
Abstract
Pathology is an evolving specialty, and the advancement of knowledge has led to the introduction of new concepts and diagnoses, and the disappearance of others. Just as in clinical medicine, the evolution of the field is rarely due to presence of perfect data. Classification systems evolve and adapt, with the integration of new discoveries. This is particularly true of endometrial cancer, where a number of new molecular factors have been identified in the past few years.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


1 Introduction
Pathology is an evolving specialty, and the advancement of knowledge has led to the introduction of new concepts and diagnoses, and the disappearance of others. Just as in clinical medicine, the evolution of the field is rarely due to presence of perfect data. Classification systems evolve and adapt, with the integration of new discoveries. This is particularly true of endometrial cancer, where a number of new molecular factors have been identified in the past few years.
This chapter presents an overview of the histopathological classification of endometrial carcinomas, as defined by the 2014 World Health Organization Classification of Tumors of Female Reproductive Organs [1, 2]. The chapter progresses from a discussion of precursor lesions to the histological carcinoma subtypes and finally to the genomic characterization of endometrial cancer by the Cancer Genome Atlas (TCGA). Where applicable, controversies are discussed under the relevant diagnoses. Finally, at the end of the chapter, a discussion of two simplified molecular classification systems based upon the TCGA is presented; first the ProMisE system, developed at the University of Vancouver and then the PORTEC system, developed at the University of Leiden. These two systems attempt to recapitulate the genomic classification of the TCGA using methods that are readily available in a modern clinical pathology lab.
2 Precursor Lesions
The diagnosis of precancers of endometrioid carcinoma has been controversial for a number of years. The hyperplasia classification has been in use for several decades. It is based on defining the complexity of gland architecture (the degree of fusing and branching of glands) as well determining if cytologic atypia is present. This results in a subgrouping with four different histological patterns: simple hyperplasia, complex hyperplasia, simple atypical hyperplasia, and complex atypical hyperplasia. This system has some advantages and it initially promised to be a good predictor of the risk of progression to cancer. However, the hyperplasia system has several weaknesses: throughout the years the criteria for gland complexity and cell atypia have been defined, redefined, and reorganized leading to confusion of pathologists, gynecologists, and oncologists. Additionally, studies have shown poor reproducibility and difficulties with molecular correlation [2].
In the late 1990s the EIN system (Endometrial, changed to “endometrioid” by the WHO, Intraepithelial Neoplasia) has been developed. This system initially used objective morphometric data to assess a “D-score” but later formal morphometry was dropped. The current system only uses routine microscopy. The system is based on assessing three factors: the stroma-to-gland ratio, size of the focus, and nuclear pleomorphism (which is assessed by comparing nuclei in the crowded gland areas to nuclei in the “background”). The system’s great advantage is a better reproducibility among pathologists and a relative ease of use in clinical pathology while also showing a close relationship with early molecular events (such as PAX2 inactivation, PTEN, and KRAS mutation). The EIN system was recently endorsed in an opinion paper by the American College of Obstetrics and Gynecology [3]. However, it made several changes to the diagnosis of “atypia,” which have been difficult for supporters of the hyperplasia system to accept.
In the latest edition of the WHO 2014 the two systems have been combined into “Atypical hyperplasia/Endometrioid intraepithelial neoplasia (AH/EIN). ” The combined system has retained the traditional definition of nuclear atypia while noting that the assessment of atypia can be facilitated by comparing crowded gland cells to adjacent normal gland cells. The EIN classification’s increased gland-to-stroma ratio (area of gland exceeds that of stroma) was incorporated fully in the current WHO classification.
It should be noted that the above discussion refers to precancerous lesions of endometrioid carcinoma, which are common. There is, however, a second precursor lesion, named “Serous endometrial intraepithelial carcinoma (SEIC). ” SEICs are rare, and are the immediate precursors of invasive serous carcinoma. It is characterized by an underlying p53 mutation. Because SEIC spreads by exfoliation of malignant cells into the uterine cavity, it can be associated with extra-uterine spread even without invasion. Therefore its clinical risk is similar to that of the fully developed carcinoma, and hence is discussed under serous carcinoma (below) [4].
