
Abstract
Diseases caused by protozoan parasites have a major impact on world health. These early branching eukaryotes cause significant morbidity and mortality in humans and livestock. During evolution, protozoan parasites have evolved toward complex life cycles in multiple host organisms with different nutritional resources. The conservation of functional metabolic pathways required for these successive environments is therefore a prerequisite for parasitic lifestyle. Nevertheless, parasitism drives genome evolution toward gene loss and metabolic dependencies (including strict auxotrophy), especially for obligatory intracellular parasites. In this chapter, we will compare and contrast how protozoan parasites have perfected this metabolic adaptation by focusing on specific auxotrophic pathways and scavenging strategies used by clinically relevant apicomplexan and trypanosomatid parasites to access host’s nutritional resources. We will further see how these metabolic dependencies have in turn been exploited for therapeutic purposes against these human pathogens.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Parasitic protozoa are an evolutionarily divergent group of unicellular eukaryotes that are responsible of a wide range of human and veterinary diseases. The most notorious and clinically relevant protozoan parasites are the apicomplexans Plasmodium spp. and Toxoplasma gondii (the causative agents of malaria and toxoplasmosis, respectively) and the trypanosomatids Trypanosoma brucei, Trypanosoma cruzi, and Leishmania spp., which cause African sleeping sickness, Chagas’ disease, and leishmaniasis, respectively. Malaria is one of the leading causes of death and morbidity worldwide, especially in the developing countries. Despite recent success of malaria control programs in reducing incidence and mortality rates to more than 20%, there is an estimated 429,000 malaria deaths in 2015 that mostly occurred in the African region (World Malaria Report 2016). T. gondii parasites are found in around a third of human population and can be fatal in immunocompromised individuals, while the trypanosomatids can be classified as the second most deadly parasites after Plasmodium, causing approximately 150,000 deaths annually (Nussbaum et al. 2010). Resistance to the frontline antiparasitic drugs, together with the lack of an effective vaccine, poses today considerable risks to global health (Fairlamb et al. 2016). There is therefore an urgent need to identify new drug targets for therapeutic alternatives. With the exception of T. brucei that remains extracellular during all its life cycle, apicomplexan and trypanosomatid parasites are readily adapted to intracellular lifestyle. For instance, P. falciparum develops within the gut of female Anopheles mosquitoes and is transmitted to vertebrate hosts during the blood meal, where they first invade hepatocytes and then enter the bloodstream to infect red blood cells (RBC). In this nutritionally complex environment, Plasmodium relies on the host cell cytosol and hemoglobin digestion to fulfill its nutrient requirements (Krugliak et al. 2002). Leishmania parasites are transmitted to vertebrate hosts through sand fly bites and are internalized by macrophages where they differentiate into amastigotes inside the phagolysosome. The survival of Leishmania amastigotes in this acidic environment is consistent with their optimal metabolism at acid pH (Mukkada et al. 1985). In contrast, T. gondii and T. cruzi are both able to invade a wide range of nucleated mammalian cells.
Before genome sequencing, all our knowledge about protozoan parasite metabolism came essentially from biochemical studies carried out on specific life cycle stages that could be readily cultured in vitro. These stages are usually those found in insect vectors in which parasites are living extracellularly but with a metabolism that is not necessarily similar to that found in mammal host and inside host’s cells. During the 2000s, the access to the genomes of most of the human protozoan parasites suddenly disclosed a global view of the parasite’s metabolic capacities. Detailed metabolic maps were drawn from the genome-derived sequences, highlighting new putative targets for drug discovery. These include parasite-specific metabolic pathways but also those for which parasites are auxotrophic. In opposition to prototrophy, auxotrophy defines the inability of an organism to synthetize a particular organic compound required for its growth. Protozoan parasites are auxotrophic for a wide range of amino acids, purines, vitamins, lipids, and other metabolites that must be imported at sufficient levels to sustain parasite growth. For a therapeutic perspective, auxotrophic pathways are very attractive targets to starve and impair parasite proliferation in its vertebrate host, with potentially high specificity.
Through the course of evolution, parasitic organisms have gained or lost specific metabolic pathways to optimize their life cycles in respect to their host environment(s). This generally concerns the loss of unnecessary or redundant functional pathways and gain of salvage routes to import and assimilate metabolic intermediates. The situation is even more complex for dixenous parasites that alternate between an insect vector and a vertebrate host, offering different nutritional resources that obligate parasites to behave as metabolically and phenotypically different microorganisms. The conservation of metabolic pathways required in these successive host environments is therefore essential for protozoan parasite life cycle completion. In that context, it can be puzzling to consider that parasites evolved toward genome compaction and metabolic auxotrophy at the expense of their metabolic autonomy. Moreover, how can metabolic dependency cause a fitness benefit? For all these questions, auxotrophy should not be considered as a metabolic “laziness” but more likely as a consequence of a fine-tuned biology. Experimental acquisition of metabolic dependencies in protozoan parasites is lacking, but this phenomenon is well documented in bacteria (D’Souza et al. 2014). For example, amino acid auxotrophic genotypes can emerge in less than 2000 generations for E. coli bacteria populations evolving in amino acid-rich environments. Interestingly, these auxotrophic mutants gained a significant fitness benefit over the evolutionary ancestor during competition experiments (D’Souza and Kost 2016). In Legionella pneumophila, a parasite of amoebae (and an accidental pathogen of humans causing severe pneumonia), the continuous passages inside mouse macrophages for hundreds of generations have fixed mutations in multiple steps of the lysine biosynthesis pathway resulting in lysine auxotrophy (Ensminger et al. 2012). Conversely, macrophage-adapted bacteria show a reduced capacity to replicate in amoebae. These results demonstrate that host restriction can rapidly modify pathogen fitness and support therefore that host cycling drives the conservation of metabolic pathways that are deleterious for growth in only specific hosts.
For space limitation, this chapter will focus on selected auxotrophic pathways and metabolic dependencies that have been validated as true or promising therapeutic targets against apicomplexan and trypanosomatid parasites. A general feature that characterizes parasite auxotrophies is the existence of multiple routes of synthesis and interconversion, combined to scavenging possibilities at different metabolic steps. These sophisticated bypass systems are readily used by parasites to circumvent the inhibition of specific enzymes. Nevertheless, such alteration of basic metabolisms can have a major impact on parasite virulence in vivo and, as detailed below, few examples of success stories exist.
2 What Genome Sequencing Can Tell Us About Parasites and Parasitism?
The release of the complete genome sequences of several important protozoan parasites over a decade ago provides a rich resource for understanding the biology and evolution of these pathogens. Comparative genomic analysis raises central questions on gene repertoires, gain/loss of genes, and genome organization and provides a global view of the metabolic potential of these parasites, irrespective of the life cycle stage. The comparison of the TriTryp genomes (T. brucei/T. cruzi/Leishmania major) has highlighted that both gene order and gene repertoire (synteny) are broadly conserved despite a divergence estimated between 200 and 500 million years ago, predating the divergence of mammals (Berriman et al. 2005; El-Sayed et al. 2005a, b; Ivens et al. 2005). This conserved core genome of trypanosomatids comprises about 6200 genes (∼70% of the genome) and reflects strong selective constraints on genome structure. Additional comparative genomic analysis from clinically relevant Leishmania species (L. major, L. infantum, and L. braziliensis causing human cutaneous, visceral, and mucocutaneous leishmaniasis, respectively) confirmed a greater extend of synteny with gene loss and pseudogene formation being the principal factors that shaped Leishmania species genomes through evolution (Peacock et al. 2007).
In addition to Plasmodium and Toxoplasma, the phylum Apicomplexa contains several pathogens of medical and veterinary importance including Cryptosporidium, Eimeria, Theileria, and Babesia. Genome content of these organisms revealed a massive gene loss in both nuclear and organellar genomes compared to most model eukaryotes. About 50% of the genes found in Plasmodium species have no orthologs in other apicomplexan, while only 85% of the genome content is conserved between P. falciparum and rodent malaria parasites (Kooij et al. 2006). The apicomplexan genome is highly dynamic with significant genome rearrangement between lineages. In contrast to trypanosomatids, syntenic regions are rare between lineages and appear to be totally absent across the phylum. It seems therefore that there are different criteria governing genome evolution within the Apicomplexa phylum relative to other unicellular eukaryotes and notably trypanosomatids (DeBarry and Kissinger 2011).
