
Abstract
It is well recognized that arsenic trioxide (ATO) is an effective agent for the treatment of acute promyelocytic leukemia (APL). Use of single agent ATO in the treatment of APL leads to remissions which are durable in the majority, although few cases of primary resistance have been reported. The drug has been used both as a single agent and in combination with other conventional drugs to treat APL. Use of ATO is the accepted standard of care in the management of relapsed APL, where it is often used effectively as a bridge to a stem cell transplant. Currently, the combination of all-trans retinoic acid (ATRA) and ATO is considered the standard of care for newly diagnosed low- and intermediate-risk APL. ATO probably has multiple mechanisms of action. Better understanding of its mechanisms of action/s and synergy with other agents will likely lead to a more rationale use of this agent or its derivatives, either alone or in combination. There is limited data on the kinetics of leukemia clearance and normal hematopoietic recovery after the administration of single agent ATO for the treatment of APL, but preliminary data suggests that it is likely to be different from conventional therapy. There have been a number of concerns of the potential short- and long-term toxicity of this agent. Most such concerns arise from the toxicity profile noted in people exposed to long-term arsenic in the environment. The overall toxicity profile has been favorable with the therapeutic doses and schedules of administration of ATO in the treatment of malignancies. In a resource-constrained environment, the use of a single agent ATO-based regimen is a realistic and acceptable option to treat almost all patients with APL. In the developed world, it has the potential in combination with other agents to improve the clinical outcome with reduction of dose intensity of chemotherapy and remains an option for patients who would not tolerate conventional therapy. This chapter will focus on the use of single agent ATO and ATO-based non-myelosuppressive regimens as a treatment for APL, especially in a developing country.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Acute promyelocytic leukemia
- Arsenic trioxide
- Developing country
- Cost-effective therapy
- ATO-based treatment
- ATO toxicity
Introduction
Significant and rapid advances in the management of acute promyelocytic leukemia (APL) over the last few decades have transformed it from a leukemia with the worst to one with the best prognosis [1]. With current diagnostic and treatment strategies, it is reasonable to expect greater than 90–95% remission rates along with long-term survival and possible cure exceeding 80%, even in high-risk APL [1]. Remarkably, most of these improvements in clinical outcome occurred without intensification of conventional chemotherapy but with the combination of non-myelotoxic differentiating agents such as all-trans retinoic acid (ATRA) and arsenic trioxide (ATO). This in turn was facilitated and paralleled by the detailed understanding of the cellular and molecular pathogenesis of this leukemia, and as a result APL has become a model of bench to bedside and back scientific progress. In the developed world, challenges remain in the management of patients with high-risk APL and a small subset of patients with relapsed APL. There also remain significant challenges with early mortality in newly diagnosed patients. In developing countries, there are even more fundamental challenges related to access to proper diagnosis and therapy primarily dictated by the high cost of treating acute myeloid leukemia (AML) and APL with conventional myelosuppressive regimens. However, an advantage in developing countries is the ease of access to low-cost generics which can offset the high costs of conventional therapy.
Challenges in Treating Myeloid Leukemia in a Developing Country
That the financial cost of treating AML is very high is a well-recognized fact [2]. However, there is limited data that has systematically addressed this issue [2,3,4]. Available data would suggest that the cost of treating AML in a developed country varies from US$ 80,000 to >100,000/patient [2,3,4,5]. With conventional treatment of AML, it is recognized that most of this cost is not related to the individual chemotherapeutic drugs used to treat this condition, but rather from the high cost of supportive care measures during repeated cycles of prolonged neutropenia induced by conventional chemotherapy. In contrast, with ATO-based regimens in APL, when the so-called expensive patented and innovator ATO was used, the 3-year direct pharmacy cost of drugs was higher with an ATO + ATRA regimen compared with a conventional ATRA + chemotherapy regimen (Euro 46,700 vs. 6700), though the cost of supportive care was a third of conventional therapy [6]. In spite of these costs, the combination of ATO + ATRA was found to be cost-effective compared to conventional ATRA + chemotherapy in an analysis done in the United States (US) [7].
It is important to place all the above cost analysis in the context that 70% of the countries in the world, contributing >70% of the world’s population, have a gross national income (GNI)/capita of less than US$10,000 [8] and that in many of these countries in the absence of universal health-care insurance, for most patients, these expenses are “out-of-pocket” self-paid expenses. As a result, it is estimated that approximately 39 million people in India alone (a figure which is greater than the entire population of Australia) will fall below the poverty line every year [9, 10].
Even in developed countries with well-established cancer registries, such as the US “Surveillance, Epidemiology, and End Results (SEER)-Medicare database,” there is up to 50% underreporting of the diagnosis of AML [11]. In developing countries, in the absence of such registries, there are limited available records of the actual incidence or prevalence of patients with myeloid leukemia. However, it is recognized that outside the major metropolis in many of these countries, the access to even basic diagnostic tests such as flow cytometry and molecular studies is not available [12, 13].
While significant advances have been made in the management of APL, the majority of these advances are based on well-controlled clinical trials from countries with universal health-care access which tend to have a significant bias in the patients enrolled being younger and having better performance scores and less comorbidities than the average patient with this diagnosis. There is limited real-world data of clinical outcomes with conventional ATRA + chemotherapy. In AML and in APL, when such data is available, the observations and conclusions often seem at odds with published clinical trial data [14].
Advantage and Access to Generic Pharmaceutical Agents in a Developing Country
The importance of access to generic pharmaceutical agents in reducing the cost of medical therapy is well recognized, even in developed countries [15, 16]. This is even more relevant in developing economies and countries due to the combination of the absence of health insurance and a predominantly self-pay system. In India, a country which fulfills the above criteria, data suggests that the use of generic chemotherapy drugs results in an annual savings of approximately US$ 843 million [17]. Whether ATO should have been patented or not has been a controversy that has been extensively debated in the past [18, 19]. Regardless of which side of the argument one favors, the reality is that there is a significant difference in the cost of ATO depending on whether the patent applies in a particular country or not. At the author’s center in 1998, the actual costing of in-house manufacturing of 10 mg of ATO was approximately 20 Indian rupees, which at that time was the equivalent of US50 cents. Subsequently, the manufacturing was transferred to a local pharmaceutical company (INTAS Ltd., Ahmedabad, India), and the current cost for a 10 mg vial is 450 Indian rupees (~US$ 7). Currently the cost of ATO in North America and Europe is US$676 and 393 Euros/vial, respectively. As a result, the use of ATO is considered very expensive in many developed countries, while it is considered the least expensive option in many developing countries, when costing analysis is limited to drug costs.
