
Abstract
The introduction of all-trans-retinoic acid (ATRA) and arsenic trioxide (ATO) as the mainstay therapy of acute promyelocytic leukemia (APL) has drastically changed the outcome of this hematologic malignancy into one of the first to receive a targeted treatment. Using frontline treatment strategies including these agents in combination with standard cytotoxic drugs has provided outstanding therapeutic results in most patients. In spite of the achievement of brilliant results in the majority of patients, some special situations still require the implementation of changes from the conventional therapeutic approach. In this chapter, we will review and discuss the management of APL in older and frail patients, children and pregnant women, as well as those with therapy-related leukemia, extramedullary relapse, and the extremely rare situation of the genetic variants.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
Therapeutic strategies in acute promyelocytic leukemia (APL) have changed greatly since the introduction of all-trans-retinoic acid (ATRA) and, more recently, arsenic trioxide (ATO) in the treatment of this disease, improving enormously the outcome of these patients. Several treatment strategies using these agents, alone or usually in combination with chemotherapy, have provided excellent therapeutic results, in fit patients whose clinical situation does not generate special difficulty for the administration of conventional therapy. However, there are conditions that prevent partially or completely the administration of ATRA, ATO, or chemotherapy, complicating the management of these patients. In this chapter, we review some of these peculiar clinical situations which, differently from the standard of care, require more careful attention. Thus, we will discuss the management of APL in older and other frail patients, children and pregnant women, as well as those with therapy-related leukemia, extramedullary relapse and the extremely rare situation of the genetic variants of APL.
Management of Special Situations
Older and Other Fragile Patients
APL is uncommon in older patients. This group tends to have a worse outcome, even if stratified as lower risk at baseline, due to a higher therapy-related toxicity leading to an increased treatment-related mortality, not only during induction but also during consolidation, maintenance, and even off therapy. The reported mortality rate in complete remission (CR) ranges from less than 1% in patients younger than 60 to 19% in patients older than 70 years [1]. It should be noted, however, that lower rates of relapse are observed in patients over 70 years of age receiving ATRA and moderately reduced anthracycline-based chemotherapy [1,2,3,4]. Therefore, it is reasonable to design less intensive therapeutic strategies aiming to reduce morbidity and mortality in this group. With the aim of decreasing the rate of treatment-related deaths, the Gruppo Italiano Malattie Ematologiche Maligne dell’Adulto (GIMEMA) group amended the AIDA protocol and reduced the number of consolidation courses from three to only one, which resulted in an improved overall survival (OS) [5]. The outcome with ATO-based treatments in elderly patients has not specifically been reported, except for a small series of 33 patients aged 60 years or older [6]. This study showed 88% CR rate and a 10-year cumulative incidence of relapse, OS, and disease-free survival (DFS) of 10.3%, 69.3%, and 64.8%, respectively, which are comparable to those reported in younger APL patients. The main reported side effect was leukocytosis, and no increased rate of secondary malignancies was reported after long-term follow-up. Based on these results, ATO could be considered as a first-line treatment option for elderly patients with APL, but more data is needed to really turn it into the standard of care for patients unfit for conventional therapy in this age group. The APL0406 trial reported similar efficacy in patients aged 60–70 years when compared to younger patients, with an improved EFS and toxicity rate [7]. Gemtuzumab ozogamicin (GO) has also been tested in the elderly, with efficacy reported also at lower doses [8, 9]. Unfortunately, the drug was temporarily withdrawn from the market in 2010 and reapplied for US and EU approval in early 2017. Therefore, we can conclude that an ATRA-ATO regimen may be appropriate in the elderly setting, but further data are still required to validate its use. See Chap. 15, “APL in the Elderly,” for more details.
Similar to the outlined approach for older patients, in younger patients who are not candidates for first-line intensive chemotherapy due to certain comorbidities (in particular, severe cardiac impairment or other organ dysfunctions), there are alternative treatment approaches which minimize the use of cytotoxic agents. These would be based on the use of ATRA in combination with ATO, with minimal or no chemotherapy. The outcome in this particular setting is not sufficiently documented. It should be noted that any therapeutic strategy used in these patients should aim to achieve molecular remission and guide the need for additional therapy with minimal residual disease (MRD) monitoring.