One of the most intriguing cellular features of the phylum Apicomplexa is the presence of a non-photosynthetic secondary plastid, termed the apicoplast. This organelle derived by secondary endosymbiosis of an alga and retention of the algal plastid. During their evolution toward parasitism, most members of Apicomplexa, including Plasmodium and Toxoplasma, lost the ability to photosynthesize while maintaining in the apicoplast the biosynthesis of fatty acids, isoprenoids, iron-sulfur clusters, and heme (Lim and McFadden 2010). One striking feature of the apicoplast is that it is essential for parasite growth and development and therefore a validated target for drug therapy. In Plasmodium, the sequencing of the small circular apicoplast genome (35 kb encoding less than 50 proteins) brought little insight into its function. Most of the apicoplast-encoded genes are ascribed to “house-keeping” functions such as transcription and translation (Wilson et al. 1996). To find out possible functions of the apicoplast, Waller et al. used publicly available T. gondii EST sequences and preliminary sequence data for P. falciparum to identify nuclear-encoded apicoplast proteins based on sequence similarity to proteins of known function in plant and algal systems (Waller et al. 1998). The identification of an apicoplast-targeting signal sequence has facilitated the prediction of proteins residing into apicoplast and the reconstruction of metabolic pathways found in this organelle (Zuegge et al. 2001; Foth et al. 2003).
All the parasite genome sequences and their functional annotation are available in the portal Eukaryotic Pathogen Database Resource (EuPathDB, http://eupathdb.org) (Aurrecoechea et al. 2017) that offers the access to large genomic-scale data set from diverse eukaryotic pathogens including Plasmodium (http://plasmodb.org), Toxoplasma (http://toxodb.org), and trypanosomatids (http://tritrypdb.org). Reconstruction of parasite metabolic network at genome-scale was first realized in an automated fashion and further improved by manual refinement and curation, some being implemented with metabolomics data sets. Among the most up-to-date metabolic resources are the “Malaria Parasite Metabolic Pathways” (MPMP), the “Library of Apicomplexan Metabolic Pathways” (LAMP) (Shanmugasundram et al. 2013), and the T. brucei TrypanoCyc database (Shameer et al. 2015).
With the increasing number of published complete genome sequences, including not only new parasite strains and subspecies but also early free-living relatives, we are now able to delineate an accurate picture regarding how parasites have evolved from free-living organisms (Janouskovec and Keeling 2016). Bodo saltans is the closest known nonparasitic relative of trypanosomatids. It lives in water habitats and feeds on bacteria by phagocytosis. Comparing the genomes of B. saltans and parasitic trypanosomatids reveals that the transition from a free-living to a parasitic lifestyle has resulted in the loss of approximately 50% of protein-coding genes. Gene loss is notably marked in macromolecular digestion and ion transport, while parasite innovations (gene gains) mostly concern cell-surface gene families and membrane transporters (Jackson et al. 2016). B. saltans does not contain however any complete metabolic pathways that are absent in parasitic trypanosomatids (Opperdoes et al. 2016). Interestingly, some similar observations can be made from comparative phylogenomic analyses between apicomplexan parasites and their closest free-living and photosynthetic relatives called chrompodellids (Woo et al. 2015; Janouskovec et al. 2015). In this case, however, chrompodellids harbor a wider range of metabolic capacities than apicomplexan parasites, notably those involved in photosynthesis.
Overall, comparative phylogenomic analyses support that the emergence of parasitism may not be determined by acquisition of novel components, but rather by loss and modification of characteristics already present in their free-living ancestors (Janouskovec and Keeling 2016).
3 Protozoan Parasite Auxotrophies
3.1 Purines
The nucleotide metabolism is considered as promising therapeutic targets against protozoan parasites since almost four decades and has consequently been the focus of considerable scientific investigations. Pyrimidines and purines are essential for the biosynthesis of DNA, RNA, and sugar nucleotides. In addition, certain of these molecules and related derivatives are particularly important for energy production (ATP, GTP), vitamin-derived cofactors synthesis (NAD+, FAD, FMN, SAM, or acetyl-CoA), and cell signaling involving intracellular second messengers (cAMP, cGMP). The purine pathway has garnered extensive attention because, unlike their vertebrate hosts, all protozoan parasites lack the capacity to synthesize the purine ring de novo. Most of them synthetize however pyrimidine de novo, similarly to mammalian cells. In order to survive and proliferate, purine auxotrophic parasites developed diverse strategies to salvage purines through an array of interconnected and seemingly redundant pathways. Marked differences in the salvage pathways and the preferred substrates between parasites exist however, reflecting host metabolic specificities. This critical participation of purine/pyrimidine nucleotides in many aspects of parasite function and replication makes the nucleotide metabolism an “Achilles’ heel” for drug intervention.
Trypanosomatids
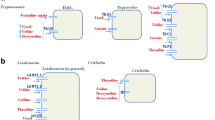
Leishmania and Trypanosoma lack nine of the ten genes required to make IMP from phosphoribosyl pyrophosphate (PRPP) and are thus unable to synthetize purines de novo. To salvage purines from the host, Leishmania possess a group of membrane and secreted nucleotidase/nuclease hydrolases (NH) that mediate the degradation of extracellular nucleosides, nucleotides, and nucleic acids with low substrate specificity. Several purine transporters are encoded in the Leishmania genome. The first parasite purine transporters identified are the nucleoside transporters (NTs) LdNT1 from Leishmania donovani (specific for adenosine and the pyrimidine nucleosides) (Vasudevan et al. 1998) and LdNT2 (for inosine, guanosine, xanthosine) (Carter et al. 2000b). Two other purine nucleobase transporters (NT3 and NT4) for hypoxanthine, xanthine, adenine, and guanine are also present in Leishmania, the latter having an optimal transport activity at acidic pH which correspond to the conditions found by parasites inside the parasitophorous vacuole (Ortiz et al. 2009). Once internalized, nucleosides and nucleobase are interconverted by a complex network of purine salvage enzymes (Fig. 9.1a). The genetic dissection of this pathway using reverse genetic tools revealed functionally redundant routes for purine acquisition and pinpointed the preferred purine source for Leishmania parasite. Metabolic flux and gene inactivation studies reported indeed that despite a superficial complexity, the majority of purine flux into nucleotides in Leishmania appeared limited to two main routes: HGPRT and XPRT (Boitz et al. 2012). Further studies showed that Leishmania is able to sense variations in extracellular purine concentration by upregulating the translation of purine transporters and salvage enzymes in a coordinated manner (Carter et al. 2010; Ortiz et al. 2010; Martin et al. 2014). Following purine removal from the culture media, Leishmania upregulate primarily purine transporters (NT1-3) and nucleases (NH), while other key purine salvage components, notably HGPRT and XPRT, are upregulated later and to a lesser extent (Martin et al. 2014). The purine salvage pathways of Leishmania and Trypanosoma are remarkably similar with one notable exception: while Leishmania has two distinct routes of adenine metabolism (AAH and APRT, see Fig. 9.1a) and the former being the preferred one, both T. brucei and T. cruzi lack AAH ortholog enzyme (Boitz and Ullman 2013). Adenosine, the preferred source of purine in T. brucei, is then successively cleaved to adenine by specific nuclease and phosphorylated to AMP by APRT (Fig. 9.1a). Remarkably, the adenosine/adenine transporter of T. brucei (TbAT1) is also the transporter of the arsenical melarsoprol and pentamidine, two important drugs currently used for the treatment of African sleeping sickness (Baker et al. 2013). In contrast to Leishmania, trypanosomes encode a complex family of at least 12 closely related nucleoside and nucleobase transporters resulting from a series of gene duplication events (Landfear et al. 2004; de Koning et al. 2005). The reason for the expansion of this gene family in trypanosomes compared to Leishmania is still not clear.

Simplified illustration of the purine salvage pathway and interconversion enzymes in trypanosomatid (a) and apicomplexan (b) protozoan parasites. Asterisks correspond to enzymes found in specific parasite species (see text for details). Abbreviations: ADE adenine, HYP hypoxanthine, GUA guanine, XAN xanthine, ADO adenosine, INO inosine, GUO guanosine, XAO xanthosine, AAH adenine aminohydrolase, GDA guanine deaminase, NH nucleoside hydrolase, AK adenosine kinase, APRT adenine phosphoribosyltransferase, HGPRT hypoxanthine-guanine phosphoribosyltransferase, XPRT xanthine phosphoribosyltransferase, HGXPRT hypoxanthine-guanine-xanthine phosphoribosyltransferase, ADSS adenylosuccinate synthetase, ASL adenylosuccinate lyase, AMPDA AMP deaminase, IMPDH inosine monophosphate dehydrogenase, GMPS GMP synthase, GMPR GMP reductase, NH nucleoside hydrolase, PNP purine nucleoside phosphorylase
Apicomplexans
Marked differences in purine scavenging abilities can be noticed between Toxoplasma and Plasmodium parasites. Three transporters involved in the uptake of purine bases and nucleosides have been identified in T. gondii. TgAT1 is specific to adenosine and inosine; TgAT2 possesses broad substrate specificity, while TgNBT1 transports hypoxanthine, xanthine, and guanine. In the case of Plasmodium, purine nucleosides and nucleobases are transported across the plasma membrane by a unique transporter PfNT1 (Carter et al. 2000a). In contrast to trypanosomatids, apicomplexans share a single phosphoribosyltransferase enzyme (HGXPRT) to catalyze the synthesis of IMP, XMP, and GMP from imported hypoxanthine, xanthine, and guanine, respectively (Fig. 9.1b). Toxoplasma mostly produces AMP from adenosine using adenosine kinase (AK), which is the most active enzyme of this pathway (Krug et al. 1989). Surprisingly, this enzyme is absent in P. falciparum, and AMP generation in this parasite strictly depends on adenylosuccinate synthase and lyase activities from IMP, the common precursor for the synthesis of all purine nucleotides in Plasmodium. The Plasmodium purine salvage pathway is actually highly simplified and relies on the activity of the three enzymes HGXPRT, purine nucleoside phosphorylase (PNP), and adenosine deaminase (ADA) (Cassera et al. 2011). These enzymes ensure the sequential conversion of exogenous adenosine to inosine and hypoxanthine, which will be then phosphoribosylated to form IMP (Fig. 9.1b). It has been suggested however that hypoxanthine is the major purine precursor incorporated into parasite during the intraerythrocytic stage of Plasmodium (Gero and O’Sullivan 1990). This implies that HGXPRT should be considered as the most promising drug target of the purine pathway in P. falciparum.
