
Abstract
In 1999, a polyglutamine expansion was identified in the transcription factor TATA-binding protein (TBP) in a patient with ataxia with negative family history. Subsequently, CAG/CAA repeat expansions in the TBP gene were identified in families with spinocerebellar ataxia (SCA), establishing this repeat expansion as the underlying mutation in SCA type 17 (SCA17). There are several characteristic differences between SCA17 and other polyglutamine diseases. First, SCA17 shows a complex and variable clinical phenotype, in some cases overlapping that of Huntington’s disease. Second, compared to the other SCA subtypes caused by expanded trinucleotide repeats, anticipation in SCA17 kindreds is rare because of the characteristic structure of the TBP gene. And thirdly, SCA17 patients often have diagnostic problems that may arise from non-penetrance. Because the gap between normal and abnormal repeat numbers is very narrow, it is difficult to determine a cutoff value for pathologic CAG repeat number in SCA17. Herein, we review the clinical, genetic and pathologic features of SCA17.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
10.1 Clinical Features
SCA17 is an autosomal dominant cerebellar ataxia caused by abnormal expansion of a CAG/CAA repeat encoding a polyglutamine (polyQ) tract in the TATA-box binding protein (TBP) gene on chromosome 6q [1,2,3,4]. Though the genetic abnormalities are mostly observed as a hereditary trait, de novo mutations have also been reported [2, 5, 6].
The clinical symptoms of SCA17 are mainly ataxia and/or dementia , as is the case for other forms of autosomal dominant SCA [7]. However, SCA17 is a more complex disease with extensive phenotypic variability, and the age at onset spans several decades. Clinical heterogeneity can be observed even within the same family [8]. In the present literature review of SCA17 patients (Table 10.1), the age at onset ranged from 3 to 60 years, and about half of the patients developed ataxia as the initial symptom. The age at onset appeared to correlate weakly with the number of repeats (Fig. 10.1). During the disease course, most of the patients (>90%) developed ataxia, which was manifested as gait instability, and slurred speech. Cognitive dysfunction and memory disturbance have also been recognized as an initial symptom [9]; dementia is the second most common symptom (73%) during the disease course. In childhood, mental deterioration may occur instead of dementia . Psychiatric symptoms , such as aggression [10], paranoia [1], euphoria [11] and depression [12, 13] are observed frequently. Behavior or personality changes as initial symptoms may indicate the presence of psychiatric disorders.

Correlation between age at onset (years) and CAA/CAG repeat number on the expanded allele
Involuntary movement is one of the characteristic features of SCA17 [14, 15]. As chorea is a well-known symptom of SCA 17, the clinical phenotype sometimes overlaps that of Huntington’s disease (HD), being characterized by the triad of movement disorder, psychiatric manifestations and cognitive impairment [16]. In many cases of clinically suspected HD, patients lack the CAG repeat expansion that causes HD. Such individuals are said to have HD phenocopy syndromes or HD-like disorders [17,18,19,20,21,22,23]. SCA17 has therefore also been termed Huntington’s disease -like 4 (HDL4; OMIM #607136) [24]. Wild et al. [21] identified gene abnormalities in 285 HD phenocopy patients in the United Kingdom. Among the patients, five (1.8%) were found to have expansions in TBP causing SCA 17. One patient (0.4%) had a 6-octapeptide insertion in the prion protein gene (PRNP) [25], and one had HDL2 caused by a pathogenic expansion in the janctofillin gene (JPH3) [26]. In addition, one patient was diagnosed later as having Friedreich’s ataxia with homozygous expansion in the flataxin gene (FXN) [27]. Moss et al. [28] recently reported that ten (1.95%) of 514 HD phenocopy patients had an expanded hexanucleotide repeat in the C9orf72 gene. If patients with HD-like disease have no mutations in huntingtin , the TBP and C9orf72 genes should be examined.
We have previously examined the relationship between repeat number and clinical symptoms (Toyoshima et al. 1993), and found that more than 75% of patients with a CAG/CAA repeat size of 43–50 had intellectual deterioration; in some individuals, intellectual problems and involuntary movements were the only signs. Psychiatric problems or dementia , parkinsonism and chorea , a clinical constellation resembling Huntington disease, are observed more frequently in individuals with CAG/CAA repeats in this range than in those with larger repeats. All individuals showing a CAG/CAA repeat size of 50–60 have ataxia and 75% have reduced intellectual function. Pyramidal signs (e.g., increased deep tendon reflexes) and dystonia are more common in these individuals than in those with smaller repeats. These features were also confirmed in the present literature review (Table 10.1). Two children with over 60 repeats have been reported. One, a familial example, with a CAG/CAA expansion of 66 repeats developed gait disturbance at the age of 3 years followed by spasticity, dementia and psychiatric symptoms [29]. The other, with a de novo CAG/CAA expansion of 63 repeats, developed ataxia and intellectual deterioration at the age of 6 years, followed later by spasticity [2].