3 Endometrioid Carcinoma
Endometrioid carcinomas are the most common epithelial tumors of the endometrium [1, 5]. Microscopically, in their most well-differentiated form, these tumors resemble proliferative phase endometrial mucosa, with columnar cells containing an abundant cytoplasm and oval nuclei (Fig. 2.1a). However, these carcinomas display an architectural complexity that is absent in benign and hyperplastic mucosa. This complexity is seen as either cribriform, solid, villoglandular, or papillary growth [5].

Representative images of endometrioid carcinoma. (a) An area of glandular growth, typical of FIGO grade 1 tumors, (b) squamous differentiation, both immature (open arrows) and more mature (closed arrows), (c) mucinous differentiation with intracytoplasmic mucin (closed arrow), (d) an area of solid growth, consistent with a FIGO grade 3 tumor
Endometrioid carcinomas are further characterized by the frequent presence of altered cell differentiation (i.e. metaplasia). Note that the term “differentiation” is typically used for changes of cell type in precancers and carcinomas, while the term “metaplasia” is reserved for similar changes in benign endometrial epithelia. Squamous differentiation is common, and so is mucinous, tubal, and secretory (Fig. 2.1b, c). These changes can confirm the diagnosis of endometrioid carcinoma, but can also make the diagnosis challenging, especially when the majority of the tumor is affected.
Grading of endometrioid carcinomas uses the FIGO grading system , presented in Chap. 1 [6]. It has been proposed that this three-grade system be combined into a two-grade system, where grades one and two are combined into a “low-grade” group and grade 3 is synonymous with “high-grade” [7]. Endometrioid FIGO grade 3 tumors are characterized by >50% solid growth, but should show areas of typical endometrioid differentiation, either by demonstrating the correct microscopic appearance of the cells, or by the presence of altered cell differentiation (Fig. 2.1d).
The molecular aberrations identified in endometrioid carcinomas vary with the grade of the tumor. Low-grade tumors are characterized by frequent mutation or inactivation of PTEN (>50%), PIK3CA, PIK3R1, and ARID1A [1]. FIGO grade 3 tumors can show mutation or inactivation of TP53. The presence of a TP53 mutation is sufficiently associated with poor prognosis and aggressive behavior that it should essentially exclude a FIGO grade 1–2 endometrioid carcinoma [8, 9].
Controversial areas within the diagnosis of endometrioid carcinoma include the distinction of FIGO 3 tumors from serous carcinomas, the correct identification of lymphovascular space invasion (LVSI) , and the clinical significance of the microcystic, elongated, and fragmented (MELF) growth pattern . Within each of these there are variations in diagnosis between labs. The distinction of high-grade tumors from each other is one area where molecular methods, discussed below, may have significant impact. The presence of LVSI is used in several risk stratification models, including the European joint guidelines for risk stratification [10].
LVSI assessment can be quite difficult, and endometrioid tumors often show “retraction artifacts” in hysterectomy specimens which can mimic true LVSI. Immunohistochemical markers for endothelial cells (CD31, CD34, ERG) and Elastin-stains for Elastin fibers in vessel walls often aid the assessment.
The MELF-pattern has been linked to increased risk of deep myometrial invasion, LVSI, and above all lymph node metastasis [11]. In broad terms it should be a straightforward diagnosis and is usually found unexpectedly in preoperatively low-risk patients. The morphology is of a low-grade endometrioid cancer with distinct widely scattered microcystic glands that deeply invade the myometrium without a desmoplastic reaction. At the deep invasive front there are usually only a few elongated glands and LVSI. Problems arise when trying to assess this morphologic pattern in “nonclassical” cases; the most common problem being that only a part of the tumor shows the MELF-pattern morphology. It is therefore difficult to clearly define how extensive the MELF patterns should be to define a cancer as MELF. It also invites a highly subjective assessment (and therefore low reproducibility) as clear and objective definitions for MELF are missing.
4 Serous Carcinoma
Serous carcinomas are typically seen in association with endometrial polyps and an atrophic endometrial mucosa. Of note, these carcinomas can grow by replacing the endometrial epithelium, leading to an appearance that has been called “serous endometrial intraepithelial carcinoma” (the preferred term) or, alternatively, “serous carcinoma in-situ” or “early serous carcinoma” [1]. Whatever the term, it is vitally important that treating surgeons and oncologists realize that serous carcinomas spread by exfoliation of cells directly into the uterine cavity and, via the fallopian tubes, to the peritoneal cavity and omentum. Thus, even in the absence of invasion, SEIC has a risk of metastasis to extra-uterine sites [12].
