension: implications for the treatment of hypertension. J Am Soc Hypertens. 2013;7:3–13.
Bussien JP, Waeber B, Nussberger J, Schaller MD, Gavras H, Hofbauer K, et al. Does vasopressin sustain blood pressure of normally hydrated healthy volunteers? Am J Physiol. 1984;246(1 Pt 2):H143–7.
Waeber B, Nussberger J, Hofbauer KG, Nicod P, Brunner HR. Clinical studies with a vascular vasopressin antagonist. J Cardiovasc Pharmacol. 1986;8(Suppl 7):S111–6.
Gross F. Renin and hypertension, physiological or pathological agents? Klin Wochenschr. 1958;36:693–706.
Weir MR, Dzau VJ. The renin-angiotensin-aldosterone system: a specific target for hypertension management. Am J Hypertens. 1999;12(12 Pt 3):205–13.
Tomaschitz A, Pilz S, Ritz E, Obermayer-Pietsch B, Pieber TR. Aldosterone and arterial hypertension. Nat Rev Endocrinol. 2010;6:83–93.
Vasan RS, Evans JC, Larson MG, Wilson PWF, Meigs JB, Rifai N, et al. Serum aldosterone and the incidence of hypertension in nonhypertensive persons. N Engl J Med. 2004;351:33–41.
Connell JMC, Davies E. The new biology of aldosterone. J Endocrinol. 2005;186(1):1–20.
Schrier RW, Masoumi A, Elhassan E. Aldosterone: role in edematous disorders, hypertension, chronic renal failure, and metabolic syndrome. Clin J Am Soc Nephrol. 2010;5:1132–40.
Briet M, Schiffrin EL. Treatment of arterial remodeling in essential hypertension. Curr Hypertens Rep. 2013;15:3–9.
Lemarié CA, Paradis P, Schiffrin EL. New insights on signaling cascades induced by cross talk between angiotensin II and aldosterone. J Mol Med. 2008;86:673–8.
Baumann M, Megens R, Bartholome R, Dolff S, van Zandvoort M, Smits J, Sruijker-Boudier HA, De Mey J. Prehypertensive renin-angiotensin-aldosterone system blockade in spontaneously hypertensive rats ameliorates the loss of long-term vascular function. Hypertens Res. 2007;30:853–61.
Unger T, Steckelings UM, dos Santos RAS, editors. The protective arm of the renin angiotensin system—functional aspects and therapeutic implications. 1st ed. London: Academic Press, Elsevier; 2015.
Iwai M, Horiuchi M. Devil and angel in the renin-angiotensin system: ACE-angiotensin II AT1 receptor axis vs. ACE2 angiotensin-(1-7)-Mas receptor axis. Hypertens Res. 2009;32:533–6.
Rentzsch B, Todiras M, Iliescu R, Popova E, Campos LA, Oliveira ML, et al. Transgenic angiotensin-converting enzyme 2 overexpression in vessels of SHRSP rats reduces blood pressure and improves endothelial function. Hypertension. 2008;52:967–73.
Santos RA, Ferreira AJ, Verano-Braga T, Bader M. Angiotensin-converting enzyme 2, angiotensin-(1–7) and Mas: new players of the renin-angiotensin system. J Endocrinol. 2013;216:R1–R17.
Sampaio WO, Souza dos Santos RA, Faria-Silva R, da Mata Machado LT, Schiffrin EL, Touyz RM. Angiotensin-(1-7) through receptor Mas mediates endothelial nitric oxide synthase activation via Akt-dependent pathways. Hypertension. 2007;49:185–92.
Bodiga S, Zhong JC, Wang W, Basu R, Lo J, Liu GC, et al. Enhanced susceptibility to biomechanical stress in ACE2 null mice is prevented by loss of the p47(phox) NADPH oxidase subunit. Cardiovasc Res. 2011;91:151–61.
Durik M, Sevá Pessôa B, Roks AJ. The renin-angiotensin system, bone marrow and progenitor cells. Clin Sci (Lond). 2012;123:205–23.
Santos RA, Simoes e Silva AC, Maric C, Silva DM, Machado RP, de Buhr I, et al. Angiotensin-(1–7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci U S A. 2003;100:8258–63.
Kostenis E, Milligan G, Christopoulos A, Sanchez-Ferrer CF, Heringer-Walther S, Sexton PM, et al. G-protein-coupled receptor Mas is a physiological antagonist of the angiotensin II type 1 receptor. Circulation. 2005;111:1806–13.