Purine-Based Chemotherapy
Different strategies have been developed to specifically target the purine synthesis in parasitic protozoans. Basically, the two main approaches consist of either inhibiting specific enzymes using substrates analogs generally derived from anticancer or antiviral drugs or using subversive substrates that will be activated to toxic products by a parasite-specific purine salvage enzyme. For comprehensive reviews of the best purine targets and the different class of purine inhibitors that have been tested against protozoan parasites, see references (Kouni 2003; Berg et al. 2010). Due to the high redundancy of the interconversion pathways in protozoan purine metabolism, it is thought that two or more inhibitors might be required for an effective chemotherapy against these parasites. Additional in vivo experiments are required to determine whether parasites can readily bypass the inhibition of a specific scavenging route of purine acquisition without loss of fitness and virulence.
3.2 Polyamines and Arginase
Polyamines defined small cationic molecules with more than two amine groups and are found in all living organisms. In eukaryotes, the most common polyamines are putrescine, spermidine, and spermine. Due to the wide range of cellular processes in which polyamines are involved, it is still difficult to delineate the precise functions of these metabolites that comprise gene regulation, cell proliferation, and stress response (Miller-Fleming et al. 2015). The polyamine biosynthetic pathway is ubiquitous and highly conserved among eukaryotes. Marked differences exist however in protozoan parasites (Birkholtz et al. 2011). For trypanosomatids, polyamine biosynthesis plays an essential role for the synthesis of the trypanosomatid-specific thiol trypanothione (TSH2) that controls redox regulation and defense against oxidative stress damages. The essential role of polyamines in cell proliferation and their association with cancer has led to the development of drug compounds with antitumoral properties. While being not enough satisfying for human application, these polyamine-related molecules were shown to be highly effective against protozoan parasites, notably T. brucei (see below). The canonical polyamine pathway in eukaryotes consists of five enzymes: arginase (ARG), ornithine decarboxylase (ODC), S-adenosylmethionine decarboxylase (AdoMetDC), spermidine synthase (SpdS), and spermine synthase (SpmS). Trypanosomatids and Plasmodium parasites lack the final SpmS enzyme. ARG, the first step in polyamine biosynthesis, converts L-arginine (Arg) to L-ornithine (Orn) that is subsequently metabolized to the diamine putrescine by the catalytic action of ODC. Putrescine is then converted into spermidine (Spd) and spermine (Spm) by the addition of aminopropyl groups provided by decarboxylated S-adenosylmethionine (dcSAM). This addition is mediated by SpdS and SpmS, respectively, while dcSAM results from the activity of AdoMetDC on S-adenosylmethionine (SAM) (Fig. 9.2).

Polyamine metabolism and transporters in trypanosomatid (a) and apicomplexan (b) parasites. Key parasite enzymes are boxed in black. The backward polyamine biosynthesis pathway of T. gondii is indicated by the presence of T. gondii spermine/spermidine N-acetyltransferase (SSAT), spermidine N-acetyltransferase (SAT), and polyamine oxidase (PAO) enzymes. Abbreviations: ARG arginase, ODC ornithine decarboxylase, SpdS spermidine synthase, SAM, S-adenosylmethionine, dcSAM decarboxylated SAM, AdoMetDC adenosylmethionine decarboxylase, ROS reactive oxygen species
Trypanosomatids
The redox system in trypanosomatids is essentially based on a specific dithiol called trypanothione (TSH2) that is composed of two molecules of glutathione (GSH) joined by a spermidine linker (Krauth-Siegel and Comini 2008) (Fig. 9.2a). Given the role of spermidine as a major component of the trypanothione synthesis, polyamine inhibition is believed to be highly efficient against trypanosomatid parasites. Actually, the last approved drug to treat African trypanosomiasis caused by T. b. gambiense is α-difluoromethylornithine (DFMO; eflornithine), an inhibitor of ODC initially developed as an anticancer agent. This is the only antitrypanosomal drug whose molecular target is known. Concomitant with its action on ODC activity, eflornithine reduces the intracellular levels of putrescine, spermidine, and trypanothione in bloodstream form of T. brucei (Fairlamb et al. 1987). The reasons why DFMO is more efficient against T. brucei than the other trypanosomatids or even mammalian ODC are multiple. The main explanation is that T. brucei parasites are directly in contact to the drug in the bloodstream and are not able to import exogenous polyamines in enough quantity. Finally, TbODC has a relatively slow turnover as compared to the human ortholog. Recently, two high-affinity Orn transporters that belong to the amino acid transporter family (TbAAT10-1, TbAAT2-4) have been described in T. brucei (Macedo et al. 2017) (Fig. 9.2a). Downregulation of these transporters renders parasites hypersensitive to DFMO (taken up by a distinct TbAAT6 transporter). Surprisingly, ODC gene is missing in T. cruzi genome, making this parasite totally dependent on putrescine uptake to synthetize spermidine and trypanothione. In contrast to T. brucei, polyamine transport in T. cruzi is indeed highly efficient and essentially mediated by the TcPOT1 transporter (also named TcPAT12). This transporter has a high affinity for putrescine (but not for spermidine), and deletion of TcPOT1 gene impairs the ability of T. cruzi parasites to sustain a robust infection in mammalian cells (Hasne et al. 2016). The Leishmania ortholog of TcPOT1 is LmPOT1, the first eukaryotic polyamine transporter cloned in eukaryotes. It is selective for putrescine and spermidine uptake but does not appear to be expressed in the amastigote stage in mammal host (Hasne and Ullman 2005). The Leishmania polyamine biosynthesis diverges from that of other trypanosomatids by the fact that Leishmania can use ARG for the de novo production of Orn from Arg. Interestingly, Leishmania parasites are able to sense changes in the external Arg concentration. Indeed, to fulfill its polyamine needs following macrophage infection, intracellular Leishmania parasites activate a coordinated “Arg deprivation response” (ADR) by rapidly increasing the expression of the high-affinity parasite Arg transporter (LdAAP3) (Goldman-Pinkovich et al. 2016). Double knockout of the ARG gene in several Leishmania species showed a significant reduction of infectivity in mice as compared to wild-type parasites. Recent data support however that inside the parasitophorous vacuole of macrophages, ARG is not essential for parasite survival. L. donovani amastigotes prefer indeed salvaging host Orn, and to a lesser extend host spermidine pools, while host putrescine levels appear too limited to support robust infection (Boitz et al. 2017).
Apicomplexans
The most distinctive feature of polyamine biosynthesis in Plasmodium parasites is the presence of a bifunctional AdoMetDC/ODC enzyme where the active site of both ODC and AdoMetDC are located on a single polypeptide (Fig. 9.2b). This bifunctional AdoMetDC/ODC enzyme appears to be important for the regulation of the polyamine pools in the parasite (Birkholtz et al. 2004). Deletion of the AdoMetDC/ODC gene in Plasmodium yoelii significantly reduced blood-stage parasitemia in mice and completely abolished Anopheles transmission (Hart et al. 2016). Furthermore, a single SpdS enzyme ensures the synthesis of both spermidine and spermine (Fig. 9.2b). Arg uptake and transformation to polyamines by its own ARG and AdoMetDC/ODC enzymes also appear to be determinant for Plasmodium development in the liver (Meireles et al. 2017). The apicomplexan Arg transporter has been recently characterized in T. gondii (TgNPT1) and P. berghei (PbNPT1), and gene deletion experiments have confirmed their role in parasite’s survival and virulence (Rajendran et al. 2017). Studies on polyamine biosynthesis in T. gondii are more limited. Surprisingly it appears that T. gondii is devoid of ODC and ARG enzymes and can only synthetize putrescine from host spermine in backward reactions through the combined action of Toxoplasma spermine/spermidine N-acetyltransferase (SSAT) and polyamine oxidase (PAO) (Cook et al. 2007) (Fig. 9.2b). Additionally, T. gondii parasites possess a high-affinity putrescine transporter and can also scavenge Orn from the culture medium (Cook et al. 2007), while none of these transporters have been identified up to now. Therefore, while polyamine supply in Plasmodium mainly depends on biosynthesis from arginine to spermidine, Toxoplasma can only rely on polyamines scavenging and interconversion.