Children
APL is a rare disease in children, with a reported median age of 9–12 years and a female prevalence. At presentation, there is a reported higher rate of hyperleukocytosis (40% versus 20–25% in adults) and of the variant morphologic hypogranular (M3v) subtype, with an increased incidence of the bcr2/bcr3 transcript type [10]. It has been suggested that the difference in the WBC count is mainly observed in children under the age of 12 years old [10]. Apart from two relatively small pediatric series from the German-Austrian-Swiss group [11] and the European APL group [12], as well as two larger series from the GIMEMA [13] and PETHEMA [14] groups, there is a recent analysis from the European APL group [15] that reports the outcome in different age groups of children and adolescents. In this analysis, children under 4 years old presented the highest relapse rate (52% versus 18% in children age 5–12 years old), a new finding owing to the lack of studies with different age groups in children. This observation was not attributed to a higher WBC count or other high-risk features. The treatment strategy with the simultaneous combination of ATRA and chemotherapy derives from adult trials. The GIMEMA and PETHEMA groups reported the larger cohorts of patients, with a CR rate higher than 90% and OS ranging between 71 and 89% [13, 14]. In children with APL, a reduction in the dose of ATRA, from 45 to 25 mg/m2/day, is recommended to reduce the incidence of severe headache and pseudotumor cerebri (PTC) , still maintaining excellent therapeutic results [16]. The study by Castaigne et al. [17] showed no difference in terms of pharmacokinetics, therapeutic efficacy, triggering of hyperleukocytosis, or differentiation syndrome with ATRA at the reduced dose as compared to the standard dose. Headache is a relatively common complication during ATRA therapy in children, but it is always necessary to rule out PTC, CNS leukemia, and bleeding. PTC diagnosis is based on increased intracranial pressure with normal cerebrospinal fluid (CSF) composition and negative cerebral imaging studies, usually accompanied by papilledema at the fundoscopic exam. This latter evidence is not a requirement for the diagnosis of PTC but is helpful in the differential diagnosis [18]. Sometimes, the symptoms of PTC resolve with the initial “diagnostic” lumbar puncture. If this occurs, no further medical treatment is required. If symptoms persist, temporary discontinuation or dose reduction of ATRA and analgesics and administration of steroids and acetazolamide are the mainstays of the medical treatment of this neurological complication. Acetazolamide is administered in an initial dose of 25 mg/kg/day and progressively increased until clinical response is attained (maximum dose 100 mg/kg/day). Electrolytes must be monitored for an early detection of hypokalemia and acidosis, common side effects during acetazolamide treatment. If this diuretic treatment is ineffective, then prednisone can be given at a dose of 2 mg/kg/day for 2 weeks followed by a 2-week taper.
Given the long life span in children cured from this disease, there is a wide concern about the potential long-term cardiac toxicity that high-dose anthracyclines can produce. Therefore, there have been some attempts to simply reduce the exposure to these agents without any additional treatment modifications, which has resulted in worse outcomes in the past [19]. The Japanese group tried to minimize anthracycline exposure with a single administration in second induction and consolidation courses: only two deaths were reported, but no cardiac adverse events or treatment-related deaths were observed in subsequent phases [20]. Similarly to older patients, other therapeutic options are being studied in order to reduce the dose of cytotoxic agents, with ATO being one of the most promising alternatives. Indeed, ATO appears to be effective in pediatric APL [21, 22], similarly to adults, but there is still very limited data. See Chap. 14, “APL in Children,” for more details.
Pregnancy
The diagnosis of APL during pregnancy is uncommon, and most reports are based on individual cases or very small series. The risk of thrombo-hemorrhagic complications and infections may be higher during pregnancy, whereas identification of differentiation syndrome (DS) could be more difficult. This is a challenging situation in which decision-making must be carried out with a multidisciplinary perspective, involving the patient, hematologist, obstetrician, and neonatologist. With this approach, there is a higher chance for a successful outcome for both mother and baby, as was highlighted in the guidelines on the management of AML in the United Kingdom [23]. A recent systematic analysis showed 43 articles with 71 patients with new-onset APL during pregnancy [24]. The results suggested that in case of pregnancy, the start of treatment should not be delayed, or it could compromise the chance for a successful remission: pregnancy in APL must be considered a medical emergency. The key problem in this special situation is the teratogenic potential of chemotherapy, ATRA, and ATO on the fetus. Therefore, the gestational age is key in this situation, and management should be adapted accordingly.