Villela D, Leonhardt J, Patel N, Joseph J, Kirsch S, Hallberg A, et al. Angiotensin type 2 receptor (AT2R) and receptor Mas: a complex liaison. Clin Sci. 2015;128:227–34.
Singh Y, Singh K, Sharma PL. Effect of combination of renin inhibitor and Mas-receptor agonist in DOCA-salt-induced hypertension in rats. Mol Cell Biochem. 2013;373:189–94.
Patel JM, Martens JR, Li YD, Gelband CH, Raizada MK, Block ER. Angiotensin IV receptor-mediated activation of lung endothelial NOS is associated with vasorelaxation. Am J Physiol. 1998;275(6 Pt 1):L1061–8.
Vinh A, Widdop RE, Drummond GR, Gaspari TA. Chronic angiotensin IV treatment reverses endothelial dysfunction in ApoE-deficient mice. Cardiovasc Res. 2008;77:178–87.
De Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev. 2000;52:415–72.
Zhang H, Han GW, Batyuk A, Ishchenko A, White KL, Patel N, et al. Structural basis for selectivity and diversity in angiotensin II receptors. Nature. 2017;544:327–32.
Gohlke P, Pees C, Unger T. AT2 receptor stimulation increases aortic cyclic GMP in SHRSP by a kinin-dependent mechanism. Hypertension. 1998;31:349–55.
Fischer TA, Singh K, O’Hara DS, Kaye DM, Kelly RA. Role of AT1 and AT2 receptors in regulation of MAPKs and MKP-1 by ANG II in adult cardiac myocytes. Am J Physiol. 1998;275:H906–16.
Nouet S, Nahmias C. Signal transduction from the angiotensin II AT2 receptor. Trends Endocrinol Metab. 2000;11:1–6.
AbdAlla S, Lother H, Abdel-tawab AM, Quitterer U. The angiotensin II AT2 receptor is an AT1 receptor antagonist. J Biol Chem. 2001;276:39721–6.
Gao J, Zhang H, Le KD, Chao J, Gao L. Activation of central angiotensin type 2 receptors suppresses norepinephrine excretion and blood pressure in conscious rats. Am J Hypertens. 2011;24:724–30.
Inagami T, Eguchi S, Numaguchi K, Motley ED, Tang H, Matsumoto T, Yamakawa T. Cross-talk between angiotensin II receptors and the tyrosine kinases and phosphatases. J Am Soc Nephrol. 1999;10(Suppl 11):S57–61.
Wiemer G, Schölkens BA, Wagner A, Heitsch H, Linz W. The possible role of angiotensin II subtype AT2 receptors in endothelial cells and isolated ischemic rat hearts. J Hypertens Suppl. 1993;11:S234–5.
Liu YH, Yang XP, Sharov VG, Nass O, Sabbah HN, Peterson E, Carretero OA. Effects of angiotensin-converting enzyme inhibitors and angiotensin II type 1 receptor antagonists in rats with heart failure. Role of kinins and angiotensin II type 2 receptors. J Clin Invest. 1997;99:1926–35.
Kraehling JR, Sessa WC. Contemporary approaches to modulating the nitric oxide-cGMP pathway in cardiovascular disease. Circ Res. 2017;120:1174–82.
Siragy HM, Inagami T, Ichiki T, Carey RM. Sustained hypersensitivity to angiotensin II and its mechanism in mice lacking the subtype-2 (AT2) angiotensin receptor. Proc Natl Acad Sci U S A. 1999;96:6506–10.
Tsutsumi Y, Matsubara H, Masaki H, Kurihara H, Murasawa S, Takai S, et al. Angiotensin II type 2 receptor overexpression activates the vascular kinin system and causes vasodilation. J Clin Invest. 1999;104:925–93.
Batenburg WW, Garrelds IM, Bernasconi CC, Juillerat-Jeanneret L, van Kats JP, Saxena PR, Danser AH. Angiotensin II type 2 receptor-mediated vasodilation in human coronary microarteries. Circulation. 2004;109:2296–301.
Savoia C, Touyz RM, Volpe M, Schiffrin EL. Angiotensin type 2 receptor in resistance arteries of type 2 diabetic hypertensive patients. Hypertension. 2007;49:341–6.
Widdop RE, Jones ES, Hannan RE, Gaspari TA. Angiotensin AT2 receptors: cardiovascular hope or hype? Br J Pharmacol. 2003;140:809–24.
Steckelings UM, Paulis L, Namsolleck P, Unger T. AT2 receptor agonists: hypertension and beyond. Curr Opin Nephrol Hypertens. 2012;21:142–6.
