
Abstract
Advances in genomics technologies, coupled with the availability of several high-throughput genotyping and sequencing platforms during recent years, provided a kick start to the adoption of modern breeding approaches to develop climate-resilient crops. Chickpea is the most important grain legume crop for global food and nutritional security in the context of population explosion and climate vagaries. During last ten years, it has transformed from orphan legume to genomics resource-rich legume like any other model legume plants. There has been a paradigm shift in the outlook of the scientific community in translating the genomic resources including the genome sequence and re-sequence information for developing superior lines with enhanced resistance or tolerance to important abiotic and biotic stresses. In addition, pan-genome and re-sequencing information of several germplasm lines will enable tailoring climate smart chickpeas. In addition, efforts to broaden the genetic base and enhanced utilization of the available trait-specific germplasm lines, multi-parent advanced generation inter-cross (MAGIC), nested association mapping (NAM) populations in breeding programs will accelerate the genetic grains at a faster pace.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

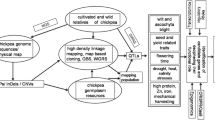

Keywords
- Chickpea
- Multi-parent Advanced Generation Inter-cross (MAGIC)
- Nested Association Mapping (NAM)
- Germplasm Lines
- Modern Breeding Approaches
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Chickpea (Cicer arietinum L.) is a cool season legume cultivated by resources-poor farmers in South Asia and sub-Saharan Africa. Despite its economic importance, productivity is lower than 1 ton per hectare because the crop is exposed to several biotic and abiotic stresses. Genomics research has accelerated the crop improvement in crops like rice, maize. In case of chickpea until 2005, about 150 SSR markers and sparse genetic maps were available which were of limited usefulness for trait dissection and implementing them in breeding programs. During last decade, efforts of chickpea research community especially at ICRISAT in collaboration with several partners across the globe developed >3,000 SSRs (Nayak et al. 2010; Thudi et al. 2011; Agarwal et al. 2015), transcriptomic resources (Hiremath et al. 2011; Kudapa et al. 2014), millions of SNPs and structural variations (Varshney et al. 2013a; Thudi et al. 2016a, b). Both desi and kabuli draft genomes have been decoded (Varshney et al. 2013a; Jain et al. 2013). In addition, several genetic maps, a physical map, consensus maps and high-density genetic maps have been made available for trait dissection (Gujaria et al. 2011; Millan et al. 2010; Varshney et al. 2014b, c; Gaur et al. 2015; Jaganathan et al. 2015; Kale et al. 2015). Furthermore, the genomic regions responsible for abiotic stress (Vadez et al., 2012; Varshney et al. 2014c; Purushothaman et al. 2015; Pushpavalli et al. 2015), biotic stresses (Sabbavarapu et al. 2013) and agronomically important traits like early flowering (Mallikarjuna et al. 2017; Samineni et al. 2016), protein content (Jadhav et al. 2015) have been identified. Thus, the availability of several genomics resources and draft genomes has transformed chickpea from orphan legume to “genomics resource rich” legume crop (Varshney 2016). This provided new opportunities for accelerating genetics research and use of these resources in breeding applications for faster genetic gains.
Recent climate changes, availability of irrigation facilities encouraged farmers in north India for cultivating commercial crops such as paddy and wheat. As a result, chickpea cultivation has expanded in the southern part of India that has been exposed to more frequent droughts and thus contributing to yield loses. Chickpea is being important for food and nutritional security, development of improved lines and cultivars that adapt to new niches in the context of climate change is a prerequisite. This chapter focusses on strategies and issues that need to utilize available genomic tools together with genetic resources for enhancing the chickpea yields to meet the future demands.
12.1 Germplasm Lines Re-sequencing and Pan-genomes
The availability of draft genome sequence of both kabuli and desi chickpea genomes (Varshney et al. 2013a; Jain et al. 2013) offers novel opportunities for understanding the genome architecture and identification of genes for crop improvement. Following the draft genomes, in recent years, efforts were also made to improve the genome assemblies using sequence data from flow cytometry isolated chromosomes to identify misplaced contigs (Ruperao et al. 2014). In addition, an improved version of desi genome assembly was reported (Parween et al. 2015) and draft genome assembly of Cicer reticulatum, the wild progenitor of chickpea, has also become available (Gupta et al. 2017). As a single genome sequence may not be enough to explain the variation existing in >93,000 chickpea, germplasm accessions being conserved in genebanks across the world. Hence, re-sequencing of diverse germplasm lines is a necessary task ahead to understand the genome wide variations and harnessing the existing variations for designing new strategies for chickpea improvement. Towards this direction, 90 elite lines, 35 parental genotypes of mapping populations, 129 released varieties were re-sequenced (Varshney et al. 2013a; Thudi et al. 2016a, b) and efforts are underway at ICRISAT to re-sequence 3,000 germplasm lines, the composite collection.