Polyamine-Based Chemotherapy
The success story of DFMO (eflornithine) in African trypanosomiasis treatment supports the legitimacy of developing inhibitors interfering with polyamine homeostasis in parasites. Unfortunately, DFMO efficiency against other protozoan parasites is far from being satisfying, mainly because of their capacity to bypass ODC enzymatic inhibition by increasing the uptake of preformed polyamines from the host. This dual source of polyamines (biosynthesis and scavenging) is certainly the key issue for future development of drugs against ODC or the different enzymes of the polyamine biosynthesis pathway (i.e., ARG, AdoMet, SpdS). Recent reviews on polyamine-based therapeutic strategies against protozoan parasites can be found in Birkholtz et al. (2011), Roberts and Ullman (2017), and Jagu et al. (2017).
3.3 Cofactors and Vitamins
Vitamins define organic compounds and vital nutrients that are not synthetized by an organism but that are required in limited amounts. They are used as precursors of cofactors for many enzymes involved in different metabolisms. Thirteen vitamins are universally recognized at present, but the evolution of vitamin-related pathways has resulted in profound differentiation of auxotrophy among species, including parasites. The most striking example is vitamin C (ascorbate), which is a vitamin for humans but not for most other organisms that can synthetize it de novo. By contrast, microorganisms such as protozoan parasites can synthesize certain vitamins de novo, either completely or partially. The complex pattern of vitamin auxotrophy across the eukaryotic tree of life is intimately connected with the interdependence between organisms (Helliwell et al. 2013). Genomic analysis revealed that trypanosomatids are auxotrophs for thiamine (B1), riboflavin (B2), nicotinic acid (B3), pantothenic acid (B5), pyridoxamine (B6), biotin (B7), and folic acid (B9) (Klein et al. 2013). Interestingly, nonpathogenic trypanosomatids harboring endosymbionts are only auxotrophs for thiamine, biotin, and nicotinic acid, most of the genes required for the synthesis of other vitamins being present in the symbiont genome. Apicomplexans possess the ability to synthetize several vitamins de novo including vitamins B1, B6, and B9. Some divergences in their biosynthesis pathway exist however between Plasmodium and Toxoplasma (Müller and Kappes 2007). The inability of human to synthetize vitamins implies therefore that the inhibition of parasite vitamin biosynthetic pathways might be a good therapeutic strategy to selectively interfere with parasite development. This is the case for the folate pathway that has proven to be a valuable target for drug intervention in Plasmodium (see below). Conversely, the lack of a specific de novo vitamin biosynthetic pathway in parasites also offers the possibility to block precursor assimilation and conversion to active cofactors. This is notably the case for vitamin B3, the unique precursor of NAD+ in protozoan parasites.
3.3.1 Folate Metabolism (Vitamin B9)
The term folates designs several structurally similar compounds that are used as cofactors in a variety of “one-carbon” (C1) transfer reactions. In their reduced tetrahydrofolate (THF) form, folates are notably involved in the biosynthesis of thymidine (DNA synthesis), the conversion of serine to glycine, methionine biosynthesis (DNA and protein methylation reactions), histidine catabolism, and purine biosynthesis. For these reasons, folate pathway has long been considered to be an attractive pathway to develop anti-parasite drugs. Folates are composed of a pterin ring linked to para-aminobenzoate (pABA) and L-glutamate (Fig. 9.3a). Inside the cell, they are further polyglutamylated to enhance cellular retention. Plasmodium and Toxoplasma parasites possess both biosynthetic and salvage pathways and are then able to synthetize pterin and pABA compounds and to perform folate assembly de novo. Pterin is synthesized from GTP (guanosine 5′-triphosphate), whereas pABA is obtained from chorismate. Instead, trypanosomatids are auxotrophs for both folates and pterins and must import them from exogenous sources.

Simplified illustration of folate metabolism in protozoan parasites. (a) Structure of folate composed of three building blocks: pterin, para-aminobenzoate (pABA), and glutamic acid (Glu) (from https://commons.wikimedia.org/wiki/File:Folic_acid_structure.svg). (b) Folate transport and reduction in trypanosomatids mediated by DHFR-TS (the target of the drug methotrexate (MTX)) and PTR1 enzymes. (c) De novo synthesis of folates in apicomplexans. Drugs targeting DHPS and DHFR-TS enzymes (boxed in black) are indicated in red. Abbreviations: BT1 biopterin transporter 1, FTs folate transporters, DHFR-TS dihydrofolate reductase-thymidylate synthase, PTR1 pterin reductase 1, DHF dihydrofolate, THF tetrahydrofolate, GTPCH GTP cyclohydrolase 1, PTPS 6-pyruvoyl-tetrahydropterin synthase, HPPK hydromethyldihydropteridine pyrophosphokinase, DHPS dihydropteroate synthase, DHFS dihydrofolate synthase
Trypanosomatids
Most of our knowledge about folate metabolism in Leishmania and other trypanosomatids came from studies using the anti-DHFR cancer drug methotrexate (MTX) (Ouellette et al. 2002; Vickers and Beverley 2011). Leishmania can acquire folates (and MTX) by at least three members of the folate-biopterin transporter (FBT) family named FT1, FT5, and BT1, the latter being a biopterin transporter that can also import folates but not MTX. This Leishmania FBT family comprises in total 14 members, while only 7 are present in T. brucei. A genome-wide RNA interference experiment has recently identified a tandem array of folate transporters (TbFT1–3) as major contributors of folates and antifolate drugs uptake in T. brucei (Dewar et al. 2016). The enzymatic reduction of folate to tetrahydrofolate (THF) in trypanosomatids is catalyzed by a bifunctional dihydrofolate reductase-thymidylate synthase (DHFR-TS). Interestingly, a similar reduction can also be carried out by an alternative enzyme called pteridine reductase 1 (PTR1) (Fig. 9.3b). PTR1 can reduce both unconjugated pteridines and folates. This gene was first discovered in MTX-resistant Leishmania parasites in which both PTR1 and BT1 genes were found amplified in a common extrachromosomal element (Ouellette et al. 2002). PTR1 was shown to contribute to 10% of the reduction of folates in wild-type parasite and is 2000-fold less susceptible to inhibition by MTX than DHFR-TS (Nare et al. 1997). This PTR1 bypass of DHFR-TS reduces the effectiveness of classical antifolate drugs in Leishmania parasites and supports therefore that both enzymes have to be targeted simultaneously. Despite numerous efforts, such an inhibitor has not been identified up to now.
Apicomplexans
Apicomplexans can both synthetize folates de novo and salvage them from their host. Nevertheless, antifolates have a long history of successful application in malaria chemotherapy, and despite widespread parasite resistance, the antifolates still play an important role in malaria control (Müller and Hyde 2013). The biosynthesis pathway starts by converting GTP to 6-hydroxymethyl-7,8-dihydropterin pyrophosphate by three successive enzymatic steps (Fig. 9.3c). The next dihydropteroate synthase enzyme (DHPS) catalyzes the formation of 7,8-dihydropteroate from pABA and the product of the hydromethyldihydropteridine pyrophosphokinase (HPPK) reaction. DHPS is the target of sulfa drugs (sulfadoxine), the first class of Plasmodium antifolates drugs that are structural analogues of pABA. The de novo synthesis of folates is completed by the action of the dihydrofolate synthase (DHFS) that converts 7,8-dihydropteroate to dihydrofolate (DHF) by conjugating L-Glu residue to pABA. The final reduction of DHF to THF is mediated by a bifunctional DHFR-TS enzyme, which is the second target of antifolate drugs like pyrimethamine and proguanil. Since the plasmodial DHFR-TS is highly susceptible to these inhibitors, pyrimethamine in combination with sulfadoxine has long been used in the treatment of malaria until drug-resistant parasites with mutations in both DHF and DHFR genes have emerged. Two folate transporters (PfFT1 and PfFT2) have been also characterized in Plasmodium and can mediate folate as well as pABA uptake from the host (Salcedo-Sora et al. 2011). This ability of Plasmodium to salvage folates can directly alter the efficacy of antifolate drugs in pregnant woman taking folate supplementation treatment (Nzila et al. 2014). Currently, antifolate drugs such as pyrimethamine, proguanil, and sulfonamides are still part of the treatment of uncomplicated malaria in combination with other drugs such as artemisinin or amodiaquine (Müller and Hyde 2013). Nevertheless, several enzymes of the folate pathway are still considered as promising targets for drug development against Plasmodium parasites (Salcedo-Sora and Ward 2013).