Management of APL During the First Trimester of Pregnancy
A conventional therapeutic approach is not possible during the first trimester of pregnancy, due to the highly teratogenic side effects of ATRA [25]. Five out of the nine reported APL cases diagnosed in early pregnancy ended in abortion (four induced and one spontaneous). The remaining four pregnant women delivered two healthy infants, one with transient dilated cardiomyopathy and one with low birthweight, jaundice, and respiratory distress syndrome at birth [24, 26]. In a Spanish series, all early pregnancies terminated in abortion (four induced and one spontaneous) [26]. During the first trimester, the only option is anthracycline-based chemotherapy. The use of daunorubicin is usually preferred over idarubicin, probably due to a wider experience with the former drug and because idarubicin is more lipophilic and can favor an increased placental transfer [27]. Even with chemotherapy, there is an increased risk of fetal malformations, abortion, and low birthweight [28]. Therefore, the first decision that should take place when APL is diagnosed in the first trimester is whether or not to continue with the pregnancy. Women who, after receiving all the accurate information, wish to continue with their pregnancy should receive anthracycline chemotherapy alone; in case of pregnancy interruption, they can receive conventional treatment with ATRA and cytotoxic agents. It should be taken into account that chemotherapy alone can increase the risk of hemorrhage due to release of procoagulants and plasminogen activators from malignant promyelocytes. If remission is achieved with chemotherapy and the pregnancy is progressing normally, ATRA could be safely administered during the second and third trimesters. Although ATO is an alternative treatment in other groups of patients, it is not an option during pregnancy due to a high potential for teratogenicity; in fact, this drug cannot be recommended at any stage of pregnancy. Human data are very limited and restricted to people exposed to arsenic from drinking water, working in, or living near metal smelters. Low birthweight, spontaneous abortion, and stillbirth were reported in this population [29]. Taking the above into account, female APL patients who are receiving conventional treatment should be advised against becoming pregnant during exposure to ATRA and/or ATO. On the other hand, it appears that fertility is maintained after ATO treatment is finalized [30].
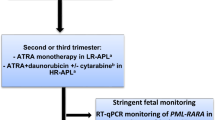
Management of APL During the Second and Third Trimesters of Pregnancy
During the second and third trimesters of pregnancy, conventional treatment with ATRA and chemotherapy is a reasonable option, even though the literature on this subject is extremely scarce. The maternal outcome seems to be the same as in nonpregnant women when conventional therapy is used. ATRA does not seem to be teratogenic past the first trimester [24, 26, 28]. This agent can be safely administered, preferably monitoring the cardiac function, due to some reports of reversible fetal arrhythmias and other cardiac complications at birth. On the other hand, although chemotherapy does not seem to cause congenital malformation, it increases in some cases the risk of abortion, premature delivery, low birthweight, neonatal neutropenia, and sepsis. Two different possible approaches could be adopted:
-
(a)
Sequential use of ATRA and chemotherapy. This approach implies the administration of ATRA alone until CR, delaying the administration of chemotherapy until elective delivery is possible. A gestational age of at least 32 weeks is considered relatively safe when appropriate neonatal care is provided [31]. For deliveries before 36 weeks of gestation, antenatal corticosteroids are recommended to reduce the risk of respiratory distress syndrome [32]. ATRA alone induces similar CR rates as compared to ATRA plus chemotherapy, but it can have an unfavorable impact on the risk of relapse and a possible increased rate of DS (approximately 25%) [33, 34]. If this strategy is followed, the administration of chemotherapy should not be delayed excessively to avoid resistance and disease recurrence, and post-remission therapy should be reinforced.
-
(b)
Simultaneous administration of ATRA and chemotherapy. This approach provides the best chances of cure for the pregnant women and is a reasonable option for high-risk patients with hyperleukocytosis and for those in which appropriate RQ-PCR monitoring is not possible. Daunorubicin is preferred to idarubicin, as mentioned previously. Vaginal delivery is preferred, due to its association with a reduced risk of bleeding. Caesarean section should only take place if it is required for other reasons [28]. After delivery, breastfeeding is contraindicated if chemotherapy or ATO is needed. The rest of management does not differ from nonpregnant women with APL.