Experience with Arsenic Trioxide in Treating Acute Promyelocytic Leukemia
Arsenical compounds were used as a medicine as early as 2000 BC [20] and were familiar to the early physicians (Hippocrates (460–377 BC) and Aristotle (384–322 BC)). Paracelsus (1493–1541 AD) used arsenical compounds extensively and was quoted saying what we now consider a universal truth for most therapeutic drugs, “All substances are poisons; the right dose differentiates a poison from a remedy” [20].
The prominence of ATO in the treatment of APL historically followed the studies with a traditional Chinese recipe called “Ailing 1 .” These early studies were conducted by Chinese investigators at Harbin Medical University, and they labeled this native preparation 713 (for the year and month that the study was initiated). Using this agent, more than 1000 patients with various malignancies were evaluated [21]. They soon noted that this agent worked best in the treatment of patients with APL. Two subsequent Chinese studies confirmed the benefit of this agent in APL [22, 23].
It was subsequently reported that a dual effect of ATO was seen on promyelocytic cell lines. At low doses (0.1–0.5 μmol/L), there was partial differentiation, and at higher doses, there was preferential apoptosis (0.5–2 μmol/L) [24]. This has been subsequently demonstrated by a number of other groups independently [25, 26]. The differentiation with ATO is incomplete and usually proceeds only until the myelocyte stage, following which it appears that apoptosis is the predominant mode of action [24]. More recent data suggests that ATO, and not ATRA, can eliminate the leukemia-initiating compartment in APL [27, 28]. This could partly explain why single agent ATO, but not ATRA, is able to induce durable remissions in the clinic. Since then, numerous reports on the use of ATO in the treatment of relapsed and newly diagnosed cases of APL have been published and summarized in a recent review [29].
Pharmacokinetics, Pharmacogenomics, Dose, and Schedule of Arsenic Trioxide
The lethal dose recorded in the literature is a single dose of more than 100 mg [11]. The dose of arsenic trioxide in the initial published study by Zhi-Xiang et al. [30] was 10 mg/day for adults until complete hematological remission (CR) was achieved. Subsequently a break of 30 days was given and a second course of 28 days administered. It is important to recognize that this dosing was based on earlier experience with doses used in native Chinese medicine and not on conventional phase I clinical trials addressing dose-limiting toxicity. The study reported by Soignet et al. [31] used a similar dose for adults but used a dose of 0.15 mg/kg/day for children with a maximum dose of 10 mg. From their experience, they noted that ATO is active in APL at a dose ranging from 0.06 to 0.2 mg/kg. Within this range, no difference in efficacy was noted. Subsequent studies have used similar dosages of ATO. Pharmacokinetic studies done at this dosage demonstrated that mean peak plasma levels of 6.85 μmol/L (range, 5.54–7.30) were achieved. The plasma half-life was 12.13 ± 3.31 h. Importantly, these parameters did not change with continuous administration [30]. Reports of daily urinary excretion in the literature vary from 1–8% to 32–65% of the daily administered dose and more importantly are continued even after the drug administration had been stopped [30, 32]. There is limited data on the dose and scheduling of ATO in the event of significant renal failure or for patients on dialysis [33]. While the cumulative level of arsenic increases in the body (as demonstrated in hair and nail analysis) with continuous administration, the urinary excretion continues even after the ATO administration has stopped leading to a gradual decrease in the cumulative amount of the drug in the body. In our own experience, there was no significant difference in the ATO content from patients and normal control hair and nail samples during long-term follow-up [34]. This was the rationale for giving the 4-week intervals between the courses of ATO in our regimen [35], since these intervals were intended to reduce the cumulative dose significantly.
This pattern of ATO exposure is very different from that seen with environmental exposure where there is a slow but constant accumulation of arsenic leading to a toxicity profile that is different from that seen when ATO is used in therapy at the currently recommended doses and schedules. Extrapolating and anticipating the toxicity profile seen with chronic environmental exposure to the potential toxicity with currently used dosage schedules of ATO are unfair, unwarranted, and without any scientific basis. In the absence of a dose-finding phase I clinical trial, there is insufficient data on the upper limit of a safe therapeutic dose. It is of interest to note that in our initial cohort we noted a decreased risk of relapse among patients who had hepatotoxicity versus those who did not follow treatment with ATO [36]. This would suggest that there is either a significant interindividual variation in biotransformation of this agent, and as a result some patients were receiving less than an optimal dose, or that there were yet unknown variables that resulted in this association [36]. There is a need to revisit what is the optimal dose of ATO to treat APL in a large clinical trial. Furthermore, there is very limited data on the optimal duration of administration of ATO as a single agent in the management of APL. Based on the general consensus that maintenance was required in the management of APL, at our center, we arbitrarily opted for a 6-month duration of maintenance [35]. Recently published data from Iran suggests that four courses of ATO were significantly better than two [37]. Zhou et al. reported treating children with ATO for prolonged periods of up to 3 years with very good efficacy and without significant toxicity [38]. However, the optimal schedule and cumulative dose of ATO remain to be defined.