Serous carcinomas are rare in the endometrium, in contrast to the ovary. These tumors resemble high-grade serous carcinomas of the ovary, with high-grade nuclear atypia, a brisk mitotic rate, and single-cell necrosis (see Fig. 2.2a, b) [8, 13] Additionally, just like ovarian high-grade serous carcinomas, these tumors show a wide variety of growth patterns, such as solid, cribriform, and gland-like, in addition to the classic papillary and micropapillary growth. Micropapillary growth is commonly seen in serous carcinomas but is not required for the diagnosis. Thus, the name “seropapillary carcinoma” should be avoided.

Representative images of serous carcinoma. (a) Low-power and (b) high-power images of the prototypical papillary growth pattern. (c) p53 immunohistochemistry consistent with a TP53 gene mutation. (d) The so-called null-pattern staining, which is also consistent with a TP53 mutation
One characteristic feature of serous carcinomas is the near ubiquitous presence of a deletion or mutation of the TP53 gene. This mutation leads to a characteristic immunohistochemical pattern, with approximately 90% of tumors showing strong nuclear positivity in over 80% of tumor cells (Fig. 2.2c) [14]. The remaining 10% can show a completely negative staining result, which has been called the “null staining pattern” (Fig. 2.2d). The sensitivity and specificity of immunohistochemistry is high but not 100%, and so in discrepant cases consensus histology, or even TP53 sequencing, may be necessary. Serous carcinomas are not graded, as in the endometrium they are all high-grade.
Beyond the near ubiquity of TP53 mutations , serous carcinomas can show mutations in PIK3CA, FBXW7, and PPP2R1A [1]. There is some data indicating that germline BRCA1/2 mutations are associated with the development of endometrial serous carcinomas [15].
5 Clear Cell Carcinoma
Clear cell carcinomas are among the rarest of subtypes, making up roughly 2% of endometrial carcinomas [1, 2, 16]. Microscopically, these tumors consist of round to polygonal tumor cells with an abundant clear to granular cytoplasm and a typically central round to polygonal nucleus. The tumor cells contain abundant glycogen, which can be demonstrated using special stains. The characteristic feature of these tumors is the presence of papillary, tubulocystic, and solid growth patterns and the presence of myxoid or hyalinized stroma (Fig. 2.3a, b). Hobnail cells are the most common cell seen [16].

Clear cell carcinoma. (a) Low-power and (b) high-power images of clear cell carcinoma
Immunohistochemistry can be useful in the diagnosis of these tumors, where they are characteristically ER and PR negative, and can show expression of Napsin A [17]. Approximately 30% of cases can show a mutation in p53, as detected by immunohistochemistry [18].
Molecular studies have demonstrated a variety of mutations in these tumors, such as mutations in PTEN, TP53, ARID1A, and PIK3CA [19].
6 Undifferentiated and Dedifferentiated Carcinoma
There is increasing recognition of undifferentiated carcinomas as a distinct tumor type, separate from other high-grade carcinomas, such as FIGO 3 endometrioid tumors and carcinosarcomas. In the pure form, where no other tumor component is seen, they are called undifferentiated carcinomas. In a dedifferentiated carcinoma, the undifferentiated component is seen in combination with a FIGO1-2 endometrioid carcinoma. The identification of dedifferentiated carcinomas implies that biologically the tumor represents a dedifferentiation, or transformation, of the lower-grade endometrioid tumor to the high-grade undifferentiated component.
Microscopically these tumors are made of solid sheets of high-grade tumor cells showing no particular differentiation. In practice, this means a lack of endometrioid or serous type growth patterns, a lack of variant differentiation (e.g. squamous differentiation). The tumor cells are typically highly dyscohesive and thus can resemble a high-grade lymphoma (Fig. 2.4a). These tumors typically show a reduction in staining with epithelial markers such as keratin, but epithelial membrane antigen is typically retained.