Therapy-Related APL
The true incidence of therapy-related APL (tAPL) is still a matter of discussion since these patients are less likely to be enrolled into clinical trials. Available data is based on retrospective series [35, 36] or experience of single referral centers [37, 38]. The incidence reported in these studies ranges from less than 5–22% of all APL cases. The incidence of tAPL has increased over the last few years due to the use of topoisomerase II-targeted drugs in both malignant and nonmalignant diseases. Breast carcinoma is the most frequent previous cancer, followed by lymphoma, with a large predominance of non-Hodgkin lymphoma, whereas other tumor types have a lower incidence [35]. Epirubicin and mitoxantrone are the most common implicated drugs in tAPL, but a number of cases have been reported after exposure to radiotherapy alone [39,40,41,42]. Cases of secondary APL occurring in patients whose primary tumor was treated only by surgery, without chemotherapy or radiotherapy, have also been reported [35, 36]. The latency period between chemotherapy exposure and onset of tAPL is usually relatively short (less than 3 years) and occurs without a previous myelodysplastic phase. Hematologic findings are similar to de novo APL, as also previously reported for other tAML with a specific karyotype [43, 44]. Cases of tAPL are increasingly reported in patients treated for nonmalignant diseases, in particular in patients affected by aggressive forms of multiple sclerosis treated with mitoxantrone [45]. The risk of developing this complication has been estimated at approximately 1 in 400 patients with multiple sclerosis treated with this topoisomerase II inhibitor [45]. In these patients, specific genomic loci were identified, such as PML intron 6 and RUNX1 intron 5, containing preferential sites of topoisomerase II-mediated DNA cleavage [46, 47].
In the past, a higher incidence of early death during treatment was reported [35, 48]. However, a more precise knowledge on the outcome of patients with tAPL treated with state-of-the-art therapy should be prospectively established. At present, there is no specific reason to manage these patients differently from those with de novo APL. However, in a significant number of patients with tAPL, the use of anthracycline-based regimens is limited by previous exposure to topoisomerase II inhibitors. In such situations, ATO in combination with ATRA provides an option for consolidation following standard induction therapy or as first-line treatment using schedules such as those published by the MD Anderson and GIMEMA groups [49]. See Chap. 19, “Therapy-Related APL,” for more details.
Genetic Variants of APL
Less than 1% of APL cases are due to rare variant translocations, which typically involve RARA [50]. No APL variants with PML involvement alone have been identified to date; thus, RARA is assumed to have a key role in the pathogenesis of APL. Several variant translocations have been identified, including ZBTB16/RARA (previously named PLZF-RARA), NMP/RARA, NUMA/RARA, STAT5B/RARA, PRKAR1a/RARA, BCOR/RARA, and FIP1L1/RARA [51]. APL with complex, cryptic, or variant translocations usually present with the same clinical features of typical APL. There are no specific guidelines for rare genetic variants of APL, because the available evidence is mostly based on single case reports. Nevertheless, it is a general rule that patients with ATRA-sensitive variants (NuMA-RARA, NPM1-RARA, BCOR/RARA, PRKAR1a/RARA, and FIP1L1-RARA) should be treated with standard protocols involving ATRA combined with anthracycline-based chemotherapy, while those with ATRA-resistant variants (ZBTB16/RARA, STAT5b-RARA) should be managed with AML-like approaches [51]. For those relatively ATRA resistant (PLZF-RARA), it is reasonable to combine ATRA with AML-like chemotherapy. Sensitivity to ATO has not been documented outside PML-RARA-positive APL, except for PLZF-RARA-positive APL that has been shown to be resistant [52]. See Chap. 20, “Rare APL Variants,” for more details.
Central Nervous System and Other Extramedullary Relapses
Central nervous system (CNS) and other extramedullary relapses are uncommon in APL. In particular, CNS involvement can occur as an isolated event or associated with bone marrow involvement at first relapse or, more frequently, after two or more hematological relapses. The reported incidence of CNS relapses in APL ranges from 0.6 to 2% [53, 54]. Hyperleukocytosis at presentation is a predisposing factor, and the optimal management of such patients is still controversial [55]. The literature on this subject is scarce, but it seems pragmatic to manage these cases just like an extramedullary relapse of acute lymphoblastic leukemia or non-APL AML. In this regard, treatment of CNS relapse should consist of weekly triple intrathecal therapy (ITT) with methotrexate, hydrocortisone, and cytarabine until complete clearance of blasts in the CSF, followed by six to ten more spaced-out ITT treatments as consolidation [56]. Since CNS disease is almost invariably accompanied by hematologic or molecular relapse in the marrow, systemic treatment should also be given. Chemotherapy regimens with high CNS penetrance (e.g., high-dose cytarabine) have been used in this situation, and, in patients responding to treatment, allogeneic or autologous transplant should be the consolidation approach of choice with appropriate craniospinal irradiation. In case of any extramedullary localization (peculiar localizations include the ear, scalp, and skin [53, 54, 57]), local radiation and intensive systemic therapy should be considered. See Chap. 12, “Treatment of Refractory and Relapsed APL,” for more details.