Less common symptoms reported are epilepsy [1,2,3, 13, 29,30,31] (20%), autonomic symptoms [4, 11, 31, 32] (9%), apraxia [31] (7%) and peripheral nerve symptoms [12, 13, 29] (3%). Lin et al. [11] reported a patient who developed ophthalmoplegia with parkinsonism, and Rolfs et al. [31] reported a patient showing hypogonadism.
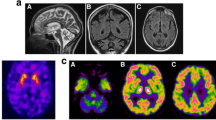
Neuroradiological examination has been reported to demonstrate several characteristic features. In early reports, some degree of cerebellar and cerebral atrophy was shown on MRI [1, 3]. Later, putaminal rim hyperintensity on T2-weighted images was also reported [33]. Striatal hyperintensity on MRI has been reported in a range of disease conditions, including SCA2 [34], dentatorubral-pallidoluysian atrophy (DRPLA) [35], neuroacanthocytosis [36], mitochondrial cytopathy [37] and multiple system atrophy (MSA) [38, 39]. Functional imaging using radioisotopes is helpful for understanding the pathophysiological conditions in the basal ganglia. Günther et al. [40] measured striatal pre-synaptic dopamine transporter (DAT) availability and striatal dopamine D2 receptor (D2R) expression using [123I]FP-CIT and [123I]IBZM, respectively, applying a brain-dedicated single-photon emission computed tomography (SPECT) system; [18F]Fluordeoxyglucose positron emission tomography scanning (PET) was also used for measurement of glucose metabolism. They found that the striatum had a reduction in the availability of presynaptic dopamine transporters, although postsynaptic dopamine D2 receptor binding capacity was reduced only slightly, whereas marked reduction of glucose metabolism was evident in the basal ganglia. Lin et al. [11] reported a patient with parkinsonism partially responsive to L-dopa administration, in whom presynaptic degeneration of the nigrostriatal dopaminergic system was manifested by a marked decrease of dopa uptake in the striatum , as demonstrated by [18F]-6-fluorodopa PET.
10.2 Genetic Cause and Penetrance of SCA17
SCA17 is caused by abnormal expansion of the TBP gene on chromosome 6q. TBP is a transcription factor with a polyglutamine tract encoded by the CAG/CAA sequence [1,2,3]. The normal repeat range reported is from 25 up to 42–45 units, varying among studies. The highest number of repeats reported to date is 66 [29]. Expansions of between 41 and 49 repeats may constitute an intermediate range with incomplete penetrance. In our previous review [41], the pathologic number of repeats was 43 or more. However, there have been reports of patients with 41 repeats, presenting with progressive cerebellar ataxia and/or involuntary movements [12, 32, 42], late-onset chorea and psychiatric symptoms [43] and parkinsonism with chorea [44]. On the other hand, healthy individuals with 45 [45] and 49 repeats [46] have also been recognized. Because the gap between normal and abnormal numbers of repeats is very narrow, it is difficult to determine the cutoff value for the pathologic number of CAG repeats in SCA17. Shin et al. [47] reported very interesting results from large genetic study conducted in Korea. They examined 2099 patients (classified by dominant clinical phenotype: parkinsonism, n = 1706; ataxia, n = 345; chorea , n = 37; and dystonia , n = 11) and 522 normal controls, and reported that 64 patients had 42 repeats or less (3%) in the TBP gene. Forty-five repeats were the greatest number in normal populations. They recommended that 41 through 45 repeats should be considered as the intermediate range, requiring cautious interpretation.
Compared to the other SCA subtypes caused by expanded trinucleotide repeats, anticipation [48] is rare in SCA17 kindreds because interruption of CAA within the CAG repeat configuration of the TBP gene stabilizes the microsatellite [49, 50]. The allele basic structure is (CAG)3 (CAA)3 (CAG)n CAA CAG CAA (CAG)n CAA CAG. However, when the basic structure is broken, anticipation may be observed [50]. Maltecca et al. [29] reported a family showing marked anticipation. PCR analysis of the CAG repeat region within the TBP gene in the third generation showed a 300-base-pair (bp) band (53 CAG/CAA repeats), whereas the patient in the fourth generation showed a band of ~330 bp (66 repeats). The allele structure of the father was (CAG)3 (CAA)4 (CAG)44 CAA CAG, and that of the daughter was (CAG)3 (CAA)4 (CAG)57 CAA CAG. The basic allele structure was not conserved in patients whose alleles had a simplified pattern with loss of CAA interruptions, possibly leading to a reduction of repeat stability.
We previously reported a patient having 48 CAA/CAG repeats as a homozygous state [15]. The clinical features of that patient essentially did not differ from those of heterozygotes, although homozygosity might have exerted some influence on the severity and progression of dementia . Zühlke et al. [51] also reported a patient who had 47 homozygous glutamine residues caused by apparent partial isodisomy 6. Compared with the heterozygote, the patient had no apparent differences in clinical features.