It has been noted that there is considerable interindividual variation in susceptibility to ATO-induced toxicity, which is probably related to differences in the in vivo biotransformation of arsenic. This in turn could be a result of age, nutritional status, comorbid conditions, environmental factors, and genetic polymorphisms [39]. In addition to a poorly characterized arsenic methyltransferase , a number of other enzyme systems and polymorphisms have been shown to have an effect on arsenic methylation [39, 40]. Among these, polymorphism in the methylenetetrahydrofolate reductase (MTHFR) gene , which results in MTHFR deficiency in 10–20% of the Caucasian population, has been reported to be associated with increased arsenic-related neurotoxicity [41]. The glutathione S-transferases (GST) are a family of proteins that conjugate glutathione (GSH) to various electrophiles [42]. Chiou et al. reported that genetic polymorphisms of GST M1 and GST T1 altered the methylation of arsenic [43]. GSTs catalyze the GSH-dependent reduction of hydroperoxides to their corresponding alcohols and help prevent propagation of free radicals. It is conceivable that genetic polymorphisms in these genes could alter the biotransformation of ATO, which in turn could have an impact on the efficacy and toxicity profile of this drug. We had earlier reported that the hepatotoxicity profile in a cohort of patients with newly diagnosed APL treated at our center with a single agent ATO regimen was significantly associated with the homozygous mutant of MTHFR 1298 (C/C) (RR = 8.75, P = 0.004), and there was a trend toward an increased risk of hepatotoxicity associated with the GST M1 null genotype (RR = 3.28, P = 0.06) [36]. We had hypothesized then that alteration in biotransformation possibly leads to quantitative and qualitative differences in the methylated intermediaries that are generated; these differences could have an effect on the efficacy and toxicity profile of ATO. A recent study, in part, validates this hypothesis by suggesting that dimethylarsinous acid is more toxic than inorganic ATO and monomethylarsinic acid [44], which are some of the methylated intermediaries produced in vivo in humans and animals. It is possible to consider in future the use of pure or combination of methylated ATO derivatives with optimal therapeutic and toxicity profiles.
Clinical Experience with the Use of ATO in APL
The earliest clinical data available on the use of ATO in the treatment of APL was from two Chinese publications [45, 46]. In these studies, the CR rates varied from 65.6 to 84%, and long-term survival (>10 years) was seen in 9/32 patients in one of these studies [30]. Most of the early trials involved relapsed cases of APL. There is limited data on the use of this as a single agent in the management of newly diagnosed cases of APL. Even when used as a single agent for induction chemotherapy, the subsequent consolidation therapy varies making comparisons between the published data difficult to interpret.
Our early experience with ATO consisted of two patients who were treated in the early 1990s with what was then considered standard of care regimens, one with ATRA and one without. Both these patients relapsed and were sent on palliative care considering that therapeutic options were limited, very expensive, and associated with poor clinical outcome. These patients subsequently took treatment from an “Ayurvedic” (indigenous Indian medicine) practitioner and went into durable remissions. We were aware that the agent used by the practitioner contained ATO, but we were not sure of the dose used. The therapeutic “Ayurvedic” mix was administered continuously in these cases and for more than 5 years in one case. One of these patients developed severe arsenic keratosis and died of a secondary squamous epithelial carcinoma [47].
It was only after the publication in 1997 by Shen et al. that we had a sense of the dose of pure ATO that could be used in humans [30]. In 1998, we initiated a study using single agent ATO to treat APL, with intravenous ATO being manufactured in-house in our hospital pharmacy with appropriate quality control measures. Due to legal-related issues, we transferred this manufacturing process to the industry in 2001 (INTAS Pharmaceuticals Ltd., Matoda, Gujarat, India). Our observation was that there was no significant difference with the agent prepared by us or that subsequently manufactured by industry in terms of infusion-related toxicity or efficacy (unpublished data).
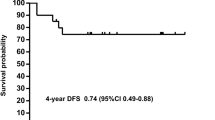
From January 1998 to December 2004, 72 newly diagnosed cases of APL were treated with a regimen of single agent ATO at our center. The details of the regimen were as previously reported [34, 35]. Overall 62 (86.1%) achieved a hematological remission, and a total of 13 patients relapsed. As previously reported by us, at a median follow-up of 60 months, the 5-year estimate of event-free survival (EFS), disease-free survival (DFS), and overall survival (OS) were 69 ± 5.5%, 80 ± 5.2%, and 74.2 ± 5.2%, respectively (Fig. 18.1) [35]. This data has since been validated by a subsequent multicenter trial in India involving seven centers across the country [48].

Five-year Kaplan-Meier product limit estimate of (a) overall survival (n = 72) and (b) event-free survival (n = 72)
However, until very recently, large multicenter randomized controlled clinical trials (RCT) to validate such low toxicity ATO-based regimes were not available to challenge the conventional ATRA plus chemotherapy regimens. The first large multicenter phase II study that reported such an approach and relied predominantly on the synergistic effect of ATRA and ATO combination with a conventional anthracycline administration being limited to induction therapy came from the Australasian Leukaemia and Lymphoma Group (ALLG) APML4 study. This was initially published in 2012 and was followed up with long-term data in 2016 [49]. Along with a similar approach but in a RCT design and with a regimen structure that was completely eliminated, all anthracyclines and other chemotherapeutic agents were reported by the combined Gruppo Italiano Malattie Ematologiche dell’Adulto, German-Austrian Acute Myeloid Leukaemia Study Group, and Study Alliance Leukaemia (GIMEMA-AMLSG-SAL) APL 0406 study in 2013 [50]. The APL 0406 study was limited to low- and intermediate-risk patients which account for two third to three quarters of patients with APL. This study demonstrated for the first time that the combination of ATRA and ATO without chemotherapy in induction and consolidation (four courses) and without maintenance therapy was superior to a conventional ATRA plus chemotherapy induction with repeated cycles of myelotoxic chemotherapy and 2 years of maintenance therapy. A similar RCT was reported by the UKMRC AML-17 trial which again demonstrated the superiority of ATO + ATRA over conventional chemotherapy in all risk groups [51]. A significant difference in the MRC-AML17 trial was the addition of gemtuzumab ozagomycin (GO) in the treatment of high-risk cases.
Based on the published literature [34, 52, 53] and experience, the data would suggest that it would be possible to cure APL in about 40–60% of patients with single agent ATO alone, and for the remaining low- and intermediate-risk patients, probably a combination of ATRA and ATO would suffice as demonstrated in the GIMEMA-AMLSG-SAL-APL 0406 study, without maintenance therapy in either of these hypothetical subsets. This approach would be truly beneficial for patients with APL in developing countries since with generic ATO, the cost of treatment would be very low in the absence of significant cytopenias and requirement of supportive care, especially post induction therapy. For the truly high-risk patients, the addition of an anthracycline, at least, in induction is probably warranted, and the role of maintenance therapy remains to be further evaluated. The data from major studies using either single agent ATO or ATO as a major component in frontline therapy [37, 38, 49,50,51, 54,55,56] is summarized in Table 18.1.