The allelic variations available in a gene of interest that may lead to desirable phenotype within a species are quite limited. Hence, Tattelin et al. (2005) proposed the concept of “pan-genome” to capture the complete gene set from different species of genera. The pan-genome is essential to fully understand the genetic control of phenotypes. Further, understanding the interconnection of genome and phenome is essential for achieving faster genetic gains in crop improvement programs. Insights into pan-genomes of several crop plants are now available for soybean (Li et al. 2014), maize (Hirsch et al. 2014; Lu et al. 2015), Brassica oleracea (Golicz et al. 2016), hexaploid wheat (Montenegro et al. 2017) and a pan-genome browser was developed in case of rice (Sun et al. 2016). The draft genomes and/or re-sequence information in any species is not of much use if no biological sense is made out of the data. It is also a herculean task to store as well as to analyse the huge amount of data. The tools available for pan-genome analysis have been extensively discussed by Xiao et al. (2015).
12.2 Functional Genomics
Plant stress responses are complex and form a coordinated response network with every gene involved from recognition to signaling to direct involvement. Functional genomics facilitates understanding the stress response at the genomic level and to characterize specific genes involved in resistance to biotic and abiotic stresses in chickpea. Functional genomics approaches such as suppression subtractive hybridization (SSH), super serial analysis of gene expression (SuperSAGE), microarray and EST sequencing have been performed to identify the abiotic stress-responsive transcripts in chickpea (Molina et al. 2008; Varshney et al. 2009; Buhariwalla et al. 2005; Garg et al. 2016). In addition, sequencing and de novo assembly of chickpea transcriptome using short reads have been reported in chickpea (Garg et al. 2011a, b). Since gene expression is post-transcriptionally regulated by microRNAs, recent studies used high-throughput small RNA sequencing approach to discover tissue-specific and stress-responsive expression profile of chickpea microRNAs (Jain et al. 2014; Kohli et al. 2014). The availability of next-generation sequencing technologies accelerated the development of gene expression profiles at the whole genome level (Jain 2012; O’Rourke et al. 2014) and transcriptome sequencing as well as NGS-based large-scale discovery and high-throughput genotyping of informative markers like simple sequence repeat (SSR), single nucleotide polymorphism (SNP) in chickpea (Garg et al. 2014; Hiremath et al. 2012; Jhanwar et al. 2012; Agarwal et al. 2012; Kudapa et al. 2014; Pradhan et al. 2014; Parida et al. 2015).
12.3 Next Generation Mapping Populations
Linkage mapping studies use family-based populations like F2, recombinant inbred lines (RILs), near isogenic lines (NILs) and double haploid populations, but alleles in these mapping populations come from only two parental lines. Hence, specialized mapping populations with a broad genetic base such as multi-parent advanced generation inter-cross (MAGIC) and nested association mapping (NAM) populations need to be developed and used. MAGIC population is generated from multiple parents of diverse origin, and the genome of the founder parents is reshuffled in different combinations (Huang et al. 2015). It serves as an important resource for high-resolution mapping and identification of target genomic regions, besides useful in the breeding programmes. A MAGIC population comprising of 1136 RILs using eight parental genotypes has been developed in chickpea. Nested association mapping (NAM), which combines the benefits of both linkage analysis and association mapping approaches, is used for high-resolution mapping of target traits. Development of NAM population is underway in chickpea to generate new breeding material with enhanced diversity. In addition, some other next-generation multi-parental populations like multiline cross inbred lines and recombinant inbred advanced intercross lines can also be developed in chickpea.
12.4 High-Resolution Mapping for Must Have Traits
Chickpea is cultivated under a wide range of agro-climatic conditions around the world and is adversely affected by diseases, insect pests, soil and environmental stresses. In addition, climatic variability and change in cultivation niches also have further implications on the cultivation of chickpea in different regions. Hence, future varieties must be able to withstand adverse and more variable conditions. Making of genetic adjustments of chickpea is needed to increase adaptation to drought, heat stress in semi-arid areas, cold stress tolerance in the Mediterranean region, resistance to biotic stresses like Fusarium wilt, Ascochyta blight and pod borer.