Almost all SCA17 patients have a family history and the disease is inherited as an autosomal dominant trait. However, a few cases develop de novo mutations in the TBP gene. One example is the child with 63 repeats mentioned above [2]. In addition, [5] reported a 33-year-old woman presenting with a HD-like phenotype with a de novo 54 CAG/CAA repeat expansion, and Wu et al. (2004) described a de novo 55 CAG repeat expansion in a patient with a Pakinson’s disease phenotype. The presence of such cases indicates that even in the absence of a positive family history, genetic testing for SCA17 should be considered in patients with a HD-like, or PD-like, phenocopy.
10.3 Pathological Findings
Several reports have described the pathological findings in SCA17 patients [1, 52]. We previously reported a homozygous patient focusing on the histopathological findings; the patient did not show earlier onset of the disease than heterozygotes reported with similar CAA/CAG-repeat sizes, and the pathological features appeared to be very similar, if not identical, to those described for heterozygotes [15].
The patient died at the age of 49 years, about 6 years after disease onset. The main symptoms were dementia and choreic movement. At autopsy, the brain showed mild atrophy in the caudate nucleus and putamen (Fig. 10.2). Histologically, mild neuronal loss and gliosis were observed in the cerebral cortex, especially the deep layers. Neuronal loss was moderate in the neostriatum, affecting both small and large neurons, and in the Purkinje cell layer with Bergmann gliosis (Fig. 10.3). Mild neuronal loss was also evident in the cornu ammonis region 1 (CA1) of Ammon’s horn, subiculum, parahippocampal gyrus, substantia nigra , brainstem reticular formation, and inferior olivary nucleus. It is known that the formation of neuronal intranuclear inclusions (NIIs) is a common hallmark of the CAG repeat diseases [53]. In the above SCA17 patient, immunohistochemistry showed that expanded polyQ stretches had accumulated in the neuronal nuclei in a diffuse pattern [54, 55] (Fig. 10.4a), and no labeling was detected in their cytoplasm or in the neuropil. NII formation was rare (<1%) in affected neurons and was restricted to brain regions such as the cerebral cortex (Fig. 10.4b), putamen, and midbrain reticular formation. The intranuclear diffuse accumulation of polyQ involved many neurons in a wide range of CNS regions far beyond the lesion distribution assessed by neuronal loss. Regions in which more than 40% of neurons were 1C2- immunoreactive included the sixth layer of the cerebral cortex (Fig. 10.5a), neostriatum, hippocampal formation (CA1 and subiculum) (Fig. 10.5b) and Purkinje cell layer. Of note was the high frequency (>60%) of nuclear 1C2 immunoreactivity in the large neurons of the neostriatum. Brain regions showing a frequency of 20–40% included the cerebral cortical layers III and VI, subthalamic nucleus, and inferior olive. In the white matter, a few glial nuclei also were immunolabeled for 1C2. No positive staining was observed in the visceral organs.

Coronal section of the patient (a) showing mild atrophy of the caudate nucleus (*) and putamen (p). (b) Age matched control

There is moderate loss of Purkinje cells with Bergmann gliosis in the cerebellum . The granule cells are well preserved. Hematoxylin and eosin. Scale bar = 50 μm

The neuronal nuclei are often stained diffusely by 1C2 (a). There are a few neurons showing intra nuclear inclusion (b, arrow). a putamen. b brainstem reticular formation. Immunohistochemistry with a monoclonal antibody (1C2). Scale bar = 10 μm

There are many 1C2-positive nuclei in the subiculum of the hippocampal formation (a). 1C2-positive neuronal nuclei in the neurons (*) and an oligodendrocyte (arrow) (b, frontal cortex). Immunohistochemistry with 1C2. Scale bar = a 50 μm, b 20 μm
Intranuclear accumulation of mutant proteins has been recognized in many polyQ-disease brains of humans [56, 57] and transgenic mice [54]. The wide neuronal intranuclear distribution of the mutant proteins in the CNS far beyond the lesions assessed by neuronal loss must be important when considering the clinical and pathological correlations in polyQ diseases, including SCA17 [54, 56, 57].
10.4 Pathophysiological Reviews
The molecular mechanisms responsible for the pathogenesis of the polyQ diseases have not yet been completely explained [58, 59]. PolyQ expansion renders the protein more prone to aggregation and formation of inclusion bodies that are a pathological hallmark of polyglutamine diseases [60, 61]. Early discussions focused on whether the tendency of mutant polyQ proteins to aggregate was responsible for the disease-associated neurodegeneration (gain of function). Yet, several studies have indicated that disease severity can be disassociated from the presence of inclusions [62,63,64]. Moreover, there are data indicating that inclusions may be protective, perhaps as a result of sequestration of the mutant protein [62, 65, 66].