References
Sanz MA, Vellenga E, Rayon C, Diaz-Mediavilla J, Rivas C, Amutio E, et al. All-trans retinoic acid and anthracycline monochemotherapy for the treatment of elderly patients with acute promyelocytic leukemia. Blood. 2004;104(12):3490–3.
Mandelli F, Latagliata R, Avvisati G, Fazi P, Rodeghiero F, Leoni F, et al. Treatment of elderly patients (> or =60 years) with newly diagnosed acute promyelocytic leukemia. Results of the Italian multicenter group GIMEMA with ATRA and idarubicin (AIDA) protocols. Leukemia. 2003;17(6):1085–90.
Lengfelder E, Hofmann WK, Nolte F. Management of elderly patients with acute promyelocytic leukemia: progress and problems. Ann Hematol. 2013;92(9):1181–8.
Ades L, Chevret S, De Botton S, Thomas X, Dombret H, Beve B, et al. Outcome of acute promyelocytic leukemia treated with all trans retinoic acid and chemotherapy in elderly patients: the European group experience. Leukemia. 2005;19(2):230–3.
Latagliata R, Breccia M, Fazi P, Vignetti M, Di Raimondo F, Sborgia M, et al. GIMEMA AIDA 0493 amended protocol for elderly patients with acute promyelocytic leukemia. Long-term results and prognostic factors. Br J Haematol. 2011;154(5):564–8.
Zhang Y, Zhang Z, Li J, Li L, Han X, Han L, et al. Long-term efficacy and safety of arsenic trioxide for first-line treatment of elderly patients with newly diagnosed acute promyelocytic leukemia. Cancer. 2013;119(1):115–25.
Lo Coco F, Avvisati G, Vignetti M, Thiede C, Orlando SM, Iacobelli S, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. 2013;369(2):111–21.
Lo Coco F, Cimino G, Breccia M, Noguera NI, Diverio D, Finolezzi E, et al. Gemtuzumab ozogamicin (Mylotarg) as a single agent for molecularly relapsed acute promyelocytic leukemia. Blood. 2004;104(7):1995–9.
Breccia M, Cimino G, Diverio D, Gentilini F, Mandelli F, Lo Coco F. Sustained molecular remission after low dose gemtuzumab-ozogamicin in elderly patients with advanced acute promyelocytic leukemia. Haematologica. 2007;92(9):1273–4.
Testi AM, D’Angiò M, Locatelli F, Pession A, Lo Coco F. Acute promyelocytic leukemia (APL): comparison between children and adults. Mediterr J Hematol Infect Dis. 2014;6(1):e2014032.
Mann G, Reinhardt D, Ritter J, et al. Treatment with all-trans retinoic acid in acute promyelocytic leukemia reduces early deaths in children. Ann Hematol. 2001;80(7):417–22.
de Botton S, Coiteux V, Chevret S, et al. Outcome of childhood acute promyelocytic leukemia with all-trans-retinoic acid and chemotherapy. J Clin Oncol. 2004;22(8):1404–12.
Testi AM, Biondi A, Lo Coco F, et al. GIMEMA-AIEOPAIDA protocol for the treatment of newly diagnosed acute promyelocytic leukemia (APL) in children. Blood. 2005;106(2):447–53.
Ortega JJ, Madero L, Martin G, et al. Treatment with all-trans retinoic acid and anthracycline monochemotherapy for children with acute promyelocytic leukemia: a multicenter study by the PETHEMA Group. J Clin Oncol. 2005;23(30):7632–40.
Bally C, Fadlallah J, Leverger G, et al. Outcome of acute promyelocytic leukemia (APL) in children and adolescents: an analysis in two consecutive trials of the European APL Group. J Clin Oncol. 2012;30(14):1641–6.
Abla O, Ribeiro RC. How I treat children and adolescents with acute promyelocytic leukemia. Br J Haematol. 2014;164(1):24–38.