Advances in chickpea genomics and availability of genome sequences (Jain et al. 2013; Varshney et al. 2013a; Gupta et al. 2017) and re-sequencing data from hundreds of germplasm lines in chickpea have offered a different kind of marker genotyping platforms. For instance, large-scale SSR markers (Nayak et al. 2010; Thudi et al. 2011), VeraCode assays (Roorkiwal et al. 2013) and KASPar assays (Hiremath et al. 2012) have become available for genotyping germplasm collections and mapping populations. Genotyping of different populations with above-mentioned marker systems, however, is an expensive and time-consuming business. Furthermore, for undertaking association mapping, there is a need to genotype populations with high-density markers. In this direction, Axiom® arrays comprising 50 K single nucleotide polymorphism (SNP) markers have been developed in chickpea. These arrays have been proven very useful for generating large-scale polymorphisms in bi-parental mapping populations (Roorkiwal et al. unpublished). In addition, genotyping by sequencing and skim sequencing-based bin mapping approaches were adopted for fine mapping the traits (Jaganathan et al. 2015; Kale et al. 2015). Nevertheless, unlike genotyping the entire population, approaches like sequencing bulk segregant analysis (BSA-Seq) and QTL-Seq approaches have been deployed to identify the causal SNPs and candidate genes in legumes including chickpea (Singh et al. 2015, 2016; Pandey et al. 2017). We believe that in coming years trait mapping can be faster by using QTL-Seq approaches and use of MAGIC population, NAM with high-density arrays like Axiom® will help fine map the QTLs.
12.5 Next Generation Breeding
Development of large-scale genomic resources in chickpea (Varshney et al. 2012) and availability of pedigree information combined with optimized precision phenotyping methods make it possible to undertake new generation of breeding approaches in chickpea. Some of the genomics assisted breeding approaches like marker-assisted backcrossing (MABC) have been successfully employed to introgress disease resistance (Varshney et al. 2014a) and drought tolerance (Varshney et al. 2013b) into elite cultivars of chickpea. Marker-assisted recurrent selection (MARS) is another breeding approach proposed for pyramiding of superior alleles at different loci/QTLs in a single genotype (Bernardo and Charcosset 2006) is also being initiated to assemble favourable alleles for drought tolerance in chickpea (Thudi et al. 2014b). In addition, Advanced backcross (AB-QTL) analysis is another useful approach to introgress desired QTL or a gene especially from wild/exotic species (Tanksley and Nelson 1996) that can be developed in chickpea.
12.6 Genomic Selection
Genomic selection (GS) is a novel approach that predicts the breeding values of a line based on historical phenotyping data and the genotyping data. For addressing complex traits controlled by many small effect QTLs, genome-enabled selection of genotypes based on their breeding value (i.e. the genomics estimated breeding values) has potential relevance (Meuwissen et al. 2001). GS utilizes genome wide markers data along with phenotypic data to increase the accuracy of the prediction of breeding and genotypic values. This has become feasible due to the availability of a large number of SNP discovered by various NGS approaches and cost-effective genotyping platforms available in chickpea (Hiremath et al. 2012; Varshney et al. 2012). Genomic selection has been successfully used in animal breeding for predicting breeding values (Hayes et al., 2009) and also in crop plants like oil palm (Wong and Bernardo, 2008) and maize (Zhao et al. 2012). Recent study showed that genomic-enabled prediction as a promising avenue for improving yield in chickpea (Roorkiwal et al. 2016).
In addition to the above, we believe that diagnostic markers associated with must have traits can be used in an early generation in chickpea breeding programs which we call as “early generation selection (EGS)”. Right now, diagnostic markers are being used in EGS for drought tolerance, Fusarium wilt and Ascochyta blight in chickpea. We believe that in coming years, we will have more markers for must have traits and all loci. In summary, we need to adopt MABC approach for elite varieties deficient of one or two traits. For normal breeding, we propose to use diagnostic markers for EGS for target trait improvement and genomics selection approach for multiple traits. We envisage the use of a combination of EGS, GS and genome editing in chickpea in coming years.
12.7 Conclusion
As evident from different chapters of the book, we got large-scale germplasm and genomic resources for trait mapping, etc. It is high time to use the markers in regular breeding programs. We believe that combination of EGS and GS will accelerate genetic gains in breeding programs.