Toxicity Profile of Arsenic Trioxide
The toxicity profile in most studies reported to date was mild as illustrated in a publication from our center [35]. Our experience with single agent ATO was that there were no infusion-related toxicities, alopecia, or evidence of exacerbation of coagulopathy. Post induction, almost all patients for the rest of the duration of treatment had ECOG performance scores of 0 or 1. The non-hematological toxicities in most studies were mild, were frequently reverted on continuing ATO, and in the rest were reversible on discontinuing the drug for an interval of 1–2 weeks [35]. There were no sudden deaths attributable to a cardiac event in this series of patients, and during long-term follow-up, there were no cases with clinical cardiac dysfunction. There were no documented second malignancies in any of the long-term follow-up cohorts to date that could be attributed to the use of ATO. With the exception of some early reports of increased hepatic and cardiac toxicity [57,58,59,60], the majority of subsequent reports using ATO in newly diagnosed cases is similar to our experience [37, 38, 54,55,56].
There have been periodical major concerns raised about the administration of ATO. Very early there was a concern about cardiac arrhythmia-related sudden deaths in patients with APL who were treated with ATO. Almost all these deaths occurred during induction in previously heavily treated patients [57,58,59]. There have been no such deaths reported when ATO was used for treating a number of other malignancies, albeit in stable patients. Similarly, sudden death does not seem to occur when ATO is administered to APL patients who are in remission (none reported in the literature). The role of QTc interval prolongation and limitations of the corrected QTc interval value generated with tachycardia due to any cause such as infection have been reviewed previously, and it is increasingly recognized and accepted that QTc prolongation is an electrocardiographic phenomenon with little clinical significance in the majority of patients [61]. This does not mean we should not monitor it or ignore it, though response should be judicious and clinically appropriate. It has been reported that in more than 2900 cases treated by US FDA-approved ATO, there have been no arrhythmia-related deaths [61]. Secondly, there was a suggestion that acute hepatic failure and death from hepatotoxicity occurred with ATO [60]. There have been no other major reports since this initial publication about two decades ago. This has definitely not been our experience with more than 250 newly diagnosed and relapsed APL patients treated to date at our center.
There has always been a concern of second malignancies with the use of ATO. This is based on in vitro experiments suggesting oxidative DNA damage [62] and clinical observations from cases with long-term environmental exposure. This theoretical concern is in contrast to the available clinical data. In early reports of investigators from China, it was noted that there was no increase in second malignancies in patients followed up for 10 years [30]. A similar observation was made in 1982, in a cohort of 479 patients who had been treated with Fowler’s solution [potassium arsenite] for duration varying from 2 weeks to 12 years during the period 1945–1969. The median cumulative dose in this cohort was 448 mg. There was a marginal increase in fatal and nonfatal skin cancers but no increase in the incidence of internal malignancies [63].
There have been concerns raised about embryo toxicity based on animal models and some data from cases with environmental exposure [64]. Again, this is not based on data in humans exposed to what is currently defined as a therapeutic dose of ATO, and for obvious ethical reasons, this data is unlikely to be ever generated. However, in our series, seven of the patients (four women and three men) have had eight normal babies delivered after completing treatment with ATO [34], though all happened after completion of therapy. In this relatively young cohort, there were no reports of abortions, fetal abnormalities, or stillbirths in any couple. While we did not actively evaluate fertility, there were no reports of couples requesting evaluation for sterility [34].
In studies looking at long-term accumulation of ATO in the body by studying hair and nail levels, there was no significant difference in the ATO retention in controls and patients who had completed therapy at least 2 years earlier [34]. The median levels, even among the patients who had just completed therapy, were below the lower limit of the normal range described for normal controls by the Agency for Toxic Substances and Disease Registry (ATSDR based in Atlanta, Georgia, USA) (http://www.atsdr.cdc.gov/) [34].
Pattern and Timing of Hematopoietic Recovery Following Treatment with Single Agent ATO
In our initial series, the median time to achieve CR was 42 days (range, 24–70) [35]. However, this figure does not reflect the entire details of the kinetics of leukemia clearance and pattern of normal hematopoietic recovery. As reported initially by us, about two thirds of patients have a leukocytosis response after initiation of single agent ATO, while in about a third, there is prolonged leucopenia prior to gradual normalization [65]. The leukocytosis can at times be very rapid and alarming, and based on our early observations, we had introduced hydroxyurea to control it with a recommended sliding scale to adjust the dose depending on the WBC count [35]. We also noted that this was at times not adequate; thus, we allowed the use of an anthracycline in induction if there was rapid rise in the WBC counts after initiation of therapy at predefined levels and time points [35]. Some cases of leukocytosis were followed by a second leukopenic phase (variable duration) and then recovery to normal values [65] (a triphasic response; Fig. 18.2). Unlike ATRA plus chemotherapy schedules, the WBC count remains high (in two thirds) or very low (in one third), with a low platelet count and significant circulating promyelocytes for the first 2 weeks as illustrated in Fig. 18.3. At this time point, there is often a concern raised, among those not familiar with this agent, as to whether ATO is doing anything at all to the disease, and a consideration to change protocol or add on additional drugs is discussed. However, if the diagnosis is correct, with adequate support during this period and continuing ATO, all patients will go on to achieve CR. Another common observation in some cases is a clinically stable patient in the fourth week of therapy, with a normal platelet count but very low WBC count, and a consideration to stop ATO is made based on the argument that the ATO is causing a myelosuppressive effect. Our experience would suggest that ATO can be safely continued for the intended duration, and one would probably be compromising treatment efficacy by prematurely stopping therapy at this time point.