Castaigne S, Lefebvre P, Chomienne C, et al. Effectiveness and pharmacokinetics of low-dose all-trans retinoic acid (25 mg/m2) in acute promyelocytic leukemia. Blood. 1993;82(12):3560–3.
Wang SJ, Silberstein SD, Patterson S, Young WB. Idiopathic intracranial hypertension without papilledema: a case-control study in a headache center. Neurology. 1998;51(1):245–9.
Powell BL, Moser B, Stock W, et al. Arsenic trioxide improves event-free and overall survival for adults with acute promyelocytic leukemia: North American Leukemia Intergroup Study C971. Blood 2010; 116(19): 3751–3757.
Takahashi H, Watanabe T, Kinoshita A, et al. High event-free survival rate with minimum-dose-anthracycline treatment in childhood acute promyelocytic leukaemia: a nationwide prospective study by the Japanese Paediatric Leukaemia/Lymphoma Study Group. Br J Haematol. 2016;174(3):437–43.
Fox E, Razzouk BI, Widemann BC, et al. Phase 1 trial and pharmacokinetic study of arsenic trioxide in children and adolescents with refractory or relapsed acute leukemia, including acute promyelocytic leukemia or lymphoma. Blood. 2008;111(2):566–73.
George B, Mathews V, Poonkuzhali B, Shaji RV, Srivastava A, Chandy M. Treatment of children with newly diagnosed acute promyelocytic leukemia with arsenic trioxide: a single center experience. Leukemia. 2004;18(10):1587–90.
Milligan DW, Grimwade D, Cullis JO, et al. Guidelines on the management of acute myeloid leukaemia in adults. Br J Haematol. 2006;135(4):450–74.
Verma V, Giri S, Manandhar S, Pathak R, Bhatt VR. Acute promyelocytic leukemia during pregnancy: a systematic analysis of outcome. Leuk Lymphoma. 2016;57(3):616–22.
Lammer EJ, Chen DT, Hoar RM, et al. Retinoic acid embryopathy. N Engl J Med. 1985;313(14):837–41.
Sanz MA, Montesinos P, Casale MF, et al. Maternal and fetal outcomes in pregnant women with acute promyelocytic leukemia. Ann Hematol. 2015;94(8):1357–61.
Cardonick E, Iacobucci A. Use of chemotherapy during human pregnancy. Lancet Oncol. 2004;5(5):283–91.
Culligan DJ, Merriman L, Kell J, et al. The management of acute promyelocytic leukemia presenting during pregnancy. Clin Leuk. 2007;1:183–91.
U.S. Environmental Protection Agency. Arsenic compounds. http://www.epa.gov. Accessed 8 Jan 2008.
Breccia M, Molica M, Efficace F, et al. Pregnancy in acute promyelocytic leukemia after frontline therapy with arsenic trioxide and all-trans retinoic acid. Br J Haematol. 2014;167(3):428–30.
Slattery MM, Morrison JJ. Preterm delivery. Lancet. 2002;360(9344):1489–97.
Royal College of Obstetricians and Gynecologists. Antenatal corticosteroids to reduce respiratory distress syndrome. Guideline No. 7. http://www.rcog.org.uk/resources/Public/pdf/Antenatal_corticosteroids_No7.pdf. Accessed 1 Apr 2007.
Fenaux P, Chastang C, Chevret S, et al. A randomized comparison of all transretinoic acid (ATRA) followed by chemotherapy and ATRA plus chemotherapy and the role of maintenance therapy in newly diagnosed acute promyelocytic leukemia. The European APL Group. Blood. 1999;94(4):1192–200.
Tallman MS, Andersen JW, Schiffer CA, et al. All-trans-retinoic acid in acute promyelocytic leukemia. N Engl J Med. 1997;337(15):1021–8.
Pulsoni A, Pagano L, Lo Coco F, et al. Clinicobiological features and outcome of acute promyelocytic leukemia occurring as a second tumor: the GIMEMA experience. Blood. 2002;100(6):1972–6.
Beaumont M, Sanz M, Carli PM, et al. Therapy-related acute promyelocytic leukemia. J Clin Oncol. 2003;21(11):2123–37.
Mattson JC. Acute promyelocytic leukemia. From morphology to molecular lesions. Clin Lab Med. 2000;20(1):83–103, ix.
Pollicardo N, O’Brien S, Estey EH, et al. Secondary acute promyelocytic leukemia. Characteristics and prognosis of 14 patients from a single institution. Leukemia. 1996;10(1):27–31.