References
Agarwal G, Jhanwar S, Priya P, Singh VK, Saxena MS, Parida SK, Garg R, Jtayagi AK, Jain M (2012) Comparative analysis of kabuli chickpea transcriptome with desi and wild chickpea provides a rich resource for development of functional markers. PLoS ONE 7(12):e52443. doi:10.1371/journal.pone.0052443
Agarwal G, Sabbavarapu MM, Singh VK, Thudi M, Sheelamary S, Gaur PM and Varshney RK (2015) Identification of a non-redundant set of 202 in silico SSR markers and applicability of a select set in chickpea (Cicer arietinum L.). Euphytica 205:381–394
Bernardo R, Charcosset A (2006) Usefulness of gene information in marker assisted recurrent selection: a simulation appraisal. Crop Sci 46:614–621
Buhariwalla HK, Jayashree B, Eshwar K, Crouch JH (2005) Development of ESTs from chickpea roots and their use in diversity analysis of the Cicer genus. BMC Plant Biol 5:16
Garg R, Patel RK, Tyagi AK, Jain M (2011a) De novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res 18:53–63
Garg R, Patel RK, Jhanwar S, Priya P, Bhattacharjee A, Yadav G, Bhatia S, Chattopadhyay D, Tyagi AK, Jain M (2011b) Gene discovery and tissue-specific transcriptome analysis in chickpea with massively parallel pyrosequencing and web resource development. Plant Physiol 156:1661–1678
Garg R, Bhattacharjee A, Jain M (2014) Genome-scale transcriptomic insights into molecular aspects of abiotic stress responses in chickpea. Plant Mol Biol Rep 33:388–400
Garg R, Shankar R, Thakkar B, Kudapa H, Krishnamurthy L, Mantri N, Varshney RK, Bhatia S, Jain M (2016) Transcriptome analyses reveal genotype-and developmental stage-specific molecular responses to drought and salinity stresses in chickpea. Sci Rep. doi:10.1038/srep19228
Gaur R, Jeena G, Shah N, Gupta S, Pradhan S, Tyagi AK, Jain M, Chattopadhyay D, Bhatia S (2015) High density linkage mapping of genomic and transcriptomic SNPs for synteny analysis and anchoring the genome sequence of chickpea. Sci Rep 5:13387
Golicz AA, Bayer PE, Barker GC, Edger PP, Kim H, Martinez PA, Chan CKK, Severn-Ellis A, McCombie WR, Parkin IA, Paterson AH (2016) The pangenome of an agronomically important crop plant Brassica oleracea. Nat Commun 7. doi:10.1038/ncomms13390
Gujaria N, Kumar A, Dauthal P, Dubey A, Hiremath P, Bhanu Prakash A, Farmer A, Bhide M, Shah T, Gaur PM Upadhyaya HD, Bhatia S, Cook DR, May GD, Varshney RK (2011) Development and use of genic molecular markers (GMMs) for construction of a transcript map of chickpea (Cicer arietinum L.) Theor Appl Genet 122:1577–1589
Gupta S, Nawaz K, Parween S, Roy R, Sahu K, Pole AK, Khandal H, Srivastava R, Parida SK, Chattopadhyay D (2017) Draft genome sequence of Cicer reticulatum L., the wild progenitor of chickpea provides a resource for agronomic trait improvement. DNA Res 24:1–10
Hayes BJ, Bowman PJ, Chamberlain AJ, Goddard ME (2009) Invited review: genomic selection in dairy cattle: progress and challenges. J Dairy Sci 92:433–443
Hiremath PJ, Farmer A, Cannon SB, Woodward J, Kudapa H, Tuteja R, Kumar A, BhanuPrakash A, Mulaosmanovic B, Gujaria N, Krishnamurthy L, Gaur PM, KaviKishor PB, Shah T, Srinivasan R, Lohse M, Xiao Y, Town CD, Cook DR, May GD, Varshney RK (2011) Large-scale transcriptome analysis in chickpea (Cicer arietinumL.), an orphan legume crop of the semi-arid tropics of Asia and Africa. Plant Biotechnol J 9:922–931
Hiremath PJ, Kumar A, Penmetsa RV, Farmer A, Schlueter JA, Chamarthi SK, Whaley ma, Garcia NC, Gaur PM, Upadhyaya HD, Kavi Kishor PB, Shah TM, Cook D, Varshney RK (2012) Large-scale development of cost-effective SNP marker assays for diversity assessment and genetic mapping in chickpea and comparative mapping in legumes. Plant Biotechnol J 10:716–732
Hirsch CN, Foerster JM, Johnson JM, Sekhon RS, Muttoni G, Vaillancourt B, Peñagaricano F, Lindquist E, Pedraza MA, Barry K, de Leon N (2014) Insights into the maize pan-genome and pan-transcriptome. The Plant Cell 26(1):121–135
Huang BE, Verbyla KL, Verbyla AP, Raghavan C, Singh VK, Gaur PM, Leung H, Varshney RK, Cavanagh CR (2015) MAGIC populations in crops: current status and future prospects. Theor Appl Genet 128:999–1017
Jadhav AA, Rayate SJ, Mhase LB, Thudi M, Chitikineni A, Harer PN, Jadhav AS, Varshney RK, Kulwal PL (2015) Marker-trait association study for protein content in chickpea (Cicer arietinum L.)