3.3.2 Niacin (Vitamin B3) and NAD+
Niacin or nicotinate (Na), together with its amide form nicotinamide (Nam), defines the group of vitamin B3 complex. They are all precursors of the nicotinamide adenine dinucleotide (NAD+), an essential cofactor in all living cells that carries electrons from one reaction to another in redox reactions. By its redox function, NAD+ and its derived phosphorylated form (NADP+) are involved in hundreds of reactions in many metabolic pathways including glycolysis, TCA cycle, pentose phosphate pathway, or fatty acid biosynthesis. In the last decades, non-redox roles have been also attributed to NAD+. NAD+ is consumed by a number of important cellular enzymes that participate in various cellular functions such as epigenetic regulation, DNA repair, or signal transduction. In mammals, those enzymes include class III protein deacetylases (sirtuins), poly(ADP)-ribose polymerases (PARPs), or cADP-ribose synthases (CD38 or CD157). These NAD+-dependent signaling processes involve the release of Nam and thus require constant replenishment of cellular NAD+ pools. The role of NAD+ in host-pathogen interactions and its potential for new treatments of infectious diseases have been recently reviewed (Mesquita et al. 2016). The characterization of this metabolism in protozoan parasites is quite recent. Apicomplexan and trypanosomatid parasites cannot synthesize NAD+ de novo and must salvage NAD+ precursors from host or culture medium. The different enzymes involved in NAD+ salvage pathway have been notably characterized in Leishmania and Plasmodium parasites (Gazanion et al. 2011; O’Hara et al. 2014). In contrast, human can synthetize NAD+ either by a salvage pathway or by de novo synthesis from tryptophan, both pathways converging on nicotinic acid phosphoribosyltransferase (NaMN) (Fig. 9.4a). Briefly, Na enters the Preiss-Handler pathway and is converted to NAD+ in three enzymatic steps by the action of nicotinate phosphoribosyltransferase (NaPRT), nicotinate mononucleotide adenylyltransferase (NMNAT), and NAD+ synthase (NADS). In lower eukaryotes including protozoan parasites, Nam is converted to Na by a highly conserved enzyme called nicotinamidase (pnc1) (Fig. 9.4b). This enzyme is absent in humans in which Nam is first converted to Nam mononucleotide (NMN) through the activity of a Nam phosphoribosyltransferase (NAMPT), and then adenylated to NAD+ by NMNAT (Fig. 9.4a).

NAD+ metabolism in human cells (a) and protozoan parasites (b). Abbreviations: Na nicotinic acid, Nam nicotinamide, NR nicotinamide riboside, NaMN nicotinic acid mononucleotide, NaAD nicotinic acid dinucleotide, NMN nicotinamide mononucleotide, NAD+ nicotinamide adenine dinucleotide, NADP+ nicotinamide adenine dinucleotide phosphate, NaPRT nicotinamide phosphoribosyltransferase, NMNAT nicotinamide mononucleotide adenylyltransferase, NADS NAD+ synthase, NADK NAD+ kinase, NAMPT nicotinamide phosphoribosyltransferase, NRK nicotinamide riboside kinase, Trp tryptophan, pnc1 nicotinamidase
Trypanosomatids
Leishmania parasites are NAD+ auxotrophs and can import Na, Nam, or NR to ensure intracellular NAD+ synthesis (Gazanion et al. 2011). The Leishmania nicotinamidase has been recently biochemically and functionally characterized in L. infantum. Deletion of this gene led to obtain NAD+ depleted parasites that cannot sustain infection in mice model nor in sand fly vector (Gazanion et al. 2011; Gazanion et al. 2012). This enzyme is therefore a validated drug target against Leishmania and possibly other trypanosomatid parasites (Michels and Avilán 2011). Based on comparative genomic analysis, all Trypanosoma and Leishmania species possess a canonical NAD+ salvage pathway. Other enzymes of this pathway, notably NMNAT, have recently been characterized in L. braziliensis and T. cruzi (Contreras et al. 2015; Sánchez-Lancheros et al. 2016).
Apicomplexans
Some members of apicomplexans including P. falciparum or T. gondii have a NAD+ salvage pathway similar to that of trypanosomatids, although with subtle differences. In contrast to the cytosolic localization of the Leishmania nicotinamidase, the Plasmodium nicotinamidase concentrates mainly in the nucleus (O’Hara et al. 2014). The same study showed that PfNMNAT can be a valuable drug target for drug development and lead compounds were proposed based on previously identified bacterial NMNAT inhibitors. Surprisingly, other apicomplexans such as Cryptosporidium, Babesia, and Theileria species are not able to synthesis NAD+ from Na and Nam. They all possess NAD+ kinase orthologs, suggesting that they can directly salvage NAD+ from host and convert it to NADP+, but no NAD+ transporter has been identified to date, and no homologs of validated vitamin B3 transporters can be found in apicomplexans or trypanosomatids genome databases. How NAD+ or vitamin B3 precursors are imported into protozoan parasites remains to be determined, and such transporters might constitute new promising drug targets for chemical inhibition.
3.3.3 Other Vitamins and Vitamin-Based Chemotherapy
In addition to the two examples cited above, biosynthesis of other vitamins in protozoan parasites has been well documented for their suitability to identify new anti-parasite drugs. Most of the vitamin biosynthesis pathways are indeed absent in humans and constitute therefore a valuable reservoir of drug targets. While folate metabolism has been largely exploited for drug design, other vitamins such as thiamine (B1), riboflavin (B2), pyridoxamine (B6), or pantothenate (B5) constitute promising alternatives (Müller and Kappes 2007; Müller et al. 2010; Kronenberger et al. 2013). Pantothenate (B5), for example, is required for the synthesis of coenzyme A (CoA), an essential cofactor for both energy metabolism and in fatty acid biosynthesis. T. gondii parasites possess all four genes required for de novo synthesis of pantothenate from valine while these genes are absent in P. falciparum. Once taken up by the parasites, pantothenate is phosphorylated by the pantothenate kinase (PanK), the first enzyme and the rate-limiting step in the biosynthesis of CoA. Even if this enzyme has four isoforms in humans, the unique Plasmodium PanK is highly sensitive to pantothenate analogues. Resistant lines to pantothenate analogues with mutations in PfPanK1 gene can be generated however in vitro (Tjhin et al. 2017).
Riboflavin (B2) is the vital precursor of both flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD) used as cofactors by flavoenzymes. The synthesis of FMN and FAD in trypanosomatids is effective but dependent on efficient riboflavin uptake. The first family of protist riboflavin transporters (RibJ) has recently been identified in trypanosomatids and might constitute new attractive trypanosomatid targets (Balcazar et al. 2017).
3.4 Heme Synthesis
Heme, or iron protoporphyrin IX, is a ubiquitous cofactor composed of four pyrrole rings interconnected to each other and to a Fe2+ ion (Fig. 9.5a). Heme iron can exist in either a reduced (Fe2+) or oxidized state (Fe3+), and this transitional property is exploited by heme-dependent proteins to carry out diverse redox conversions within the cell. For parasites, heme is notably required for the synthesis of hemoproteins such as cytochromes a–c or P450 and is therefore essential for cellular respiration via the electron transport chain, the desaturation of fatty acids, and sterol synthesis (Kořený et al. 2013). Heme is also a central source of iron for parasites, and, as a component of parasite peroxidases and catalases, it helps parasites to deal with reactive oxygen species (ROS) and oxidative burst produced by the host cell. However, due to its reactive nature, free heme is also a potentially toxic molecule that can generate ROS and induce lipid peroxidation. Protozoan parasites have evolved therefore robust pathways to synthesize heme de novo or scavenge it from their environment together with efficient heme detoxification pathways (Kořený et al. 2013; Sigala and Goldberg 2014). In most eukaryotes, heme biosynthesis involves eight enzymatic steps that begin with the formation of aminolevulinic acid (ALA) in the mitochondria by the condensation of glycine and succinyl-CoA by ALA synthase (ALAS). The next enzymatic steps continue in the cytosol (or the apicoplast for apicomplexans parasites) and finally return back to mitochondria to finalize heme synthesis by ferrochelatase enzyme (FC) (Fig. 9.5).