Wan Y, Wallinder C, Plouffe B, Beaudry H, Mahalingam AK, Wu X, et al. Design, synthesis, and biological evaluation of the first selective nonpeptide AT2 receptor agonist. J Med Chem. 2004;47:5995–6008.
Paulis L, Becker STR, Lucht K, Schwengel K, Slavic S, Kaschina E, et al. Direct angiotensin II type 2 receptor stimulation in Nω-nitro-L-arginine-methyl ester-induced hypertension: the effect on pulse wave velocity and aortic remodeling. Hypertension. 2012;59:485–92.
Rehman A, Leibowitz A, Yamamoto N, Rautureau Y, Paradis P, Schiffrin EL. Angiotensin type 2 receptor agonist compound 21 reduces vascular injury and myocardial fibrosis in stroke-prone spontaneously hypertensive rats. Hypertension. 2012;59(2):291–9.
Hrenák J, Arendášová K, Rajkovičová R, Aziriová S, Repová K, Krajčírovičová K, et al. Protective effect of captopril, olmesartan, melatonin and compound 21 on doxorubicin-induced nephrotoxicity in rats. Physiol Res. 2013;62(Suppl 1):S181–9.
Matavelli LC, Huang J, Siragy HM. Angiotensin AT2 receptor stimulation inhibits early renal inflammation in renovascular hypertension. Hypertension. 2011;57:308–13.
Begorre MA, Dib A, Habchi K, Guihot AL, Bourreau J, Vessieres E, et al. Microvascular vasodilator properties of the angiotensin 2 receptor in a mouse model of type 1 diabetes. Sci Rep. 2017;7:45625.
Sales VL, Sukhova GK, Lopez-Ilasaca MA, Libby P, Dzau VJ, Pratt RE. Angiotensin type 2 receptor is expressed in murine atherosclerotic lesions and modulates lesion evolution. Circulation. 2005;112:3328–36.
Nakajima M, Hutchinson HG, Fujinaga M, Hayashida W, Morishita R, Zhang L, et al. The angiotensin II type 2 (AT2) receptor antagonizes the growth effects of the AT1 receptor: gain-of-function study using gene transfer. Proc Natl Acad Sci U S A. 1995;92:10663–7.
Iwai M, Chen R, Li Z, Shiuchi T, Suzuki J, Ide A, et al. Deletion of angiotensin II type 2 receptor exaggerated atherosclerosis in apolipoprotein E-null mice. Circulation. 2005;112:1636–43.
Kaschina E, Scholz H, Steckelings UM, Sommerfeld M, Kemnitz UR, Artuc M, Schmidt S, Unger T. Transition from atherosclerosis to aortic aneurysm in humans coincides with an increased expression of RAS components. Atherosclerosis. 2009;205:396–403.
Meltzer JI. A specialist in clinical hypertension critiques the TROPHY trial. Am J Hypertens. 2006;19:1098–100.
Julius S, Kaciroti N, Egan BM, Nesbitt S, Michelson EL. Trial of Preventing Hypertension (TROPHY) Investigators. TROPHY study: outcomes based on the Seventh Report of the Joint National Committee on Hypertension definition of hypertension. J Am Soc Hypertens. 2008;2:39–43.
Julius S, Kjeldsen SE, Weber M, Brunner HR, Ekman S, Hansson L, et al. Outcomes in hypertensive patients at high cardiovascular risk treated with regimens based on valsartan or amlodipine: the VALUE randomised trial. Lancet. 2004;363(9426):2022–31.
Kjeldsen SE, Narkiewicz K, Hedner T. An American TROPHY in the prevention of hypertension. Blood Press. 2006;15:132–4.
Qureshi AI, Suri MF, Kirmani JF, Divani AA. Prevalence and trends of prehypertension and hypertension in United States: National Health and Nutrition Examination Surveys 1976 to 2000. Mred Sci Monit. 2005;11:CR403–9.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Kaschina, E., Unger, T. (2019). Prehypertension and the Renin-Angiotensin-Aldosterone System. In: Zimlichman, R., Julius, S., Mancia, G. (eds) Prehypertension and Cardiometabolic Syndrome. Updates in Hypertension and Cardiovascular Protection. Springer, Cham. https://doi.org/10.1007/978-3-319-75310-2_22
Download citation
DOI: https://doi.org/10.1007/978-3-319-75310-2_22
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-75309-6
Online ISBN: 978-3-319-75310-2
eBook Packages: MedicineMedicine (R0)