Jaganathan D, Thudi M, Kale S, Azam S, Roorkiwal R, Gaur PM, Kavi Kishor PB, Nguyen H, Sutton T, Varshney RK (2015) Genotyping-by-sequencing based intra-specific genetic map refines a “QTL-hotspot” region for drought tolerance in chickpea. Mol Genet Genomics 290:559–571
Jain M (2012) Next-generation sequencing technologies for gene expression profiling in plants. Briefings in Funct Genomics 11:63–70
Jain M, Misra G, Patel RK, Priya P, Jhanwar S, Khan AW, Shah N, Singh VK, Garg R, Yadav M, Kant C, Sharma P, Bhatia S, Tyagi AK, Chattopadhya D (2013) A draft genome sequence of the pulse crop chickpea (Cicer arietinum L.). Plant J 74:715–729
Jain M, Chevala VV, Garg R (2014) Genome-wide discovery and differential regulation of conserved and novel microRNAs in chickpea via deep sequencing. J Exp Bot 65:5945–5958
Jhanwar S, Priya P, Garg R, Parida SK, Tyagi AK, Jain M (2012) Transcriptome sequencing of wild chickpea as a rich resource for marker development. Plant Biotechnol J 10:690–702
Kale SM, Jaganathan D, Ruperao P, Chen C, Punna R, Kudapa H, Thudi M, Roorkiwal M, Katta AVKS Mohan, Doddamani D, Garg V, Kavi Kishor PB, Gaur PM, Nguyen HT, Batley J, Edwards D, Sutton T, Varshney RK (2015) Prioritization of candidate genes in “QTL-hotspot” region for drought tolerance in chickpea (Cicer arietinum L.). Sci Rep 5:15296. doi:10.1038/srep15296
Kohli D, Joshi G, Deokar AA, Bhardwaj AR, Agarwal M, Katiyar-Agarwal S, Srinivsan R, Jain PK (2014) Identification and characterization of wilt and salt stress-responsive microRNAs in Chickpea through high-throughput sequencing. PLoS ONE 9(10):e108851. doi:10.1371/journal.pone.0108851
Kudapa K, Azam S, Sharpe AG, Taran B, Li R, Deonovic B, Cameron C, Farmer AD, Cannon SB, Varshney RK (2014) Comprehensive transcriptome assembly of chickpea (Cicer arietinum L.) using Sanger and next generation sequencing platforms: development and applications. PLoS ONE 9:e86039. doi:10.1371/journal.pone.0086039
Li YH, Zhou G, Ma J, Jiang W, Jin LG, Zhang Z, Guo Y, Zhang J, Sui Y, Zheng L, Zhang SS (2014) De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nat Biotechnol 32(10):1045–1052
Lu F, Romay MC, Glaubitz JC, Bradbury PJ, Elshire RJ, Wang T, Li Y, Li Y, Semagn K, Zhang X, Hernandez AG (2015) High-resolution genetic mapping of maize pan-genome sequence anchors. Nat Commun 6:6914. doi:10.1038/ncomms7914
Mallikarjuna BP, Samineni S, Thudi M, Sajja SB, Kahn AW, Patil A, Viswanatha KP, Varshney RK, Gaur PM (2017) Molecular mapping of flowering time major genes and QTLs in chickpea (Cicer arietinum L.). Front Plant Sci 8:1140
Meuwissen TH, Hayes BJ, Goddard ME (2001) Prediction of total genetic value using genome wide dense marker maps. Genetics 157:1819–1829
Millan T, Winter P, Jüngling R, Gil J, Rubio J, Cho S, Cobos MJ, Irulea M, Rajesh PN, Tekeoglu M, Kahl G, Muehlbauer FJ (2010) A consensus genetic map of chickpea (Cicer arietinum L.) based on 10 mapping populations. Euphytica 175:175–189
Molina C, Rotter B, Horres R, Udupa SM, Besser B, Bellarmino L, Baum M, Matsumura H, Terauchi R, Kahl G, Winter P (2008) Super SAGE: the drought stress-responsive transcriptome of chickpea roots. BMC Genomics 9:553
Montenegro JD, Golicz AA, Bayer PE, Hurgobin B, Lee H, Chan CKK, Visendi P, Lai K, Doležel J, Batley J, Edwards D (2017) The pangenome of hexaploid bread wheat. The Plant J. doi:10.1111/tpj.13515
Nayak SN, Zhu H, Varghese N, Datta S, Choi HK, Horres R, Jüngling R, Singh J, Kishor P B, Sivaramakrishnan S, Hoisington DA, Kahl G, Winter P, Cook DR, Varshney RK (2010) Integration of novel SSR and gene-based SNP marker loci in the chickpea genetic map and establishment of new anchor points with Medicago truncatula genome. Theor Appl Genet 120(7):1415–1441