Several disease models, including cells [67, 68], Drosophila [59, 69, 70] and rodents [71,72,73,74], have been generated to clarify the pathomechanisms of SCA17. Overexpression of full-length-mutant TBP and truncated-mutant TBP lacking the DNA-binding domains (DBDs) was found to cause formation of inclusions , suggesting that insoluble aggregates are causative factors and that the neurotoxicity of mutant TBP is independent of DNA binding [69]. Thus, the pathogenesis of SCA17 seems similar to that of other polyQ diseases [75]. On the other hand, whether or not expanded polyQ tracts affect the function of TBP has yet to be comprehensively addressed. TBP is a general transcription factor [76, 77] essential for formation of the transcription preinitiation complex and transcription of RNA polymerases I, II and III (Pol I, II and III). Aberrant TBP activity is expected to profoundly affect normal cellular function. Inactivation of TBP in mice causes downregulation of Pol I and Pol III transcription, growth arrest and cell death [78]. Friedmann et al. [79] have reported that polyQ expansion reduces in vitro binding of TBP to DNA. Furthermore, they observed that polyQ-expanded TBP fragments, which were incapable of binding DNA, formed nuclear inclusions and caused a severe neurological phenotype in transgenic mice. Huang et al. [80] reported muscle dysfunction in a knock-in mouse model that had long polyQ repeats. They considered that decreased interaction between mutant TBP and MyoD, a muscle-specific transcription factor [81, 82], might affect the association between MyoD and the DNA promoter, thus reducing its transcriptional activity. Hsu et al. [69] recently reported that deactivation of TBP may contribute to SCA17 pathogenesis. They generated novel Drosophila models for SCA17 that overexpressed polyQ-expanded TBP, and demonstrated neurotoxic aggregates , the mutant TBP sequestering wild-type TBP in the neuroblasts of the flies. Moreover, they generated Drosophila mutants with loss of TDP (dTbp) to examine whether the neurodegeneration was the same as that of the SCA-17 model flies. They confirmed that loss of TDP function caused age-associated neurodegeneration in Drosophila. Interestingly, they reported that dTbp expression exacerbated retinal degeneration induced in polyQ-expanded SCA3 and Huntington’s disease fly models. These findings suggest that dysfunction of TBP may play a universal role in polyQ-induced neurodegeneration . Therefore, it is very significant to study the pathophysiology of SCA17 to clarify the causes of other polyglutamine diseases .
References
Fujigasaki H, De Martin JJ, Deyn PP, Camuzat A, Deffond D, Stevanin G, Van Dermaut B, Broeckhoven C, Durr A, Brice A (2001) CAG repeat expansion in the TATA box-binding protein gene causes autosomal dominant cerebellar ataxia. Brain 124:1939–1947
Koide R, Kobayashi S, Shimohata T, Ikeuchi T, Maruyama M, Saito M, Yamada M, Takahashi H, Tsuji S (1999) A neurological disease caused by an expanded CAG trinucleotide repeat in the TATA-binding protein gene: a new polyglutamine disease? Hum Mol Genet 8:2047–2053
Nakamura K, Jeong SY, Uchihara T, Anno M, Nagashima K, Nagashima T, Ikeda S, Tsuji S, Kanazawa I (2001) SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet 10:1441–1448
Zuhlke C, Hellenbroich Y, Dalski A, Kononowa N, Hagenah J, Vieregge P, Riess O, Klein C, Schwinger E (2001) Different types of repeat expansion in the TATA-binding protein gene are associated with a new form of inherited ataxia. Eur J Hum Genet 9:160–164
Bech S, Petersen T, Norremolle A, Gjedde A, Ehlers L, Eiberg H, Hjermind LE, Hasholt L, Lundorf E, Nielsen JE (2010) Huntington’s disease-like and ataxia syndromes: identification of a family with a de novo SCA17/TBP mutation. Parkinsonism Relat Disord 16:12–15
Shatunov A, Fridman EA, Pagan FI, Leib J, Singleton A, Hallett M, Goldfarb LG (2004) Small de novo duplication in the repeat region of the TATA-box-binding protein gene manifest with a phenotype similar to variant Creutzfeldt-Jakob disease. Clin Genet 66:496–501