(a) Undifferentiated carcinoma showing solid growth of dyscohesive cells lacking in differentiation. (b) Carcinosarcoma showing high-grade mesenchymal (upper left) and epithelial (lower right) components
Molecularly these tumors appear to be associated with mutation of members of the SWI/SNF family of genes, as well as loss of functional mismatch repair, as demonstrated by immunohistochemistry for the proteins MLH1, PMS2, MSH2, and MSH6.
7 Mixed Carcinoma
Mixed carcinomas are defined in the WHO 2014 as a tumor composed of a mixture of two tumor types, where at least one of them must be a “Type 2” tumor. The two types must be readily recognizable in routine hematoxylin- and eosin-stained sections. The minimum percentage of the secondary component has been arbitrarily set to 5%, and the behavior of the tumor clinically is expected to follow the most high-grade component. Indeed, research has shown that as little as 5% serous carcinoma can adversely affect outcome [20]. Immunohistochemistry can be used to further support a diagnosis of a mixed carcinoma.
8 Neuroendocrine Tumors
Neuroendocrine tumors range from low-grade neuroendocrine tumor (carcinoid tumor) to high-grade neuroendocrine carcinoma (small cell and large cell neuroendocrine carcinoma). These tumors share a characteristic neuroendocrine morphology, and the neuroendocrine differentiation should be confirmed using immunohistochemistry.
Low-grade neuroendocrine tumors of the endometrium are extremely rare; they have been described only in a few case reports. Clearly a metastasis from a low-grade neuroendocrine tumor outside the uterus needs to be excluded before a primary endometrial tumor can be considered. This exclusion must be done with careful clinical and radiological correlation. Microscopically, low-grade neuroendocrine tumors show a variety of growth patterns and a characteristic “salt and pepper” chromatin of the nuclei.
High-grade neuroendocrine tumors can be divided into small cell and large cell types. Small cell neuroendocrine carcinomas resemble the tumor of the same name seen in the lung, with poorly cohesive cells with minimal cytoplasm, nuclear molding, high mitotic rate, karyorrhexis, and the common presence of “crush” artifact. Large cell neuroendocrine carcinomas should only be diagnosed if they show the characteristic growth patterns of well-demarcated nests, trabeculae, and cords, with peripheral palisading. Immunohistochemistry with chromogranin, synaptophysin and CD56, can be used to confirm neuroendocrine differentiation.
9 Carcinosarcoma
These tumors are defined as biphasic tumors consisting of high-grade carcinomatous (i.e. epithelial) and sarcomatous (i.e. mesenchymal) components. They were previously called “Malignant mixed Müllerian tumor, ” and this term, as well as its abbreviation MMMT, is still commonly in use.
Microscopically there is typically an intimate mixture of the two components (Fig. 2.4b). The carcinomatous component is usually either endometrioid or serous. The sarcomatous component is typically high-grade and nonspecific (i.e. showing no particular diagnostic features of a more specific sarcoma type); however, tumors can show rhabdomyosarcoma, chondrosarcoma, and even osteosarcoma differentiation. Regardless of the type of sarcomatous differentiation it is believed that the origin of these tumors is from the carcinoma, which is why they have been included in this section. Immunohistochemistry is not helpful in the diagnosis and the immunophenotype can be more confusing than helpful.
The Tumor Cancer Genome Atlas recently sequenced 57 untreated patients with carcinosarcoma. The tumors had extensive copy-number alterations and highly recurrent somatic mutations. Frequent mutations were seen in TP53, PTEN, PIK3CA, PPP2R1A, FBXW7, and KRAS, also often found in endometrioid and serous carcinomas [21].
10 The Cancer Genome Atlas (TCGA) Endometrial Carcinoma Analysis
In 2013 the TCGA completed its integrated genomic characterization of 373 endometrial cancers, including low- and high-grade endometrioid and serous tumors [22]. Tumors were studied by a comprehensive series of methods, including somatic copy-number alterations, exome sequencing, mRNA expression, protein expression, microRNA expression, and DNA methylation. Given the wealth of data, and the number of different methods applied, a custom-built clustering algorithm called “SuperCluster” was developed to derive overall subtypes across all methods. The SuperCluster data indicates the limitations of current diagnostic methods. Within tumors diagnosed “endometrioid” (based on routine light microscopy) are multiple molecular subtypes. Four molecular subtypes were identified, including ultramutated “POLE,” hypermutated “MSI,” copy-number low “endometrioid-like,” and copy-number high “serous-like,” as described in (Table 2.1).