O’Rourke JA, Bolon YT, Bucciarelli B, Vance CP (2014) Legume genomics: understanding biology through DNA and RNA sequencing. Ann Bot 113:107–1120
Pandey MK, Khan AW, Singh VK, Vishwakarma MK, Shasidhar Y, Kumar V, Garg V, Bhat RS, Chitikineni A, Janila P, Guo B, Varshney RK (2017) HYPERLINK “http://oar.icrisat.org/9956/” QTL-seq approach identified genomic regions and diagnostic markers for rust and late leaf spot resistance in groundnut (Arachis hypogaea L.). Plant Biotechnol J 15(8):927–941
Parida, SK. Verma M, Yadav SK, Ambawat S, Das S, Garg R, Jain M (2015) Development of genome-wide informative simple sequence repeat markers for large-scale genotyping applications in chickpea and development of web resource. Frontiers Plant Sci 6:645
Parween S, Nawaz K, Roy R, Pole AK, Venkata Suresh B, Misra G, Jain M, Yadav G, Parida SK, Tyagi AK, Bhatia S, Chattopadhyay D (2015). An advanced draft genome assembly of a desi type chickpea (Cicer arietinum L.). Sci Rep 5:12806
Pradhan S, Bandhiwal N, Shah N, Kant C, Gaur R, Bhatia S (2014) Global transcriptome analysis of developing chickpea (Cicer arietinum L.) seeds. Frontiers. Plant Sci 5:698
Purushothaman R, Thudi M, Krishnamurthy L, Upadhyaya HD, Kashiwagi J, Gowda CLL, Varshney RK (2015) Association of mid-reproductive stage canopy temperature depression with the molecular markers and grain yields of chickpea (Cicer arietinum L.) germplasm under terminal drought. Field Crops Res 174:1–11
Pushpavalli R, Krishnamurthy L, Thudi M, Gaur PM, Rao MV, Siddique KHM, Colmer TD, Turner NC, Varshney RK, Vadez V (2015) two key genomic regions harbour QTLs for salinity tolerance in ICCV 2 × JG 11 derived chickpea (Cicer arietinum L.) recombinant inbred lines. BMC Plant Biol 15:124
Roorkiwal M, Sawargaonkar SL, Chitikineni A, Thudi M, Saxena RK, Upadhyaya HD, Isabel Vales M, Riera-Lizarazu O, Varshney RK (2013) Single nucleotide polymorphism genotyping for breeding and genetics applications in chickpea and pigeonpea using the BeadXpress platform. Plant Genome 6:2. doi:10.3835/plantgenome2013.05.0017
Roorkiwal M, Rathore A, Das RR, Singh MK, Jain A, Srinivasan S, Gaur PM, Chellapilla B, Tripathi S, Li Y, Hickey JM, Lorenz A, Sutton T, Crossa J, Jannink J-L, Varshney RK (2016) Genome-enabled prediction models for yield related traits in chickpea. Frontiers in Plant Sci 7:1666
Roorkiwal M, Jain A, Kale SM, Doddamani D, Chitikineni A, Thudi M, Varshney RK (2017) Development and evaluation of high density SNP array (Axiom®CicerSNP Array) for high resolution genetic mapping and breeding applications in chickpea. Plant Biotechnol J. doi:10.1111/pbi.12836
Ruperao P, Chan KCK, Azam S, Karafiátová M, Hayashi S, Čížková J, Saxena RK, Šimková H, Song C, Vrána J, Chitikineni A, Visendi P, Gaur PM, Millán T, Singh KB, Taran B, Wang J, Batley J, Doležel J, Varshney RK, Edwards D (2014) A chromosomal genomics approach to assess and validate the desi and kabuli draft chickpea genome assemblies. Plant Biotechnol J 12:778–786
Sabbavarapu MM, Sharma M, Chamarthi SK, Swapna N, Thudi M, Gaur PM, Pande S, Singh S, Kaur L, Varshney RK (2013) Molecular mapping of QTLs for resistance to Fusarium wilt (race 1) and Ascochyta blight in chickpea (Cicer arietinum L.). Euphytica 193:121–133
Samineni S, Kamatam S, Thudi M, Varshney RK, Gaur PM (2016) Vernalization response in chickpea is controlled by a major QTL. Euphytica 207:453–461