Schols L, Bauer P, Schmidt T, Schulte T, Riess O (2004) Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol 3:291–304
Koutsis G, Panas M, Paraskevas GP, Bougea AM, Kladi A, Karadima G, Kapaki E (2014) From mild ataxia to huntington disease phenocopy: the multiple faces of spinocerebellar ataxia 17. Case Rep Neurol Med 2014:643289
Nielsen TT, Mardosiene S, Lokkegaard A, Stokholm J, Ehrenfels S, Bech S, Friberg L, Nielsen JK, Nielsen JE (2012) Severe and rapidly progressing cognitive phenotype in a SCA17-family with only marginally expanded CAG/CAA repeats in the TATA-box binding protein gene: a case report. BMC Neurol 12:73
van de Schneider SA, Warrenburg BP, Hughes TD, Davis M, Sweeney M, Wood N, Quinn NP, Bhatia KP (2006) Phenotypic homogeneity of the Huntington disease-like presentation in a SCA17 family. Neurology 67:1701–1703
Lin IS, Wu RM, Lee-Chen GJ, Shan DE, Gwinn-Hardy K (2007) The SCA17 phenotype can include features of MSA-C, PSP and cognitive impairment. Parkinsonism Relat Disord 13:246–249
Herrema H, Mikkelsen T, Robin A, LeWitt P, Sidiropoulos C (2014) SCA 17 phenotype with intermediate triplet repeat number. J Neurol Sci 345:269–270
Mariotti C, Alpini D, Fancellu R, Soliveri P, Grisoli M, Ravaglia S, Lovati C, Fetoni V, Giaccone G, Castucci A, Taroni F, Gellera C Di, Donato S (2007) Spinocerebellar ataxia type 17 (SCA17): oculomotor phenotype and clinical characterization of 15 Italian patients. J Neurol 254:1538–1546
Stevanin G, Fujigasaki H, Lebre AS, Camuzat A, Jeannequin C, Dode C, Takahashi J, San C, Bellance R, Brice A, Durr A (2003) Huntington’s disease-like phenotype due to trinucleotide repeat expansions in the TBP and JPH3 genes. Brain 126:1599–1603
Toyoshima Y, Yamada M, Onodera O, Shimohata M, Inenaga C, Fujita N, Morita M, Tsuji S, Takahashi H (2004) SCA17 homozygote showing Huntington’s disease-like phenotype. Ann Neurol 55:281–286
Walker FO (2007) Huntington’s disease. Lancet 369:218–228
Bauer P, Laccone F, Rolfs A, Wullner U, Bosch S, Peters H, Liebscher S, Scheible M, Epplen JT, Weber BH, Holinski-Feder E, Weirich-Schwaiger H, Morris-Rosendahl DJ, Andrich J, Riess O (2004) Trinucleotide repeat expansion in SCA17/TBP in white patients with Huntington’s disease-like phenotype. J Med Genet 41:230–232
Kambouris M, Bohlega S, Al-Tahan A, Meyer BF (2000) Localization of the gene for a novel autosomal recessive neurodegenerative Huntington-like disorder to 4p15.3. Am J Hum Genet 66:445–452
Margolis RL, O’Hearn E, Rosenblatt A, Willour V, Holmes SE, Franz ML, Callahan C, Hwang HS, Troncoso JC, Ross CA (2001) A disorder similar to Huntington’s disease is associated with a novel CAG repeat expansion. Ann Neurol 50:373–380
Schneider SA, Walker RH, Bhatia KP (2007) The Huntington’s disease-like syndromes: what to consider in patients with a negative Huntington’s disease gene test. Nat Clin Pract Neurol 3:517–525
Wild EJ, Mudanohwo EE, Sweeney MG, Schneider SA, Beck J, Bhatia KP, Rossor MN, Davis MB, Tabrizi SJ (2008) Huntington’s disease phenocopies are clinically and genetically heterogeneous. Mov Disord 23:716–720
Wild EJ, Tabrizi SJ (2007) Huntington’s disease phenocopy syndromes. Curr Opin Neurol 20:681–687
Xiang F, Almqvist EW, Huq M, Lundin A, Hayden MR, Edstrom L, Anvret M, Zhang Z (1998) A Huntington disease-like neurodegenerative disorder maps to chromosome 20p. Am J Hum Genet 63:1431–1438
Stevanin G, Brice A (2008) Spinocerebellar ataxia 17 (SCA17) and Huntington’s disease-like 4 (HDL4). Cerebellum 7:170–178
Moore RC, Xiang F, Monaghan J, Han D, Zhang Z, Edstrom L, Anvret M, Prusiner SB (2001) Huntington disease phenocopy is a familial prion disease. Am J Hum Genet 69:1385–1388
Holmes SE, O’Hearn E, Rosenblatt A, Callahan C, Hwang HS, Ingersoll-Ashworth RG, Fleisher A, Stevanin G, Brice A, Potter NT, Ross CA, Margolis RL (2001) A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease-like 2. Nat Genet 29:377–378