Average WBC count among patients with a leukocytic response and who achieved complete remission (n = 6), illustrating the triphasic response

The mean WBC and platelet count ± 1SE over time among patients treated on single agent ATO regimen. (a) WBC response among those with leukocytosis (n = 40). (b) WBC response among those without leukocytosis (n = 18). (c) Platelet count recovery (n = 60)
Impact of Additional Cytogenetic and Molecular Markers Such as FLT3 Mutations on Clinical Response Following Treatment with ATO
The presence of cytogenetic abnormalities at diagnosis remains an important prognostic variable in patients with newly diagnosed AML [66]. Secondary cytogenetic changes have been reported to have an adverse impact in some subsets of AML, though in patients with APL treated with conventional chemotherapy, a similar adverse effect was not reported [67, 68]. At our center, we initially reported a small series of newly diagnosed patients with APL treated with single agent ATO in which there was no significant adverse impact of the presence of an additional karyotypic abnormality at diagnosis [69]. However, more recent analysis of our data (larger cohort) does suggest that there may be an adverse impact of an additional cytogenetic finding at diagnosis in newly diagnosed patients, though it was not significant in a multivariate analysis (unpublished data).
Fms-like tyrosine kinase 3 (FLT3) is a member of the class III receptor tyrosine kinase family and is expressed on hematopoietic progenitors [70, 71]. Mutations in the FLT3 receptors have been reported to be associated with a poor prognosis in both adult and pediatric patients with AML [70]. Mutations in the FLT3 receptor are commonly seen in patients with APL [70]. The common activating mutations of FLT3 in leukemia include the FLT3 internal tandem duplication (FLT3-ITD) and a point mutation in the activation loop (D835V) [70]. A recent gene expression profiling study reported that patients with APL could be segregated into those with and without a FLT3-ITD mutation, suggesting that these groups were biologically different [72]. A retrospective analysis of the impact of FLT3 mutations in patients with APL, treated with conventional ATRA plus chemotherapy regimens, reported a higher incidence of induction deaths in one study [73], while another study reported a trend toward a shorter OS [74]. More recently Chillon et al. [75] analyzed the Spanish cooperative group data and showed that patients with increased ITD mutant/wild-type ratio or longer ITD size displayed shorter 5-year relapse-free survival (RFS) (P = 0.048 and P < 0.0001, respectively), though patients with D835 mutations did not show differences in RFS or OS. In our series, we found that FLT3-ITD mutation in 21% and its presence did not impact the clinical outcome of patients treated with ATO [69]. We did however note a longer time to achieve molecular remission among those who were FLT3 mutation positive [69].
ATO for the Treatment of Relapsed APL
Patients who relapse following an ATRA/chemotherapy-based regimen can achieve a second CR in 60–95% of cases with chemotherapy, although the toxicity with such a regimen in this population approaches that seen with high-dose chemotherapy for AML [61]. There is a high incidence of ATRA resistance in this population especially if the relapse occurred within a year of completing an ATRA/chemotherapy protocol. In this setting, ATO is extremely effective in inducing molecular remissions in the majority of patients without the toxicity profile of combination chemotherapy and does not have cross-resistance with ATRA [61]. This is the only indication for which it is approved by the US Food and Drug Administration (FDA). Achieving molecular remission prior to a consolidation with an autologous hematopoietic stem cell transplant (HSCT), the preferred option in this setting, has a significant effect on long-term outcome. The use of single agent ATO as consolidation therapy after achieving molecular remission was less effective in this population with a 2-year OS of 41% in one series [61] and an EFS of 33% in another [60]. In our own series, we reported a significantly better clinical outcome in patients who were consolidated with an autologous HSCT versus those consolidated with ATO or ATO + ATRA following treatment of a frank hematological relapse of APL [76]. Based on the available data, it would be reasonable in patients with a hematological relapse to induce molecular remission with ATO and consolidate with an autologous HSCT in those who achieve molecular remission and consider an allogeneic HSCT for those who fail to achieve a molecular remission. We recently demonstrated the considerable synergy between ATO and a proteasome inhibitor. With in vitro studies, in vivo animal models, and preliminary clinical data, we have shown that the combination of bortezomib with ATO and ATRA is comparable to the effect of anthracycline with ATRA and ATO on malignant promyelocytes [77, 78]. In the evolving strategy of de-escalation of therapy in APL [79], the addition of bortezomib with ATO along with ATRA has the potential to further de-escalate the therapy in high-risk and relapsed APL by replacing the myelotoxic anthracycline with a relatively non-myelotoxic proteasome inhibitor.
Conclusions
There is no doubt as to the efficacy of ATO in the management of APL, and its position in the treatment algorithm of this condition has been recently re-defined as it can be considered standard of care for newly diagnosed cases. Based on the available data, it is clear that as a single agent, it is the most effective drug in the management of APL. For patients who have relapsed following conventional ATRA plus chemotherapy regimens, ATO is the established agent of choice to induce a second molecular remission. Preliminary concerns of fatal toxicity profile appear to be related more to the associated comorbid conditions than the drug itself, as noted by their absence when used in patients with newly diagnosed APL without comorbid conditions and in other malignant conditions. The ongoing concerns about potential long-term toxicity are not based on significant data. Better understanding of its in vivo biotransformation and the effect of the different methylated derivatives that are generated in this process might help further reduce its toxicity profile while enhancing its efficacy. This could be achieved by better methods to predict toxicity or efficacy, based on genetic polymorphisms that have an impact on biotransformation pathways, or by the use of specific methylated derivatives for therapy rather than the native compound. More research may potentially demonstrate that these derivatives have a more favorable therapeutic profile. In the developing world where the cost of generic ATO is low, the absence of grade III/IV neutropenia and mucositis along with the ability to administer the regimen in the outpatient setting post remission induction significantly reduces the cost of treating this condition in comparison to a standard ATRA plus chemotherapy regimen.
References
Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood. 2008;111(5):2505–15.
Redaelli A, Botteman MF, Stephens JM, Brandt S, Pashos CL. Economic burden of acute myeloid leukemia: a literature review. Cancer Treat Rev. 2004;30(3):237–47.
Uyl-de Groot CA, Gelderblom-den Hartog J, Huijgens PC, Willemze R, van Ineveld BM. Costs of diagnosis, treatment, and follow up of patients with acute myeloid leukemia in the Netherlands. J Hematother Stem Cell Res. 2001;10(1):187–92.