Singh VK, Khan AW, Saxena RK, Kumar V, Kale SM, Sinha P, Chitikineni A, Lekha PT, Garg V, Sharma M, Sameer Kumar CV, Parupalli S, Vechalapu S, Patil S, Muniswamy S, Ghanta A, Yamini KN, Dharmaraj PS, Varshney RK (2015) Next-generation sequencing for identification of candidate genes for Fusarium wilt and sterility mosaic disease in pigeonpea (Cajanus cajan). Plant Biotechnol J. doi:10.1111/pbi.12470
Singh VK, Khan AW, Jaganathan D, Thudi M, Roorkiwal M, Takagi H, Garg V, Kumar V, Chitikineni A, Gaur PM, Sutton T, Terauchi R, Varshney RK (2016) QTL-seq for rapid identification of candidate genes for 100-seed weight and root/total plant dry weight ratio under rainfed conditions in chickpea. Plant Biotechnol J. doi:10.1111/pbi.12567
Sun C, Hu Z, Zheng T, Lu K, Zhao Y, Wang W, Shi J, Wang C, Lu J, Zhang D. and Li Z (2016) RPAN: rice pan-genome browser for ∼ 3000 rice genomes. Nucleic Acids Res p.gkw958
Tanksley SD, Nelson JC (1996) Advanced backcross QTL analysis: a method for simultaneous discovery and transfer of valuable QTL from unadapted germplasm into elite breeding lines. Theoretical Appl Genet 92:191–203
Tattelin H. Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, Angiuoli SV, Crabtree J, Jones AL, Durkin AS, Deboy RT, Davidsen TM, Mora M, Scarselli M, Margarit y Ros I, Peterson JD, Hauser CR, Sundaram JP, Nelson WC, Madupu R, Brinkac LM, Dodson RJ, Rosovitz MJ, Sullivan SA, Daugherty SC, Haft DH, Selengut J, Gwinn ML, Zhou L, Zafar N, Khouri H, Radune D, Dimitrov G, Watkins K, O’Connor KJ, Smith S, Utterback TR, White O, Rubens CE, Grandi G, Madoff LC, Kasper DL, Telford JL, Wessels MR, Rappuoli R, Fraser CM (2005) Genomic analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial pan-genome. Proc Natl Acad Sci USA 102:13950–13955
Thudi M, Bohra A, Nayak SN, Varghese N, Shah TM, Varma RP, Nepolean T, Gudipati S, Gaur PM, Kulwal PL, Upadhyaya HD, Kavikishor PB, Winter P, Kahl G, Town CD, Kilian A, Cook DR, Varshney RK (2011) Novel SSR markers from BAC-End sequences, DArT arrays and a comprehensive genetic map with 1,291 marker loci for chickpea (Cicer arietinum L.). Plos One 6(11):e27275
Thudi M, Upadhyaya HD, Rathore A, Gaur PM, Krishnamurthy L, Roorkiwal M, Nayak SN, Chaturvedi SK, Gangarao NVPR, Fikre A, Kimurto P, Sharma PC, Sheshashayee MS, Tobita S, Kashiwagi J, Ito O, Varshney RK (2014a) Genetic dissection of drought and heat tolerance in chickpea through genome-wide and candidate gene-based association mapping approaches. PLoS ONE 9(5):e96758
Thudi M, Gaur PM, L Krishnamurthy L, Mir RR, Kudapa H, Fikre A, Kimurto P, Tripathi S, Soren KR, Mulwa R, Bharadwaj C, Datta S, Chaturvedi SK, Varshney RK (2014b) Genomics-assisted breeding for drought tolerance: a dream comes true in chickpea!. Funct Plant Biol 41:1178–1190
Thudi M, Chitikineni, Liu X, He W, Roorkiwal M, Yang W, Jian J, Doddamani D, Gaur PM, Rathore A, Samineni S, Saxena RK, Xu D, Singh NP, Chaturvedi SK, Zhang G, Wang J, Datta SK, Xu X, Varshney RK (2016a). Recent breeding programs enhanced genetic diversity in both desi and kabuli varieties of chickpea (Cicer arietinum L.). Sci Rep 6:38636
Thudi M, Khan AW, Kumar V, Gaur PM, Katta AVSK, Garg V, Roorkiwal M, Samineni S, Varshney RK (2016b) Whole genome re-sequencing reveals genome wide variations among parental lines of mapping populations in chickpea (Cicer arietinum). BMC Pant Biol 16(1):10
Vadez V, Krishnamurthy L, Thudi M, Anuradha C, Colmer TD, Turner NC, Siddique KHM, Gaur PM, Varshney RK (2012) Assessment of ICCV 2 × JG 62 chickpea progenies shows sensitivity of reproduction to salt stress and reveals QTLs for seed yield and yield components. Mol Breeding 30(1):9–21
Varshney RK, Hiremath PJ, Lekha P, Kashiwagi J, Balaji J, Deokar AA, Vadez V, Xiao Y, Srinivasan R, Gaur PM, Siddique KH, Town CD, Hoisington DA (2009) A comprehensive resource of drought-and salinity-responsive ESTs for gene discovery and marker development in chickpea (Cicer arietinum L). BMC Genomics 10:523