Schematic representation of the heme metabolism in protozoan parasites. (a) Chemical structure of heme B (from https://commons.wikimedia.org/wiki/File:Heme_b.svg). (b) Heme auxotrophy in trypanosomatids parasites and the presence of the last three mitochondrial enzymes involved in heme synthesis for Leishmania spp. (boxed in black). (c) Heme biosynthesis in P. falciparum taking place into mitochondrion, apicoplast, and cytoplasm. Key enzymes are boxed in black. Abbreviations: ALAS aminolevulinic acid synthase, ALA 5-aminolevulinic acid, ALAD aminolevulinic acid dehydratase, PBGD porphobilinogen deaminase, UROS uroporphyrinogen synthase, UROD uroporphyrinogen decarboxylase, CPOX coproporphyrinogen oxidase, PPOX protoporphyrinogen oxidase, FC ferrochelatase
Trypanosomatids
The evolution of the heme synthetic pathway in trypanosomatids is highly unusual. All trypanosomatid parasites are heme-auxotrophs and lost the ability to synthesize heme before acquiring a parasitic lifestyle since the closest nonparasitic relatives of trypanosomatids (bodonids) lack heme synthesis pathway (Kořený et al. 2010). To satisfy their heme requirements, T. brucei and T. cruzi take up heme directly from their hosts. In Leishmania spp., however, this pathway was partially rescued by the presence of three genes encoding enzymes for the last steps of heme synthesis (coproporphyrinogen oxidase (CPOX), protoporphyrinogen IX oxidase (PPOX), and FC). These genes were supposedly obtained from bacteria by lateral gene transfer (Kořený et al. 2010; Fig. 9.5b). The signification of this acquisition is not clear, but it may translate the ability of Leishmania parasites to use heme precursors from their hosts when the heme concentration is too low (Chang and Chang 1985; Zwerschke et al. 2014). In the bloodstream, haptoglobin-hemoglobin (HpHb) complexes are the major source of extracellular circulating heme. The bloodstream stage of T. brucei scavenges heme in hemoglobin via a specific HpHb receptor localized in the flagellar pocket (Dostert et al. 2008). In the tsetse fly however, the HpHb receptor is not expressed, and heme importation is mediated by the recently characterized TbHrg transporter (Horáková et al. 2017). In Leishmania, two independent pathways of heme uptake have been described. One involves a specific heme transporter named LHR1 (Huynh et al. 2012). The second one is mediated by hemoglobin endocytosis, lysosomal degradation, and heme salvage and involves the ATP-binding cassette protein LABCG5 (Campos-Salinas et al. 2011). Leishmania and some other trypanosomatids are currently the only known eukaryotes possessing a bacterial-type PPOX enzyme (Zwerschke et al. 2014). This enzyme, together with the characterized heme transporters, may constitute promising drug targets for trypanosomatid parasites.
Apicomplexans
Plasmodium parasites require heme for growth and have access to abundant host-derived heme within infected red blood cells, the most heme-rich cells in the human body. Blood-stage Plasmodium parasites import up to 80% of the erythrocytes’ hemoglobin into its digestive vacuole to fulfill its nutrient requirements. During hemoglobin digestion, malaria parasites scavenge amino acids and peptides but also generate millimolar concentration of host-derived heme. Nevertheless, Plasmodium also expresses a complete, although unusual, heme biosynthetic pathway that spans between the mitochondria, the plantlike non-photosynthetic apicoplast, and the cytosol (Fig. 9.5c). Heme is essential for malaria parasites as a precursor for cytochrome production in the mitochondrial electron transport chain. However, de novo heme synthesis pathway is dispensable during the blood stage of the parasite life cycle since the genes coding for ALAS and FC, the first and the last enzymes involved in heme biosynthesis, respectively, can be inactivated without effect on parasite growth (Nagaraj et al. 2013; Ke et al. 2014). These results support that Plasmodium can scavenge host-derived heme, either released from the digestive vacuole or derived from a partial heme synthesis pathway present in RBC (Sigala et al. 2015). No homologs of heme transporters identified in trypanosomatids and other eukaryotes can be found however in Plasmodium genome sequence. The de novo heme synthesis pathway is nonetheless essential for parasite development during mosquitoes and liver stages, justifying why this pathway is maintained in these parasites (Nagaraj et al. 2013; Ke et al. 2014). While being useful for many cellular processes, free heme is also a highly toxic molecule. Numerous heme detoxification mechanisms have been identified in blood-feeding parasites including heme degradation by heme oxygenases (HO), heme containment by physical barriers, or heme conversion into inert crystalline structure called hemozoin (Toh et al. 2010). Plasmodium parasites sequester heme derived from hemoglobin digestion into crystals of insoluble hemozoin inside the parasite digestive vacuole. This heme detoxification process has proved to be an exploitable drug target against malaria parasites. Chloroquine (CQ) and other quinolone drugs are known to enter the food vacuole and interfere with the formation of hemozoin, resulting in increased free heme concentration and oxidative stress in the parasite. While the mode of action of CQ is not fully understood, parasite resistance is now widespread and generally involves the increase of drug efflux by mutations in several classes of efflux pumps (Fairlamb et al. 2016). Importantly, there is evidence that heme is also directly implicated in the activation of the most potent antimalarial drug artemisinin (Zhang et al. 2015).
Heme-Based Chemotherapy
Pathways of heme biosynthesis, uptake, detoxification, and breakdown have provided several potential important targets for drug development in protozoan parasites. In Plasmodium, heme detoxification is intimately connected to the mode of action of several antimalarial drugs used in the clinic. Heme synthesis is only present in apicomplexans, and functional studies have shown that it is not essential for parasite growth during blood-stage infection. In the case of Leishmania parasites harboring a partial heme synthesis pathway, the last three enzymes, and notably the bacterial-type PPOX, might be attractive targets for drug inhibition. Interestingly, the enzymatic activity of PPOX can be exploited for photodynamic therapy (PDT), an alternative therapeutic approach to conventional drugs that uses light-activated molecules. PPOX ensures the formation of protoporphyrin IX (PP-IX) in mitochondria, which upon exposure to light generates cytotoxic ROS and triggers oxidative damage. This approach has been successfully tested against Plasmodium parasites (Sigala et al. 2015). The authors showed that infected RBC actively import exogenous ALA resulting in the accumulation of PP-IX through the concomitant action of parasites and vestigial human enzymes. Once photoactivated by nontoxic chemicals, PP-IX will selectively kill malaria parasites while leaving uninfected cells intact. The PDT strategy using light-excitable properties of porphyrins present clear advantages among which a low toxicity and the inability of treated pathogens to develop resistance mechanisms (Wainwright et al. 2017).
4 Concluding Remarks
Understanding the metabolic interactions of protozoan parasites with their hosts during their complex multistage life cycle has the potential to identify new key biological processes that could lead to new and improved antiparasitic drugs. It is generally admitted that the metabolic optimization of parasites culminates with the complete, or partial, loss of redundant metabolic pathways to reduce energy costs and increase fitness, two conditions required to be a good parasite. This loss of metabolic autonomy is usually compensated by the acquisition of a complex system of nutrients import and interconversion that have only been partially uncovered in protozoan parasites of medical importance. This chapter provides a non-exhaustive list of essential metabolic compounds for which protozoan parasites are totally, or partially, dependent to their host. Interestingly, it seems that while most of these parasites need to scavenge similar host-derived compounds, many differences exist in the way they are acquired and assimilated. For instance, all mentioned protozoan parasites are auxotrophs for purines, but marked differences exist within trypanosomatid and apicomplexan parasites in the routes of purine acquisition and the presence of enzymes that mediate purine interconversion. In the same way, polyamine biosynthesis in trypanosomatids is directly connected to their specific thiol metabolism, and if ODC enzyme is a validated drug target in T. brucei, this enzyme is absent in the related parasite T. cruzi. To fulfill their polyamine needs, Plasmodium parasites use a unique bifunctional AdoMetDC/ODC enzyme, whereas Toxoplasma only relies on backward reactions from host spermine, the final product of polyamine biosynthesis. Finally, if apicomplexans synthetize folates de novo and can further scavenge host folate intermediates, trypanosomatid parasites are auxotrophs for folates and pterins but can rely on a large family of folate transporters and can bypass the need of DHFR-TS by PTR1 enzymatic activity. With the exception of the NAD+ synthesis pathway that appears to be highly conserved between the main trypanosomatid and apicomplexan parasites, all other metabolic pathways presented here possess notable parasite-specific singularities that can be explained by the highly different environments in which these parasites evolve during their complex life cycle.
Until now, the polyamine biosynthesis pathway in T. brucei, the synthesis of folates, and the mechanism of heme detoxification in Plasmodium have shown to be genuine drug targets for the treatment of African sleeping sickness and malaria. Additional investigations are required to evaluate the druggability of alternative metabolic pathways for which protozoan parasites are also dependent to their host. The recent improvement of metabolomics analyses together with the new powerful reverse genomic tools available in these parasites (i.e., CRISPR/Cas9) will undoubtedly rapidly increase our comprehension of the different mechanisms used by protozoan parasites to hijack essential nutrients from their host, one of the most fascinating aspects of parasite’s biology.