Ultramutated “POLE” Group
One of the most fascinating finding of the TCGA classification was the identification of an ultramutated tumor type with an extremely favorable prognosis. These tumors show high mutation rates (232 × 10−6 mutation per Mb) and an increased C to A transversion frequency. All of these tumors show mutations in the exonuclease domain of POLE, the catalytic subunit of polymerase epsilon, which is involved in nuclear DNA replication and repair. Mutation rates seen in these tumors exceed those found in any other tumor lineage.
Prognosis of these tumors appears to be extremely favorable [22, 24,25,26,27]. The TCGA showed a progression-free survival of 100%. Subsequent studies have confirmed this finding. A European study using the PORTEC-1 and -2 trial cohorts (n = 788) identified 48 POLE tumors (6.1%) [25]. There was a strong association of POLE mutation status with high tumor grade; however, none of the patients with high-grade POLE tumors experienced progression or death. These results have been confirmed in a number of subsequent studies. The conclusion of these studies is that POLE tumors of all grades display excellent prognosis, independent of other known prognostic factors. In vitro studies showed that POLE mutated cells had resistance toward cisplatin, suggesting that the good outcome is not secondary to the response to chemotherapy [23, 28]. The ultra-mutated status of these tumors produces a strong immunogenic reaction, this is seen in intra- and peritumoral lymphocyte infiltration, expression of PD-1 and PD-L1, as also additional T cell markers thus being a possible target for checkpoint inhibitors [29,30,31,32].
Hypermutated “MSI” Group
These tumors show an intermediate mutation frequency (18 × 10−6 mutations per Mb) and were associated with MLH1 promoter methylation. These tumors showed microsatellite instability, few somatic copy-number alterations, and frequent nonsynonymous KRAS mutations.
Copy-Number Low “Endometrioid-Like” Group
These tumors have a low mutation rate (2.9 × 10−6 mutations per Mb). This group consists primarily of microsatellite stable endometrioid cancers. These tumors showed an unusually high frequency of CTNNB1 mutations (52%).
Copy-Number High “Serous-Like” Group
These tumors also have a low mutation rate (2.3 × 10−6 mutation per Mb), but they have extensive somatic copy-number alterations. These tumors had a significantly worse progression-free survival than the endometrioid groups. Of note, 25% of cases diagnosed by light microscopy as “high-grade endometrioid” cancer had a genomic profile matching the serous-like group. Several potential therapeutic copy-number alterations were detected, including 15q26.2 (amplification in IGF1R) and ERBB2, FGFR1 and FGFR3, and LRP1B deletion. A subset of serous-like endometrial cancers may in fact be derived from the fallopian tube [33]. Tubal serous carcinomas are treated differently than uterine, but this distinction cannot be made without molecular testing. These similarities were seen in the TCGA analysis, where uterine serous cancers show demonstrated similarities to both ovarian serous cancers basal-like breast carcinoma, including high frequencies of TP53 and PTEN mutations. Differences included the higher frequency of PIK3CA, FBXW7 and PPP2R1A1 in uterine serous carcinomas.
11 ProMisE (Proactive Molecular Risk Classifier for Endometrial Cancer)
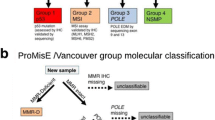
Researchers at the University of British Columbia have developed and studied a classification system , with the goal of recapitulating the TCGA genomic classifier, but using readily available methods such as immunohistochemistry and gene sequencing [34, 35]. Here markers such as POLE mutation, p53 IHC and TP53 mutation, PTEN, MMR IHC (MLH1, MSH2, MSH6, and PMS2), and FISH for three specific loci (FGFR (4p16.3), SOX17 (8q11.23), and MYC (8q24.12) to determine the copy-number groups, were tested. The result is a molecular classifier called ProMisE, which divides patients with endometrial cancer into MMR abnormal, POLE-mutated, and p53 abnormal or wild-type (Fig. 2.5). These groups correlate with the TCGA subgroups concerning outcomes , and could be a base for developing clinical studies on treatment (Table 2.2).