Varshney RK, Kudapa H, Roorkiwal M, Thudi M, Pandey KM, Saxena RK, Chamarthi SK, Murali Mohan S, Mallikarjuna N, Upadhyaya HD, Gaur PM, Krishnamurthy L, Saxena KB, Nigam SN, Pande S (2012) Advances in genetics and molecular breeding of three legume crops of semi-arid tropics using next-generation sequencing and high-throughput genotyping technologies. J Biosci 37:811–820
Varshney, RK Song C, Saxena RK, Azam S, Yu S, Sharpe A, Cannon S, Baek J, Rosen BD, Tar’an B, Millan T, Zhang X, Ramsay LD, Iwata A, Wang Y, Nelson W, Farmer AD, Gaur PM, Soderlund C, Penmetsa RV, Xu C, Bharti AK, He W, Winter P, Zhao S, Hane JK, Garcia NC, Condie JA, Upadhyaya HD, Luo MC, Thudi M, Gowda CLL, Singh NP, Lichtenzveig J, Gali KK, Rubio J, Nadarajan N, Dolezell J, Bansal KC, Xu X, Edwards D, Zhang G, Kahl G, Gil J, Singh KB, Datta SK, Jackson SA, Wang J, Cook DR (2013a). Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat Biotechnol 31:240–246
Varshney RK, Gaur PM, Chamarthi SK, Krishnamurthy L, Tripathi S, Kashiwagi J, Samineni S, Singh VK, Thudi M, Jaganathan D (2013b) Fast-track introgression of for root traits and other drought tolerance traits in JG 11, an elite and leading variety of chickpea. The Plant Genome 6(3)
Varshney RK, Mohan SM, Gaur PM, Chamarthi SK, Singh VK, Srinivasan S, Swapna N, Sharma M, Singh S, Kaur L, Pande S (2014a) Marker-assisted backcrossing to introgress resistance to Fusarium Wilt race 1 and Ascochyta blight in C 214, an elite cultivar of chickpea. The plant Genome 7(1):1–11
Varshney RK, Mir RR, Bhatia S, Thudi M, Hu Y, Azam S, Zhang Y, Jaganathan D, You FM, Gao J, Riera-Lizarazu O, Luo M-C (2014b) Integrated physical, genetic and genome map of chickpea (Cicer arietinum L.). Funct Integrative Genomics 14:59–73
Varshney RK, Thudi M, Nayak SN, Gaur PM, Kashiwagi J, Krishnamurthy L, Jaganathan D, Koppolu J, Bohra A, Tripathi S, Rathore A, Jukanti AK, Jayalakshmi V, Vemula A, Singh SJ, Yasin M, Sheshshayee MS, Viswanatha KP (2014c) Genetic dissection of drought tolerance in chickpea (Cicer arietinum L.). Theor Appl Genet 127:445–462
Varshney RK, Thudi M, Upadhyaya HD, Dwivedi SL, Udupa S, Furman B, Baum M, Hoisington DA (2014d) A SSR kit to study genetic diversity in chickpea (Cicer arietinum L.). Plant Genet Res: Utilization and Charact. doi:10.1017/S1479262114000392
Varshney RK (2016) Exciting journey of 10 years from genomes to fields and markets: Some success stories of genomics-assisted breeding in chickpea, pigeonpea and groundnut. Plant Sci 242:98–107
Wong CK, Bernardo R (2008) Genome-wide selection in oil palm: increasing selection gain per unit time and cost with small populations. Theor Appl Genet 116:815–824
Xiao J, Zhang Z, Wu J, Yu J (2015) A brief review of software tools for pangenomics. Genomics, Proteomics & Bioinformatics 13(1):73–76
Zhao Y, Gowda M, Liu W, Würschum T, Maurer HP, Longin FH, Ranc N, Reif JC (2012) Accuracy of genomic selection in European maize elite breeding populations. Theor Appl Genet 124:769–776
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter

Cite this chapter
Varshney, R.K., Thudi, M., Muehlbauer, F.J. (2017). Future Prospects for Chickpea Research. In: Varshney, R., Thudi, M., Muehlbauer, F. (eds) The Chickpea Genome. Compendium of Plant Genomes. Springer, Cham. https://doi.org/10.1007/978-3-319-66117-9_12
Download citation
DOI: https://doi.org/10.1007/978-3-319-66117-9_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-66115-5
Online ISBN: 978-3-319-66117-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)