Leunis A, Blommestein HM, Huijgens PC, Blijlevens NM, Jongen-Lavrencic M, Uyl-de Groot CA. The costs of initial treatment for patients with acute myeloid leukemia in the Netherlands. Leuk Res. 2012;37(3):245–50.
Stalfelt AM, Brodin H. Costs over time in conventional treatment of acute myeloid leukaemia. A study exploring changes in treatment strategies over two decades. J Intern Med. 1994;236(4):401–9.
Kruse M, Wildner R, Barnes G, Martin M, Mueller U, Lo-Coco F, et al. Budgetary impact of treating acute promyelocytic leukemia patients with first-line arsenic trioxide and retinoic acid from an Italian payer perspective. PLoS One. 2015;10(8):e0134587.
Tallman M, Lo-Coco F, Barnes G, Kruse M, Wildner R, Martin M, et al. Cost-effectiveness analysis of treating acute promyelocytic leukemia patients with arsenic trioxide and retinoic acid in the United States. Clin Lymphoma Myeloma Leuk. 2015;15(12):771–7.
Wikipedia. List of countries by GNI (nominal, Atlas method) per capita: http://en.wikipedia.org/wiki/List_of_countries_by_GNI_(nominal,_Atlas_method)_per_capita; 2014 [updated 10/06/201410/15/2014]. http://en.wikipedia.org/wiki/List_of_countries_by_GNI_(nominal,_Atlas_method)_per_capita.
Balarajan Y, Selvaraj S, Subramanian SV. Health care and equity in India. Lancet. 2011;377(9764):505–15.
Garg CC, Karan AK. Reducing out-of-pocket expenditures to reduce poverty: a disaggregated analysis at rural-urban and state level in India. Health Policy Plan. 2009;24(2):116–28.
Craig BM, Rollison DE, List AF, Cogle CR. Underreporting of myeloid malignancies by United States cancer registries. Cancer Epidemiol Biomark Prev. 2012;21(3):474–81.
Philip C, George B, Korula A, Srivastava A, Balasubramanian P, Mathews V. Treatment rates of paediatric acute myeloid leukaemia: a view from three tertiary centres in India—response to Gupta et al. Br J Haematol. 2016;175(2):347–9.
Philip C, George B, Ganapule A, Korula A, Jain P, Alex AA, et al. Acute myeloid leukaemia: challenges and real world data from India. Br J Haematol. 2015;170(1):110–7.
Juliusson G, Lazarevic V, Horstedt AS, Hagberg O, Hoglund M. Acute myeloid leukemia in the real world: why population-based registries are needed. Blood. 2012;119(17):3890–9.
Kerr D. Generic drugs: their role in better value cancer care. Ann Oncol. 2013;24(Suppl 5):v5. https://doi.org/10.1093/annonc/mdt321.
Experts in Chronic Myeloid Leukemia. The price of drugs for chronic myeloid leukemia (CML) is a reflection of the unsustainable prices of cancer drugs: from the perspective of a large group of CML experts. Blood. 2013;121(22):4439–42.
Lopes GL. Cost comparison and economic implications of commonly used originator and generic chemotherapy drugs in India. Ann Oncol. 2013;24(Suppl 5):v13–v6.
Cyranoski D. Arsenic patent keeps drug for rare cancer out of reach of many. Nat Med. 2007;13(9):1005.
Warrell RP Jr. Reply to ‘Arsenic patent keeps drug for rare cancer out of reach for many’. Nat Med. 2007;13(11):1278.
Jolliffe DM. A history of the use of arsenicals in man. J R Soc Med. 1993;86(5):287–9.
Zhang TD. Treatment of acute granulocytic leukemia with “Ai ling No. 1”—clinical analysis and experimental research. Zhong Xi Yi Jie He Za Zhi. 1984;4(1):19–20.
Zhang PWS, Hu LH, Shi FD, Giu FQ, Hong GJ, et al. Treatment of acute promyelocytic leukemia with arsenic trioxide injection (713): clinical observations and study of action mode. Chin J Hematol. 1996;17:58–60.
Sun HDML, Hu XC, Zhang TD. Thirty two cases of treating acute promyelocytic leukemia by Ailing I therapy combined with syndrome differentiation treatment of traditional Chinese medicine. Chin J Comb Trad Chin Med West Med. 1992;1996(12):170–1.
Chen GQ, Shi XG, Tang W, Xiong SM, Zhu J, Cai X, et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): I. As2O3 exerts dose-dependent dual effects on APL cells. Blood. 1997;89(9):3345–53.
Tang W, Chen G, Shi G. Double effects of arsenic trioxide (As2O3) on acute promyelocytic leukemic cell line. Zhonghua Yi Xue Za Zhi. 1997;77(7):509–12.
Chen Z, Wang ZY, Chen SJ. Acute promyelocytic leukemia: cellular and molecular basis of differentiation and apoptosis. Pharmacol Ther. 1997;76(1–3):141–9.
Zheng X, Seshire A, Ruster B, Bug G, Beissert T, Puccetti E, et al. Arsenic but not all-trans retinoic acid overcomes the aberrant stem cell capacity of PML/RARalpha-positive leukemic stem cells. Haematologica. 2007;92(3):323–31.
Ablain J, de The H. Revisiting the differentiation paradigm in acute promyelocytic leukemia. Blood. 2011;117(22):5795–802.
Mathews V, Chendamarai E, George B, Viswabandya A, Srivastava A. Treatment of acute promyelocytic leukemia with single-agent arsenic trioxide. Mediterr J Hematol Infect Dis. 2012;3(1):e2011056.
Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM, Qiu QY, et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood. 1997;89(9):3354–60.
Soignet SL, Maslak P, Wang ZG, Jhanwar S, Calleja E, Dardashti LJ, et al. Complete remission after treatment of acute promyelocytic leukemia with arsenic trioxide. N Engl J Med. 1998;339(19):1341–8.
Wang Z, Zhou J, Lu X, Gong Z, Le XC. Arsenic speciation in urine from acute promyelocytic leukemia patients undergoing arsenic trioxide treatment. Chem Res Toxicol. 2004;17(1):95–103.
Sweeney CJ, Takimoto C, Wood L, Porter JM, Tracewell WG, Darwish M, et al. A pharmacokinetic and safety study of intravenous arsenic trioxide in adult cancer patients with renal impairment. Cancer Chemother Pharmacol. 2010;66(2):345–56.