A schematic diagram showing the application of the ProMisE , molecular subtyping
12 PORTEC
Researchers at the Leiden University Medical Center have developed and tested a molecular classification, using some additional methods when compared to ProMisE [36, 37]. This classification uses microsatellite instability testing, sequencing for hotspot mutations in 14 genes (including POLE), and immunohistochemistry for a number of biomarkers (Table 2.2). The sub-analysis for early-stage endometrial cancer gave an indication that applying the MSI, POLE and p53 analyses, and also stratifying into favorable and nonfavorable by using other markers such as L1CAM, LVSI, CTNNB1 [36]. The use of these markers can help to identify subgroups eligible to targeted treatment. However, in the same cohort, the identification of p53, POLE mutations, and MSI status alone lead to an identification of four subgroups similar to those proposed by the TCGA. The clinical utility of these groups will be assessed in the prospective PORTEC-4 study .
13 Outlook and Future Directions
The validation of the proposed molecular classification systems is an important next step to proving their sustainability and applicability to different cohorts in clinically different settings. Prospective studies will be necessary to evaluate adaptations of adjuvant treatment in different subgroups. The evolution towards combining known pathological risk factors with newer molecular markers is a large step towards personalized medicine , with associated improvements in treatment selection.
References
Kurman RJ, Carcangiu ML, Herrington CS, Roung RH, IARC. WHO classification of tumours of female reproductive organs. Lyon: IARC Press; 2014.
Zaino RJ, Kauderer J, Trimble CL, Silverberg SG, Curtin JP, Lim PC, Gallup DG, Mackey D. Reproducibility of the diagnosis of atypical endometrial hyperplasia: a gynecologic oncology group study. Cancer. 2006;106(4):804–11. https://doi.org/10.1002/cncr.21649.
ACOG. Endometrial Intraepithelial Neoplasia Committee Opinion No. 631. 2015.
Owings RA, Quick CM. Endometrial intraepithelial neoplasia. Arch Pathol Lab Med. 2014;138(4):484–91. https://doi.org/10.5858/arpa.2012-0709-RA.
Malpica A. How to approach the many faces of endometrioid carcinoma. Mod Pathol. 2016;29(S1):S29–44. https://doi.org/10.1038/modpathol.2015.142.
FIGO. Announcements. Gynecol Oncol. 1989;35(1):125–7. https://doi.org/10.1016/0090-8258(89)90027-9.
Alkushi A, Abdul-Rahman ZH, Lim P, Schulzer M, Coldman A, Kalloger SE, Miller D, Gilks CB. Description of a novel system for grading of endometrial carcinoma and comparison with existing grading systems. Am J Surg Pathol. 2005;29(3):295–304. https://doi.org/10.1097/01.pas.0000152129.81363.d2.
Gatius S, Matias-guiu X. Practical issues in the diagnosis of serous carcinoma of the endometrium. Mod Pathol. 2016;29(S1):S45–58. https://doi.org/10.1038/modpathol.2015.141.
Soslow RA. Endometrial carcinomas with ambiguous features. Semin Diagn Pathol. 2010;27:261–73. https://doi.org/10.1053/j.semdp.2010.09.003.
Colombo N, Creutzberg C, Amant F, Bosse T, González-Martín A, Ledermann J, Marth C, et al. ESMO-ESGO-ESTRO consensus conference on endometrial cancer: diagnosis, treatment and follow-up. Ann Oncol. 2016;27(1):16–41. https://doi.org/10.1093/annonc/mdv484.
Cole AJ, Quick CM. Patterns of myoinvasion in endometrial adenocarcinoma: recognition and implications. Adv Anat Pathol. 2013;20(3):141–7. https://doi.org/10.1097/PAP.0b013e31828d17cc.
Hui P, Kelly M, O’Malley DM, Tavassoli F, Schwartz PE. Minimal uterine serous carcinoma: a clinicopathological study of 40 cases. Mod Pathol. 2005;18(1):75–82. https://doi.org/10.1038/modpathol.3800271.