References
Aurrecoechea C, Barreto A, Basenko EY et al (2017) EuPathDB: the eukaryotic pathogen genomics database resource. Nucleic Acids Res 45:D581–D591
Baker N, de Koning HP, Mäser P, Horn D (2013) Drug resistance in African trypanosomiasis: the melarsoprol and pentamidine story. Trends Parasitol 29:110–118
Balcazar DE, Vanrell MC, Romano PS et al (2017) The superfamily keeps growing: Identification in trypanosomatids of RibJ, the first riboflavin transporter family in protists. PLoS Negl Trop Dis 11:e0005513–e0005522
Berg M, Van der Veken P, Goeminne A et al (2010) Inhibitors of the purine salvage pathway: a valuable approach for antiprotozoal chemotherapy? Curr Med Chem 17:2456–2481
Berriman M, Ghedin E, Hertz-Fowler C et al (2005) The genome of the African trypanosome Trypanosoma brucei. Science 309:416–422
Birkholtz L-M, Wrenger C, Joubert F et al (2004) Parasite-specific inserts in the bifunctional S-adenosylmethionine decarboxylase/ornithine decarboxylase of Plasmodium falciparum modulate catalytic activities and domain interactions. Biochem J 377:439–448
Birkholtz L-M, Williams M, Niemand J et al (2011) Polyamine homoeostasis as a drug target in pathogenic protozoa: peculiarities and possibilities. Biochem J 438:229–244
Boitz JM, Ullman B (2013) Adenine and adenosine salvage in Leishmania donovani. Mol Biochem Parasitol 190:51–55
Boitz JM, Ullman B, Jardim A, Carter NS (2012) Purine salvage in Leishmania: complex or simple by design? Trends Parasitol:1–8
Boitz JM, Gilroy CA, Olenyik TD et al (2017) Arginase is essential for survival of Leishmania donovani promastigotes but not intracellular amastigotes. Infect Immun 85:e00554–16
Campos-Salinas J, Cabello-Donayre M, García-Hernández R et al (2011) A new ATP-binding cassette protein is involved in intracellular haem trafficking in Leishmania. Mol Microbiol 79:1430–1444
Carter NS, Ben Mamoun C, Liu W et al (2000a) Isolation and functional characterization of the PfNT1 nucleoside transporter gene from Plasmodium falciparum. J Biol Chem 275:10683–10691
Carter NS, Drew ME, Sanchez M et al (2000b) Cloning of a novel inosine-guanosine transporter gene from Leishmania donovani by functional rescue of a transport-deficient mutant. J Biol Chem 275:20935–20941
Carter NS, Yates PA, Gessford SK et al (2010) Adaptive responses to purine starvation in Leishmania donovani. Mol Microbiol 78:92–107
Cassera MB, Zhang Y, Hazleton KZ, Schramm VL (2011) Purine and pyrimidine pathways as targets in Plasmodium falciparum. Curr Top Med Chem 11:2103–2115
Chang CS, Chang KP (1985) Heme requirement and acquisition by extracellular and intracellular stages of Leishmania mexicana amazonensis. Mol Biochem Parasitol 16:267–276
Contreras LE, Neme R, Ramírez MH (2015) Identification and functional evaluation of Leishmania braziliensis nicotinamide mononucleotide adenylyltransferase. Protein Expr Purif 115:26–33
Cook T, Roos D, Morada M et al (2007) Divergent polyamine metabolism in the Apicomplexa. Microbiology 153:1123–1130
D’Souza G, Kost C (2016) Experimental evolution of metabolic dependency in bacteria. PLoS Genet 12:e1006364
D’Souza G, Waschina S, Pande S et al (2014) Less is more: selective advantages can explain the prevalent loss of biosynthetic genes in bacteria. Evolution 68:2559–2570
de Koning HP, Bridges DJ, Burchmore RJS (2005) Purine and pyrimidine transport in pathogenic protozoa: from biology to therapy. FEMS Microbiol Rev 29:987–1020
DeBarry JD, Kissinger JC (2011) Jumbled genomes: missing apicomplexan synteny. Mol Biol Evol 28:2855–2871
Dewar S, Sienkiewicz N, Ong HB et al (2016) The role of folate transport in antifolate drug action in Trypanosoma brucei. J Biol Chem 291:24768–24778
Dostert C, Pétrilli V, Van Bruggen R et al (2008) Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320:674–677
El-Sayed NMA, Myler PJ, Bartholomeu DC et al (2005a) The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science 309:409–415
El-Sayed NMA, Myler PJ, Blandin G et al (2005b) Comparative genomics of trypanosomatid parasitic protozoa. Science 309:404–409
Ensminger AW, Yassin Y, Miron A, Isberg RR (2012) Experimental evolution of Legionella pneumophila in mouse macrophages leads to strains with altered determinants of environmental survival. PLoS Pathog 8:e1002731–12
Fairlamb AH, Henderson GB, Bacchi CJ, Cerami A (1987) In vivo effects of difluoromethylornithine on trypanothione and polyamine levels in bloodstream forms of Trypanosoma brucei. Mol Biochem Parasitol 24:185–191
Fairlamb AH, Gow NAR, Matthews KR, Waters AP (2016) Drug resistance in eukaryotic microorganisms. Nat Microbiol 1:16092
Foth BJ, Ralph SA, Tonkin CJ et al (2003) Dissecting apicoplast targeting in the malaria parasite Plasmodium falciparum. Science 299:705–708
Gazanion E, Garcia D, Silvestre R et al (2011) The Leishmania nicotinamidase is essential for NAD(+) production and parasite proliferation. Mol Microbiol 82:21–38
Gazanion E, Seblova V, Votýpka J et al (2012) Leishmania infantum nicotinamidase is required for late-stage development in its natural sand fly vector, Phlebotomus perniciosus. Int J Parasitol 42:323–327
Gero AM, O’Sullivan WJ (1990) Purines and pyrimidines in malarial parasites. Blood Cells 16:467–484. discussion 485–98
Goldman-Pinkovich A, Balno C, Strasser R et al (2016) An arginine deprivation response pathway is induced in Leishmania during macrophage invasion. PLoS Pathog 12:e1005494–18
Hart RJ, Ghaffar A, Abdalal S et al (2016) Plasmodium AdoMetDC/ODC bifunctional enzyme is essential for male sexual stage development and mosquito transmission. Biology Open 5:1022–1029
Hasne M-P, Ullman B (2005) Identification and characterization of a polyamine permease from the protozoan parasite Leishmania major. J Biol Chem 280:15188–15194
Hasne M-P, Soysa R, Ullman B (2016) The Trypanosoma cruzi diamine transporter is essential for robust infection of mammalian cells. PLoS One 11:e0152715–17
Helliwell KE, Wheeler GL, Smith AG (2013) Widespread decay of vitamin-related pathways: coincidence or consequence? Trends Genet 29:469–478
Horáková E, Changmai P, Vancová M et al (2017) The Trypanosoma brucei TbHrg protein is a heme transporter involved in the regulation of stage-specific morphological transitions. J Biol Chem 292:6998–7010
Huynh C, Yuan X, Miguel DC et al (2012) Heme Uptake by Leishmania amazonensis is mediated by the transmembrane protein LHR1. PLoS Pathog 8:e1002795
Ivens AC, Peacock CS, Worthey EA et al (2005) The genome of the kinetoplastid parasite, Leishmania major. Science 309:436–442
Jackson AP, Otto TD, Aslett M et al (2016) Kinetoplastid phylogenomics reveals the evolutionary innovations associated with the origins of parasitism. Curr Biol 26:161–172
Jagu E, Pomel S, Pethe S et al (2017) Polyamine-based analogs and conjugates as antikinetoplastid agents. Eur J Med Chem 139:982–1015
Janouskovec J, Keeling PJ (2016) Evolution: causality and the origin of parasitism. Curr Biol 26:R174–R177
Janouskovec J, Tikhonenkov DV, Burki F et al (2015) Factors mediating plastid dependency and the origins of parasitism in apicomplexans and their close relatives. Proc Natl Acad Sci 112:10200–10207
Ke H, Sigala PA, Miura K et al (2014) The heme biosynthesis pathway is essential for Plasmodium falciparum development in mosquito stage but not in blood stages. J Biol Chem 289:34827–34837
Klein CC, Alves JMP, Serrano MG et al (2013) Biosynthesis of vitamins and cofactors in bacterium-harbouring trypanosomatids depends on the symbiotic association as revealed by genomic analyses. PLoS One 8:e79786
Kooij TWA, Janse CJ, Waters AP (2006) Plasmodium post-genomics: better the bug you know? Nat Rev Microbiol 4:344–357
Kořený L, Lukes J, Oborník M (2010) Evolution of the haem synthetic pathway in kinetoplastid flagellates: an essential pathway that is not essential after all? Int J Parasitol 40:149–156
Kořený L, Oborník M, Lukes J (2013) Make it, take it, or leave it: heme metabolism of parasites. PLoS Pathog 9:e1003088
Kouni el MH (2003) Potential chemotherapeutic targets in the purine metabolism of parasites. Pharmacol Ther 99:283–309
Krauth-Siegel RL, Comini MA (2008) Redox control in trypanosomatids, parasitic protozoa with trypanothione-based thiol metabolism. Biochim Biophys Acta 1780:1236–1248