Mathews V, George B, Chendamarai E, Lakshmi KM, Desire S, Balasubramanian P, et al. Single-agent arsenic trioxide in the treatment of newly diagnosed acute promyelocytic leukemia: long-term follow-up data. J Clin Oncol. 2010;28(24):3866–71.
Mathews V, George B, Lakshmi KM, Viswabandya A, Bajel A, Balasubramanian P, et al. Single-agent arsenic trioxide in the treatment of newly diagnosed acute promyelocytic leukemia: durable remissions with minimal toxicity. Blood. 2006;107(7):2627–32.
Mathews V, Desire S, George B, Lakshmi KM, Rao JG, Viswabandya A, et al. Hepatotoxicity profile of single agent arsenic trioxide in the treatment of newly diagnosed acute promyelocytic leukemia, its impact on clinical outcome and the effect of genetic polymorphisms on the incidence of hepatotoxicity. Leukemia. 2006;20(5):881–3.
Ghavamzadeh A, Alimoghaddam K, Rostami S, Ghaffari SH, Jahani M, Iravani M, et al. Phase II study of single-agent arsenic trioxide for the front-line therapy of acute promyelocytic leukemia. J Clin Oncol. 2011;29(20):2753–7.
Zhou J, Zhang Y, Li J, Li X, Hou J, Zhao Y, et al. Single-agent arsenic trioxide in the treatment of children with newly diagnosed acute promyelocytic leukemia. Blood. 2010;115(9):1697–702.
Vahter M. Genetic polymorphism in the biotransformation of inorganic arsenic and its role in toxicity. Toxicol Lett. 2000;112–113:209–17.
Goering PLAH, Mass MJ, et al. The enigma of arsenic carcinogenesis: role of metabolism. Toxicol Sci. 1999;49:5–14.
Brouwer OF, Onkenhout W, Edelbroek PM, et al. Increased neurotoxicity of arsenic in MTHFR deficiency. Clin Neurol Neurosurg. 1992;94:307–10.
Pickett CB. Glutathione S-transferases: gene structure, regulation and biological function. Annu Rev Biochem. 1989;58:743–64.
Chiou HY, Hsueh YM, Hseih LL, et al. Arsenic methylation capacity, body retention and null genotypes of glutathione S-transferases M1 and T1 among current arsenic exposed residents of Taiwan. Mutat Res. 1997;386(3):197–207.
Naranmandura H, Carew MW, Xu S, Lee J, Leslie EM, Weinfeld M, et al. Comparative toxicity of arsenic metabolites in human bladder cancer EJ-1 cells. Chem Res Toxicol. 2011;24(9):1586–96.
Zhang PWS, Hu LH, Shi FD, Giu FQ, Hong GJ, et al. Arsenic trioxide treated 72 cases of acute promyelocytic leukemia. Chin J Hematol. 1996;2:58.
Sun HDML, Hu XC, Zhang TD. Ai-Lin I treated 32 cases of acute promyelocytic leukemia. Chin J Integrat Chin West Med. 1992;12:170.
Treleaven J, Meller S, Farmer P, Birchall D, Goldman J, Piller G. Arsenic and ayurveda. Leuk Lymphoma. 1993;10(4–5):343–5.
Mathews V, George B, Jijina F, Ross C, Nair R, Apte S, et al. Final analysis of a multi-center randomized controlled trial (IAPLSG04) to study the optimal duration of arsenic trioxide maintenance therapy in the treatment of newly diagnosed acute promyelocytic leukemia. ASH Annual Meeting Abstracts. 2011;118(21):426.
Iland HJ, Bradstock K, Supple SG, Catalano A, Collins M, Hertzberg M, et al. All-trans-retinoic acid, idarubicin, and IV arsenic trioxide as initial therapy in acute promyelocytic leukemia (APML4). Blood. 2012;120(8):1570–80; quiz 752.
Lo-Coco F, Avvisati G, Vignetti M, Thiede C, Orlando SM, Iacobelli S, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. 2013;369(2):111–21.
Burnett AK, Russell NH, Hills RK, Bowen D, Kell J, Knapper S, et al. Arsenic trioxide and all-trans retinoic acid treatment for acute promyelocytic leukaemia in all risk groups (AML17): results of a randomised, controlled, phase 3 trial. Lancet Oncol. 2015;16(13):1295–305.
Ghavamzadeh A, Alimoghaddam K, Rostami S, Ghaffari SH, Jahani M, Iravani M, et al. Phase II study of single-agent arsenic trioxide for the front-line therapy of acute promyelocytic leukemia. J Clin Oncol. 2011;29(20):2753–7.
Estey E, Koller C, Cortes J, Reed P, Freireich E, Giles F, et al. Treatment of newly-diagnosed acute promyelocytic leukemia with liposomal all-trans retinoic acid. Leuk Lymphoma. 2001;42(3):309–16.
Ravandi F, Estey E, Jones D, Faderl S, O’Brien S, Fiorentino J, et al. Effective treatment of acute promyelocytic leukemia with all-trans-retinoic acid, arsenic trioxide, and gemtuzumab ozogamicin. J Clin Oncol. 2009;27(4):504–10.
Gore SD, Gojo I, Sekeres MA, Morris L, Devetten M, Jamieson K, et al. Single cycle of arsenic trioxide-based consolidation chemotherapy spares anthracycline exposure in the primary management of acute promyelocytic leukemia. J Clin Oncol. 2010;28(6):1047–53.
Hu J, Liu YF, Wu CF, Xu F, Shen ZX, Zhu YM, et al. Long-term efficacy and safety of all-trans retinoic acid/arsenic trioxide-based therapy in newly diagnosed acute promyelocytic leukemia. Proc Natl Acad Sci U S A. 2009;106(9):3342–7.
Ohnishi K, Yoshida H, Shigeno K, Nakamura S, Fujisawa S, Naito K, et al. Prolongation of the QT interval and ventricular tachycardia in patients treated with arsenic trioxide for acute promyelocytic leukemia. Ann Intern Med. 2000;133(11):881–5.