Sherman ME, Bitterman P, Rosenshein NB, Delgado G, Kurman RJ. Uterine serous carcinoma. A morphologically diverse neoplasm with unifying clinicopathologic features. Am J Surg Pathol. 1992;16(6):600–10.
Köbel M, Piskorz AM, Lee S, Lui S, LePage C, Marass F, Rosenfeld N, Mes Masson A-M, Brenton JD. Optimized p53 immunohistochemistry is an accurate predictor of TP53 mutation in ovarian carcinoma. J Pathol Clin Res. 2016;2(4):247–58. https://doi.org/10.1002/cjp2.53.
Lavie O, Ben-Arie A, Segev Y, Faro J, Barak F, Haya N, Auslender R, Gemer O. BRCA germline mutations in women with uterine serous carcinoma--still a debate. Int J Gynecol Cancer. 2010;20(9):1531–4. https://doi.org/10.1111/IGC.0b013e3181cd242f.
Fadare O, Zheng W, Crispens M a, Jones HWI, Khabele D, Gwin K, Liang SX, et al. Morphologic and other clinicopathologic features of endometrial clear cell carcinoma: a comprehensive analysis of 50 rigorously classified cases. Am J Cancer Res. 2013;3(1):70–95.
Fadare O, Desouki MM, Gwin K, Hanley KZ, Jarboe EA, Liang SX, Quick CM, Zheng W, Parkash V, Hecht JL. Frequent expression of napsin A in clear cell carcinoma of the endometrium: potential diagnostic utility. Am J Surg Pathol. 2014;38(2):189–96. https://doi.org/10.1097/PAS.0000000000000085.
Fadare O, Gwin K, Desouki MM, Crispens MA, Jones HW, Khabele D, Liang SX, et al. The clinicopathologic significance of p53 and BAF-250a (ARID1A) expression in clear cell carcinoma of the Endometrium. Mod Pathol. 2013;26(8):1101–10. https://doi.org/10.1038/modpathol.2013.35.
Hoang LN, Mcconechy MK, Meng B, Mcintyre JB, Ewanowich C, Gilks CB, Huntsman DG, Köbel M, Lee CH. Targeted mutation analysis of endometrial clear cell carcinoma. Histopathology. 2015;66(5):664–74. https://doi.org/10.1111/his.12581.
Quddus M, Ruhul CJS, Zhang C, Dwayne Lawrence W. Minor serous and clear cell components adversely affect prognosis in mixed-type endometrial carcinomas: a clinicopathologic study of 36 stage-I cases. Reprod Sci. 2010;17(7):673–8. https://doi.org/10.1177/1933719110368433.
Cherniack AD, Shen H, Walter V, Stewart C, Murray BA, Bowlby R, Hu X, et al. Integrated molecular characterization of uterine carcinosarcoma. Cancer Cell. 2017;31(3):411–23. https://doi.org/10.1016/j.ccell.2017.02.010.
Network, Cancer Genome Atlas Reserach. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497(53):67–73. https://doi.org/10.1038/nature12113.
Bellone S, Bignotti E, Lonardi S, Ferrari F, Centritto F, Masserdotti A, Pettinella F, et al. Polymerase ε (POLE) ultra-mutation in uterine tumors correlates with T lymphocyte infiltration and increased resistance to platinum-based chemotherapy in vitro. Gynecol Oncol. 2017;144(1):146–52. https://doi.org/10.1016/j.ygyno.2016.11.023.
Billingsley CC, Cohn DE, Mutch DG, Stephens JA, Suarez AA, Goodfellow PJ. Polymerase?? (POLE) mutations in endometrial cancer: clinical outcomes and implications for lynch syndrome testing. Cancer. 2015;121(3):386–94. https://doi.org/10.1002/cncr.29046.
Church DN, Briggs SEW, Palles C, Enric Domingo SJ, Kearsey JM, Grimes MG, et al. DNA polymerase {varepsilon} and δ exonuclease domain mutations in endometrial cancer. Hum Mol Genet. 2013;22(14):2820–8. https://doi.org/10.1093/hmg/ddt131.
Hussein YR, Britta Weigelt DA, Levine JKS, Dao LN, Balzer BL, Liles G, et al. Clinicopathological analysis of endometrial carcinomas harboring somatic POLE exonuclease domain mutations. Mod Pathol. 2015;28(4):505–14. https://doi.org/10.1038/modpathol.2014.143.