Kronenberger T, Schettert I, Wrenger C (2013) Targeting the vitamin biosynthesis pathways for the treatment of malaria. Future Med Chem 5:769–779
Krug EC, Marr JJ, Berens RL (1989) Purine metabolism in Toxoplasma gondii. J Biol Chem 264:10601–10607
Krugliak M, Zhang J, Ginsburg H (2002) Intraerythrocytic Plasmodium falciparum utilizes only a fraction of the amino acids derived from the digestion of host cell cytosol for the biosynthesis of its proteins. Mol Biochem Parasitol 119:249–256
Landfear SM, Ullman B, Carter NS, Sanchez MA (2004) Nucleoside and nucleobase transporters in parasitic protozoa. Eukaryotic Cell 3:245–254
Lim L, McFadden GI (2010) The evolution, metabolism and functions of the apicoplast. Philos Trans R Soc Lond B Biol Sci 365:749–763
Macedo JP, Currier RB, Wirdnam C et al (2017) Ornithine uptake and the modulation of drug sensitivity in Trypanosoma brucei. FASEB J 31(10):4649–4660. fj.201700311R–17
Martin JL, Yates PA, Soysa R et al (2014) Metabolic reprogramming during purine stress in the protozoan pathogen Leishmania donovani. PLoS Pathog 10:e1003938
Meireles P, Mendes AM, Aroeira RI et al (2017) Uptake and metabolism of arginine impact Plasmodium development in the liver. Sci Rep 7:849–812
Mesquita I, Varela P, Belinha A et al (2016) Exploring NAD(+) metabolism in host-pathogen interactions. Cell Mol Life Sci 73:1225–1236
Michels PAM, Avilán L (2011) The NAD(+) metabolism of Leishmania, notably the enzyme nicotinamidase involved in NAD(+) salvage, offers prospects for development of anti-parasite chemotherapy. Mol Microbiol 82:4–8
Miller-Fleming L, Olín-Sandoval V, Campbell K, Ralser M (2015) Remaining mysteries of molecular biology: the role of polyamines in the cell. J Mol Biol 427:3389–3406
Mukkada AJ, Meade JC, Glaser TA, Bonventre PF (1985) Enhanced metabolism of Leishmania donovani amastigotes at acid pH: an adaptation for intracellular growth. Science 229:1099–1101
Müller IB, Hyde JE (2013) Folate metabolism in human malaria parasites—75 years on. Mol Biochem Parasitol 188:63–77
Müller S, Kappes B (2007) Vitamin and cofactor biosynthesis pathways in Plasmodium and other apicomplexan parasites. Trends Parasitol 23:112–121
Müller IB, Hyde JE, Wrenger C (2010) Vitamin B metabolism in Plasmodium falciparum as a source of drug targets. Trends Parasitol 26:35–43
Nagaraj VA, Sundaram B, Varadarajan NM et al (2013) Malaria parasite-synthesized heme is essential in the mosquito and liver stages and complements host heme in the blood stages of infection. PLoS Pathog 9:e1003522–e1003513
Nare B, Hardy LW, Beverley SM (1997) The roles of pteridine reductase 1 and dihydrofolate reductase-thymidylate synthase in pteridine metabolism in the protozoan parasite Leishmania major. J Biol Chem 272:13883–13891
Nussbaum K, Honek J, Cadmus CMCVC, Efferth T (2010) Trypanosomatid parasites causing neglected diseases. Curr Med Chem 17:1594–1617
Nzila A, Okombo J, Molloy AM (2014) Impact of folate supplementation on the efficacy of sulfadoxine/pyrimethamine in preventing malaria in pregnancy: the potential of 5-methyl-tetrahydrofolate. J Antimicrob Chemother 69:323–330
O’Hara JK, Kerwin LJ, Cobbold SA et al (2014) Targeting NAD+ metabolism in the human malaria parasite Plasmodium falciparum. PLoS One 9:e94061
Opperdoes FR, Butenko A, Flegontov P et al (2016) Comparative metabolism of free-living Bodo saltans and parasitic trypanosomatids. J Eukaryot Microbiol 63:657–678
Ortiz D, Sanchez MA, Koch HP et al (2009) An acid-activated nucleobase transporter from Leishmania major. J Biol Chem 284:16164–16169
Ortiz D, Valdes R, Sanchez MA et al (2010) Purine restriction induces pronounced translational upregulation of the NT1 adenosine/pyrimidine nucleoside transporter in Leishmania major. Mol Microbiol 78:108–118
Ouellette M, Drummelsmith J, El-Fadili A et al (2002) Pterin transport and metabolism in Leishmania and related trypanosomatid parasites. Int J Parasitol 32:385–398
Peacock CS, Seeger K, Harris D et al (2007) Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat Genet 39:839–847
Rajendran E, Hapuarachchi SV, Miller CM et al (2017) Cationic amino acid transporters play key roles in the survival and transmission of apicomplexan parasites. Nat Commun 8:1–13
Roberts S, Ullman B (2017) Parasite polyamines as pharmaceutical targets. Curr Pharm Des 23:1–17
Salcedo-Sora JE, Ward SA (2013) The folate metabolic network of falciparum malaria. Mol Biochem Parasitol 188:51–62
Salcedo-Sora JE, Ochong E, Beveridge S et al (2011) The molecular basis of folate salvage in Plasmodium falciparum: characterization of two folate transporters. J Biol Chem 286:44659–44668
Sánchez-Lancheros DM, Ospina-Giraldo LF, Ramírez-Hernández MH et al (2016) Nicotinamide mononucleotide adenylyltransferase of Trypanosoma cruzi (TcNMNAT): a cytosol protein target for serine kinases. Mem Inst Oswaldo Cruz 111:670–675
Shameer S, Logan-Klumpler FJ, Vinson F et al (2015) TrypanoCyc: a community-led biochemical pathways database for Trypanosoma brucei. Nucleic Acids Res 43:D637–D644
Shanmugasundram A, Gonzalez-Galarza FF, Wastling JM et al (2013) Library of apicomplexan metabolic pathways: a manually curated database for metabolic pathways of apicomplexan parasites. Nucleic Acids Res 41:D706–D713
Sigala PA, Goldberg DE (2014) The peculiarities and paradoxes of Plasmodium heme metabolism. Annu Rev Microbiol 68:259–278
Sigala PA, Crowley JR, Henderson JP, Goldberg DE (2015) Deconvoluting heme biosynthesis to target blood-stage malaria parasites. eLife 4:50
Tjhin ET, Spry C, Sewell AL et al (2017) Mutations in the pantothenate kinase of Plasmodium falciparum confer diverse sensitivity profiles to antiplasmodial pantothenate analogues. bioRxiv:137182
Toh SQ, Glanfield A, Gobert GN, Jones MK (2010) Heme and blood-feeding parasites: friends or foes? Parasit Vectors 3:108
Vasudevan G, Carter NS, Drew ME et al (1998) Cloning of Leishmania nucleoside transporter genes by rescue of a transport-deficient mutant. Proc Natl Acad Sci 95:9873–9878
Vickers TJ, Beverley SM (2011) Folate metabolic pathways in Leishmania. Essays Biochem 51:63–80
Wainwright M, Maisch T, Nonell S et al (2017) Photoantimicrobials-are we afraid of the light? Lancet Infect Dis 17:e49–e55
Waller RF, Keeling PJ, Donald RGK et al (1998) Nuclear-encoded proteins target to the plastid in Toxoplasma gondii and Plasmodium falciparum. Proc Natl Acad Sci 95:12352–12357
Wilson RJ, Denny PW, Preiser PR et al (1996) Complete gene map of the plastid-like DNA of the malaria parasite Plasmodium falciparum. J Mol Biol 261:155–172
Woo YH, Ansari H, Otto TD et al (2015) Chromerid genomes reveal the evolutionary path from photosynthetic algae to obligate intracellular parasites. eLife 4:441
World Malaria Report 2016: Summary. World Health Organization, Geneva; 2017 (WHO/HTM/GMP/2017.4). Licence: CC BY-NC-SA 3.0 IGO.
Zhang C-J, Chia WN, Loh CCY et al (2015) Haem-activated promiscuous targeting of artemisinin in Plasmodium falciparum. Nat Commun 6:1–11
Zuegge J, Ralph S, Schmuker M et al (2001) Deciphering apicoplast targeting signals – feature extraction from nuclear-encoded precursors of Plasmodium falciparum apicoplast proteins. Gene 280:19–26
Zwerschke D, Karrie S, Jahn D, Jahn M (2014) Leishmania major possesses a unique HemG-type protoporphyrinogen IX oxidase. Biosci Rep 34:391–400
Funding Statement
This work was supported by the IRD (Institut de Recherche pour le Développement) institutional funding. E.G. is a recipient of the FRM (Fondation pour la Recherche Médicale) postdoctoral fellowship (ARF2015093409).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

{kind=link}
Cite this chapter
Gazanion, E., Vergnes, B. (2018). Protozoan Parasite Auxotrophies and Metabolic Dependencies. In: Silvestre, R., Torrado, E. (eds) Metabolic Interaction in Infection. Experientia Supplementum, vol 109. Springer, Cham. https://doi.org/10.1007/978-3-319-74932-7_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-74932-7_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-74931-0
Online ISBN: 978-3-319-74932-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)