Westervelt P, Brown RA, Adkins DR, Khoury H, Curtin P, Hurd D, et al. Sudden death among patients with acute promyelocytic leukemia treated with arsenic trioxide. Blood. 2001;98(2):266–71.
Unnikrishnan D, Dutcher JP, Varshneya N, Lucariello R, Api M, Garl S, et al. Torsades de pointes in 3 patients with leukemia treated with arsenic trioxide. Blood. 2001;97(5):1514–6.
Niu C, Yan H, Yu T, Sun HP, Liu JX, Li XS, et al. Studies on treatment of acute promyelocytic leukemia with arsenic trioxide: remission induction, follow-up, and molecular monitoring in 11 newly diagnosed and 47 relapsed acute promyelocytic leukemia patients. Blood. 1999;94(10):3315–24.
Douer D, Tallman MS. Arsenic trioxide: new clinical experience with an old medication in hematologic malignancies. J Clin Oncol. 2005;23(10):2396–410.
Hei TK, Liu SX, Waldren C. Mutagenicity of arsenic in mammalian cells: role of reactive oxygen species. Proc Natl Acad Sci U S A. 1998;95(14):8103–7.
Cuzick J, Evans S, Gillman M, Price Evans DA. Medicinal arsenic and internal malignancies. Br J Cancer. 1982;45(6):904–11.
Sanz MA, Grimwade D, Tallman MS, Lowenberg B, Fenaux P, Estey EH, et al. Management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood. 2009;113(9):1875–91.
Mathews V, Chandy M, Srivastava A. Arsenic trioxide in the management of acute promyelocytic leukaemia. Natl Med J India. 2001;14(4):215–22.
Mrozek K, Heinonen K, de la Chapelle A, Bloomfield CD. Clinical significance of cytogenetics in acute myeloid leukemia. Semin Oncol. 1997;24(1):17–31.
Slack JL, Arthur DC, Lawrence D, Mrozek K, Mayer RJ, Davey FR, et al. Secondary cytogenetic changes in acute promyelocytic leukemia—prognostic importance in patients treated with chemotherapy alone and association with the intron 3 breakpoint of the PML gene: a Cancer and Leukemia Group B study. J Clin Oncol. 1997;15(5):1786–95.
De Botton S, Chevret S, Sanz M, Dombret H, Thomas X, Guerci A, et al. Additional chromosomal abnormalities in patients with acute promyelocytic leukaemia (APL) do not confer poor prognosis: results of APL 93 trial. Br J Haematol. 2000;111(3):801–6.
Mathews V, Thomas M, Srivastava VM, George B, Srivastava A, Chandy M. Impact of FLT3 mutations and secondary cytogenetic changes on the outcome of patients with newly diagnosed acute promyelocytic leukemia treated with a single agent arsenic trioxide regimen. Haematologica. 2007;92(7):994–5.
Gilliland DG. FLT3-activating mutations in acute promyelocytic leukaemia: a rationale for risk-adapted therapy with FLT3 inhibitors. Best Pract Res Clin Haematol. 2003;16(3):409–17.
Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002;100(5):1532–42.
Marasca R, Maffei R, Zucchini P, Castelli I, Saviola A, Martinelli S, et al. Gene expression profiling of acute promyelocytic leukaemia identifies two subtypes mainly associated with Flt3 mutational status. Leukemia. 2006;20(1):103–14.
Gale RE, Hills R, Pizzey AR, Kottaridis PD, Swirsky D, Gilkes AF, et al. Relationship between FLT3 mutation status, biologic characteristics, and response to targeted therapy in acute promyelocytic leukemia. Blood. 2005;106(12):3768–76.
Callens C, Chevret S, Cayuela JM, Cassinat B, Raffoux E, de Botton S, et al. Prognostic implication of FLT3 and Ras gene mutations in patients with acute promyelocytic leukemia (APL): a retrospective study from the European APL Group. Leukemia. 2005;19(7):1153–60.
Chillon MC, Santamaria C, Garcia-Sanz R, Balanzategui A, Maria Eugenia S, Alcoceba M, et al. Long FLT3 internal tandem duplications and reduced PML-RARalpha expression at diagnosis characterize a high-risk subgroup of acute promyelocytic leukemia patients. Haematologica. 2010;95(5):745–51.
Thirugnanam R, George B, Chendamarai E, Lakshmi KM, Balasubramanian P, Viswabandya A, et al. Comparison of clinical outcomes of patients with relapsed acute promyelocytic leukemia induced with arsenic trioxide and consolidated with either an autologous stem cell transplant or an arsenic trioxide-based regimen. Biol Blood Marrow Transplant. 2009;15(11):1479–84.
Ganesan S, Alex AA, Chendamarai E, Balasundaram N, Palani HK, David S, et al. Rationale and efficacy of proteasome inhibitor combined with arsenic trioxide in the treatment of acute promyelocytic leukemia. Leukemia. 2016;30(11):2169–78.
Mathews V, Korula A, Kulkarni U, Ganesan S, David S, Alex AA, Nisham PN, Abraham A, Srivastava A, Lakshmi KM, Balasubramanian P, George B. Management of relapsed acute promyelocytic leukemia post ATO upfront therapy: open-labeled phase II study evaluating role of proteasome inhibition. Blood. 2016;128(22):446.
Mathews V. De-escalation of treatment for acute promyelocytic leukaemia? Lancet Haematol. 2015;2(9):e348–9.
Powell BL, Moser B, Stock W, Gallagher RE, Willman CL, Stone RM, et al. Arsenic trioxide improves event-free and overall survival for adults with acute promyelocytic leukemia: North American Leukemia Intergroup Study C9710. Blood. 2010;116(19):3751–7.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter
Cite this chapter
Mathews, V. (2018). APL in Developing Countries: ATO-Based Approach. In: Abla, O., Lo Coco, F., Sanz, M. (eds) Acute Promyelocytic Leukemia . Springer, Cham. https://doi.org/10.1007/978-3-319-64257-4_18
Download citation
DOI: https://doi.org/10.1007/978-3-319-64257-4_18
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-64256-7
Online ISBN: 978-3-319-64257-4
eBook Packages: MedicineMedicine (R0)