Meng B, Hoang LN, McIntyre JB, Duggan MA, Nelson GS, Lee CH, Köbel M. POLE exonuclease domain mutation predicts long progression-free survival in grade 3 endometrioid carcinoma of the endometrium. Gynecol Oncol. 2014;134(1):15–9. https://doi.org/10.1016/j.ygyno.2014.05.006.
Santin AD, Bellone S, Buza N, Choi J, Schwartz PE, Schlessinger J, Lifton RP. Regression of chemotherapy-resistant polymerase ε (POLE) ultra-mutated and MSH6 hyper-mutated endometrial tumors with nivolumab. Clin Cancer Res. 2016;22(23):5682–7.
Bellone S, Centritto F, Black J, Schwab C, English D, Cocco E, Lopez S, et al. Polymerase?? (POLE) ultra-mutated tumors induce robust tumor-specific CD4 + T cell responses in endometrial cancer patients. Gynecol Oncol. 2015;138(1):11–7. https://doi.org/10.1016/j.ygyno.2015.04.027.
Eggink FA, Van Gool IC, Leary A, Pollock PM, Crosbie EJ, Mileshkin L, Jordanova ES, et al. Immunological profiling of molecularly classified high-risk endometrial cancers identifies POLE -mutant and microsatellite unstable carcinomas as candidates for checkpoint inhibition. OncoImmunology. 2017;6(2):e1264565. https://doi.org/10.1080/2162402X.2016.1264565.
Van Gool IC, Eggink FA, Freeman-Mills L, Stelloo E, Marchi E, De Bruyn M, Palles C, et al. POLE proofreading mutations elicit an antitumor immune response in endometrial cancer. Clin Cancer Res. 2015;21(14):3347–55. https://doi.org/10.1158/1078-0432.CCR-15-0057.
Mehnert JM, Panda A, Zhong H, Hirshfield K, Damare S, Lane K, Sokol L, et al. Immune activation and response to pembrolizumab in POLE-mutant endometrial cancer. J Clin Investig. 2016;126(6):2334–40. https://doi.org/10.1172/JCI84940.
Jarboe E, Folkins A, Nucci MR, Kindelberger D, Drapkin R, Miron A, Lee Y, Crum CP. Serous carcinogenesis in the fallopian tube. Int J Gynecol Pathol. 2008;27(1):1–9. https://doi.org/10.1097/pgp.0b013e31814b191f.
Talhouk A, McAlpine JN. New classification of endometrial cancers: The development and potential applications of genomic-based classification in research and clinical care. Gynecol Oncol Res Pract. 2016;3(1):14. https://doi.org/10.1186/s40661-016-0035-4.
Talhouk A, McConechy MK, Leung S, Li-Chang HH, Kwon JS, Melnyk N, Yang W, et al. A clinically applicable molecular-based classification for endometrial cancers. Br J Cancer. 2015;113(2):299–310. https://doi.org/10.1038/bjc.2015.190.
Stelloo E, Nout RAA, Osse EMM, rgenliemk-Schulz IJJ, Jobsen JJJ, Lutgens LCC, van der Steen-Banasik EMM, et al. Improved risk assessment by integrating molecular and clinicopathological factors in early-stage endometrial cancer - combined analysis of PORTEC cohorts. Clin Cancer Res. 2016;22(16):4215. https://doi.org/10.1158/1078-0432.CCR-15-2878.
Stelloo E, Bosse T, Nout RA, MacKay HJ, Church DN, Nijman HW, Leary A, et al. Refining prognosis and identifying targetable pathways for high-risk endometrial cancer; a TransPORTEC initiative. Mod Pathol. 2015;28(6):836–44. https://doi.org/10.1038/modpathol.2015.43.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Imboden, S., Nastic, D., Carlson, J.W. (2020). Controversies in Pathology and Advances in Molecular Diagnostics. In: Mirza, M. (eds) Management of Endometrial Cancer. Springer, Cham. https://doi.org/10.1007/978-3-319-64513-1_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-64513-1_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-64512-4
Online ISBN: 978-3-319-64513-1
eBook Packages: MedicineMedicine (R0)