Mistry AR, Felix CA, Whitmarsh RJ, et al. DNA topoisomerase II in therapy-related acute promyelocytic leukemia. N Engl J Med. 2005;352(15):1529–38.
Andersen MK, Pedersen-Bjergaard J. Therapy-related MDS and AML in acute promyelocytic leukemia. Blood. 2002;100(5):1928–9; author reply 1929.
Pedersen-Bjergaard J. Insights into leukemogenesis from therapy-related leukemia. N Engl J Med. 2005;352(15):1591–4.
Smith SM, Le Beau MM, Huo D, et al. Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: the University of Chicago series. Blood. 2003;102(1):43–52.
Pedersen-Bjergaard J, Philip P, Larsen SO, Jensen G, Byrsting K. Chromosome aberrations and prognostic factors in therapy-related myelodysplasia and acute nonlymphocytic leukemia. Blood. 1990;76(6):1083–91.
Quesnel B, Kantarjian H, Bjergaard JP, et al. Therapy-related acute myeloid leukemia with t(8;21), inv(16), and t(8;16): a report on 25 cases and review of the literature. J Clin Oncol. 1993;11(12):2370–9.
Ledda A, Caocci G, Spinicci G, Cocco E, Mamusa E, La Nasa G. Two new cases of acute promyelocytic leukemia following mitoxantrone treatment in patients with multiple sclerosis. Leukemia. 2006;20(12):2217–8.
Hasan SK, Mays AN, Ottone T, et al. Molecular analysis of t(15;17) genomic breakpoints in secondary acute promyelocytic leukemia arising after treatment of multiple sclerosis. Blood. 2008;112:3383–90.
Ottone T, Hasan SK, Montefusco E, et al. Identification of a potential “hotspot” DNA region in the RUNX1 gene targeted by mitoxantrone in therapy-related acute myeloid leukemia with t(16;21) translocation. Genes Chromosomes Cancer. 2009;48:213–21.
Ghalie RG, Mauch E, Edan G, et al. A study of therapy-related acute leukaemia after mitoxantrone therapy for multiple sclerosis. Mult Scler. 2002;8(5):441–5.
Lo-Coco F, Avvisati G, Vignetti M, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. 2013;369(2):111–21.
Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood. 2008;111(5):2505–15.
Adams J, Nassiri M. Acute promyelocytic leukemia: a review and discussion of variant translocations. Arch Pathol Lab Med. 2015;139(10):1308–13.
Koken MH, Daniel MT, Gianni M, et al. Retinoic acid, but not arsenic trioxide, degrades the PLZF/RARalpha fusion protein, without inducing terminal differentiation or apoptosis, in a RA-therapy resistant t(11;17)(q23;q21) APL patient. Oncogene. 1999;18(4):1113–8.
Specchia G, Lo Coco F, Vignetti M, et al. Extramedullary involvement at relapse in acute promyelocytic leukemia patients treated or not with all-trans retinoic acid: a report by the Gruppo Italiano Malattie Ematologiche dell’adulto. J Clin Oncol. 2001;19:4023–8.
De Botton S, Sanz M, Chevret S, et al. Extramedullary relapse in acute promyelocytic leukemia treated with all-trans retinoic acid and chemotherapy. European APL group; PETHEMA Group. Leukemia. 2006;20:35–41.
Breccia M, Carmosino I, Diverio D, et al. Early detection of meningeal localization in acute promyelocytic leukemia patients with high presenting leukocyte count. Br J Haematol. 2003;120:266–70.
Sanz MA, Grimwade D, Tallmann MS, et al. Guidelines on the management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood. 2009;113:1875–91.
Breccia M, Petti MC, Testi AM, et al. Ear involvement in acute promyelocytic leukemia at relapse: a disease-associated sanctuary? Leukemia. 2002;16(6):1127–30.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter
Cite this chapter
Breccia, M., Iacoboni, G., Sanz, M.A. (2018). Special Situations in APL. In: Abla, O., Lo Coco, F., Sanz, M. (eds) Acute Promyelocytic Leukemia . Springer, Cham. https://doi.org/10.1007/978-3-319-64257-4_16
Download citation
DOI: https://doi.org/10.1007/978-3-319-64257-4_16
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-64256-7
Online ISBN: 978-3-319-64257-4
eBook Packages: MedicineMedicine (R0)