Hanna MG, Davis MB, Sweeney MG, Noursadeghi M, Ellis CJ, Elliot P, Wood NW, Marsden CD (1998) Generalized chorea in two patients harboring the Friedreich’s ataxia gene trinucleotide repeat expansion. Mov Disord 13:339–340
Hensman Moss DJ, Poulter M, Beck J, Hehir J, Polke JM, Campbell T, Adamson G, Mudanohwo E, McColgan P, Haworth A, Wild EJ, Sweeney MG, Houlden H, Mead S, Tabrizi SJ (2014) C9orf72 expansions are the most common genetic cause of Huntington disease phenocopies. Neurology 82:292–299
Maltecca F, Filla A, Castaldo I, Coppola G, Fragassi NA, Carella M, Bruni A, Cocozza S, Casari G, De Servadio A, Michele G (2003) Intergenerational instability and marked anticipation in SCA-17. Neurology 61:1441–1443
Belluzzo M, Musho-Ilbeh S, Monti F, Pizzolato G (2012) A case of nocturnal frontal lobe epilepsy in a patient with spinocerebellar ataxia type 17. Seizure 21:805–806
Rolfs A, Koeppen AH, Bauer I, Bauer P, Buhlmann S, Topka H, Schols L, Riess O (2003) Clinical features and neuropathology of autosomal dominant spinocerebellar ataxia (SCA17). Ann Neurol 54:367–375
Doherty KM, Warner TT, Lees AJ (2014) Late onset ataxia: MSA-C or SCA 17? A gene penetrance dilemma. Mov Disord 29:36–38
Loy CT, Sweeney MG, Davis MB, Wills AJ, Sawle GV, Lees AJ, Tabrizi SJ (2005) Spinocerebellar ataxia type 17: extension of phenotype with putaminal rim hyperintensity on magnetic resonance imaging. Mov Disord 20:1521–1523
Payami H, Nutt J, Gancher S, Bird T, McNeal MG, Seltzer WK, Hussey J, Lockhart P, Gwinn-Hardy K, Singleton AA, Singleton AB, Hardy J, Farrer M (2003) SCA2 may present as levodopa-responsive parkinsonism. Mov Disord 18:425–429
Imamura A, Sugai K, Watanabe S, Hamada F, Kurashige T, Takashima S (1994) High intensity in the globus pallidus on proton and T2-weighted MRI in a case of dentato-ruburo-pallido-luysian atrophy of myoclonus epilepsy type. Acta Paediatr Jpn 36:527–530
Tanaka M, Hirai S, Kondo S, Sun X, Nakagawa T, Tanaka S, Hayashi K, Okamoto K (1998) Cerebral hypoperfusion and hypometabolism with altered striatal signal intensity in chorea-acanthocytosis: a combined PET and MRI study. Mov Disord 13:100–107
Wray SH, Provenzale JM, Johns DR, Thulborn KR (1995) MR of the brain in mitochondrial myopathy. AJNR Am J Neuroradiol 16:1167–1173
Feng JY, Huang B, Yang WQ, Zhang YH, Wang LM, Wang LJ, Zhong XL (2015) The putaminal abnormalities on 3.0T magnetic resonance imaging: can they separate parkinsonism-predominant multiple system atrophy from Parkinson’s disease? Acta Radiol 56:322–328
Schrag A, Kingsley D, Phatouros C, Mathias CJ, Lees AJ, Daniel SE, Quinn NP (1998) Clinical usefulness of magnetic resonance imaging in multiple system atrophy. J Neurol Neurosurg Psychiatry 65:65–71
Gunther P, Storch A, Schwarz J, Sabri O, Steinbach P, Wagner A, Hesse S (2004) Basal ganglia involvement of a patient with SCA 17—a new form of autosomal dominant spinocerebellar ataxia. J Neurol 251:896–897
Toyoshima Y, Onodera O, Yamada M, Tsuji S, Takahashi H (2005) Spinocerebellar ataxia type 17. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, Stephens K (eds) GeneReviews(R). Seattle (WA)
Nanda A, Jackson SA, Schwankhaus JD, Metzer WS (2007) Case of spinocerebellar ataxia type 17 (SCA17) associated with only 41 repeats of the TATA-binding protein (TBP) gene. Mov Disord 22:436
Alibardi A, Squitieri F, Fattapposta F, Missori P, Pierelli F, Trompetto C, Curra A (2014) Psychiatric onset and late chorea in a patient with 41 CAG repeats in the TATA-binding protein gene. Parkinsonism Relat Disord 20:678–679
Park H, Jeon BS, Shin JH, Park SH (2016) A patient with 41 CAG repeats in SCA17 presenting with parkinsonism and chorea. Parkinsonism Relat Disord 22:106–107
Oda M, Maruyama H, Komure O, Morino H, Terasawa H, Izumi Y, Imamura T, Yasuda M, Ichikawa K, Ogawa M, Matsumoto M, Kawakami H (2004) Possible reduced penetrance of expansion of 44 to 47 CAG/CAA repeats in the TATA-binding protein gene in spinocerebellar ataxia type 17. Arch Neurol 61:209–212
Zuhlke C, Dalski A, Schwinger E, Finckh U (2005) Spinocerebellar ataxia type 17: report of a family with reduced penetrance of an unstable Gln49 TBP allele, haplotype analysis supporting a founder effect for unstable alleles and comparative analysis of SCA17 genotypes. BMC Med Genet 6:27
Shin JH, Park H, Ehm GH, Lee WW, Yun JY, Kim YE, Lee JY, Kim HJ, Kim JM, Jeon BS, Park SS (2015) The pathogenic role of low range repeats in SCA17. PLoS ONE 10:e0135275
Duyao M, Ambrose C, Myers R, Novelletto A, Persichetti F, Frontali M, Folstein S, Ross C, Franz M, Abbott M et al (1993) Trinucleotide repeat length instability and age of onset in Huntington’s disease. Nat Genet 4:387–392
Dorsman JC, Bremmer-Bout M, van Pepers B, Den Ommen GJ, Dunnen JT (2002) Interruption of perfect CAG repeats by CAA triplets improves the stability of glutamine-encoding repeat sequences. Biotechniques 33:976–978
Gao R, Matsuura T, Coolbaugh M, Zuhlke C, Nakamura K, Rasmussen A, Siciliano MJ, Ashizawa T, Lin X (2008) Instability of expanded CAG/CAA repeats in spinocerebellar ataxia type 17. Eur J Hum Genet 16:215–222
Zuhlke CH, Spranger M, Spranger S, Voigt R, Lanz M, Gehlken U, Hinrichs F, Schwinger E (2003) SCA17 caused by homozygous repeat expansion in TBP due to partial isodisomy 6. Eur J Hum Genet 11:629–632
Bruni AC, Takahashi-Fujigasaki J, Maltecca F, Foncin JF, Servadio A, Casari G, D’Adamo P, Maletta R, Curcio SA De, Michele G, Filla A El, Hachimi KH, Duyckaerts C (2004) Behavioral disorder, dementia, ataxia, and rigidity in a large family with TATA box-binding protein mutation. Arch Neurol 61:1314–1320
Yamada M, Tsuji S, Takahashi H (2000) Pathology of CAG repeat diseases. Neuropathology 20:319–325
Sato T, Miura M, Yamada M, Yoshida T, Wood JD, Yazawa I, Masuda M, Suzuki T, Shin RM, Yau HJ, Liu FC, Shimohata T, Onodera O, Ross CA, Katsuki M, Takahashi H, Kano M, Aosaki T, Tsuji S (2009) Severe neurological phenotypes of Q129 DRPLA transgenic mice serendipitously created by en masse expansion of CAG repeats in Q76 DRPLA mice. Hum Mol Genet 18:723–736
Yamada M, Sato T, Tsuji S, Takahashi H (2008) CAG repeat disorder models and human neuropathology: similarities and differences. Acta Neuropathol 115:71–86
Yamada M, Hayashi S, Tsuji S, Takahashi H (2001) Involvement of the cerebral cortex and autonomic ganglia in Machado-Joseph disease. Acta Neuropathol 101:140–144
Yamada M, Wood JD, Shimohata T, Hayashi S, Tsuji S, Ross CA, Takahashi H (2001) Widespread occurrence of intranuclear atrophin-1 accumulation in the central nervous system neurons of patients with dentatorubral-pallidoluysian atrophy. Ann Neurol 49:14–23
Lupton CJ, Steer DL, Wintrode PL, Bottomley SP, Hughes VA, Ellisdon AM (2015) Enhanced molecular mobility of ordinarily structured regions drives polyglutamine disease. J Biol Chem 290:24190–24200
Xu Z, Tito AJ, Rui YN, Zhang S (2015) Studying polyglutamine diseases in Drosophila. Exp Neurol 274:25–41
Ross CA, Margolis RL, Becher MW, Wood JD, Engelender S, Cooper JK, Sharp AH (1998) Pathogenesis of neurodegenerative diseases associated with expanded glutamine repeats: new answers, new questions. Prog Brain Res 117:397–419
Trottier Y, Lutz Y, Stevanin G, Imbert G, Devys D, Cancel G, Saudou F, Weber C, David G, Tora L et al (1995) Polyglutamine expansion as a pathological epitope in Huntington’s disease and four dominant cerebellar ataxias. Nature 378:403–406
Cummings CJ, Reinstein E, Sun Y, Antalffy B, Jiang Y, Ciechanover A, Orr HT, Beaudet AL, Zoghbi HY (1999) Mutation of the E6-AP ubiquitin ligase reduces nuclear inclusion frequency while accelerating polyglutamine-induced pathology in SCA1 mice. Neuron 24:879–892
Klement IA, Skinner PJ, Kaytor MD, Yi H, Hersch SM, Clark HB, Zoghbi HY, Orr HT (1998) Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell 95:41–53
Saudou F, Finkbeiner S, Devys D, Greenberg ME (1998) Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95:55–66
Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S (2004) Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431:805–810
Bowman AB, Lam YC, Jafar-Nejad P, Chen HK, Richman R, Samaco RC, Fryer JD, Kahle JJ, Orr HT, Zoghbi HY (2007) Duplication of Atxn1l suppresses SCA1 neuropathology by decreasing incorporation of polyglutamine-expanded ataxin-1 into native complexes. Nat Genet 39:373–379
Reid SJ, van Rees MI, Roon-Mom WM, Jones AL, MacDonald ME, Sutherland G, During MJ, Faull RL, Owen MJ, Dragunow M, Snell RG (2003) Molecular investigation of TBP allele length: a SCA17 cellular model and population study. Neurobiol Dis 13:37–45
Schaffar G, Breuer P, Boteva R, Behrends C, Tzvetkov N, Strippel N, Sakahira H, Siegers K, Hayer-Hartl M, Hartl FU (2004) Cellular toxicity of polyglutamine expansion proteins: mechanism of transcription factor deactivation. Mol Cell 15:95–105
Hsu TC, Wang CK, Yang CY, Lee LC, Hsieh-Li HM, Ro LS, Chen CM, Lee-Chen GJ, Su MT (2014) Deactivation of TBP contributes to SCA17 pathogenesis. Hum Mol Genet 23:6878–6893
Ren J, Jegga AG, Zhang M, Deng J, Liu J, Gordon CB, Aronow BJ, Lu LJ, Zhang B, Ma J (2011) A Drosophila model of the neurodegenerative disease SCA17 reveals a role of RBP-J/Su(H) in modulating the pathological outcome. Hum Mol Genet 20:3424–3436
Friedman MJ, Shah AG, Fang ZH, Ward EG, Warren ST, Li S, Li XJ (2007) Polyglutamine domain modulates the TBP-TFIIB interaction: implications for its normal function and neurodegeneration. Nat Neurosci 10:1519–1528
Huang S, Ling JJ, Yang S, Li XJ, Li S (2011) Neuronal expression of TATA box-binding protein containing expanded polyglutamine in knock-in mice reduces chaperone protein response by impairing the function of nuclear factor-Y transcription factor. Brain 134:1943–1958
Kelp A, Koeppen AH, Petrasch-Parwez E, Calaminus C, Bauer C, Portal E, Yu-Taeger L, Pichler B, Bauer P, Riess O, Nguyen HP (2013) A novel transgenic rat model for spinocerebellar ataxia type 17 recapitulates neuropathological changes and supplies in vivo imaging biomarkers. J Neurosci 33:9068–9081
Lee GC, Lin CH, Tao YC, Yang JM, Hsu KC, Huang YJ, Huang SH, Kung PJ, Chen WL, Wang CM, Wu YR, Chen CM, Lin JY, Hsieh-Li HM, Lee-Chen GJ (2015) The potential of lactulose and melibiose, two novel trehalase-indigestible and autophagy-inducing disaccharides, for polyQ-mediated neurodegenerative disease treatment. Neurotoxicology 48:120–130
Koshy BT, Zoghbi HY (1997) The CAG/polyglutamine tract diseases: gene products and molecular pathogenesis. Brain Pathol 7:927–942
Burley SK (1996) The TATA box binding protein. Curr Opin Struct Biol 6:69–75
Sharp PA (1992) TATA-binding protein is a classless factor. Cell 68:819–821
Martianov I, Viville S, Davidson I (2002) RNA polymerase II transcription in murine cells lacking the TATA binding protein. Science 298:1036–1039
Friedman MJ, Wang CE, Li XJ, Li S (2008) Polyglutamine expansion reduces the association of TATA-binding protein with DNA and induces DNA binding-independent neurotoxicity. J Biol Chem 283:8283–8290
Huang S, Yang S, Guo J, Yan S, Gaertig MA, Li S, Li XJ (2015) Large polyglutamine repeats cause muscle degeneration in SCA17 mice. Cell Rep 13:196–208
Di Marco S, Mazroui R, Dallaire P, Chittur S, Tenenbaum SA, Radzioch D, Marette A, Gallouzi IE (2005) NF-kappa B-mediated MyoD decay during muscle wasting requires nitric oxide synthase mRNA stabilization, HuR protein, and nitric oxide release. Mol Cell Biol 25:6533–6545
Guttridge DC, Mayo MW, Madrid LV, Wang CY, Baldwin AS Jr (2000) NF-kappaB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science 289:2363–2366
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter

Cite this chapter
Toyoshima, Y., Takahashi, H. (2018). Spinocerebellar Ataxia Type 17 (SCA17). In: Nóbrega, C., Pereira de Almeida, L. (eds) Polyglutamine Disorders. Advances in Experimental Medicine and Biology, vol 1049. Springer, Cham. https://doi.org/10.1007/978-3-319-71779-1_10
Download citation
DOI: https://doi.org/10.1007/978-3-319-71779-1_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-71778-4
Online ISBN: 978-3-319-71779-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)